生物可降解的树枝状结构、方法及其用途与流程
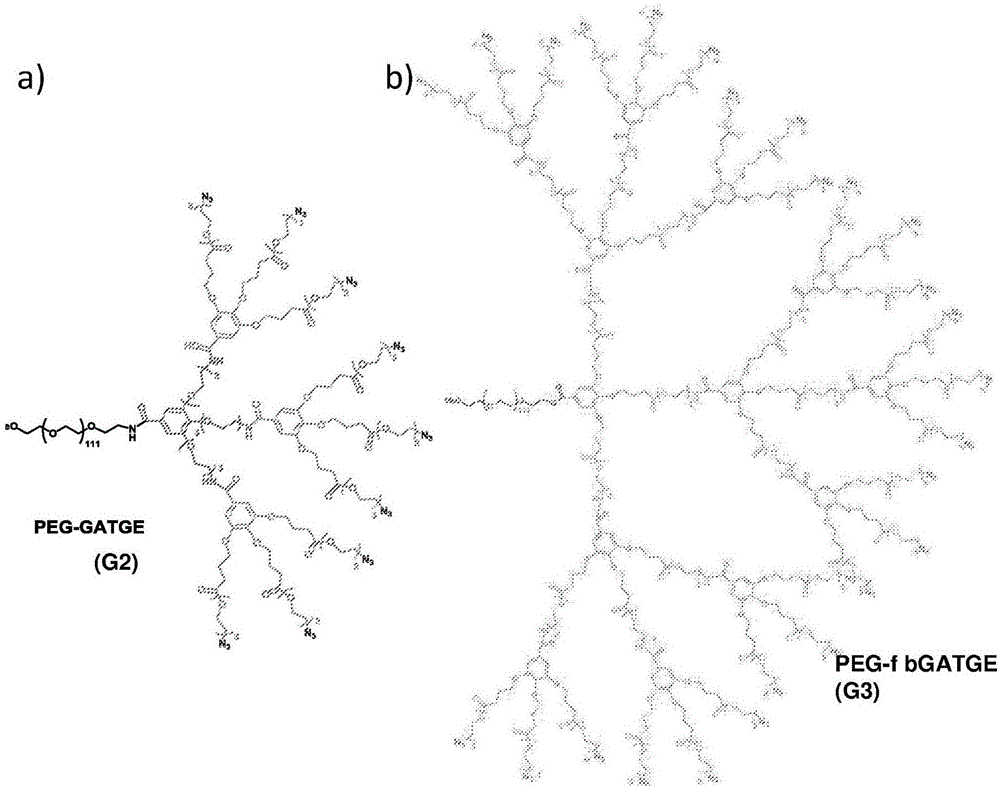
本发明公开涉及生物可降解且生物相容的树枝状重复单元/支架(bru),涉及生物可降解且生物相容的树枝状重复单元/支架(bru)的合成方法并且涉及生物可降解且生物相容的树枝状重复单元/支架(bru)的生物医学应用。该bru可用作支架以合成完全生物可降解的树枝状聚合物和/或“混合”或“杂化(杂合,混杂,hybrid)”生物可降解树枝状聚合物,从而提供生物可降解的壳和已现有的树枝状系统的水解稳定/非可降解的核。本文所述的发明公开还涉及基于bru的杂化和完全生物可降解、生物相容且无毒的peg-树枝状嵌段共聚物家族的合成以及不同胺部分对它们的官能化。这种胺官能化的实例使得能够复合sirna,并因此使得能够研究它们作为这种核酸载体的官能性评价。将生物可降解树枝状结构的开发设想为防止或避免来源于不可降解的合成材料在细胞和/或组织中的积累的细胞毒性的方法。此外,还在设计“智能”受控递送系统的背景中提出了生物可降解树枝状结构,在“智能”受控递送系统中,一个目标在于通过控制其载体的降解谱(降解特性,降解曲线,降解简况,degradationprofile)来引发和/或维持治疗剂的释放。
背景技术:
:由于独特的结构特征:球形、良好限定且多分枝的结构、低多分散性和可调尺寸,树枝状纳米结构被认为是不同生物活性分子有前景的载体。1树枝状外周上化学柄(chemicalhandles)的存在还允许配体和/或药物以特异和可控的方式对它们进行官能化以模拟多种生物系统中存在的多价性。另外,树枝状聚合物(树形分子,dendrimers)还可以通过形成非共价相互作用稳定的纳米尺寸和致密结构来运输不同的生物活性物质(如核酸(na)、不同类型的化学药物、蛋白质、生长因子)。即使在对生物医学应用具有改善的功能的树枝状结构的设计和使用中取得了进展,但是大部分目前使用的树枝状家族在生理条件下是不可降解的,其可以致使产生通过不可降解的合成材料在细胞内部或组织中的积累引起的细胞毒性。2为了克服这些障碍,一些研究小组关注生物可降解树枝状结构的设计。3的确,预期在生理条件下随时间降解成较小片段(其可以被排泄或者通过代谢途径消除)的生物可降解材料克服了与不可降解的合成材料在组织中的长期存在有关的并发症的风险。4另外,一些作者提议了可以克服长期gd(iii)组织积累的作为大分子造影剂的可降解树枝状结构的使用。5另外,还在设计“智能”受控递送系统的背景中提出了生物可降解树枝状聚合物的开发,在“智能”受控递送系统中,一个目标在于通过控制其载体的降解谱来引发和/或维持治疗剂的释放。然而,由于在合成、纯化以及后续官能化和处理步骤期间不希望的主链降解,生物可降解纳米载体的制备是困难的。6这解释了对于生物医学中的特定功能所报道的有关生物可降解树枝状聚合物的出版物的数目仍较少3,以及专利申请较少(cn103055328,wo2005003260)的原因。5a,7最近,jianbin,t等人公开了可降解树枝状大分子磁共振造影剂及其制备方法(cn103055328)。5a另外,leinweber,d.等人表明烷氧基化树枝状聚合物作为生物可降解破乳剂用于破坏油/水乳液的使用(wo2005003260)。7然而,这不是生物医学应用。因此,仍期待生物可降解的树枝状结构。技术实现要素:在本发明中,公开了新的生物可降解树枝状结构,即生物相容且叠氮化物封端的gatge树枝状重复单元,其基于没食子酸(鞣酸,五倍子酸,gallicacid,ga)核和三甘醇(三乙二醇,triethyleneglycol,tg)丁酸酯臂且在树枝状分枝处引入了生物可降解酯键(e)。最近,c.tyler和v.zubkova报道了用于制备生物不可降解树枝状聚合物的化合物,这些化合物用于制备树枝状聚合物的用途和制备所述化合物的方法(wo2014084743,us2015291522)。8与这些化合物相比,本发明公开提供了明显新的和具有优势的生物可降解性。因此,将gatge单元用作构建单元/支架以合成“混合”或“杂化”生物可降解树枝状聚合物和/或完全生物可降解树枝状聚合物(fbgatge树枝状聚合物)。“杂化”树枝状聚合物将包含具有已现有的树枝状系统(例如聚(酰胺-胺)(pamam)、聚(丙烯亚胺)(ppi)、聚(l-赖氨酸)(pll)、gatg(没食子酸三甘醇)树枝状聚合物等)的生物不可降解且耐受性的核(core)的生物可降解的壳。但是完全生物可降解的gatge树枝状系统将由完全生物可降解的组分/层(具体地从核到壳)组成。本文所提供的杂化和完全生物可降解的gatge树枝状结构两者提供了外周叠氮化物,其易于通过cu(i)-催化的huisgen环化加成(cuaac,点击化学)可官能化以具有多种配体,因此这些新的生物可降解的gatge基(基于gatge的,gatge类)树枝状纳米材料易于可调以用作不同生物医学应用的通用载体。本发明公开涉及gatge重复单元,并且将在下文中详细描述。本发明公开的另一个方面描述了peg-gatge树枝状嵌段共聚物的新家族。从gatge构建单元合成它们并作为基因疗法应用中核酸的非病毒载体评价它们的生物功能性。在调控基因表达的这些策略中,在临床环境中,通过小干扰rna(sirna)介导的rna干扰(rnai)对蛋白质生产的下调已证明了优良的治疗潜力。9然而,即使其早期成功,但是rnai疗法的广泛应用仍然需要开发临床适合、安全且有效的并且具有挤压和保护裸露sirna的能力的递送载体。目前已测试的大部分sirna非病毒载体主要基于先前开发用于质粒dna(pdna)递送的阳离子系统,如细胞穿透肽、脂质、天然和合成聚合物,以及更近期的树枝状聚合物。已报道阳离子pamam、ppi和pll树枝状聚合物等作为有前景的pdna和sirna载体。10用于掩蔽所得树枝状复合物(dendriplex)(树枝状聚合物-na复合物)的特征正电荷和改善它们在血流中的生物相容性和循环时间的常规方法是连接聚(乙二醇)(peg)链。11当在树枝状聚合物的中心点进行peg化时,获得peg-树枝状嵌段共聚物。12一些研究小组已报道了peg-树枝状嵌段共聚物用于递送pdna的用途。直到最近才使用4代(g4)peg-pamam对sirna描述了相同的策略。13然而,在na递送领域中,如通常在生物医学领域中所发生的,所使用的树枝状家族是生物不可降解的。仅有的生物可降解的情况限制于使用胺封端的双hmpa[2,2-双(羟甲基)丙酸]树枝状分子合成子(dendron)封装dna的少见实例。14,15与在同一结构中不受保护的伯/仲胺基(获得用于复合na的阳离子电荷所需的)与可水解(并因此亲电)键两者的组合有关的困难向工程设计生物可降解的树枝状载体的已有困难工作增添了额外的限制。在一个实施方式中,为了显示现在公开的gatge重复单元的用途和应用,开发了杂化和完全生物可降解、生物相容、无毒且聚乙二醇化的树枝状系统,并将其命名为peg-gatge和peg-fbgatge(图1)。peg-gatge合成至2代(g2),而对于完全生物可降解的peg-fbgatge合成了三代(g1、g2和g3)。此外,作为其它实例,提供了这些基于gatge的纳米材料作为sirna的有效载体的适合性,包括低细胞毒性谱以及保护sirna不被核酸内切酶降解和转染哺乳动物细胞的能力。出乎意料地,gatge中的酯键确保了树枝状复合物(树枝状聚合物-sirna复合物)中更有效的sirna释放,并因此与相应的水解稳定的peg-gatg共聚物相比,提高了转染效率/沉默。此外,peg-gatge和peg-fbgatge代表了开发用于基因疗法应用的基于生物可降解酯的peg-树枝状嵌段共聚物的第一实例。gatge是基于没食子酸(ga)核和三甘醇(tg)丁酸酯臂的生物可降解且生物相容的叠氮化物封端的树枝状重复单元。由于定位在树枝状分枝的酯键(e)的存在,因此新依赖于它们的生物可降解性特性。这种生物可降解重复单元作为合成“杂化”生物可降解树枝状聚合物(图1a)和/或合成完全生物可降解树枝状聚合物(fbgatge树枝状聚合物)(图1b)的构建单元/支架是有用的。“杂化”树枝状聚合物将由以下组成:其它树枝状系统(如pamam、ppi、gatg等)(以下将详细描述gatg的情况)的耐受性核和生物可降解壳。然而在完全生物可降解的gatge树枝状系统中,所有它们的构建单元是对水解敏感的gatge单元,因此它们将表现出完全生物可降解特性。两类gatge树枝状系统可以示出外周叠氮基,其使得能够通过cuaac用多种配体对它们有效且容易地修饰。这种官能化的可能性使得这些树枝状聚合物成为用于不同生物医学应用的适合的纳米载体。在一个实施方式中,合成了2代生物可降解、生物相容且无毒的peg-gatge树枝状嵌段共聚物,其由水解稳定的gatg核和生物可降解的gatge壳组成。在一个实施方式中,合成了1、2和3代完全生物可降解、生物相容且无毒的peg-fbgatge树枝状嵌段共聚物,其完全基于生物可降解gatge单元。在两种情况下,出于获得具有低毒性、溶解度提高且循环时间更长的树枝状载体的目的,在树枝状嵌段的中心点连接了5kda的peg链(在实施例24和25中,10kda的peg)。在其它实施方式中,peg链的连接可以表示新的可能的官能化位点的引入,其允许将目标部分连接至所述共聚物,同时确保它们在相应树枝状纳米颗粒表面上的暴露。在一个实施方式中,本文所述的方法克服了先前提及的认为在同一结构中不受保护的伯/仲胺基与可水解的(并因此亲电)酯键两者的组合的困难,这是因为:i)通过酰胺键成功实现了树枝状部分/嵌段的生长,其需要不受保护的胺作为每一代的端部官能团以通过选择性攻击gatge构建单元的(亲电)中心点(羧酸)而不攻击它们的酯键来获得下一代,和ii)使用不受保护的烷基化胺通过cuaac成功实现了对这些生物可降解树枝状纳米材料的表面官能化(定量获得)。由于不需要保护/脱保护步骤,因此设想不受保护的胺基的存在和/或引入具有巨大优势。在一个实施方式中,胺官能化使得能够作为它们生物功能性的实例来研究peg-gatge作为na载体的功能。使用用于sirna递送的水解稳定的peg-gatg共聚物对应物的初步研究致使了仅23%阳性细胞(数据未显示)的非常有限的内化效率,这可能与树枝状复合物的稳定性缺陷有关。在本发明公开中,已表明在用烷基化的1,3-丙二胺(13)和苯甲胺(14)配体的cuaac官能化后(图3a),阳离子peg-gatge共聚物使得能够有效复合sirna并将其递送至细胞。二胺(13)的使用旨在通过提高正多价性来提高树枝状聚合物-sirna的结合强度。苯甲基胺(14)的使用设法进一步提高系统的疏水性。由于丁酸酯间隔臂所造成的gatge构建单元的额外的疏水性特性赋予这些可水解树枝状系统极大的复合、保护和介导sirna细胞内化的能力。此外,与它们水解稳定的peg-gatg共聚物对应物相比,发现降解位点在接近于所复合的sirna的树枝状外周的定位对于核酸从纳米颗粒的释放是至关重要的。在一个实施方式中,对于有关这种生物可降解性方面的更好结果,基于gatge的树枝状纳米材料提供了改善的降解谱。这些新的树枝状结构足够稳定以允许生物活性输送,以有利于其释放的特定百分比降解,同时在实现它们的生物学功能之后断裂成较小的片段。这致使它们清除,避免长期积累,这对于体内应用是特别重要的。引入gatge构建单元的生物可降解树枝状结构在生物医学领域具有巨大潜力,这是因为如通过烷基化胺所示,外周叠氮化物在它们表面上的存在将允许使用替代官能团和配体通过cuaac进行有效且容易的修饰:a)带正电或带负电的基团。连接胺或羧酸部分(例如,戊炔酸或苯甲酸),以允许复合作为生长因子的蛋白质(例如,bdnf)并且研究出于再生目的作为载体的bgatge。b)疏水基团,如胆固醇或脂族链(脂肪链,aliphaticchain),将允许包封亲脂性/不溶性药物并使用这些树枝状聚合物作为药物递送载体。c)造影剂(例如,gd(iii)-dtpa、gd(iii)-dota、gd(iii)-do3a),用于研究它们出于诊断目的作为生物可降解的树枝状mri造影剂的使用。因此,亲水性(三甘醇)、疏水性(没食子酸和丁酸酯间隔臂)、生物可降解性特性(酯键)和官能化可能性(外周叠氮化物)在一个支架(骨架,scaffold)中的组合赋予gatge构建单元巨大的多样性/功能性。因此,gatge树枝状结构是通用且适合的纳米载体,其不仅用于核酸递送,而且还在药物递送、诊断、疫苗、组织工程等中用于更广泛的应用,从而最终产生了新的纳米医学策略。本发明公开涉及基于没食子酸(ga)核和三甘醇(tg)丁酸酯臂,在树枝状分枝处引入生物可降解酯键(e)的新生物可降解、生物相容且叠氮化物封端的gatge树枝状重复单元。本发明公开还涉及相应的生物可降解、生物相容且叠氮化物封端的peg-gatge树枝状嵌段共聚物。本发明公开还涉及合成和表征生物可降解且生物相容的树枝状重复单元/支架(bru)的方法。以及合成和表征部分和完全生物可降解且生物相容的基于bru的peg-树枝状嵌段共聚物的方法。此外,本发明公开还提供了生物可降解且生物相容的树枝状结构的生物医学应用的实例。bru作为合成“混合”或者“杂化”生物可降解树枝状聚合物(其具有生物可降解的壳和已现有的树枝状系统的水解稳定/非可降解的核)的支架以及作为合成完全生物可降解树枝状聚合物(它的组分或层(核、中间层和壳)是完全生物可降解的)的支架是有用的。与生物不可降解的其它化合物相比,基于gatge的树枝状结构表现出作为巨大优势的生物可降解性。将gatge单元用作构建单元/支架以获得“混合”或“杂化”的生物可降解树枝状聚合物。这些“杂化”的树枝状聚合物将由以下组成:具有已现有的树枝状系统(如聚(酰胺-胺)(pamam)、聚(丙烯亚胺)(ppi)、聚(l-赖氨酸)(pll)、gatg(没食子酸三甘醇)树枝状聚合物等)的生物不可降解且耐受性的核的生物可降解的壳;和/或合成完全生物可降解树枝状聚合物(fbgatge树枝状聚合物),其将由以下组成:完全生物可降解的组分/层(从核到壳)。杂化和完全生物可降解的基于gatge的树枝状系统两者提供了外周叠氮化物,其易于通过cu(i)-催化的huisgen环化加成(cuaac,点击化学)用多种配体可官能化,因此这些新的生物可降解的基于gatge的树枝状纳米材料易于可调以用作不同生物医学应用的通用载体。本发明公开涉及生物可降解的树枝状结构,其包含g树枝状代(dendriticgenerations)和连接至树枝状结构的中心点的聚亚烷基聚醇,其中至少一代包含:有机酸;聚醚基和酯基,其中树枝状1代包含连接至聚亚烷基聚醇和连接至聚醚基的有机酸,和至少一个其它树枝状代,其包含与前代聚醚基连接和与间隔臂链(spacerchain)连接的额外的有机酸(进一步的有机酸,其它有机酸,furtherorgnanicacid),其中间隔臂链包含至少一个酯基。在一个实施方式中,树枝状1代包含有机酸,其可以连接至聚亚烷基聚醇和连接至3个聚醚基,并且其它树枝状代可以包含3个间隔臂链,其中每个间隔臂连接至有机酸。在一个实施方式中,树枝状1代还可以包含间隔臂链,其中间隔臂链包含至少一个酯基。在一个实施方式中,间隔臂链可以选自乙酸酯、丙酸酯、丁酸酯、戊酸酯、己酸酯、庚酸酯或其混合物,具体地,树枝状结构1代可以包含丁酸酯,具体地2-(2-乙氧基乙氧基)丁酸乙酯。在一个实施方式中,树枝状g代可以是树枝状2-4代,优选地,树枝状2-3代。在一个实施方式中,间隔臂链的酯还可以在其端部连接至官能团,官能团选自由以下组成的列表(清单,名单,list):胺基(氨基,amine)、叠氮基(叠氮化物)、羟基、硫醇基、羧基、烯基或炔基。在一个实施方式中,间隔臂链的酯还可以连接至2、3或其它代的有机酸,其中连接包含选自由以下组成的列表中的官能团:胺基、羟基或硫醇基。在一个实施方式中,胺基可以是丙二胺基、苯甲胺基或其它亚烷基多胺、芳香族胺、胍基、叔胺、咪唑、组氨酸或其混合物。在一个实施方式中,有机酸可以是3,5-二羟基苯甲酸、3,4-二羟基苯甲酸、2,4,6-三羟基苯甲酸、没食子酸和其衍生物,具体地是没食子酸。在一个实施方式中,聚醚基可以选自以下列表:乙二醇、二甘醇、三甘醇、四甘醇、五甘醇或其混合物,优选地三甘醇。在一个实施方式中,聚亚烷基聚醇可以选自以下列表:聚乙二醇、聚丙二醇、其混合物,优选地聚乙二醇。在一个实施方式中,聚亚烷基聚醇可以在其其它端部含有胺基、羟基、硫醇基、羧酸、叠氮基、炔基、烯基。在一个实施方式中,聚乙二醇可以包含2000-15000g/mol,具体地2000-10000g/mol的分子量。在一个实施方式中,生物可降解的树枝状结构还可以包含配体。在一个实施方式中,生物可降解的树枝状结构还可以包含靶标配体,其中所述配体为疏水基团-脂族链、芳基-荧光标签、化学药物或造影剂或者生物分子,具体地,肽、蛋白质、单糖、多糖、抗体、适体(适配子,aptamer)、糖胺聚糖(粘多糖,glycosaminoglycan)、有利于受体识别的试剂、内化试剂、核定位试剂或其混合物。在一个实施方式中,生物分子可以是核酸、药物、蛋白质、生长因子,具体地sirna。在一个实施方式中,靶标配体可以是生物分子,具体地,肽、蛋白质、多糖、抗体、糖胺聚糖、有利于受体识别的试剂、内化试剂、从细胞内涵体中生物分子逃避剂和生物分子释放试剂、稳定剂或其混合物。在一个实施方式中,靶标配体和/或配体可以结合至树枝状结构分枝或者结合至聚亚烷基聚醇。因此,现在所公开的生物可降解树枝状结构是式i所示的生物可降解树枝状结构:其中n为40-350;x为0-4;r1选自n、o或s;r2选自:z为1-9;r3为r2,其条件是在树枝状结构端部,r3为其中a选自胺基、酰胺基、叠氮基、羟基、硫醇基、羧基、异氰酸酯基、烯基或炔基;y为1-9;r4或r5为c1-c6烷基链;a、r1、r2、r3、r4、r5、n、x、y和z独立地选自彼此。在一个实施方式中,n可以是45-250,优选地70-150,更优选地90-120。在一个实施方式中,x可以为1-4,优选地1-3,更优选地1-2。在一个实施方式中,y可以为2-7,优选地2-5,更优选地3-4。在一个实施方式中,z可以为2-7,优选地2-5,更优选地3-4。在一个实施方式中,r4或r5为c2-c5烷基链、c3-c4烷基链。在一个实施方式中,a可以是胺基,其中胺基为1,3-丙二胺基、1,2-乙二胺基或其它亚烷基多胺基、苯甲胺基或其它芳族胺基、胍基、叔胺基、咪唑基、组氨酸基团或其混合物。在一个实施方式中,现在所公开的生物可降解树枝状结构还可以包含胺基、羟基、硫醇基、羧酸基、异氰酸酯基、叠氮基、炔基、环辛炔基(环甲基异辛烯胺,cyclooctin)、烯基、丙烯酸酯基,其中所述基团替代och3基团。在一个实施方式中,生物可降解树枝状结构可以包含2500-75000g/mol,具体地2500-25000g/mol或2500-27000g/mol的分子量。在一个实施方式中,生物可降解树枝状结构还可以包含配体,其中所述配体为疏水基团、脂族链、芳基、荧光标签、化学药物、造影剂、细胞内涵体生物分子逃避剂、核定位剂和生物分子释放剂、稳定剂或生物分子。在一个实施方式中,配体选自1,3-丙二胺、苯甲胺、苯甲酸、胆固醇、2,2’,2”-(10-(2-(甲基氨基)-2-氧乙基)-1,4,7,10-四氮杂环十二烷-1,4,7-三基)-三乙酸钆(iii)复合物或其混合物。在一个实施方式中,生物可降解树枝状结构可以包含作为配体的生物分子,其中生物分子为蛋白质、生长因子、核酸,具体地sirna。在一个实施方式中,生物可降解树枝状结构还可以包含靶标配体,其中所述靶标配体为生物分子,具体地,肽、蛋白质、多糖、抗体、适体、糖胺聚糖、有利于受体识别的试剂、内化试剂或其混合物。在一个实施方式中,靶标配体和/或配体结合至生物可降解树枝状结构。在一个实施方式中,生物可降解树枝状结构可以是化合物n.5、n.9、n.10、n.11、n.12、n.15、n.16、n.17、n.18、n.21、n.22、n.23、n.24、n.24或者n.25。在一个实施方式中,根据任何上述权利要求所述的生物可降解树枝状结构用于在药物中使用。本发明公开还涉及生物可降解树枝状结构用于在药剂中使用,具体地在癌症的治疗或预防中使用。在一个实施方式中,生物可降解树枝状结构可以在核酸递送、药物递送或者再生医学或基因疗法中使用。在一个实施方式中,生物可降解树枝状结构可以用作显像剂,即用于放射疗法或光疗。本发明公开还涉及包含如本文所定义的生物可降解树枝状结构和药用载体、佐剂、赋形剂或其混合物的药物组合物。在一个实施方式中,可以通过局部、口服、肠胃外或注射施用药物组合物。此外,本发明公开还涉及包含本文所公开的生物可降解树枝状结构的疫苗。本发明公开还涉及现在所公开的生物可降解树枝状结构作为施用药物和/或诊断试剂的载体的用途或者作为显像剂,即用于磁共振成像或计算机断层成像的用途。附图说明以下附图提供了用于说明描述内容的优选实施方式并且不应将其视作对本发明范围的限制。图1-a)g2生物可降解peg-gatge嵌段共聚物(peg-b[g2]-n3)的结构。b)g3完全生物可降解peg-fbgatge嵌段共聚物(peg-fb[g3]-n3)的结构。图2-a)生物可降解gatge重复单元5的合成。b)通过1hnmr,不同pd下的单元6中酯水解动力学。图3-a)烷基化丙二胺(13)和苯甲胺(14)配体的结构。b)胺封端的bd(15)和bb(16)的1hnmr光谱(400mhz,d2o)结构。c)水解稳定的hsd(19)和hsb(20)的结构。图4-a)来自:水解稳定的peg-gatg(hsd和hsb)和生物可降解peg-gatge(bd和bb)的sirnami树枝状复合物在不同n/p下的聚丙烯酰胺凝胶保留测定(page)。b)rt下的拒染测定。将单因素方差分析检验用于统计分析。显著差异:*p<0.05,**p<0.01。n/p之间的显著差异:hsb20对hsb160(p<0.05)。图5-a)不同n/p比值下通过dls测量的sirnami树枝状复合物的粒径分布(n=3,±sd)。n/p之间的显著差异:bd40对bd160(p<0.05)。b)不同n/p比值下通过dls测量的sirnami树枝状复合物的多分散指数(pdi)(n=3,±sd)。n/p之间无显著差异。c)使用dls测量的sirnami树枝状复合物(n/p80)的代表性尺寸:hsd(z均:141nm:0.38);bd(z均:134nm:0.38);hsb(z均:168nm:0.24);bar(z均:163nm:0.26)。d)不同n/p比值下,所有开发的树枝状复合物的ζ电位值。n/p之间的显著差异:bd20对bd80(p<0.05),hsd20对hsd40/80/160(p<0.01),hsb20对hsb160(p<0.01)。e)n/p80和160下sirnami树枝状复合物的tem图像:hsd、hsb、bd和bb。显著差异:*p<0.05,**p<0.01和***p<0.001。图6-u2os细胞与:(a)peg-gatge和peg-gatg树枝状共聚物:叠氮化物封端和胺封端一起温育24h时所确定的使用未处理的细胞作为参考的相对代谢活性(刃天青测定)。(b)n/p80和160时的树枝状复合物(分别等价于约0.5和1mg/ml的共聚物浓度)。显著差异:*p<0.05(单因素方差分析检验)。图7-在与dna酶i(dnasei,脱氧核糖核酸酶i)温育0、5、15、30和60min后,通过page确定的sirnami降解。裸露的sirnami。n/p160树枝状复合物与:hsd、hsb、bd和bb。图8-树枝状复合物的细胞结合。将含有cy5标记的sirnami(cy5-sirnami)的树枝状复合物与u2os细胞一起在0.1μm的最终sirnami浓度下温育24h。根据生产商的说明,将2000(l2k)用作对照。a)不同n/p下的流式细胞术表征。高亮区域对应于具有高相对fl的细胞群体。b)在n/p160下,通过cy5-sirnamibb树枝状复合物的成像流式细胞术采集扩大景深的场图像(红色)。灰色背景:明场图像;黑色背景:通道5成像(cy-5)。比例尺:10μm。显示了每一类的代表性图像:低斑点计数、中等斑点计数和高斑点计数。c)n/p160下,bd和bb的共聚焦显微镜图像。使用hoechst33342染色的核(蓝色)。表达egfpluc的细胞(绿色)。cy5-sirnami树枝状复合物(红色)。图9-对于:(a)不同n/p下,抗-egfpsirna/bd树枝状复合物和l2k。(b)n/p160下,非编码sirna/bd树枝状复合物(不存在(-)和存在(+)cq的情况下的实验)。(c)n/p160下,水解稳定的和生物可降解的抗-egfpsirna/peg-gatg树枝状复合物:hsd、bd、hsb、bb(不存在(-)和存在(+)cq的情况下的实验)转染后72h的荧光素酶活性百分比。显著差异:*p<0.05,**p<0.01和***p<0.001(单因素方差分析检验)。对于每个n/p,符号#表示不存在和存在cq的情况下的实验之间的显著差异。图10-叔丁基没食子酸酯(2)的1h和13cnmr光谱。图11-2-[2-(2-叠氮基乙氧基)乙氧基]乙基4-溴丁酸酯(4)、三{2-[2-(2-叠氮基乙氧基)乙氧基]乙基}4,4’,4”-{[5-(叔丁氧羰基)苯-1,2,3-三基]三(氧)}三丁酸酯的1h和13cnmr光谱。图12-三{2-[2-(2-叠氮基乙氧基)乙氧基]乙基}4,4',4”-{[5-(叔丁氧羰基)苯-1,2,3-三基]三(氧)}三丁酸酯的1h和13cnmr光谱。图13-3,4,5-三(4-{2-[2-(2-叠氮基乙氧基)乙氧基]乙氧基}-4-氧丁氧基)苯甲酸(5)(gatge构建单元)的1h和13cnmr光谱图14-peg-[g1]-n3(8)的1hnmr光谱。图15-peg-[g1]-nh2的1hnmr光谱图16-peg-b[g2]-n3(9)的1h和13cnmr光谱。图17-peg-fb[g1]-n3(10)的1h光谱。图18-peg-fb[g2]-n3(11)的1h光谱。图19-peg-fb[g3]-n3(12)的1h光谱。图20-bd(15)的1h和13cnmr光谱。图21-bb(16)的1hnmr光谱。图22-fbd(17)的1h光谱。图23.fbb(18)的1h光谱。图24-hsd(19)的1hnmr光谱和信号归属(assignment)。图25-hsd(19)的13cnmr光谱。图26-hsb(20)的1hnmr光谱和信号归属。图27-hsb(20)的13cnmr光谱。图28-peg-b[g2]-n3(9)的maldi-tof光谱。图29-peg-b[g2]-n3(9)的ftir透射光谱(kbr)。图30-peg-b[g2]-n3(9)、bd(15)、bb(16)、hsd(19)和hsb(20)的ftir透射光谱(atr)。图31-可降解性研究。不同时间点(0、1、7、14和30天),胺封端的bd(15)和bb(16)的1hnmr光谱(600mhz,d2o,pbs3×,pd7.4)。通过虚线矩形框突出显示了光谱上观察到的最显著改变。图32-树枝状复合物在不同介质中的稳定性。生物可降解树枝状复合物在具有20%胎牛血清(fbs)的1×pbs中的稳定性。a)bd(n/p80);b)bd(n/p160);c)bb(n/p80);d)bb(n/p160)。以n/p80和160形成树枝状复合物,在pbs+20%fbs中稀释2倍,并在37℃温育1h(红色)、4h(蓝色)和8h(棕色)。然后,通过dls确定平均粒度。采集具有fbs且无树枝状复合物的pbs谱(空白曲线,绿色)以区分蛋白质-相关聚集体。图33-生物可降解树枝状复合物在:a)ph5.0和b)ph7.4的稳定性。a.1)bd(n/p80);a.2)bd(n/p160);a.3)bb(n/p80);a.4)bb(n/p160);b.1)bd(n/p80);b.2)bd(n/p160);b.3)bb(n/p80);b.4)bb(n/p160)。以n/p80和160形成树枝状复合物,在10mmnaoac+137mmnaclph5.0和1×pbsph7.4中稀释2倍,并在37℃温育1h(蓝色)、4h(绿色)和8h(棕色)。然后,通过dls确定平均粒度。红色曲线代表未稀释的树枝状复合物。图34-a)bd和b)bb树枝状复合物的肝素解离测定。将n/p160的bd和bb树枝状复合物在37℃在生理学盐和ph条件下与提高浓度的肝素温育2h。然后,使用page运行样品以验证sirnami从树枝状复合物的解离程度。在sirnami泳道中,加载与用于树枝状复合物制备相同的量的游离sirnami。图35-树枝状复合物降解研究。a)bd和b)bbsirna树枝状复合物的降解研究。将n/p160的bd和bb在酸(ph5.0)和生理学ph(ph7.4)条件下温育1、24和48h。然后,在37℃将树枝状复合物与肝素温育2h(对于bd和bb树枝状复合物,最终肝素浓度分别为0.010mg/ml和0.025mg/ml),从而以间接方式确定随时间释放的sirna的量。图36-每个细胞的树枝状复合物负载的小泡(囊泡,载体,vesicles)(dlv)。a)hsd、b)hsb、c)bd、d)bb。左列:细胞掩蔽(cellmask)的斑点计数。右列:细胞质掩蔽(cytoplasmmask)的斑点计数。绿色:cy-5阳性细胞区(膜和细胞质);低:低斑点计数细胞(细胞质)区;橙色:中等斑点计数细胞(细胞质)区;红色:高斑点计数细胞(细胞质)区。图37-方案1.peg-gatge共聚物9的合成和1hnmr光谱(400mhz,cd2cl2)。图38-方案2.完全生物可降解的peg-fbgatge共聚物10(g1)、11(g2)和12(g3)的合成。图39-具有聚乙二醇5000的g2生物可降解的peg-gatge嵌段共聚物(peg-b[g2]-n39)的结构。图40-具有聚乙二醇5000的g1完全生物可降解的peg-fbgatge嵌段共聚物(peg-fb[g1]-n310)的结构。图41-具有聚乙二醇5000的g2完全生物可降解的peg-fbgatge嵌段共聚物(peg-fb[g2]-n311)的结构。图42-具有聚乙二醇5000的g3完全生物可降解的peg-fbgatge嵌段共聚物(peg-fb[g3]-n312)的结构。图43-具有聚乙二醇5000且1,3-丙二胺封端的g2生物可降解的peg-gatge嵌段共聚物(bd15)的结构。图44-具有聚乙二醇5000且苯甲基胺封端的g2生物可降解的peg-gatge嵌段共聚物(bb16)的结构。图45-具有聚乙二醇5000且1,3-丙烯胺封端的g3完全生物可降解的peg-gatge嵌段共聚物(fbd17)的结构。图46-具有聚乙二醇5000且苯甲基胺封端的g3完全生物可降解的peg-gatge嵌段共聚物(fbb18)的结构。图47-具有聚乙二醇5000且苯甲酸封端的g2生物可降解的peg-gatge嵌段共聚物(bbz21)的结构。图48-具有聚乙二醇5000且胆固醇封端的g2生物可降解的peg-gatge嵌段共聚物(bch22)的结构。图49-具有聚乙二醇5000且do3a-gd封端的g2生物可降解的peg-gatge嵌段共聚物(bdo3a-gd23)的结构。(do3a-gd=2,2',2”-(10-(2-(甲基氨基)-2-氧乙基)-1,4,7,10-四氮杂环十二烷-1,4,7-三基)-三乙酸酯钆(iii)复合物)。图50-具有聚乙二醇10000的g1生物不可降解的peg-gatge嵌段共聚物(peg(10000)-[g1]-n324)的结构。图51-具有聚乙二醇10000的g2生物可降解的peg-gatge嵌段共聚物(peg(10000)-b[g2]-n325)的结构。具体实施方式在一个实施方式中,如下所示进行生物可降解gatge重复单元的设计、合成和表征。图2a显示了包含没食子酸核和三甘醇-脂族酯臂的gatge单元5的合成。在一个实施方式中,现在所公开的合成路线允许通过改变包含酯基(图2中以粉红色显示)的间隔臂的长度/性质来进行结构改变和可降解性调整。从可商购的氯代三甘醇(3)、4-溴丁酸和没食子酸(1)有效合成了构建单元5。用叠氮三甘醇(得自3和nan3)16,通过dcc和dmap对4-溴丁酸进行初步处理致使产生了酯4,具体地,综合得率97%。后续用叔丁基没食子酸酯(2)(k2co3,18c6)对4进行偶联,然后水解,从而提供了所需的构建单元5,具体地,总得率82%(图2)。实施例在其中将peg-gatge共聚物设想为na载体的实施方式中,对易得的gatge单元6进行初步可降解性测试(h2,pd/c),其引入了作为阳离子替代物的端部伯胺基来模拟对na复合所设想的阳离子性质。如图2b所示,在模拟生理学和内涵体ph条件(分别为7.4和5.0;37℃)下进行降解研究,并以时间-依赖性方式提供了酯键水解。在pd7.4获得了比5.0更高的降解速率(图2b,在重水(氘化水)中进行实验)。该观察结果可以解释为在重水中,在碱性ph(pd9.8,图2b)下催化了水解反应。然而,值得提及地,在细胞内,可以预期在较低的ph下具有较高的水解速率(如在内涵体ph下的情况),这是因为在水中酸催化的降解可以以比重水(d2o)中快三倍的速率发生。17在一个实施方式中,如下所示进行了生物可降解peg-gatge嵌段共聚物的设计、合成和表征。在一个实施方式中,通过5受控的可靠合成和考虑较低g的加快的接近和提高的生物相容性,作为本发明公开的概念验证(gatge构建单元5),注意力集中在g2peg-gatge共聚物(peg-b[g2])的合成和sirna递送性质的评价。设计了杂化树枝状共聚物,其组合了位于核的经典gatg单元(蓝色,图1),核周围围绕着生物可降解gatge单元的壳(绿色,图1)。在一个实施方式中,将降解位点的定位限定在树枝状外周并与生物活性物质(在本实施例中为sirna)紧密接触,与水解稳定的peg-gatg对应物相比,这将提高生物活性物质的胞内释放和生物效率。因此,按照从一甲基醚peg氨基(peg-nh2·hcl,mn=5079g/mol,mw=5113g/mol,pdi=1.007)和gatg单元(7)开始的歧化策略,进行peg-gatge共聚物的制备(方案1)。16以这种方式,容易地获得了嵌段共聚物peg-[g1]-n3(8),具体地,在通过沉淀纯化后,得率93%(方案1)。18在一个实施方式中,8中的端部叠氮化物催化氢化,然后用5(edc,hobt)对所得三胺进行处理致使产生了所期望的peg-b[g2]-n3(9),具体地,得率86%(方案1)。在一个实施方式中,通过1h和13cnmr(1d和2d)、maldi-tofms和ftir表征嵌段共聚物9(图16、28、29)。在一个实施方式中,通过1h-nmr光谱(cd2cl2),通过跟踪丁酸酯间隔臂信号(质子h、i、j)和邻近于叠氮基的那些(l和l':3.31-3.35ppm)(方案1)并且通过对应于邻近于胺基的亚甲基质子的信号的消失(2.72-2.78ppm)来监控树枝状的生长(图15)。在一个实施方式中,9的maldi-tof光谱显示出与通过对应于所期望的peg寡聚物的44da间隔的,具有钠的嵌段共聚物加合物有关的峰值的高斯分布(图28)。实验分子量(mp)、分子量(mw)和数均分子量(mn)与计算值一致。ftir光谱显示在9中,在1736和2110cm-1存在特征峰,其对应于酯和叠氮基(图29)。在一个实施方式中,如下所示进行了完全生物可降解的peg-gatge嵌段共聚物的设计、合成和表征。在一个实施方式中,公开了peg-gatge树枝状嵌段共聚物的完全生物可降解性。设计了完全生物可降解的树枝状共聚物并合成至g3(peg-fb[g3],图1b)。peg-fb[g3]完全基于生物可降解构建单元5,因此它是完全生物可降解的。在一个实施方式中,在全部树枝状结构(包括peg和树枝状部分之间新的可降解键—参见图1b中以红色显示的键)中限定40个降解位点的定位将致使断裂为非常小的片段,这些片段将更容易从生物中排泄出,并且与杂化生物可降解peg-gatge对应物相比,还预期更好的生物活性物质胞内释放和生物效率。因此,按照从聚(乙二醇)甲醚(peg(5000)-oh,mw=5000g/mol)和gatg单元(5)开始的歧化策略,进行完全生物可降解的peg-gatge共聚物的制备(方案2)。16以这种方式,容易地获得了嵌段共聚物peg-fb[g1]-n3(10),具体地,在通过沉淀纯化后,得率91%(方案2)。在一个实施方式中,在酸性介质中,10中的端部叠氮化物催化氢化,然后用5(edc,hobt,et3n)对所得的质子化的三胺进行处理产生了所期望的peg-fb[g2]-n3(11),具体地,得率81%(方案2)。在一个实施方式中,以与获得peg-fb[g2]-n3(11)相似的方式,在酸性介质中,11中的端部叠氮化物后续催化氢化,然后用5(edc,hobt,et3n)对所得的质子化的三胺进行连续处理产生了所期望的peg-fb[g3]-n3(12),具体地,得率75%(方案2)。在一个实施方式中,通过nmr表征完全生物可降解的嵌段共聚物10、11和12(图17、18和19)。在一个实施方式中,通过1h-nmr光谱(cd2cl2),通过跟踪邻近于叠氮基的信号(约3.32-3.35ppm)并且通过对应于邻近于胺基的亚甲基质子的信号的消失(2.72-2.78ppm)来监控树枝状的生长。还通过生长代对芳香族信号裂分的影响:在peg-fb[g1]-n3的1hnmr光谱中出现位于7.25ppm处的尖锐单峰(图17),在peg-fb[g2]-n3和peg-fb[g3]-n3的光谱中发现了位于7.02和7.25ppm的两个单峰(分别为图18和19)。作为生物可降解peg-gatge共聚物的多价官能化的实施,通过cuaac用不受保护的胺进行官能化。在一个实施方式中,如下所示实施了通过cuaac用不受保护的胺对生物可降解的peg-gatge共聚物的多价官能化。通常利用生理学ph下胺的阳离子性质来使得能够在树枝状复合物内进行na的离子缩合和保护。先前已提议胺封端的gatg树枝状聚合物和它们的聚乙二醇化的共聚物作为pdna的载体。然而,相同共聚物用于sirna递送的使用致使了非常有限的内化效率,仅有23%的阳性细胞,这可能与树枝状复合物的稳定性缺陷有关。有关尺寸、形态、灵活性和电荷的pdna和sirna分子之间的根本差异可以致使不太有效的相互作用和对后者较少的保护。19因此,对于基因递送常用的阳离子载体不必需致使产生最优的sirna载体。20在本发明中,已表明在用烷基化的丙二胺(13)和苯甲基胺(14)配体的cuaac官能化后(图3a),阳离子peg-gatg和peg-gatge共聚物使得能够有效复合sirna并将其递送至细胞。二胺(13)的使用旨在通过提高正多价性来提高树枝状聚合物-sirna的结合强度。苯甲基胺(14)的使用设法进一步提高系统的疏水性。先前已观察到来源于peg-gatg共聚物的具有该芳香部分的聚离子复合物(pic)胶束作为蛋白质载体表现出较高的稳定性。21在一个实施方式中,使用无n-保护基的烷基化的游离胺进行peg-b[g2]-n3(9)和peg-fb[g3]-n3(12)的官能化,其简化了方法的步骤数目。作为替代,将它们的铵盐13和14用作掩盖它们的亲核性和避免通过与酯基的副反应的降解的方式。此外,该策略允许已具有复合核酸所必需的正电荷。在一个实施方式中,通过以下方法进行cuaac,具体地,在1∶1的dmf:h2o中,通过每叠氮化物5mol%的cuso4和每叠氮化物25mol%的抗坏血酸钠,具体地,在室温下,对于g2进行12h,对于g3进行24h。在通过超滤纯化后,以定量得率容易地获得了所得二胺和苯甲基胺封端的共聚物bd(15)和bb(16)(图3b)。通过1hnmr(d2o)(邻近于叠氮基的亚甲基质子的信号,图3b)和ftir(图30中的叠氮化物条带(2101cm-1))监控复合的完成。还在1hnmr中通过出现:对应于三唑质子的8.20ppm周围的信号(m和m')、由于对于三唑基处于α位的质子所造成的4.5-4.7ppm周围的多重峰(l和l')以及对于胺配体的特征信号(对于15的n、o、p和q和对于16的r、s和t)确认成功偶联(图3b)。按照相似的反应条件,还合成了来源于完全生物可降解的peg-fb[g3](12)的胺封端的共聚物fbd(17)、fbb(18)。以同样的方式,还作为对照合成了来源于水解稳定的g2peg-gatg共聚物的hsd(19)、hsb(20)以用于sirna递送测定(图3c)。有关这四种胺-树枝状共聚物(17、18、19和20)的合成和表征的详细信息可见于本发明文档(图22-27)。类似地,对于gatge单元6所实施的研究,对生物可降解共聚物bd(15)和bb(16)实施了可降解性测试。定性nmr数据分析还表明了这些树枝状共聚物在pd7.4随时间的降解(图31),这是因为可以在光谱上清楚地观察到显著的变化,其表明了在模拟生理条件下所提议的树枝状共聚物的不稳定性/可降解性。这些变化包括信号强度和/或形状的变化以及对应于降解产物和新片段的质子的信号出现和/或消失。具体gatge构建单元5的树枝状结构的生物医学应用的实施例:peg-gatge共聚物作为sirna载体的生物功能性评价。在一个实施方式中,如下所示进行树枝状复合物的制备和表征:研究了胺封端的共聚物15、16、19和20与sirna的结合并且评价了所得树枝状复合物的物理化学性质。对于其中不评价生物活性的实验,由于其易于合成和以较高得率和纯度获得的可能性,作为抗增强型绿色荧光蛋白sirna(抗egfpsirna),将具有完全相同序列的双链dna用于模拟sirna(sirnami)。在一个实施方式中,确定了sirna的结合能力。一开始,通过聚丙烯酰胺凝胶阻滞测定(page)评价了共聚物与sirnami的相互作用强度,与常规琼脂糖凝胶相比,其简要来说,na提供了更高的分辨率。在凝胶中迁移的游离sirnami的量随着所使用的共聚物的量的增加而降低(n/p电荷比在20至160的范围内)(图4a)。电荷比(n/p)定义为树枝状共聚物中可质子化的伯胺的最大数目与sirna或sirnami中带负电的磷酸盐的数目之间的比值。通过核酸染料可接近性测定研究共聚物的复合效率。复合的sirnami的量随n/p而提高(图4b)。对于所有共聚物,在所分析的全部n/p范围内,复合≥70%,在n/p≥40时,该值>80%。如所期望的,page和测定两者均显示由于二胺的二价性质,二胺封端的(d)是保留和复合sirnami最有效的基团。就树枝状骨架而言,由于有助于提高包装的疏水性间隔臂,gatge构建单元的引入产生了更有效的复合。总之,就sirnami复合而言,bd致使产生了最有效的共聚物,即使在所研究的最小n/p值,其具有约90%的复合的sirnami。在一个实施方式中,如下所示确定了树枝状复合物的尺寸和形态:就尺寸和形态,分别使用动态光散射(dls)和透射电子显微镜术(tem)表征树枝状复合物。不考虑树枝状共聚物,对于所测试的所有n/p获得了处于纳米尺度的窄颗粒尺寸分布,并且具有适合于细胞吸收的尺寸和多分散性。10e,22此外,发现尺寸和pdi两者均与n/p无关。对于二胺系列获得了平均粒度约145nm的树枝状复合物,对于苯甲基胺系列,获得了平均粒度约175nm的(图5a和5c)。对于苯甲基胺,树枝状复合物群体比二胺共聚物稍微更均一(pdi分别为约0.3和0.4,图5b)。因此,为了比较基于两个端部胺基(二胺和苯甲基胺)的树枝状复合物的稳定性,在含有20%(v/v)fbs的pbs中温育1、4和8h后测量了n/p80和160的生物可降解树枝状复合物的尺寸(图32)。所得的相对于尺寸谱的峰强度表明在存在血清的情况下bd树枝状复合物的温育引起最大峰强度的降低,峰宽增加和向增大尺寸的迁移。然而,对于bb树枝状复合物未观察到显著差异,表明该系统的高稳定性。当在不同时间点(1、4和8h),作为ph的函数研究树枝状复合物(n/p80和160)的尺寸时(图33),当分析更高强度的峰时(图33b),在ph7.4,对于所有配方未观察到显著差异。而在ph5,观察到树枝状复合物群体向增大的尺寸的轻微迁移(图33a)。对于bb树枝状复合物,这些变化不太明显,再次表明了它们更高的稳定性。在所有情况下,观察到较小群体的出现,我们将其归因于树枝状复合物降解副产物的出现。在这些条件下,由于不存在其它较大的分子,这些是可以区分的(如在先前讨论的数据的情况下,图32,其中在存在fbs的情况下进行表征)。如tem图中所示(图5e),所有树枝状复合物显示出球形和致密结构,其尺寸与通过dls所获得的那些良好相关(图5a)。转染树枝状复合物的细胞活性的另一个重要参数为一旦处于细胞内部,它们释放sirna的能力。因此,为了测试树枝状复合物形成的可逆性,将生物可降解的树枝状复合物在37℃以及在生理学盐和ph条件下与肝素(通常用于测试不稳定性和核酸从树枝状复合物释放的模式聚阴离子)一起温育。20c,23测试了bd和bb树枝状复合物(n/p比160),其中当以增加的肝素浓度测试这些时,观察到sirna的彻底释放(图34)。总的来说,根据在存在血清的情况下并且作为ph的函数的这些结果和树枝状复合物的稳定性,可以推断就胞外稳定性和胞内sirna释放而言,存在优良的平衡,特别是对于bb-基树枝状复合物。载体和na之间的疏水性相互作用的重要性似乎再次从所有这些结果中出现,其中端部芳基致使产生了更均一和稳定的树枝状复合物。在一个实施方式中,进行了ζ电位。通过激光多普勒电泳测量了树枝状复合物在水中的表面电荷(图5d)。对于所有测试的配方,树枝状复合物的净电荷为正,与苯甲基胺相比,二胺树枝状复合物表现出更高的ζ电位,这与预期的更高的正电荷密度一致。树枝状复合物降解研究。为了评价作为时间函数且在不同ph下sirna从树枝状复合物中的释放,将bd和bbsirna树枝状复合物在酸性(ph5.0)和生理学(ph7.4)ph条件下温育1、24和48h。然后,用肝素处理树枝状复合物,并通过page确定所释放的sirna的量。结果表明即使在温育1h后,已释放了大量的sirna(图35)。这在ph7.4特别显著,与对于gatge单元6(图2b)在pd7.4时所观察到的更高的降解百分比一致。作为整体观察降解研究,可以推断树枝状结构(部分3.1和图2b,部分3.2和图31)的水解速率比树枝状复合物的那些更缓慢。然而,必须考虑两个实验的缓冲液是在水的不同同位素类型中配制的:对于通过树枝状结构的nmr的可降解性研究为重水,而“正常”水用于通过page的树枝状复合物降解研究。如先前所讨论的,由于动力学同位素效应,两种介质中的催化速率可以显著不同。17此外并且不考虑同位素效应,仅每个peg-树枝状嵌段共聚物组中的“低”降解百分比可以致使(一些peg-gatge分子和一些sirna分子之间形成的)树枝状复合物的“高”不稳定性,其反过来可以致使大量释放的sirna,如图35所示。整体上,这些结果确认具有两个氨基的共聚物的官能化致使了良好限定的树枝状复合物,其具有适合于细胞吸收和sirna递送的性质。在一个实施方式中,进行了生物学性能评价。对所有共聚物评价了它们的细胞毒性、保护sirna不被核酸内切酶降解的能力和转染效率。在一个实施方式中,进行了细胞代谢活性/细胞毒性。考虑到毒性可以代表在生物医学中实现大分子体系的障碍,在人骨肉瘤u2os细胞中评价了共聚物和树枝状复合物的细胞毒性。通过基于刃天青的测定,就细胞代谢活性变化评价了细胞毒性。对于游离的叠氮化物封端和胺封端的共聚物(9、15、16、19和20),所评价的浓度在0.25至1.5mg/ml之间的范围内(图6a)。在所有情况下,温育24h后,细胞代谢活性高于85%,其表明即使在用带正电的端部基团官能化后,这些共聚物具有低细胞毒性谱。随后,还测试了对细胞具有潜在更强毒性的树枝状复合物(n/p比为80和160)的毒性。再次,在所有情况下,获得了大于90%的存活力(图6b)。核酸内切酶保护。保护sirna抵抗内源核酸酶是开发新na载体中的关键参数。为此,将从四种共聚物和sirnami以n/p160所制备的树枝状复合物与核酸内切酶温育不同时间段(5-60min)。然后,在用十二烷基硫酸钠(sds)从复合物中替换sirnami之后,通过page分析样品(图7)。裸露的na在5min内完全降解,而对于复合物,随时间观察到不同程度的sirnami保护。bb显示出最高的保护能力,即使在温育60min后观察,仍具有显著的不受影响的sirnami水平。这再次表明通过芳香族苯甲基胺(b)以及gatge中的疏水性间隔臂一起提供的额外的疏水性贡献为na提供抵抗降解的非常好的保护的相关性。先前研究显示胺封端的peg-gatg不能保护pdna抵抗超过5min的时间段的降解。24这些结果表明了通过这些氨化基团的外周官能化的益处。在一个实施方式中,细胞结合/吸收。评价了所有开发的树枝状复合物hsd结合和/或穿过细胞膜的能力。将稳定表达融合蛋白egfp-荧光素酶(u2os/egfpluc细胞)的u2os细胞与具有cy5标记的sirnami的树枝状复合物在37℃下温育24h。通过荧光激活细胞分类术(facs)和共聚焦荧光显微术分析细胞。对于用树枝状复合物处理的所有细胞,facs数据(图8a)显示由于树枝状复合物的细胞结合/内化,与未处理的细胞相比,向更高荧光强度(fl)的迁移。更高的fl值与n/p比的提高相关,这可以通过树枝状复合物中更高的na保护来解释。对于所有载体,阳性细胞百分比始终大于95%(表1)。bb(n/p80和160)显示出最高的内化效率,其fl值更接近于用于体外转染的金标准试剂2000(l2k)(图8a)。表1.通过流式细胞术具有结合的树枝状复合物的细胞(阳性细胞)的百分比还定量了树枝状复合物的内化,并且由于成像流式细胞术使得能够将仅结合至细胞膜的纳米颗粒与存在于细胞的细胞质(内化)中的纳米颗粒相区分,因此通过该技术进行表征。阳性细胞百分比始终大于95%(表2),从而确认接触24h后,树枝状复合物已完全内化。对于所有共聚物,确定了每个细胞中树枝状复合物负载的小泡(dlv)的百分比(表3和图36)。在一些不同平面中在z轴上采集细胞图像,并产生图像投影。确定了限定每个细胞中具有低(<1.5)、中(1.5-5.5)或高(>5.5)dlv数目的细胞的三个组(图8b以及表3和图36)。发现生物可降解共聚物显示出比它们水解稳定的对应物更高的相对内化效率。bb为具有最高百分比(44%)的具有高dlv数目的细胞的共聚物(图8b和表3)。表2.通过成像流式细胞术以n/p160具有内化树枝状复合物的细胞(阳性细胞)的百分比处理阳性细胞%nt0.4l2k98.7hsd99.0bd94.6hsb99.8bb99.8表3.树枝状复合物负载的小泡(dlv)。此外,u2os细胞的共聚焦显微镜图像显示了点状cy5荧光图案,其指示了树枝状复合物的内涵体吸收(图8b)。bb树枝状复合物中通过gatge单元和苯甲基赋予的提高的na保护能力可以以两种方式证明通过该系统介导的最高吸收:i)更好的sirna保护方式,更多完整的标记的cy5-sirna将能够进入细胞;ii)通过gatge赋予的额外的疏水性可以改善细胞膜相互作用并辅助内化。在一个实施方式中,转染效率如下所示。在u2os/egfpluc细胞中测试了树枝状复合物介导基因沉默的能力。将细胞与含有抗egfpsirna的树枝状复合物(图9a和9c)和复合了非编码sirna的树枝状复合物(阴性对照,图9b)一起在37℃温育24h。通过相对于未处理的细胞的荧光素酶活性的降低来评价转染效率。当用复合了非编码sirna的bd树枝状复合物(n/p160)处理细胞时,观察到荧光素酶活性升高(图9b),这是可以归因于载体对启动子活性的影响的作用。25基于处于一些n/p的bd,通过抗egfpsirna树枝状复合物实施的实验提供了荧光素酶活性的小幅降低(图9a),这与先前来自其它组的报道一致。13,26为了研究这些结果是否表明了内涵体逃逸过程—sirna递送中的关键问题—的损害,在转染期间加入氯喹(cq,内涵体小泡的破坏剂)。在所应用的条件下,cq将引起反离子在内涵体中的积累,从而致使内涵体膨胀和破裂。27在存在cq的情况下,在n/p≥40观察到荧光素酶活性的显著降低,在n/p160,其占据了高达80%的沉默。当在存在该试剂的情况下测试了恒定n/p下所有树枝状载体的转染效率时,除水解稳定的hsb共聚物外,在所有情况下均观察到沉默(图9c);其中生物可降解系列(bd和bb)介导了最高的转染效率。这种功能作用可以与gatge重复单元的可降解性有关,其具有接近于sirna结合位点的降解位点。因此,揭示可降解性是从这些颗粒中释放sirna的重要特征,相对于水解稳定的peg-gatg对应物,其有助于改善转染效率。有趣地,bb比bd更高的内化sirna的能力(图8)不会以优良的沉默效率转化。这种作用可能是因为其较高的na保护损害了有关bd的后续胞内sirna释放。由于当通过内涵体破坏分子辅助时,树枝状复合物显示出优良的潜力,因此具有这些内涵体溶解性质的树枝状复合物的能力如我们先前所研究的。28在一个实施方式中,无水ch2cl2购自prolabo。无水dmf、et3n、没食子酸、叔丁醇、草酸、nahco3、na2so4、4-溴丁酸、n,n'-二环己基碳二亚胺(dcc)、二甲基氨吡啶(dmap)、k2co3、18-冠-6、pd/c、羟基苯并三唑(hobt)、抗坏血酸钠和聚(乙二醇)甲醚(peg-oh;mw5000g/mol)购自sigma。一甲基醚peg(5000)氨基(peg-nh2·hcl,mn=5079da,mw=5113,mw/mn=1.007)和一甲基醚peg(10000)氨基(peg-nh2·hcl,mn=10083da,mw=10153,mw/mn=1.007)购自jenkemtechnologyusa。n1-(丙-2-炔-1-基)丙烷-1,3-二胺·2hcl和4-乙炔基-苯甲胺·hcl购自amatekchemical。所有溶剂为hplc级并且使用时无需进一步纯化。氘化溶剂购自cortecnetsas。在一个实施方式中,使用230-400目硅胶进行柱色谱。在二氧化硅60/f-254铝背板(e.merck)上进行薄层色谱(tlc)。在具有1kda膜的amicon搅拌池上进行超滤。超滤膜购自millipore。nanopure水(18mω·cm)得自milli-q水过滤系统,milliporecorp。未标记的sirnami/sirna和在有义链的5'端标记的sirnami/sirna双螺旋购自integrateddnatechnologies。无核酸酶的水购自qiagen。荧光素酶测定系统购自promega。dmem和opti-mem购自gibco。胎牛血清(fbs)购自gibco。细胞培养板购自bdbiosciences。hoecsht33342购自lifetechnologies。edvardsmith教授(karolinskainstitute,sweden)友善地赠送了u2os/gfpluc细胞。按照先前报道的规程制备了2-[2-(2-叠氮基乙氧基)乙氧基]乙醇(叠氮三甘醇)、3,4,5-三-(2-(2-(2-叠氮基乙氧基)乙氧基)乙基)苯甲酸(gatg构建单元7)、[4,10-双羧甲基-7-[(2-丙炔基胺甲酰基)-甲基]-1,4,7,10-四氮杂-环十二烷-1-基]-乙酸酯钆(iii)复合物(炔基-do3a-gd)以及1和2代水解稳定的peg-gatg树枝状嵌段共聚物。16,18,29按照文献中已知的程序制备4-乙炔基苯甲酸。30将k2co3在65℃减压干燥。使用brukeravanceiii400mhz和brukeravanceiiihd600mhz光谱仪在d2o、cd2cl2或cdcl3中记录nmr光谱。以ppm(δ单位)报告化学位移并且参考残余溶剂信号(cd2cl2,cdcl3)或hod信号(d2o)进行比较。使用通过ltqtuneplus2.5.5和xcalibur2.1.0控制的ltqorbitrapxl质谱仪进行si-ms分析。电喷雾电离(esi)的毛细管电压设置为3000v。毛细管温度为250℃。鞘气(保护气,鞘流气,sheathgas)流速(氮气)设置为5(软件设置的任意单位)。毛细管电压为16v,管透镜电压为80v。使用ftir-ramanperkinelmer2000光谱仪(kbr)和配备有金刚石晶体的ft-irperkinelmerspectrumtwo(atr)记录ir光谱。对于kbr技术,通过将2mgpeg-树枝状嵌段共聚物(在45℃真空干燥24h)与200mgkbr(在105℃干燥24h)共混来制备每个颗粒。在用n2对样品室吹扫5min后,立即通过背景扣除,在400至4000cm-1的范围内以4cm-1的光谱分辨率的200个干涉图积累来记录ir光谱。对于atr技术,直接使用peg-树枝状嵌段共聚物而无需进一步制备。通过背景扣除,在400至4000cm-1的范围内以4cm-1的光谱分辨率的20个干涉图积累来记录ir光谱。在以正线性模式运行的具有ndyag激光器的brukerautoflexiii上进行maldi-tofms。将样品以2×10-4m的浓度溶于meoh并与基质(反式-2-[3-(4-叔丁基苯基)-2-甲基-2-丙叉基]丙二腈,dctb,10mg/ml,ch2cl2中的溶液)以基质:样品4∶1的比例混合。将nai用作阳离子化试剂。在perkin-elmeroptima4300-dv上进行电感耦合等离子体发射光谱(icp-oes)测量。对所有标准品和样品以2mg/l的最终浓度加内标。在setaramsetsysevolutiontga上进行热重分析(tga)测量。在一个实施方式中,如下所述进行叔丁基没食子酸酯(2)的合成和表征:将edc·hcl(1180mg,6.17mmol)以小部分缓慢加入至没食子酸(1000mg,5.88mmol)在无水tbuoh(35ml)中的混悬液中。在惰性气氛(ar)下,将反应混合物在rt下磁力搅拌48h。然后,加入et2o(25-40ml)和草酸(970mg,0.29mmol)。将所得混合物过滤并用0.3mnahco3清洗滤液、干燥(na2so4)并浓缩以提供作为浅黄色固体的2(980mg,74%)。1hnmr(300mhz,cdcl3)δ:1.54(s,9h),5.22(brs,3h),7.20(s,2h)。13cnmr(75mhz,cdcl3)δ:31.0,81.4,109.7,123.1,136.6,143.9,166.6。c11h13o5-的esi-ms计算值:225.08412。[m-h]-的实验值:225.07795。在一个实施方式中,如下所述进行2-[2-(2-叠氮基乙氧基)乙氧基]乙基4-溴丁酸酯(4)的合成和表征:将2-[2-(2-叠氮基乙氧基)乙氧基]乙醇(在文中称为“叠氮化三甘醇”)(504mg,2.88mmol)、4-溴丁酸(720mg,4.31mmol)、dcc(890mg,4.31mmol)和4-二甲基氨吡啶(dmap)(35mg,0.29mmol)溶于无水ch2cl2(5.7ml)。将混悬液在rt磁力搅拌12h,然后加入et3n(1.0ml,7.19mmol)并搅拌1h。将混悬液蒸发并将所得残余物再悬浮于et2o中并过滤。将滤液蒸发并通过柱色谱(己烷/乙酸乙酯[2∶1])纯化所得黄色油以获得作为浅黄色油的4(906mg,97%)。1hnmr(400mhz,cdcl3)δ:2.08(五重峰,j=6.8hz,2h),2.44(t,j=7.1hz,2h),3.30(t,j=4.6hz,2h),3.38(t,j=6.5hz,2h),3.57-3.64(m,8h),4.16(t,j=4.7hz,2h)。13cnmr(100mhz,cdcl3)δ:27.4,32.1,32.4,50.3,63.3,68.8,69.8,70.3,70.4,172.1。c10h18brn3nao4+的esi-ms理论值:346.03784。[m+na]+的实验值:346.03663。在一个实施方式中,如下所述进行3,4,5-三(4-{2-[2-(2-叠氮基乙氧基)乙氧基]乙氧基}-4-氧丁氧基)苯甲酸(5)(gatge构建单元)的合成和表征:在ar下,将叔丁基没食子酸酯2(97mg,0.43mmol)、无水k2co3(596mg,4.31mmol)和18-冠-6(11.4mg,0.04mmol)顺序加入至4(559mg,1.72mmol)在无水dmf(0.86ml)中的溶液中。然后,将反应混合物在磁力搅拌下在80℃加热12h。冷却至rt后,蒸发溶剂并过滤所得粗产物以除去固体残余物。浓缩滤液并通过柱色谱法(己烷/乙酸乙酯[1∶2])纯化所得残余物以获得作为浅黄色油的三{2-[2-(2-叠氮基乙氧基)乙氧基]乙基}4,4',4”-{[5-(叔丁氧羰基)苯-1,2,3-三基]三(氧)}三丁酸酯(336mg,82%)。1hnmr(400mhz,cdcl3)δ:1.57(s,9h),1.98-2.16(dm,6h),2.58(dt,j=28.6hz,j=7.4hz,6h),3.37(t,j=5.0hz,6h),3.63-3.71(m,24h),4.03(dt,j=8.5hz,j=6.1hz,6h),4.29-4.33(m,6h),7.18(s,2h)。13cnmr(100mhz,cdcl3)δ:24.6,25.5,28.1,30.5,30.6,50.6,63.4,63.5,67.9,69.1,69.2,70.0,70.6,72.2,81.1,108.0,127.0,141.4,152.3,165.3,173.0,173.3。ei-ms:c41h65n9nao17+的理论值:978.43961;[m+na]+的计算值:978.43760。在一个实施方式中,将三{2-[2-(2-叠氮基乙氧基)乙氧基]乙基}4,4',4”-{[5-(叔丁氧羰基)苯-1,2,3-三基]三(氧)}三丁酸酯(336mg,0.35mmol)溶于无水ch2cl2/三氟乙酸(1∶1)(3.5ml)的混合物中并在ar下搅拌1.5h,然后浓缩至干以提供作为浅黄色油的3,4,5-三(4-{2-[2-(2-叠氮基乙氧基)乙氧基]乙氧基}-4-氧丁氧基)苯甲酸5(316mg,100%)。1hnmr(400mhz,cdcl3)δ:2.09(dm,6h),2.60(dt,j=29.0hz,j=7.3hz,6h),3.38(t,j=5.0hz,6h),3.65-3.72(m,24h),4.07(t,j=6.1hz,6h),4.23-4.27(m,6h),7.31(s,2h)。13cnmr(100mhz,cdcl3)δ:24.4,25.4,30.4,30.5,50.5,63.3,63.5,67.8,69.0,69.0,69.9,70.4,70.5,72.2,108.4,108.5,124.2,142.0,142.3,152.3,170.3,172.8,173.2。ei-ms:c37h57n9nao17+的理论值:922.37646;[m+na]+的实验值:922.37809。在一个实施方式中,如下所述进行peg-b[g2]-n3(9)的合成和表征:将_pd/c(33mg,10%w/w)加入至peg-[g1]-n3(167mg,0.03mmol)在meoh(10ml)中的溶液中。将所得混合物在h2(1atm)下剧烈搅拌5h。然后,通过过滤除去催化剂并将滤液浓缩并干燥。将hobt(18mg,0.13mmol)和edc·hcl(26mg,0.13mmol)加入至上述残余物和5(121mg,0.13mmol)在无水ch2cl2(1ml)中的溶液中。将所得溶液在惰性气氛(ar)下在室温下搅拌48h,然后将其浓缩并在ch2cl2/iproh中沉淀以提供作为白色粉末的peg-b[g2]-n3(208mg,86%)。ftir(kbr):3434,2889,2110,1736,1112cm-1。1hnmr(400mhz,cd2cl2)δ:1.94-2.10(m,18h),2.48-2.60(m,18h),3.31-3.35(m,21h),3.39-3.78(m,~554h),3.96-4.03(m,18h),4.11-4.20(m,24h),7.04-7.07(m,8h)。13cnmr(100mhz,cd2cl2)δ:24.1,25.1,29.9,30.1,39.5,50.3,50.3,58.1,62.9,63.1,67.5,68.6,68.7,69.3,69.4,69.5,70.0,71.4,71.7,105.3,105.3,129.4,139.8,151.9,152.1,166.3,172.4,172.7。maldi-tofms(dctb+nai,线性模式,m/z):mp8092.0([m+na]+),mn8045.5,mw8062.3。理论值:mp8222.4([m+na]+),mn8269.0。在一个实施方式中,如下所述进行peg-fb[g1]-n3(10)的合成和表征:将edc·hcl(8mg,0.04mmol)和催化剂dmap(0.3mg,2μmol)加入至peg-oh(100mg,0.02mmol)和gatge单元5(35mg,0.04mmol)在ch2cl2(1.2ml)中的溶液中。将所得溶液在ar下在室温下搅拌12h,然后将其浓缩并在ch2cl2/iproh中沉淀以提供作为白色粉末的纯peg-fb[g1]-n3(108mg,91%)。1hnmr(400mhz,cd2cl2,me4si)δ:1.96-2.14(dm,6h),2.56(dt,j=23.2,j=7.4,6h),3.32(s,3h),3.34(t,j=5.0,6h),3.39-3.79(m,~552h),4.04(m,6h),4.19(m,6h),4.40(dd,j=5.1,j=4.8,2h),7.25(s,2h)。在一个实施方式中,如下所述进行peg-fb[g2]-n3(11)的合成和表征:将_pd/c(22mg,10%w/w)和1mhcl在甲醇中的溶液(109μl,0.11mmol)加入至peg-fb[g1]-n3(108mg,0.02μmol)在meoh(4ml)中的溶液中。将所得混合物在h2(1atm)下剧烈搅拌3h。然后,通过过滤除去催化剂并将滤液浓缩并干燥。将hobt(15mg,0.11mmol)、edc·hcl(21mg,0.11mmol)和et3n(15μl,0.11mmol)加入至上述残余物和5(98mg,0.11mmol)在无水ch2cl2(546μl)中的溶液中。将所得溶液在惰性气氛(ar)下在rt下搅拌48h,然后将其浓缩并在ch2cl2/iproh中沉淀以提供作为白色粉末的peg-fb[g2]-n3(169mg,81%)。1hnmr(400mhz,cd2cl2)δ:1.95-2.13(dm,24h),2.56(dt,j=25.6,j=7.3,24h),3.32(s,3h),3.34(m,18h),3.39-3.78(m,~552h),4.02(m,24h),4.19(m,24h),4.38-4.41(m,2h),6.69-6.79(m,3h),7.01(s,4h),7.25(s,4h)。在一个实施方式中,如下所述进行peg-fb[g3]-n3(12)的合成和表征:将_pd/c(13mg,10%w/w)和1mhcl在甲醇中的溶液(203μl,0.20mmol)加入至peg-fb[g2]-n3(64mg,8μmol)在meoh(3ml)中的溶液中。将所得混合物在h2(1atm)下剧烈搅拌7h。然后,通过过滤除去催化剂并将滤液浓缩并干燥。将hobt(18mg,0.13mmol)、edc·hcl(26mg,0.13mmol)和et3n(19μl,0.13mmol)加入至上述残余物和5(121mg,0.13mmol)在无水ch2cl2(1.5ml)中的溶液中。将所得溶液在惰性气氛(ar)下在rt下搅拌48h,然后将其浓缩并在ch2cl2/iproh中沉淀以提供作为白色粉末的peg-fb[g3]-n3(91mg,75%)。1hnmr(400mhz,cd2cl2)δ:1.95-2.12(dm,78h),2.50-2.60(dm,78h),3.32(s,3h),3.32-3.35(m,54h),3.39-3.78(m,~552h),3.95-4.05(m,78h),4.17-4.21(m,78h),4.38-4.40(m,2h),6.58(brs,8h),6.82(brs,4h),7.02(s,16h),7.24(s,10h)。在一个实施方式中,通过cuaac,使用烷基化的胺配体对peg-gatge-n3、peg-fb-gatge-n3和peg-gatg-n3的多价官能化的一般程序如下所述:将peg-gatg树枝状嵌段共聚物(peg-b[g2]-n3、peg-fb[g3]-n3和peg-[g2]-n3)溶于dmf/h2o(1∶1)中以提供0.1m最终浓度的端部叠氮化物。然后,加入烷基化铵盐10和11(每个叠氮化物200mol%)和0.1mcuso4·5h2o水溶液(每个叠氮化物5mol%)和0.1m抗坏血酸钠水溶液(每个叠氮化物25mol%)。将所得溶液在室温下搅拌24h,并通过对用0.1medta(ph6)、0.6mnacl和h2o清洗的浓缩的反应混合物超滤(1,000mwco)分离产物。在一个实施方式中,bd(15)的合成和表征如下所示:从peg-b[g2]-n3(92mg,11.4μmol)、10(38mg,0.21mmol)、0.1m抗坏血酸钠(257μl)和0.1mcuso4·5h2o(51μl)在dmf(514μl)/h2o(206μl)中的溶液中,作为白色泡沫固体获得了bd(111mg,100%)。1hnmr(400mhz,d2o)δ:1.98-2.07(m,18h),2.14(五重峰,j=7.8hz,18h),2.56-2.65(m,18h),3.14(t,j=7.8hz,~18h),3.21(t,j=7.3hz,~18h),3.42(s,3h),3.57-3.86(m,~536h),3.95-4.09(m,42h),4.23-4.25(m,~18h),4.40-4.41(m,~18h),4.64-4.67(m,~18h),7.08-7.11(m,~8h),8.21(s,6h),8.22(s,3h)。13cnmr(100mhz,d2o)δ:24.8,25.0,25.8,31.3,31.4,37.6,40.5,40.7,42.5,45.0,51.0,59.0,64.7,64.8,68.8,69.3,69.6,69.8,69.8,70.0,70.3,70.5,71.9,73.5,106.3,127.3,130.1,130.2,139.3,140.1,140.2,140.5,152.7,152.9,169.6,176.2,176.3。通过对1hnmr光谱中适当信号(k、l、n、o、p和q)积分,确定12的分子内降解高达5%(图3b)。在一个实施方式中,bb(16)的合成和表征如下所示:从peg-b[g2]-n3(45mg,5.57μmol)、11(17mg,0.10mmol)、0.1m抗坏血酸钠(124μl)和0.1mcuso4·5h2o(25μl)在dmf(249μl)/h2o(100μl)中的溶液中,作为白色泡沫固体获得了bb(53mg,100%)。1hnmr(400mhz,d2o)δ:1.73-1.88(m,18h),2.33-2.39(m,18h),3.42(s,3h),3.55-3.95(m,~578h),4.08-4.14(m,36h),4.52-4.55(m,18h),6.88-6.92(m,8h),7.34-7.43(m,18h),7.66-7.85(m,18h),8.21-8.23(m,9h)。13cnmr(100mhz,d2o)δ:24.4,25.2,30.7,40.0,40.2,43.2,50.5,,58.5,,64.3,64.3,67.7,68.1,68.6,68.8,69.1,69.4,69.5,70.0,70.4,71.4,72.2,72.5,72.7,105.9,122.9,,126.3,129.9,130.6,133.4,139.7,146.9,,152.3,152.3,168.7,175.4,175.5。在一个实施方式中,fbd(17)的合成和表征如下所示:从peg-fb[g3]-n3(23mg,1.4μmol)、10(14mg,0.08mmol)、0.1m抗坏血酸钠(97μl)和0.1mcuso4·5h2o(19μl)在dmf(194μl)/h2o(78μl)中的溶液中并且按照上述一般步骤,作为白色泡沫固体获得了peg-fb[g3]-d(29mg,97%)。1hnmr(400mhz,d2o)δ:1.97-2.19(m,132h),2.46-2.66(m,78h),3.04-3.25(~108h),3.43(s,3h),3.56-3.79(m,~552h),3.92-4.10(m,~132h),4.21-4.25(m,~78h),4.40-4.43(m,~54h),4.64-4.68(m,~54h),7.12-7.26(m,26h),8.21-8.22(m,27h)。通过对1hnmr光谱中适当信号积分,确定17的分子内降解高达5%(图22)。在一个实施方式中,fbb(18)的合成和表征如下所示:从peg-fb[g3]-n3(25mg,1.5μmol)、11(14mg,0.08mmol)、0.1m抗坏血酸钠(102μl)和0.1mcuso4·5h2o(20μl)在dmf(204μl)/h2o(82μl)中的溶液中并且按照上述一般步骤,作为白色泡沫固体获得了bb(31mg,98%)。1hnmr(400mhz,d2o)δ:1.75-1.94(m,78h),2.33-2.45(m,78h),3.42(s,~3h),3.42-3.93(m,~684h),4.06-4.17(m,~132h),4.50-4.56(m,~54h),6.93-7.06(m,26h),7.43-7.47(m,~54h),7.65-7.72(m,~54h),8.19-8.26(m,27h)。通过对1hnmr光谱中适当信号积分,确定18的分子内降解高达2%(图23)。在一个实施方式中,hsd(19)的合成和表征如下所示:从peg-[g2]-n3(45mg,6.09μmol)、10(20mg,0.11mmol)、0.1m抗坏血酸钠(132μl)和0.1mcuso4·5h2o(27μl)在dmf(264μl)/h2o(105μl)中的溶液中并且按照上述一般步骤,作为白色泡沫固体获得了hsd(52mg,94%)。1hnmr(400mhz,d2o)δ:2.06-2.14(m,18h),3.13(t,j=7.7hz,36h),3.44(s,3h),3.56-3.94(m,~536h),3.97-4.01(m,18h),4.16-4.3(m,24h),4.28-4.33(m,18h),4.66(t,j=4.8hz,18h),7.14-7.17(m,8h),8.16-8.23(m,9h)。13cnmr(100mhz,d2o)δ:24.4,37.1,40.1,42.0,44.5,50.5,58.5,68.8,69.1,69.1,69.3,69.5,70.0,71.4,72.5,106.8,126.8,129.9,139.0,140.1,152.3,169.5。在一个实施方式中,hsb(20)的合成和表征如下所示:从peg-[g2]-n3(g2)(52.5mg,7.11μmol)、11(21mg,0.13mmol)、0.1m抗坏血酸钠(160μl)和0.1mcuso4·5h2o(32μl)在dmf(320μl)/h2o(128μl)中的溶液中并且按照上述一般步骤,作为白色泡沫固体获得了peg-[g2]-b(62mg,98%)。1hnmr(400mhz,d2o)δ:3.43(s,3h),3.57-3.80(m,~560h),3.88-3.94(m,18h),4.02-4.07(m,18h),4.52-4.57(m,18h),6.72-6.80(m,8h),7.22-7.33(m,18h),7.46-7.83(m,18h),8.16-8.17(m,9h)。13cnmr(100mhz,d2o)δ:39.9,40.1,43.1,50.5,58.5,68.5,68.6,69.0,69.0,69.4,69.5,69.7,69.9,70.0,70.2,70.4,71.4,72.2,72.3,72.4,106.3,106.2,122.9,126.0,129.4,129.7,130.3,133.1,139.7,146.8,151.5,151.7,151.8,168.6,168.7。在一个实施方式中,进行了可降解性研究。模拟生理学ph条件,在37℃,研究了铵盐6和树枝状共聚物bd(15)和bb(16)的降解。将样品(1mg/ml)在37℃在磷酸盐缓冲盐水(pbs3×,pd7.4)和/或在乙酸盐缓冲液盐水(30mmnaoac+420mmnacl,pd5.0)中温育。在氧化氘中制备缓冲剂。此外,对于铵盐6的情况,出于积分的目的,缓冲剂补充丙酮-d6(d2o/丙酮-d6,85∶15)以改善溶解度和nmr光谱的分辨率。通过不同时间点的1h-nmr分析样品。在一个实施方式中,进行树枝状复合物的制备。通过将sirna(20μm)添加至不同体积的树枝状共聚物在无核酸酶(nf)的水中的溶液(6mg/ml)中制备了n/p比(其中n=结合物(缀合物)中伯胺的个数;p=rna主链中磷酸基团的个数)在20至160的范围内的树枝状共聚物/sirna复合物。然后,将树枝状复合物溶液涡旋10秒,并且在实验前使其在室温下温育30min。对于其中不评价生物活性的实验,由于其易于合成和以高得率和纯度获得的可能性,将具有与抗增强型绿色荧光蛋白sirna(抗egfpsirna)完全相同序列的双链dna用于模拟sirna(sirnami)。sirna序列:正义seq.i.d.1:5’-gcugacccugaaguucaucugcacc-3’。在一个实施方式中,聚丙烯酰胺凝胶电泳迁移测定。在tris/硼酸盐/edta缓冲液中制备聚丙烯酰胺凝胶(4%的浓缩胶和15%的分离胶)。如前所述,制备不同n/p比的树枝状复合物溶液,其中差异在于使用sirnami(其中rna核苷酸被dna替代)来替代sirna。将对应于12pmolsirnami的树枝状复合物的量与6μl加样缓冲液混合并在100v进行凝胶电泳。通过sirnami在电泳场中缺少迁移(保留在孔中)来显示树枝状复合物/sirnami的结合。在一个实施方式中,进行了sybrgold嵌入测定。如前所述制备了树枝状复合物/sirnami纳米颗粒,然后在rt下,在黑色96孔板中,在nf水(qiagen)中与2μl1∶100sybrgold(invitrogen)溶液(在tae缓冲液中)(最终体积200μl)一起温育10min。温育后,使用酶标仪(synergymx)测量荧光(λexc=485nm,λem=540nm)。结果表示为复合百分比,其中100%代表全部sirna复合。所提供的数据表示为三个独立样品测量的平均值±sd。在一个实施方式中,根据生产商的说明,在动态光散射(dls)仪(zetasizernanozs,malverninstruments,uk)上在633nm测量树枝状复合物的尺寸、多分散指数(pdi)和ζ电位(zp)。在rt,以的检测角确定尺寸和pdi。以的检测角进行ζ电位测量。对于尺寸和pdi测量,以80μl的最终体积,以不同的n/p比制备树枝状复合物,并且不稀释进行分析,或者对于稳定性研究来说,在具有20%(v/v)胎牛血清(fbs)的1×pbs、在1×pbsph7.4和/或在10mmnaoac+137mmnaclph5.0中稀释2倍分析。对于zp测量,以250μl的最终体积制备树枝状复合物并在测量前,在milliq水中稀释至750μl。将smoluchowski模型应用于ζ电位确定,并且将累积分析用于平均粒度确定。所提供的数据表示为三个独立样品测量的平均值±sd。对于肝素引起的树枝状复合物的解离的分析,以n/p160制备这些,然后在含有不同肝素浓度(先前也在1×pbs中稀释)的1×pbs中稀释等份。然后,将树枝状复合物和肝素在37℃温育2h。温育后,将树枝状复合物(对应于6pmolsirna)装载(加载,上样,loaded)到4-15%(w/v)的聚丙烯酰胺-tbe凝胶上并随后用染色。在一个实施方式中,进行透射电子显微术。如前所述,以n/p比80和160制备树枝状复合物。将样品封固在具有聚醋酸甲基乙烯脂(formvar)和碳载体膜的200目ni网格(未辉光放电)上并用2%(w/v)乙酸双氧铀(ua)溶液染色。用滤纸移除过量染色剂,并在成像前干燥网格。使用在80kv下运行的jeoljem1400对样品成像。使用imagej软件(nih,usa)处理图像。树枝状复合物降解研究。如前所述以n/p比160制备生物可降解的树枝状复合物,并且将其在乙酸盐缓冲溶液(60mmnaoac,ph5.0)和在磷酸盐缓冲液(60mmnah2po4,ph7.4)中进一步稀释2倍,并保持1、24和48h。然后,在37℃将树枝状复合物与肝素温育2h(对于bd和bb树枝状复合物,最终肝素浓度分别为0.010mg/ml和0.025mg/ml)。温育后,将树枝状复合物(对应于3pmolsirna)装载到4-15%的聚丙烯酰胺-tbe凝胶上并随后用染色。在一个实施方式中,进行核酸酶保护测定。如前所述制备n/p比为160的树枝状复合物,并将其与0.1u的dna酶i/0.2μgsirnami(模拟sirna的退火的正义和反义dna链)一起在rt下温育5、15、30和60min。对于dna酶失活,用edta(0.05m最终浓度)处理样品,加热至65℃,10分钟并进一步在室温下稳定30min。用sds处理混合物至0.1%的最终浓度,并另外温育30min。最终,将1pmolsirnami在无核酸酶(nf)的水(最终体积10μl)中稀释,与(lb)混合并进一步装载到聚丙烯酰胺凝胶(10%)。将裸露的sirnami和未处理的树枝状复合物用作对照。空的孔中装有等量浓度的盐、sds和edta以使得均一条带迁移。先前使用裸露的sirnami,通过凝胶电泳确定最优的dna酶i浓度。在一个实施方式中,将骨肉瘤细胞系u2os在补充有10%(v/v)fbs和40μg/ml庆大霉素的dmem培养基(gibco)中在37℃,5%co2下,在细胞温育箱中培养。在一个实施方式中,进行了细胞毒性研究。作为树枝状共聚物/树枝状复合物类型和浓度的函数评价细胞存活力。将细胞以3.75×104个细胞/cm2的密度接种到96孔板中。转染时,用未添加的dmem替换培养基。在转染后24h之后,用含有10%(v/v)fbs和10%刃天青的新鲜培养基替换培养基并温育另外3h。在酶标仪(synergymx,biotek)中测量荧光(λex=530nm,λem=590nm)。将暴露于树枝状共聚物的细胞存活力表示为未处理细胞存活力的百分比。在一个实施方式中,使用流式细胞术进行细胞膜结合。将细胞以2.6×104个细胞/cm2的密度接种到24孔板中并在添加的dmem培养基中,在37℃,5%co2下温育24h,并在转染前生长至70-80%汇合。转染时,用未添加的dmem替换培养基。如前所述,使用cy-5标记的sirna制备树枝状复合物。然后,使用50μl树枝状复合物,在300μl的最终体积中,具体地0.1pmol/μl的sirna浓度来转染细胞。温育24h后,用1×pbs漂洗细胞两次,胰蛋白酶化、离心、在1×pbs2%fbs中再悬浮并通过facs(facscalibur,bdbiosciences)分析。分别将未处理的细胞和用2000(lifetechnologies)转染的细胞用作阴性和阳性对照。使用flowjo软件(8.3.7版)分析数据。在一个实施方式中,使用成像流式细胞术进行细胞吸收。将u2os细胞以2.6×104个细胞/cm2的密度接种到24孔板中并在添加的dmem培养基中,在37℃,5%co2下温育24h,并在转染前生长至70-80%汇合。转染时,用未添加的dmem替换培养基。使用cy-5标记的sirnami制备n/p160的sirnami树枝状复合物。然后,用350μl的最终体积(0.1pmol/μl的最终sirnami浓度)转染细胞。温育后24h,用pbs1×漂洗细胞1次并胰蛋白酶化。然后,将细胞转移到eppendorfs管中并以1200rpm,在4℃离心5min。用pbs1×清洗后,将细胞离心(以1200rpm,在4℃离心5min),随后用4%多聚甲醛(pfa)固定15分钟。固定后,用pbs1×清洗细胞两次。在一个实施方式中,使用成像流式细胞仪(amnis,edmmillipore,darmstadt,germany),以40×的放大倍数采集细胞图像。将488nm波长激光器用于激发cy-5标记的sirnami。使用660-745nm光谱检测通道收集荧光图像。使用延伸景深(edf)滤光镜采集图像。随后,使用ideasv6.1图像分析软件(amniscorporation,edmmillipore)分析数据。在一个实施方式中,将每个细胞分成两个区域—外部(膜)和内部(细胞质)。应用覆盖整个细胞的第一遮罩。通过膜厚度所扩大的遮罩来确定外部区域。通过整个细胞的遮罩减去细胞膜遮罩来限定内部区域。然后,将遮罩归因于cy5通道的强度。将强度遮罩与细胞质(内部)遮罩合并,这使得能够对于阳性内化过滤细胞。为了确定小泡负载的树枝状复合物的数目,产生鉴别荧光斑点的遮罩。使用ideasv6.1软件的斑点计数功能(spotcountfeature)对细胞中单个小泡的数目计数,并以频率柱状图作图(图23)。基于表现最差的共聚物(hsd)定义三个区(低、中和高斑点计数)。将中等斑点计数区定义为hsd的平均斑点计数(3.5)±其相应的标准偏差(2)。低斑点计数区低于3.5-2,而高斑点区高于3.5+2。在一个实施方式中,如下所述进行共聚焦显微镜检查:将细胞以2×104个细胞/cm2的细胞密度接种到μ-slide8孔ibitreat(ibidi)上,并在添加的dmem培养基中,在37℃,5%co2下温育24h,并在转染前生长至50-60%汇合。转染时,用未添加的dmem替换培养基。使用cy5标记的sirnami制备n/p比为160的peg-bgatg-ar/sirnami和peg-bgatg-d/sirnami树枝状复合物。然后,使用50μl树枝状复合物,在300μl的最终体积中(0.1pmol/μl的sirna)转染细胞。24h后,用pbs清洗转染细胞三次并与1∶20000稀释的hoechst33342(10mg/ml,lifetechnologies)的溶液一起温育10min(rt)以用于核染色。然后,用pbs清洗细胞,并在显微镜检查前将opti-mem(无酚红)加入至细胞。用leicatcssp2aobs共聚焦显微镜对细胞成像。使用imagej软件捕获和处理立体z-堆叠。在一个实施方式中,如下所述进行沉默研究:将细胞以2.6×104个细胞/cm2的密度接种在24孔板中并在添加的dmem培养基中,在37℃,5%co2下温育24h,并在转染前生长至70-80%汇合。转染时,用未添加的dmem替换培养基。如前所述制备n/p比为160的树枝状复合物。然后,使用50μl树枝状复合物,在300μl的最终体积中(0.1pmol/μl的sirna)转染细胞。温育24h后,用氯喹(100nm)处理细胞并进一步温育4h。然后,用新鲜的添加的dmem替换培养基并温育另外44h。用pbs漂洗细胞两次,然后在冰上与0.15%tritonx-100hkr缓冲液一起温育5min。将细胞裂解液以400×g离心5min,用promega的荧光素酶测定试剂进一步分析上清液的荧光素酶活性。使用酶标仪测量发光性。使用bsa蛋白质测定试剂盒(pierce)测量细胞裂解液中的蛋白浓度。将处理的细胞的荧光素酶活性表示为相对于未处理细胞的荧光素酶活性的百分比。将相同规程用于使用不同n/p比的bd/sirna树枝状复合物的实验。在一个实施方式中,bbz(21)的合成如下所示:将peg-b[g2]-n3(47mg,5.8μmol)溶于dmf(262μl)。然后,加入4-乙炔基苯甲酸(每个叠氮化物200mol%,15mg,0.10mmol)和新鲜制备的0.1mcuso4·5h2o的水溶液(每个叠氮化物5mol%,52μl)和0.1m抗坏血酸钠的水溶液(每个叠氮化物25mol%,262μl)。将所得的端部叠氮化物最终浓度为0.1m的dmf-h2o(1∶1)中的溶液在rt搅拌48h,并通过对用0.1medta(ph6)、饱和nahco3和h2o清洗的浓缩的反应混合物超滤(1,000mwco)来分离产物。将最终的水溶液冻干,并作为浅黄色泡沫获得bbz(21)(54mg,98%)并进行表征。在一个实施方式中,bch(22)的合成如下所示:将peg-b[g2]-n3(55mg,6.8μmol)溶于dmf(461μl)/h2o(31μl)/ch2cl2(10μl)中。然后,加入o-炔丙基胆固醇(每个叠氮化物200mol%,52mg,0.12mmol)和新鲜制备的0.1mcuso4·5h2o的水溶液(每个叠氮化物5mol%,61μl)和0.5m抗坏血酸钠的水溶液(每个叠氮化物25mol%,61μl)。将所得溶液在rt下搅拌48h,并在ch2cl2/二乙醚中沉淀和对用meoh-h2o(3∶2)、0.1medta(ph6)和h2o清洗的浓缩的反应混合物超滤(1,000mwco)后分离产物。将最终的水溶液冻干,并作为白色泡沫获得bch(22)(74mg,91%)并进行表征。在一个实施方式中,bdo3a-gd(23)的合成如下所示:将peg-b[g2]-n3(50mg,6.2μmol)溶于dmf(279μl)。然后,加入alk-do3a-gd(每个叠氮化物150mol%,50mg,0.08mmol)和新鲜制备的0.1mcuso4·5h2o的水溶液(每个叠氮化物10mol%,55.8μl)和0.1m抗坏血酸钠的水溶液(每个叠氮化物50mol%,279μl)。将所得的端部叠氮化物最终浓度为0.1m的dmf-h2o(1∶1)中的溶液在rt搅拌48h,并通过对用0.1medta(ph6)和h2o清洗的浓缩的反应混合物超滤(1,000mwco)来分离产物。将最终的水溶液冻干以提供作为白色固体的bdo3a-gd(23)(72mg,87%)。通过icpoes和tga确定gd(iii)的含量,其占理论值的80%,与先前文献结果一致。31(gd%)的理论值为10.7,实验值为8.6+-0.2。在一个实施方式中,peg(10000)-[g1]-n3(24)的合成和表征如下所示:将edc·hcl(4mg,0.02mmol)和hobt加入至peg(10000)-nh3cl(100mg,9.92μmol)和水解稳定的gatg单元7(13mg,0.02mmol)在无水ch2cl2(300μl)中的溶液中。将所得溶液在ar下在室温下搅拌12h,然后将其浓缩并在ch2cl2/iproh中沉淀以提供作为白色粉末的纯peg(10000)-[g1]-n3(99mg,94%)。在一个实施方式中,如下所示进行peg(10000)-b[g2]-n3(25)的合成和表征:将_pd/c(20mg,10%w/w)加入至peg(10000)-[g1]-n3(99mg,9.37μmol)在meoh(3ml)中的溶液。将所得混合物在h2(1atm)下剧烈搅拌3h。然后,通过过滤除去催化剂并将滤液浓缩并干燥。将hobt(6mg,0.04mmol)和edc·hcl(8mg,0.04mmol)加入至上述残余物和5(38mg,0.04mmol)在无水ch2cl2(281μl)中的溶液中。将所得溶液在惰性气氛(ar)下在rt下搅拌48h,然后将其浓缩并在ch2cl2/iproh中沉淀以提供作为白色粉末的peg(10000)-b[g2]-n3(108mg,88%)。ftir(kbr):3428,2883,2109,1734,1115cm-1。1hnmr(400mhz,cd2cl2)δ:1.90-2.11(m,18h),2.45-2.58(m,18h),3.30-3.35(m,21h),3.39-3.78(m,~974h),3.94-4.05(m,18h),4.09-4.19(m,24h),7.05-7.08(m,8h)。在一个实施方式中,在1h和13cnmr光谱中,在光谱中将溶剂峰标记为*。在一个实施方式中,采集peg-b[g2]-n3(9)的maldi-tof光谱。mn=8068.49;mw=8085.27;pdi=1.002。在一个实施方式中,获得了peg-b[g2]-n3(9)、bd(15)、bb(16)、hsd(19)和hsb(20)的ftir透射光谱(atr)。在一个实施方式中,从gatge构建单元合成生物可降解的peg-gatge树枝状嵌段共聚物并作为基因疗法应用中核酸的非病毒载体评价它们的生物功能性。测试了使用不同胺部分对它们的官能化。这种胺官能化使得能够复合sirna,并因此使得能够研究它们作为这种核酸载体的官能性评价。但是,以相同方式/使用相同规程,可以用不同的官能团容易地官能化生物可降解的树枝状重复单元。因此,来源于该bru的树枝状结构将不仅能够作为适合的载体用于核酸递送,而且还能够用于更广泛的药物递送、磁共振成像(mri)、疫苗、组织工程等中的应用,从而最终致使产生新的纳米医学策略。这些纳米材料可以用作多种重要生物分子的载体,如核酸、药物、生长因子、mri试剂等。每当在本文档中使用时,术语“包含”旨在表示特征、整体、步骤或组件的存在,但是不排除一种或多种其它特征、整体、步骤或组件或它们的组的存在或添加。本领域那些技术人员将理解除非在本文中另外说明,否则步骤的具体顺序仅是说明性的并且可以在不背离本发明公开的情况下改变。因此,除非另有说明,否则步骤是无序的,从而表示当可能的情况下,可以以任何方便或期望的顺序进行步骤。不应以任何方式将本发明公开视为受限于实施方式,并且本领域的常规技术人员将预见到对其进行改变的多种可能性。上述实施方式是可组合的。随附权利要求进一步描述了本发明公开的具体实施方式。参考文献(1)a)skrifvars,m.,tulisalo,j.,rissanen,k.,andnummelin,s.maleimidemodifiedpolypropyleneiminedendrimersandaprocessfortheirpreparation.wo01/38423al,2001;b)baker,j.r.,andzhang,y.hydroxyl-terminateddendrimers.wo2011/053618a2,2011;c)tomalia,d.a.,andmajoros,i.j.biocompatibledendrimers.us2004/0151689al,2004;d)svenson,s.,andtomalia,d.(2005)dendrimersinbiomedicalapplications-reflectionsonthefield.advdrugdeliverrev57,2106-2129;e)rolland,o.,turrin,c.o.,caminade,a.m.,andmajoral,j.p.(2009)dendrimersandnanomedicine:multivalencyinaction.newj.chem.33,1809-1824;f)medina,s.h.,andel-sayed,m.e.(2009)dendrimersascarriersfordeliveryofchemotherapeuticagents.chem.rev.109,3141-57;g)tekade,r.k.,kumar,p.v.,andjain,n.k.(2009)dendrimersinoncology:anexpandinghorizon.chem.rev.109,49-87;h)mintzer,m.a.,andgrinstaff,m.w.(2011)biomedicalapplicationsofdendrimers:atutorial.chem.soc.rev.40,173-90;i)leiro,v.,santos,s.d.,lopes,c.d.f.,andpêgo,a.p.(2017.dol:10.1002/adfm.201700313)dendrimersaspowerfulbuildingblocksincentralnervoussystemdisease:headedforsuccessfulnanomedicine.advancedfunctionalmaterials.(2)a)barth,r.f.,adams,d.m.,soloway,a.h.,alam,f.,anddarby,m.w.(1994)boronatedstarburstdendrimer-monoclonalantibodyimmunoconjugates:evaluationasapotentialdeliverysystemforneutroncapturetherapy.bioconjugatechemistry5,58-66;b)malik,n.,evagorou,e.,andduncan,r.(1999)dendrimer-platinate:anovelapproachtocancerchemotherapy.anticancerdrugs10,767-776;c)malik,n.,wiwattanapatapee,r.,klopsch,r.,lorenz,k.,frey,h.,weener,j.w.,meijer,e.w.,paulus,w.,andduncan,r.(2000)dendrimers:relationshipbetweenstructureandbiocompatibilityinvitro,andpreliminarystudiesonthebiodistributionof125i-labelledpolyamidoaminedendrimersinvivo.journalofcontrolledrelease65,133-148;d)jain,k.,kesharwani,p.,gupta,u.,andjain,n.k.(2010)dendrimertoxicity:let′smeetthechallenge.internationaljournalofpharmaceutics394,122-142.(3)leiro,v.,garcia,j.p.,tomas,h.,andpego,a.p.(2015)thepresentandthefutureofdegradabledendrimersandderivativesintheranostics.bioconjugatechemistry26,1182-1197.(4)a)labieniec,m.,ulicna,o.,vancova,o.,glowacki,r.,sebekova,k.,bald,e.,gabryelak,t.,andwatala,c.(2008)pamamg4dendrimerslowerhighglucosebutdonotimprovereducedsurvivalindiabeticrats.internationaljournalofpharmaceutics364,142-149;b)li,c.,liu,h.,sun,y.,wang,h.,guo,f.,rao,s.,deng,j.,zhang,y.,miao,y.,guo,c.,meng,j.,chen,x.,li,l.,li,d.,xu,h.,wang,h.,li,b.,andjiang,c.(2009)pamamnanoparticlespromoteacutelunginjurybyinducingautophagiccelldeaththroughtheakt-tsc2-mtorsignalingpathway.journalofmolecularcellbiology1,37-45;c)jones,c.f.,campbell,r.a.,brooks,a.e.,assemi,s.,tadjiki,s.,thiagarajan,g.,mulcock,c.,weyrich,a.s.,brooks,b.d.,ghandehari,h.,andgrainger,d.w.(2012)cationicpamamdendrimersaggressivelyinitiatebloodclotformation.acsnano6,9900-9910.(5)a)jianbin,a.,youqing,s.,tingting,l.,andmeihua,s.degradabledendriticmacromoleculemagneticresonancecontrastagentandpreparationmethodthereof.cn103055328a,2013;b)ye,m.,qian,y.,shen,y.,hu,h.,sui,m.,andtang,j.(2012)facilesynthesisandinvivoevaluationofbiodegradabledendriticmricontrastagents.jmaterchem22,14369-14377.(6)a)guillaudeu,s.j.,fox,m.e.,haidar,y.m.,dy,e.e.,szoka,f.c.,andfréchet,j.m.j.(2008)pegylateddendrimerswithcorefunctionalityforbiologicalapplications.bioconjugatechemistry19,461-469;b)vanderpoll,d.g.,kieler-ferguson,h.m.,floyd,w.c.,guillaudeu,s.j.,jerger,k.,szoka,f.c.,andfréchet,j.m.(2010)design,synthesis,andbiologicalevaluationofarobust,biodegradabledendrimer.bioconjugatechemistry21,764-773.(7)leinweber,d.,feustel,m.,wasmund,e.,andrausch,h.alkoxyltateddendrimersandusethereofasbiodegradabledemulsifiers.wo2005003260a12005.(8)tyler,p.c.,andzubkova,o.v.dendriticcorecompounds.us2015/0291522al,2015.(9)castanotto,d.,androssi,j.j.(2009)thepromisesandpitfallsofrna-interference-basedtherapeutics.nature457,426-433.(10)a)yiyun,c.,andmingming,w.dendrimergenecarriermodifiedbyfluorine-containingaromaticringcompoundaswellaspreparationmethodandapplicationthereofcn103881108a,2014;b)kang,c.,pu,p.,yuan,x.,li,f.,andzhong,y.functionaldendriticpolymergenevectorsystemoftargetedmalignantcerebroma.cn101337076a,2009;c)santoyo,f.,morales,j.,megia,a.,hernandez,f.,giron,m.d.,andsalto,r.dendrimersbasedonpamamderizatizedwithalkylsulfonylgroups.wo2012/049338al,2012;d)dufes,c.,uchegbu,l.,andschatzlein,a.(2005)dendrimersingenedelivery.advdrugdeliverrev57,2177-2202;e)mintzer,m.a.,andsimanek,e.e.(2009)nonviralvectorsforgenedelivery.chemicalreviews109,259-302;f)biswas,s.,andtorchilin,v.p.(2013)dendrimersforsirnadelivery.pharmaceuticals6,161-183;g)santos,j.l.,oliveira,h.,pandita,d.,rodrigues,j.,pêgo,a.p.,granja,p.l.,andtomás,h.(2010)functionalizationofpoly(amidoamine)dendrimerswithhydrophobicchainsforimprovedgenedeliveryinmesenchymalstemcells.journalofcontrolledrelease144,55-64;h)leiro,v.,santos,s.d.,andpêgo,a.p.(2017.)deliveringsirnawithdendrimers:invivoapplicationscurrentgenetherapy.dol:10.2174/1566523217666170510160527.(11)thakur,s.,kesharwani,p.,tekade,r.k.,andjain,n.k.(2015)impactofpegylationonbiopharmaceuticalpropertiesofdendrimers.polymer59,67-92.(12)sousa-herves,a.,riguera,r.,andfernandez-megia,e.(2012)peg-dendriticblockcopolymersforbiomedicalapplications.newj.chem.36,205-210.(13)reyes-reveles,j.,sedaghat-herati,r.,gilley,d.r.,schaeffer,a.m.,ghosh,k.c.,greene,t.d.,gann,h.e.,dowler,w.a.,kramer,s.,dean,j.m.,anddelong,r.k.(2013)mpeg-pamam-g4nucleicacidnanocomplexes:enhancedstability,rnaseprotection,andactivityofspliceswitchingoligomerandpolyi:crna.biomacromolecules14,4108-4115.(14)barnard,a.,posocco,p.,pricl,s.,calderon,m.,haag,r.,hwang,m.e.,shum,v.w.,pack,d.w.,andsmith,d.k.(2011)degradableself-assemblingdendronsforgenedelivery:experimentalandtheoreticalinsightsintothebarrierstocellularuptake.jamchemsoc135,20288-20300.(15)a)welsh,d.j.,jones,s.p.,andsmith,d.k.(2009)“on-off”multivalentrecognition:degradabledendronsfortemporaryhigh-affinitydnabinding.angewandtechemie,internationaledition48,4047-4051;b)barnard,a.,calderon,m.,tschiche,a.,haag,r.,andsmith,d.k.(2012)effectsofapegadditiveonthebiomolecularinteractionsofself-assembleddendronnanostructures.organic&biomolecularchemistry10,8403-8409;c)barnard,a.,posocco,p.,fermeglia,m.,tschiche,a.,calderon,m.,pricl,s.,andsmith,d.k.(2014)double-degradableresponsiveself-assembledmultivalentarrays-temporarynanoscalerecognitionbetweendendronsanddna.organic&biomolecularchemistry12,446-455;d)movellan,j.,gonzalez-pastor,r.,martin-duque,p.,sierra,t.,delafuente,j.m.,andserrano,j.l.(2015)newlonicbis-mpaandpamamdendrimers:astudyoftheirbiocompatibilityanddna-complexation.macromol.biosci.15,657-67.(16)amaral,s.p.,fernandez-villamarin,m.,correa,j.,riguera,r.,andfernandez-megia,e.(2011)efficientmultigramsynthesisoftherepeatingunitofgallicacid-triethyleneglycoldendrimers.orglett13,4522-4525.(17)hurrell,s.,milroy,g.e.,andcameron,r.e.(2003)thedegradationofpolyglycolideinwateranddeuteriumoxide.part1:theeffectofreactionrate.polymer44,1421-1424.(18)fernandez-megia,e.,correa,j.,andriguera,r.(2006)″clickable″peg-dendriticblockcopolymers.biomacromolecules7,3104-3111.(19)gary,d.j.,puri,n.,andwon,y.y.(2007)polymer-basedsirnadelivery:perspectivesonthefundamentalandphenomenologicaldistinctionsfrompolymer-baseddnadelivery.journalofcontrolledrelease121,64-73.(20)a)mintzer,m.a.,merkel,o.m.,kissel,t.,andsimanek,e.e.(2009)polycationictriazine-baseddendrimers:effectofperipheralgroupsontransfectionefficiency.newjchem33,1918-1925;b)merkel,o.m.,mintzer,m.a.,sitterberg,j.,bakowsky,u.,simanek,e.e.,andkissel,t.(2009)triazinedendrimersasnonviralgenedeliverysystems:effectsofmolecularstructureonbiologicalactivity.bioconjugchem20,1799-806;c)merkel,o.m.,mintzer,m.a.,librizzi,d.,samsonova,o.,dicke,t.,sproat,b.,garn,h.,barth,p.j.,simanek,e.e.,andkissel,t.(2010)triazinedendrimersasnonviralvectorsforinvitroandinvivornai:theeffectsofperipheralgroupsandcorestructureonbiologicalactivity.molpharm7,969-83.(21)fernandez-villamarin,m.,sousa-herves,a.,porto,s.,guldris,n.,martinez-costas,j.,riguera,r.,andfernandez-megia,e.unpublishedresults.(22)akin,a.,andgiuseppe,b.(2013)exploitingendocytosisfornanomedicines.coldspringharborperspectivesinbiology5.(23)merkel,o.m.,zheng,m.,mintzer,m.a.,pavan,g.m.,librizzi,d.,maly,m.,hoffken,h.,danani,a.,simanek,e.e.,andkissel,t.(2011)molecularmodelingandinvivoimagingcanidentifysuccessfulflexibletriazinedendrimer-basedsirnadeliverysystems.journalofcontrolledrelease:officialjournalofthecontrolledreleasesociety153,23-33.(24)delafuente,m.,m.,sousa-herves,a.,correa,j.,riguera,r.,fernandez-megia,e.,sánchez,a.,andalonso,m.j.(2012)exploringtheefficiencyofgallicacid-baseddendrimersandtheirblockcopolymerswithpegasgenecarriers.nanomedicine7,1667-1681.(25)a)merkel,o.m.,beyerle,a.,beckmann,b.m.,zheng,m.,hartmann,r.k.,stoger,t.,andkissel,t.h.(2011)polymer-relatedoff-targeteffectsinnon-viralsirnadelivery.biomaterials32,2388-98;b)beyerle,a.,irmler,m.,beckers,j.,kissel,t.,andstoeger,t.(2010)toxicitypathwayfocusedgeneexpressionprofilingofpei-basedpolymersforpulmonaryapplications.molpharm7,727-37.(26)mahesh,l.p.,min,z.,oleh,t.,olga,b.g.,huixin,h.,andtamara,m.(2009)internallycationicpolyamidoaminepamam-ohdendrimersforsirnadelivery:effectofthedegreeofquaternizationandcancertargeting.biomacromolecules10,258-266.(27)a)erbacher,p.,roche,a.c.,monsigny,m.,andmidoux,p.(1996)putativeroleofchloroquineingenetransferintoahumanhepatomacelllinebydnalactosylatedpolylysinecomplexes.expcellres225,186-194;b)ciftci,k.,andlevy,r.j.(2001)enhancedplasmiddnatransfectionwithlysosomotropicagentsinculturedfibroblasts.intjpharm218,81-92.(28)moreira,c.,oliveira,h.,pires,l.r.,simoes,s.,barbosa,m.a.,andpego,a.p.(2009)improvingchitosan-mediatedgenetransferbytheintroductionofintracellularbufferingmoietiesintothechitosanbackbone.actabiomaterialia5,2995-3006.(29)fernandez-trillo,f.,pacheco-torres,j.,correa,j.,ballesteros,p.,lopez-larrubia,p.,cerdan,s.,riguera,r.,andfernandez-megia,e.(2011)dendriticmricontrastagents:anefficientprelabelingapproachbasedoncuaac.biomacromolecules12,2902-7.(30)yashima,e.,matsushima,t.,andokamoto,y.(1997)chiralityassignmentofaminesandaminoalcoholsbasedoncirculardichroisminducedbyhelixformationofastereoregularpoly((4-carboxyphenyl)acetylene)throughacid-basecomplexation.jamchemsoc119,6345-6359.(31)langereis,s.,lussanet,q.g.d.,vangenderen,m.h.p.,backes,w.h.,andmeijer,e.w.(2004)multivalentcontrastagentsbasedongadolinium-diethylenetriaminepentaaceticacid-terminatedpoly(propyleneimine)dendrimersformagneticresonanceimaging.macromolecules37,3084-3091.序列表<110>国家生物医学工程研究所(institutonacionaldeengenhariabiomédica)<120>生物可降解的树枝状结构、方法及其用途<130>p420.6wo<150>pt109408<151>2016-05-23<160>1<170>bissap1.3.6<210>1<211>25<212>rna<213>合成构建体<400>1gcugacccugaaguucaucugcacc25当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1