PD‑1H激动剂或拮抗剂的应用的制作方法
本发明涉及一种用于调节受试者中诱导型调节性t细胞数量水平的方法。本发明还涉及一种用于治疗受试者中t细胞免疫介导的疾病的方法。本发明还涉及pd-1h激动剂或拮抗剂在制备用于调节受试者的诱导型调节性t细胞数量水平的药物组合物中的应用。本发明还涉及pd-1h激动剂或拮抗剂在制备用于治疗受试者中t细胞免疫介导的疾病的药物组合物中的应用。
背景技术:
调节性t细胞(treg)是cd4+t细胞的一个亚群,其具有从维持自身耐受性到调节免疫反应程度的广泛功能。treg不是终末分化的细胞,并且可以在炎症期间被转化为其他cd4+t细胞亚群(包括th1和th17)。研究表明,转录因子foxp3在功能性定向调节性t细胞谱系的建立中起重要作用。foxp3+treg细胞可以通过tgf-β分为胸腺衍生的天然treg细胞(ntreg)和诱导型treg细胞(itreg),tgf-β调节itreg细胞的分化和胸腺衍生的ntreg稳定。在外周,itreg细胞的分化主要是由微环境驱动的。例如,炎性细胞因子ifn-γ和il-4抑制tgf-β诱导的itreg细胞,而il-6在tgf-β存在下引导th17细胞分化。因此,treg细胞的可塑性可决定正在进行的免疫反应的方向并控制炎症,如在几种小鼠模型(包括结肠炎模型、急性移植物抗宿主病(gvhd)模型和哮喘模型)中所显示的。
pd-1h(也称为gi24、dies1、b7-h5、vista和dd1)是具有免疫调节功能的细胞表面免疫球蛋白超家族分子,其还在调节成骨细胞、脂肪细胞和胚胎干细胞的分化以及细胞凋亡中扮演多种作用。pd-1h在造血细胞(如t细胞、nk细胞、单核细胞、nk细胞和dc)上组成型表达(b细胞除外)。不同于ctla-4敲除(ko)小鼠(其快速发展淋巴增殖性表型和致命的全身性自身免疫性疾病),pd-1h缺乏症具有更加温和的表型:pd-1hko幼鼠具有正常数量的t细胞、nk细胞、b细胞、巨噬细胞和单核细胞,而更老的小鼠经历自发的t细胞活化,当小鼠老龄化时,观察到记忆细胞水平增加和脾脏增大。此外,如加速的cona诱导的急性肝炎和gvhd中所示,pd-1h缺陷型小鼠对急性炎症和对抗原的免疫应答更敏感。pd-1h已在几个体外和体内研究中被证明分别作为配体或受体在专职抗原呈递细胞(apc)和t细胞中起作用。与这些发现一致的是,pd-1h激动型单克隆抗体已被证明是各种类型的对抗原的免疫应答的免疫抑制剂,而拮抗性mab显示为免疫刺激剂。虽然pd-1h的反受体尚未被鉴定,但最近的一项研究表明,pd-1h/dd1α可以通过血友病性相互作用(hemophilicinteraction)介导其作用。
早期研究表明,pd-1h在treg上组成型表达,其后几项研究表明其在treg功能调节中的作用。pd-1hig融合蛋白在体外能够在tgf-β存在下促进小鼠和人cd4+t细胞中的foxp3+itreg的诱导。在b16-ova肿瘤模型中,pd-1hmab的给药降低了肿瘤抗原特异性itreg细胞的分化。该结果被解释为通过该mab阻断pd-1h与其推定的反受体之间的相互作用。然而,一个不同的pd-1h激动型mab(mh5a)被证明可以在体外促进tgf-β诱导的treg细胞,并且输注mh5a可抑制小鼠模型中gvhd的进展,并且伴随着itreg的扩增。虽然这些数据表明pd-1h在treg诱导和功能中的可能作用,但尚未阐明pd-1h是否对treg细胞具有直接作用。更重要的是,pd-1h对treg细胞调节作用的机制尚不清楚。
技术实现要素:
在本发明中,发明人发现小鼠中pd-1h的遗传消融阻断了初始t细胞向foxp3+诱导型treg细胞(itreg)的分化,淋巴器官中itreg明显减少。pd-1h的这种效应对于itreg是高度特异性的,因为在肠相关组织中的天然产生的itreg和tgf-β体外诱导的itreg都降低,而天然treg(ntreg)的生成保持正常。然而,itreg和ntreg的抑制功能不受pd-1h缺失的影响。除了生成量降低,pd-1h缺陷型itreg还可以在炎症环境中迅速转化为cd4+t辅助细胞1或t辅助细胞17。这些结果表明,通过促进其分化并防止其转化为其他cd4+t细胞亚群,pd-1h可维持itreg细胞数量水平。这些发现可能对操纵treg来控制炎症具有重要意义。
在本发明的一个方面,本发明提供了一种用于调节受试者中诱导型调节性t细胞的细胞水平方法。该方法包括向有需要的受试者施用治疗有效量的pd-1h激动剂或pd-1h拮抗剂。
在本发明的另一方面,本发明提供了pd-1h激动剂或pd-1h拮抗剂在制备用于调节受试者中诱导型调节性t细胞的细胞水平的药物组合物中的用途。
在本发明的另一方面,本发明提供了治疗受试者中t细胞免疫介导的疾病的方法。该方法包括向有需要的受试者施用治疗有效量的pd-1h激动剂或pd-1h拮抗剂。
在本发明的另一方面,本发明提供了pd-1h激动剂或pd-1h拮抗剂在制备用于治疗受试者中的t细胞免疫介导的疾病的药物组合物中的用途。
在本发明的一些实施方案中,pd-1h激动剂的施用导致受试者中诱导型调节性t细胞水平升高。在本发明的一些实施方案中,pd-1h拮抗剂的施用导致受试者中诱导型调节性t细胞的水平降低。在本发明的一些实施方案中,所述施用是静脉内给药。在本发明的一些实施方案中,pd-1h激动剂是抗pd-1h的激动型单克隆抗体。在本发明的一些实施方案中,pd-1h拮抗剂选自靶向pd-1h编码多核苷酸的dsrna、sirna和shrna的反义寡聚体。在本发明的一些实施方案中,所述受试者是人。在本发明的一些实施方案中,t细胞免疫介导的疾病包括炎症、自身免疫疾病和癌症。
本发明证明pd-1h在炎性环境中抑制itreg细胞转化为th1和th17细胞,至少部分是由于其在维持foxp3表达和itreg表型中的作用导致的。这些发现对调节treg生长和功能具有重要意义。同时,如先前所示,pd-1h抑制初始t细胞的激活以限制t细胞介导的免疫应答的启动,它促进免疫应答期间itreg的生长和转化。除了调节早期t细胞活化外,pd-1h似乎通过调节treg水平参与t细胞耐受性的调节。因此,pd-1h途径可能作为一种新的靶标而被用来在炎症、自身免疫疾病和癌症中控制和操纵的t细胞介导的免疫。
附图说明
图1.pd-1h在foxp3+itreg细胞的新生成中的作用。(a)从wtot-ii或pd-1hkoot-ii小鼠纯化的初始t细胞首先用5μmcfse标记,随后以2×106/小鼠i.v.转移至b6小鼠。24小时后,在饮用水中喂食1.5%ova,喂养5天。通过流式细胞仪在代表性小鼠中分析门控cd4+cfse+vβ5.1/5.2tcr+上的foxp3频率。(b,c)实验概要,每个符号代表单个小鼠(n=5)。所显示的数据代表3个独立实验。
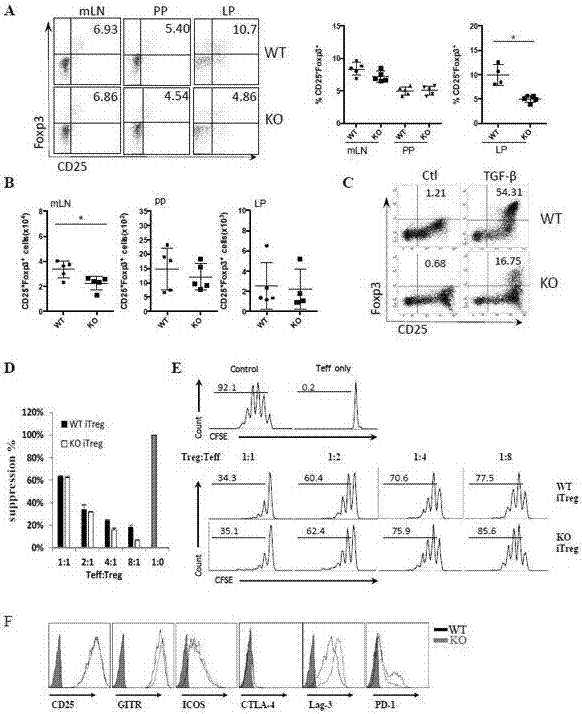
图2.pd-1h对itreg的转化和功能的影响。(a)肠道相关淋巴器官中itreg的天然发育中的pd-1h缺失。通过在流式细胞术中用特异性抗体来表征细胞表面cd25和细胞内foxp3表达,从而测定肠系膜淋巴结(mln)、payer氏集合淋巴结(pp)和固有层(lp)中cd25+foxp3+treg细胞的百分比。pd-1hko小鼠及其同窝wt仔鼠(每组n=5)用于分析。左图表示来自每组的配对单个小鼠,右图是一个代表性实验的总结(每组中n=5)。(b)肠道相关淋巴器官中cd25+foxp3+treg的绝对数。(c)itreg的体外诱导。在存在或不存在5ng/mltgf-β的情况下,用抗-cd3/cd28刺激来自wt和pd-1hko小鼠的cd4+cd25-cd62lhi初始t细胞3-5天。通过细胞内染色测定cd25+foxp3+细胞的频率。(d,e)itreg抑制功能的体外评价。如上所述,诱导来自wt或pd-1hkofoxp3(gfp)小鼠的初始cd4+t细胞成为foxp3(gfp+)itreg细胞。从b6小鼠中纯化初始cd8+t细胞或cd4+t细胞,用cfse标记并与分选的gfp+itreg细胞在存在抗cd3的情况下以指定的treg/teff细胞比率共培养。通过与没有添加itreg细胞的孔进行比较来确定包含itreg细胞时csfe的减少量。所显示的数据代表至少3次独立实验。仅teff:没有抗cd3刺激的t细胞;对照:具有抗cd3的teff细胞,不含有itreg细胞。(f)通过在流式细胞仪上门控cd4+foxp3(gfp+)来测定来自wt或pd-1hkofoxp3(gfp)小鼠的itreg上的cd25、gitr、lag-3、ctla-4、icos和pd-1的表达。
图3.细胞因子环境对pd-1h介导itreg细胞转化缺陷的影响。(a)在没有pd-1h的情况下诱导itreg细胞的细胞因子谱。如上所述,将初始t细胞在体外诱导为itreg,并在第4天收集培养上清液,通过小鼠th1/th2/th17cba试剂盒测定细胞因子水平。(b)在培养开始时向培养物中加入ifn-γ和il4的中和mab以从wt或pd-1hko初始t细胞体外诱导itreg细胞。培养3-5天后评价cd25+foxp3+细胞。呈现的结果来自一对小鼠。(c)来自(b)的数据的柱状图,数据来自一组5只小鼠。所显示的数据代表至少3次独立实验。
图4.pd-1h对eae模型中itreg细胞稳定性的影响。(a)来自wt或ko的cd45.2+foxp3(gfp+)itreg细胞在体外诱导后通过细胞分选获得。将1×106个wt或pd-1hkofoxp3(gfp+)itreg细胞i.v.转移到cd45.1b6小鼠(n=4或5每组)中,再用mog35-55肽免疫。对照小鼠用pbs接种。监测eae疾病进展和严重程度作为临床评分(见方法部分)。*p<0.05,(双向abova检验)。显示的数据代表3次具有相似的结果和疾病表型的实验中的1个。(b,c)在eae诱导的第13天,门控脾脏和引流ln(dln)细胞中的cd45.2+cd4+,并分析foxp3(gfp+)itreg细胞。(d)计数来自eae小鼠的脾脏和dln中的cd45.2+foxp3+的绝对数量。(e)使用pma/离子霉素/bfa离体将第13天的eae小鼠脾脏和dln细胞再次刺激4小时。门控cd4+cd45.2+gfp+细胞,用细胞内染色分析ifn-γ+或il-17+表达。显示的数据来自代表性的一对小鼠。(f)在4只或5只小鼠组中显示的(e)中的数据的图表。所显示的数据代表至少3次独立实验。
图5.pd-1h促进itreg细胞的定型。(a)在第13天分析来自eae模型中各组的所转移的itreg细胞(cd45.2+cd4+门控)。用具有特异性mab的pstat3或pstat5对脾脏细胞和dln细胞进行细胞内染色。(b)与a相同,但转移的itreg细胞上stat3和stat5的磷酸化以绘图值显示。(c)通过亚硫酸氢盐测序确定cns2区域的dna甲基化状态。每条线代表一个克隆(一个dna链);空心圆,非甲基化引物;实心圆,甲基化引物。
图6.pd-1h影响itreg细胞的从头分化。(a)将wt和koot-iit细胞(cd25-t细胞)分别转移到宿主小鼠中。转移的ot-iit细胞在转移前用cfse标记。该图显示了cfse的稀释和foxp3诱导。(b)wt(cd45.1/cd45.2)初始ot-iit细胞和ko(cd45.2)ot-ii初始t细胞以1:1的比例混合并共转移到宿主小鼠(cd45.1)。用1.5%ova口服饲养宿主小鼠后,分析mln和pp,并通过细胞内染色测定foxp3的频率。(c)计数所示器官中的foxp3+t细胞的绝对数量。
图7.pd-1h调节淋巴细胞环境中itreg细胞的分化。(a)将来自wt小鼠(cd45.2)和pd-1hko小鼠(cd45.1)的初始cd4+cd62lhit细胞以1:1的比例混合,将总共2百万个细胞转移到rag1ko小鼠(n=4)并在20天后测定foxp3上调。该图显示转移前后wt和kocd4+t细胞比例的变化。通过细胞内染色测定foxp3+细胞的频率。(b)计数来自rag1ko小鼠的所示器官中的cd25+foxp3+t细胞的绝对数量。(c)分析转移的初始cd4+t细胞的细胞因子的产生。在pma/离子霉素/bfa存在下,将来自所示器官的细胞体外活化4小时,然后通过细胞内染色进行分析。所显示的数据代表2次独立的实验。
图8.pd-1h对ntreg细胞的产生和抑制功能起着重要作用。(a)使用foxp3的细胞内染色和cd25的细胞表面染色,通过流式细胞术测定脾脏和ln中cd25+foxp3+ntreg的百分比。使用来自wt和pd-1hko小鼠的六周龄同窝出生小鼠(n=5)。计数脾脏和ln中foxp3+cd25+t细胞的绝对数量。(b)通过流式细胞仪上门控cd4+foxp3(gfp+)测定来自wt或pd-1hkofoxp3(gfp)小鼠的ntreg上的cd25、gitr、lag-3、ctla-4、icos和pd-1的表达。(c)对来自wt或pd-1hkofoxp3(gfp)小鼠的foxp3(gfp+)ntreg进行分选,并在存在丝裂霉素c处理的脾细胞及抗cd3抗体的情况下以不同的treg/teff比率与cfse标记的cd8+teff细胞共培养3天。通过流式细胞术测定cfse稀释度。所显示的数据代表至少3次独立实验。
图9.pd-1h决定了骨髓嵌合小鼠中treg细胞的水平。(a)将来自cd45.1wt小鼠和cd45.2小鼠的总数为1000万各混合骨髓细胞转移到亚致死性辐射处理的小鼠(cd45.1/cd45.2)中。重构后10周分析小鼠。显示wt和kot细胞的门控策略。(b)通过细胞内染色测定所示器官中的foxp3频率。(c)脾脏中的foxp3频率,并显示foxp3+细胞和foxp3-细胞的绝对数量。所显示的数据是两个独立实验的其中一个。
图10.pd-1h激动型mab轻微促进itreg细胞的分化。(a)从wtfoxp3(gfp)小鼠中分离初始t细胞,随后在tgf-β存在下用对照小鼠igg或mam82用预包被的抗cd3刺激4天。通过流式细胞术分析cd25+foxp3(gfp+)itreg细胞。(b)显示不同时间点诱导的itreg细胞。显示的结果来自3个单独的实验。所显示的数据代表至少3次独立实验。
图11.在eae模型中用wt或pd-1hkoitreg细胞转染后,受试者中cd4+th1和th17细胞的分析。(a)来自wt或pd-1hkofoxp3(gfp)小鼠的初始t细胞如上所述在体外分化成itreg细胞。将ifn-γ和il-4中和mab加入到培养物中以促进itreg的产生。分离foxp3(gfp+)细胞,通过facs评估纯度。(b,c)在转移itreg细胞的eae模型中的受试者cd45.1+cd4+t细胞通过pma/离子霉素/bfa离体再刺激。通过门控cd45.2-cd4+t细胞,通过细胞内染色测定受试者cd4+t细胞的ifn-γ+或il-17+细胞。来自代表性的一对小鼠的数据显示在(b)中,显示的数据的曲线图来自4或5只小鼠的组(c)。所显示的数据代表至少3次独立实验。
图12.pd-1h缺乏的itreg细胞不能保留其抑制功能和foxp3表达。(a)单独或与来自wt或ko小鼠的cd45.2+foxp3(gfp+)itreg细胞一起转移cd25-cd45rbhicd45.1t细胞后不同时间的rag1ko宿主小鼠的重量(每组n=5)(见方法)。转移后受体小鼠的体重变化被标准化至其在转移前的初始体重。*p<0.05,(双向abova检验)。(b)诱导结肠炎后(第10周,n=5),rag1ko宿主小鼠的结肠组织h&e染色(标尺:100μm)和临床评分(c)。(d,e,f)分析来自rag1ko宿主小鼠的脾脏和dln中cd45.2treg细胞中foxp3+t细胞的百分比,并根据比例和活细胞计数foxp3+cd45.2t细胞的绝对数量。(g)流式细胞术分析来自rag1ko小鼠的所示器官中cd45.2treg细胞中ifn-γ和il-17的表达。用pma/离子霉素/bfa在体外刺激脾细胞和dln细胞4小时,并染色以测定细胞内的细胞因子。(h)(g)中的数据总结,每个点代表一只小鼠。(i)流式细胞术分析来自rag1ko宿主小鼠的所示器官中的teff细胞(cd45.1)中ifn-γ和il-17的表达。用pma/离子霉素/bfa在体外刺激脾细胞和dln细胞4小时,并染色以测定细胞内的细胞因子。显示的数据代表2个独立实验中的其中一个。
具体实施方式
定义
在本发明中,术语“治疗”是指疗法上的以及预防性的措施,其阻止或减缓对象发生不期望的生理学改变或病症,例如哮喘发作或癌症进展。有利或期望的临床效果包括,但不限于,症状的缓解、疾病程度的降低、疾病状态的稳定化(即不恶化)、疾病进展的延迟或减缓、疾病状态的减轻或缓和以及疾病的部分或全部治愈,不论上述效果是否可检测到。“治疗”也可指与不治疗相比生存期延长。需要治疗的对象包括已患有该疾病或病症的对象,以及有可能患有该疾病或病症的对象,或要预防该疾病或病症的对象。
“对象”或“患者”、“个体”是指任何期望进行诊断、预后或治疗的对象,特别是哺乳动物对象。哺乳动物包括人、家畜、农畜、动物园动物、竞技动物或宠物,例如狗、猫、几内亚猪、兔、大鼠、小鼠、马、牛、奶牛等。本文所称的对象优选是人。
本文所用的术语“有治疗需要的患者”或“有治疗需要的对象”包括因施用本发明用于例如检测、诊断和/或治疗用途的多肽或其组合物而受益的对象,如哺乳动物对象。
本文所用的术语“激动剂”是指任何能够增加pd-1h的水平和/或活性的试剂。例如,术语“激动剂”是指使能够使pd-1h的表达和/或活性增加至少10%或更多(例如10%或更多、50%或更多、100%或更多、200%或更多、500%或更多、1000%或更多)的试剂。pd-1h的激动剂的非限制性例子可包括pd-1h多肽或其激动剂片段以及编码pd-1h多肽的核酸。
如本文所用,术语“拮抗剂”是指降低pd-1h的水平和/或活性的任何试剂。拮抗剂是与特定蛋白(例如配体)竞争结合另一种蛋白质(例如受体)的化合物。这种结合通常诱导被竞争性拮抗剂阻断的特异性生物反应或作用。拮抗剂具有亲和力但对其同源结合蛋白没有功效,并且结合将破坏相互作用并抑制这种同源蛋白的功能。拮抗剂通过结合至任何同源蛋白(或在适用情况下受体)上的活性(正位体=正确的位置)位点或变构(=其他位置)位点来调节其作用,或者它们可能在通常不涉及的独特结合位点相互作用。
本发明使用反义低聚物和类似物质用于调节编码pd-1h的核酸分子的功能或作用。这通过提供与编码pd-1h的一种或多种核酸分子特异性杂交的寡核苷酸来实现。如本文所用,术语“靶核酸”和“编码pd-1h的核酸分子”被用于方便地包括编码pd-1h的dna、从该dna转录的rna(包括前mrna和mrna或其部分),以及衍生自这些rna的cdna。本发明的低聚物与其靶核酸的杂交通常称为“反义”。因此,被认为包括在本发明的一些优选实施方案中的优选机理在本文中称为“反义抑制”。这种反义抑制通常基于寡核苷酸链或链段的基于氢键的杂交,使得至少一个链或链段被切割、降解或以其它方式不可操作。在这方面,目前优选靶向特异性核酸分子及其用于这种反义抑制的功能。在一些实施方案中,反义寡聚物选自dna寡核苷酸、rna寡核苷酸(例如microrna)和嵌合寡核苷酸。例如,反义寡聚物选自dsrna、sirna和shrna。
本发明的一个方面提供一种调节对象的诱导型调节性t细胞的细胞水平的方法,包括向有此需要的对象施用治疗有效量的pd-1h激动剂或pd-1h拮抗剂或包含pd-1h激动剂或pd-1h拮抗剂的药物组合物。本发明还提供上述pd-1h激动剂或pd-1h拮抗剂在调节对象的诱导型调节性t细胞的细胞水平的方法中的应用。
在一些实施方式中,该pd-1h激动剂或pd-1h拮抗剂或其药物组合物以肠胃外方式给药,例如静脉内、肌内、经皮或皮内给药。
在某些实施方案中,pd-1h激动剂的施用导致受试者中诱导型调节性t细胞水平的增加,例如itreg细胞的扩增。在某些实施方案中,施用pd-1h拮抗剂导致受试者中诱导型调节性t细胞水平降低。在一些实施方案中,itreg细胞的水平降低伴随着th1和/或th17细胞的产生增加。
在某些实施方案中,本发明提供了用于治疗或减轻t细胞免疫介导的疾病的方法。该方法包括向有需要的受试者施用治疗有效量的pd-1h激动剂或pd-1h拮抗剂或包含pd-1h激动剂或pd-1h拮抗剂的药物组合物。本文使用的术语“t细胞免疫介导的疾病”是指与t细胞免疫相关的疾病或病症,包括炎症、自身免疫性疾病和癌症。
组合物
本发明的一个方面提供了包含治疗有效量的pd-1h激动剂或pd-1h拮抗剂和药学上可接受的载体的药物组合物。药物组合物可用于调节受试者中诱导型调节性t细胞的水平。pd-1h激动剂或pd-1h拮抗剂可以在合适的药学上可接受的载体或赋形剂中制备。
本文使用的术语“载体”包括任何或所有的溶剂、分散介质、载体、包衣、稀释剂、抗菌剂、抗真菌剂、等渗剂、吸附延缓剂、缓冲液、载体溶液、悬浮液、胶体等。这些用于药物活性成分的介质和试剂的使用在本领域是已知的。除非已知任何常规介质或试剂不能与该活性成分配伍,否则本发明预期可在组合物中使用任何上述载体。
“药学上可接受的”是指当施用至人体时不会产生过敏反应或类似的不期望的反应的分子和成分。本领域已知如何制备包含作为活性组分的蛋白的水性组合物。通常,这些组合物被制备成注射剂,例如液态溶液或悬浮液;也可以制备成适于在注射之前配制成溶液或悬浮液的固体形式。
实施例
材料和方法
小鼠:8周龄的c57bl/6(b6)小鼠购自中山大学动物供应中心。b6背景的pd-1h-ko小鼠已有报道。转基因品系cd45.1、ot-ii、foxp3(gfp)、rag1ko均购自thejacksonlaboratory。pd-1hko和野生型(对照小鼠)的同窝小鼠由pd-1h杂合子产生并保持在相同的条件下。将foxp3(gfp)小鼠和ot-ii小鼠分别与pd-1hko小鼠回交,产生pd-1h缺陷型foxp3(gfp)报道小鼠和pd-1h缺陷型ot-ii小鼠。将小鼠保持在特定的无病原体设施中,所有动物实验均按照“国家卫生研究院实验动物护理和使用指南”进行,经中山大学科学调查委员会批准(广东,中国)。
抗体、试剂盒和流式细胞术分析:对于小鼠pd-1h的细胞表面染色,使用mh5a(仓鼠抗小鼠pd-1h),之后使用抗仓鼠igg-pe(ebioscience);仓鼠igg(ebioscience)被用作同种型对照。pd-1h激动剂mab克隆mam82(小鼠抗小鼠igg1)已有描述。所有其他荧光标记的抗体包括cd4、cd25、cd44、cd69、foxp3、tcrvβ5.1/5.2、p-stat3、p-stat5、ctla-4、lag-3、gitr、iocs、cd45.1和cd45.2从ebioscience和bdpharmingen购买。对于中和试验,从r&dsystem购买抗-ifn-γ(克隆xmg1.2)、抗il-4(克隆11b11),抗il-6(克隆mp5-20f3)中和抗体。根据bd的cytofix/cytoperm试剂盒手册进行foxp3和其他细胞内细胞因子的细胞内染色。使用小鼠th1/th2/th17cba试剂盒(bdbioscience)进行细胞因子分析。小鼠pan-t分离试剂盒、cd8+t细胞分离试剂盒、cd4+t细胞分离试剂盒、cd25微珠试剂盒和初始cd4+t细胞分离试剂盒购自miltenyibiotec(cambridge,ma)。使用bdfacsverse(bdbiosciences)进行流式细胞术分析,并使用flowjo软件(treestar)分析数据。
细胞分选和纯化:收集脾脏和ln的单细胞悬浮液,使用初始cd4+t细胞分离试剂盒(miltenyibiotec)分离初始的cd4+cd25-cd44locd62hit细胞。对于foxp3(gfp)敲入小鼠,在使用cd4+t细胞分离试剂盒纯化后,通过facsaria(bdbiosciences)对cd4+cd25-foxp3(gfp)t细胞和cd4+cd25+foxp3(gfp+)t细胞进行分选。在所示的实验中,使用cd4+t细胞分离试剂盒首先富集cd4+t细胞,然后使用cd25微珠试剂盒(miltenyibiotec)去除cd25+t细胞,得到cd4+cd25-t细胞。使用该方法分选的细胞的纯度通常大于95%。
itreg细胞的体外转化:使用结合至平板的抗cd3(克隆2c11,2μg/ml,ebioscience)和可溶性抗cd28(1μg/ml)在存在或不存在重组tgf-β(5ng/ml,r&dsystems)和il-2(5ng/ml,peprotech)的情况下在体外刺激分选的cd4+cd25-foxp3(gfp-)t细胞3-5天。然后通过流式细胞仪基于gfp的表达或foxp3的细胞内染色分析foxp3+treg细胞的转化。在所示实验中,在用抗cd3(1μg/ml)包被平板之后,将细胞在包被pd-1h激动剂mam82(10μg/ml)的平板中培养。对于细胞因子中和实验,在培养开始时向孔中加入10μg/ml的il-4、il-6和ifn-γ的mab。在所示时间点收集培养的上清液进行细胞因子分析。
treg细胞的抑制试验:按之前文献所述方法进行treg细胞抑制功能试验。简言之,首先用1μmcfse(lifetechnologies)标记初始cd8+t细胞(在一些实验中使用cd4+初始t细胞),随后在存在作为饲养细胞的1×105丝裂霉素c处理的同基因脾细胞的情况下以1×105共同培养细胞。在u底96孔板中以指定的比例向培养物加入或不加入treg细胞并培养72小时,随后加入抗小鼠cd3(1μg/ml)以进行刺激。通过cfse稀释法测定t细胞的增殖。在一些实验中,向培养物中加入可溶性小鼠对照igg或pd-1h激动剂mam82(10μg/ml)。在这些情况下,饲养细胞是pd-1hko脾细胞。
口服耐受小鼠模型:根据公开的方案进行foxp3+itreg细胞的全新生成。简言之,如前所述,从ot-ii小鼠(或pd-1hkoot-ii小鼠)中分离初始cd4+cd25-t细胞,并在过继转移前用5μmcfse标记。向wtb6小鼠静脉内注射2×106个细胞。24小时后,将笼中饮用水连续5天替换为1.5%ova溶液(v级;sigma-aldrich)。在第6天,收集肠系膜ln和pp,通过流式细胞术利用foxp3的细胞内染色测定tcr特异性foxp3+treg细胞。
骨髓嵌合体:将来自wt(cd45.1)和pd-1hko小鼠(cd45.2)的胫骨和股骨的骨髓以1:1的比例混合,并将总共1×107个细胞转移到亚致死性辐射处理的(6gy)同源wt小鼠(cd45.1/cd45.2)。重构10周后分析所示器官。通过细胞内染色测定foxp3+细胞的频率。
eae疾病模型:按文献方法进行实验性自身免疫性脑脊髓炎(eae)模型。简言之,将7-8周龄雌性小鼠s.c.免疫在完全弗氏佐剂(difco)中的200μgmog35-55(lifetechnologies)。在免疫后第0天和第2天,给小鼠腹腔注射在500μlpbs中的400ng百日咳毒素(listbiologicallabs)。对于过继转移,在第0天,进行免疫前,静脉注射1×106个wtfoxp3(gfp+)itreg细胞或pd-1hkofoxp3(gfp+)itreg细胞。wt小鼠静脉注射相同体积的pbs(盐水)作为对照。每天观察小鼠,以0-5的等级确定临床评分(盲法):0=健康;1=拖尾;2=拖尾和后肢无力;3=后肢瘫痪;4=所有后肢瘫痪和前肢无力;5=垂死状态。对于从eae小鼠获得的细胞的间接体内分析,收集每组的脾和dln,并且用pma/离子霉素/bdgolgiplug再次刺激细胞悬浮液4小时。使用细胞内染色评估ifn-γ和il-17的水平,并通过facs(bdcytofix/cytoperm试剂盒)分析。
慢性结肠炎的t细胞转移模型:简言之,如上所述制备和分选来自wt和pd-1hko小鼠的foxp3+itreg细胞。通过腹膜内,将同源cd4+cd45rbhit细胞(cd45.1,4×105)和或不和2x105itreg细胞(cd45.2)一起共注射到rag1ko小鼠。每周称重一次小鼠,10周后,收集结肠组织,用h&e进行组织染色。收集脾脏和mln细胞,并使用pma/离子霉素/bfa离体活化4小时,然后分析细胞因子产量。
dna甲基化分析:分离foxp3(gfp+)itreg细胞,用dneasyblood&tissuekit(qiagen)纯化基因组dna。使用ezdna甲基化-金试剂盒(zymo研究)进行dna的亚硫酸氢盐转化。使用文献报道的引物组扩增foxp3增强子的cns2区,并将t/a克隆到pmd18-t载体(clonetech)中。对每组10个插入的质粒进行纯化和测序,并通过biqanalyzer2.0分析甲基化结果。
图表和统计分析:图形和数据分析是使用graphpad软件生成。进行非配对studentt检验的统计学分析,p值小于0.05被认为是显著的。图中的误差条表示标准误差(se)。对于疾病进展,使用双因素方差分析(*p<0.05)进行分析。所有实验重复至少3次。
结果
pd-1h对于treg细胞的全新生成是必需的
发明人首先探索了pd-1h在口服耐受模型中的作用,在该模型中口服给予鸡卵清蛋白(ova)显示能够促进itreg细胞在外周血中的扩增和全新生成。将ot-iitcr转基因小鼠与pd-1hko小鼠回交得到新的koot-ii品系。从wtot-ii或koot-ii小鼠的脾细胞中纯化cd4+t细胞,并去除cd25+t细胞(以避免ntreg污染),随后转移到wtb6小鼠中。小鼠随后在饮用水中连续5天加入1.5%ova(图1a)。与之前公开的结果一致,foxp3+v5.1/5.2+ot-ii细胞可以在包括肠系膜淋巴结(mln)和派耶氏集合淋巴结(pp)的肠道相关淋巴器官中检测到,而在脾脏、外周ln和椎板固有层(lp)中可检测到较少的细胞(数据未显示),表明肠道相关淋巴器官对ova的treg反应。在不存在pd-1h的情况下,尽管mln和pp中wt和koot-ii细胞基于cfse稀释度的分裂速度相当(图6a)但与wtot-ii相比,mln和pp中的foxp3+v5.1/5.2+ot-ii细胞显著降低(图1b和1c)。当将初始wtot-ii细胞和koot-ii细胞共转移到ova喂养小鼠中时,观察到了类似的结果(图6b和6c)。我们发现在相同的宿主小鼠中,与共转移的wtot-ii细胞相比,ot-ii细胞上的pd-1h缺乏导致foxp3+t细胞的分化受损(图6b和6c)。我们的研究结果表明pd-1h的丧失影响体内ova特异性itreg细胞的诱导。
将初始cd4+t细胞转移到淋巴球减少症小鼠可实现其稳态增殖,该作用可以上调foxp3的表达,并且这些foxp3+itreg细胞在体外获得抑制功能。为了确定pd-1h是否影响该过程,将来自wt(cd45.1)和ko(cd45.2)小鼠的初始cd4+t细胞以1:1的比例混合,并转移到rag1ko小鼠中。3周后分析指示器官中cd25+foxp3+t细胞的百分数和绝对数量(图7)。我们发现kocd4+t细胞的总数比脾脏中的wtcd4+t细胞增加了两倍多(数据未显示)。然而,与wtcd4+t细胞相比,在脾、ln和mln中发现kocd4+t细胞中的cd25+foxp3+itreg细胞的显著更低。此外,尽管在脾脏中未观察到明显变化,但cd4+t细胞上的pd-1h损失也导致在ln和mln中比wtcd4+t细胞数量少的foxp3+itreg细胞(图7b)。此外,与淋巴细胞环境中的wtcd4+t细胞相比,我们发现回收的kocd4+t细胞产生显着更高的ifn-γ和il-17细胞因子(图7c)。我们得出结论,pd-1h是foxp3+itreg细胞从头生成所必需的。
pd-1h是itreg细胞的扩增是必需的,但非其产生和功能所必需
pd-1hko小鼠在胸腺、脾脏和淋巴结中显示出正常数量的ntreg细胞。此外,pd-1hko小鼠中ntreg细胞的表型和抑制功能也与wt同窝小鼠相似(图8)。这些发现与以前的观察结果一致,即幼年pd-1hko小鼠没有明显的自身免疫样表型。因此,pd-1h似乎对于淋巴器官中ntreg的发育和功能成熟不是必需的。
因为itreg细胞主要在稳定状态或炎症的肠道中产生,我们接下来考察pd-1h的缺乏是否影响itreg的产生和功能。虽然在pd-1hko小鼠和wt同窝出生小鼠中都发现了在mln和pp中的相似比例的foxp3+treg细胞,但pd-1hko小鼠的lp中的foxp3+细胞显著更低,尽管foxp3+细胞的绝对数量在不存在pd-1h的情况下不变(图2a和2b)。当pd-1hko/wt小鼠回交到foxp3(gfp)小鼠(其中gfp基因处于foxp3启动子的控制下)时,lp中也发现了类似的结果(数据未显示)。这些数据表明在体内不存在pd-1h的情况下,itreg细胞的分化具有缺陷。为了进一步验证我们在pd-1hko小鼠中的发现,我们创建了混合骨髓嵌合体。将来自wt(cd45.1)和ko小鼠(cd45.2)的混合骨髓过继转移至被亚致死量辐射的b6小鼠。10周后分析指示器官中foxp3+细胞的频率。我们发现kot细胞总数比wtt细胞增加了近两倍(图9a)。相比之下,kofoxp3+t细胞显著低于外周器官中的wtt细胞和受体t细胞(图9b和9c)。在嵌合小鼠的脾脏中,kofoxp3+t细胞的绝对数量也低于wtt细胞,而foxp3-效应t细胞(teff)则更加剧烈地扩增(图9c)。这些数据表明pd-1h对t细胞的活化和稳态是共抑制的,但促进体内foxp3+treg细胞的发育。
接下来我们测试pd-1h的损失是否会影响体外itreg细胞的转化。从wt和pd-1hko脾细胞纯化得到初始cd4+cd25-cd62lhi细胞,并在tgf-β存在下用抗cd3/cd28刺激。从wt初始t细胞分化出的foxp3+itreg细胞的频率显著高于由ko初始t细胞分化出的foxp3+itreg细胞(图2c)。综上,我们得出结论,pd-1h对于itreg的从头分化是必需的。
为了进一步测试pd-1h损失是否影响itreg功能,如上所述在体外诱导来自wt或pd-1hkofoxp3(gfp)小鼠的itreg细胞,并通过分选进行纯化。通过抗cd3刺激纯化的cd4+cd25-cd62lhi初始t细胞产生teff。在受辐射的脾细胞(作为抗原呈递细胞)及可溶性抗cd3的存在下,将wt或koitreg细胞与效应细胞以指定的treg/teff比共同培养3天。teff在共培养前被cfse标记,并且使用降低的cfse稀释液作为treg抑制的指标。如图2d和2e所示,来自wt和ko小鼠的itreg细胞以相当的活性抑制teff的增殖。与这些发现一致,我们未在wt与koitreg中发现激活标记物cd25、gitr、lag-3、ctla-4、icos或pd-1的表达的显著差异(图2f)。因此,尽管对itreg的分化有影响,但pd-1h不影响itreg的抑制功能。
我们最后测试了pd-1h激动剂mabmam82对itreg扩增的影响。从wtfoxp3(gfp)敲入小鼠中分选初始t细胞,随后在tgf-β存在下用抗cd3/cd28刺激。在刺激下测量gfp+cd25+itreg细胞的诱导。加入mam82轻微增加foxp3+cd25+itreg扩增,尽管效果呈现中等水平(图10a和10b)。然而,这种中等水平的作用可能是由于体外t细胞的细胞表面pd-1h的快速损失导致。总而言之,我们的数据表明,pd-1h是从初始t细胞产生itreg是必需的,但不影响其抑制功能。
pd-1h通过细胞因子调节itreg分化
之前文献报道活化的pd-1hkocd4+t细胞比wtt细胞在体外产生更高水平的ifn-γ和il-17。此外,将来自wt和ko小鼠的初始t细胞共转染淋巴细胞小鼠导致ko与wtt细胞中的foxp3+t细胞减少并增加ifnγ+和il-17+细胞(图7)。因此,细胞因子可能在不存在pd-1h的情况下调节itreg细胞的分化。为了验证该推测,首先用抗cd3/cd28刺激纯化的cd4+t细胞,并收集培养的上清液用于细胞因子检测。与我们以前的发现一致,与wtcd4+t细胞相比,活化的kocd4+t细胞产生较高水平的ifn-γ和il-17(图3a)。在tgf-β存在下,这些细胞因子的产生进一步增加,而tnf-α没有变化(数据未显示)。此外,pd-1hkocd4+t细胞产生更多il-4,伴随tgf-β增加或不增加(图3a)。因为这些细胞因子是由不同的cd4+t细胞亚群产生的,所以我们的发现支持pd-1h作为t辅助细胞的泛抑制剂。
将il-4和/或ifn-γ的中和mab加入到培养物中以利用这些细胞因子在诱导itreg细胞中的作用。如图所示,与wt对照相比,中和il-4或ifn-γ部分恢复pd-1hkoitreg细胞的分化,而包含两种mab恢复了大部分活性。作为对照,单独或组合使用ifn-γ/il-4中和mab也可以增强itreg分化(图3b和3c)。这些研究结果表明,在不存在pd-1h的情况下,受损的itreg分化至少部分地是由于t细胞改变细胞因子产生而导致的。
pd-1h的损失有助于在炎症环境中itreg转化为th17
随后我们确定了pd-1h是否对已经分化的itreg细胞有直接作用。建立鼠实验性自身免疫性脑脊髓炎(eae)模型,以测试foxp3+itreg细胞的稳定性。简言之,如上所述产生wt或pd-1hkofoxp3(gfp+)itreg细胞,并通过基于gfp阳性的流式细胞术(>97%foxp3+,图11a)进行分选。浓度为1×106/小鼠的foxp3(gfp+)cd45.2+itreg细胞被静脉内转移到cd45.1+b6免疫前小鼠中,其中转移的wtitreg的数目不足以阻止eae进展。然后用髓磷脂碱性蛋白质免疫小鼠以诱导eae。在这种情况下,与对照相比,wtitreg细胞的转移稍微延迟了疾病的发病。然而,pd-1hkoitreg细胞的转移导致更严重的疾病(如临床评分所示),尽管在疾病高峰期没有发现显著差异(图4a)。有趣的是,eae小鼠脊髓的h&e染色显示kotreg转移的小鼠和wttreg转移的小鼠之间的淋巴细胞浸润具有微小差异(数据未显示)。然后我们分析了在疾病进展期间转移的treg细胞中foxp3表达的稳定性。与wt小鼠的itreg相比,koitreg的foxp3频率(gfp+)在脾脏和dln中都降低了50%以上(图4b和4c)。此外,dln中kofoxp3+细胞的绝对数量与dln中的wtfoxp3+细胞相比显著减少,尽管脾脏中foxp3+t细胞的数量相当(图4c)。因此,我们的研究结果表明了pd-1h在维持itreg表型和功能方面的作用。
在treg向其他cd4+t细胞亚群的可转换性方面,我们的研究结果表明了itreg向包括th1和th17在内的效应性t细胞亚群的转化(在eae中被认为是致病性的)。为了验证这种可能性,用pma/离子霉素在第13天刺激来自宿主小鼠的脾脏和dln细胞4小时,使用细胞内染色检查cd45.2+cd4+门控细胞的gfp(foxp3表达的指标)、ifn-γ和il-17。与用wtitreg细胞转移的小鼠相比,用pd-1hkoitreg细胞转移的小鼠的脾脏和dln中观察到转化的th1样细胞(ifn-γ+foxp3-)的比例适度增加(图4e)。然而,在这两组小鼠中,转化的th1样细胞ifn-γ+细胞的绝对数量没有显著差异(图4e),表明itreg向th1的转化非常少。然而,大部分pd-1hkoitreg细胞(脾脏中16.7%、dln中6.1%)转化为il-17+细胞,并且从wtitreg细胞到il-17+细胞的转化非常少(在脾脏和dln中均少于2%)(图4e和4f)。使用相同的策略,我们分析了受试者cd4+t细胞(门控cd45.1+),其显示与wt或pd-1hkoitreg细胞转移的两组小鼠中ifn-γ+和il-17+细胞的相当水平(图11b和11c)。这些结果表明,pd-1h的损失促进了itreg在炎性环境中转化为th17样细胞。因此,pd-1hkoitreg细胞部分促进eae进展的作用(图4a)可能是炎症环境中itreg转化为th17的结果。
发明人还评估了pd-1hkotreg对慢性结肠炎t细胞转移模型的影响。如上所述制备wt和kofoxp3(gfp+)treg(cd45.2),分别与同种cd45.1teff(cd45rbhicd25-cd4+)混合,并且随后转移到rag1ko小鼠中。在这个实验中,即使单独转移cd45rbhiteff,我们也没有观察到疾病进展期间明显的体重减轻(图12a)。然而,共转移pd-1hkotreg/teff导致大量白细胞浸润和结肠严重的组织损伤(通过h&e染色看出),而wttreg/teff的共转移显示结肠组织没有明显的损伤(图12b和12c)。为了确定pd-1hkotreg细胞减弱的抑制能力是否与foxp3表达的损失相关,我们评估了宿主rag1ko小鼠的脾脏和mln中的foxp3表达。与treg功能损失一致,与wttreg相比(脾脏17%,mln中为54%),pd-1hkotreg中foxp3表达下调(脾脏11%,mln39%)(图12d和12e)。然而,在pd-1hkotreg与wttreg共转移小鼠之间,foxp3+t细胞的绝对数量没有显著差异(图12f)。此外,用pd-1hkotreg转移的小鼠(脾脏为30.7%,mln为14.9%)比用wttreg(脾脏为7%,mln为3.95%)转移的小鼠具有显著更多的ifn-γ+细胞(图12g)。最后,pd-1hkotreg(脾脏为13.3%,mln为9.8%)转移后比wttreg(脾脏3.64%,mln为4.44%)转移后出现更多的il-17+t细胞(图12g和12h)。在pd-1hkotreg/teff转移和在wttreg/teff转移之间未观察到ifn-γ+和il-17+teff(cd45.1)在脾脏和mln中的差异(图12i)。我们的研究结果表明,treg缺乏pd-1h导致treg快速转化为其他可能促进炎症的效应cd4细胞。因此,pd-1h对于维持treg细胞在炎症下的抑制功能和foxp3的表达是必需的。
在pd-1hkoitreg细胞中stat3活性和foxp3增强子甲基化增加
stat5激活驱动treg谱系定型,而stat3抑制foxp3表达并促进th17细胞反应。为了检查pd-1h的损失是否形成了treg细胞中的stat通路,我们分析了eae模型中stat-5和stat-3的激活。pd-1hkoitreg细胞(高达47%的p-stat3)在eae小鼠的脾和dln中比wtitreg细胞表达显著更高水平的磷酸化stat-3(p-stat3)。然而,在wt和pd-1hkoitreg细胞中发现相当的p-stat5水平(图5a和5b),暗示stat3(而不是stat5)在pd-1h功能中的作用。
以前的研究表明,foxp3增强子的cns2(保守的非编码dna序列2)区域的dna甲基化状态对维持foxp3表达特别重要。我们在本发明中确定了pd-1hkoitreg细胞中cns2区域的甲基化。如图5c所示,体外诱导的wtitreg显示cns2区域的几乎一半被去甲基化(平均为40.8%)。然而,pd-1hkoitreg细胞的去甲基化显著减少(平均17.5%),foxp3增强子cns2区域的超甲基化平均为82.5%。这一观察结果解释了pd-1hkoitreg细胞在foxp3表达方面的不稳定性。总之,我们的数据表明,pd-1h缺乏对treg的分化和表型稳定性具有显著影响。
本发明的结果表明,pd-1h是一种关键的细胞表面信号分子,通过两种不同的机制控制诱导性treg的细胞数量水平。第一,从初始t细胞产生itreg需要pd-1h。这种作用主要是通过抑制炎性细胞因子(如ifn-γ、il-4和il-17)介导的。结果,响应环境刺激的itreg的数量减少。尽管pd-1h不影响itreg在每个细胞水平的抑制功能,但itreg水平的减少可能最终会影响免疫反应期间treg的整体抑制功能。第二,pd-1h信号传导阻止在炎症环境中将itreg转化为th1和th17。这种作用可能是由于pd-1h对itreg细胞上的foxp3表达的直接调节作用导致的。通过促进itreg的产生和防止itreg转化为th1和th17,pd-1h有助于在炎症期间保持恒定的itreg水平。据我们所知,这是描述pd-1h的调解机制和控制调节性t细胞的机制的第一次全面研究。
虽然pd-1h的共抑制功能已经在早期研究中得到证实,但这种作用所依赖的机制尚未阐明。wang及其同事表明,在tgf-β存在下,pd-1hig部分促进了itreg细胞的分化,这种作用可以在小鼠和人类cd4+t细胞中发现。此外,pd-1h的单克隆抗体降低肿瘤抗原特异性itreg细胞在体内的分化。在这些研究中,pd-1h被认为是一种配体,它与treg上的一个尚未鉴别的抑制性受体结合,以调节这种作用。我们使用pd-1h缺陷小鼠和pd-1h激动剂mab的结果显示pd-1h作为t细胞上的共抑制受体。还应注意,pd-1h在抑制免疫应答中的作用可能更复杂,并且可以通过多于一种单一机制起作用。我们最近的研究表明,pd-1h介导的对同种异体抗原的t细胞耐受性的抑制是由两种不同的机制介导的:早期阻止细胞增殖和晚期诱导treg以维持移植物耐受性。在本发明中,我们揭示了pd-1h在控制treg细胞数量水平中的关键作用,这可能有助于诱导t细胞反应的长期耐受
pd-1h对treg的影响是一个高度选择性的事件。虽然pd-1h对于从初始t细胞产生itreg是必不可少的,但ntreg和itreg的抑制功能不受影响。特别是pd-1h对于通过自身抗原选择的、胸腺来源的ntreg细胞的产生和抑制功能不是必需的(图8)。这个结果可能部分解释了为什么在年幼pd-1hko小鼠中没有观察到自发性自身免疫疾病或淋巴组织增生症状。然而,在初始小鼠中自发产生的itreg细胞明显受到影响(图2a),这在先前在tgf-β和视黄酸存在下在活化的cd4+t细胞中观察到过,并且该群体主要积聚在肠和皮肤中粘膜。
尽管发现foxp3+itreg细胞的频率在肠内降低,特别是在pd-1hko小鼠的lp中,但lp中foxp3+itreg的绝对数量没有变化(图2a)。这可能部分解释了为什么pd-1hko小鼠不会在肠道中发展自发性致病性疾病。我们还在pd-1hko小鼠验证了这个结论,该小鼠与foxp3(gfp)小鼠(gfp表达在foxp3启动子控制下)交配而产生pd-1hkofoxp3(gfp)小鼠(数据未显示)。使用口服耐受模型进一步证实了这些发现,如前所述,该模型用于在体内从体内产生全新itreg细胞。与pd-1hko小鼠的肠道中的发现一致,pd-1h缺乏导致在mln和pp中抗原特异性foxp3+itreg细胞的全新生成受损(图1a和1b)。此外,初始cd4+t细胞在tgf-β存在下体外转化为itreg细胞进一步证明pd-1h缺乏可减少itreg细胞的分化(图2c)。最后,与wtitreg细胞相比,itreg细胞的pd-1h缺乏表现出相似的抑制功能,尽管treg/teff比值较高时可以发现微小的差异(图2d和2e)。此外,我们发现在淋巴细胞环境中,cd4+t细胞上pd-1h的损失导致平衡增殖后外周器官中foxp3+treg数量的减少,同时产生大量的促炎细胞,这进一步阻碍了treg的体内分化(图7)。在骨髓嵌合小鼠中也发现了类似的发现,造血来源的细胞上的pd-1h的损失导致骨髓细胞(如嗜中性粒细胞、巨噬细胞)(数据未显示)的更高的恢复,因此促炎细胞因子的产生可能阻碍外周treg细胞的数量水平(图9)。因此,我们的研究结果支持pd-1h在产生itreg方面的高度选择性作用。
treg不是终末分化的,并且itreg可以通过特异性细胞因子或炎症环境转化成th1或th17亚类。treg细胞的这种可塑性使得在慢性炎症期间难以研发出治疗策略。本发明表明,pd-1h对于treg谱系的标志物foxp3的稳定性是必需的,并保持了treg的表型。pd-1h的损失导致各种体外和体内系统和模型中itreg的快速降低。我们的研究表明,在缺乏pd-1h的情况下,itreg细胞易于在炎症状态下重编程成为th17细胞,这可能解释了pd-1hkoitreg的转移不是抑制而是促进eae疾病的观察结果。在这个模型中,itreg主要转化为th17,很少转化为th1。pd-1h也可能影响itreg向th1的转化,因为在不存在pd-1h的情况下,在cd4+t细胞中检测到高水平的ifn-γ。有意思的是,我们的研究结果表明il-4水平也显着增加,这些数据暗示pd-1h在控制th2中的可能作用。pd-1h在itreg向其他t细胞亚群的调节和转化中的作用尚待阐明。
总之,本发明支持pd-1h在炎性环境中抑制itreg细胞转化为th1和th17细胞,至少部分是由于其在维持foxp3表达和itreg表型中的作用。这些发现对调节treg生长和功能具有重要意义。同时,如先前所示,pd-1h抑制初始t细胞的激活以限制t细胞介导的免疫应答的启动,它促进免疫应答期间itreg的生长和转化。除了调节早期t细胞活化外,pd-1h似乎通过调节treg细胞数量水平参与t细胞耐受性的调节。因此,pd-1h途径可能一种是在炎症、自身免疫疾病和癌症中控制和操纵t细胞介导的免疫方面有希望的靶标。
本发明虽然已针对具体实施方式做详细的描述,但应当理解的是,本领域技术人员可在所公开的实施例的基础上对本发明做出修改和改进而不违背本发明的精神,而这些改进和修改仍属于本发明的范围。
- 还没有人留言评论。精彩留言会获得点赞!