口腔护理活性剂的包封的制作方法
本发明涉及一种包封水溶性口腔护理活性剂的方法。特别地,本发明涉及用于在口中受控释放水溶性口腔护理活性剂的制剂。
背景技术:
人的口腔,特别是牙齿和牙龈,通常需要用于例如牙齿增白、牙齿健康、牙龈健康和口臭的应用中的口腔护理活性剂。考虑例如抗牙斑剂、抗牙垢剂、抗龈炎剂、抗菌剂、漂白剂等。
此类试剂通常自牙膏和/或口腔冲洗液中给药。由于口腔的典型环境,例如存在唾液,本领域的标准困难在于来自牙膏和口腔冲洗剂的活性剂将在其施用后迅速降低浓度。因此它们不能长时间保护口部,并因此它们需要每天被施用若干次。
因此,在口腔健康护理中,越来越多的注意力转到以一旦施用到口中即允许随着时间以持续和受控的方式释放它们的制剂提供口腔健康护理剂。
这方面的一个背景参考文献为WO 2013/093877。其中描述了用于漂白剂的受控释放的包封系统。所述漂白剂(特别是作为过氧化氢源的过氧化脲)被包封在包含疏水性材料的壳中。胶囊通过喷雾冷却/凝结过程制备。其中将石蜡熔化,将组分加到熔融蜡中,将所得悬浮液装入经预热的注射器中,并让其自这里滴到冷表面上。虽然该方法成功地产生了过氧化脲微粒,但是希望提供更方便的方法。另外,考虑到口腔护理活性剂的水溶性,希望提供一种方法,其可对广泛的此类水溶性口腔护理活性剂具有更通用的适用性。
由此应考虑待包封的试剂是水溶性的并且所得的微胶囊用于口腔的水性环境中。因此,为了提供如上所述方法,期望使用油基体系,并使用疏水性聚合物来制备微胶囊。这些体系需要使用有机溶剂,为此,使用常规的氯化溶剂(如二氯甲烷)、丙酮或乙醇。然而,这些溶剂不适于产生用于口腔护理活性剂的通用包封体系,因为若干重要的此类试剂(如过氧化脲和氯己定)可溶于这些溶剂中。因此期望提供一种基于油包油(O/O)体系的包封水溶性口腔护理活性剂、但避免上述溶剂的方法。
技术实现要素:
为了更好地满足上述期望,在一个方面,本发明涉及一种通过包含疏水性聚合物的壳包封水溶性口腔护理活性剂的方法,所述方法包括(a)将所述聚合物溶解于选自甲酸乙酯、乙酸乙酯、乙酸甲酯、乙酸丙酯、乙酸丁酯、乙酸异丁酯、乙酸异丙酯、乙酸异戊酯及其混合物的溶剂中以形成聚合物溶液;(b)将所述口腔护理活性剂分散或溶解于所述聚合物溶液中;(c)将所述聚合物溶液与油性液体合并以提供包含所述聚合物溶液和所述油性液体的体系;升温以从所述溶液提取溶剂,并蒸发溶剂,从而允许聚合物颗粒固化。
在另一个方面,本发明提供了可自前述方法获得的微粒。
在又一个方面,本发明提供了选自甲酸乙酯、乙酸乙酯、乙酸甲酯、乙酸丙酯、乙酸丁酯、乙酸异丁酯、乙酸异丙酯、乙酸异戊酯及其混合物的溶剂用作包含包封在疏水性聚合物的壳中的水溶性口腔护理活性剂的胶囊的制备中所述聚合物的溶剂的用途,所述方法包括提取和/或蒸发所述溶剂从而允许聚合物颗粒固化的步骤。
附图说明
图1至8均为示出如实施例中所讨论的各种释放曲线的图。
具体实施方式
在一般意义上,本发明基于正确的认识,即甲酸乙酯和有限数量的其他酯表现出溶解疏水性壳聚合物,以及允许在油性液体(如液体石蜡)的帮助下发生相分离、然后蒸发溶剂的两种恰当性质。
本发明提供的体系特别适合于水溶性口腔护理活性剂的包封。应理解,用于包封水溶性口腔护理活性剂的壳应具有足够的疏水特性。因为,如果不是这样,则口腔护理活性剂的水溶性和口腔的水性环境(即,唾液)将导致试剂从任何亲水性包封体系中过早地冲掉。
口腔护理活性剂的水溶性意味着,与疏水性壳聚合物组合,所述试剂将不可避免地形成双相体系。这又对包封提出了挑战,因为所需要提供的体系应导致在活性物周围形成壳而不仅仅是活性物与聚合物的混合物。尽管本领域中存在若干方法,如油包油(O/O)乳液溶剂蒸发和相分离或凝聚,但挑战在于找到特别适合于口腔护理活性剂的恰当体系。本发明允许形成具有所需性质的包封物,所述性质包括小于300μm且优选小于100μm的粒度,其允许水溶性活性物以受控的方式在口腔中持续释放相当长的时间段(从数分钟到数天,取决于制剂和活性物的类型)。
在本发明的胶囊中使用的聚合物(包括共聚物和聚合物的组合)是疏水的。当与快速变得水溶胀并溶解于口腔中的亲水性聚合物相比时,疏水性聚合物在与口腔中的水或唾液接触时通常相对长期地保持其结构性质。
待在本发明中用作载体材料的一种感兴趣的聚合物为例如疏水性纤维素衍生物,如乙基纤维素和乙酸丁酸纤维素(CAB)。CAB和乙基纤维素可例如从Sigma Aldrich和Eastman Chemical获得。另一感兴趣的聚合物类型为乙酸乙酯和甲基丙烯酸甲酯的共聚物系列,其还具有低含量的带季铵基团的甲基丙烯酸酯。这种类型的共聚物是已知的,例如由Evonik生产的Eudragit RS(各种等级)。另一种感兴趣的聚合物为可广泛获得的聚(甲基丙烯酸甲酯)和聚酯如聚己内酯(PCL)。也可使用聚合物的混合物。
合适的油性液体不仅有液体石蜡,而且有可与溶剂混溶且不溶解聚合物的任何液体或混合物,如庚烷。甲酸乙酯:庚烷的合适比率为1:5至1:15,优选1:10。其他合适的油性液体包括例如矿物油十二烷、十六烷、肉豆蔻酸甲酯、月桂酸甲酯及它们的混合物。
口腔护理活性剂被给予到口腔中以便在牙齿、牙龈上或口内的其他表面上发挥其作用。口腔护理活性剂可为增白剂、抗牙斑剂、抗牙垢剂、抗龈炎剂、抗菌剂及它们的组合。这些试剂是技术人员已知的,并且本发明不取决于任何特定的试剂。相反,本发明提供了一种包封广泛的不同的此类活性剂的通用方法。特别感兴趣的试剂为漂白剂,如过氧化氢和过氧碳酸钠、或过氧化氢前体,例如过氧化脲。还特别感兴趣的是抗菌剂如氯己定或其他杀菌剂如三氯生和/或抑菌剂如氯化锌或柠檬酸锌。可根据本发明包封的其他感兴趣的试剂有抗牙垢剂,例如焦磷酸盐如焦磷酸四钠(TSPP)。还有利于包封的是氟化物,其通常因其防龋的能力而存在于洁牙剂和漱口剂中。合适的氟化物有例如氟化钠或单氟磷酸钠。
活性物的负载优选为5%至50%,更优选8%至33%,制剂中活性物:载体比率优选为1:6至1:1、更优选1:4至1:2。
本发明通常可根据两种路线实现。在第一种路线中,将聚合物溶解于溶剂中以制备聚合物溶液。随后,将一种或多种待包封的活性剂添加到聚合物溶液中。接下来,将聚合物溶液乳化到油性液体中,以形成包含一种或多种活性剂、疏水性聚合物和油性液体的乳液。由此,可添加乳化剂以降低两相之间的表面张力并制备具有较小尺寸的颗粒。然而,也可不使用乳化剂而制备微粒。
然后升高乳液的温度。通常,初始温度在高于0℃至室温的范围内,如18℃,优选5℃-10℃。然后通常升高温度至室温以上,优选20℃至35℃。这将使得溶剂被提取和蒸发,其导致颗粒沉淀和固化。
在第二种路线中,如第一种路线中一样,将聚合物溶解于溶剂中以制备聚合物溶液,并向聚合物溶液中加入一种或多种口腔护理活性剂。不是如上制备乳液,在第二种路线中,在没有乳化步骤的情况下加入油性液体。由此,油性液体,特别是液体石蜡,充当反溶剂。这将导致颗粒沉淀。其后,升高温度以提取和蒸发溶剂,从而使所形成的颗粒固化。因此,初始温度通常远低于室温,例如至多10℃,并升高温度至10℃-35℃。在使用具有相对较高沸点的溶剂的情况下,第二种路线是格外感兴趣的。在这种情况下,微粒的固化可通过相对于具有较低沸点的溶剂如甲酸乙酯或乙酸乙酯,增大反溶剂的量来实现。在这种实施方案中,另一步骤可包括向所形成的凝聚液滴中加入硬化溶剂(如己烷)以帮助移除溶剂,并从而使凝聚液滴固化。
另外,可在任一路线中使用较高沸点和较低沸点溶剂的组合。
不希望受理论的束缚,本发明人相信前述路线在颗粒形成的机理方面不同。在乳液路线中,在乳液中形成含有活性物的聚合物液滴,它们然后随着有机溶剂的扩散/蒸发而硬化。在使用反溶剂时,由于液相从含有壳材料的液相中分离而形成微粒,这导致壳材料的溶解度的降低及其沉淀,从而包封核材料。
因此,本发明在上述两种路线中涉及提供一种体系,其包含疏水性聚合物、溶剂、口腔护理活性剂和油性液体,例如液体石蜡,所述溶剂选自甲酸乙酯、乙酸乙酯、乙酸甲酯、乙酸丙酯、乙酸丁酯、乙酸异丁酯、乙酸异丙酯、乙酸异戊酯及它们的混合物。在第一种路线中,首先取得分离的相(即O/O乳液),自其可通过溶剂移除实现沉淀。在另一种路线中,产生分离的相以引起沉淀。两种路线中均发生升温以迫使溶剂移除,从而导致固化。
在又一个实施方案中,可向制剂中加入可溶胀的材料或胶凝材料。这在本发明的胶囊被混合到清漆制剂中(如用在牙齿美白中)的情况下是特别感兴趣的。
可加入口腔健康护理中惯用的其他成分,例如无定形磷酸钙的前体,其能够充当脱敏剂,特别是与牙齿漂白有关。也可使用其他脱敏剂。如果加入,合适的脱敏剂可包括例如碱金属硝酸盐如硝酸钾、硝酸钠和硝酸锂;和其他钾盐如氯化钾和碳酸氢钾。
其他试剂可为,例如,抗牙斑剂、抗牙垢剂、抗龈炎剂、抗菌剂、抗龋剂及它们的组合。
优选的抗龋剂为氟化物。氟化物可作为单独的组分提供,但优选包含在钙组分或磷组分中。合适的氟化物源包括氟化钠、氟化亚锡、单氟磷酸钠、氟化锌铵、氟化锡铵、氟化钙、氟化钴铵、氟化钾、氟化锂、氟化铵、氟化锌铵、氟化锡铵、氟化钙、氟化钴铵、水溶性胺氢氟化物或它们的混合物。含氟化物的组分优选以总体系的重量的至少0.001%、更优选0.01至12%、最优选0.1至5%的量包含氟化物源。
可包含在任一或两种组分中的其他可能的口腔健康护理活性剂有例如抗菌剂。这些包括例如酚醛树脂和水杨酰胺,以及某些金属离子如锌、铜、银和亚锡离子的源,例如以盐形式如氯化锌、氯化铜和氯化亚锡及硝酸银。当使用时,它们以本领域已知的少量存在。另外,在任一或两种组分中可存在任选的添加剂。这些包括例如保湿剂、调味剂、着色剂、抗牙斑剂、防污化合物、pH调节剂、赋形剂如柔软剂、防腐剂、其他类型的稳定剂如抗氧化剂、螯合剂、张力调节剂(如氯化钠、甘露醇、山梨醇或葡萄糖)、铺展剂和水溶性润滑剂如丙二醇、甘油或聚乙二醇。各自的浓度可由本领域技术人员容易地确定。
保湿剂包括水、多元醇如甘油、山梨醇、聚乙二醇、丙二醇类、氢化的部分水解多糖等。还设想单一保湿剂或组合。它们通常以例如制剂的至多约85%、更优选例如约15%至约75%的量存在。
在根据本发明的方法的各种实施方案中,可加入抗聚集剂如硬脂酸镁(MS)、硬脂酸钙或其他颗粒。特别地,在本发明中,可将此类试剂加到分散相或非常规地加到连续相中。本发明人惊奇地发现,可仅通过改变MS加到体系中的方式来调节口腔护理活性剂的释放曲线。不希望受理论束缚,本发明人认为当在乳液形成之前将MS分散在连续相中时,形成具有更紧凑结构的微粒。
此外,根据本发明制备的胶囊可根据需要涂布具有一个或多个另外的控释材料层。待用作涂层的一种特别感兴趣的聚合物为疏水性聚合物,其可溶于庚烷并可用作酯特别是甲酸乙酯的反溶剂。优选的疏水性聚合物为聚异丁烯。聚异丁烯从压敏粘合剂和密封剂或用于透皮药物递送系统的压敏粘合剂的制造中已知。它由FDA(美国食品和药物管理机构)批准用于口香糖和医疗装置生产,高度耐氧化并对小分子(例如,甲醇、水分和气体)具有非常低的渗透性。本发明人已惊奇地发现,聚异丁烯非常适合作为根据本发明的胶囊上的控释涂层。
在另一个方面,本发明提供了可通过前述方法获得的微粒。这些微粒可结合剩余(残余)溶剂或油性液体来识别。残余溶剂结合甲酸乙酯讨论,但类似地适用于其他的酯或酯的混合物。
微粒中溶剂或油性液体的浓度可通过以下方式来监测:收集给定量的微粒(在生产的不同阶段,直至并包括最终产物)、然后用适宜的溶剂如二甲亚砜、二甲基甲酰胺、乙醇或非极性溶剂如石油醚从微粒中提取所述溶剂和油性液体、并使用配备有FID的气相色谱仪或与质谱仪联用的气相色谱仪进行分析。
为了从所产生的微粒消除油性液体,将其用油性液体可溶于其中的溶剂洗涤并真空干燥。这些溶剂包括己烷、庚烷和石油醚。最终微粒中残余溶剂的定量分析可如欧洲药典“Identification and Control of Residual Solvents(2.4.24.),2010,European Pharmacopoeia(6th ed.)European Department for the Quality of Medicines,Strasbourg”中所述进行。
干燥后微粒中的残余甲酸乙酯如下测定:使用研钵和研杵将10mg自由流动的微粒压碎,与3mL HPLC级乙醇混合,并将混合物转移到玻璃小瓶(20mL)中。研钵和研杵用3mL乙醇冲洗三次并使总提取溶液达到10mL。然后用丁基橡胶塞和铝压接(crimp)密封件封闭小瓶,并用手动压接机密封,然后用实验室旋转混合器混合2小时。用0.22μm注射过滤器过滤样品溶液,并通过配备有火焰离子化检测器的气相色谱仪和氦气作为载气来测定甲酸乙酯的含量。
未知样品中甲酸乙酯的浓度基于校准曲线计算,所述校准曲线通过对用无水乙醇稀释的不同浓度甲酸乙酯获得的已知浓度的甲酸乙酯标准物的峰面积积分来构建。
表1:微粒中的残余溶剂
平均值±S.D.;n=3。
在又一个方面,本发明还涉及如上文所定义的溶剂作为胶囊制备中疏水性聚合物的溶剂的用途,所述胶囊包含包封在聚合物的壳中的水溶性口腔护理活性剂。所述溶剂因此选自甲酸乙酯、乙酸乙酯、乙酸甲酯、乙酸丙酯、乙酸丁酯、乙酸异丁酯、乙酸异丙酯、乙酸异戊酯及它们的混合物,并优选为甲酸乙酯或更优选乙酸乙酯。在实施这些溶剂的这种新用途时,制备胶囊的方法优选为根据如上所述的包封水溶性口腔护理活性剂的方法的一个或多个实施方案的方法。在该用途中,此方法包括蒸发溶剂从而允许聚合物颗粒固化的步骤,特别是在上述两种路线中的任一者中。
虽然已在附图和前面的描述中详细示意和描述了本发明,但是这样的示意和描述应视为示意性或示例性的而非限制性的;本发明不限于所公开的实施方案。
例如,可在一个实施方案中运行本发明,其中一部分水溶性口腔护理活性剂通过乳化步骤包封,而另一部分通过允许油性液体充当反溶剂来包封。
通过研究附图、公开内容和附随的权利要求,本领域技术人员在实践所要求保护的发明时可理解和实现所公开的实施方案的其他变型。在权利要求中,词语“包含”不排除其他要素或步骤,并且不定冠词“一个”或“一种”不排除多个或多种。在彼此不同的从属权利要求中述及本发明的某些特征的纯粹事实并不表示不能有利地使用这些特征的组合。
总之,我们因此公开了通过包含疏水性聚合物的壳包封水溶性口腔护理活性剂的方法。描述了两种路线,二者均涉及提供一种体系,其包含疏水性聚合物、溶剂、口腔护理活性剂和油性液体,所述溶剂选自甲酸乙酯、乙酸乙酯、乙酸甲酯、乙酸丙酯、乙酸丁酯、乙酸异丁酯、乙酸异丙酯、乙酸异戊酯及它们的混合物,所述油性液体例如为液体石蜡。在第一种路线中,首先取得分离的相(即O/O乳液),自其可通过溶剂移除实现沉淀。在另一种路线中,产生分离的相以引起沉淀。两种路线中均发生升温以迫使溶剂移除,从而导致固化。
下面结合实施例和附图进一步解释本发明。这些将例示本发明,但不限制本发明。
实施例1
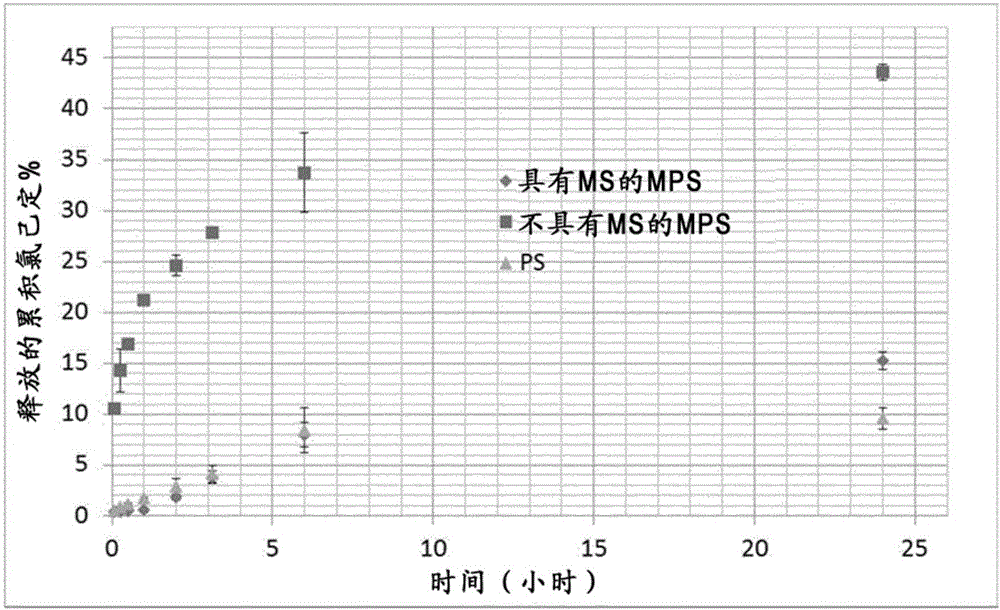
通过在室温下在玻璃密封容器中将6g乙酸纤维素溶解于40ml甲酸乙酯中过夜(14小时)来制备聚合物溶液。然后将氯己定二乙酸酯(1.5g)分散到预冷却聚合物溶液中,将其置于冰上,同时用磁力搅拌器以300rpm搅拌10分钟,然后将悬浮液倒入连接到循环水浴的夹套反应器中以将温度控制在5℃。基于重量计,活性物/聚合物比率为1/4。将通过用高剪切转子定子混合器(Silverson L4RT)在8000rpm下共混2分钟、同时用冰浴冷却该体系而制备的包含硬脂酸镁(抗聚集和释放速率控制剂(MS,1g))和聚甘油聚蓖麻酸酯乳化剂(PGPR,0.5g)的反溶剂液体石蜡(200mL)泵送(6mL/min)至先前制备的活性物-聚合物悬浮液中,同时用安装到IKA EUROSTAR顶置式搅拌器上的斜叶片叶轮于600rpm下混合,以促进聚合物在活性物周围沉淀。然后将反应器温度升至20℃并将体系保持在600RPM下搅拌2小时以进一步提取和蒸发甲酸乙酯,从而导致微粒固化。
此方法在本文中称为标准相分离法(PS)。微粒也通过改进的相分离法(MPS)制备,其中液体石蜡被分成2部分:第一部分80mL,含具有或不具有MS的PGPR,另一部分(120mL)由纯的液体石蜡组成。将含有活性物的预冷却聚合物溶液装入注射器中并加到含有由15%(v/v)甲酸乙酯(12mL)在5℃下饱和的第一部分反溶剂的反应器中。甲酸乙酯的该百分数基于溶剂在液体石蜡中计算出的溶解度来选择(表3)。混合物用斜叶片于600rpm下搅拌20分钟,然后将剩余的120mL纯液体石蜡泵入反应器中。然后如上所述将反应器的温度升至20℃,并通过在3000rpm下离心4分钟将所得微粒与液体石蜡分离。将油性微粒装载到烧结盘过滤漏斗中,用冷的正己烷洗涤三次并真空干燥过夜。得到自由流动的微粒粉末。
为测定氯己定的释放曲线,将50mg微粒装载到茶袋(t-sac,GmbH,德国汉诺威)中并置于250ml瓶中。将在37℃下预调节的100mL去离子水加到瓶中并置于在150rpm、37℃下的定轨振荡器(HT INFORS)中。在预定的时间取出5mL液体,用保持在37℃下的等效体积的新鲜介质替换,并使用分光光度计在255nm下参考标准曲线确定等分试样中存在的氯己定的量。
图1为示出生产方法和硬脂酸镁对氯己定的释放曲线的影响的图。“PS”指MS(硬脂酸镁)在制剂中的相分离。“具有MS的MPS”和“不具有MS的MPS”对应于通过改进的相分离制得的CAB微粒。反溶剂的泵送速率对于MPS方法为3mL/min,对于PS方法为6mL/min,因为较低的泵送速率导致聚合物团聚,这是由于在向反应器中加入足够的反溶剂之前,随着甲酸乙酯过早地蒸发,体系的粘度增大。
甲酸乙酯在石蜡油中的溶解度的测定
将12mL甲酸乙酯(0.917g/cm3)加到玻璃瓶中的18mL液体石蜡(密度0.830g/cm3)中。然后用丁基橡胶塞和铝压接密封件封闭该瓶并用手动压接机密封,然后用涡旋器将体系剧烈混合2分钟。将含混合物的瓶在温度受控的夹套烧杯中浸没24小时并允许形成两个不同的层,上层由甲酸乙酯和液体石蜡组成,下层仅由甲酸乙酯组成。用针头从上层取样,过滤并使用具有火焰离子化检测器(GC-FID)的气相色谱仪在表2中所示的条件下分析。混合物中甲酸乙酯的量从用无水乙醇稀释的不同浓度的甲酸乙酯获得的校准曲线推断。
表2:通过GC-FID量化甲酸乙酯的操作条件
表3:不同温度下甲酸乙酯在液体石蜡中的溶解度
实施例2
将4g乙酸纤维素溶解在20ml甲酸乙酯中。通过向70ml含0.35gPGPR乳化剂的液体石蜡中加入0.85g MS并用高剪切转子定子混合器(Silverson L4RT)于8000rpm下混合2分钟、同时用冰浴冷却该体系来制备连续相。然后向聚合物溶液中加入给定量的氯己定,置于冰上,以在搅拌下达到特定的活性物/聚合物比率(1/2;1/3)。用磁力搅拌器将混合物于300rpm下进一步搅拌10分钟,然后将其加到含有先前制备的连续相的反应器中,用30%(v/v)甲酸乙酯(即,21mL)饱和并冷却至5℃。使用六斜叶Rushton叶轮在400rpm下将混合物磁力搅拌2小时以产生乳液。将该油包油乳液逐渐加热至20℃,在此温度下搅拌16小时以允许甲酸乙酯向液体石蜡中扩散及其通过空气/液体界面的蒸发,从而导致微粒固化。通过在3000rpm下离心4分钟来从液体石蜡分离微粒,用冷的正己烷洗涤三次并真空干燥过夜。当MS被加到分散相中时,将乙酸丁酸纤维素溶解在15ml甲酸乙酯中,并使用含冰的超声浴、用另外5mL来分散MS(0.85g)达30分钟。然后如其中MS在连续相中的情况下那样获得乳液和微粒。
图2为示出硬脂酸镁(MS)在连续或分散相中对氯己定释放曲线的影响的图。MS-D和MS-C分别对应于硬脂酸镁在分散相和连续相中。1:3和1:2分别为1:3和1:2的活性物/聚合物比率。
实施例3
根据具有三个因子的二水平因素设计(表4)通过乳液溶剂蒸发制备负载有NaF的微粒,所述三个因子为:聚合物浓度(10%和20%,重量/体积)、溶剂蒸发温度(10℃和20℃)和连续相中的溶剂浓度(即,乳化开始时液体石蜡中甲酸乙酯的量,15%(10.5mL)和30%(21mL),体积/体积)。根据实施例2中描述的程序制备样品,其中MS在分散相中。向含1g或0.5g NaF的CAB溶液中加入用含冰的超声浴分散在5mL甲酸乙酯中达30分钟的0.85g硬脂酸镁以达到1:4的活性物聚合物比率。通过分别将2g和4g聚合物溶解于15mL甲酸乙酯中来制备总甲酸乙酯中含10%和20%聚合物的聚合物溶液。在加入MS之前将NaF分散到聚合物溶液中达7分钟并使用磁力搅拌器进一步搅拌混合物达3分钟以形成分散相。同时,在5℃下将由0.35gPGPR和70mL液体石蜡制成的连续相转移到反应器中并向体系中加入10.5mL或21mL冷的甲酸乙酯以达到15体积/体积%或30体积/体积%的浓度。然后将分散相加到连续相中并在5℃的温度下于600rpm下搅拌两小时以产生O/O乳液,随后如实施例2中那样将温度升高至20℃以便微粒硬化。样品也在相同的条件下制备,但NaF:CAB的比率为1:2(2g NaF,样品9)。
在从微粒提取分析包封的NaF之后根据公式1和2计算包封效率(EE)和负载效率(LE)。为此,向用研杵在玻璃研钵中压碎的25mg负载有NaF的微粒中加入50mL去离子水。用研杵不时地(4次)搅拌混合物以提取活性物一小时,然后用亲水性聚醚砜过滤器单元(Millex-GP,0.22μm)过滤提取物并弃去前15ml。将4.4mL滤液与4.4mL TISAB III缓冲液混合并使用氟化物选择性电极计(Eutech Instruments PTE LTD)结合标准曲线测定氟化物的浓度。
用粒度分析仪(Mastersizer Hydro 2000SM,Malvern Instrument Ltd,UK)测定微粒的粒度。
EE(%)=(实际活性物含量/理论活性物含量)X 100 公式1
LE(%)=(微球中活性物的质量/微球的质量)X 100 公式2
表4:聚合物浓度、连续相中溶剂体积和溶剂蒸发温度对负载有NaF的CAB微粒的包封效率和粒度的影响
通过O/O乳液溶剂蒸发制备微粒,活性物/聚合物比率为1/4,叶轮速度为600rpm。MS加到分散相中。
NaF微粒的释放研究在37℃下进行24小时或一周。将50mg微粒装载到茶袋(t-sac,GmbH,德国汉诺威)中并置于250ml瓶中。将在37℃下预调节的100mL去离子水加到瓶中并置于在150rpm、37℃下的定轨振荡器(HT INFORS)中。在预定的时间取出5mL释放介质,用5mL去离子水替换,并用经2.2ml去离子水和4.4ml TISAB III缓冲溶液稀释的2.2ml样品如上所述测定等分试样中存在的氟化物的量。
图3A为示出24小时内氟化物的释放的图,其关于聚合物浓度、连续相中溶剂体积和溶剂蒸发温度对氟化物释放曲线的影响。
图3B为类似的图,示出了一周内氟化物的释放。
实施例4
通过改进的相分离(样品Z18)或乳液溶剂蒸发(样品Z19和Z21)或(Z18)制备负载有二乙酸锌(ZnAc)的微粒。对于Z18,将6g CAB溶解在甲酸乙酯中达14小时(过夜)并将1.5g ZnAc分散在聚合物溶液中达3分钟,然后加入1g在5mL甲酸乙酯中的MS悬浮液。用磁力棒于300rpm下在冰上进一步搅拌该混合物,然后倒入含40mL液体石蜡、0.5g PGPR和6mL甲酸乙酯的冷反应器中。整个体系于5℃下共混20分钟,然后向反应器中泵入160mL液体石蜡(泵送速率6mL/min),将反应器温度升至20℃并搅拌2小时以允许如实施例1中那样实现溶剂提取/蒸发和微粒形成。Z19和Z21通过将1g和1.5g ZnAc分散到通过溶解3g CAB于20mL甲酸乙酯中达3分钟所制得的CAB溶液中来制备。然后将0.85g MS在5mL甲酸乙酯中的悬浮液加到混合物中并再搅拌两分钟。将混合物倒入含0.35g PGPR和15mL甲酸乙酯的70mL液体石蜡中并在600rpm、5℃下搅拌2小时以制备O/O乳液。将反应器的温度升至20℃,保持15小时以允许溶剂的蒸发和微粒的固化。然后如实施例3中那样获得负载有ZnAc的自由流动的微粒。
在与针对氯己定和氟化物相同的条件下进行锌负载微粒的释放研究,并在用释放介质适当稀释后用原子吸收光谱仪(AAnalyst 200,PerkinElmer)分析锌。
图4为示出锌从通过MPS和乳液溶剂蒸发制备的CAB微粒的释放曲线的图。
实施例5
如实施例2中所述制备负载有氯己定的聚己内酯微粒,其中MS在连续相中。将1g氯己定分散在通过将4g PCL溶解于15ml甲酸乙酯中所制得的聚己内酯(PCL)溶液中。连续相由70ml液体石蜡、0.35g PGPR表面活性剂和0.55g MS、0.25g MS或无MS的用高剪切混合器(Silverson L4RT)共混制成。然后如实施例2中那样获得乳液和负载有氯己定二乙酸酯的自由流动的微粒。
图5为示出氯己定从PCL微粒的释放曲线的图。
实施例6
通过溶剂蒸发和改进的溶剂蒸发如实施例1中所述的那样制备氯己定负载的CAB微粒。
将1.5g或3g氯己定分散在通过溶解6g CAB于40mL甲酸乙酯中达14小时(过夜)所制得的CAB溶液中以分别产生1/4和1/2的活性物/聚合物比率。首先在约70℃下将PIB溶解于液体石蜡(200mL)中,然后冷却至工作温度。向该液体石蜡中加入0.25g MS并用高剪切转子定子混合器(Silverson L4RT)于8000rpm下将混合物共混2分钟,同时用冰浴冷却体系,然后泵送(6mL/min)到活性物-聚合物悬浮液(1/4)中以如实施例1中那样制备微粒。还用Silverson如上共混40mL含0.25g MS的液体石蜡,并在如针对标准改进相分离那样在用6mL甲酸乙酯饱和之后于冷的反应器中与活性物-CAB(1/2)的悬浮液混合20分钟。然后将含有0.25g MS的160mL液体石蜡泵送(3mL/min)到反应器中,将反应器温度升至20℃并搅拌2小时以如实施例1中那样允许溶剂提取/蒸发和微粒形成。
图6为示出通过相分离(PS)或改进的相分离方法(MPS)在溶解于反溶剂中的PIB的存在下制得的负载有氯己定的CAB微粒在一周内的释放曲线的图。对于PS和MPS,活性物:CAB比率分别为1/4和1/2。
实施例7
使用丙酮或甲酸乙酯作为溶剂和Eudragit RS作为载体,根据表4中示出的制剂如实施例3中所述,通过乳液溶剂蒸发制备过氧化脲(CP)负载微粒。使用Precirol ATO 5(甘油二硬脂酸酯)或MS作为抗聚集剂并且在使用丙酮作为溶剂时不向液体石蜡中加入溶剂。
对于包封效率,将25ml HPLC级预冷却乙醇加到用研杵在置于冰上预冷却的研钵中压碎的25mg样品中。用研杵不时地(4次)搅拌混合物以提取过氧化氢(H2O2)。提取物用亲水性聚醚砜过滤器单元(Millex-GP,0.22μm)过滤,并用分光光度法在351nm下测定H2O2的量。
在释放介质(磷酸盐缓冲盐水(PBS),pH 7.4,在37℃下)中用100mg的每种CP负载微粒进行H2O2释放研究,其设计为模拟当接触牙齿时过氧化氢从颗粒的释放。将微粒分散在20mL释放介质中,置于150rpm下的定轨振荡器中。在预定的时间取出1mL等分试样,用释放介质适当地稀释,并用分光光度法在351nm下定量H2O2的量。立即用保持在相同温度下的等效体积的新鲜介质替换取出的体积。
图7在7A和7B中示出了使用丙酮(A)和甲酸乙酯作为溶剂(B)通过O/O乳液制备的Eudragit微粒的释放曲线的比较。图7A的样品分别用2重量/体积%、4重量/体积%和9重量/体积%的在丙酮中的MS配制1.5重量/重量%MS、3重量/重量%MS和7重量/重量%MS的制剂。图7B的那些用4重量/体积%的在甲酸乙酯中的MS配制。
表4:以Eudragit RS作为载体的CP负载微粒的不同制剂(样品编号#)的组成
表5:贮存在5℃下的冰箱中、使用丙酮(A)和甲酸乙酯作为溶剂(E2和E6)通过O/O乳液制备的Eudragit RS微粒的稳定性研究的实例
实施例8
使用甲酸乙酯作为溶剂和CAB作为载体,根据表6中示出的制剂(样品编号#)如实施例3中所述,通过乳液溶剂蒸发制备过氧化脲(CP)负载微粒。
表6(实施例8):负载有CP的CAB微粒的组成
图8为示出过氧化氢从负载有CP的CAB微粒的释放曲线的图。
- 还没有人留言评论。精彩留言会获得点赞!