受体靶向构造物和其使用的制作方法
本申请请求Lali K.MEDINA-KAUWE等人于2014年1月17日提交的且名称为“c-MET TARGETING CONSTRUCT AND USES THEREOF”的美国临时申请系列号61/928,903,该专利的全部公开内容(包括附图)以引用的方式并入本文。
政府权利
本文公开的主题是在国家健康研究所/国家癌症研究所授予的基金CA140995和CA129822下由政府支助所进行。政府对所公开主题具有某些权利。
发明领域
本发明涉及生物技术的领域。特定而言,本发明涉及将治疗剂递送至靶细胞(诸如癌细胞)的组合物。
发明背景
抵抗临床中所用的当前靶向治疗或获得对该其靶向治疗的耐受性的许多肿瘤显示出蛋白质(诸如c-Met)的表面水平升高。例如,肺癌获得对EGF-R抑制剂(诸如Tarceva)的耐受性。诸如Tarceva之类抑制剂旨在阻断受体酪氨酸激酶的活性(称为酪氨酸激酶抑制剂,或TKI's),但大多数情况对TK抑制不响应。这些肿瘤的特征在于细胞表面蛋白质(诸如c-Met)水平升高,并且因此变成用于本文所述治疗方法的极好候选者,该方法可靶向过表达蛋白质并且渗透肿瘤细胞。
本领域中,目前正试图研发出旨在阻断通过c-Met的信号的c-Met抗体或抑制剂。然而,过往历史表明大多数情况将对信号阻断抗体或小分子无响应,因为该肿瘤采用替代性方式来连续增生,而不论信号抑制如何。
本文所述组合物规避了使用细胞表面受体(例如c-Met)作为入口来将毒性分子递送至肿瘤细胞中并且从内部杀死肿瘤以便阻断信号的需要。配体导向递送允许靶向结合至对特定细胞表面受体(例如c-Met)呈阳性的肿瘤,并且递送分子中的膜渗透结构域允许在细胞表面受体介导的胞吞作用之后跨体内膜进行渗透和溶解。递送蛋白经改性以通过(例如)离子相互作用来与某些治疗性分子组装并且运送所述某些治疗性分子。
发明概述
本文公开的是药物递送分子,其包括:配体,其靶向细胞表面分子;膜渗透结构域;和有效负载结合结构域;以及包含所述药物递送分子的药物组合物。还公开了在有需要的受试者中治疗癌症、抑制癌症的进展、预防癌症转移、和向脑部递送治疗性化合物,所述方法包括:鉴别需要该方法的受试者;提供包含本文公开的药物递送分子的组合物;以及向受试者施用有效量的所述组合物。
附图简述
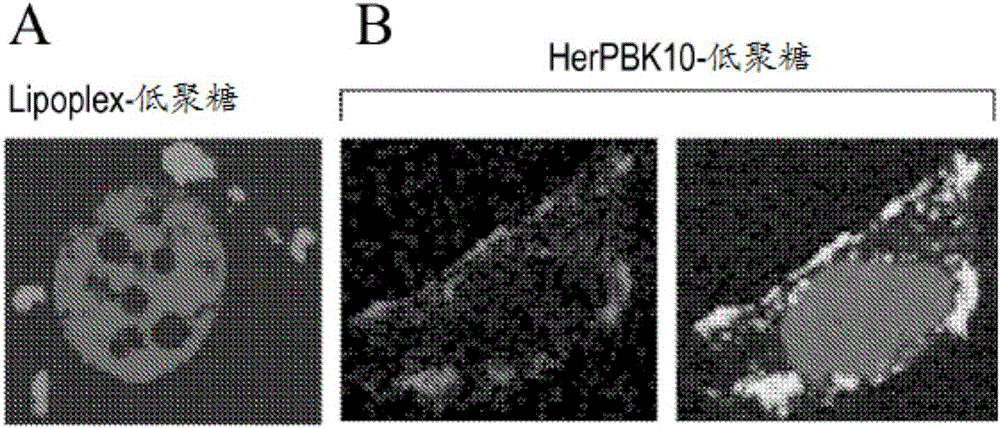
图1示出使用作为对照物的Lipofectamine(图1A)以及HerPBK10(图1B)来摄取单链低聚核苷酸的结果。
图2示出PBK10可递送编码GFP的合成mRNA。图2A提供本实验中所用的mRNA的概要。图2B示出来自由Lipofectin递送的mRNA(右)和与其比较的由Lipofectin递送的GFP表达质粒(左)的GFP表达。图2C是PBK10-mRNA复合物的示意图。图2D示出用于PBK10-mRNA复合物的细胞结合数据。图2E示出细胞结合复合物的摄取的结果。图2F示出GFP表达的结果的图像,而图2G以图表形式描绘相同数据。
图3是以下的示意图:(A)Inlb321,其涵盖用于c-MET受体结合的最小结构域;(B)InlB321,其结合至c-MET的胞外结构域。
图4是示出重组基因构造物被组装以编码新融合蛋白(InlB-PBK10)的示意图。A.pRSET-InlB-PBK10的构造。B.pRSET-GFP-InlB的构造。C.通过限制性消化证实克隆插入物。
图5是Western印迹的结果,其表明在细菌中产生了重组蛋白InlB、InlB-PBK10和GFP-InlB。
图6是示出在不同肿瘤和非肿瘤细胞系之间c-MET的表面水平变化的图表。示出了通过以96孔格式执行的细胞表面ELISA来测得的在非透化细胞表面上的相对受体水平。ELISA结果表明H1993(肺癌细胞系)和MDA-MB-231(乳腺癌细胞系)是具有c-MET的最高细胞表面水平的细胞。RANKL(前列腺癌细胞系)和MDA-MB-435(乳腺癌细胞系)显示中等水平,而LN-GFP(前列腺癌细胞系)和Cos-7(非洲绿猴肾成纤维细胞)显示细胞表面c-MET的较低水平。
图7示出实验结果,该结果表明InlB衍生肽识别出c-MET。A.InlB321肽展示出对c-MET阳性(而不是低c-MET)细胞的优先结合。使用荧光激化细胞分选(FACS)来测量结合至c-MET阳性细胞的InlB(由免疫荧光识别)的相对水平。FACS数据示出与低c-MET细胞(LN GFP)相比,结合至高c-MET细胞(H1993)的InlB的水平相对更高。B.通过竞争性抑制证实c-MET结合。当在结合至细胞之前,采用来源于c-MET(MET)的胞外配体结合结构域的可溶解肽来预培育游离InlB时,可抑制InlB向H1993结合。InlB和MET在1:1摩尔比率(MET:InlB)下培育,如果InlB对MET具有特异性,则预测受体结合降低50%。C.通过InlB-PBK10的细胞结合与c-MET水平成比例。InlB-PBK10表明:如通过细胞表面ELISA所测量,与表达相对低c-MET水平的细胞(Cos-7)相比,具有更高c-MET细胞表面表达的细胞(MDA-MB-231)的结合水平更高。D.通过竞争性配体抑制对c-MET+细胞的结合。当浓度增加的InlB被预结合至细胞时,在添加InlB-PBK10之前,经1h在冰上使浓度逐步升高的游离InlB配体预结合至MDA-MB-231细胞。E.InlB-PBK10在混悬液中经历向c-MET+细胞的受体特异性结合。混悬液中的MDA-MB-435细胞用浓度增加的游离InlB配体来培育,并且在移除未结合InlB之后,用InlB-PBK10培育细胞。选择游离InlB配体的浓度,以使得InlB-PBK10与InlB的摩尔比率为1:1、1:5和1:10。执行Western印迹以测量与细胞团块共沉淀的相对InlB-PBK10水平。免疫印迹条带的密度计测量表明,InlB-PBK10结合的水平随InlB的浓度增加而降低,这与向c-MET结合的InlB-PBK10相一致。
图8示出InlB-PBK10内化入c-MET+细胞中。
图9示出InlB-PBK10可向c-MET+细胞递送毒性分子。A.制备InlB-PBK10-Ga颗粒。示意图示出在将InlB-PBK10与Ga-咔咯(Ga-corrole)混合以促进非共价组装之后通过超滤作用分离颗粒的程序。B.InlB-PBK10-Ga颗粒的DLS。C.InlB-PBK10介导咔咯(corrole)有效负载的细胞溶质入口。D.I-Dox降低c-MET阳性肿瘤细胞的存活率。E.游离InlB抑制I-Dox毒性。
图10示出在携带皮下双侧胁腹MDA-MB-435肿瘤的nu/nu小鼠中全身性(尾静脉)递送之后,InlB-PBK10的生物分布的Xenogen Spectum图像。A.在尾静脉注射之后所示时间点处的整个小鼠的图像。蓝色箭头指向肾。白色箭头指向肿瘤。B.从4h时间点之后处死的相同小鼠中收获的肿瘤和组织的图像。
图11描绘HerMn的组装。A.HerPBK10蛋白的示意图,突出显示了功能域。B.Mn-咔咯的化学结构(S2Mn)。C非共价组装的示意图。D.溶液中HerMn颗粒的TEM(插入物)和动态光散射(DSL)测量。
图12是一组图表,其示出HerPBK10结合至HER3并且未受患者血清抑制。A.对HerPBK10结合至固定化HER3(人类ErbB3胞外结构域;Prospec)的ELISA-/+用可溶解HER3肽作为竞争性抑制剂(HER3阻断)来预培育。Un∶无HerPBK10。B.对HerPBK10结合至HER2+细胞的ELISA-/+用1x和10x摩尔比率的可溶解HER3肽、可溶解HER4肽(ERBB4肽,Abnova)、β细胞素(10μg/mL)、或帕妥珠单抗(Pz)作为竞争性抑制剂进行预培育。C.在来自5位HER2+患者和年龄匹配对照物(HER2-)的血清中,对HerPBK10结合到HER2+(MDA-MB-435)细胞的ELISA。对照样品在牛血清中结合,并且通过与重组体heregulin配体(+Her)的竞争性抑制进行受体结合检验。N=3.*,p<0.05,与对照物相比(-Her:无竞争性抑制剂)。
图13示出HerPBK10结合至小鼠HER3的结果。A.人类和小鼠HER3的结构域I-II(aa 20-239,heregulin结合结构域)的氨基酸序列比对。蓝色残基指示氨基酸差异。B.使用与人类和小鼠HER3交叉反应的抗HER3抗体(1B2E;细胞信号技术)通过ELISA(无透化作用)检测的相对HER3水平。C.HerPBK10向4T1小鼠乳房肿瘤细胞的结合。N=3.*,p<0.05,与单独HerPBK10相比
图14是示出HerMn毒性对人类HER2+和HER2-肿瘤细胞的数据的图表。
图15是示出HerMn细胞毒性的机制的图片。A.共焦荧光图像,示出通过MDA-MB-435细胞中的HerMn使线粒体膜电势降低。B.共焦荧光图像,通过HerMn示出肌动蛋白(红色)和微管蛋白(绿色)的超氧化物介导瓦解。
图16示出S2Ga与TSPO相互作用的数据。A-B.通过测量吸光率和荧光光谱,针对TSPO结合咔咯的存在,评估渗余物。C-D.HerGa与TSPO原位相互作用的证据。当积累到线粒体中时,C中所用的绿色荧光JC-1染料发红色荧光。D是对C中红色荧光的定量。*,p<0.05。
图17示出荷瘤小鼠中的生物分布。在尾静脉注射之后对Alexa680标记HerMn、曲妥单抗(Tz)和BSA(12nmol ea)进行Xenogen成像和定量。图表平均荧光-/+SEM。
图18示出HerMn的治疗效力的数据。A.雌性裸小鼠中的HER2+MDA-MB-435肿瘤生长,该雌性裸小鼠接受HerMn或S2Mn的每日IV(经由尾静脉)注射(5nmole咔咯/注射),每日一次,持续6个连续日。对照组接受与HerMn等同浓度的盐水或HerPBK10。治疗在约200mm3平均肿瘤体积时开始。在试剂注射之前(第1天),在试剂注射期间(第3天),以及在试剂注射之后(第8天、第15天和第22天),测量肿瘤体积。N=8-10肿瘤/组。*p<0.05(单向ANOVA)。B.在持续2天(实线)或5天(虚线)暴露于HerMn、S2Mn、HerPBK10、或阿霉素(Dox)期间,人类CDC生活力。N=3per conc,来自3次单独实验
图19示出在无肿瘤的小鼠中HerPBK10的组织分布。
实施方案的详述
定义∶
为了方便起见,在此收集了在说明书、实施例和附加权利要求书中采用的某些术语。除非另行指出或从上下文暗示,下列术语和短语包括下面提供的含义。除非另外明确说明或从上下文明显可见,以下术语和短语不排除术语或短语在其所述的领域获取的含义。提供定义是为了有助于描述特定实施方案,并且不旨在限制要求保护的发明,因为本发明的范围仅受权利要求书限制。除非另外定义,否则本文中所用的所有技术和科学术语均具有与本发明所属领域一般技术人员通常所理解含义相同的含义。
“有益的结果”可以包括,但决不限于,减轻或缓和该疾病状态的严重程度、预防该疾病状态恶化、治愈该述疾病状态、预防该疾病状态发展、降低患者发展该疾病状态的几率以及延长患者寿命或预期寿命。在一个实施方案中,疾病状态是癌症。在一些实施方案中,疾病状态为自身免疫疾病。
“癌症”和“癌性”是指或描述哺乳动物的特征通常在于细胞生长不受调控的生理病状。癌症的实例包括但不限于:白血病、骨髓瘤、B细胞淋巴瘤(霍奇金淋巴瘤和/或非霍奇金淋巴瘤)、脑癌、乳腺癌、结肠直肠癌、肺癌、肝细胞癌、肾癌、胃癌、胰癌、宫颈癌、卵巢癌、肝癌、膀胱癌、尿道癌、甲状腺癌、肾脏癌、癌肿、黑素瘤、头颈癌、脑癌、前列腺癌、雄激素依赖性前列腺癌、和雄激素非依赖性前列腺癌。
如本文所用的“化学治疗药物”或“化学治疗剂”是指用于治疗癌症的药物,所述药物包括但不限于:白蛋白结合紫杉醇(nab-紫杉醇)、放线菌素、阿利类视色素(Alitretinoin)、全反式视黄酸、阿扎胞苷、硫唑嘌呤、贝伐珠单抗、贝沙罗汀(Bexatotene)、博莱霉素、硼替佐米、卡铂、卡培他滨、西妥昔单抗、顺铂、苯丁酸氮芥、环磷酰胺、阿糖胞苷、柔红霉素、多西他赛、去氧氟尿苷、阿霉素、表柔比星、埃博霉素、埃罗替尼、依托泊苷、氟尿嘧啶、吉非替尼、吉西他滨、羟基脲、伊达比星、伊马替尼、易普利单抗(Ipilimumab)、伊立替康、氮芥、美法仑、巯基嘌呤、甲氨蝶呤、米托蒽醌、阿仑单抗(Ocrelizumab)、奥法木单抗、奥沙利铂、紫杉醇、帕尼妥单抗(Panitumab)、培美曲塞、利妥昔单抗、他氟泊苷(Tafluposide)、替尼泊苷、硫鸟嘌呤、托泊替康、类视色素、戊柔比星、威罗菲尼、长春花碱、长春新碱、长春地辛、长春瑞滨、伏立诺他、罗米地辛、5-氟尿嘧啶(5-FU)、6-巯基嘌呤(6-MP)、克拉屈滨、氯法拉滨、氟尿苷、氟达拉滨、喷司他丁、丝裂霉素、伊沙匹隆、雌莫司汀或其组合。
“受试者”或“个体”或“动物”或“患者”或“哺乳动物”意指任何受试者,具体是哺乳动物受试者,希望对它们进行诊断、预后或治疗。在一些实施方案中,受试者患有癌症。在一些实施方案中,受试者受试者在该受试者寿命中某一时间点患有癌症。在各种实施方案中,受试者的癌症处于缓解期,是复发的或是非复发的。
如本文所用"哺乳动物"是指哺乳纲的任何成员,其包括但不限于:人类,家畜,农场动物,动物园动物,运动会动物,宠物动物,诸如狗、猫、豚鼠、兔子,大鼠,小鼠,马,牛,奶牛;灵长类动物,诸如类人猿、猴子、猩猩和黑猩猩;犬科动物,诸如狗和狼;猫科动物,诸如猫、狮子、和老虎;马科动物,诸如马、驴和斑马;食品动物,诸如奶牛、猪和羊;有蹄类动物,诸如鹿和长颈鹿;啮齿动物,诸如小鼠、大鼠、仓鼠和豚鼠;等等。在某些实施方案中,哺乳动物为人类受试者。所述术语不指示特定年龄或性别。因此,成年和新生受试者以及胎儿无论雄性或雌性都意欲包括在这个术语的范围内。
如本文所用的“治疗(Treatment/treating/therapy)”或“治疗性(therapeutic)是指治疗性治疗和预防性或防止性措施,其中目的是预防或减缓(减轻)针对的病理性病状、预防该病理性病状、追求或获得有益的结果或降低个体发展所述病状的几率,即使治疗最终不成功。需要治疗的对象包括已经患有病状的对象,以及易患所述病状的对象或要预防所述病状的对象。具体的程序或施用被认为是治疗性的,即便在施用之后受试者未感觉更佳。因此,认为受试者疾病状态的任何改善、或疾病进展的任何减缓是治疗性的。癌症治疗的实例包括但不限于:主动监测,观察,手术介入,化学疗法,免疫疗法,辐射疗法(诸如外线束辐射,立体定位辐射手术(伽马刀),和分次立体定向辐射治疗(FSR)),局部疗法,全身性疗法,疫苗疗法,病毒疗法,分子靶向疗法,或其组合。
如本文所用的“肿瘤”是指所有赘生性细胞生长和增殖(无论恶性或良性)以及所有癌前和癌性细胞和组织。
如本文所用的“治疗剂”是指用于(例如)治疗、抑制、预防、减轻疾病的影响,降低疾病的严重程度,降低疾病发展的可能性,减缓疾病进展和/或治愈疾病的药剂。治疗剂靶向的疾病包括但不限于:癌肿、肉瘤、淋巴瘤、白血病、生殖细胞肿瘤、胚细胞瘤各种免疫者细胞上表达的抗原以及在与各种血液疾病、自身免疫疾病和/或炎性疾病相关的细胞上表达的抗原。
在本公开的背景下,“约”某一值意指本公开涵盖所列值±25%,或者所列值±15%,或者所列值±10%,或者所列值±5%。因此,例如,“约50个氨基酸”意指50±25%氨基酸(即,37至63个氨基酸的范围),或者50±15%氨基酸(即,42至58个氨基酸的范围),或者50±10%氨基酸(即,45至55个氨基酸的范围),或者50±5%氨基酸(即,47至53个氨基酸的范围)。
药物递送分子
本文所述的是药物递送分子。药物递送分子包括靶向细胞表面分子、膜渗透结构域和有效负载结合结构域的配体。药物递送分子中的配体将分子递送至靶细胞,诸如癌细胞。膜渗透结构域介导靶细胞的细胞溶质渗透。有效负载结合结构域形成具有治疗剂的复合物。在具有治疗性分子的复合物中的药物递送分子将治疗剂递送至靶细胞,诸如癌细胞。在各种实施方案中,所述癌细胞以下中的任何一种或多种:白血病、骨髓瘤、B细胞淋巴瘤(霍奇金淋巴瘤和/或非霍奇金淋巴瘤)、脑癌、乳腺癌、结肠直肠癌、肺癌、肝细胞癌、肾癌、胃癌、胰癌、宫颈癌、卵巢癌、肝癌、膀胱癌、尿道癌、甲状腺癌、肾脏癌、癌肿、黑素瘤、头颈癌、脑癌、前列腺癌、雄激素依赖性前列腺癌、和雄激素非依赖性前列腺癌。
在一些实施方案中,由此公开的药物递送分子形成直径为约5nm至约50nm的纳米颗粒。在一些实施方案中,纳米颗粒患有约10至约30nm的大小。有效负载(即,治疗剂)随后结合至该纳米颗粒。在一个实施方案中,纳米颗粒包含金属化咔咯,例如锰(Mn)、铁(Fe)、或镓(Ga)咔咯。在其他实施方案中,纳米颗粒包含蛋白质或蛋白质片段。在一些实施方案中,治疗剂向纳米颗粒的结合是通过选自由以下组成的组的方法进行:静电相互作用、疏水性相互作用、亲水性相互作用、氢键以及共价键。一旦纳米颗粒治疗剂组合进入细胞,则纳米颗粒与治疗剂之间的结合断裂,这要么是因为该细胞破坏纳米颗粒与治疗剂的缔合,要么是因为这两者间的共价键被酶水解。
在一些实施方案中,细胞表面分子是涉及到导致细胞程序死亡减少或消除的信号转导路径的受体。在本文所述药物递送分子中可能被配体靶向的细胞表面分子的实例包括但不限于以下中的任何一种或多种:4-1BB、5T4、腺癌抗原、α-胎儿球蛋白、BAFF、B淋巴瘤细胞、C242抗原、CA-125、碳酸酐酶9(CA-IX)、c-MET、CCR4、CD152、CD19、CD20、CD200、CD22、CD221、CD23(IgE受体)、CD28、CD30(TNFRSF8)、CD33、CD4、CD40、CD44v6、CD51、CD52、CD56、CD74、CD80、CEA、CNTO888、CTLA-4、DR5、EGFR、EpCAM、CD3、FAP、纤连蛋白额外结构域-B、叶酸盐受体1、GD2、GD3神经节苷脂、糖蛋白75、GPNMB、HER2/neu、肝细胞生长因子(HGF)、人类分散因子受体激酶、IGF-1受体、IGF-I、IgGl、L1-CAM、IL-13、IL-6、胰岛素样生长因子I受体、整联蛋白α5β1、整联蛋白αvβ3、MORAb-009、MS4A1、MUC1、粘蛋白CanAg、N-羟乙酰神经氨酸、NPC-1C、PDGF-Rα、PDL192、磷脂酰丝氨酸、前列腺癌肿细胞、RANKL、RON、ROR1、SCH 900105、SDC1、SLAMF7、TAG-72、腱生蛋白C、TGFβ2、TGF-β、TRAIL-R1、TRAIL-R2、肿瘤抗原CTAA16.88、VEGF-A、VEGFR-1、VEGFR2、波形蛋白或其组合。对癌症特异性的其他靶向分子或颗粒将对本领域技术人员显而易见,并且可结合本发明的替代性实施方案来使用。在实施方案中,配体靶向癌细胞上的c-MET。在一个实施方案中,配体是HGF。在实施方案中,配体是内化蛋白B或其片段或其变型。在另一实施方案中,配体是细菌侵袭素(Inv)蛋白质。
因此,在一些实施方案中,配体是天然结合配偶体,或可结合至细胞表面分子的分子。在一些实施方案中,配体是蛋白质、蛋白质片段、多肽、或低聚肽。在其他实施方案中,配体是抗体或抗体片段。在某些其他实施方案中,配体是结合至细胞表面分子的有机小分子。在一些实施方案中,有机小分子模仿用于靶细胞表面分子的天然结合配偶体的结构,并且竞争性结合至该靶细胞表面分子,而在其他实施方案中,该有机小分子非竞争性结合至靶细胞表面分子。
在一些实施方案中,膜渗透结构域是蛋白质、蛋白质片段、多肽、或低聚肽。在某些实施方案中,膜渗透结构域是具有约3至约35个氨基酸的多肽。
在实施方案中,所述膜渗透结构域是五邻体基底蛋白或其得自腺病毒的片段。五邻体基底蛋白通常在感染早期期间介导腺病毒(例如腺病毒血清型5)的细胞结合、进入和细胞溶质渗透。五邻体基底可包括RGD基元(Arg-Gly-Asp)。如本文所用,“PB”是指五邻体基底片段。
在一些实施方案中,有效负载结合结构域是蛋白质、蛋白质片段、多肽、或低聚肽。在某些实施方案中,膜渗透结构域是具有约3至约35个氨基酸的多肽。
在一个实施方案中,有效负载结合结构域是十赖氨酸基元,也称为“K10”。十赖氨酸基元包含十个赖氨酸残基。
在一些实施方案中,结合至有效负载结合结构域的有效负载结合的是核酸。在一些实施方案中,核酸选自由以下组成的组:双链脱氧核糖核酸(dsDNA),单链脱氧核糖核酸(ssDNA),核糖核酸(RNA),信使核糖核酸(mRNA),转移核糖核酸(tRNA),核糖体核糖核酸(rRNA),小干扰核糖核酸(siRNA),单链核糖核酸(ssRNA),和低聚核苷酸(无论单链或双链)。
在某些实施方案中,有效负载结合结构域向有效负载的结合是通过选自由以下组成的组的方法进行:静电相互作用、亲电子相互作用、亲水性相互作用(范德瓦耳斯力)、氢键或共价键。
在各种实施方案中,药物递送分子中的配体靶向癌细胞上的细胞表面分子。在实施方案中,细胞表面分子是癌细胞上的受体。
在实施方案中,本文所述的是药物递送分子,其包括靶向细胞表面分子的配体、膜渗透结构域和有效负载结合结构域,其中所述配体是内化蛋白B(InlB)或其片段或其变型,所述膜渗透结构域是五邻体基底蛋白或其片段并且所述有效负载结合结构域是十赖氨酸基元。内化蛋白B靶向细胞表面蛋白质c-Met。c-Met的天然配体是肝细胞生长因子(HGF)。HGF形成四聚物并且需要二硫键形成。得自核细胞增生利斯特氏菌(Listeria monocytogenes)的内化蛋白B也识别并且结合c-MET,但不四聚化或不需要二硫键形成。内化蛋白B可表达为融合蛋白质,并且该融合蛋白质也结合至c-Met。InlB未与HGF竞争。在一些实施方案中,包含InlB、五邻体基底蛋白和十赖氨酸基元的药物递送分子可能还包含细胞毒素剂,诸如咔咯化合物。咔咯化合物是卟啉样分子。这些化合物可螯合大量不同金属(诸如铁、镓、锰等等),非共价结合载体蛋白质,是细胞毒素,并且在无载体蛋白质情况下不能渗透细胞。
在其他实施方案中,配体靶向细胞表面分子CD4,或者CD19或CD20。在其他实施方案中,配体靶向人类表皮生长因子受体(HER)中的一个,例如HER2或HER3。在另一实施方案中,配体靶向整联蛋白。
在各种实施方案中,药物递送分子还包含治疗剂。治疗剂形成具有有效负载结合结构域的复合物。在各种实施方案中,治疗剂包括但不限于烷基化剂、抗代谢物、抗肿瘤抗菌素、有丝分裂抑制剂、皮质类固醇、细胞毒素剂或其组合。治疗剂与有效负载结合结构域之间的复合物可能是共价的或非共价的。在一些实施方案中,非共价复合物可能是经由范德瓦耳斯力、氢键、静电相互作用、疏水性/亲水性相互作用来形成。在一些实施方案中,治疗剂与有效负载结合结构域之间的相互作用可通过联结子介导。在一些实施方案中,治疗剂是阿霉素或咔咯化合物。
本文还描述了用于在有需要的受试者中治疗癌症的方法。所述方法包括鉴别需要治疗的受试者和提供包括药物递送分子的组合物以及向受试者施用有效量的组合物以便治疗癌症。在各种实施方案中,药物递送分子包括靶向细胞表面上的受体的配体,膜渗透结构域以及有效负载结合结构域,并且还包括本文所述的治疗剂。
本文还描述了用于在有需要的受试者中抑制癌症进展的方法。所述方法包括鉴别需要治疗的受试者和提供包括药物递送分子的组合物以及向受试者施用有效量的组合物以便抑制癌症进展。在各种实施方案中,药物递送分子包括靶向细胞表面上的受体的配体,膜渗透结构域以及有效负载结合结构域,并且还包括本文所述的治疗剂。
本文还描述了用于在有需要的受试者中预防癌症转移的方法。所述方法包括鉴别需要预防的受试者和提供包括药物递送分子的组合物以及向受试者施用有效量的组合物以便预防癌症转移。在各种实施方案中,药物递送分子包括靶向细胞表面上的受体的配体,膜渗透结构域以及有效负载结合结构域,并且还包括本文所述的治疗剂。
本文还提供了用于治疗、抑制或预防药物抗性癌症的转移(例如,抗EGFR酪氨酸激酶抑制剂的癌症)。所述方法包括鉴别需要治疗的受试者和提供包括药物递送分子的组合物以及向受试者施用有效量的组合物以便治疗、抑制或预防药物抗性癌症的转移。在各种实施方案中,药物递送分子包括靶向细胞表面上的受体的配体,膜渗透结构域以及有效负载结合结构域,并且还包括本文所述的治疗剂。在实施方案中,药物抗性癌症过表达选自由c-Met、HER2、CD4和CD20组成的组的受体。
本文还提供了向脑部递送治疗性化合物的方法。所述方法包括鉴别需要此类递送的受试者和提供包括药物递送分子的组合物以及向受试者施用有效量的组合物。在各种实施方案中,药物递送分子包括靶向细胞表面上的受体的配体,膜渗透结构域以及有效负载结合结构域,并且还包括本文所述的治疗剂。本文所述组合物令人意外地越过血脑屏障并且向脑部中细胞递送有效负载。因此,这些组合物独一无二适合于治疗脑部中的癌症(例如转移性癌症)。
在本文所述治疗性方法的各种方面中,组合物中的药物递送分子包括靶向细胞表面上受体的配体、膜渗透结构域和有效负载结合结构域,其中该配体是内化蛋白B(InlB)或其片段或其变型,该膜渗透结构域是五邻体基底蛋白或其片段并且该有效负载结合结构域是十赖氨酸基元。药物递送分子还包含治疗剂,诸如阿霉素或咔咯化合物。
疗法
本发明的另一方面涉及通过施用有效量的组合物来治疗癌症(例如白血病、骨髓瘤、B细胞淋巴瘤(霍奇金淋巴瘤和/或非霍奇金淋巴瘤)、脑癌、乳腺癌、结肠直肠癌、肺癌、肝细胞癌、肾癌、胃癌、胰癌、宫颈癌、卵巢癌、肝癌、膀胱癌、尿道癌、甲状腺癌、肾脏癌、癌肿、黑素瘤、头颈癌、脑癌、前列腺癌、雄激素依赖性前列腺癌和雄激素非依赖性前列腺癌),所述组合物包括与本文所述治疗剂复合的药物递送分子。在一些实施方案中,治疗剂可以是适用于治疗特定类型癌症的任何化学治疗药物。治疗剂可以是有机分子、生物分子(例如肽或核酸)、或其组合。在各种实施方案中,治疗剂包括但不限于烷基化剂、抗代谢物、抗肿瘤抗菌素、有丝分裂抑制剂、皮质类固醇、细胞毒素剂或其组合。在一个实施方案中,治疗剂是咔咯化合物。在实施方案中,治疗剂是siRNA分子。
在一些实施方案中,将包含有效量的与治疗剂复合的药物递送分子的组合物与诸如本文所述那些的一种或多种化学治疗剂一起施用。有效量的组合物和化学治疗剂可以顺序地或同时地施用。
在一些实施方案中,施用是全身性的。在一些实施方案中,施用是局部的。向受试者施用组合物的各种方式是本领域技术人员已知的。在本发明的所有实施方案的一些方面中,组合物是通过包括眼部、经口、胃肠外、静脉内、肌内、皮下、经皮肤、气道(气溶胶)、肺部、皮肤、局部、或注射施用的途径来施用。
可以与包含有效量的与治疗剂复合的药物递送分子的组合物一起使用来治疗癌症的另外的疗法包括但不限于外科手术、辐射、免疫疗法、疫苗或其组合。可以与包括施用有效量的包含有效量的与治疗剂复合的药物递送分子的组合物的疗法顺序地或同时地,施用所述另外的疗法来治疗癌症(例如,黑素瘤或卵巢癌肿)。
在一些实施方案中,化学治疗剂可以选自以下中任何一种或多种:细胞毒素抗菌素、抗代谢物、抗有丝分裂剂、烷基化剂、砷化合物、DNA局部异构酶抑制剂、紫杉烷、核苷类似物、植物生物碱和毒素;以及其合成衍生物。示例性化合物包括但不限于:烷基化剂∶苏消安,和曲洛磷胺;植物生物碱∶长春花碱,紫杉醇,多西紫杉醇;DNA局部异构酶抑制剂∶阿霉素,表柔比星,依托泊苷,喜树碱,托泊替康,依立替康,替尼泊苷,克立那托,和丝裂霉素;抗叶酸剂∶氨甲喋呤,霉酚酸,和羟基脲;嘧啶模拟物∶5-氟尿嘧啶,去氧氟尿苷,和阿糖胞苷;嘌呤模拟物∶巯基嘌呤和硫鸟嘌呤;DNA抗代谢物∶2'-脱氧-5-氟尿苷,阿非迪霉素甘氨酸盐,和吡唑并咪唑;和抗有丝分裂剂∶软海绵素,秋水仙碱,和根霉素。还可以使用包含一种或多种化学治疗剂(例如FLAG、CHOP)的组合物。FLAG包含氟达拉滨、阿糖胞苷(Ara-C)和G-CSF。CHOP包含环磷酰胺、长春新碱、阿霉素和强的松。在另一实施方案中,使用PARP(例如PARP-1和/或PARP-2)抑制剂,并且此类抑制剂是本领域众所周知的(例如,Olaparib,ABT-888,BSI-201,BGP 15(N-Gene Research Laboratories,Inc.);INO-1001(Inotek Pharmaceuticals Inc.);PJ34(Soriano等人,2001;Pacher等人,2002b;3-氨基苯甲酰胺(Trevigen);4-氨基-1,8-萘酰亚胺;(Trevigen);6(5H)-菲啶酮(Trevigen);苯甲酰胺(美国专利Re.36,397);和NU1025(Bowman等))。
如本文所述,在各种实施方案中,疗法包括例如辐射疗法。辐射疗法中所用辐射可能是电离辐射。辐射疗法还可以是伽马射线、X射线、或质子束。辐射疗法的实例包括但不限于:外线束辐射疗法、放射性同位素(I-125、钯、铱)的间质植入、诸如锶-89的放射性同位素、胸部辐射疗法、腹膜内P-32辐射疗法,和/或全腹部和骨盆辐射疗法。对于辐射疗法的总体概述,参见Heilman,Chapter 16:Principles of Cancer Management:Radiation Therapy,第6版,2001,DeVita等人编,J.B.Lippencott Company,Philadelphia。辐射疗法可作为外线束辐射或远距疗法来施用,其中所述辐射是从远端源引导出。辐射治疗还可以作为内部疗法或短距疗法来施用,其中辐射源被放置在身体内部接近于癌细胞或肿瘤块。还涵盖了使用光动力疗法,所述光动力疗法包括施用光敏剂,诸如血卟啉和其衍生物,维替泊芬(Vertoporfin)(BPD-MA),酞菁,光敏剂Pc4,去甲氧基-竹红菌甲素;和2BA-2-DMHA。
如本文所述,在各种实施方案中,疗法包括例如免疫疗法。免疫疗法可以包括例如使用癌症疫苗和/或敏化的抗原递呈细胞。免疫疗法可涉及用于对宿主进行短期保护的被动免疫,其通过施用针对癌症抗原或疾病抗原的预形成抗体来实现(例如,向肿瘤抗原施用任选地连接至化学治疗剂或毒素的单克隆抗体)。免疫疗法还可以集中于使用癌细胞系的细胞毒素淋巴细胞识别表位。
如本文所述,在各种实施方案中,,疗法包括例如激素疗法。激素治疗性治疗可包括例如:激素激动剂,激素拮抗剂(例如氟他米特、比卡鲁胺、三苯氧胺、雷洛昔芬、乙酸亮脯利特(LUPRON)、LH-RH拮抗剂),激素生物合成和加工的抑制剂,和类固醇(例如地塞米松、类视色素、类三角醇(deltoid)、倍他米松、考的索、考的松、强的松、去氢睾酮、糖皮质激素、盐皮质激素、雌激素、睾酮、孕酮),维生素A衍生物(例如全反式视黄酸、(ATRA));维生素D3模拟物;抗促孕激素(例如米非司酮、奥那司酮),或抗雄激素(例如,乙酸环丙孕酮)。
用抗癌疗法进行治疗的持续时间和/或剂量可能根据具体抗癌剂或其组合来变化。具体癌症治疗剂的恰当治疗时间将是熟练技术人员所了解的。本发明涵盖对各癌症治疗剂的最佳治疗日程表的连续评价,其中由本发明方法确定的受试者的癌症的基因签字是确定最佳治疗剂量和日程表的因子。
在各种实施方案中,用于确定抗癌疗法的预测效力的受试者是哺乳动物(例如小鼠,大鼠,灵长类,非人类哺乳动物,家畜,诸如狗、猫、母牛、马),并且优选是人类。在本发明方法的另一实施方案中,受试者未经历化学疗法或辐射疗法。在替代性实施方案中,受试者经历化学疗法或辐射疗法(例如,诸如采用顺铂、卡铂、和/或紫杉烷)。在相关实施方案中,受试者未暴露于超过受试者物种一般或平均遭遇的辐射或化学毒性剂的水平。在某些实施方案中,受试者必须采用外科手术来移除癌性或癌前组织。在其他实施方案中,癌性组织没有被移除,例如,癌性组织可能位于身体的不宜手术区域,诸如在生命所必需的组织中,或处于手术操作将对患者造成相当的伤害风险的区域中,或例如,所述受试者在移除癌性组织之前被给予抗癌疗法。
药物组合物
在各种实施方案中,本发明提供药物组合物,所述药物组合物包括药学上可接受的赋形剂以及治疗有效量的本文所述药物递送分子,以便治疗癌症(例如白血病、骨髓瘤、B细胞淋巴瘤(霍奇金淋巴瘤和/或非霍奇金淋巴瘤)、脑癌、乳腺癌、结肠直肠癌、肺癌、肝细胞癌、肾癌、胃癌、胰癌、宫颈癌、卵巢癌、肝癌、膀胱癌、尿道癌、甲状腺癌、肾脏癌、癌肿、黑素瘤、头颈癌、脑癌、前列腺癌、雄激素依赖性前列腺癌以及雄激素非依赖性前列腺癌)。在各种实施方案中,药物递送分子包括靶向细胞表面上的受体的配体,膜渗透结构域和有效负载结合结构域。药物递送分子还包括与本文所述药物递送分子复合的治疗剂。在各种实施方案中,药物递送分子递送治疗剂以靶向癌细胞。
“药学上可接受的的赋形剂”是指适用于制备药物组合物的通常安全、无毒且合乎需要的赋形剂,并且包括可为兽医学用途以及人医药用途所接受的赋形剂。此类赋形剂可为固体、液体、半固体,或在气溶胶组合物的情况下可为气体。
在各种实施方案中,根据本发明的药物组合物可被配制来通过任给药途径进行递送。“施用途径”可指本领域中已知的任何施用途径,包括但不限于气溶胶、经鼻、经口、经粘膜、经皮肤、胃肠外或肠内。“胃肠外”是指通常与注射相关的施用途径,包括眶内、眼内、输注、动脉内、囊内、心内、皮内、肌内、腹膜内、肺内、脊椎内、胸骨内、鞘内、子宫内、静脉内、蛛网膜下、囊下、皮下、经粘膜或经气管。通过胃肠外途径,组合物可呈用于输注或用于注射的溶液或混悬液形式,或呈冻干粉末形式。通过胃肠外途径,组合物可呈用于输注或用于注射的溶液或混悬液形式。通过经肠途径,药物组合物可呈片剂、凝胶胶囊、糖包衣片剂、糖浆、混悬液、溶液、粉末、颗粒剂、乳剂、允许控释的微球或纳米球或脂质囊泡或聚合物囊泡形式。通常,组合物通过静脉内地或腹膜内地注射来施用。对于本领域的技术人员而言,这些施用的方法使已知的。
根据本发明的药物组合物还可含有任何药学上可接受的载体。如本文所用的“药学上可接受的载体”是指涉及自身体的一种组织、器官或部分携带或运送目标化合物至身体的另一组织、器官或部分的药学上可接受的物质、组合物或媒介物。举例而言,载体可为液体或固体填充剂、稀释剂、赋形剂、溶剂或包封物质或其组合。载体的各组分必须是“药学上可接受的”,因为它必须与配方的其他成分相容。它还必须适用于与它可能与之接触的任何组织或器官接触,这意味着它必须不携有毒性、刺激、过敏反应、免疫原性或过度超过其治疗益处的任何其他并发症的风险。
根据本发明的药物组合物也可被包载、制片或以乳剂或糖浆形式制备以供口服施用。可添加药学上可接受的固体或液体载体以增强或稳定组合物,或有助于制备组合物。液体载体包括糖浆、花生油、橄榄油、甘油、盐水、醇和水。固体载体包括淀粉、乳糖、硫酸钙、二水合物、白土、硬脂酸镁或硬脂酸、滑石粉、果胶、阿拉伯胶、琼脂或明胶。载体还可以包括持续释放物质,如单独或与蜡组合的单硬脂酸甘油酯或二硬脂酸甘油酯。
药物制剂是遵循常规制药技术制备的,所述技术涉及研磨、混合、粒化和必要时压制(针对片剂形式);或研磨、混合和填充(针对硬质明胶胶囊形式)。当使用液体载体时,制剂将呈糖浆、酏剂、乳剂或水性或非水性混悬液形式。此类液体制剂可直接经口施用或填充至软质明胶胶囊中。
根据本发明的医药组合物可以以治疗有效量递送。精确的治疗有效量是就在给定受试者中的治疗疗效而言,组合物将产生最有效的结果的那个量。这个量将取决于多种因子而变化,所述因子包括但不限于治疗化合物的特征(包括活性、药物动力学、药效学和生物利用度)、受试者的生理状况(包括年龄、性别、疾病类型和阶段、一般身体状况、对给定剂量的反应性和药物类型)、制剂中一种或多种药学上可接受的载体的性质以及施用途径。临床和药理学领域的技术人员将能够通过常规实验,例如通过监测受试者对施用化合物的反应以及相应调整剂量来确定治疗有效量。对于其他指导,参见Remington:The Science and Practice of Pharmacy(Gennaro编,第20版,Williams&Wilkins PA,USA)(2000)。
在施用到受试者之前,可将制剂组分添加到药剂。液体制剂可能是优选的。例如,这些制剂组分可包括油、聚合物、维生素、碳水化合物、氨基酸、盐、缓冲剂、白蛋白、表面活性剂、增量剂或其组合。
碳水化合物制剂组分包括糖或糖醇,诸如单糖、二糖或多糖,或水溶性葡聚糖。糖或葡聚糖可包括果糖、右旋糖、乳糖、葡萄糖、甘露糖、山梨糖、木糖、麦芽糖、蔗糖、右旋糖酐、支链淀粉、糊精、α-环糊精和β-环糊精、可溶性淀粉、羟乙基淀粉和羧甲基纤维素,或其混合物。“糖醇”定义为具有--OH基团的C4-C8烃,并且包括半乳糖醇、肌醇、甘露糖醇、木糖醇、山梨糖醇、甘油、和阿拉伯糖醇。上述这些糖或糖醇可单独使用或联合使用。用量没有固定的限制,只要糖或糖醇在液体制备物中可溶。在一个实施方案中,糖或糖醇浓度在1.0重量/体积%和7.0重量/体积%之间,更优选在2.0和6.0重量/体积%之间。
氨基酸制剂组分包括肉毒碱、精氨酸和甜菜碱的左旋(L)形式;然而,可添加其他氨基酸。
在一些实施方案中,作为制剂组分的聚合物包括平均分子量在2,000和3,000之间的聚乙烯吡咯烷酮(PVP)或平均分子量在3,000和5,000之间的聚乙二醇(PEG)。
还优选在组合物中使用缓冲液以将冻干前或复水后溶液的pH变化最小化。可使用大多数任何生理缓冲液,包括但不限于柠檬酸盐、磷酸盐、琥珀酸盐和谷氨酸盐缓冲液或其混合物。在一些实施方案中,浓度为0.01至0.3摩尔。可添加到制剂的表面活性剂示于欧洲专利号270,799和268,110中。
另外,例如,组合物可通过共价轭合至聚合物来化学改性以增加其循环半衰期。优选聚合物以及将它们连接至肽的方法示于美国专利号4,766,106;4,179,337;4,495,285;以及4,609,546中,所述专利全文藉此以引用方式并入本文。优选聚合物是聚氧乙烯化多元醇和聚乙二醇(PEG)。PEG在室温下可溶于水,并且在一些实施方案中,具有1000至40,000之间、2000至20,000之间、或3,000至12,000之间的平均分子量。在一些实施方案中,PEG具有至少一个羟基基团,诸如羟端基基团。羟基基团可能被激活以与抑制剂上的游离氨基基团反应。然而,应理解,反应性基团的类型和量可能变化,从而实现本发明的共价轭合的PEG/抗体。
水溶性聚氧乙烯化多元醇也可用于本发明。它们包括聚氧乙烯化山梨糖醇、聚氧乙烯化葡萄糖、聚氧乙烯化甘油(POG)等等。POG是优选的。一个原因是因为聚氧乙烯化甘油的甘油主链是以单酸甘油酯、二酸甘油酯、三酸甘油酯天然存在于例如动物和人类中的相同主链。因此,这种支化在身体内不会必然地被视作外来作用剂。POG具有与PEG相同范围的分子量。用于POG的结构示于Knauf等人,1988,J.Bio.Chem.263:15064-15070,并且POG/IL C 2轭合物的讨论见于美国专利号4,766,106中,这两份文献皆以其全文藉此以引用方式并入本文。
液体药物组合物被制备后,可冻干以防止降解并保持无菌。用于冻干液体组合物的方法是本领域普通技术人员已知的。在使用之前,组合物可用无菌稀释剂(例如林格氏溶液,蒸馏水,或无菌盐水)复水,其可包括额外成分。复水后,使用对于本领域的技术人员已知的那些方法将组合物施用给受试者。
施用的剂量和模式将取决于个体。一般而言,施用组合物,以使得以1μg/kg至20mg/kg之间、20μg/kg至10mg/kg之间、1mg/kg至7mg/kg之间的剂量给予抗体。在一些实施方案中,其作为单次剂量给予,以将循环水平增加10至20倍并且在单次剂量之后持续4至6小时。也可在单次剂量之后使用连续输注。倘若如此,抗体可以以5μg/kg/分钟至20μg/kg/分钟、或7μg/kg/分钟至15μg/kg/分钟之间的剂量输注。
试剂盒
本发明还提供试剂盒来在有需要的受试者中治疗、抑制和/或预防癌症的转移。该试剂盒包括:组合物,该组合物包含与本文所述治疗剂复合的药物递送分子;和用于在有需要的受试者中治疗、抑制和/或预防癌症的转移的组合物的使用说明书。
试剂盒是包括至少一种本发明组合物的材料或组分的集合物。因此,在一些实施方案中,试剂盒含有包括与如上所述治疗剂复合的药物递送分子的组合物。
本发明试剂盒中配置的组分的准确性质取决于所述试剂盒的预定目的。在一个实施方案中,试剂盒是特别针对人类受试者配置的。在其他实施方案中,试剂盒被配置来用于治疗诸如但不限于农场动物、家畜和实验室动物的受试者的兽医学应用。
使用说明书可包括在试剂盒中。“使用说明书”通常包括描述在使用试剂盒的组分来实现所需结果时采用的技术的明确表述,以便治疗受试者中的中性粒细胞减少症、减轻其严重程度、抑制或预防。任选地,试剂盒还含有其它适用组分,如测量工具、稀释剂、缓冲剂、药学上可接受的载体、注射器或将易于由本领域技术人员认识到的其它适用附件。
可向从业者提供试剂盒中组装的以保持材料或组分的可操作性和效用的任何便利和适合方式储存的材料或组分。举例而言,组分可呈溶解、脱水或冻干形式;它们可提供在室温、冷藏温度或冷冻温度下。组分通常包含于合适的包装材料中。如本文所用的短语“包装材料”是指一种或多种用于容纳试剂盒的内容物(如本发明组合物等)的物理结构。包装材料是通过熟知方法构造的,优选提供无菌、无污染物的环境。如本文所用的术语“包装”是指诸如玻璃、塑料、纸、箔等能够容纳个别试剂盒组分的合适的固体基质或材料。因此,例如,包装可以是用于容纳合适数量的发明组合物,所述发明组合物含有由本文所述方法产生的具有唾液酸酶活性的催化活性抗体。包装材料通常具有指示试剂盒和/或其组分的内容物和/或目的的外部标签。
实施例
提供以下实施例以更好说明要求保护的本发明且不应解释为限制本发明的范围。就提及的特定材料而言,仅出于说明目的且不意图限制本发明。本领域技术人员可在不行使本发明的能力和不脱离本发明的范围下开发等效手段或反应物。
实施例1∶单链低聚核苷酸和合成mRNA的递送
合成mRNA的递送是对基因递送的改进替代方式。基因递送载体必须破坏细胞核以允许基因表达,而mRNA仅需要递送到用于翻译蛋白质产物的细胞质中。诸如将方法用于表达(例如)用于诱发体细胞中的多潜能的所谓山中因子(Yamanaka factor)。虽然传统“脂质转染法”可用于活体外递送mRNA,但这种系统和类似系统在活体内无效。
所公开的靶细胞渗透蛋白(HerPBK10)已证明了用于活体内核酸和药物递送的效力。还为基因和药物递送发展了相关蛋白质(PBK10)。HerPBK10经由与人类表皮生长因子受体(HER3)的相互作用来促进细胞的靶向结合和渗透,并且PBK10经由整联蛋白相互作用来促进细胞的靶向结合和渗透。
下文中,证明了HerPBK10和PBK10用于递送单链低聚核苷酸和合成mRNA的效用。
HerPBK10在MDA-MB-435细胞中运送标记的低聚核苷酸。为确定HerPBK10是否可以介导单链低聚核苷酸递送,采用HerPBK10(5μg)或Lipofectamine 2000(Invitrogen,Carlsbad,CA,USA)、商用转染试剂作为比较性对照物,在0.1M HEPES/Optimem I(Invitrogen;Carlsbad,CA,USA)中,室温下培育Cy3标记低聚核苷酸(50pmol)20分钟。将合成混合物加入MDA-MB-435细胞,其表达HER,并且在37℃下培育1h。细胞在室温下固定于4%PFA中15分钟,并且针对HerPBK10为免疫荧光法进行处理。用DAPI复染色细胞以鉴别细胞核。使用Leica SP2激光扫描共焦显微镜获得图像。正如所料,脂质体介导摄取导致低聚核苷酸定位在细胞内部(图1A)。重要地,HerPBK10在HerPBK10低聚复合物的结合和摄取期间与低聚核苷酸共存(图1B),从而HerPBK10介导低聚核苷酸向细胞中的运送。
PBK10介导mRNA递送。将编码GFP的1kb合成mRNA(图2A)用于测试PBK10介导向用于蛋白质表达的细胞中递送的能力。将来自由Lipofectin递送的mRNA的GFP表达与由Lipofectin递送的GFP表达质粒相比较。当由Lipofectin递送时,mRNA在HeLa细胞中以可由荧光显微术检测到的水平表达GFP(图2B)。PBK10和编码GFP的合成mRNA以PBK10:mRNA为20:1的体重比在室温下于HEPES-缓冲盐水(HBS)中混合约20分钟,并随后以约50至70%汇合度被添加至附着HeLa细胞。因此,PBK10形成具有mRNA的复合物,该复合物类似于先前发展PBK10来递送的基因递送复合物(图2C)。
通过首先评价在PBK10与mRNA之间制得的复合物的细胞结合,检验PBK10介导递送的能力。在HeLa细胞上在冰上培育PBK10-mRNA复合物,以促进受体结合而不是内化,随后所述细胞被洗涤以移除游离(未结合)复合物并且通过ELISA处理以使用针对PBK10的一级抗体鉴别细胞表面结合复合物。细胞表面结合的基于ELISA的检测表明复合物(PBK10+mRNA)结合至细胞(图2D)。在细胞上培育PBK10(无mRNA)之后,还为免疫荧光法单独针对PBK10处理细胞。为验证细胞结合复合物(E)的摄取,细胞如早前描述在冰上用复合物培育,洗涤,随后在37℃下温热以促进内化。细胞在温热之后指示时间点处固定,并且为免疫荧光法针对PBK10(绿色)进行处理。细胞用罗丹明鬼笔环肽与DAPI分别地复染色以鉴别肌动蛋白(红色)和细胞核(蓝色)。使用Leica SPE激光扫描共焦显微镜获得图像。该发现表明复合物经历向HeLa细胞内进行的时间依赖性内化(图2E)。在用PBK10-mRNA复合物培育细胞之后约24h时,固定一组单独的HeLa细胞并且为免疫荧光法针对GFP进行处理。该发现表明GFP表达可在PBK10介导的mRNA递送之后检测到,虽然频率很低(极少细胞)(图2D)。允许充分GFP表达(可被免疫荧光法检测)的PBK10:mRNA的最佳体重比是20(图2E)。这些发现总而言之表明有可能通过PBK10或HerPBK10来递送mRNA,因为两种蛋白质皆以类似方式与核酸相互作用。
实施例2∶靶向c-Met的蛋白质纳米构造物
受体酪氨酸激酶(RTK)、c-MET以及其内源性配体、肝细胞生长因子(HGF)有助于细胞迁移、形态发生分化、和三维管状结构的机化以及在正常组织发育期间的细胞生长和血管生成。然而,c-MET和HGF的调节异常可有助于肿瘤进展,并且在此类环境中,与大量人类癌症中的不良预后有关。
c-MET的细胞表面上升与耐药性相关,包括获得的对电信号阻断疗法的耐受性,并因此变为RTK靶向疗法的重要生物标记。然而,大多数靶向RTK的疗法被设计成抑制支持肿瘤存活的下游信号路径,初始响应此类治疗的肿瘤几乎普遍获得对信号抑制的耐受性,同时大量组体已内在地抵抗此类信号阻断疗法。
不需要信号抑制的肿瘤靶向策略可证明对c-MET阳性癌细胞更有效。可通过配体识别c-MET以触发附属治疗的的细胞摄取,从而省略对阻断信号的需要,解决这一问题。虽然HGF有潜力完成此举,但其对四聚体化和二硫键的需要对治疗学发展构成技术难题。一种用于c-MET靶向的替代性和潜在更优配体可能来源于导致食物中毒的细菌。
人类病原体(核细胞增生利斯特氏菌)结合c-MET,从而通过其称为内化蛋白的表面蛋白质来侵袭寄主细胞。特定而言,在c-Met结合之后,内化蛋白B(InlB)触发受体介导胞吞作用。InlB和HGF识别c-Met的不同区域,并且InlB不需要用于结合的四聚体化或二硫键。因此,InlB可能适宜于我们先前建立来用于靶向诸如人类表皮生长因子受体(HER)之类其他受体的用于肿瘤靶向的纳米生物策略。
PBK10是来源于腺病毒衣壳五邻体基底的重组蛋白,所述腺病毒衣壳五邻体基底可通过五邻体基底的膜渗透活性来介导进入细胞中的基因和药物递送。已表明,当融合至肿瘤特异性配体时,PBK10可靶向肿瘤细胞。在该研究中,InlB的受体-结合位点是作为与PBK10的重组融合体来产生,以产生新蛋白质(InlB-PBK10),其介导细胞毒素剂向c-MET阳性癌细胞的靶向递送。
该发现表明,InlB-PBK10可作为可溶解融合蛋白质产生,可溶解融合蛋白质识别各种肿瘤细胞系上的c-MET,并且在细胞结合之后经历迅速内化。InlB-PBK10形成具有毒性化合物(诸如咔咯)的直径约10至20nm的纳米簇,并且在细胞进入之后介导咔咯渗透入向细胞质中,导致肿瘤细胞死亡。因此,Inl-PBK10是用于毒性分子向MET表达肿瘤的靶向递送的新颖构造物。
背景
作为肿瘤生物标记的c-MET。间充质上皮转变因子或MET是首先作为激活癌基因发现的受体酪氨酸激酶(RTK)。用于MET、肝细胞生长因子(HGF)的内源性配体(又名成纤维细胞衍生细胞运动因子或分散因子(SF))通常激活MET以诱发细胞增殖、运动性、存活和分化路径。MET和HGF主要分别在上皮和间充质来源的细胞中表达。HGF和MET之间的旁分泌信号介导上皮-间充质相互作用,该相互作用调控组织生长和形态发生分化。正常组织中的HGF-MET信号有助于胚胎发生、器官发生、血管生成、伤口愈合和组织再生,而这种路径的异常信号是与肿瘤发展和进展、肿瘤细胞侵袭和转移相关。
c-MET由胺基(N)-端胞外结构域、跨膜片段和羧基(C)-端胞内激酶结构域组成。胞外区域由接近PSI结构域(存在于plexin、semaphorin和整联蛋白中)的胺基(N)-端Semaphorin(Sema)结构域以及4种免疫球蛋白(Ig)-样结构域组成,这4种免疫球蛋白(Ig)-样一起组成HGF的结合部位。通过HGF的受体结合,导致MET的二聚作用,激活了通过ERK1/2、AKT和STAT3、磷脂酰肌醇3-激酶(PI-3K)、Ras-Raf-MAPK和磷脂酶C的信号。配体触发的MET胞吞作用通过发动蛋白和网格蛋白依赖性路径发生,所述路径介导通过降解和再循环胞吞途径的运输(trafficking)。
c-MET信号的调节异常通过若干机制发生,包括有或者没有基因扩增的过表达和组成性激酶活化,激酶结构域突变,以及通过过表达HGF对c-MET的旁分泌/自分泌激活。c-MET的配体独立性激活还破坏正常HGF-MET信号,并且可由导致组成性二聚作用的突变以及缺氧条件引起。后者可激活MET的HIF-1α-诱发转录,导致蛋白水平升高,扩增HGF信号并且促进侵袭。
一般而言接受的是,跨肿瘤的各种RTK的异源表达是许多种癌症中耐受性的主要机制。旨在抑制一种特异性RTK的疗法常常因为其他RTK的上调或配体刺激而不成功,所述上调或配体刺激又维持对于细胞存活关键性的因子的信号,所述因子包括PI3K和促分裂原激活蛋白质激酶(MAPK)。研究中对这种耐受性机制的鉴别表明,对EGFR抑制剂的耐受性与非小细胞肺癌以及其他肿瘤(包括乳腺癌)中MET信号的补偿上调相关。总而言之,c-MET是治疗性干预的有效候选者,因为其与各种类型的癌症、不良预后和转移相关,并且可能是用于抗鉴别和靶向肿瘤的有用生物标记。
临床中目前靶向HGF/MET路径的疗法是由针对HGF(Ficlatuzumab)或MET(Onartuzumab)的抗体、或导向MET激酶结构域的小分子抑制剂(Tivantinib或ARQ197)组成。所有这些方法的问题在于它们对治疗效力的信号抑制的依赖,这可能造成先前提及的补偿性RTK串馈,促进耐受性的发展。存在着对使用组合疗法来阻断或破坏多个RTK、尤其EGF-R和MET的功能的日益增加的兴趣,条件是这两个受体之间有显著串馈。虽然测试这种方法的活体外研究已显示出对肿瘤细胞系诸如肺癌、乳腺癌、胃癌和结肠直肠癌的前景,但测试这个方法的临床试验尚在进行中并且还有不确定性。
细菌病原体蛋白质用作潜在c-MET靶向剂。内化蛋白B(InlB)是在人类细菌病原体(单核细胞增生性李斯特菌(L.monocytogenes;Lm))的表面上显示的蛋白质,并且通过结合至c-MET来促进Lm进入到非吞噬性细胞中。InlB是Lm内化蛋白的大家族的成员,所述Lm内化蛋白包含N端螺旋帽结构域,以及不同数目的富白氨酸重复(LRR),所述重复接近于免疫球蛋白样内重复(IR)区域。与其他内化蛋白不同,InlB结构还在C端含有B重复和3个GW模块。InlB321(InlB的能够以高亲合力结合至c-MET的片段)由称为Cap、LRR(蛋白-蛋白相互作用域)和IR区域的蛋白结构域组成。InlB321向MET的结合通过其两个结构域发生∶LRR和IR,其分别结合至MET的Ig1和Sema结构域。
InlB作为潜在肿瘤靶向配体比HGF具有若干优点。HGF是含有20个硫化物键的异源二聚蛋白,并且需要将前蛋白分裂成组装亚基,这可能使重组体生产变得复杂,尤其是作为向外源蛋白结构域的融合体时。相比之下,InlB321已经在大肠杆菌(Escherichia coli)中作为重组可溶解配体产生,并且可容忍向其他蛋白质的融合(图3A)。活体外研究已证明,InlB321肽可在MET结合之后触发受体介导内化,这有助于其作为纳米递送剂来使用。同样重要的是注意到,MET的HGF结合部位与InlB321的HGF结合部位之间不存在重叠(图3B);因此,用于结合至MET的这两个配体之间不存在竞争。
在目前研究中,通过将这种肽作为向Ad5五邻体基底(PB)的重组融合体来产生它,使其重塑,所述Ad5五邻体基底在羧基(C)端由十赖氨酸序列(K10)修饰。所得多结构域蛋白质(InlB-PBK10)包含用于以下的功能:运送阴离子装载物,如核酸或咔咯(由K10结构域介导);靶向结合和内化(由InlB321结构域介导);以及膜渗透和胞内运输(由PB结构域介导)。提供了针对以下的结果:InB-PBK10融合基因的构造,由这种重组基因编码的融合蛋白质的生产,针对该蛋白质结合至MET和进入细胞的能力进行表征,和估价其在活体外递送细胞毒素有效负载的能力。
结果
可构造重组基因以编码InlB-PBK10。产生InlB-PBK10基因构造物的策略涉及两步克隆法,所述两步克隆法必须包括InlB 321的聚合酶链式反应(PCR)扩增以克隆进入pRSET-A细菌表达质粒中,随后在InlB321序列的3’端处插入PBK10。为完成此举,设计低聚核苷酸引物以通过PCR将限制性内切位点引入InlB321中来将这种序列与PBK10接合以用于“框内”插入表达质粒pRSET-A中。
我们的正向和反向低聚核苷酸引物分别包含以下序列∶
5’-AGTGAGCTCGAGACTATCACTGTG-3’(SEQ ID NO:1)和5’-GTTGGTGACTTTCTCCCACCTTCACCACCTTCTCTAGATATCCATGGTAT-3’(SEQ ID NO:2)。
将限制性内切位点SacI引入正向引物,以将InlB321阅读框放置成与由pRSET-A编码的N端聚组氨酸序列毗连。BglII和KpnI限制性内切位点被引入反向引物中。SacI和KpnI被用于将InlB321插入pRSETA中,并且BglII被用于随后将PBK10插入InlB321编码序列的3’端。编码Gly-Gly-Ser-Gly-Gly-Ser(SEQ ID NO:3)氨基酸基元的序列被包括在反向引物中以在InlB321和PBK10之间掺入短的柔性联结子。使用高保真度聚合酶来确保不将突变引入InlB321PCR产物。
800bp InlB321PCR产物在SacI和KpnI限制性内切位点连接到pRSETA中,产生质粒构造物,pRSETA-InlB。PBK10随后被在限制性内切位点BglII和HindIII处插入pRSETA-InlB中。为此,在限制性内切位点BamHI和HindIII处从质粒构造物pRSETA-PBK10中切除PBK10,并且连接到两倍消化pRSETA-InlB中,产生pRSETA-InlB-PBK10构造物(图4A)。我们还生产了构造物pRSETA-GFP-InlB,其将InlB321编码为绿色荧光蛋白(GFP)的羧基[C]端融合以用于将来可能的活体外和活体内成像实验。该构造物通过消化先前用SacI和BglII描述的800bp InlB PCR产物来制得,并且将它连接到pRSETA-GFP质粒中,这将InlB插入物放置成与GFP的编码序列在框内(图4B)。所有构造物被两倍消化并且使用1%琼脂糖凝胶通过电泳来评估以确认预期条带的存在(图4C)。另外,所有3种构造物被测序以确认其同一性以及验证突变未被引入阅读框中。
重组蛋白InlB、InlB-PBK10和GFP-InlB可在细菌中产生。先前描述的所有3种质粒构造物(pRSET-InlB、pRSET-InlB-PBK10和pRSET-GFP-InlB)被转变成大肠杆菌菌株,BLR(DE3)pLysS,以用于随后的蛋白表达和纯化,如所述方法中描述。与更传统的(即BL21)表达菌株相反,该菌株更加容忍重复序列,因此实现获得包含C端聚赖氨酸的全长蛋白质的能力。同时,pRSET-A质粒将蛋白质编码为多组氨酸序列的N端融合。这种组氨酸(His)标记实现使用镍螯合树脂的亲合力纯化,并且通过抗His标记抗体提供用于识别的表位。
所有3种构造物在细菌中产生可溶解蛋白质(InlB、InlB-PBK10和GFP-InlB),所述可溶解蛋白质可使用镍-轭合树脂通过金属螯合物亲和色谱法分离,如所述方法中描述。抗His标记抗体识别所有3种蛋白质,其通过在约37kDa(InlB)、约100kDa(InlB-PBK10)和约63kDa(GFP-InlB)的预测分子量处使凝胶电泳变性来迁移(图5)。
c-MET的表面水平在不同肿瘤细胞系之间不同。表征InlB-PBK10的第一目标是确定该蛋白质是否识别肿瘤细胞上的c-MET。大部分针对与不同肿瘤细胞系相关的c-MET水平的文献是基于总c-MET蛋白质表达或者RNA表达,且因此不提供关于在细胞表面上显示的c-MET的水平的信息。因此,重要的是首先在可为我们所用的细胞系的图上确定c-MET的相对细胞表面水平。这经证明是最初的挑战,因为大多数可用于此类评价的抗体已针对c-MET的细胞溶质结构域被产生,并且用于评估c-MET表达和激活的机制。为了该研究,使用特别针对细胞表面c-MET研发的MET3抗体。我们使用细胞表面ELISA来测量c-MET在各种细胞系上的相对细胞表面水平。简单地说,这种方法实现我们在非透化细胞上测量表面上相对受体水平,并且可以以96孔格式执行,允许多重复制以及保存贵重试剂。我们的ELISA结果表明H1993(肺癌细胞系系)和MDA-MB-231(乳腺癌细胞系系)是具有最高的c-MET细胞表面水平的细胞系。RANKL(前列腺癌细胞系)和MDA-MB-435(乳腺癌细胞系)显示中等水平,而LN-GFP(前列腺癌细胞系)和Cos-7(非洲绿猴肾成纤维细胞)显示细胞表面c-MET的较低水平(图6)。
InlB衍生肽识别c-MET。为了评价靶向配体的受体特异性,我们使用荧光激化细胞分选(FACS)来测量InlB结合至c-MET阳性细胞的相对水平。测试两种肿瘤细胞系对InlB的结合∶一种肿瘤细胞系具有高表达的c-MET(H1993),并且另一种肿瘤细胞系具有低表达的c-MET(LN GFP)。与LN GFP相比,InlB显示出相应更高水平的对H1993细胞的结合(图7A)。为验证InlB是否特异性结合至c-MET,我们使用来源于MET的胞外、配体结合结构域的可溶解肽(MET肽)作为InlB结合的竞争性抑制剂。抑制剂与配体的等摩尔比率预测受体结合降低50%。一致同意,MET:InlB的等摩尔(1:1)浓度使InlB对H1993细胞的结合减少50%,指示c-MET选择性结合。(图7B)。
InlB-PBK10的细胞结合与c-MET水平相关,并且被游离配体竞争性抑制。在上述部分中,我们展示了InlB具有通过c-MET结合至c-MET阳性肿瘤细胞系的能力。下一个目标是测试当具体化为融合蛋白质的一部分时,通过InlB的受体结合是否将被影响。为评估此状况,我们使用细胞表面ELISA评价InlB-PBK10对表达高c-MET(MDA-MB-231)和低c-Met(Cos-7)的细胞系的结合。与表达相对低c-MET水平的细胞(Cos-7)相比,InlB-PBK10展示出对具有更高c-MET细胞表面表达(MDA-MB-231)的细胞的更高结合水平(图7C)。为进一步确认InlB-PBK10识别c-MET,将游离InlB配体用作竞争性抑制剂。在增加浓度的InlB存在下,InlB-PBK10展示出对MDA-MB-231细胞的结合减少,表明InlB-PBK10通过c-MET结合这种细胞(图7D)。
InlB-PBK10在悬浮液中显示对细胞上c-MET的结合。作为对c-MET识别的进一步确认,我们测试InlB-PBK10是否可在悬浮液中结合至细胞(与评价结合至附着细胞的上述试验相反)。我们执行拉下实验,其中我们在作为竞争性抑制剂的增加浓度的游离InlB配体存在下,在具有InlB-PBK10的悬浮液中培育c-MET阳性细胞系(MDA-MB-435)。选择游离InlB配体的浓度,以使得InlB-PBK10:InlB的摩尔比率为1:1、1:5和1:10。与细胞团块共沉淀的InlB-PBK10水平随后通过Western印迹使用对InlB-PBK10特异性的抗体(Ad5抗体,其识别五邻体基底)来评价。Inl-PBK10结合的水平随InlB的浓度增加而降低,其中结合几乎被完全抑制,从而验证InlB-PBK10在悬浮液中可识别和结合c-MET(图7E)。
InlB-PBK10内化到c-MET+细胞中。为检验细胞摄取之后InlB-PBK10的存活率和检测性,我们首先在4℃下培育具有3种不同细胞类型(MDA-MB-231、MDA-MB-435和H1993)的InlB-PBK10以促进受体结合而不是内化。为诱发胞吞作用,我们随后在37℃下经历指示时间培育细胞并且在特定时间点固定所述细胞至多30分钟。免疫荧光着色和共焦显微术表明在所有3种细胞系中,InlB-PBK10(以绿色示出)聚集在细胞膜上,并且在结合的首个5分钟内集合到细胞膜上的焦点中。InlB-PBK10随后经30分钟积累在核周区域中。这些数据表明InlB-PBK10可在细胞结合之后30分钟内在细胞内部内在化和积累(图8)。细胞在特定时间点固定至多30分钟,并且通过激光扫描共焦显微术成像。红色,肌动蛋白;蓝色,细胞核。比例尺,约10微米。
InlB-PBK10向c-MET+细胞递送毒性分子。为评估InlB-PBK10的内涵体溶解能力,我们评价其是否可介导自身先天不能够渗透细胞膜的细胞毒素剂进行细胞质进入。包含镓(III)(S2Ga或Ga-咔咯)的磺化咔咯是强荧光化合物,其可自发地与蛋白质组装。在没有膜渗透载体来将这些化合物递送到细胞中以触及毒性的细胞质靶标的情况下,所述化合物不可能跨越细胞膜。20-22Medina-Kauwe实验室先前已显示单独的Ga-咔咯是无毒的,但当通过HerPBK10递送到HER2+肿瘤细胞中时可促进细胞死亡。
为了该研究,将InlB-PBK10与Ga-咔咯混合以促进非共价组装,并且所得InlB-PBK10-Ga复合物通过透射电子显微术(TEM)和动态光散射来表征(图9A和9B)。TEM(图9A的右图)显示,与单独的InlB-PBK10(-S2Ga)相比,当Ga-咔咯(+S2Ga)添加至InlB-PBK10时,形成球形组装物。该图像显示所述蛋白和咔咯形成10至20nm直径的簇。用于测量粒度的动态光散射显示,单独的InlB-PBK10形成平均直径8.4nm的颗粒,而InlBPBK10-Ga直径为约16.4nm。
为检验InlB-PBK10-Ga是否可渗透细胞以及诱发c-MET阳性癌细胞系中的咔咯介导毒性,我们将MDA-MB-435细胞暴露于1μM、5μM和10μM的以下各者:InlB-PBK10、S2Ga和InlB-PBK10-Ga。在1μm浓度下,InlB-PBK10-Ga使细胞存活率减少60%,而1μm和更高浓度的单独S2Ga和单独InlB-PBK10不影响细胞存活率。这些数据确认InlB-PBK10具有内涵体溶解能力,并且表明该构造物能够递送诸如Ga-咔咯的细胞毒素剂到表达细胞表面c-MET的细胞中(图9C)。
为进一步评估InlB-PBK10的治疗性能力,评价该蛋白是否可用于递送更多主流化学治疗分子。阿霉素(Dox)是FDA批准的用于大量癌症类型的细胞毒素剂。如果此类药物可主要地靶向肿瘤细胞,则其就减少副作用而言将是有益的。在该方法中,Dox首先被插入到双链低聚中以形成DNA-Dox,其随后可结合至InlB-PBK10的聚赖氨酸(经由与DNA磷酸盐主链的电荷相互作用)并且形成我们称为I-Dox的复合物。I-Dox复合物(相对于Dox浓度5μM)是在所述方法中描述的条件下在指示细胞系上培育。InlB-PBK10是在与I-Dox复合物等同的浓度下培育。细胞存活率是通过在治疗之后24h的代谢(MTT)分析来评价。当与具有低c-MET表达的细胞(LAPC4)相比时,I-Dox复合物诱发具有高c-MET表达的细胞(H1993)显著的毒性(图9D)。H1993细胞首先用游离InlB配体处理以阻断c-MET,随后在如上相同条件下暴露于I-Dox。单独的InlB-PBK10似乎稍微而不是显著地减少细胞数量,表明细胞死亡的主要机制是通过Dox的递送发生作用。另外,游离InlB配体通过I-Dox抑制细胞毒性,表明I-Dox-介导的细胞毒性通过用于递送Dox的c-MET结合和进入来发生(图9E)。
实验程序
细胞。人类乳腺癌细胞系(MDA-MB-435*和MDA-MB-231)是得自国家癌症研究所(National Cancer Institute)。人类肺癌(H1993)和非洲绿猴肾成纤维细胞(Cos-7)细胞系是得自ATCC。人类卵巢癌细胞(A2780)是得自Sigma-Aldrich。前列腺癌细胞系(LNCaPNeo/RANKL,LNCaPNeo)由Leland Chung博士友情提供(Cedars-Sinai医疗中心)。MDA-MB-435和MDA-MB-231被维持在DMEM中(5%CO2下,10%v/v胎牛血清(FBS,Sigma-Aldrich),和1%v/v青霉素/链霉素(Sigma-Aldrich))。H1993和A2780细胞被维持在RPMI中(5%CO2下,10%v/v FBS和1%v/v青霉素/链霉素)。Cos-7细胞维持在DMEM/F12培养基中(5%CO2下,20%v/v非加热钝化胎牛血清(ATCC 30-2020)和1%v/v青霉素/链霉素(Sigma-Aldrich))。LNCaPNeo/RANKL、LNCaPNeo被维持在RPMI培养基中(10%v/v FBS,和1%v/v青霉素/链霉素和200μg G418重硫酸盐溶液(Sigma Aldrich G8168))。将200μg/mL潮霉素(GEMINI生物产物400-123)添加至LNCaPNeo/RANKL细胞的培养基以维持RANKL表达质粒。为了维持细胞系之间的等同汇合度,在某些分析中,根据其生长率来涂覆它们。
DNA构建体。靶向构造物(InlB-PBK10)是通过两步克隆法产生(图8A中概括),其中编码InlB和PBK10的序列顺序地一起被连接到蛋白表达质粒pRSET-A(Invitrogen,Carlsbad,CA,USA)中。编码内化蛋白B的aa 36-321的质粒构造物(pMET-30-InlB321)(CeBiTec),Bielefeld University,Germany)被用作聚合酶链式反应(PCR)扩增的模板,使用分别包含序列5’-AGTGAGCTCGAGACTATCACTGTG-3’(SEQ ID NO:1)和5’-GTTGGTGACTTTCTCCCACCTTCACCACCTTCATCTAGATATCCATGGTAT-3’(SEQ ID NO:2)的正向和反向低聚核苷酸引物。SacI限制性内切位点被引入正向引物中以用于框内插入p RSET-A中。BglII和KpnI限制性内切位点被引入反向引物以用于P BK10随后框内插入InlB编码序列的3’。反向引物还包含位于InlB和PBK10序列中间的编码柔性联结子(GlyGlySerGlyGlySer)(SEQ ID N O:3)。800bp InlB PCR产物用SacI和KpnI消化以连接到pRSET-A中。所得构造物pRSETA-InlB随后用BglII和HindIII消化以容纳与I nlB在框内的PBK10的插入。使用BamHI和HindIII限制酶从质粒p RSET-PBK10中切除PBK10,并且插入pRSETA-InlB的BglII-HindII I位点中,产生pRSET-InlB-PBK10构造物。
通过用SacI和BglII消化先前描述的800bp InlB PCR产物并且将其连接到pRSETA-GFP质粒中以用于InlB321的框内克隆,制得pRSETA-GFP-InlB构造物,其将InlB321编码为绿色荧光蛋白(GFP)的羧基[C]端融合。
细菌的蛋白表达和纯化。将BLR(DE3)pLysS(Novagen,Madison WI,USA)细菌转化体的过夜培养物1:50接种于包含0.5mg/ml氨苄西林和0.034mg/mL氯霉素和0.0125mg/ml四环素的LB中。当培养物在600nm光密度波长(OD 600)下达到0.6的吸光率读数时,培养物用0.4mM IPTG诱发并且在37℃下伴随振动进一步生长3小时。收获培养物并且压成丸。细胞团块再悬浮于溶解缓冲液(50mM Tris,pH 8.0,50mM NaCl,2mM EDTA,pH 8.0)中,并且通过添加0.1%Triton X-100和一个循环的凝固-解冻将其溶解,其中1mM苯基甲基磺酰氟化物(PMSF)在解冻期间添加。解冻之后,溶解产物接受10mM MgCl2,和0.01mg/mL DNase,并且溶解产物在室温下摇晃10分钟以允许在溶解产物恢复至冰之前消化基因组DNA,此后它们接受300mM NaCl和10mM咪唑。溶解产物转移至预冷却离心管,配平,并且在4℃下于预冷却转子中以39,000x g离心1小时。回收上清液,添加至用MCAC-10(50mM NaH2PO4,300mM NaCl,10mM imidazole,和1%Triton X-100,pH 8.0)预平衡的Ni-NTA树脂(Qiagen,Valencia,CA,USA),并且在冰上培育1小时。将包含结合蛋白的树脂洗涤,一次用20mL MCAC-10缓冲液在冰上10分钟摇晃,三次用MCAC-20(50mM NaH2PO4,500mM NaCl,20mM咪唑,和10%甘油,pH 8.0),随后用50mM Na-磷酸盐(pH 8.0)、300mM NaCl、250mM咪唑和10%甘油的2mL溶液洗提。蛋白质同时地缓冲交换到低盐缓冲液中并且通过超滤作用浓缩(Amicon Ultra Centrifugal Filters-Ultracel-50K(Millipore,Bedford,MA,USA),并且使用BioRad蛋白定量分析(BioRad Laboratories,Hercules,CA,USA)测量其浓度。
蛋白质检测。在本领域已知的不连续凝胶缓冲系统中执行变性聚丙烯酰胺凝胶电泳。使用设定在恒压(20V)下的BioRad半干转移细胞中的192mM甘氨酸、25mM Tris和20%甲醇,历时40分钟,将蛋白质电学转移到硝化纤维上。印迹用Tris-缓冲盐水中的3%牛血清白蛋白(10mM Tris,pH 7.5,150mM NaCl)阻断。印迹以阻断缓冲液中以1:1500稀释用抗RGS-His Tag抗血清(Qiagen)过夜培育。抗体-抗原复合物通过以下方式检测:用辣根过氧化酶(HRP)轭合二级抗体(Sigma,St Louis,MO USA)培育,随后与HRP底物和化学发光检测试剂(Thermo Fisher)反应,以及暴露于膜(Hyperfilm ECL;Amersham Pharmacia Biotech)。
用于c-MET细胞表面表达的基于ELISA的分析。为确定细胞系上的c-MET水平,使用以下细胞数量将细胞涂覆于96孔板中:每孔8x103MDA-MB-231、MDA-MB-435、A2780、LNCaPNeo/RANKL、LNCaPNeo以及9x103H1993和LAPC4。48小时后培养基被吸出,并且细胞简单用包含1mM MgCl2和1mM CaCl2(PBS+)的磷酸盐缓冲盐水(PBS)洗涤,随后在室温下(RT)用12分钟固定于PBS中的4%PFA内,随后用PBS+(200μL每孔)洗涤3次,然后在阻断溶液中(3%BSA/PBS,100μL每孔)在室温下培育1小时。抗c-MET抗体(小鼠单克隆抗c-MET,按0.87μg/mL使用;Knudson博士)一式三孔地添加(100μL每孔)并且在室温下培育1小时。细胞用PBS+洗涤3次,并且在室温下用HRP-轭合二级抗体以1:2000稀释培育1小时。细胞用PBS+洗涤3和用蒸馏水洗涤一次,并且根据制造商指示,将100μL TMB(eBioscience)溶液添加至各孔。所述板用底物在黑暗中培育30分钟(或直至看就蓝色显色),并且通过添加100μL的1N HC1来终止。在450nm处于Spectra MaxM2板式读数器(Molecular Devices Corp.)中测量吸光率。随后执行结晶紫分析以确定相对细胞数量。简单地说,细胞用PBS+(200μL/孔)洗涤一次并且用0.1%结晶紫(100μL/孔)在室温下培育15分钟。细胞用PBS+(200μL/孔)彻底洗涤4次,并且在室温下用95%乙醇(100μL/孔)培育10分钟。在490nm处测量吸光率。
细胞结合分析。为确定不同浓度InlB-PBK10对细胞的结合,按以下浓度将细胞涂覆于96孔板中∶每孔8x103LNCaPNeo/RANKL、LNCaPNeo和9x103H1993和LAPC4。在48小时后,培养基被吸出并且细胞简单用100μL缓冲液A(无血清DMEM,20mM HEPES,pH 7.4,2mM MgCl2,3%BSA)洗涤。细胞用包含指示浓度的InlB-PBK10的50μL缓冲液A在4℃下在冰上伴随搅动培育1小时。细胞用PBS+(200μL/孔)洗涤一次,并且经受先前描述的细胞表面ELISA(用于c-MET的细胞表面表达ELISA),并作以下更改∶为检测InlB-PBK10,板用一级抗体(RGS-His;Qiagen)以1:1500稀释过夜培育,并且用二级抗体(山羊抗小鼠,1:2000稀释)在室温下培育1小时。
为了竞争性抑制分析,在添加InlB-PBK10之前,使指示浓度的InlB在细胞上培育。特定而言,按先前描述涂覆的细胞系简单地用100μL缓冲液A洗涤,随后在包含1μM、5μM或10μM InlB的50μL缓冲液A中在4℃下伴随搅动培育1小时。细胞用缓冲液A(100μL/孔)洗涤一次,并随后用包含1μM InlB-PBK10的50μL缓冲液A以4℃下伴随搅动在冰上培育1小时。细胞用PBS+(200μL/孔)洗涤一次,并且处理以用于先前描述的表面结合的免疫检测,不同的是板用识别InlB-PBK10的五邻体基底域的抗体(Ad5抗体;Abcam)以1:5000稀释培育。板用二级抗体(山羊抗兔,1:2000稀释)在室温下培育1小时。如解释针对c-MET细胞表面表达ELISA解释的那样检测结合。
细胞存活率分析。MDA-MB-435细胞以8x103细胞每孔涂覆于96孔板中。48小时后的培养基被吸出,并且用包含不同浓度的InlB-PBK10-Ga和S2Ga(1、5和10μM)的30至50μL培养基在5%CO2培育箱中37℃下在摇摆器上培育4小时。4小时培育周期之后,添加额外的培养基来使每孔总容量达到100μL,并且细胞在不摇晃的情况下培育大约24小时。使用Promega CellTiter 96水性非放射性细胞增殖分析来测量细胞生活力。移除培养基,并且将100μL新鲜培养基添加至孔。根据制造商指示,将2.0mL MTS溶液与100μL PMS溶液混合并且添加20μL至各孔。板在37℃下经5%CO2培育,并且在添加MTT试剂之后1小时和2小时,在490nm处读取吸光率。随后执行结晶紫着色以确定相对细胞数量。简单地说,细胞用包含1mM Ca2+和1mM Mg2+的PBS+洗涤,随后用0.1%结晶紫(100μl/孔)在室温下培育15分钟并随后用PBS+(200μL/孔)洗涤4次。板用95%乙醇(100μL/孔)在室温下培育10分钟,并且在590nm处测量吸光率。
InlB-PBK10摄取/胞内运输分析。为了InlB-PBK10摄取分析,细胞以1x105细胞/孔涂覆在12孔盘中的盖片上并且生长2天。在治疗当天,将盘转移在冰上并且细胞针对所示实验根据以下方法进行处理。为研究摄取的时间过程,细胞用冷PBS洗涤两次,并随后将含8μg的InlB-PBK10的0.4mL缓冲液A添加至各孔。在冰上培育盘1小时并在4℃下摇晃以促进受体结合而不是内化,并随后用冷PBS洗涤细胞以移除未结合蛋白质。孔随后接受预温热完全培养基,并且以37℃下以5%CO2培育盘至指示时间点。个别盖片随后被移除,用PBS/1%MgCl2洗涤3次,随后在室温下用4%多聚甲醛固定于PBS中15分钟。盖片用PBS洗涤3次,随后在室温下,用PBS中50mM氯化铵培育5分钟,用PBS中0.1%Triton X-100培育5分钟,并随后用PBS中1%BSA阻断30分钟。盖片上细胞用一级抗体在4℃下过夜培育,用PBS洗涤3次并且在室温下在黑暗中用二级抗体培育1小时。在有指示的情况下,针对肌动蛋白和细胞核而复染色的细胞在二级抗体培育期间用Texas Red x-鬼笔环肽(Invitrogen T7471)以1:100稀释培育,随后在DAPI中以300nM最终浓度培育5分钟。鬼笔环肽、DAPI和一级和二级抗体全部烯释于PBS中1%BSA内。在二级抗体和DAPI治疗之后,盖片上细胞用PBS洗涤3次并且在Prolong Antifade封固剂(Molecular Probes,Eugene,OR,USA)中封固。为检测InlB-PBK10,以1:150稀释使用RGS-His抗体(Qiagen),并且将荧光团轭合山羊抗小鼠(FITC-轭合)以1:500稀释用于在488nm处荧光检测InlB-PBK10,在405nm处(DAPI)荧光检测细胞核以及在532nm处(Texas red)荧光检测F-Actin。
InlB-PBK10-Dox(I-Dox)组装。病毒衣壳衍生融合蛋白质InlB-PBK10遵循严格建立的程序与阿霉素组装。简单地说,通过将等于摩尔浓度的30碱基低聚核苷酸、LLAA-5(5'-CGCCTGAGCAACGCGGCGGGCATCCGCAAG-3')(SEQ ID NO:4)和其对应反向补充物LLAA-3混合到一起来制备互补低聚核苷酸双链。混合物沸腾5分钟并且历时30分钟冷却至室温以促进低聚核苷酸的退火。双链低聚糖以1:10摩尔比率(DNA:Dox)与Dox混合,并且在室温下(RT)培育30分钟,随后用InlB-PBK10以6:1摩尔比率的InlB-PBK10:DNA-Dox于HBS中培育,并且使用50k MW截止(mwco)过滤膜进行超滤(其通过用10%甘油预培育2小时至过夜来制备)以从不完全组装的组分中分离I-Dox。复合物以4000x g离心15分钟或直至体积减少≥80%。I-Dox中的Dox浓度通过依照Dox吸光率或荧光校准曲线(SpectraMax,Molecular Devices,USA)推断480nm处的测得吸光率或590nm处的荧光来确定(Ex:480nm)。Dox的浓度通过应用Beer-Lambert公式来计算∶(在λ最大/Dox消光系数下的吸光率)×稀释因子=浓度(M),使用的Dox系数为11,500M-1cm-1。实验中所用I-Dox剂量是基于I-Dox中Dox浓度计。
InlB-PBK10-Ga组装。通过用InlB-PBK10在黑暗中4℃下伴随轻轻搅动培育10x摩尔过量咔咯达1h,使InlB-PBK10与磺化镓咔咯(S2Ga)非共价地组装。通过50K MW截止旋转柱过滤器(Millipore Corporation,Billerica,MA,USA)根据制造商的过滤程序进行超滤来移除未结合的咔咯,并且复合物用PBS洗涤,直至滤液澄清。渗余物在过滤法中保持其亮绿色(指示咔咯颜料)。渗余物再悬浮于PBS中并且在咔咯的λ最大处测量吸光率以获得先前描述的咔咯浓度。然而,当结合至蛋白质时,S2Ga的λ最大(例如)从424nm移动至429nm,这未显著地改变对复合物中咔咯浓度的估计。
FACS分析。为评估InlB321的结合,将H1993和Ln GFP细胞培养48小时。在用PBS洗涤两次之后,通过在2mM EDTA中37C下培育5至10分钟分离细胞。将包含0.01%MgCl2和CaCl2(0.01%PBS+)的PBS添加至所述细胞,并且以2000rpm旋转4分钟。细胞再用0.01%PBS+洗涤3次,再悬浮,计数,并且分入Eppendorf管中。所述管以300x g旋转2分钟,并且用包含指示浓度InlB的PBS中3%牛奶再悬浮,并且在低温室中在冰上培育1小时。细胞随后用PBS洗涤4次并且在室温下用PBS中3%BSA内烯释150倍的RGS-His抗体培育30分钟。细胞洗涤4次,然后用二级抗体抗体(Alexa Fluor647、山羊抗小鼠IgG(H+L))在3%BSA中再悬浮,并且在室温下培育30分钟。细胞洗涤4次,用0.1%PFA培育15分钟,并随后再洗涤4次,并且用Beckman Cyan ADP FACS机器分析。
为验证由InlB321进行的细胞结合是通过c-MET进行,InlB321和MET肽(YCP2247SPEED BioSystems.Rockville,MD)在冰上在1xPBS中3%牛奶内培育30至40分钟,然后结合至细胞。结合分析如以下所述进行。
细胞拉下实验。使用1x PBS中2mM EDTA在37℃下伴随搅动5至10分钟,提升生长48小时的MDA-MB-435细胞。在以300x g将细胞旋转5分钟之后,将细胞洗涤一次并且再悬浮于缓冲液A+3%BSA并且分入4个Eppendorf管(2x 106细胞/管)。细胞再旋转并且再悬浮到500μL的缓冲液A+3%BSA中。在3个管中,添加0.4μM、2μM和4μM,并且未添加蛋白质至第四管;所有管皆在冰上培育1小时。在洗涤和旋转所有细胞以移除未结合InlB之后,将500ul缓冲液A中0.4μM的InlB-PBK10添加至所有4个管,随后在冰上摇晃2小时以促进受体结合而不是内化。如先前描述,细胞洗涤并且旋转3次。团块随后悬浮在50μL SDS-PAGE样本缓冲液中,并且通过SDS-PAGE/Wesrten印迹使用对InlB-PBK10特异性的抗体(Ad5抗体,其识别五邻体基底)以1:5000稀释来评估。
实施例3∶携带双侧胁腹MDA-MB-435肿瘤的nu/nu小鼠中的Inl-PBK10生物分布
背景
如图6中名称为“靶向c-Met的蛋白质纳米构造物”的先前实例所示,MDA-MB-435细胞显示出细胞表面上c-MET的标记水平。此外,如图7E所示,c-MET靶向蛋白质构造物InlB-PBK10通过被c-MET配体InlB竞争性抑制的相互作用来结合这些细胞,表明该构造物可通过c-MET结合这些细胞。图8示出InlB-BK10在受体结合之后进入这些细胞,并且图9C示出该蛋白质可介导膜不可渗透毒性分子(镓金属化咔咯)递送到这些细胞中,导致细胞死亡(从而还验证Inl-PBK10能够体内膜分裂)。
结果
在此,我们评估了用MDA-MB-435肿瘤评估Inl-PBK10在小鼠中的生物分布,以评价其是否能够在活体内在这些肿瘤上积累。为此,携带MDA-MB-435肿瘤的双侧胁腹异种移植的雌性nu/nu小鼠接受单次尾静脉注射Alexa680标记InlB-PBK10(2nmol蛋白质),并且使用640nm激发和700nm发射过滤器,通过在注射之后所示时间点的Xenogen成像来成像。整体动物成像显示出在注射之后4小时,动物中的荧光完全清除(图10A)。4小时时间点之后,将小鼠处死并且移除肿瘤和组织用于进一步成像。虽然肿瘤显示一些荧光累积,但显著比例的注入材料经4小时被排除至肾脏,同时可在肝脏检测到一些荧光(图10B)。剩余组织,包括心脏、脾、肺、脑和骨骼肌,未显示可检测荧光(图10B)。进一步研究将必需在较早和较晚时间点的组织收获来确定向肾的递送是否是迅速清除的结果,以及确定在竞争性抑制剂情况下确定生物分布以验证肿瘤递送经由c-MET发生。
实施例4∶脑转移性乳腺肿瘤的纳米生物靶向
人类表皮生长因子受体亚基3(HER3)的细胞表面水平升高与转移性乳腺肿瘤相关,包括扩散至脑部的那些乳腺肿瘤。然而,大量靶向疗法当前是使用来与周围性乳腺肿瘤斗争,这些分子向脑转移的递送受血脑屏障(BBB)限制。这通过转移至脑部的HER2+乳腺肿瘤来例证∶这些肿瘤虽然在中枢神经系统(CNS)外可由HER2抗体(诸如曲妥单抗)靶向,但不能由这些相同抗体触及,因为HER2亚基虽然存在于脑部内皮上,但不介导跨血管壁的抗体转胞吞。
在另一方面,HER3在配体结合时经历跨脑部内皮的迅速转胞吞,所述迅速转胞吞通常发生来介导对神经调节蛋白生长因子的递送以用于神经生长和维持。我们已研发出自组装纳米生物学颗粒HerMn,其使用HER3作为入口以用于毒性分子向肿瘤细胞中的靶向进入。HerMn是10至20nm直径的血清稳定颗粒,其由受体靶向细胞渗透蛋白HerPBK10组成。所述HerPBK10与磺化锰(III)咔咯(S2Mn或Mn-咔咯)非共价组装(图11)。HerPBK10的靶向结构域来源于配体(调蛋白α),其特异性地与人类表皮生长因子受体亚基3(HER3)相互作用并且诱发迅速受体介导胞吞作用。因为HER3是HER2的优选二聚作用配偶体,并且HER2-3异源二聚体在HER2+上很普遍,我们先前已展示HerPBK10可将治疗性和成像分子靶向至小鼠中的HER2+肿瘤,与化学疗法药剂(阿霉素)相比,在>10x的更低剂量下介导肿瘤生长消融,同时节约心脏和肝脏组织,并且没有可检测的免疫原性。HerMn的肿瘤靶向毒性通过线粒体膜分裂和对细胞骨架的超氧化物介导损害来发生。由于咔咯的顺磁性质,HerMn还可以通过磁共振成像(MRI)引出肿瘤选择性检测。
HerMn似乎在全身性注射于小鼠之后分布至脑部,另外显示出优先归巢和对皮下HER2+肿瘤的毒性。有趣地,由于其在正常组织上的抗氧化剂活性,锰咔咯已知显示出神经保护作用。在支持中,HerMn支持人类心脏病细胞活体外存活率。总而言之,有趣的是猜测HerMn可能具有对脑转移性乳腺肿瘤的靶向毒性,同时节约脱靶组织,因为其具有靶向能力和向正常组织诸如脑部和心脏提供有益保护效应的能力。
背景
脑转移是严重临床问题。乳腺癌转移至脑部的患者平均幸存小于1年。虽然已出现不断增长的靶向疗法用于治疗周围性肿瘤,但大多数都不适合于治疗脑转移,因为它们不能跨越血脑屏障(BBB)。对此的实例为使用单克隆抗体(曲妥单抗(Tz)),其是针对人类表皮生长因子受体亚基2(HER2)用于治疗HER2+乳腺癌。HER2+癌症与侵袭性肿瘤、对标准疗法的不顺应性、转移、和死亡率增加相关。转移至脑部的HER2+肿瘤不可能用Tz治疗,因为HER2虽然存在于脑内皮上,但不跨血管壁胞吞转运(transcytose)。三阴性乳腺癌(TNBC)是HER2-转移性以及脑转移性的,并且因为缺乏特异性细胞表面生物标记而具有甚至更少的选择。虽然拉帕替尼(Lapatinib;Lp)(HER2和EGFR的小分子酪氨酸激酶抑制剂(TKI))可以容易地透过细胞膜以靶向这两种肿瘤类型,但这些肿瘤可能部分地由于HER3升高而抗此类抑制剂。
结果
HerPBK10对HER3具有特异性。HerPBK10包含HER3配体的受体结合区域,调蛋白-α(氨基酸35-239,包含Ig样和EGF样结构域),融合至来源于腺病毒五邻体基底衣壳蛋白质的膜渗透部分(图11A)。HerPBK10初始设计为非病毒基因转移载体,在单个多畴蛋白质分子内包含有效负载组装、靶向、内化和体内渗透功能。因为HER3是HER2的优选二聚作用配偶体,并且HER2-3异源二聚体在HER2+上很普遍,已表明HerPBK10可将治疗性和成像分子靶向至小鼠中的HER2+肿瘤,与化学疗法药剂(阿霉素)相比,在>10x的更低剂量下介导肿瘤生长消融,同时节约心脏和肝脏组织,并且没有可检测的免疫原性。
HerPBK10对HER3的特异性由其结合至包含人类HER3的胞外结构域的固定化肽的能力支持(图12A)。这受到在活体外游离HER3肽情况下HerPBK10的预吸附的抑制(图12A)。相同肽还抑制对人类(图12B)和小鼠(图13B-C)HER3+细胞两者的结合。特别地,小鼠和人类HER3的配体结合结构域享有很高(94%)序列同一性(图13A)。虽然HER2-3异源二聚体在HER2+肿瘤细胞上很普遍,但异源二聚化抑制抗体(帕妥珠单抗(Pz))不阻止HerPBK10对细胞HER3的结合(图12B),从而表明HER2-3二聚作用并非HerPBK10结合所必要的。结合也不受HER4肽或β细胞素(其阻断HER4)抑制(图12B),从而表明HerPBK10是HER3特异性的。
在培养物中,来自HER2+患者和年龄匹配对照物的血清未阻止HerPBK10对HER2+细胞的结合(并且表明彼此之间无显著差异),与添加过量重组配体(+Her)用作竞争性抑制剂形成对比(图12C)。先前研究已表明HerPBK10在免疫能力小鼠中的重复剂量未产生针对该蛋白质的可检测中和抗体形成。此外,在培养物中,可识别HerPBK10的针对整体腺病毒产生的多克隆抗体未阻止受体结合至细胞。
HerPBK10与Mn-咔咯自组装以形成HerMn纳米颗粒。HerPBK10的羧基[C]-端包含十赖氨酸尾部(图11A),其可以介导对阴离子分子(包括咔咯)的亲电子结合。咔咯是与卟啉具有结构相似性的大环分子,并且同样地可以包含金属配体(图11B)。磺化咔咯是两亲的,可溶于生理溶液,并且可以通过非共价相互作用(包括亲电子和疏水性相互作用)自发地结合蛋白质。因为受带负电细胞膜的排斥,阴离子磺酸基团阻止非特异性细胞进入,从而实现经由载体蛋白质将咔咯引导到靶细胞中的可能性。我们已将HerPBK10与磺化锰(III)咔咯(S2Mn或Mn-咔咯)组合以形成称为HerMn的迅速自组装10-20nm直径颗粒(图11C-D)。这些颗粒包含结合至单个蛋白质(25-35咔咯/蛋白质)的多个咔咯分子,并且可以经得起高速超滤作用。这些发现与我们先前的研究和我们合作者的研究相一致,表明咔咯可以以可忽略解离结合于蛋白质袋(pockets)中,并且一旦结合至HerPBK10,则抵抗转移至血清蛋白质。
HerMn靶向HER3+肿瘤。在培养物中,HerMn显示出对HER2+/HER3+而不是HER2-/HER3-肿瘤细胞的靶向毒性,同时Mn-咔咯对细胞存活率无影响。数据示于图14中,其中各细胞系接受指示浓度的HerMn或S2Mn,并且在24小时后经由结晶紫(CV)着色评价存活率。每个浓缩物的N=3,来自3个单独实验。已表明,HerPBK10的靶向配体诱发受体结合之后的迅速胞吞作用。因为磺化咔咯不可能自主破坏体内膜,HerPBK10的五邻体基底部分实现胞吞摄取之后的有效膜分裂,以及向细胞质中的进入。
在一个实验中,细胞接受10μM S2Mn或HerMn,随后在24小时后接受HBSS中TMRM(30nM)。结果示于图15A中,其中对照物是PBS治疗。共焦荧光图像(图15B)示出在MDA-MB-435细胞上培育24小时之后,肌动蛋白(红色)和微管蛋白(绿色)通过HerMn(5μM)而超氧化物介导瓦解。S2Mn(5μM)、HerPBK10(与HerMn等同的蛋白质浓度)和PBS充当对照物。另外的细胞接受Tiron(5mM)1小时,然后HerMn治疗。蓝色,细胞核。比例尺=10μm
一旦处于细胞质中,HerMn瓦解(collapse)线粒体膜电势(图15A)以及通过超氧化物升高和对细胞骨架(图15B)的氧化损害使这些细胞骨架瓦解,与镓(III)咔咯(S2Ga或Ga-咔咯)一致。
人们发现,Ga-咔咯直接结合线粒体外部膜蛋白TSPO。图16中展示结果。可溶解重组TSPO蛋白用S2Ga在室温下以等同摩尔浓度(1μM)培育约20分钟,随后超滤以移除游离、未结合S2Ga。通过测量吸光率和荧光光谱,针对TSPO蛋白结合咔咯的存在,评估渗余物。在有指示的情况下,将PK11195用作用于TSPO上卟啉结合部位的竞争性抑制剂(图16A-B)。存在HerGa与TSPO原位相互作用的证据。在细胞用HerGa处理并且检验HerGa-介导线粒体分裂之前24小时,MDA-MB-435细胞用表达外源TSPO的质粒(作为内源性TSPO结合的竞争性抑制剂)转染,证据为线粒体中的红色荧光染料累积减少并且细胞质中绿色荧光积累(图16C-D)。
TSPO移位卟啉和其他代谢物到线粒体中以用于处理,与线粒体磁导率转变核孔复合物的组分相互作用,并且有助于细胞体内平衡。Ga-咔咯特异性识别TSPO上的卟啉结合部位(图16A-B),因为咔咯结合可以受PK11195竞争性抑制,这抑制卟啉结合至TSPO。重组可溶解TSPO于MDA-MB-435细胞中的过表达阻止线粒体膜电势的咔咯-介导分裂(图16C-D),表明咔咯与TSPO原位相互作用。因为Mn-咔咯显示与Ga-咔咯类似的线粒体膜分裂,很可能TSPO是Mn-咔咯的分子靶标。在异种移植小鼠肿瘤模型中,HerMn在全身性递送之后活体内归巢至肿瘤,绕过包括心脏在内的大多数正常组织(图17),并且以极低药理学剂量(0.008mg/kg)消融肿瘤生长(图18A)。Mn-咔咯还是顺磁的,从而可用于MRI。
已发现,除在小鼠中全身性递送之后积累于肿瘤中以外,与曲妥单抗(Tz)相反,HerMn可以分布至脑部(图17)注入无肿瘤小鼠的尾静脉中的HerPBK10还示出脑定位,而PBK10的全身性递送(其缺乏HER3靶向配体)未示出可检测的脑部递送(图19)。已发现,HerMn不但对人类心球衍生细胞(CDC)无毒,而且在逐步升高剂量与延长暴露的情况下,增加培养物中CDC存活率,这与阿霉素(其具有已知的心肌毒性)以及单独的HerPBK10相反,所述HerPBK10对肿瘤细胞或CDC不具有毒性或生长促进作用(图18B)。这支持在活体外和活体内的视神经病变模型中的先前发现,表明Mn-咔咯可以是神经保护性的。这似乎对Mn-咔咯是独特的,因为Ga-咔咯未展示此类神经保护作用。
一同地,这些发现表明HerMn给予正常组织有益作用,同时使毒性靶向肿瘤组织,尤其靶向具有升高HER3的脑转移。HerMn的靶向特异性使得这一点不那么相关,因为全身性颗粒的少数似乎分布至非肿瘤组织(图17)。但是,因为相对低的咔咯水平提供神经保护,以及肿瘤毒性(图18A),这种潜在双活性值得探索。
应用
HerMn具有递送毒性至脑转移性乳腺肿瘤同时节约脱靶组织的能力,因为其具有靶向能力以及向正常组织诸如脑部和心脏提供保护的能力。
实施例5∶使用小肠结肠炎耶尔森氏菌(Yersinia enterocolitica)侵袭素衍生肽靶向β-1整联蛋白
小肠结肠炎耶尔森氏菌是细菌病原体,其侵袭肠内上皮,尤其是肠内壁的集合淋巴结(Peyer’s patch),以及导致食物中毒。对肠内上皮的结合和进入是通过细菌侵袭素(Inv)蛋白与β-1整联蛋白的相互作用来发生,所述β-1整联蛋白主要表达在覆盖集合淋巴结的上皮细胞上。
包含编码侵袭素的核苷酸序列的质粒(pHIT123-Inv)被用作PCR模板,并且将低聚核苷酸引物设计来扩增编码β-1整联蛋白结合部位的最小序列(约600bp),同时将用于框内克隆的限制性内切位点引入到用于靶向递送的外源肽中。所用引物包括以下序列∶5’-ACAGAGCTCATAACCGGCATTAACGTGAAT-3’(SEQ ID NO:6);5’-CTGTGTGCGGAGCCGCAATAGGAATTCATC-3’(SEQ ID NO:7);和5’-GATGAATTCCTATTGCGGCTCCGCACACAG-3’(SEQ ID NO:8)。这些引物被用于向Inv插入物中扩增和添加SacI和EcoRI限制性内切位点。另外的引物包括以下序列∶5’-GTCCAAAACCAAGAAAAGGCGGAGCAGCTGTAC-3’(SEQ ID NO:9);5’-ACAATGACGCGTGTACAGCTGCTCCGCCTTTTCTTGGTTTTGGAC-3’(SEQ ID NO:10);5’-CCGCTGTGTGCGGAGCCGCAAGGAGGAACGCGTACACAC-3’(SEQ ID NO:11);5’-GTGTGTACGCGTTCCTCCTTGCGGCTCCGCACACAGCGG-3’(SEQ ID NO:12);和5’-ACACACGGATCCTTGCGGCTCCGC ACACAGCGG-3’(SEQ ID NO:13)。这些引物引入MluI或BamHI限制性内切位点以用于克隆到鼓起(Knob)或全长纤维构造物中。
外源肽编码腺病毒(Ad)衣壳纤维和鼓起蛋白质。该纤维勾划出从Ad衣壳伸出的各个触角样突出物,所述突出物介导一级细胞结合以开始感染,并且包含N端长纤维轴结构域以及随后的C端球状鼓起域。鼓起域与细胞表面受体特异性相互作用,并且作为在不存所述轴的情况下作为可溶解蛋白质产生。当融合至PBK10时,Inv是有效配体。
使用由Inv替换鼓起亚域的两种策略来将Inv序列插入该鼓起中,产生重组鼓起构造物:ABCJ-Inv和AGJ-Inv。重组蛋白是在细菌中从表达这些构造物的pRSET载体中产生,如N端GFP标记蛋白(GFP-ABCJ-Inv和GFP-AGJ-Inv)和未标记蛋白。在第三策略中,Inv被插入该鼓起的DG环中并且用于体外翻译中以产生S35标记蛋白。非变性凝胶电泳示出该重组蛋白维持形成类似于野生型可溶解鼓起的低聚物(二聚物和三聚物)(从而表明该Inv插入物未混淆融合蛋白质的三級结构)。在所有情形下,产生全长可溶解蛋白质。
还产生其中Inv将全长纤维蛋白质的鼓起替换的构造物。使用杆状病毒载体来在细菌中表达该蛋白质。产生第二纤维构造物以在全长纤维蛋白质的轴和鼓起域中间表达Inv,并且FactorX分裂位点被引入Inv和鼓起之间。
实施例6∶CD4的配体
来源于人类免疫缺陷病毒(HIV)gp120包膜蛋白的CD4结合结构域的氨基酸序列TITLPCRIKQFINMWQEVGKAMYAPPISGQIRCSS NITGLLLTR(SEQ ID No:5)足以结合至CD4。使用标准分子生物学技术获得该序列。编码SEQ ID NO:5的核酸序列(约132nt)被克隆到pBluescript(PSK)质粒中。溴化乙锭着色电泳凝胶示出通过BamH I-EcoRI消化从载体(约3kb)上切除了插入物(约132nt)。所得产物随后随后与诸如PBK10的外源肽框内插入以用于将此类递送蛋白靶向至表达CD4的细胞(即,“助手”T-细胞)。
- 还没有人留言评论。精彩留言会获得点赞!