一种靶向表达EGFR肿瘤细胞的前抗体偶联药物及其应用的制作方法
本发明属于生物技术领域,更具体地,本发明公开了一种靶向表达EGFR肿瘤细胞的前抗体偶联药物及其应用。
背景技术:
表皮生长因子受体(EGFR)是一种跨膜受体酪氨酸激酶,在肿瘤进展中有着重要作用。过度的EGFR激活和肿瘤的发生及转移相关。据报道,60%-80%的结肠癌有EGFR的过度表达。另外,EGFR也是预测结直肠癌和乳腺癌的重要标志物。更重要的是EGFR位于癌细胞表面,因此是开发靶向抗体的理想靶点。抗体可以特定地结合到EGFR的胞外区III结构域,不仅阻止了配体的结合, 而且阻止了在结构域II上延伸结构域的二聚体化(Li S, et al. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab[J]. Cancer Cell, 2005, 7(4):301-311.;Iida M, et al. Sym004, a novel EGFR antibody mixture, can overcome acquired resistance to cetuximab[J]. Neoplasia, 2013, 15(10):1196-1206.)经FDA批准的靶向EGFR的抗体包括西妥昔单抗和帕妥木单抗。它们被用于在临床上治疗KRAS野生型的进展性结肠癌。
表皮生长因子受体(即EGFR)也称作c-erbB l/Herl,其家族成员都是生长因子受体酪氨酸激酶,它们在细胞表面与特异的生长因子或天然配体相互作用,如与EGF或TGFa相互作用,由此活化受体酪氨酸激酶。已发现的该家族第一个成员是表观分子量为 165KD 的糖蛋白。
EGFR 在调节肿瘤细胞的生长、修复和生存、新生血管生成、侵袭和转移中具有重要的作用,同时在相当一部分的人类肿瘤中都有表达。在很多恶性肿瘤中,EGFR的表达往往与较差的预后和较低的生存率相关。由此可知,如果有药物能阻断EGFR 的活性,将会抑制其磷酸化和信号传导,从而起到多重途径的抗肿瘤作用,同时也能增加化疗和放疗的抗肿瘤疗效。在一些研究中,EGFR抑制剂与多种化疗药物和放疗药物联合作用于一些肿瘤细胞株时,表现出累加和协同作用。
EGFR抑制剂主要包括单克隆抗体、酪氨酸激酶抑制剂、喹唑啉pyralo-/吡咯并-/吡啶并嘧啶、配体-毒素和免疫毒素联结物,以及反义核苷酸和EGFR/配体主导的疫苗等。一些体内和体外实验显示抗 EGFR 抗体成功的抑制了表达 EGFR 的肿瘤细胞株的生长。在实体瘤的治疗中,一些抗 EGFR 单克隆抗体单独应用或与传统治疗方法合并应用的治疗结果令人鼓舞。现有技术中已经描述了多种小鼠的抗 EGFR 单克隆抗体,可与EGFR高特异的结合,这些抗体大致可分为两类,一类是可与受体结合但不抑制 EGF结合的抗体,另一类是可与受体结合并且还抑制 EGF结合的抗体。但是患者对鼠源抗体产生的免疫应答限制了这些抗体在人类治疗和体内诊断中的应用。患者会产生人抗小鼠抗体反应(HAMA),从而限制了抗体到达其抗原靶标的能力,使抗体的效能降低。
一些研究人员为了减少HAMA 反应,研制出了人鼠嵌合的抗体,其中小鼠可变区(V)与人恒定区(C)连接。该抗体证明在临床上好于鼠源抗体,但是嵌合抗体的小鼠V 区中仍然含有使患者产生免疫原性的成分,限制了其应用。
目前已有全人源抗体Panitumumab(WO9850433)被应用于治疗生长因子过度表达的疾病中。Panitumumab(E7.6.3,ABX-EGF,AMG954,商品名:Vectibix,中文名:帕尼单抗)为全人源单克隆抗体,其结构为IgG2κ,其作用靶点为表皮生长因子受体(EGFR,ErbB-1或HER1) ,该抗体主要用于治疗表达表皮生长因子受体的转移性结直肠癌。Panitumumab是利用XenoMouse技术制备的全人源抗体,不良免疫应答的发生几率很低。
通过结合到EGFR的胞外区,单克隆抗体介导了直接的效应机制,导致了封闭配体结合,受体下调,细胞周期阻滞及诱导凋亡的作用,并且引起了针对癌细胞的免疫调节机制,例如ADCC效应及CDC效应。然而并非所有的抗体都可以介导ADCC效应,IgG1比IgG2有更强的引发ADCC效应的能力。尽管EGFR是抗癌治疗中一个有效的靶点,但针对EGFR的抗体也可以靶向其他正常组织,如:肝脏,皮肤和胃肠道等正常组织,这可能形成剂量限制性的副反应及不同类型的毒副作用。
帕尼单抗是一种全人的IgG2型单克隆抗体,可以特异性地结合EGFR,且有较高的亲和力。使用帕尼单抗进行治疗具有免疫原性低及超敏反应发生率低的优势。Vogit等的实验结果表明了帕尼单抗在EGFR上的结合位点与西妥昔并非完全一致(Voigt M, Functional dissection of the epidermal growth factor receptor epitopes targeted by panitumumab and cetuximab. Neoplasia 2012; 14:1023-31),但对西妥昔耐药的患者对帕尼治疗敏感(西妥昔耐药:第492位丝氨酸突变为精氨酸),与上述结论相一致,这解释了对西妥昔耐药的患者对帕尼治疗敏感的原因(Montagut C, Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med 2012; 18:221-3)然而,作为一种IgG2型抗体,它缺乏了巨噬细胞和NK-细胞介导的经典的ADCC效应。最近的研究结果表明,缺乏ADCC效应可能会限制帕尼单抗在体内发挥功能效应。在此之前,帕尼单抗选择了IgG2的亚型被认为可以尽量降低潜在的因ADCC效应及CDC效应引起的皮肤毒性(Kasper S, Oncogenic RAS simultaneously protects againstanti-EGFR antibody-dependent cellular cytotoxicity and EGFR signaling blockade. Oncogene 2012)。因此,考虑到增强这些效应可能增加的皮肤毒性,尚未有通过改造帕尼单抗增加其ADCC效应的报道。
包括帕尼单抗在内的抗-EGFR的抗体引起皮肤毒性的主要特征是红疹,痤疮,瘙痒及皮肤干燥。尽管由EGFR抑制剂导致的皮肤毒性的机制尚不清楚,越来越多研究都认为在角质细胞中的EGFR信号通路对于皮肤炎症反应的发生起到了关键的作用。因此,识别肿瘤特异性EGFR表位的抗体有可能消除潜在的相关副作用。
最近的研究显示了在临床前研究中使用的新型probody技术能够在一定程度上限制EGFR抗体对皮肤的毒副作用。probody技术(WO2010096838)是由美国Cytomx 生物公司开发的一种“抗体前药”或称作“基于抗体的前药”技术。其通过对传统抗体的改造使之具有在人的正常组织保持“沉默”,即减少其对正常组织表达的相应靶点的结合;而在抗体达到肿瘤部位后借助肿瘤微环境中特异性表达的蛋白酶如:uPA(尿激酶型纤溶酶原激活物)激活而成为和改造前抗体等效的治疗用药物。它在抗肿瘤治疗中可以提高抗体的靶向性,减少抗体对正常组织形成的皮肤毒性等副反应,因此是一种有潜力的新型抗体药物。
尽管靶向EGFR的抗体已经被用于临床治疗,然而正常细胞的EGFR通路受到抑制后形成的靶向性毒性往往会导致治疗的中断或停止(Lacouture M E. Mechanisms of cutaneous toxicities to EGFR inhibitors[J]. Nat Rev Cancer, 2006, 6(10):803-812;Li T, Skin toxicities associated with epidermal growth factor receptor inhibitors[J]. Target Oncol, 2009, 4(2):107-119)。正如相关研究所述,EGFR抑制剂可以通过诱导皮肤炎症反应和天然宿主防御而影响角质细胞(Mascia F, et al. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation[J]. Am J Pathol, 2003, 163(1):303-312;Lichtenberger B M, et al. Epidermal EGFR controls cutaneous host defense and prevents inflammation[J]. Sci Transl Med, 2013, 5(199):199ra111)。前抗体(或称蛋白酶激活型抗体)是一种在正常组织中保持惰性状态而在肿瘤组织中可被特异性蛋白酶剪切激活的抗体药物,将其用于肿瘤治疗时副作用较小(Sandersjoo L. A new prodrug form of Affibody molecules (pro-Affibody) is selectively activated by cancer-associated proteases[J]. Cell Mol Life Sci, 2015, 72(7):1405-1415;Baumann A, et al. New challenges and opportunities in nonclinical safety testing of biologics[J]. Regul Toxicol Pharmacol, 2014, 69(2):226-233.)。最近,Desnoyers等在西妥昔基础上开发的前抗体,将有望在不影响其临床疗效的前提下,提高使用时的安全性(Desnoyers L R, et al. Tumor-specific activation of an EGFR-targeting probody enhancestherapeutic index[J]. Sci Transl Med, 2013, 5(207):207ra144)。
抗体偶联药物是将靶向性抗体与具有活性的细胞毒性药物联接而得的药物制剂。整体而言,抗体偶联药物由三部分组成:靶向可内化的细胞表面分子的抗体、高效毒性化合物及连接前两者的连接物。许多研究表明,美登醇DM1(美登素的衍生物)作为一种有效的微管聚合抑制剂类细胞毒性药物,是开发抗体偶联药物的理想选择。此外,抗体-DM1的偶联物已经在临床前和临床评估中显示了理想的结果(Lopus M. Antibody-DM1 conjugates as cancer therapeutics[J]. Cancer Lett, 2011, 307(2):113-118)。作为EGFR家族成员,HER2是成熟的发展抗体偶联药物的靶点。FDA批准的靶向HER2的抗体偶联药物T-DM1,由曲妥珠和DM1构成,用以治疗HER2阳性的乳腺癌。之前的研究表明,在和靶向EGFR的抗体帕妥木单抗孵育后,EGFR将会很快地内化入胞。最近,靶向EGFR的抗体偶联药物IMGN289正在进行I期临床试验。靶向EGFR的抗体偶联药物是有潜力的治疗药物,然而它仍然存在有伴随着严重副反应的风险。
技术实现要素:
本发明提供了一种靶向EGFR的新型前抗体偶联药物(PDC),将其命名为PanP-DM1。在此之前,我们已经构建并表征了具有肿瘤选择性的前抗体PanP ,其重链具有SEQ ID NO:2所示的氨基酸序列,所述前抗体轻链具有SEQ ID NO:4所示的氨基酸序列(Yang Y, et al. Generation and characterization of a target-selectively activated antibody against epidermal growth factor receptor with enhanced anti-tumor potency[J]. MAbs, 2015, 7(2):440-450;专利申请号为:201410724477.4)。
本发明中,通过稳定的非还原硫醚键将美登素衍生物(DM1)连接到PanP上。在体外和体内肿瘤模型中,对其抗肿瘤效果进行评估。进一步,本发明证实PanP-DM1可以内化及诱导细胞周期阻滞。另外,在BALB/c小鼠和荷瘤小鼠上进行了基本毒性试验的评估。本发明提供的一种靶向表达EGFR肿瘤细胞的前抗体偶联药物PDC,其具有高效能及肿瘤的选择性,且经实验证明前抗体偶联药物具有较小的对正常组织的毒副作用,解决现有技术中肿瘤治疗药物对正常组织细胞的杀伤,引起较大毒副作用的缺点。
本发明公开了:
1、一种靶向表达EGFR肿瘤细胞的前抗体偶联药物,其特征在于,含有蛋白酶激活型抗体和细胞毒性药物。
2、上述一种靶向表达EGFR肿瘤细胞的前抗体偶联药物,其特征在于,所述的前抗体重链具有SEQ ID NO:2所示的氨基酸序列,所述前抗体轻链具有SEQ ID NO:4所示的氨基酸序列。
3、上述一种靶向表达EGFR肿瘤细胞的前抗体偶联药物,其特征在于,所述细胞毒性药物为美登素衍生物。
4、上述一种靶向表达EGFR肿瘤细胞的前抗体偶联药物,其特征在于,所述的美登素衍生物为美登醇。
5、一种靶向表达EGFR肿瘤细胞的前抗体偶联药物的应用,其特征在于,所述偶联药物用于治疗肿瘤。
6、上述一种靶向表达EGFR肿瘤细胞的前抗体偶联药物的应用,其特征在于,所述肿瘤包含头颈部鳞癌、鼻咽癌、乳腺癌、结直肠癌、肺癌、卵巢癌以及膀胱癌。
附图说明
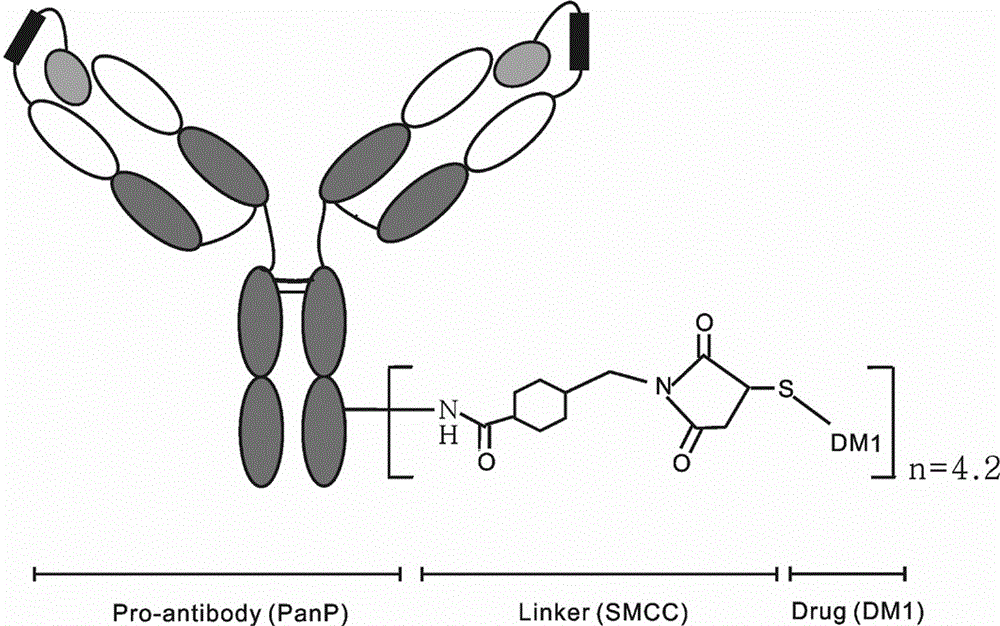
图1、靶向表达EGFR肿瘤细胞的前抗体偶联药物PDC的结构示意图
图2、PDC结构SDS-PAGE鉴定图谱
图3、PDC结构ESI-TOF-MS分析图谱
图4、PDC活性检测流式细胞检测图谱
图5、PDC活性检测ELISA检测图谱
图6、PDC对DiFi细胞体外细胞毒性试验图谱
图7、PDC对H292细胞体外细胞毒性试验图谱
图8、PDC对A431细胞体外细胞毒性试验图谱
图9、激活型PDC与PDC肿瘤抑制活性比较图谱
图10、PDC对BT-474细胞体外细胞毒性试验图谱
图11、PDC对MCF-7细胞体外细胞毒性试验图谱
图12、激活型PDC内化入A431细胞图谱
图13、PDC对A431细胞周期作用图谱
图14、PDC对A431细胞周期作用柱状图谱
图15、PanP和PDC延缓A431肿瘤生长图谱
图16、PDC抗肿瘤效应图谱
图17、PDC治疗期A431荷瘤小鼠体重变化图谱
图18、PDC治疗期H292荷瘤小鼠体重变化图谱
图19、不同剂量PDC对小鼠毒性试验结果。
具体实施方式
以下实施例、实验例对本发明进行进一步的说明,不应理解为对本发明的限制。实施例不包括对传统方法的详细描述,如那些用于构建载体和质粒的方法,将编码蛋白的基因插入到这样的载体和质粒的方法或将质粒引入宿主细胞的方法。这样的方法对于本领域中具有普通技术的人员是众所周知的,并且在许多出版物中都有所描述。
人表皮癌细胞系A431,人乳腺癌细胞系MCF-7和BT-474以及人肺癌细胞系NCI-H292都来自美国菌种保藏中心(ATCC)。人结肠癌细胞系DiFi来自MD安德森医学中心。抗EGFR的抗体Pan和PanP都产自抗体药物与靶向治疗国家重点实验室,重组的uPA购自义翘神州公司,DM1-SMCC购自联宁制药公司,六周左右的雌性裸鼠和BALB/c小鼠都购自中科院上海实验动物中心。
实施例1 、前抗体偶联药物的制备
按照专利US 2011/0003969所述,通过将DM1-SMCC与抗体PanP的赖氨酸残基偶联制得PanP-DM1。主要过程如下:将5 mg/mL的抗体与6.4 mg/mL的DM1-SMCC混合,反应缓冲液为含有2mM EDTA和50 mM Nacl的磷酸盐缓冲液(50mM, PH=7.5),反应时间为2小时。按照上述方法,也将Pan与DM1-SMCC偶联构成了Pan-DM1。
另外,将抗TNFα的抗体与DM1偶联,制得了对照组抗体偶联药物。根据专利US 2010/0092495,利用紫外分光光度法在252nm和280nm测定了抗体偶联药物的吸光值后,计算药物抗体的偶联比率。利用反相超高效液相色谱串联质谱系统(RP-UPLC/ESI-MS)测定药物的分布。在还原和非还原条件下,将制得的PDC在10%SDS-PAGE上进行分析,考马斯亮蓝染色分析。
(1)PanP-DM1的表征分析
按照材料和方法所描述的,利用非还原的硫酯键的连接物SMCC将DM1连接到抗体的赖氨酸残基。PanP-DM1(PDC)的结构如图1所示,利用SDS-PAGE对其进行鉴定,结果如图2所示。在还原条件下,PanP-DM1的重链有着比PanP更高的分子量,表明了DM1-SMCC优先选择连接到重链的赖氨酸残基上。另外,在非还原条件下,仅有少量的聚体出现。经测定,其含量小于3%。此外,利用紫外/可见光分光光度法测得PanP-DM1的DAR为4.2左右,与商品化的T-DM1相似。
(2)去糖基化的PanP-DM1的RP-UPLC/ESI-MS分析
利用反相层析串联ESI-TOF-MS进行样品的分析。首先,将100 µgPanP-DM1与7.14 µg糖苷内切酶Endo F2在37度孵育4 h。将大约2 µg的脱糖的PanP-DM1上样到Waters C4色谱柱(2.1 x 50 mm, 1.7 µm),以含有25-35%乙腈的洗脱液(含0.1%的甲酸)洗脱,流速为0.4 mL/min,时间为6分钟,色谱柱温度为60度。最后,使用Biopharmalynx软件分析数据。
为进一步评药物的分布,利用ESI-TOF-MS分析去糖基化的PanP-DM1(PDC)。质谱检测结果见图3。图谱中显示了8个主峰,分别对应了连接了0到7个DM1分子的抗体。
综合上述实验结果,数据表明成功构建PanP-DM1(PDC)。
实验例1 、PDC活性检测
通过ELISA和流式细胞术测定EGFR的结合
将EGFR-Fc包被到96孔板上,用含有10%脱脂奶粉的PBS封闭。洗涤后加入指定浓度的抗体,室温孵育1小时。洗涤后,加入HRP-标记的抗人F(ab’)2,于37度孵育30min,最后加入TMB底物溶液。在450nm读取吸光度,利用4参数曲线进行拟合。
在进行流式细胞术分析时,过表达EGFR的A431细胞与待测抗体在FACS缓冲液(含1%的胎牛血清的PBS缓冲液)中混合孵育1小时。洗涤后,加入FITC标记的抗人二抗,孵育30分钟。接下来,再次用FACS缓冲液洗涤,并在BD的FACSCalibur系统上进行分析。
PanP-DM1显示了与PanP相似的酶激活特性。
为进一步评估uPA-激活型PanP-DM1对细胞表面EGFR的结合,使用了流式细胞术分析技术。通过将重组的uPA与PanP-DM1共同孵育得到激活型PanP-DM1,激活型PanP-DM1和激活型PanP在饱和浓度(10 µg/ml)和亚饱和浓度(1 µg/ml)下都显示了对过表达EGFR的A431细胞同等程度的结合,这表明交联DM1后未影响uPA介导的激活,结果见图4。
另外,在将PanP-DM1与uPA孵育后,利用ELISA检测到其结合活性的显著增加,与Pan-DM1相似,结果见图5。结果表明,在uPA激活前,PanP-DM1保持了对EGFR的低结合活性,此特点与PanP相似。
实验例2、PDC体外细胞毒性试验
利用CCK-8试剂盒评估PanP和PanP-DM1偶联物对肿瘤细胞活率的影响。将含有1%小牛血清培养的细胞平铺到96孔细胞培养板中(BT-474, MCF-7和DiFi细胞为每孔5000个;H292细胞为每孔3000个;A431细胞为每孔2000个),过夜培养使其贴壁。第二天,将相应浓度的激活型PanP,激活型PanP-DM1或作为对照组的抗TNFα-DM1抗体添加到每孔中,孵育不同时间(A431细胞为48小时,其他细胞为72小时)。利用CCK-8试剂盒测定细胞活率。
按照下列公式计算存活细胞比率:[(实验组的A450值-背景组的A450值)/(未处理组的A450值-背景组的A450值)]*100。
对比PanP-DM1,激活型PanP-DM1及激活型PanP对三株过表达EGFR的肿瘤细胞系(A431,H292及DiFi细胞)的抑制活性。在48小时或72小时的孵育后,激活型PanP-DM1显示了对DiFi,H292及A431细胞的生长抑制。PDC对DiFi细胞体外细胞毒性试验结果见图6,PDC对H292细胞体外细胞毒性试验结果见图7,PDC对A431细胞体外细胞毒性试验结果见图8。
重要的是,在1 µg/ml的浓度下,激活型PanP-DM1显示了较之激活型PanP的显著更强的抑制效应(P<0.001)。另外如预期一样,在低浓度条件下(0.1µg/mL)激活型PanP-DM1较之非激活型PanP-DM1有更强的抑制效应,这也反映了完整PDC药物的功能性“隐蔽”效果。
因此,综上所述,PanP-DM1有着较之PanP更强的体外抗肿瘤效果。PanP-DM1有较之PanP更强的体外抗肿瘤活性。
另外,我们在低表达EGFR的乳腺癌细胞BT-474和MCF-7中评估PanP-DM1的非靶向性抑制效果。结果见图9,激活型PanP-DM1在这些细胞中仅显示了极微弱的剂量依赖性抑制效应,这与对照组抗-TNFα ADC相似。另外,仅高浓度的PanP-DM1 (1 µg/ml)抑制BT-474或MCF-7细胞,显示了非特异性的抑制效应。PDC对BT-474细胞体外细胞毒性试验结果见图10,PDC对MCF-7细胞体外细胞毒性试验结果见图11。
实验例3、PDC对A431 细胞周期分析
将激活型PanP (1 µg/mL)、激活型PanP-DM1(1 µg/mL)或PanP-DM1(1 µg/mL)与细胞(1 × 105/mL)在37度孵育3小时,洗涤后用不含药物的培养基在37度继续培养18个小时。用1 mL70%的甲醇固定细胞,利用溴化乙锭(PI)染色后,流式细胞仪检测。最后用Flowjo 7.6软件分析流式数据。
PanP-DM1对A431细胞周期的效应
抗体偶联药物的内化测定:在4度下将A431细胞与饱和浓度下的激活型PanP-DM1偶联物混合孵育30分钟。通过洗涤洗去未结合的偶联物。接下来,继续在4度或37度孵育。在不同的时间点,利用FITC标记的羊抗人IgG检测细胞表面结合的抗体偶联药物。
为进一步阐释PanP-DM1的作用机制,分析偶联物对A431细胞周期阻滞的效应。首先在A431细胞中评估了激活型PanP-DM1的内化,内化的过程对于DM1入胞内有着重要的意义。图12表明了在37度条件下孵育90分钟后,细胞表面激活型PanP-DM1的数目明显下降,这表明其已经内化入A431细胞中。
接下来,利用流式细胞术评估激活型PanP-DM1对细胞周期的影响。激活型PanP-DM1处理会导致发生明显的G2/M期阻滞,这可能直接导致细胞凋亡,结果见图13及图14。在激活型PanP-DM1处理后的细胞中,G2/M期细胞比例从19.69 ± 1.809%上升至30.92 ±1.097%,而在PanP-DM1处理后的细胞中G2/M期细胞比例从19.69 ± 1.809%上升至25.92 ± 1.092 % 。这些结果表明,未激活的PanP-DM1较之激活型PanP-DM1有着更弱的诱导A431细胞发生G2/M期阻滞的能力(P<0.05),这可能要归结于未激活PanP-DM1的减少的抗原结合活性。另外,与之前的研究结果相似,激活型PanP诱导了G0/G1期阻滞,这也是西妥昔单抗和帕妥木单抗发挥效应的主要机制。
总结来说,这些数据表明PanP-DM1可以通过靶向递送DM1及诱导G2/M期阻滞而发挥增强的抗肿瘤效应。
实验例4、PDC体内疗效分析
在鼠的背部右侧注射1 × 106个A431细胞或1 × 106个NCI-H292细胞。当肿瘤长至100 mm3(对于A431细胞)或130 mm3时,将鼠分为三组,每组10只。分别腹腔注射PanP (35 or 20 mg/kg),PanP-DM1偶联物(35 or 20 mg/kg)或人IgG对照组(35 or 20 mg/kg),注射次数为一次或两次(如图中所示)。肿瘤体积的测算公式=长度*宽度2*0.5。利用CO2窒息的方式对鼠施以安乐死。
PanP-DM1在A431和H292异种移植肿瘤模型中的治疗效果
接下来,我们在裸鼠上建立异种移植的A431和H292肿瘤以评估PanP-DM1和PanP的治疗效果。正如之前文章所述,此两种细胞系皆可表达并分泌uPA。正如图15所示,PanP和PanP-DM1皆可有效延缓A431肿瘤的生长。值得注意的是,注射PanP-DM1后7天,肿瘤已经基本消退。相比之下,PanP治疗组小鼠的荷瘤仅部分消退。在观察终点时(第21天),所有接受了PanP-DM1处理小鼠的肿瘤皆完全消失,然而接受了PanP处理的小鼠的肿瘤大小达到了555.8 ± 70.58 mm3。明显地是,在A431肿瘤模型中,PanP-DM1显示了强于PanP的抑瘤效能。
接下来,我们在H292异种移植肿瘤模型中验证了PanP-DM1的抗肿瘤效应,结果如图16所示。单次的腹腔注射PanP-DM1(20 mg/kg)使所有的荷瘤小鼠肿瘤消退(n=10)。值得注意的是,在H292异种移植模型中,PanP-DM1显示了较之PanP更强的抑瘤效果,尽管这种差异没有在A431肿瘤模型中明显。因此,与体外结果一致的是,在A431和H292异种移植模型中,PanP-DM1也显示了优于PanP的抑瘤效应。
实验例5、PDC 体内毒性分析
通过对BALB/c小鼠单次注射0, 25, 50, 75 or 100 mg/kg的PanP-DM1的方式评估PDC的抗原非依赖性毒性。另外,在A431细胞和NCI-H292细胞异种移植的裸鼠身上评估剂量依赖性的毒性。观察时间为15-20天。通过观察鼠体重的减少进行毒性评估。
PanP-DM1对小鼠的毒性评估
为评估PanP-DM1的治疗相关的非特异性毒性,在PanP-DM1治疗期间测定了A431和H292荷瘤小鼠的体重变化。A431荷瘤小鼠对于PanP-DM1治疗是耐受的且在第二次注射导致的较小体重减轻后体重明显地恢复, PDC治疗期A431荷瘤小鼠体重变化结果见图17。相似的结果也在接受了PanP-DM1的H292荷瘤小鼠上被发现。在整个观察过程中(约18天),这些小鼠体重没有明显下降且行为正常,C治疗期H292荷瘤小鼠体重变化结果见图18。
因为PanP无法识别鼠的EGFR, 通过BALB/c小鼠体重的减轻评估了多个剂量下的PanP-DM1的抗原非依赖性效应。在测试的剂量下,0, 25及50mg/kg的PanP-DM1剂量并未导致小鼠体重减轻。然而,75或100 mg/kg的PanP-DM1注射导致了小鼠体重在前三天的快速下降(-8%或-12%),这也表明了PanP-DM1的使用剂量在75 mg/kg以上时会对BALB/c鼠有毒性效应,不同剂量下的作用结果见图19。体而言,PanP-DM1在多个剂量下都显示出了良好的耐受。
- 还没有人留言评论。精彩留言会获得点赞!