具有高的负载物与表面活性剂比率的剥离了表面活性剂的胶束组合物的制作方法
关于联邦赞助研究的声明
本发明是在由国家卫生研究院授予的批准文号5dp5od017898的政府支持下进行的。政府对本发明具有一定权利。
相关申请的交叉引用
本申请要求于2014年7月2日提交的申请号为62/020,249的美国临时专利申请、以及2014年7月2日提交的申请号为62/020,233的美国临时专利申请的优先权,其公开内容通过引用并入本文中。
背景技术:
用于在生物系统内运输的疏水性分子的包封或螯合已成为广泛关注和研究的主题。疏水性药物包含相当大比例的目前使用的所有药物化合物。这些药物在水中的溶解度有限。对于需要精确给药的应用,口服递送可导致可变的生物利用度。在一些情况下,肠胃外给药是优选的途径。在一些情况下,通过改变溶液ph或通过添加适当的盐类来可以将疏水性药物弄成水溶液。在其它情况下,少量的增溶赋形剂如糊精或脂质是足够的。然而,对于仍更难溶解的许多化合物,需要其它赋形剂策略。这些通常涉及由表面活性剂和非水溶剂形成的制剂。非离子表面活性剂如聚氧乙烯蓖麻油(cremophorel)和聚山梨酯-80,通常用于母方(parentalformulation),但可能诱导包括过敏性超敏反应和神经毒性的不利副作用。非水溶剂具有引起溶血的可能性,并且在油的情况下具有引起肺微栓塞的可能性。对于可注射制剂,优选中性ph、等渗的水溶液。绕过这些问题的新药物递送系统有产生下一代制剂的可能,并且已经逐渐向临床开放。然而,迄今为止,描述的许多药物递送系统本身由相对大量的赋形剂形成,其本身可能携带副作用以及未知的长期安全性特点。因此,与表面活性剂溶液相比,当前纳米颗粒递送系统的药物与赋形剂的质量比或摩尔比可能不会明显更佳,并且通常为接近1:10的质量比(药物:赋形剂)。由于制剂复杂性和低药物负载能力,替代药物递送系统的临床应用受到限制。
疏水性分子也常用作成像剂。胃肠道的成像用于诊断中。然而,基于x射线辐射、磁共振和超声的用药程式受到了在安全性、可达性或缺乏足够的对比度方面的限制。例如,使用现有方法,动态肠道过程的功能性肠成像是不切实际的。
技术实现要素:
本公开基于我们的观察,当疏水剂与表面活性剂(如嵌段共聚物(例如,泊洛沙姆,例如可以由商品名
基于我们的研究,本公开提供了与已经剥离了表面活性剂的胶束制剂相关的组合物和方法,所述表面活性剂不是负载疏水剂的胶束的一部分。疏水剂在本文中也称为疏水性负载物。在所述组合物中,基本上所有的表面活性剂(例如泊洛沙姆)存在于胶束中,并且存在很少或不存在游离的表面活性剂。本公开还提供了制备所述组合物的方法和使用所述组合物的方法。
疏水剂可以是期望在生物系统中运输的任意疏水性分子。例如,疏水剂可以被递送到期望的位点(用于在位点释放),也可以在没有释放的情况下(例如当用于成像目的时)被运输通过位点。在一个实施方案中,疏水剂是药物。在一个实施方案中,疏水剂是光学成像造影剂。在各种实施方案中,组合物包含负载疏水药物的胶束和/或负载疏水光学造影染料的胶束,基本上由负载疏水药物的胶束和/或负载疏水光学造影染料的胶束组成,或由负载疏水药物的胶束和/或负载疏水光学造影染料的胶束组成。
低温处理导致去除了基本上所有未缔合的泊洛沙姆(当温度降低时泊洛沙姆与负载疏水剂的胶束不缔合)。例如,去除85%以上、90%以上、99%以上或99.9%以上的起始表面活性剂。剩余的泊洛沙姆存在于胶束中。
这些组合物表现出高稳定性和高负载。最终组合物中的疏水剂:泊洛沙姆摩尔比为3:1以上。例如,在疏水剂是药物的情况下,我们观察到疏水剂:泊洛沙姆摩尔比高达55:1,比使用其它增溶赋形剂的现有临床制剂的数量级高,其通常在1:10。在一个实例中,其中疏水剂是药物,我们观察到药物:泊洛沙姆摩尔比为7:1。在一个实施方案中,本发明组合物的疏水剂:泊洛沙姆比为60:1。
适于制备本发明胶束组合物的表面活性剂包括嵌段共聚物(例如泊洛沙姆)。在一个实施方案中,胶束由泊洛沙姆作为唯一的表面活性剂分子形成,并且所述胶束含有疏水剂。在一个实施方案中,表面活性剂是嵌段共聚物,所述嵌段共聚物至少包含亲水嵌段和疏水嵌段。在一个实施方案中,所述嵌段共聚物是三嵌段共聚物,如泊洛沙姆。所述组合物基本上不含未缔合的表面活性剂分子。术语“未缔合的表面活性剂分子”或描述特定的表面活性剂(例如“未缔合的泊洛沙姆分子”或“未缔合的泊洛沙姆127分子”)的相应术语意在表示一旦温度降低(如低于室温,例如至10℃至-20℃),不属于胶束的一部分的表面活性剂分子。未缔合的表面活性剂分子可以是非嵌合形式的表面活性剂分子,彼此松散地缔合(统称为“游离”表面活性剂),或形成空胶束,即其中没有疏水剂分子掺入的胶束。通过诸如膜过滤或透析的方法将胶束与小得多的非嵌合表面活性剂分离,并通过本领域已知的标准分析方法(如用于泊洛沙姆检测的比色硫氰酸钴方法)可以检测未缔合的表面活性剂。
这些胶束可以在用于肠胃外施用的溶液中制备而没有其它赋形剂。可选地,这些胶束可以存在于含有ph缓冲剂(例如柠檬酸盐或磷酸盐)和用于控制张力的成分(如盐水或蔗糖)的溶液中。胶束可以储存在水或含有高达4mnacl的高渗盐水溶液中。高渗盐水可在给药前稀释。胶束组合物可以与另外的药学上可接受的载体一起配制,所述药学上可接受的载体包括糖类、淀粉类、鲸蜡醇、纤维素、粉末状黄蓍胶、麦芽、明胶、滑石、油类、二醇类、单油酸甘油酯、多元醇类、聚乙二醇、乙醇、另外的乳化剂等。
本公开还提供了制备剥离了非缔合表面活性剂的组合物的方法。该方法包括将溶解在有机溶剂如氯仿或二氯甲烷或其它有机溶剂(包括例如乙醇,甲醇,四氢呋喃等)中的疏水剂分子与表面活性剂分子(如泊洛沙姆分子)接触以形成胶束,其中至少一些具有掺入其中的疏水剂分子。随后是有机溶剂的蒸发或部分蒸发(主动或被动),并且随后去除不包含于负载疏水剂的胶束中的表面活性剂分子。在一个实施方案中,通过降低温度来除去未缔合的表面活性剂分子,使得所有或基本上所有的未缔合的表面活性剂分子成为未嵌合的,随后除去未嵌合的表面活性剂分子(例如通过过滤方法如膜过滤方法)。可以根据需要重复或继续低温处理,直到除去所有可检测到的非缔合表面活性剂。由于没有更多的表面活性剂可以从胶束中去除,因此胶束基本上缺少未缔合的表面活性剂分子。所得组合物包含在本文中称为剥离表面活性剂诱导的“冷冻”胶束(“ss-infroms”)的胶束。组合物可以原样使用或可以浓缩。例如,负载疏水剂的胶束可以浓缩至高达150mg/ml的试剂。
在一些情况下,利用用高渗盐溶液、维生素e或辅酶q10共负载,或利用目的药物的脂肪酯化以使其具有足够的疏水性可以形成负载疏水剂的胶束。维生素e或辅酶q10共负载涉及将这些试剂掺入胶束中以改善稳定性或另一负载的疏水剂。高渗盐水的作用是使胶束外的溶液更具离子性,这具有将疏水性分子驱动到胶束中的作用。
术语剥离表面活性剂诱导的冷冻胶束“ss-infroms”、纳米颗粒或胶束可以互换使用。在一个实施方案中,其中胶束负载有疏水光学造影剂染料,所述ss-infroms也称为纳米粒子(nanonaps)。
在一个实施方案中,本公开的组合物包含多个胶束,其具有15至250nm(以及其间的所有整数纳米值)的尺寸(指胶束的直径)。在一个实施方案中,所述尺寸为20-120nm。在一个实施方案中,胶束中至少80-90%(以及其间的所有整数百分比值)在20-100nm或20-120nm(以及其间的所有整数纳米值)的范围内。
根据疏水性负载物选择,这些组合物可用于各种应用,包括例如药物递送和成像。在施用给小鼠后,本公开的胶束组合物在体内表现出安全性和功效。
在一个实施方案中,本公开提供了一种包含胶束的水性组合物,所述胶束包含泊洛沙姆并在其中掺入疏水剂,由此形成负载疏水剂的泊洛沙姆胶束,其中组合物中疏水剂:泊洛沙姆的摩尔比为至少2:1或3:1,并且其中组合物中至少90%或95%的泊洛沙姆形成负载疏水剂的胶束。在一个实施方案中,构成胶束的唯一表面活性剂是一种以上类型的泊洛沙姆(例如407、338或188;也分别称为普朗尼克(pluronic)f127、f108或f68),并且在这里分别称为f127、f108或f68),并且胶束中掺入了疏水剂作为负载物。泊洛沙姆可以是单一类型的泊洛沙姆,也可以是多种类型的泊洛沙姆。在一个实施方案中,制剂中至少96%、97%、98%或99%的泊洛沙姆分子作为胶束存在,其中掺入了作为负载物的疏水剂。泊洛沙姆在胶束中的掺入可以通过将温度降低至-20至10℃来量化,其导致未缔合的泊洛沙姆成为未缔合的,通过膜分离技术分离负载疏水剂的胶束,并对未缔合的泊洛沙姆进行定量。当疏水剂是药物(例如治疗药物)时,药物:泊洛沙姆摩尔比可以为7:1至55:1或7:1至60:1。当疏水剂负载物为成像造影染料时,染料:泊洛沙姆的摩尔比可以为3:1至10:1。在一个实施方案中,疏水剂以具有至少3、或3至11的辛醇-水分配系数(logp值)为特征。
本公开提供了制备本发明组合物的方法,其包括:将溶解在有机溶剂中的疏水剂(如x摩尔)与泊洛沙姆(如y摩尔)的水溶液接触,由此形成负载疏水剂的泊洛沙姆胶束;使没有形成负载疏水剂的胶束的泊洛沙姆分子成为单一泊洛沙姆单位。x和y可以根据需要选择。在一个实施方案中,x:y的比例为0.1:1至2:1。单位泊洛沙姆分子的形成可通过使组合物处于或低于泊洛沙姆的cmt的温度诱导。在各种实施方案中,降低的温度为0℃至25℃、0℃至20℃、0℃至15℃、0℃至10℃、或对于高渗盐水溶液为-20℃至0℃,并去除单位泊洛沙姆单位以得到负载剥离了泊洛沙姆的疏水剂的胶束组合物,其中泊洛沙姆分子起始量的至少85%被除去,疏水剂:泊洛沙姆摩尔比为3:1至55:1,以及组合物中90%以上的(例如95、86、97、98或99%或99.5、99.9或100%)泊洛沙姆存在于负载疏水剂的胶束中。组合物可以新鲜使用或储存备用。组合物可以以粉末或冷冻干燥形式储存,并且随后可以用水介质复原。
本公开还提供了药物递送的方法,其包括:制备如本文所述的负载疏水性的药物的胶束组合物,其基本上不含未缔合的泊洛沙姆(即,至少90%的泊洛沙姆作为负载疏水性负载物的胶束存在),并将该组合物施用于个体,使其被输送到期望的位置。在一个实施方案中,本公开提供了成像方法(例如胃肠道),包括:制备如本文所述的负载疏水性造影染料的胶束组合物,其基本上不含未缔合的泊洛沙姆(即,至少90%的泊洛沙姆作为负载疏水性染料的胶束存在),将组合物施用于个体,使得其被运输至并通过胃肠道,并且当组合物通过胃肠道运输时对胃肠道进行成像。成像可以在给药后立即进行,并且可以持续所需的时间段,或者可以在给药后一定时间后开始。对于药物递送目的,例如,组合物可以通过静脉内、腹膜内、肌内、局部、皮下或粘膜递送施用。为了成像目的如胃肠道的成像,组合物可以通过口服途径施用,并且为了其它成像目的,组合物可以通过静脉内、瘤内、腹膜内、皮下、皮内或肌内递送施用。
附图说明
图1:离心过滤洗涤后表面活性剂保留。在指定温度下旋转10%(w/v)的表面活性剂溶液,并使用1,6-二苯基-1,3,5-己三烯荧光法和标准曲线评估保留物中的表面活性剂。
图2:低温离心过滤后保留和溶解的八丁氧基-萘酞菁的染料吸光度。使用10%(w/v)的表面活性剂溶液溶解染料,然后进行3次离心过滤洗涤。在860nm处测量可溶性保留物的吸光度。
图3:维生素kss-infroms的产生。a)将维生素k溶解于二氯甲烷中并加入到10%(w/v)的f127溶液中。在有机溶剂蒸发后,对溶液进行低温离心过滤洗涤,并测定保留物中药物和pluronic的量。b)维生素kinforms的吸收光谱和照片(插图)。c)维生素kinfroms的粒径分布。d)与f68、f127形成的纯化维生素kinforms以及市场上混合胶束临床制剂的摩尔比的比较。
图4:盐辅助的环孢霉素ass-infroms的生成。a)将环孢霉素a溶解于二氯甲烷中,并加入到含有所示量的盐类的10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定药物产量。b)在洗涤步骤期间(在1mnacl中)环孢霉素和pluronic保留。c)环孢霉素informs的粒径分布。d)环孢霉素informs的吸收光谱。
图5:氟维司群ss-infroms的产生。a)将氟维司群溶解于二氯甲烷中并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定每个洗涤步骤的药物和pluronic的量。b)氟维司群informs的吸收光谱。
图6:胺碘酮ss-infroms的产生。a)将胺碘酮溶解在二氯甲烷中,并加入到含有所示量nacl的10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定药物产量。b)在洗涤步骤期间(在1mnacl中)胺碘酮和pluronic的保留。c)胺碘酮infroms的吸收光谱。d)胺碘酮infroms的粒径分布。
图7:伊维菌素ss-infroms的产生。a)将伊维菌素溶解于二氯甲烷中并加入10%(w/v)的溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定药物产量。b)伊维菌素informs的吸收光谱。b)伊维菌素informs的粒径分布。
图8:十一酸睾酮ss-infroms的产生。a)将十一酸睾酮溶解于二氯甲烷中并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定滞留物中药物和f127的量。b)十一酸睾酮ss-infroms的吸收光谱。
图9:胆钙化醇ss-infroms的产生。a)将胆钙化醇溶解于二氯甲烷中并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定滞留物中药物和f127的量。b)胆钙化醇ss-infroms的吸收光谱。
图10:视黄醇棕榈酸酯ss-infroms的产生。a)将视黄醇棕榈酸酯溶解于二氯甲烷中并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定滞留物中药物和f127的量。b)视黄醇棕榈酸酯informs的吸收光谱。将100mg视黄醇棕榈酸酯溶解在1ml二氯甲烷(dcm)中,并加入到含有2mnacl的10ml10%(w/v)f127中,并搅拌直至有机溶剂蒸发。通过用装配有蠕动泵(masterflexl/s)和管道(masterflex6434-16)的膜过滤(sartoriusvivaflow,1501008vs)进行未掺入的f127去除过程。在-7℃下进行去除过程,并将2mnacl溶液用于透析过滤溶液。为了使f127去除百分比最大化,将膜组件、管道和用于洗涤的溶液浸入乙二醇和乙醇(v/v=9:1)的混合物中,并使用干冰作为冷却剂。
图11:西罗莫司脂化物ss-infroms的产生。a)将西罗莫司脂化物溶解于二氯甲烷中并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定滞留物中药物和f127的量。b)西罗莫司脂化物informs的吸收光谱。
图12:米非司酮ss-infroms的产生。
a)将十一酸睾酮溶解于二氯甲烷中并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定滞留物中药物和f127的量。
b)十一酸睾酮ss-informs的吸收光谱。
图13:视黄醇ss-infroms的产生。a)将视黄醇溶解于二氯甲烷中并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定滞留物中药物和f127的量。b)视黄醇ss-informs的吸收光谱。
图14:辅酶q10ss-infroms的产生。a)将辅酶q10溶解于二氯甲烷中并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,将溶液进行低温离心过滤洗涤,并测定滞留物中药物和f127的量。b)辅酶q10ss-informs的吸收光谱。
图15:与维生素e共负载的紫杉烷inform形成的增强。将多西他赛或紫杉醇与所示量的维生素e(α-生育酚)一起溶解于二氯甲烷中,并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,对溶液进行离心以测定溶解的紫杉烷的吸光度。
图16:与辅酶q共负载的紫杉烷inform形成的增强。将多西他赛或紫杉醇与所示量的辅酶q一起溶解于二氯甲烷中,并加入到10%(w/v)f127溶液中。在有机溶剂蒸发后,对溶液进行离心以测定溶解的紫杉烷的吸光度。
图17:pluronic家族的表面活性剂适用于onc疏水染料的低温(4℃)洗涤,导致表面活性剂剥离以产生高度浓缩的oncss-infroms。
图18:a)在低温渗滤期间,游离的f127被去除,而维生素k1被完全保留。b)维生素k1ss-infroms的透射电子显微照片。c)差示扫描量热法测量显示表面活性剂剥离过程除去了所有由胶束化过程中传递的热指示的游离f127。d)基于少量与维生素k1胶束共负载的疏水性荧光团的forster共振能量转移(fret)测定揭示,负载物被锁定在动力学冷冻胶束中,而没有表面活性剂剥离前后的胶束间交换。e)维生素k1ss-infroms与临床制剂相比表现出高的药物与增溶剂摩尔比。f)基于凝血时间,维生素k1ss-infroms有效地作用抵抗口服华法林对小鼠的影响。
图19:在维生素k1洗涤过程中,基于在滤液中不存在任何可检测到的表面活性剂,从保留物中除去所有游离f127。
图20:a)高渗盐水增强了负载环孢菌素a的胶束的产率,b)f127可以在较低温度下有效剥离,c)剥离了表面活性剂的环孢菌素a胶束与临床制剂相比表现出高摩尔比,d)剥离了负载环孢菌素a的表面活性的胶束有效地用作免疫抑制剂。
图21:a)高渗盐水通过盐提高了负载十一酸睾酮胶束的产率,b)十一酸睾酮ss-infroms与临床制剂相比表现出更高的摩尔比。
图22:a)盐增强卡巴他赛informs的产量,b)辅酶q10改善ctxinfroms在稀释时的稳定性,c)ctxss-infroms与临床制剂相比表现出高摩尔比,d)剥离了负载ctx的表面活性剂的胶束有效地治愈了裸鼠皮下miapaca-2肿瘤,其中在第0天和第4天两次静脉内注射30mg/kg(由箭头标记)。
图23:不可交换的f127-萘酞菁冷冻胶束的形成。a)具有不同疏水性的染料加入到10%(w/v)f127的水溶液中,然后对20mm胆酸盐透析24小时后染料的保留。mb=亚甲基蓝,qr=喹哪啶红,6g=罗丹明6g,ir780=ir780碘化物。b)所用的萘酞菁的化学结构。bnc:x1=2h;x2=t-bu;x3,x4=h。vbnc:x1=v=o;x2=t-bu;x3,x4=h。znbnc:x1=zn;x2=t-bu;x3,x4=h;onc:x1=2h;x2=h;x3,x4=o-(ch2)3ch3。酞菁不含外苯:bpc:x1=2h;x2=t-bu;x3,x4=h。vbpc:x1=v=o;x2=t-bu;x3=n(ch3)2,x4=h。
图24:温度介导的cmc转换产生不含表面活性剂的纳米粒子(nanonaps)。a)纯化的纳米粒子的产生。f127peo嵌段和ppo嵌段显示为链,并且nc染料显示为填充的封闭结构。b)f127保留作为在4℃(黑色)和25℃(红色)下的离心过滤洗涤的函数。对于n=3,得到平均值±标准差。c)f127-溶解的染料保留作为在4℃下对于nc(黑色)和亚甲基蓝(红色)的离心过滤洗涤的函数。对于n=3,得到平均值±标准差。d)通过水中动态光散射的纳米粒子粒径分布。e)干燥纳米粒子的负染透射电子显微照片。比例尺,50nm。f)将由2mgonc形成的纳米颗粒冻干后,来自浓缩的重构的纳米粒子(黑色)或脂质体(红色,1:19摩尔比nc:脂质)的等效吸光度。插图显示放大的脂质体吸光度。
图25:在超高光密度下多光谱纳米粒子没有峰值波长漂移。a)将由bpc(蓝色)、znbnc(深绿色)、bnc(浅绿色)或onc(青铜色)形成的纳米粒子的吸光度标准化。b)纳米粒子在水中的照片。从左到右:bpc、znbnc、bnc和onc。c)在高光密度下的吸收峰值波长漂移。在10μm路径长度比色皿中测量浓缩溶液,并与在水中1000倍稀释液相比较。将指示的纳米粒子与吲哚菁绿(icg)和亚甲基蓝(mb)进行比较。对于n=3,得到平均值±标准差。
图26:纳米粒子口服后安全通过肠道。a)在37℃下在模拟胃液(红色)或模拟肠液(黑色)中透析的onc纳米粒子的保留。对于n=3,得到平均值±标准差。b)在粪便(黑色)和尿液(红色)中排出100光密度的onc纳米粒子(“od”-一个od定义为用标准1cm路径长度测量在1ml溶液中产生1个吸光度所需的纳米颗粒的量)。对于n=3只小鼠,得到平均值±标准差。c)在粪便(黑色)和尿液(红色)中排出100od的亚甲基蓝(mb)。对于n=3只小鼠,得到平均值±标准差。d)对照小鼠(左)或灌胃100od的onc纳米粒子24小时后的小鼠(右),用苏木精和曙红染色的肠部分。绒毛和隐窝是完整的,没有炎症细胞的流入。比例尺,100μm。
图27:使用纳米粒子对肠道进行无创解剖性和功能性pa成像。a)利用单个换能器pa系统灌胃100od的znbnc纳米粒子后,纳米粒子的pa最大强度投影(mip)。红色箭头显示纳米颗粒运输。b)深度编码的pamip的肠可视化znbnc纳米粒子。c)在灌胃100od的onc纳米粒子后,具有pa信号(颜色)以及同时的us(灰色)采集的实时多模式小鼠肠横断面。d)肠中纳米粒子运动。黑色箭头显示流入,并且白色箭头显示流出。e)目的肠道区域分析。一阶导数零交叉提供最大纳米粒子流入(黑色三角形)和流出点(灰色三角形)的时间。f)随时间绘制从该区域收缩运动的速率。g)共注册us用于纳米粒子的解剖绘图。膀胱(b)和肾脏(k)位于us(灰色),而纳米粒子pa信号以颜色显示。h)us(灰色)/pa(彩色)横切片的mip显示onc纳米粒子随时间通过肠。mip用于在目的单个切片(左下)内定向pa信号。参考两个其它的在“b”(蓝色)和“c”(灰色)中保持稳定的纳米粒子内容物,示出了区域“a”(红色)中的纳米粒子的随时间的流出定量。“a”的波动是由于纳米粒子的收缩性流入和流出。i)us/pa检测肠梗阻。将小鼠进行十二指肠结扎或假手术。给予3.4mg(对应于100od860)onc纳米粒子,并且1小时后对小鼠成像。顶部显示膀胱上方2.4cm的横切片,显示梗阻的小鼠中肿胀的胃。底部显示us/pamip。在假手术组中,纳米粒子的无阻塞流动是清楚的。虚线表示近似的手术切口部位,图像宽度对应于2.4cm。每组n=3的代表图像。实心标尺,指示5mm。
图28:用64cu标记的无缝纳米粒子对gi肠道的全身pet成像。a)使用64cu标记纳米粒子。f127peo块显示为蓝色,ppo块显示为黑色,nc染料显示为红色并且64cu显示为放射性黄色圆圈。b)在37℃孵育的模拟胃液(红色)、模拟肠液(蓝色)和水(黑色)中放射性标记的纳米粒子中64cu螯合的保留稳定性。对于n=3,得到平均值±标准差。c)在灌胃100od的onc纳米点后24小时后,小鼠中onc纳米粒子和螯合的64cu的粪便清除。利用γ计数评估64cu并利用吸收评估纳米粒子。对于n=3到4只小鼠,得到平均值±标准差。d)灌胃后24小时64cu和纳米粒子的生物分布。对于一些器官,由于没有测量而没有获得数据(“n.d.”)。对于n=3到4只小鼠,得到平均值±标准差。e)纳米粒子的代表性pet成像。灌胃100od的64cu标记的onc纳米粒子,并在指定的时间点对小鼠成像。比例尺,1厘米。f)在灌胃后3小时,贯穿小鼠的代表性的0.8mm厚的冠状切片。
图29:作为初始f127浓度的函数的纳米粒子的产量。在不同pluronicf127浓度的溶液中纳米粒子形成后的纳米粒子产量。由于溶液粘度增加超过该浓度,选择10%(w/v)pluronicf127用于纳米粒子制剂。对于n=3,得到平均值±标准差。
图30:用于在离心清洗期间测定游离f127浓度的校准曲线。pluronicf127和硫氰酸钴形成深蓝色复合物(在623nm处的吸光度)。所呈现的数据说明稀释因子。对于n=3,得到平均值±标准差。
图31:洗涤循环的接触角分析。基于接触角分析确定除去游离f127所需的洗涤次数(角度如图所示)。纳米粒子在10%(w/v)的f127(“洗涤前”)样品的溶液中形成,并且在采用离心洗涤步骤的cmc转换后除去游离f127。
图32:nc染料在二氯甲烷和水性纳米粒子形式中的标准化吸收光谱。bpc、znbnc、bnc、vbpc、vbnc、onc、nionc分别用蓝色、深绿色、黄绿色、紫色、粉红色、琥珀色和黄色表示。成功形成的纳米粒子相比于二氯甲烷光谱的偏移吸收光谱表明nc染料在纳米粒子中的密集排列改变了一些电子性质。
图33:znbnc纳米粒子的自淬灭荧光发射。在水中的吸收匹配的znbnc纳米粒子和在二氯甲烷(dcm)中游离znbnc的荧光。
图34:冻干znbnc纳米粒子和纯znbnc的x射线衍射光谱。尽管与pluronicf127相比,znbnc(灰色线)表现出弱的结晶性质,但小的峰指示一些结晶取向(7°)。然而,这些在纳米粒子形成后消失(黑线)。在纳米粒子中观察到的两个大峰是由于基于peo结晶度的特征性pluronicf127晶体图案。
图35:znbnc、bnc和onc纳米粒子的光声光谱。将所示纳米粒子的最大吸光度调节至10,并在vevolazr上的pe20管中进行pa光谱扫描。
图36:纳米粒子在宽ph范围内保持接近中性ζ电位。将纳米粒子稀释到ph经调节的磷酸盐缓冲液中,并记录ζ电位。对于n=3,得到平均值±标准差。
图37:浓度匹配纳米粒子和金纳米棒的光声光谱。将onc纳米粒子和金标准化为1.2mg/ml浓度,并且在vevolazr上记录光声光谱。纳米棒质量仅基于金。三个独立试验的代表性试验。
图38:与纳米粒子或亚甲基蓝孵育后的caco-2细胞活力。将浓缩的onc纳米粒子和亚甲基蓝溶液稀释到caco-2细胞培养基中,最终的nir吸光度作为指示。将细胞与染料在含有20%血清的dmem培养基中在37℃下孵育24小时,然后采用xtt测定法评估存活率。对于n=6,得到平均值±标准差。基于单因素方差分析(one-wayanova),在对照组和任何纳米粒子治疗组之间发现无统计学显著性差异。对于亚甲基蓝,基于单因素anova和图基事后分析(tukey’sposthocanalysis),星号标记区别于未经处理的对照的统计学显著的组(p<0.001)。
图39:50,000od860/kg纳米粒子是安全的口服纳米粒子剂量。a)在灌胃1000od剂量后的小鼠体重。在2周的时间内,小鼠没有表现出痛苦或异常行为的迹象。对于每个雄性(+/-纳米点)和雌性(+/-纳米点)组,n=5只小鼠,得到平均值±标准差。在研究完成后(基于双尾学生t检验,p>0.05),在治疗和对照小鼠的质量之间没有观察到统计学上的显著差异。b)来自治疗或对照小鼠的h&e染色的肝、脾、肾、肺和心。没有观察到系统毒性的迹象。c)基于h&e染色,包括食道、胃、小肠和大肠的胃肠道器官的组织学,显示没有明显损伤。所有比例尺表示200μm。
图40:鸡胸模型中znbnc和onc纳米粒子的信噪比。将吸光度匹配(~400的吸光度)的znbnc和onc纳米粒子置于鸡体模型中,并通过逐步添加鸡乳腺组织监测光声信号。能量脉冲密度在710nm和860nm下分别为2mj/cm2和1.5mj/cm2。
图41:铜标记不影响纳米粒子ζ电位或大小。将onc纳米粒子(100od)在0、0.01、0.1、1、10mm冷cucl2中在37℃下温育30分钟,同时持续摇动。将标记的纳米粒子以离心过滤洗涤4次以除去过量的铜,并测量ζ电位。对于n=3,得到平均值±标准差。示出了对于10mm标记条件的尺寸。
图42:表示用示例性疏水剂形成的ss-infroms的性质的表。logp是指通过alogps算法预测的试剂的疏水性的性质。用动态光散射法评价最终制剂的尺寸和pdi(多分散指数)。
图43:示出纳米粒子光学参数的表。
图44:示出用64cu标记的纳米粒子的表:使用每mci64cu不同量的纳米点的放射性标记产率。除了作为单次实验的最大剂量,对于三次重复试验,数据表示平均值±sd。
发明内容
本公开提供了用于在生物系统中运输疏水剂分子的组合物和方法。所述组合物包含负载疏水剂的胶束(本文中也称为纳米颗粒)。纳米颗粒由表面活性剂分子(如泊洛沙姆)组成并且其中掺入了疏水剂。纳米颗粒可以存在于载体中,例如水性载体。本文所用的术语“掺入”是指疏水剂存在于胶束的疏水域中。
在一个实施方案中,本公开提供了递送疏水性药物的组合物和方法。本文所用的术语“药物”是指为了治疗、诊断或监测生理功能的目的而递送的任何试剂。在一个实施方案中,本公开提供了用于运输疏水性造影剂的组合物和方法。
在一个实施方案中,本公开提供了粉末形式的包含多个负载疏水剂的胶束的组合物。胶束可以是冷冻干燥的。所述组合物基本上或完全不含未缔合的表面活性剂。例如,所述组合物基本上或完全不含未缔合的泊洛沙姆。
在一个实施方案中,从组合物中除去构成胶束的表面活性剂(例如,泊洛沙姆)的起始量的85%以上。剩余的泊洛沙姆形成胶束,其负载有疏水剂。在各种实施方案中,从组合物中除去高达90%、95%、99%或99.9%的起始量的泊洛沙姆。
在一个实施方案中,本公开提供了包含一种以上泊洛沙姆和一种以上类型的疏水剂(例如药物或造影染料)分子的胶束。胶束可以是冻干形式。冷冻干燥的组合物基本上不含任何未缔合的表面活性剂(即,为未嵌合形式或可以在低温处理后呈现未嵌合形式的表面活性剂,例如空胶束、或任何非嵌合形式的泊洛沙姆、或其中单体彼此松散地缔合,但是没有将药物分子并入其中)。
胶束含有可能密集填充但不结晶的疏水剂。由于低表面活性剂含量,胶束可以容易地通过例如过滤(如膜过滤)被进一步浓缩。负载疏水剂的胶束组合物的剩余表面活性剂不是分散剂,而是形成胶束。
在一个实施方案中,所述组合物在合适的缓冲液中(如含有或不含有ph缓冲剂的糖溶液或盐水溶液中,如柠檬酸盐、磷酸盐、组氨酸或谷氨酸盐)含有胶束,并且基本上不含未缔合的泊洛沙姆分子。
本公开的表面活性剂分子能够溶解疏水性药物,并且药物表面活性剂复合物能够形成胶束。在一个实施方案中,用于本公开的表面活性剂是包含至少疏水性和亲水性嵌段的嵌段共聚物。在一个实施方案中,表面活性剂是三嵌段共聚物,例如泊洛沙姆。泊洛沙姆是具有不同分子量的聚氧化乙烯(peo)-聚氧化丙烯(ppo)-聚氧化乙烯三嵌段共聚物。例如,泊洛沙姆由中间疏水链聚丙烯(聚(氧化丙烯))组成,其侧面是两个亲水链聚氧乙烯(聚(氧化乙烯))。泊洛沙姆是可商购的,例如商品名为
在一个实施方案中,本发明组合物包含胶束,其包含选自泊洛沙姆氏f127、f68、f108及其组合的表面活性剂,以及一种以上疏水剂。在一个实施方案中,存在于胶束中的唯一表面活性剂是泊洛沙姆。在一个实施方案中,胶束中唯一的表面活性剂是f127、f68和/或f108。在一个实施方案中,没有其它表面活性剂存在于包含胶束的组合物中,所述胶束包含、基本上由或由泊洛沙姆表面活性剂组成并且具有掺入其中的疏水性负载物分子。
本公开的药物可以是为了诊断或监测生理功能或改善、治疗、预防、诊断或监测病理状况的目的施用于个体所需的任何疏水性分子。因此,治疗性和非治疗性疏水剂可以通过该方法递送。
用于本公开的药物或造影染料通常是疏水性的。在一个实施方案中,辛醇-水分配系数(例如logp值,用alogps算法预测)为至少2。在一个实施方案中,辛醇-水分配系数为2至11。在一个实施方案中,辛醇-水分配系数为3至11。在各种实施方案中,其为3、4、5、6、7、8、9、10和11。
在一些实施方案中,所述疏水性药物为α-生育酚、阿巴芬净、胺碘酮、阿奇霉素二水合物、苄普地尔、β-胡萝卜素、布地奈德、卡巴他赛、卡马西平、钙化醇、卡维地洛、氯喹、氯丙嗪、胆钙化醇、克霉唑、辅酶q10、可替宁、赛克利嗪、环孢霉素a、地西泮、多西他赛、益康唑、维生素d2、依托泊苷、芬太尼、非诺贝特、非那雄胺、氟维司群、氟哌啶醇、安度利可、伊曲康唑、伊维菌素、拉贝洛尔、拉坦前列素、美洛昔康、咪康唑、米非司酮、霉酚酸酯、尼莫地平、紫杉醇、苯妥英、吡罗昔康、孕烯醇酮、孕烯醇酮醋酸酯、孕酮、异丙酚、利血平、视黄醇、视黄醇棕榈酸酯、舍他康唑、西布曲明、辛伐他汀、西罗莫司、角鲨烯、他克莫司、他莫昔芬、西罗莫司脂化物、睾酮、环戊丙酸睾酮、丙酸睾酮、十一酸睾酮、替拉那韦、曲伏前列素、去炎松、维生素k1及其组合。
在一些实施方案中,疏水剂是造影染料,例如发色团。用于本公开的发色团可以是适于成像的任何疏水性造影剂。合适的发色团的实例包括四吡咯类及其类似物和衍生物,包括卟啉及其衍生物、二氢卟吩及其衍生物(包括叶绿素a、脱镁叶绿素a和相关化合物)、酞菁及其衍生物、萘酞菁及其衍生物、菌绿素及其衍生物、细菌叶绿素及其衍生物。合适的发色团的特征是:在适于生物体内成像的光谱区域中高光吸收。这通常包括在600-1000nm范围内的近红外吸收。在一个实施方案中,染料是酞菁或萘酞菁衍生物。合适的染料包括但不限于2,11,20,29-四-叔-丁基-2,3-萘酞菁(bnc)、2,11,20,29-四-叔-丁基-2,3-萘酞菁锌(znbnc)、5,9,14,18,23,27,32,36-八丁氧基-2,3-萘酞菁(onc)、5,9,14,18,23,27,32,36-八丁氧基-2,3-萘酞菁镍(nionc)、2,11,20,29-四-叔-丁基-2,3-萘酞菁氧钒(vbnc)、2,9,16,23-四-叔-丁基-29h,31h-酞菁(bpc)、3,10,17,24-四-叔-丁基-1,8,15,22-四(二甲氨基)-29h,31h-酞菁氧钒(vbpc)。也包括这样的染料的衍生物和类似物,其以四吡咯结构和疏水性为特征,使得辛醇-水分配系数(通过测量或通过alogps算法预测确定)为至少2。
疏水剂与泊洛沙姆的起始摩尔比可以在0.02:1至3:1的范围内,并且在制备如本文所述的胶束组合物的方法之后,疏水剂:泊洛沙姆比可以高达55:1。在一个实施方案中,疏水剂是药物,并且起始药物:泊洛沙姆的摩尔比为0.1:1至3:1,而最终摩尔比为7:1至55:1。在一个实施方案中,疏水剂是光学造影染料,并且起始染料:泊洛沙姆的摩尔比为0.02:1至1:1,而最终摩尔比为3:1至10:1。
在一个实施方案中,所述组合物包含含有药物和表面活性剂分子的胶束,并且基本上不含未缔合的表面活性剂分子。在一个实施方案中,所有(或基本上所有)疏水剂分子以掺入到泊洛沙姆胶束中的形式存在,并且不存在未掺入胶束中的疏水剂分子(或小于1%)。在各种实施方案中,存在少于0.5%或0.1%(以及其间至小数第十位的所有百分比值)的未掺入胶束中的疏水剂分子。因此,组合物具有疏水剂分子掺入其中的胶束,但基本上缺乏空的胶束,即不具有掺入其中的疏水剂分子或未嵌合的或松散缔合的表面活性剂分子的胶束。在一个实施方案中,本公开的组合物包含至少90%的所有存在于其中掺入了疏水剂分子的胶束中的表面活性剂分子。在各种实施方案中,所述组合物包含至少91、92、93、94、95、96、97、98、99%的存在于其中掺入了疏水剂分子的胶束中的表面活性剂分子。在一个实施方案中,所述组合物包含100%的存在于其中掺入了疏水剂分子的胶束中的表面活性剂分子,使得不存在可检测到的未缔合的表面活性剂分子。因此,各种实施方案提供了这样的组合物,所述组合物中存在的10%以下的总表面活性剂分子不与负载疏水剂的胶束缔合。在各种实施方案中,组合物具有9%以下、8%以下、7%以下、6%以下、5%以下、4%以下、3%以下、2%以下、1%以下或小于1%的总表面活性剂分子不与负载疏水剂的胶束缔合。
本发明的组合物还可以含有合适量的其它组分,例如盐(nacl、kcl或其它盐类)、糖、ph缓冲剂等,包括用于施用于个体的制剂中使用的任何其它组分。例如,盐浓度可以高达4m。
本发明的胶束组合物很好地分散,并且没有明显的胶束聚集。在一个实施方案中,通过亚微米过滤技术和/或动态光散射技术和/或通过目视检查(表现为浑浊的外观),没有检测到可检测到的聚集。在一个实施方案中,组合物包含高度均匀并且是单分散的(基于动态光散射的动态光散射多分散指数小于0.5)的纳米颗粒。在一个实施方案中,多分散指数为0.05至0.5。在各种实施方案中,纳米颗粒具有0.4以下、0.35以下、0.3以下、0.2以下、0.1以下或0.05以下的多分散指数。在各种实施方案中,纳米颗粒具有0.4至0.05、0.35至0.05、0.3至0.05、0.2至0.05或0.1至0.05的多分散指数。
本公开的组合物具有高疏水剂:表面活性剂摩尔比。在一个实施方案中,该比例为0.5:1至50:1(以及它们之间的所有比例和范围)。在一个实施方案中,所述比例为1:1至55:1。例如,所述比例为1:1、10:1、15:1、20:1、25:1、30:1、35:1、40:1、45:1、50:1或55:1。在实施方案中,所述比例为3:1至10:1、10:1至50:1、10:1至55:1、10:1至60:1(以及它们之间的所有比例)。在一个实施方案中,疏水剂是药物,并且组合物中药物:泊洛沙姆摩尔比为至少10:1,可以高达50:1,高达55:1或高达60:1(以及它们之间的所有比例)。例如,在实施方案中,药物:泊沙沙姆比为10:1、15:1、20:1、25:1、30:1、35:1、40:1、45:1、50:1、55:1或60:1。在一个实施方案中,疏水剂是造影剂(染料),并且组合物中的染料:泊洛沙姆摩尔比为至少3:1,并且可以高达10:1(以及它们之间的所有比例)。例如,染料:泊洛沙姆比为3:1、3.5:1、4:1、4.5:1、5:1、5.5:1、6:1、6.5:1、7:1、7.5:1,8:1、8.5:1、9:1、9.5:1或10:1。
胶束具有10至250nm之间的尺寸(直径)。在一个实施方案中,胶束具有15至250nm(以及其间的所有整数纳米值)的尺寸。在一个实施方案中,至少90%的胶束在15-250或15-100nm范围内。在一个实施方案中,平均尺寸为20-100nm直径。在一个实施方案中,平均尺寸为20-120nm。在各种实施方案中,其为20、30、40和50、60、70、80、90、100、110或120nm。在一个实施方案中,胶束中至少80-90%(以及其间的所有整数百分比值)在20-100nm(以及其间的所有整数纳米值)的范围内。在一个实施方案中,至少80-90%(以及其间的所有整数百分比值)的胶束在20-120nm(以及其间的所有整数纳米值)的范围内。在一个实施方案中,超过90%的胶束在20-100nm范围内或在20-120nm范围内。在一个实施方案中,91、92、93、94、95、96、97、98、99或100%的胶束在20-100nm范围内或在20-120nm范围内。
在一个实施方案中,本公开的组合物用于成像应用,并包含多个平均尺寸为15至40nm(以及其间所有整数纳米值)的冷冻胶束。在一个实施方案中,平均尺寸为20-30nm(直径)。在各种实施方案中,其为20、21、22、23、24、25、26、27、28、29或30nm。在一个实施方案中,至少80-90%(以及其间的所有整数百分比值)的胶束在20-30nm(以及其间所有整数纳米值)的范围内。在一个实施方案中,超过90%的胶束在20-30nm范围内。在一个实施方案中,91、92、93、94、95、96、97、98、99或100%的胶束在20-30nm范围内。在另一个实施方案中,至少90%的胶束在15-40nm范围内。
组合物可以根据如下制备。将疏水剂溶解在有机溶剂中,例如以10mg/ml至200mg/l的试剂浓度,并加入到泊洛沙姆水溶液中,例如5、10或15%w/v的泊洛沙姆,并使有机溶剂蒸发(主动或被动方式)。通过过滤或离心除去较大的聚集体(如果有的话)。去除未掺入的泊洛沙姆(即,与疏水剂分子未缔合的泊洛沙姆)。在一个实施方案中,去除是通过改变条件而促进的,使得形成空胶束的表面活性剂、或与胶束松散地或外周缔合的表面活性剂改变为单体(未嵌合的形式)。当这样完成时,空胶束或松散缔合的表面活性剂成为未嵌合的,然后变得更容易除去。在一个实施方案中,这是通过临界胶束浓度(cmc)切换实现的,即通过将温度降低到或低于cmt的温度,从而使得胶束变成未嵌合形式。在一个实施方案中,形成未嵌合形式的温度可以是从30℃至0℃(以及其间至小数第十位的所有温度值)的任何温度。在一个实施方案中,降温温度为从室温(25℃)至0℃。在一个实施方案中,降温温度为从25℃至1℃或从22℃至1℃。在一个实施方案中,降温温度为从20℃至1℃(以及其间至小数第十位的所有温度值)。在一个实施方案中,降温温度为从10℃至-20℃(以及其间至小数第十位的所有温度值)。在一个实施方案中,不需要降低温度,并且可以通过使用其它溶剂或盐条件通过其它方式实现从空胶束到单体的转化。在一个实施方案中,降温温度为从0℃至-20℃(以及其间至小数第十位的所有温度值)。
在一个实施方案中,例如,将澄清溶液(将疏水剂(在溶剂中)加入到泊洛沙姆水溶液中并使溶剂蒸发后得到)在冰上冷却,并使用在4℃下以500至5000g持续通常10至100分钟的时间进行离心过滤,直到保留显著体积的溶液(例如100至1000ul)。用于过滤的离心力可以为2,000g以上。例如,离心力可以为2,000g至4,000g。离心可以在1℃至室温、或1℃至10℃、或4℃至10℃下进行。在一个实施方案中,在1至10℃下以3,500g持续25分钟来进行离心。可以将水加回到浓缩物中。将保留物进行一次或多次洗涤和离心过滤。因此,可以根据需要重复洗涤和过滤。在一个实施方案中,洗涤和过滤程序重复2至8次(以及其间的所有整数)。在一个实施方案中,重复3次。在另一个实施方案中,采用渗滤以连续方式进行洗涤,而不是离散步骤。在一个实施方案中,洗涤和过滤程序使得通过洗涤除去最初用于制备制剂的表面活性剂的至少60%。在各种实施方案中,至少70%、至少80%、至少85%、至少90%、至少95%、至少99%和所有所述未缔合的表面活性剂被除去。可以通过确定洗涤液中出现的表面活性剂来检查表面活性剂的所需量是否被除去。在一个实施方案中,将组合物进行洗涤,直到在洗涤液(或滤液)中没有发现可检测到的表面活性剂。例如,我们观察到通常在三次或四次洗涤后,在进一步洗涤中没有可检测到的表面活性剂。表面活性剂可以用标准方法如比色硫氰酸钴方法检测。
组合物的物理和光学性质可以通过标准技术测定。粒度测量和均匀性也可以通过标准技术例如透射电子显微镜等来确定。稳定性可以通过相对于相关流体的透析来评估。
在一个实施方案中,一种以上共负载剂(例如疏水性分子,如维生素e和/或辅酶q)与药物一起使用以制备胶束。观察到使用维生素e共负载药物导致胶束中药物负载的协同增加。在一个实施方案中,紫杉醇与维生素e或辅酶q一起使用以形成胶束。在一个实施方案中,多西他赛与维生素e一起使用以形成胶束。共负载剂相对于目的药物的摩尔比可以在0.1:1至10:1的范围内。
本发明的组合物可以新鲜使用,或者可以储存为冷冻水溶液或储存在室温或其间任何温度(如25℃至0℃)下。组合物也可以冷冻干燥并干燥储存。因此,组合物可以储存,然后以比可获得的先前组合物更浓缩的形式重构。
如果疏水性酞菁或萘酞菁用作疏水剂,则可将所得溶液浓缩至具有至少高达500个吸光度单位的近红外吸光度。在一个实施方案中,纳米颗粒可以被可检测地标记。例如,通过与疏水剂形成金属络合物,如在染料的大环内,对纳米粒子进行放射性标记或磁性标记。在一个实施方案中,纳米颗粒用64cu标记。它们可以用mn标记用于mri检测。
对于使用本发明的组合物,可以通过任何合适的给药途径进行给药。例如,组合物可以口服、静脉内、皮内、肌内、粘膜、瘤内、局部或任何其它给药方式给药。
所述组合物可用于成像技术,如光学成像(包括光声成像和荧光成像),以及全身技术,如正电子发射断层摄影(pet)成像,磁共振成像(mri)等。我们观察到,当负载染料的胶束用于成像时,胶束可以承受胃和肠环境的恶劣条件,避免全身吸收,并为光声成像产生良好的光学对比度。用于成像应用的负载染料的胶束在本文中称为纳米粒子。
在一个实施方案中,胶束具有可调并且大的近红外吸收值(>1000)。例如,吸光度是用相同染料制备的传统脂质体制剂的吸光度的500至1000倍大。在一些实施方案中,纳米颗粒具有约650至约1000nm的峰值发射。
与常规发色团不同,纳米粒子在超高光密度下显示出非移动光谱,并且在小鼠口服给药后安全通过胃肠道。在一个实施方案中,非侵入性、非电离光声技术可用于显示纳米粒子肠分布,具有低背景和0.5厘米深度的分辨率。可以通过改进的pat技术进行更深的成像。这允许实时肠功能成像以及超声影像融合。在一个实施方案中,可以使用其它成像技术,例如正电子发射断层显像。本公开提供了使用放射性标记的纳米粒子的pet的数据,允许补充全身成像。
为了用于成像,将本发明组合物口服给予个体或以其它方式递送至胃肠道。个体可以是人或非人动物。在临床前研究中,我们给出了100od的剂量,并且这导致通过光声成像的强信号检测。可以执行高分辨率扫描以及实时成像。例如,在灌胃100od的组合物后,可以监测消化系统中纳米粒子的移动。术语od代表“光密度”并且是不依赖于体积的吸光度测量量(“od”-一个od被定义为在用标准1cm路径长度测量的1ml溶液中产生1个吸光度所需的纳米颗粒的量)。这允许评价目的区域并且还分析蠕动、肠梗阻等。另外,可以通过使用放射性标记的纳米粒子(例如用64cu标记的纳米粒子)进行成像技术,如pet扫描。然后可以执行图像重建。
光声(pa)成像是比其它光学方法具有更深的穿透的非电离模式。仪器成本低,并且系统小且模块化,可以广泛地用于常规临床探查慢性和急性gi症状。pa成像是数据丰富、固有的实时模态,适合于成像动态肠道过程,如蠕动和分割,无需空间分辨率牺牲。此外,pa成像是安全的,非侵入性和非电离模式,其匹配gi成像的优选特征,特别是在儿科患者的情况下。pa技术对成像外源近红外(nir,650-1000nm)造影剂特别有用。由于它们对身体示出可忽略的全身吸收,本发明的组合物可用于这种模式。由于随后造影剂从肠的损失将导致信号减少,干扰定量测量并引入毒性问题,这是重要的。本发明组合物的纳米颗粒在胃和肠的苛刻的化学和消化环境中也不降解。
本发明的组合物可以通过其它途径施用,包括静脉内、肌内、皮内或任何其它途径以到达感兴趣的区域。本发明的组合物还可以用于其它器官和系统的成像。例如,所述组合物可在局部区域或更普遍的用于淋巴系统的成像,以及用于静脉内给药后的血液血管成像。为了使淋巴结成像,可以将其注射到淋巴系统中。这些系统的成像可以以与用于对胃肠道成像的描述类似的方式进行。
提供以下实施例以说明本发明。它们不旨在以任何方式限制。
实施例1
该实施例描述了胶束的制备及其特征。除非另有说明,材料得自sigma公司。
材料和方法
pluronicf127(sigma公司,p2443)、pluronicf68(sigma公司,412325)、cremophorel(sigma公司,c5135)、cremophorrh40(sigma公司,07076)、二氯甲烷(fisher公司)、叶绿醌(维生素k1,vwr公司,aaal10575-03)、环孢霉素a(vwr公司,89156-334)、2,6-二异丙基苯酚(异丙酚,vwr公司,aaal06841-14)、氟维司群(biorbyt公司,orb62178)、胺碘酮盐酸盐(vwr公司,aaj60456-03)、伊维菌素(vwr公司,aaj62777-03)、十一酸睾酮(matrix公司,099258)、胆钙化醇(vwr公司,tcc0314)、视黄醇棕榈酸酯(vwr公司,ic15652125)、西罗莫司脂化物(lclabs公司,t-8040)、米非司酮(vwr公司,tcm1732)、视黄醇(kracker公司,45-t3634)、辅酶q10(kracker公司,45-c9538)、多西他赛(lclabs公司,d-1000)、紫杉醇(lclabs公司,p-9600)、卡巴他赛(proactivemolecularresearch公司)、角鲨烯(sigma公司)以及5,9,14,18,23,27,32,36-八丁氧基-2,3-萘酞菁(onc,得自sigma公司)、2,11,20,29-四叔丁基-2,3-萘酞菁(得自sigma的bnc)、2,9,16,23-四-叔-丁基-29h,31h-酞菁、锌2,11,20,29-四-叔-丁基-2,3-萘酞菁(得自sigma的zn-bnc)。
通过旋转4ml10%(w/v)普朗尼克(pluronic)或克列莫佛(cremophor)水溶液,然后将溶液置于离心过滤管(fisher公司,#ufc810024)中并在4℃或25℃下用3500g旋转25分钟进行表面活性剂保留实验(图1)。在将水加回到保留物至4ml之后,将溶液在3500g下进行第二次离心10分钟。将蒸馏水加入到最终保留物中至4ml,并且通过比色法和1,6二苯基-1,3,5-己三烯(dph)探针法测定pluronic和cremophor浓度。具体地,通过首先通过将0.3g硝酸钴六水合物和1.2g硫氰酸铵溶解在3ml水中制备的硫氰酸钴试剂来测定普朗尼克浓度。然后将100μl硫氰酸钴溶液、40μl浓度范围在0-7.5wt%的f127溶液(更浓缩的f127溶液稀释至适合的范围)、200μl乙酸乙酯和80μl乙醇合并。将混合物轻轻涡旋并在14000×g离心1分钟。除去蓝色上清液,用乙醚洗涤蓝色沉淀几次(~5)次,直到上清液变为无色。然后将沉淀溶解在1ml丙酮中以测量在623nm处的吸光度。对于dph探针法,将50μl0.4mmdph的甲醇储备溶液加入1ml浓度为0-1%(wt)的cremophor溶液中。在黑暗中平衡至少3小时后,记录356nm处的uv-vis吸收强度。
药物吸收保留实验(图2)开始于溶解onc药物的表面活性剂溶液形成。简言之,将2ml的溶解onc的dcm溶液(浓度:0.4mgonc/mldcm)滴加到6ml的10%pluronic或cremophor水溶液中。搅拌至少4小时以使dcm蒸发后,将所得溶液以3500g离心10分钟,然后将1ml上清液在3500g下低温离心15分钟,重复三次;在每次离心之前,将蒸馏水加入到起始溶液或浓缩物中,体积为4ml。在约863nm测量uv-vis吸收。
药物infroms形成开始于疏水性药物溶解到f127溶液中。将100ul储备溶液(50mg药物/mldcm)(对于紫杉烷药物共负载实验,将指定量的维生素e或coq10与紫杉烷药物一起溶解在储备溶液中)滴加入将1ml10%(w/v)f127溶液(对于环孢霉素a为具有0.5、1、2、3mnacl或kcl的10%f127溶液;对于丙酚和胺碘酮为水或具有0.15、0.5、1mnacl的10%f127),同时搅拌3小时。然后将所得溶液进行几次低温离心洗涤。对于大规模infroms形成,将30mg药物溶解于150mldcm中,将所得溶液逐滴加入750ml10%(w/v)f127/f68溶液中。相反,通过使用单模块vivaflow200(sartorius)的透滤法除去过量的f127/f68。在perkinelmerxls上使用具有1cm路径长度的石英比色杯测量吸光度。在nanobrook90pluspals机器上测量尺寸。对于保留在共负载实验中od的定量,平行进行维生素e和coq10单独(无药物)infroms对照,并减去药物特征峰处的吸光度。对于摩尔比测定,将浓缩的infroms冻干。然后测定infroms粉末的质量,将粉末溶解在二氯甲烷中以确定药物质量。基于总冻干质量的差异确定f127或f68的质量。
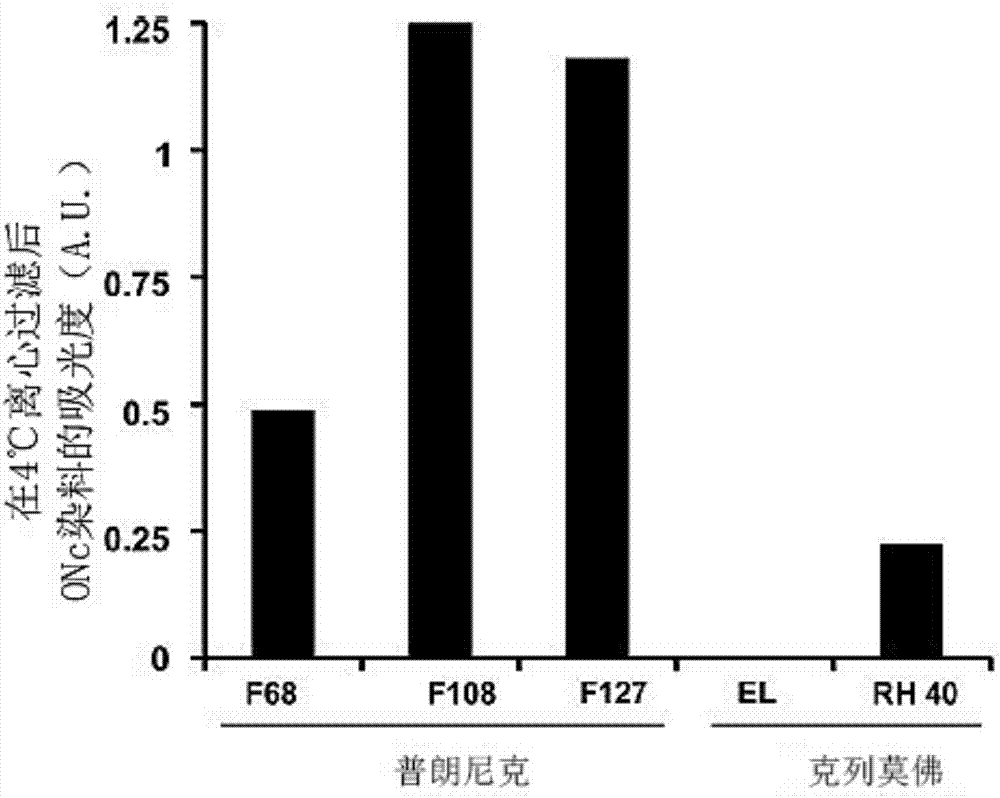
首先,我们在4℃和25℃的离心过滤期间检查了几种普朗尼克表面活性剂以及cremophorel和rh40的10%(w/v)溶液的保留。当处于胶束形式时,表面活性剂不容易通过过滤膜中的孔。如图1所示,由于其温度敏感的临界胶束浓度(cmc),pluronicf127保持在较高温度,但在低温(4℃)下除去。然而,都具有较高cmc的f68和f108可以在25℃和4℃下利用离心过滤除去。我们检查的cremophors不能通过离心过滤去除。我们接下来检查表面活性剂是否可以与疏水萘酞菁形成冷冻胶束,其将具有更大的尺寸并因此在离心过滤期间保留。从二氯甲烷溶液中加入萘酞菁并滴入表面活性剂的搅拌溶液中。使有机溶剂蒸发,然后将药物在4℃下进行离心过滤。在除去所有普朗尼克的条件下,大量的萘酞菁药物通过冷冻胶束保持溶解。(图2)。在另一个实施例中,使用在水中包含10%w/w的表面活性剂的扩展组来重复该洗涤过程,包括pluronicf127、pluronicf108、pluronicf68;聚山梨酯20、聚山梨酯40、聚山梨酯80;cremophorel、cremophorrh40;tergitolnp9、tergitolnp10、tergitolnp40;聚氧乙烯脂肪醇醚(brij)97、brij35、brijl23、brijo20;并且4℃洗涤重复三次。如图17所示,只有普朗尼克类可以在洗涤过程中产生高吸光度。
实施例2
评估维生素k1是否适合形成诱导的冷冻胶束。维生素k1是涉及血液凝固的疏水性分子,有时静脉内给予。如图3a所示,在pluronicf127中溶解后,维生素k1诱导的冷冻胶束形成,离心过滤可以除去大多数表面活性剂,留下纯化的维生素k1informs,其具有特征吸收光谱(图3b)。这些ss-infroms的大小为100nm(图3c)。重要的是,当与f68形成时,发现其具有高达20:1的药物:表面活性剂,其是临床制剂超过两个数量级高(图3d)。在目前只能利用甘氨胆酸表面活性剂的混合胶束形式之前,维生素k1之前是与cremophor表面活性剂临床配制的。这种添加剂可以取代胆红素,并不总是建议患有晚期肝病的患者使用。
实施例3
在维生素k1的另一个实施例中,使用透滤法。将150mg维生素k1溶解于1.5ml二氯甲烷(dcm)中,并加入15ml10%(w/v)f127并搅拌直至有机溶剂蒸发。将溶液用水稀释至75ml体积,在4℃进行膜过滤(sartoriusvivaflow公司,1501008vs),以除去未掺入的f127,并收集5份滤液级分(每次200ml)。维生素k1的保留通过吸光度测量定量,而f127通过硫氰酸钴方法定量。如图18a所示,维生素k1在洗涤过程中保留,而f127被除去。如图18b所示,基于透射电子显微照片,这产生尺寸小于100nm的纳米颗粒。如图42所示,药物:f127的摩尔比为39.5:1;典型的浓缩溶液可达到150mg/ml的维生素k1,尺寸为74nm,多分散指数为0.25。当使用差示扫描量热法探测起始f127溶液时,观察到超过2j/g的胶束化焓,峰值在20℃附近(图18c)。在维生素k1添加后,焓峰变小约50%,显示大部分游离f127保留在溶液中。然而,在洗涤过程后,在ss-infroms中没有观察到可检测的胶束化焓。如图18d所示,维生素kinfroms掺杂有1%的2,9,16,23-四-叔-丁基-29h,31h,酞菁(bpc)、用于znbpc(锌,2,22,20,20-四-叔-丁基-2,3-萘酞菁)的fret供体。通过将0.5mgbpc、49.5mg维生素k1溶解在500μl二氯甲烷中制备供体dcm溶液。受体二氯甲烷溶液通过将5mgznbnc和45mg维生素k1溶解在500μl二氯甲烷中制备。由0.5mgbpc、5mgznbnc和44.5mg维生素k1在500μl二氯甲烷中制备预混合供体和受体二氯甲烷溶液。将上述三种二氯甲烷溶液分别加入3个分别的5ml10%(w/v)f127中,然后搅拌至有机溶剂蒸发。通过在搅拌后将供体溶液和受体溶液(1:1,v/v)组合制备后混合供体和受体。然后在荧光计上测量荧光。当分别由fret供体和受体形成infroms,然后随后组合时,即使在洗涤过程后也没有发生明显的能量转移。当将fret供体和受体在有机溶剂中混合以产生infroms时,观察到大量的fret。这些观察结果证实了ss-infroms的动力学冷冻性质。使用渗滤方法,基于nmr分析,观察到超过40:1的标准维生素k1ss-infroms的高表面活性剂-药物摩尔比,是现有制剂的几阶数量级大(图18e)。如图18f所示,当对小鼠静脉内给药时,维生素k1ss-infroms可有效地抵抗华法林给药的作用。在维生素k1informs静脉内注射之前,将6周龄雌性icr小鼠(harlan)喂饲华法林钠溶液24小时。小鼠(n=6)静脉注射维生素kss-informs剂量0、1、2、5mg/kg。其余组用作对照,不服用华法林或任何注射。24小时后,取小鼠血液,通过coagucheckxs系统(roche)测定小鼠血液的inr值。
当在维生素k1infroms的洗涤滤液中测量未缔合和剥离的f127的量时,在充分洗涤后,不能检测到进一步的f127(图19)。这表明ss-infroms已经去除了未缔合普朗尼克。
实施例4
评估环孢霉素a是否适合形成诱导的冷冻胶束。环孢霉素a是一种免疫抑制药物,有时在cremophor溶液中静脉内给予。通过将盐添加到普朗尼克溶液中可以增强环孢霉素inform形成(图4a)。我们假设的原因是盐使溶液更离子化和亲水,导致更加稳定的疏水性负载物分配到冷冻的胶束核心。可以从informs中洗去多余的普朗尼克(图4b)。尺寸接近100nm,并且溶液具有特征吸收峰(图4c、d)。
实施例5
在环孢霉素a的另一个实施例中,将10mg环孢霉素a溶解在1ml二氯甲烷中,并加入含有0、1、2、3mnacl的10ml10%(w/v)f127溶液。搅拌3小时后,将溶液在0℃(对于0m和1m)或-10℃(对于2m和3m)下进行离心过滤,直至保持~200μl的溶液或保留物体积保持不变,将相应的盐溶液加回到浓缩物中,洗涤步骤进行三次。将保留物通过0.45μm过滤器,然后使用高效液相色谱(hplc)定量环孢霉素a的浓度。为了将pluronicf127的去除百分比定量为不同温度下盐浓度的函数,保存滤液,并使用硫氰酸钴方法。如图42所示,药物:f127的摩尔比为15:1;典型的浓缩溶液可以达到7mg/ml的环孢霉素a,尺寸为165nm,多分散指数为0.34。盐对在ss-infroms中的环孢霉素a的产量的影响如图20a所示。高渗盐水增加产量。图20b显示通过低温洗涤(-10℃)可以在高渗盐水中有效地除去游离的普朗尼克。如图20c所示,在ss-infroms中环孢霉素a的摩尔比是现有的临床制剂的几个数量级高。当环孢霉素a的ss-infroms在注射羊红细胞之前施用于小鼠时,其有效抑制免疫系统反应,如对免疫抑制药物所预期的那样(图20d)。
实施例6
通过将100mg药物溶解在1ml二氯甲烷(dcm)中并将其添加到10ml10%(w/v)f127溶液(含或不含nacl)中并搅拌直到有机溶剂蒸发而产生许多其它疏水药物的ss-infroms。然后去除未掺入的f127,包括:1)离心过滤f127剥离方法:在低温(0℃,4℃或-10℃)下对溶液进行离心过滤(fisher#ucf9-100-24)直到保留约200μl的溶液(或保留物的体积不变)。将水(或nacl溶液)加回到浓缩物中,并将洗涤过程重复三次。2)渗滤过滤方法:对于大规模(>15ml)或高盐(>2或3m)溶液,通过用装配有蠕动泵(masterflexl/s)和管道(masterflex6434-16在低温(对于2m为-7℃,对于3m为-12℃和对于4m为-16℃)下)的膜过滤(sartoriusvivaflow,1501008vs)进行去除过程。为了达到更低的温度并使f127去除百分比最大化,将膜组件、管道和待洗涤的溶液浸入乙二醇和乙醇(v/v=9:1)的混合物中,并使用干冰作为冷却剂。评估氟维司群是否适合形成诱导的冷冻胶束。氟维司群是一种可注射激素化疗药物。如图5a所示,加入pluronic后形成氟维司群胶束,然后可以洗掉过量的pluronic,而氟维司群保留在氟维司群informs中。溶液具有特征吸收光谱(图5b)。
实施例7
评估胺碘酮是否适合形成诱导的冷冻胶束。胺碘酮是一种可注射的心脏药。如图6a所示,氯化钠大大增强了胺碘酮inform的形成。可以洗掉过量的pluronic,而保留胺碘酮(图6b)。informs具有特征吸收光谱和接近30nm的窄粒径分布(图6c和d)。
实施例8
评估伊维菌素是否适合形成诱导的冷冻胶束。伊维菌素是一种抗寄生虫药物,其注射形式的主要应用是治疗牲畜,但也用于人类。如图7a所示,可以形成伊维菌素informs,这允许洗去过量的pluronic而保留伊维菌素。伊维菌素informs在240nm具有接近40nm的特征吸收峰(图7b、c)。将100mg伊维菌素溶解在1ml二氯甲烷中并加入10ml10%(w/v),然后搅拌直至有机溶剂蒸发。为了除去未掺入的f127,在0℃下对溶液进行离心过滤(fisher#ucf9-100-24),直至保留约200μl的溶液。将水加回到浓缩物中,洗涤过程重复三次。如图42所示,药物:f127的摩尔比为45:1;典型的浓缩溶液可以达到79mg/ml的伊维菌素,尺寸为39nm,并且多分散指数为0.03。
实施例9
评估十一酸睾酮是否适合形成诱导的冷冻胶束。十一酸睾酮是睾酮的酯化形式,其为主要的雄激素,已被用于激素替代以及探索男性避孕。它通常在植物油中以肌内注射施用。如图8a所示,可以形成十一酸睾酮informs,这允许洗去过量的pluronic而保留十一酸睾酮。十一酸睾酮informs在235nm处具有特征吸收峰(图8b)。十一酸睾酮informs具有40:1的药物:普朗尼克比。在另一个实施例中,以小规模搅拌十一酸睾酮,并将10mg药物溶解在100μl的dcm中,并加入到含有0、1、2、3、4mnacl的1ml10%(w/v)f127水溶液中,然后搅拌3小时,直到dcm完全蒸发。然后溶液以5,000×g离心10分钟。弃去上清液,将沉析物溶于1ml乙醇中,测量240nm(对于十一酸睾酮)和230nm(对于卡巴他赛)的吸光度以定量未掺入的药物。将100mg十一酸睾酮溶解在1ml二氯甲烷(dcm)中,并加入到含有4mnacl的10ml10%(w/v)f127中,搅拌直至有机溶剂蒸发。通过用装配有蠕动泵(masterflexl/s)和管道(masterflex6434-16)的膜过滤(sartoriusvivaflow,1501008vs)进行未掺入的f127去除过程。在-16℃下进行去除过程,并将4mnacl溶液用于透析过滤溶液。为了使f127去除百分比最大化,将膜组件、管道和待洗涤的溶液浸入乙二醇和乙醇(v/v=9:1)的混合物中,并使用干冰作为冷却剂。如图42所示,药物:f127的摩尔比为9:1;典型的浓缩溶液可以达到16mg/ml的十一酸睾酮,尺寸为112nm,并且多分散指数为0.19。如图21a所示,高渗盐水至4m可以极大地防止十一酸睾酮的聚集。与溶解在油中的现有制剂相比,ss-infroms具有高得多的药物与增溶剂摩尔比(图21b)。
实施例10
评估胆钙化醇是否适合形成诱导的冷冻胶束。胆钙化醇是维生素d的一种形式。如图9a所示,可以形成胆钙化醇informs,这允许这允许洗去过量的pluronic而保留胆钙化醇。胆钙化醇informs在270nm具有特征吸收峰(图9b)。将100mg钙钙石溶解在1ml二氯甲烷(dcm)中,并加入到含有2mnacl的10ml10%(w/v)f127中,搅拌直至有机溶剂蒸发。通过用装配有蠕动泵(masterflexl/s)和管道(masterflex6434-16)的膜过滤(sartoriusvivaflow,1501008vs)进行未掺入的f127去除过程。在-7℃下进行去除过程,并将2mnacl溶液用于透析过滤溶液。为了使f127去除百分比最大化,将膜组件、管道和待洗涤的溶液浸入乙二醇和乙醇(v/v=9:1)的混合物中,并使用干冰作为冷却剂。如图42所示,使用高渗盐水用于形成,并进行洗涤,药物:f127的摩尔比为9:1;典型的浓缩溶液可以达到75mg/ml的胆钙化醇,尺寸为44nm,并且多分散指数为0.16。
实施例11
评估视黄醇棕榈酸酯是否适合形成诱导的冷冻胶束。视黄醇棕榈酸酯是酯化的维生素a前体。如图10a所示,可以形成视黄醇棕榈酸酯informs,这允许洗去过量的pluronic而保留视黄醇棕榈酸酯。视黄醇棕榈酸酯informs在320nm具有特征吸收峰(图10b)。将100mg视黄醇棕榈酸酯溶解在1ml二氯甲烷(dcm)中,并加入到含有2mnacl的10ml10%(w/v)f127中,并搅拌直至有机溶剂蒸发。通过用装配有蠕动泵(masterflexl/s)和管道(masterflex6434-16)的膜过滤(sartoriusvivaflow,1501008vs)进行未掺入的f127去除过程。在-7℃下进行去除过程,并将2mnacl溶液用于透析过滤溶液。为了使f127去除百分比最大化,将膜组件、管道和待洗涤的溶液浸入乙二醇和乙醇(v/v=9:1)的混合物中,并使用干冰作为冷却剂。如图42所示,使用高渗盐水用于形成,并进行洗涤,药物:f127的摩尔比为54:1;典型的浓缩溶液可以达到38mg/ml的视黄醇棕榈酸酯,尺寸为114nm,并且多分散指数为0.1625。
实施例12
评估西罗莫司脂化物是否适合形成诱导的冷冻胶束。西罗莫司脂化物是一种免疫抑制药物,在某些情况下静脉内给予。如图11a所示,可以形成西罗莫司脂化物informs,这允许洗去过量的pluronic而保留西罗莫司脂化物。西罗莫司脂化物informs在275nm具有特征吸收峰(图112b)。
实施例13
评估米非司酮是否适合形成诱导的冷冻胶束。米非司酮是一种通常用作堕胎剂的类固醇化合物。它不经常通过注射给予。如图12a所示,可以形成米非司酮informs,这允许完全洗去过量的pluronic而保留米非司酮。米非司酮informs在310nm具有特征吸收峰(图12b)。
实施例14
评估视黄醇是否适合形成诱导的冷冻胶束。视黄醇是维生素a的一种形式。如图13a所示,可以形成视黄醇informs,这允许完全洗去过量的pluronic而保留视黄醇。视黄醇informs在接近300nm具有特征吸收峰(图13b)。
实施例15
接着评估辅酶q10是否适合形成诱导的冷冻胶束。辅酶q10是一种必需的维生素。如图14a所示,可以形成辅酶q10informs,这允许完全洗去过量的pluronic而保留辅酶q10。辅酶qinforms在290nm附近具有特征吸收峰(图14b)。将100mg辅酶q10溶解在1ml二氯甲烷(dcm)中,并加入到含有4mnacl的10ml10%(w/v)f127中,并搅拌直至有机溶剂蒸发。通过用装配有蠕动泵(masterflexl/s)和管道(masterflex6434-16)的膜过滤(sartoriusvivaflow,1501008vs)进行未掺入的f127去除过程。在-16℃下进行去除过程,并将4mnacl溶液用于透析过滤溶液。为了使f127去除百分比最大化,将膜组件、管道和待洗涤的溶液浸入乙二醇和乙醇(v/v=9:1)的混合物中,并使用干冰作为冷却剂。如图42所示,药物:f127的摩尔比为30:1;典型的浓缩溶液可以达到42mg/ml的辅酶q,尺寸为115nm,并且多分散指数为0.28。
实施例16
接下来,我们研究了紫杉烷inform制剂。紫杉烷是作用于癌细胞中微管的常用化疗剂。多西他赛和紫杉醇是两种最常见的紫杉烷。即使在3mnacl中,inform形成也是无效的(图15)。然而,以等摩尔比添加维生素e,多西他赛和紫杉醇inform的形成被大大增强。同样,添加辅酶q具有显著提高多西他赛和紫杉醇inform形成功效的相同效果(图16)。高渗盐水对提高卡巴他赛(ctx)形成f127ss-infroms的溶解度的影响示于新图6a中,并且使用3或4mnacl以基本上防止聚集。
实施例17
在另一个实施例中,将10mg卡巴他赛(ctx)与不同质量比的辅酶q10(ctx:coq=10:0;10:0.5;10:1;10:2)溶解在100μldcm中,并加入到含有3.5mnacl的1ml10%(w/v)f127水溶液中,然后搅拌5小时(直到溶剂蒸发,溶液变得澄清)。发现高渗盐水在胶束形成期间防止聚集(图22a)。然后,将1份溶液稀释在15份水中,置于室温。在不同的时间点(1小时、2小时、3小时、4小时、5小时、6小时),溶液以5,000×g离心5分钟;数据在图22b中示出),并在6小时聚集。弃去澄清和黄色的上清液,加入1ml水以冲洗白色沉淀,并重复旋转过程。弃去上清液后,将ctx沉淀溶解在1ml乙醇中,测量吸光度以对药物的量定量。这些结果在图22b中示出,并且添加质量比为10:1或10:2的ctx:coq以防止在稀释到水中后的聚集。如图22c所示,相对于当前临床制剂,ctxss-infroms具有高得多的药物与增溶剂摩尔比。如图42所示,药物:f127的摩尔比接近8:1;典型的浓缩溶液可以达到41mg/ml的ctx,尺寸为62nm,并且多分散指数为0.1。如图22d所示,当向具有4-5mm直径的皮下miapaca-2肿瘤的无胸腺裸小鼠以30mg/kg卡巴他赛剂量在第0天和第4天静脉内施用时,ss-infroms可以根除肿瘤。
实施例18
将100mgα-生育酚溶解在1ml二氯甲烷(dcm)中,并加入到含有2mnacl的10ml10%(w/v)f127中,并搅拌直至有机溶剂蒸发。通过用装配有蠕动泵(masterflexl/s)和管道(masterflex6434-16)的膜过滤(sartoriusvivaflow,1501008vs)进行未掺入的f127去除过程。在-7℃下进行去除过程,并将2mnacl溶液用于透析过滤溶液。为了使f127去除百分比最大化,将膜组件、管道和待洗涤的溶液浸入乙二醇和乙醇(v/v=9:1)的混合物中,并使用干冰作为冷却剂。如图42所示,药物:f127的摩尔比为21:1;典型的浓缩溶液可以达到58mg/ml的α-生育酚,尺寸为86nm,并且多分散指数为0.26。
实施例19
将100mg维生素d2(ergocalciferol)溶解在1ml二氯甲烷(dcm)中,并加入到含有2mnacl的10ml10%(w/v)f127中,并搅拌直至有机溶剂蒸发。通过用装配有蠕动泵(masterflexl/s)和管道(masterflex6434-16)的膜过滤(sartoriusvivaflow,1501008vs)进行未掺入的f127去除过程。在-7℃下进行去除过程,并将2mnacl溶液用于透析过滤溶液。为了使f127去除百分比最大化,将膜组件、管道和待洗涤的溶液浸入乙二醇和乙醇(v/v=9:1)的混合物中,并使用干冰作为冷却剂。如图42所示,药物:f127的摩尔比为9:1;典型的浓缩溶液可以达到64mg/ml的维生素d2,尺寸为112nm,并且多分散指数为0.31。
实施例20
将100mg角鲨烯溶解在1ml二氯甲烷(dcm)中,并加入到含有3mnacl的10ml10%(w/v)f127中,并搅拌直至有机溶剂蒸发。通过用装配有蠕动泵(masterflexl/s)和管道(masterflex6434-16)的膜过滤(sartoriusvivaflow,1501008vs)进行未掺入的f127去除过程。在-12℃下进行去除过程,并将3mnacl溶液用于透析过滤溶液。为了使f127去除百分比最大化,将膜组件、管道和待洗涤的溶液浸入乙二醇和乙醇(v/v=9:1)的混合物中,并使用干冰作为冷却剂。如图42所示,药物:f127的摩尔比为43:1;典型的浓缩溶液可以达到80mg/ml的角鲨烯,尺寸为81nm,并且多分散指数为0.28。
实施例21
将2mg2,9,16,23-四-叔-丁基-29h,31h-酞菁溶解在1ml二氯甲烷中,并加入到10ml10%(w/v)中,并搅拌直至有机溶剂蒸发。为了除去未掺入的f127,在4℃下对溶液进行离心过滤(fisher#ucf9-100-24),直至保留约200μl的溶液。将水加回到浓缩物中,洗涤过程重复三次。如图42所示,药物:f127的摩尔比为5:1;典型的浓缩溶液可以达到19mg/ml,尺寸为18nm,并且多分散指数为0.15。
实施例22
将2mg锌2,11,20,29-四-叔-丁基-2,3-萘酞菁溶解在1ml二氯甲烷中,并加入到10ml10%(w/v)中,并搅拌直至有机溶剂蒸发。为了除去未掺入的f127,在4℃下对溶液进行离心过滤(fisher#ucf9-100-24),直至保留约200μl的溶液。将水加回到浓缩物中,洗涤过程重复三次。如图42所示,药物:f127的摩尔比为4:1;典型的浓缩溶液可以达到30mg/ml,尺寸为20nm,并且多分散指数为0.16。
实施例23
将2mg5,9,14,18,23,27,32,36-八丁氧基-2,3-萘酞菁溶解在1ml二氯甲烷中,并加入到10ml10%(w/v)中,并搅拌直至有机溶剂蒸发。为了除去未掺入的f127,在4℃下对溶液进行离心过滤(fisher#ucf9-100-24),直至保留约200μl的溶液。如图42所示,药物:f127的摩尔比为3:1;典型的浓缩溶液可以达到13mg/ml,尺寸为20nm,并且多分散指数为0.16。
实施例24
这个实施例,包括方法和结果部分,描述了纳米粒子的准备和用于gi成像的纳米粒子的使用。除非另有说明,材料获自sigma公司。
方法
具有不同疏水性的染料的溶解和保留:使用在vcclab.org托管的alogps2.1程序评价logp值。将2mg亚甲基蓝、喹钠啶红、罗丹明6g、ir780、2,11,20,29-四-叔-丁基-2,3-萘酞菁(bnc)、2,11,20,29-四-叔-丁基-2,3-萘酞菁锌(znbnc)、5,9,14,18,23,27,32,36-八丁氧基-2,3-萘酞菁(onc)、5,9,14,18,23,27,32,36-八丁氧基-2,3-萘酞菁镍(nionc)、2,11,20,29-四-叔-丁基-2,3-萘酞菁氧钒(vbnc)、2,9,16,23-四-叔-丁基-29h,31h-酞菁(bpc)、3,10,17,24-四-叔-丁基-1,8,15,22-四(二甲氨基)-29h,31h-酞菁氧钒(vbpc)溶解在1ml二氯甲烷或甲醇中用于mb,然后滴加到10%w/v的pluronicf127(sigma#p2443)。将溶液在通风橱中在室温下(或对于mb为80℃)搅拌4小时以蒸发有机溶剂。在4000×g离心5分钟以除去任何大的聚集体后,将100μl上清液稀释在3ml的20mm胆酸钠溶液中。记录吸光度后,将溶液置于透析管(fisher,#21-152-16;标称分子量截止值为12,000-14,000道尔顿中,并在室温下相对于500ml20mm胆酸钠缓冲液透析。4小时后更换缓冲液。24小时后,再次测量透析管中溶液的吸光度,以确定染料保留百分比。
按如下制备胶束。简言之,将2mgnc或pc染料溶解在1ml二氯甲烷中,滴加到10mlf127(10%,w/v)的水溶液中。选择二氯甲烷,因为所有染料都发现是可溶的(>10mg/ml),而甲醇的溶解度小于0.1mg/ml。将悬浮液在通风橱中在室温下搅拌4小时以蒸发二氯甲烷。在4000×g离心5分钟以除去聚集体后,将上清液用于cmc转换纯化。为了除去未掺入的f127,将上清液在冰上冷却,然后在4℃下在具有100,000mwco(fisher#ufc9-100-24)的amiconultra-15离心过滤装置中离心,直到200μl溶液保留在过滤装置中。储存滤液用于测定f127和染料浓度。将水加回到过滤装置中,并且重复洗涤过程至少三次。
为了定量掺入的f127,收集收集的滤液,并通过先前报道的比色测定方法(稍作修改)测定f127浓度。简言之,首先通过将0.3g六水合硝酸钴和1.2g硫氰酸铵溶解在3ml水中制备硫氰酸钴试剂。然后将100μl硫氰酸钴溶液、40μl浓度范围为0-7.5wt%的f127溶液(将更浓缩的f127溶液稀释至适合的范围)、200μl乙酸乙酯和80μl乙醇合并。将混合物轻轻涡旋并在14000×g离心1分钟。除去蓝色上清液,用乙醚洗涤蓝色沉淀几次(约5次),直到上清液变为无色。然后将沉淀溶解在1ml丙酮中以测量623nm处的吸光度(图30示出了硫氰酸钴-f127复合物的标准曲线和f127的浓度)。通过称重质量并通过比色测定法测定质量百分比来计算每次洗涤后的f127保留百分比。染料的浓度通过测量吸光度来确定。
对于重构研究,通过相同的方案使用2mgonc染料制备纳米粒子。通过将2mgonc和19.9mgdmpc(对应于95摩尔%dmpc)溶解在小体积氯仿中制备dmpc脂质体。在通过氮气吹扫蒸发溶剂后,将薄膜置于真空下1小时,然后用1.5ml蒸馏水再水合并超声处理30分钟。然后将onc纳米粒子和脂质体冷冻干燥过夜(labconcofreezone)。然后将粉末再悬浮在最小体积的水(50μl)中,并记录吸光度。将样品简单离心以除去干扰吸收基线的大的不溶性聚集体。
纳米粒子的物理和光学性质的表征:使用动态光散射用nanozs90zetasizer(malverninstruments)进行尺寸和ζ电位测量。使用jem-2010电子显微镜进行透射电镜术以确定用1%醋酸铀阴性染色的纳米粒子的水分散体的形态。除了使用10μm路径长度比色皿的高浓度光谱移动分析外,使用lambda35uv/vis分光光度计(perkinelmer)在室温下使用具有1cm光程长度的比色皿测量吸光度。
在rigakuultimaiv上,用40kv、44ma和1.76kw的操作条件,用冷冻干燥的样品进行x射线衍射粉末图。使用的衍射仪的源是具有单色仪滤波器的
使用荧光计(photontechnologyinternational)评估散射和荧光性质。为了检查纳米粒子(峰值吸收在707nm处的znbnc纳米粒子)和具有700nm峰值吸收的金纳米棒(nanopartz#a12-10-700)的散射性质,并且在水中700nm下将消光标准化为0.05。在具有2nm狭缝宽度的在600nm和800nm之间同时激发和发射扫描的荧光计上记录共振散射。记录缓冲液散射背景空白并从纳米颗粒测量量中减去。通过用纳米粒子形式或直接溶解于具有4nm激发和发射狭缝宽度的二氯甲烷中的吸光度匹配的稀释znbnc的300nm激发测量发射光谱来进行标准化荧光测量。
为了确定光学参数,将具有已知吸光度的浓缩纳米粒子冻干。测定纳米粒子粉末的质量,然后将一部分粉末溶解在二氯甲烷中以确定染料的浓度和质量。然后基于总冻干质量的差异确定f127的质量。为了计算纳米颗粒光学性质,由于疏水性染料可以漂浮/悬浮在水中并且f127的密度为1.05g/cm3,假定染料的密度为1g/cm3。使用动态光散射测量均匀球形的纳米粒子的直径,并且对于bpc、znbnc、bnc和onc分别发现为17nm、20nm、26nm和20nm。纳米粒子体积假定从其内部排除水。基于纳米粒子的平均密度和体积,可以估计每个颗粒的质量和随后的每个颗粒的染料数。
为了评估纳米粒子在模拟胃液(sgf)和模拟肠液(sif)中的稳定性,将纳米粒子相对于200mlsgf(ricca,#7108-32)透析,加入胃蛋白酶和含胰蛋白酶的sif(ricca#7109-32)。浓缩纳米粒子用sgf和sif稀释,使吸光度接近1,然后在37℃下透析。
纳米粒子清除研究:根据布法罗大学动物护理和使用委员会的规定进行动物实验。将6-8周龄雌性balb/c小鼠(harlan实验室)饥饿过夜,自由饮水。灌胃后引入食物。灌胃100odonc纳米粒子(3.42mg)或亚甲基蓝后,将小鼠转移到代谢笼中,分别收集粪便和尿。在0、2、4、8和24小时收集粪便和尿液,称重并在分析前保持在4℃。为了测定回收百分比,直接测量尿液和血清样品的吸光度。将组织或粪便(约50mg)溶解于2ml氯仿(用于回收亚甲基蓝的甲醇)中,并使用tissuetearor匀浆器(型号985-370)破碎30秒或直至染料完全溶解。将溶液在3000×g离心3分钟以除去碎片,测量含氯仿的染料的吸光度以测定回收率。为了校准纳米粒子形式和氯仿中染料的吸光度差异,将纳米粒子冷冻干燥过夜,并溶解在相同体积的氯仿中并测量吸光度。
纳米粒子毒性:对于体外研究,将2×104个caco-2细胞(atcc)接种在96孔板中的含有20%胎牛血清的dulbecco改良的eagle培养基中(dmem)。第二天,在指定浓度下用onc纳米粒子或亚甲基蓝处理细胞。24小时后,除去培养基并加入xtt以确定在450nm处测量吸光度的存活率。对于体内研究,通过灌胃给予小鼠(harlanlabs,6周龄balb/c小鼠)每20gonc纳米粒子1000od860(在24小时内给予3次给药)或保持作为对照(n=5/每组雄性灌胃、雌性灌胃、雄性对照和雌性对照组)。每隔一天监测行为,每周测量质量。2周后,处死小鼠并收获器官。pbs用于冲洗血液和碎片。将器官浸入10%中性缓冲福尔马林(vwr#16004-114)中并固定24小时。固定的器官通过增加等级的醇处理、在二甲苯中澄清并用石蜡(tbs)渗透。随后将它们包埋、切割并用苏木精和曙红染色。最后,用单个载玻片扫描仪(aperio)扫描载玻片。
光声实验。利用使用单个元件超声换能器的定制的体积反射模式pat系统。简而言之,由泵激光器(slii-10;continuum;q-switchednd:yag;532nm)激发的opo激光器(sureliteopoplus;continuum;波长调谐范围,680至2500nm;脉冲宽度,5ns;以及脉冲重复频率,10hz)合成可调激光脉冲。将与znbnc或onc纳米粒子的相应吸收峰匹配的710nm或860nm的光学波长用于pa成像实验。产生的光通过自制的球面锥形透镜和光学聚光器,脉冲能量约为约5mj/cm2,远小于安全极限。在用于体积成像的光栅扫描期间,利用定制的水盘改进了声耦合。小鼠(6-8周龄雌性balb/c小鼠)位于水盘下方。诱导的pa信号由聚焦超声换能器(v308;olympusndt;5-mhz中心频率)捕获。vevolazrus/pa成像系统用于具有21mhz换能器频率的实时成像。在雌性balb/c小鼠中灌胃100od的纳米粒子后,对消化系统中纳米粒子的移动进行光声监测。这对应于3.4mg的onc纳米粒子和13.2mg的znbnc纳米粒子。用系统软件进行目的区域分析。通过获取目的区域强度的一阶导数(具有0.2秒分辨率)和在平均10秒窗口中量化数字零交叉(对应于收缩)来确定每分钟蠕动计算的速率。使用vevolazr(visualsonics)记录光声光谱响应,并且在纳米粒子的情况下将样品置于浸没在水中的pe20管中,并且将样品置于具有在860nm峰值吸收的浓度匹配的金纳米棒。纳米棒浓度仅基于金并由制造商提供(纳米棒llc)。使用家用光声系统,通过将含有znbnc和onc纳米粒子吸收匹配至400的管顶部上的鸡肉组织层叠来确定鸡组织中的深度反应。在710nm和860nm波长处记录2和1.5mj/cm2脉冲能量,分别用于激发znbnc和onc纳米粒子。对于肠阻塞研究,将12-14g雌性cd-1小鼠(harlan)在可以获得水的情况下禁食过夜。然后用在胃附近的1cm横向切口打开腹部,并且用尼龙缝合线(vwr#89219-096)结扎十二指肠。假手术处理的小鼠没有进行十二指肠结扎,但否则是相同的程序。再次缝合腹部皮肤,并在几小时内,通过灌胃给小鼠施用100od860剂量的onc纳米粒子。1小时后,麻醉小鼠并用vevolazr系统成像。
纳米粒子放射性标记实验。使用威斯康星-麦迪逊大学的ctirds112回旋加速器,通过64ni(p,n)64cu反应生产64cu。使用增加量的纳米粒子的初步研究显示,对每37mbq的64cu用少至1μg的纳米粒子即可以实现良好的放射性标记产率(>65%,图44)。尽管对于体内检测,pet比pat更敏感,每只小鼠使用相似量的纳米粒子以确保两个研究之间的相当的生物分布模式。
为了标记,将37mbq的64cucl2稀释在300μl的0.1m乙酸钠缓冲液(ph5.5)中,并加入400od纳米粒子。将反应混合物在37℃下在恒定摇动下温育30分钟。通过amiconultra-4离心过滤单元(millipore)用磷酸盐缓冲盐水(pbs)作为流动相来纯化64cu纳米粒子。将最终纯化的64cu纳米粒子重悬于500μl的pbs中并用于体外稳定性、口服灌胃、pet成像和生物分布研究。
对于体外螯合稳定性研究,将37mbq的64cucl2与1od的纳米粒子温育30分钟,并使用100kda的截止amicon滤器(millipore,billerica,ma)分离未偶联的64cu。之后,将一个od的64cu纳米粒子重悬于1ml的sgf或sif中,并在37℃下搅拌温育。在不同的时间点(孵育后0.5、1、2、4、8和24小时)对部分混合物(50μl)进行取样,并通过100kda截止滤器过滤。收集滤液,并通过wizard2自动γ计数器(perkin-elmer,waltham,ma)测量放射性。使用以下公式计算纳米粒子上保留的64cu的百分比:(总放射性-滤液中的放射性)/总放射性。所有实验重复三次进行。
使用inveonmicropet/microct啮齿动物模型扫描仪(siemensmedicalsolutionsusa,inc.)进行pet扫描。禁食过夜后,通过口服灌胃给每只balb/c小鼠施用月7.4mbq的64cu-纳米点(在125μlpbs中100od)。在注射后的不同时间点进行5至10分钟的静态pet扫描。使用最大后验(map)算法重建图像,没有散射校正。使用供应商软件(inveonresearchworkplace)对衰变校正的全身图像进行每个pet扫描的目的区域分析,以计算肠的每克组织的注射剂量百分比(%id/g)值。
在注射后24小时的最后pet扫描之后,对所有小鼠实施安乐死并进行生物分布研究以确认基于pet成像的定量示踪剂摄取值真实地表示小鼠中的放射性分布。收集血液和主要器官/组织并称湿重。使用γ-计数器(perkinelmer)测量组织中的放射性,并以%id/g表示。
结果:
冷冻萘酞菁胶束的形成
检查不同疏水性的发色团以确定它们在稀释成生物相容性表面活性剂后是否自发组装成稳定的纳米颗粒。选择pluronic(聚(氧乙烯)-聚(氧丙烯)-聚(氧乙烯);peo-ppo-peo)f127,因为其被美国食品和药物管理局(fda)批准用于口服。为了检查生色团-f127复合物稳定性,然后将溶液相对于胆汁表面活性剂胆酸钠透析,由于其的小胶束尺寸,其可以通过透析管。如图23a所示,基于辛醇-水分配系数(logp值,用alogps算法预测(tetko,i.v.&tanchuk,v.y.,j.chem.inf.comput.sci.42,1136–1145(2002))的非常疏水的染料在透析后显示出高保留,因此不容易与大量过量的胆酸盐胶束交换。在所评价的染料中,酞菁(pc)和萘酞菁(nc)衍生物(图23b)(其以它们的四吡咯结构和极端疏水性为特征),几乎完全保留。离心除去任何聚集体后色彩浓郁的上清液的存在,意味着形成可溶性纳米配方的萘酞菁(纳米粒子)的形成。纳米粒子的产率随着f127浓度的增加而升高(图29)。在f127的临界胶束浓度(cmc)(在室温下约1%)之前观察到纳米粒子产率没有急剧增加,这意味着与未聚合物-微团平衡无关的纳米粒子形成机制。
因为f127具有温度敏感的cmc,我们检查了降低溶液温度以将胶束转化为f127单体的效果。将温度降低至4℃不会导致任何nc聚集,这可以通过形成冷冻胶束来解释。这使得能够去除所有过量的f127的新策略(图24a)。如图24b所示,离心过滤除去在4℃所有游离的f127,但是如使用先前报道的比色测定法(图30)所检测的那样,该方法在25℃下无效。cmc切换不影响纳米粒子的自组装,其在4℃洗涤过程中定量保留(图24c)。用3个低温洗涤循环从纳米粒子除去所有游离表面活性剂,并且在额外洗涤下观察不到接触角的进一步变化(图31)。与纳米粒子不同,用于pa应用的染料亚甲基蓝(mb)在3次离心过滤洗涤后完全从保留物中除去。
纳米颗粒形成20nm球体(图24d、24e)。由于cmc转换过程除去了所有过量的f127,良好分散的纳米粒子可以浓缩成高染料与f127摩尔比(>3:1的染料:f127,参见图43)。我们使用二肉豆蔻酰磷脂酰胆碱(dmpc)以19:1的脂质:染料摩尔比在纳米粒子或脂质体制剂中制备2mg的nc染料。初始溶解后,将溶液冷冻干燥并在最小体积的水(50μl)中重构。如图24f所示,浓缩的纳米点溶解在水中,这通过大约1000的极端ncnir吸收证实。然而,在冷冻干燥的脂质体重构后,观察到一些nc再溶解,但是其数量级低于纳米点制剂。由于cmc转换显著降低了存在的f127表面活性剂的总含量,纳米粒子可以以高得多的浓度重构。nc溶解所需的磷脂量不能通过cmc转换类似地降低,并且在冷冻干燥和重构期间进一步浓缩时,磷脂浓度高于溶解度极限。在溶剂去除期间,nc的无定形沉淀可能进一步影响封装的难度。
由于纳米粒子可以由一系列疏水的pc和nc发色团产生(图23a),我们开始识别具有跨越nir窗口的光谱性质的子集。使用cmc切换方法筛选不同的市售可得的pc和nc染料以产生纯纳米粒子(补充图4)。在有机溶剂中染料消光系数范围为1.0-2.2×105m-1cm-1,而在纳米粒子形式,这些降低到0.4-1.5×105m-1cm-1(图43)。这表明纳米粒子中nc的密集排列导致改变的电子性质和分子间相互作用,这进一步由水性纳米粒子的完全荧光自淬灭(图33)所支持。冷冻干燥的样品的粉末衍射分析没有显示纳米粒子内的任何结晶nc的存在,显示染料可能嵌入f127而没有组织堆积(图34)。假定纳米粒子内部是染料和疏水f127ppo嵌段的无定形共混物。然而,由于结构研究已经显示f127ppo嵌段的回转半径仅为1.6nm,并且由于peo-ppo-peo嵌段的连续性质,纳米粒子的内部也可能含有一小部分亲水peo,其与更加疏水的nc和ppo分离。纳米粒子的面向水的壳被推测为仅由peo组成。
鉴定产生在600、707、793和863nm处具有峰的纳米粒子的1pc和3nc染料(图25a、b)。纳米粒子产生跨越nir光谱同时保持相当窄的全宽度半最大值(50-100nm)的吸收。由于pa成像可以分辨多个吸收波长,所以纳米颗粒的多波长类别是期望的。将纳米粒子的pa光谱响应与它们的吸收光谱比对(图35)。可以将纳米颗粒浓缩成具有大于1000的吸收的完全溶解的溶液。纳米粒子与游离染料相比的一个优点是在浓缩时,吸收峰位置显示可忽略的偏移(图25c)。这通过测量10μm路径长度中浓缩溶液的吸收(~1000光密度(od)/ml),然后测量在1cm路径长度中的相同溶液的1000倍稀释来评估。通常使用的pa染料mb和吲哚菁绿在浓缩溶液中表现出大的吸收偏移,这是由于在高浓度下遇到的自相互作用诱导的调制的电子性质的结果。另一方面,nc与纳米粒子矩阵中的f127共组装没有表现出改变的峰值吸收位移,表明纳米粒子防止浓度依赖性染料相互作用,否则会影响较高浓度的吸收。尽管浓度依赖性吸收偏移在pa成像中是有用的,但是与浓度无关的光学参数导致对比度运动的简化分析,如gi光声层析成像(pat)的情况。基于ζ电位测量,纳米粒子在宽的ph值范围内保持接近中性的表面电荷(图36)。
在分光光度计上测量的吸光度包括吸收和散射两者的效应。然而,仅吸收有助于光声效应。使用共振光散射来估计散射。与消光匹配的金纳米棒相比,纳米粒子显示出可忽略的散射。纳米粒子被认为没有散射分量。基于纯化纳米粒子中nc与f127的摩尔比和几何计算,我们估计每个5,9,14,18,23,27,32,36-八丁氧基-2,3-萘酞菁(onc)纳米粒子含有501个分子的nc和155个分子的f127,光学截面为2.9×10-17m2。附加的光学参数在图43中报道。虽然这个横截面是纳米棒的横截面的两个数量级分之一,但是纳米粒子的独特的可分散性使它们能够在保持溶解度的同时被浓缩到更高数量级的颗粒密度。因此,可以获得稳定的纳米颗粒溶液,其总吸收大于1000。
光声肠成像
为了评估纳米粒子用作口服给药的pa剂的适宜性,我们确定纳米粒子是否能够承受胃和肠的恶劣条件,这通常对纳米颗粒造成障碍。当在37℃下在模拟胃液(sgf)或模拟肠液(sif)中透析纳米粒子时,没有观察到明显的吸收损失,表明在苛刻的透析条件下的稳定性(图26a)。在水中,1.2mg/mlonc纳米粒子产生的光声信号是浓度匹配和波长匹配的金纳米棒超出一百倍高(图37)。
使用caco-2细胞评估onc纳米粒子的细胞毒性。然而当在具有大于1的吸光度的细胞培养基中孵育时测试mb诱导的毒性,直至测试的最高值吸光度100时,纳米粒子没有表现出任何毒性(图38)。受这些结果的鼓励,我们通过灌胃给小鼠施用100od的onc纳米粒子。纳米粒子完全排泄在粪便中(图26b)。肠吸收的缺乏可能源自20nm尺寸的纳米粒子,其防止通过膜的被动扩散,以及f127的peo特征,其防止生物吸附。为了比较,以相同的方式施用100od的mb。mb被全身吸收,并且可在尿中检测到,而大部分mb保留在体内或被代谢(图26c)。
使用组织学检查纳米粒子对肠组织的影响(图26d)。没有引起明显的炎症反应或损伤作用,并且肠绒毛和隐窝似乎是健康的。考虑到通过其定量排泄预测的纳米粒子的安全性和全身吸收的缺乏,我们接下来使用口服剂量50,000od860/kg评估纳米粒子的急性毒性。这表示10倍过量的用于成像应用的功能纳米粒子剂量。在两周的研究中,雄性或雌性小鼠没有不利的行为或体重变化(图39a)。组织学显示没有全身性(图39b)或胃肠道(图39c)毒性。
我们接下来检查了纳米粒子对在体内肠内非侵入性pat的效用。如图27a所示,使用定制的单元素扫描系统的pa成像显示纳米粒子在胃肠道中的生物分布,其具有100μm轴向分辨率。清楚地观察到2,11,20,29-四-叔-丁基-2,3-萘酞菁锌(znbnc)纳米粒子的进展。检测到可忽略的背景,使肠特征分辨率清晰并且个体小肠憩室是可区分的。深度编码分析揭示了具有深度映射到5mm的肠分布的进一步空间细节(图27b)。
对于动态成像,使用vevolazr换能器阵列系统。通过灌胃给予100od的onc纳米粒子。如图27c中的横切片所示,pa(彩色)完美地与us(灰色)重叠以显示具有最小背景的胃表面下方的肠中的纳米粒子分布。每秒5帧扫描速度使得能够详细跟踪纳米粒子在肠道运动。纳米粒子流的快速变化是显而易见的(图27d),并且详细的蠕动运动是清楚的。通过选择显示波动纳米粒子内容物的目的区域,定量分割或蠕动流。纳米粒子进入目的代表性区域的流动周期性地出现,具有不同的流入和流出运动(图27e)。图27f所示的蠕动肠液流速的计算表明收缩率接近每分钟30次。
通过检查us共注册,将肠纳米粒子的分布映射到解剖特征。如图27g所示,膀胱和肾脏用us鉴定,并且相邻肠内纳米粒子的相对位置随时间变化。从一叠扫描中产生两个us/pa最大强度投影(mip),所述扫描在30分钟内跟踪纳米粒子通过肠的运动(图27h)。mip可用于在任何给定的单个横切片中提供肠定向。所指示的目的区域实时显示纳米粒子的平面外通过肠的横切片。与含有相对恒定的纳米粒子体积的对照区“b”和“c”相比,纳米粒子在1分钟内定量地从区域“a”离开并在该过程中表现出蠕动收缩。
在美国,小肠梗阻每年导致30万次手术。为了确定us/pa成像是否可用于检测肠梗阻,我们使用外科诱导的十二指肠结扎小鼠模型。在十二指肠结扎或假手术(打开腹部,但省略结扎)后,将腹部缝合闭合。然后给小鼠施用100od860剂量的onc纳米粒子,并在灌胃后1小时成像。具有阻塞的小鼠的胃可见地肿胀至大体积。us横切片在结扎的小鼠中显示出显著的空隙胃体积,但不显示假处理的小鼠(图27i,上图)。虽然us可以区分阻塞的小鼠的胃膨胀,pa信号几乎检测不到。阻塞的小鼠的增大的胃包含可能引起pa衰减的大袋空气,并且需要进一步调查这种现象。在阻塞的小鼠中,在整个肠区域几乎检测不到任何pa信号(图27i,底部)。然而,假处理的小鼠显示强pa信号,表明纳米粒子不受抑制地进展通过肠道。因此,纳米点可用作检测小肠梗阻的诊断工具。
基于它们的高吸收,znbnc(707nm)和onc纳米粒子(860nm)都适合于低背景gipa成像。最佳纳米粒子波长的选择取决于不同病例。例如,当前在光声仪器中使用的许多可调谐激光器在707nm产生较高的激光输出,而860nm可具有较低的固有生物背景和散射。在鸡胸组织中,吸光度匹配的onc和znbnc纳米粒子都可以容易地被检测到高达2.5cm的深度,具有相似的光声信噪比(图40)。使用的脉冲能量仅为2和1.5mj/cm2,分别对应于znbnc和onc纳米粒子波长的激光安全限度的仅约1/10和约1/30。
正电子发射断层扫描
虽然pa技术正在迅速改善,深部组织(>5厘米)pa成像尚未在人类报道。由于正电子发射断层扫描(pet)临床上用于非侵入性全身成像,我们检查了基于纳米粒子的pet成像作为一种互补的技术。nc大环内的4个吡咯氮可以与铜配位以用作螯合剂,并且已经显示正电子发射体64cu可以用于方便地标记完整的基于四吡咯的纳米颗粒。因为纳米粒子由nc本身形成,所以不需要额外的螯合剂缀合步骤。
当纳米粒子与64cu的水溶液孵育时,在30分钟内用超过65%的放射性标记产率进行标记(图28a和图44)。尺寸和ζ电位不受影响(图41)。在除去游离铜后,当64cu-纳米点在37℃下在sif和sgf中孵育时,在体外螯合是稳定的(图28b)。然后灌胃100od860剂量的放射性标记的onc纳米粒子(每只小鼠7.4mbq)。99%的纳米粒子在粪便中排泄,而85%的64cu放射性标记在粪便中排泄(图28c)。这种差异可能是由于在苛刻的gi环境中一些铜从nc螯合物的置换。最小放射性保留在小鼠的任何部分,所有器官保持小于1.5%id/g的64cu(图28d)。由于它们在粪便中清除,纳米粒子本身在任何器官中都没有检测到,除了在小肠中保留少量痕量。
使用pet跟踪纳米粒子通过胃肠道的运动。在口服灌胃后,放射性存在于胃和上肠中,如在0.5小时时从pet图像可以看到的(图28e)。在给药后3小时观察到肠内64cu纳米粒子的清晰分布模式。由于pet是没有组织穿透极限的层析成像,可以获得小鼠的连续全身连续冠状切片(图28f)。断层分析在三维中显示无背景的肠可视化。
由于高nc疏水性,可以形成在肠中稳定的动力学冷冻纳米粒子,避免全身吸收,并在nir中产生极端和可调的光学吸收。它们是有机的,由fda批准的表面活性剂组装,并且完全排泄在粪便中而没有观察到毒性。使用纳米粒子的实时us/pa肠成像提供高分辨率、低背景、实时原理图的肠道解剖、病理和功能的映射。此外,直接使用纳米粒子的pet可以实现定量、灵敏、用于全身成像的具有完全组织穿透的临床建立的成像方法。pet的空间分辨率限制(几毫米)可用使用单一试剂的局部pat技术来补偿。超越gi成像,基于其多峰性质,稳定性和高于肾清除阈值的小尺寸,纳米粒子也具有用作静脉内施用造影剂的潜力。未来的研究方向可能包括修改纳米粒子表面性质以用于目标检测,并检查多色pa成像以诊断肠道疾病。
虽然通过具体实施例描述了本公开,但是常规修改对于本领域技术人员将是显而易见的,并且这样的修改旨在处于本公开的范围内。
- 还没有人留言评论。精彩留言会获得点赞!