用于靶向基因组编辑的系统和方法与流程
交叉引用部分
本专利申请要求于2017年10月17日提交的美国临时专利申请号62/573,402以及2017年7月28日提交的美国临时专利申请号62/538,213的优先权,其全部内容通过引用并入本文中。
以电子方式提交的序列表的引用
所述序列表的官方副本经由efs-web作为ascii格式的序列表以电子方式提交,文件名为“7452wopct_sequence_listing_st25”,创建于2018年7月25日,且具有33千字节大小,并与本说明书同时提交。包括在所述ascii格式的文件中的序列表是本说明书的一部分并且以其全文通过引用并入本文。
背景技术:
基因组编辑技术的最新发展已使特定序列位置的序列修饰成为可能。例如,采用crispr-cas系统的序列编辑使用与靶向dna序列互补的rna来引导cas蛋白到达特定的序列位点进行修饰,其中位点是序列或序列内的区域,所述序列是天然的或修饰的或人工核酸分子或其表示。编辑实验可以包括位点特异性核酸酶,例如crispr-cas9、talen、大范围核酸酶、靶向或栓系核酸酶、可编程的核酸酶、核糖核蛋白(rnp),并且可以涉及直接转化、生物弹道递送(biolisticdelivery)、共培养,或为了实现特异性的、定向的核酸修饰或编辑的多种递送方法。此类基因组编辑可用于递送赋予所希望表型的基因组修饰,例如改善作物物种的农艺性状。
技术实现要素:
特定的变种、近交种或种质可以使用任何方法的组合直接进行编辑,以将基因组编辑成分传递给植物或植物细胞,然后针对所需的一种或多种修饰进行富集或选择。通常,变种、近交种或种质将在整个基因组中包含dna序列变异。将两个或多个dna碱基对处的dna序列变异的每个不同模式称为单倍型。对于要进行编辑的每个品种、近交种或种质,都需要了解待修饰位置周围的单倍型,以便将指导rna或其他试剂正确地靶向编辑位点,并且还产生一种或多种所希望的序列修饰。可以使用所谓的性状渐渗(ti)或选择性育种渐渗方法,将已编辑的性状从作为终点的一个供体品种、近交种或种质转移到新品种、近交种或种质。这通常是通过有性繁殖来完成的,但不仅限于有性繁殖的作物。在ti中,富集靶向或所选渐渗的典型过程是通过回交策略进行,所述策略监控并选择目的性状或分子特征,同时或相继富集合理的最大百分比的轮回亲本(终点)基因组。了解由植物育种种群所具有的在目标基因座周围的单倍型,可以选择供体和受体亲本,从而使靶基因座的遗传差异最小化,从而促进更快速和准确的性状渐渗。可以将通过基因组编辑产生的新颖的性状、等位基因或分子特征用于所谓的正向育种应用中,其中基因组编辑的品系是与一组其他品种、近交种或种质的育种杂交中的亲本,以繁殖和增加育种群中所希望的修饰的频率。为减少靶基因座附近遗传变异的损失,可以希望的是在一组遗传实体中进行编辑,这些遗传实体均表示较大群体中靶基因座处的所有现存单倍型。这种方法将需要了解在所希望的区域内的所有序列变异。
在用于将基因组编辑部署到新颖的品种、近交种、杂交种、种质或产品中的所有可能的方法中,希望存在灵活的方法允许需要的靶向组分的靶特异性或等位基因特异性,或单倍型特异性的或其他此类背景特异性的设计的,或允许需要的靶向组分的保守的、保留的、相同的或通用的设计方法,这些灵活的方法可以在更广泛的品种、近交种、杂交种或种质或甚至跨亚种或物种边界或序列集中使用。
序列编辑技术的另一个共同问题是,有时在指导rna或靶向核酸组分或其他靶向组分对靶向位点特异性不足的情况下,它可能会将编辑引导至无意的,非靶向的(脱靶)序列区域,有时会导致不希望的结果。
因此,在本领域中存在对灵活的核酸序列编辑系统和方法的需要,这些系统和方法适应靶向特定位点或位点组的序列编辑,其考虑了等位基因的相似性或差异,并且考虑了还可以最小化无意的脱靶编辑的策略、系统和方法。
本文公开了设计指导多核苷酸的方法,所述方法使产生脱靶位点基因编辑的可能性最小化。所述方法可以包括(a)将核酸内切酶的靶位点序列与来自群体中个体的未经组装的原始核苷酸序列读数进行比较;(b)将与靶位点序列的部分或全部对齐的原始核苷酸序列读段组装成单个重叠群;(c)在来自步骤b的重叠群中选择包含单个拷贝的靶序列的靶位点序列;任选地,(d)设计针对所述靶位点序列的指导rna;和(e)使用核酸内切酶复合物中的设计的指导多核苷酸,在核酸中的靶位点处产生预期基因编辑。
本文还公开了针对在群体中发现的单倍型产生共有序列的方法。所述方法可以包括(a)对群体中不同基因型的两个或更多个个体的目的区域进行测序,以产生核苷酸序列读段;(b)将核苷酸序列读段与一个或多个主题序列进行比对,以鉴定核苷酸变异;(c)使用目的区域中的核苷酸变异来定义一个或多个单倍型;(d)将来自所述群体的至少一个个体分配给步骤(c)中的一个或多个单倍型;和(e)产生单倍型共有序列,所述单倍型共有序列从来自步骤(d)中分配的一个或多个个体的区域的核苷酸序列读段组装。
本文公开了针对在群体中发现的主题单倍型产生共有序列的方法。所述方法可以包括(a)对群体中不同基因型的两个或更多个个体的目的区域进行测序,以产生核苷酸序列读段;(b)将核苷酸序列读段与一个或多个主题序列进行比对,以鉴定核苷酸变异;(c)使用目的区域中的核苷酸变异来定义一个或多个单倍型;(d)将来自所述群体的至少一个个体分配给步骤(c)中的单倍型;(e)基于目的区域中的核苷酸变异,针对每个共同单倍型产生核苷酸变体频率的图谱,以产生共同单倍型图谱;(f)对主题单倍型中是否存在与共同单倍型图谱或其组合相对应的断点进行鉴定;(g)将由断点定义的主题单倍型的那些区域分配给相应的两个或更多个共同单倍型;和(h)针对从步骤(g)中的主题单倍型分配给的共同单倍型的区域的核苷酸序列读段组装的单倍型产生共有序列。
本文还公开了表征在群体中发现的两个或更多个单倍型的方法。所述方法可以包括:(a)对群体中两个或更多个具有不同基因型的个体中所定义的目的区域进行测序,以产生核苷酸序列读段;(b)使用在所定义的区域中的核苷酸变异来定义两个或更多个单倍型;(c)将跨不同基因型的核苷酸序列读段组装成针对所述两个或更多个单倍型的共有序列;(d)比较所述单倍型共有序列以鉴定一个或多个另外的核苷酸变异;和(e)基于目的区域中经鉴定的核苷酸变异表征每个单倍型。这些方法可以进一步包括:(f)基于所述核苷酸变异,将来自所述群体的至少一个个体分配给一个或多个单倍型;和(g)产生单倍型共有序列,所述单倍型共有序列从例如在步骤(f)中分配的一个或多个个体的区域的核苷酸序列读段组装。
附图说明
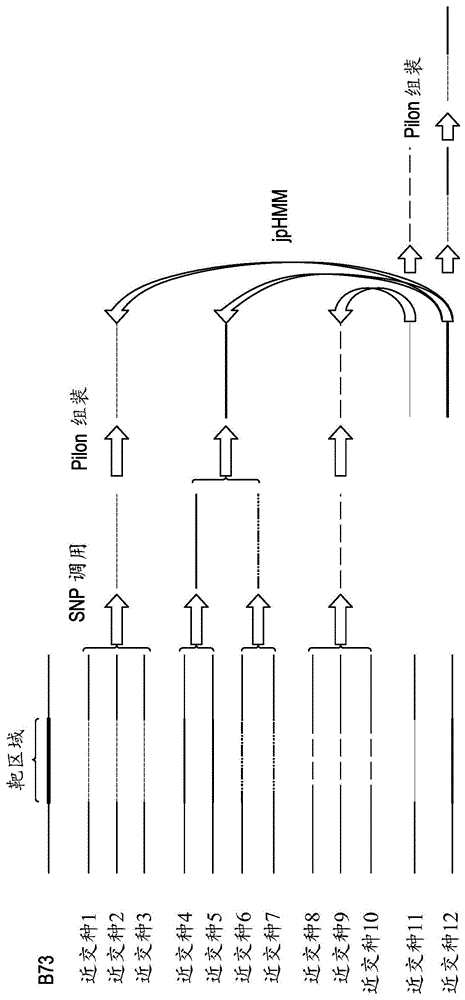
图1提供了在具有12个近交系的实例下,序列背景建模算法的概述。多个加权线和/或虚线标记了所述12个近交系的真实的单倍型关系。所述方法导致单倍型序列(在此被称为等位基因模型)的产生。
图2是针对天然丰度序列集的本发明的编辑位点选择过程方面的示意图。
图3是基于位点特异性筛选过程的参考基因组的示意图。
图4是参考游离位点特异性筛选过程的示意图。
图5示出了10个相同状态的组解析成在sss和nss杂合池内主要的等位基因模型组。
图6提供了如何将本发明的方法用于产品开发的概述。
具体实施方式
本发明包括用于确定可被序列集合内的序列编辑化合物作用的核酸序列的谱的系统和方法。本发明另外包括用于设计和/或选择核酸序列的系统和方法,所述核酸序列可以特异性地靶向待编辑的序列的区域或序列的集合(包括但不限于基因组),同时避免对非预期用于编辑的脱靶位点进行修饰。本发明进一步包括系统和方法,所述系统和方法使用前述核酸序列来指导基因组编辑系统到达待编辑的一个或多个核酸的特异性靶区域,同时最小化避免不预期用于编辑的脱靶位点。
以下描述了用于基于共享遗传信息从不同近交种、品种或种质合并序列数据,鉴定和选择编辑位点的方法,并且描述了特异性靶向待编辑的序列区域(同时最小化或避免非预期用于编辑的脱靶位点的修饰)的序列的设计。尽管此描述是针对近交种玉蜀黍系,但应理解,可以使用相同的方法来设计位点特异性靶向核酸,以靶向任何其他类型的植物、动物、微生物、序列、序列集合或任何其他基于天然或人工核酸的实体。另外,尽管本说明书的一些方面着眼于基于cas9的编辑系统作为特定但非限制性实例的用途,但应理解,这些方法也可以采用针对其他靶向序列编辑化合物的小且明显的修饰而广泛使用,所述靶向序列编辑化合物包括但不限于talen、大范围核酸酶、靶向或栓系核酸酶、可编程的核酸酶、核糖核蛋白(rnp)、归巢核酸内切酶或限制酶等。
术语“共有序列”是指如下任何核苷酸序列,所述核苷酸序列与群体中的两个或更多个个体具有在其基因组中具有预定同源性程度的相应核苷酸序列。
术语“参考序列”是指被组装为群体的基因组的至少一部分的代表性序列的任何核苷酸序列。
术语“主题序列”是指在核苷酸序列数据库中的任何核苷酸序列。
术语“单倍型”是指个体的基因组的任何部分的基因型,或个体的组的基因组的任何部分(在其基因组的所述部分中共享基本上相同的基因型)的基因型。
术语“主题单倍型”是指在单倍型数据库中的任何单倍型。
术语“共同单倍型”是指在群体中的超过预定百分比的个体中发现的单倍型。
术语“主要单倍型”是指在群体中相比任何其他单倍型,在更多个体中发现的单倍型。
术语“罕见单倍型”是指在群体中的低于预定百分比的个体中发现的单倍型。
术语“断点”是指核苷酸序列中的点,其中所述序列从与第一单倍型同源变为与第二单倍型同源。
术语“图谱”是指对具有相同单倍型的个体基因型的描述,任选地包括诸如基因型等位基因频率的信息。
在应用于一组玉蜀黍基因组序列时的一般序列编辑流程的实例
测序策略
对代表目的种质或遗传材料的近交系的集进行全基因组测序。每个近交种可以通过序列读段的变化的量或“深度”来表示。
读段比对和变体调用(variantcalling)
使用bowtie2(langmead等人.2012),将在多种测序深度(例如,30x、20x、3x)下从全基因组测序产生的测序读段与参考序列进行比对。许多其他比对程序也是可用的,并且将对本领域技术人员可用。例如,这些可以包括bwa(li和durbin2009)、bwa-mem(li2013)、novoalign(novocraft.com)、gem(marco-sola等人.2012)、soap2(li等人.2009)、cushaw2(liu和schmidt2012)、seqalto(mu等人.2012)、meta-aligner(nashta-ali等人.2017),等人。在将读段与参考序列比对后,使用samtools(li等人.2009)调用单核苷酸多态性(snp),并基于最小读段覆盖范围和来自个体内读段的等位基因的均匀性的最小速率进行过滤。可用其他流行的snp调用程序:freebayes(garrison和marth2012)、gatk软件包中unifiedgenotyper和haplotypecaller(depristo等人.2011;vanderauwera等人.2013)、platypus(rimmer等人.2014)、soapsnp(li等人.2009)以及许多其他的程序。可以使用任何合适的snp调用方法。
在一些替代方案中,可以按以下方式组织序列,所述方式将集合中序列之间共享相似性的所有点集合在一起,并例如在基于序列图的模型中标记发散位置。在这些结构的一些版本中,作为序列掺入过程的一部分,可以追踪丰度和/或可以提高序列的可靠性。
单倍型组分配
单倍型是指目的基因组区域中在多于一个dna序列变体上等位基因的组合。可以将遗传材料分配给针对序列区域的单倍型组。可以将单倍型组定义为一组遗传实体,所述遗传实体在群体中在目的区域存在的遗传变体上携带相同的等位基因。对单倍型组的优选的解释是单倍型组的成员共享所述区域的相同的dna序列。在一些方法中,可以将单倍型组解释为一组近交种,所述近交种共享基因组区域的遗传相关但不相同的dna序列。单倍型组中的遗传实体可以是分配给单个单倍型组的近交系。在一些方法中,基因材料可以是杂合的,使得一些遗传实体可以被分配给两个不同的单倍型。在这种情况下,可以使用谱系信息或群体中纯合个体的单倍型,从杂合基因型确定或估计个体单倍型组。在以下一组实例方法中,用于定义单倍型并将个体分配给单倍型组的序列集来源于玉蜀黍基因组序列,但应理解它们可以实际上是来自任何来源(天然的或其他方式)的序列的任何集合,并且所述方法类似地应用,与源序列集类型无关。单倍型组代表变体谱,以考虑有意序列修饰靶标以及序列集内脱靶位点的可能的范围。存在用于产生单倍型的多个公开的,经同行评审的方法,并对于本领域技术人员将是可用的。实例包括beagle(browning和browning2007)和shapeit(deleaneu等人.2013),等人.。可以根据特定的序列间隔定义单倍型组。在其他方法中,只要满足遗传同一性或相似性的标准,单倍型组就可以沿着基因组延伸。对遗传同一性或相似性的测量可以基于snp、插入和缺失、拷贝数变异、表观遗传学标记,或这些特征的组合,或适合于区分集合中的序列的其他序列多态性。在一些方法中,对遗传相似性或遗传同一性的测量可以基于遗传实体之间的序列特征差异。在一些方法中,该得分可以基于特征差异的计数或频率测量。一些方法可以对不同于纯合dna序列差异的杂合基因型或缺失数据进行评分。一些方法可以为缺失数据和杂合基因型的可允许的数量或频率设置阈值。一些方法可以对遗传实体的整个群体中每个等位基因的不同等位基因频率的匹配或错配的得分进行不同地加权。一些方法可以使用概率模型,从dna序列相似性估计单倍型组。在一些方法中,概率模型可以包括遗传实体的共享群体历史的模型,所述模型可以包括描述遗传实体的家族关系的谱系信息。这样的模型还可以包括有关预期的单倍型频率、连锁不平衡以及单倍型之间遗传重组的模式和比率的信息。在一些方法中,可以设置阈值以将遗传实体分配给相同的单倍型组。阈值可以基于对遗传相似性或差异的测量。阈值可以基于对根据概率模型遗传实体共享相同单倍型的概率的估计。
在一些方法中,可以在单倍型分配之前估算缺失数据。由本领域的技术人员广泛地进行估算。一些方法进行估算连同单倍型分配。其他方法在单倍型分配之前进行估算。一些方法仅使用基因组中特定遗传或物理距离内的其他变体,对遗传变体进行估算。其他方法使用在单个染色体或跨整个基因组上的所有遗传基因座进行估算。一些方法使用最近邻方法,其中估算是由在遗传距离测量情况下与所讨论的遗传实体具有最低遗传距的不同的遗传实体获知的。一些方法使用来自指定遗传距离内所有遗传实体的信息进行估算。在一些方法中,可以将遗传或核酸实体的整个群体内的等位基因频率用作用于估算的信息。在一些方法中,可以使用概率模型进行估算。在一些方法中,概率模型可以包括遗传实体的共享群体历史的模型,所述模型可以包括描述遗传实体的家族关系的谱系信息。这样的模型还可以包括有关预期的等位基因频率、单倍型频率、连锁不平衡以及单倍型之间遗传重组的模式和比率的信息。
单倍型组可以被认为是在特定基因组区域内共享相同或相似dna序列的遗传实体聚簇。单倍型聚簇的准确性在很大程度上受一个或多个靶区域中鉴定的snp的普遍性和质量的影响。为了简洁起见,其中可以使用首字母缩略词“snp”,应理解替代地可以使用如上所提及的多态性的许多其他类型。从低测序深度的样品调用的snp可以导致低snp密度和缺失数据的高水平。在本文所述的方法中,使用两轮单倍型聚簇方法来减轻该问题(图1)。将来自靶区域加上5’和3’侧翼区(默认值3kb)的高质量snp用于具有严格同一性阈值要求(默认值100%)的近交系序列的第一轮分层聚簇。如果snp的数量少于所希望的阈值(默认值20),通过增量步长(默认值1kb)将窗口延伸至侧翼区,直到满足阈值。将靶区域中具有相同单倍型的样品聚簇成单倍型组。具有少于给定来源或近交系数目(默认值为3)的单倍型组被定义为罕见单倍型组。具有成员数等于或大于来源或近交系的一定数量(默认值为3)的单倍型组被定义为主要单倍型组,并在下一步骤中用于snp调用。在该实例中,将来自相同的主要单倍型组中来源的测序读段比对合并成针对靶区域的一个bam文件。使用合并的bam文件,将pilon(walker等人.2014)和vcf工具(danecek等人.2011)用于调用针对靶区域的每个单倍型组的一组新的snp。原则上,此步骤也可以将其他snp调用方法(参见变体调用部分)与以各种格式或方法提供的序列信息一起使用。然后使用如前述单倍型分配方法的相同聚簇算法,将新的snp(多态性)集(可以包含相比在第一轮单倍型聚簇中使用的snp更多或不同的snp)用于来自以上鉴定的主要单倍型组的来源或近交系的第二轮聚簇。由于该第二组snp可能包含比初始集更多的信息,因此它可以在使用更小的基因组窗口的同时产生更准确的单倍型聚簇。
主要单倍型组的局部组装
对于针对给定目的区域所定义的给定单倍型组,可以存在按不同测序水平(例如,3x、30x、100x或更高或更低)测序的多个基因型。由于单倍型组中所有基因型针对该具体的目的区域共享相同的单倍型标识,可以将源自所述目的区域的这些基因型的序列(例如,测序读段)全部处理为在所述目的区域中该单倍型组的序列。虽然个体基因型可以具有较浅的测序深度(例如,3x),但一个单倍型组内所有基因型的所有序列的累积可以达到足够高的深度(例如,100x),从而实现该单倍型组的可靠的共有序列,所述共有序列相比从任何单一基因型推断的dna序列更完整并且具有更高的准确性。可以通过多种方法产生该单倍型共有序列,所述方法包括但不限于根据各种共有序列产生方法需要的组装和序列比对。所述共有序列在本文中被称为“等位基因模型”。
在基于组装的共有序列产生过程的实例中,通过将组中所有基因型的多种测序深度加总来计算单体型组的测序深度。当单倍型组的总测序深度超过用于实现可靠的组装的最小深度截止值(例如,30x)时,将局部组装应用于所述组。
对于具有足够测序深度的单倍型组,将通过一些标准(例如,映射质量得分)所选的映射到目的区域的序列的全部或子集进行收集,并然后输送入公共组装工具(例如,pilon)以产生共有序列。
共有序列传达了由单倍型携带的dna序列变体,并且还鉴定了其中单倍型组的序列仍然不确定或未解析的区域。在优选的方法中,合适的跨参考序列被替换为在共有序列内的任何未改进或未解析的区域。
罕见单倍型组的序列组装
罕见单倍型组(含有少量近交种的那些)可能不包含能够进行局部组装的足够的序列读段覆盖范围。为改善此类罕见单倍型的序列,一种优选的方法是使用跳跃图谱隐蔽马尔科夫模型(hmm)使罕见单倍型与主要单倍型进行区段比对。跳跃图谱hmm(schultz等人.2006;schultz等人.2009)是图谱hmm扩展为多个图谱。在该方法中,将近交种单倍型或代表每个主要单倍型组的序列的多个比对用于针对每个主要单倍型产生hmm图谱。考虑到针对目的区域的多个图谱的套件,可以使用经修改的维特比(viterbi)算法(schultz等人.2006)来确定沿着核苷酸序列的最可能的路径,通过所述路径可以由主要单倍型图谱产生罕见单倍型。所得的序列区段将罕见单倍型映射到一个或多个主要单倍型,并且将在经比对的主要单倍型图谱中的转换称为断点(图1)。可以将缺少断点证据的罕见单倍型分配给它们所映射到的最可能的主要单倍型组。具有经鉴定的断点的罕见单倍型具有侧翼于重新分配给相关主要单倍型的断点的子序列。许多其他方法可用于鉴定序列内的潜在断点,实例包括rdp(martin和rybicki2000)、simplot(lole等人.1999)、geneconv(sawyer1989),等人。
编辑位点候选者鉴定
用于编辑序列的优选方法是使用编辑化合物,所述编辑化合物可通过提供与待编辑位点具有相似性程度的指导核苷酸序列来指导编辑靶序列。以此方式操作的编辑系统包括cas9、cpf1、c2c1等。可替代的编辑化合物(例如,大范围核酸酶和talen等)可以识别特定的位点集,或具有一定组成或特征的那些位点。用于修饰的理想位点的特征根据特定编辑化合物的要求而变化。位点要求可能广泛适用于编辑化合物的给定类别或类型的成员,并且所使用的特定编辑化合物可能具有另外的或经修改的要求。例如,最初被描述为细菌的ii型crispr/cas免疫系统的单一指导rna(sgrna)系统已成功地重新用作基因组工程化工具,并且对于本领域技术人员可以使用的基因组编辑的此类型的特定编辑化合物的列表已经持续超出了最初描述的那些。此类的大多数成员对在优选长度范围内的指导序列共享相似的要求,要求修饰位置附近存在前间区序列邻近基序(pam),并且要求具有用于成功靶向的、与指导序列的一定程度的相似性。长度和基序和序列含量的具体参数在此类编辑化合物中变化,但是最近已经开发了许多指导rna(grna)设计工具,可以将其改适用于此类基因组编辑化合物。实例包括cas-offinder(bae等人,2014)、gt-scan(o’brien等人,2014)、cctop(stemmer等人,2015)、crisprdirect(naito等人,2015)、off-spotter(pliatsika和rigoutsos,2015)、crisprscan(moreno-mateos等人,2015)和breaking-cas(oliveros等人,2016)。大多数工具通过检测用户可自定义的pam基序序列并预测整个基因组序列中的脱靶来鉴定潜在的grna靶。其中,一些工具支持脱靶中的可自定义的最大错配数(例如,crisprdirect)或对脱靶提供排序(例如,breaking-cas)。然而,没有工具提供可自定义的pam基序序列、可自定义的最大错配数、经排序的脱靶的组合,并且没有一种工具提供报告具有非天然序列丰度(诸如短读段测序数据)的序列集合中的特异性、对基因组编辑化合物和系统的多种类型具有适用性的手段。以下描述了改进的方法,用于以高成功概率为给定序列或序列区域鉴定优选的潜在靶位点。
crispr相关的编辑化合物的pam位点扫描
使用优选的cas9编辑化合物,多种方法用于在玉蜀黍编辑实例中的靶向序列集之间定位编辑位点,从而为特定的玉蜀黍基因型赋予蜡状(waxy)性状表型。对靶向序列进行扫描以鉴定两条链上所有的pam位点位置。靶向序列可以包含正在分析的一组序列内有限的区域、所述组中序列的子集或包括整个序列集合。用于检测潜在pam位点的许多方法对于基因组编辑从业者是可使用的。在一些方法中,搜索pam的预期大小的窗口以与该基因组编辑化合物所需的核苷酸相匹配。在其他情况下,可以计算统计概率以鉴定与pam碱基概率图谱匹配的序列位置。同样,可以使用长度等于pam要求的短窗口来沿着序列集中的序列长度进行扫描来匹配。在其他方法中,可以将待查询的集中的序列分裂为称为kmers的子序列,并且将这些子序列用于鉴定可能的pam位置。另一个实例将使用动态编程对比方法来查找位点。又另一个实例可能依赖于使用可替代的序列集表示形式(诸如后缀数组或序列图模型)来检索包含与编辑化合物匹配要求相匹配的所有序列。对于本领域技术人员存在大批的软件工具来检测全部或部分序列匹配。
对于每个pam位点,将落入由编辑化合物有效识别的范围内(例如,对于cas9,17nt至25nt)并且在相对于特定编辑化合物需要相对于经检测的pam位点的正确定位中的靶序列定义为候选靶位点。为了用cas9进行说明,将靶序列定义为后接pam序列的grna序列。例如,如果pam是ngg,靶序列是23nt序列,其中20ntgrna后接3ntpam。在优选的实施例中,另外的要求是一个或多个经鉴定的识别序列以核苷酸g开始。这些代表候选编辑位点的池,从中将用于编辑的实际位点进行如下所述的编辑。
其他类别的编辑化合物的候选者鉴定
可以使用总结用于pam位点检测的相同的一组检测方法(只需针对给定编辑化合物的特定要求进行适当修改),对用于具有序列基序或基于组分限制的编辑化合物的候选位点进行鉴定。
对于那些需要特定序列特征进行位点识别的编辑化合物,可能需要其他检测方法。例如,如果还需要潜在修饰位点的某种结构构象,则可能需要核苷酸结构预测工具来界定具有用于编辑潜力的位置,然后来自那些位置的序列成为候选池。
对可修饰位点的物理鉴定
还可以通过许多其他的方式来鉴定适用于编辑的位点,所述方法包括但不限于:体外或体内核苷酸保护测定以及在核苷酸序列上检测编辑化合物定位的其他方法。对于一些检测方法,必须使编辑化合物失活以便于保留必要的定位。在其他方法中,可以通过对序列修饰的位点侧翼的区域进行测序来凭经验鉴定合适的位点。在其他方法中,如果存在核苷酸结构的要求,则可以使用以下方法来收集潜在的修饰靶标,所述方法对集合中具有所述基序结构类别的序列进行富集。例如,可以在靶向序列集的剪切形式进行凝胶迁移率测定。在又其他方法中,可以将引物设计成已知的识别基序,并用于通过引物结合来对靶序列集中的所有成员进行扩增和或测序。通过本领域技术人员通常使用的任何这些方法或其他方法生成的位点序列的集合成为了候选编辑位点序列池。
靶位点背景(tsc)
希望根据所希望的修饰的功效、特异性和效率来选择最佳位点进行编辑。可以按多种方式来提供用于编辑位点的背景信息,以便于确定要使用的一个或多个位点。可以对候选池的成员应用多个过滤器,以减少用于修饰的候选位点集,并根据期望它们满足特异性的所需质量、修饰效率、敏感性和易用性的期望程度应用优先级划分。
对于单一指导rna编辑化合物,优选的要求是潜在的靶位点以核苷酸g开头,并以所述编辑化合物的合适的pam结束,以实现有效的u6poliii指导序列表达。
通常,在通过序列编辑剂的识别位点需求指导下设计和产生基因组编辑产物的过程中,可以将位点长度过滤器应用于所有类型的基因组修饰剂。例如,可能需要普通cas9位点的识别序列组分以落入在17nt和25nt之间。
特异性过滤器
使用多种方法来确定序列集之间的特异性。所使用的具体方法取决于是否期望序列集来反映序列的天然丰度。例如,可以使用参考基因组序列或其他类型的未扩增序列来反映天然丰度。或,如果修饰序列集包含潜在改变的丰度,例如pcr扩增的下一代序列读段,则可以使用相应改变的序列集。使用优选的cas9编辑化合物,将这些方法应用于为特定的玉蜀黍基因型赋予蜡状性状表型的玉蜀黍编辑实例。
通常用于提高特异性的过滤器是仅报告正在被编辑的序列集合中具有唯一或罕见(例如,默认值2)序列和/或一个或多个关键子序列(例如,所谓的crispr/cas9种子序列)的那些位点。通过过滤候选编辑位点来增强功效,这些候选编辑位点在序列集中具有相似但不相同的序列或关键子序列,其中编辑距离(默认值4)在由相关编辑化合物识别的范围内。可以使用短读比对仪(例如,bowtie,bwa)或以上在pam选择部分中指示的或在由本领域技术人员通常使用的任何其他方法来检测待编辑的序列的集合中位点的存在。通过将命中序列与靶位点序列的那些比较,针对每个所检测的命中计算编辑距离。如下进行计算:每个错配碱基具有编辑距离为1,每个插入或缺失具有编辑距离为其长度。当靶位点序列或经检测的命中序列中存在歧义核苷酸(例如,iupac代码)时,它们不会被罚分,且给定编辑距离为0。
在具有经潜在修饰的丰度的序列集合中,对用于确定集内的可能的特异性的候选选择方法进行修改通常是很有用的。在确定候选修饰位点的可能特异性时,数据量可能会带来其他挑战。例如,如果靶标集作为illumina短读段数据存在,则可能存在数亿甚至数十亿个读段。另外,可能出现由于测序平台或其他原因引起的序列错误。在序列集的这些类型中对原始序列数据进行的预处理变得很有必要。在优选的实施例中,预处理包括改进序列可靠性的步骤。例如,修整衔接子序列、去除pcr重复、重叠的序列合并、序列错误校正和相同序列的折叠。这些步骤使得由于待修饰的集内序列的非天然丰度而导致的歧义对潜在脱靶命中的检测的影响最小化。在我们的优选实施例中,cutadapt(martin2011)用于修整衔接子序列;flash(magoc和salzberg2011)用于合并重叠的序列;并且bfc(li2015)用于序列错误校正。
减少序列集规模的影响的一种方法是运行以下步骤,所述步骤不依赖于在起始序列集合的整个集或子集上、同时并行地对序列集的完全了解。一些步骤(例如,序列校正的优选方法)需要访问整个数据集,因此无法进行分块,并且必须以顺序方式运行。
可替代地,这些步骤中的许多可以通过使用组织序列数据的专用方法(诸如前述序列图模型)(其某些形式将固有地减少数据集中的冗余信息并提高序列的可靠性)被替换或取代。
在序列集合并和清除后,对用于发现靶序列的经修改的数据集进行搜索,寻找与候选位点池的成员具有相似性的序列,以产生经检测的潜在位点的集,如先前针对天然丰度序列集所述。在优选的实施例中,在净化的靶序列集中、具有经检测的位点的序列由匹配的候选池位点分组。在每个组中都应用了组装,以减少错误组装的可能性,并例如使用cap3(huang和madan1999)针对位点生成共有序列背景。然后将每个组中的序列组装成重叠群,以使脱靶的独特性最大化。每个重叠群代表基因组中的脱靶基因座。将相似性截止值(例如,默认值99%同一性)用于减少序列发生过度折叠的可能性,所述序列是相似的但源自不同来源。然后使用组装的重叠群作为被靶向用于修饰的序列集来进行第二轮选择过程。图4示出了在非天然序列丰度集合中特异性筛选的过程。将组装中使用的读段的数量和重叠群中歧义碱基的数量用作对每个脱靶基因座评分中另外的过滤因子。
另外的过滤器.
在利用pam的编辑化合物的情况下,类似的序列还必须满足针对所述编辑化合物的pam要求,包括任何可替代的pam序列基序(例如,对于最初描述的酿脓链球菌(spy)cas9而言,对于ngg为nag)。
在优选的实施例中,对于每个潜在的编辑位点,报告了位点序列的许多特征及其基因组背景。这些位点的实例包括所述位点是否具有3+个连续的t、g或c来评估早期终止的可能性;所述位置处其他特征(例如,基因或其他注释特征)破坏的可能性;周围dna的重复性质;dna甲基化状态;以及如果可获得深度测序数据,则靶位点序列在待编辑的基因型中是否保守。在待编辑的序列的集合中,位点序列或其周围背景的许多其他特征对于本领域技术人员将是可用的。
候选位点评分
将加权分配给针对位点的每个过滤器结果的状态,并提供罚分以简化对如希望地准确做出的所希望的修饰的可能性的评估。在优选的实施例中,罚分加权方案如下:
·编辑距离.编辑(如果有的话)越靠近位点的最受约束的部分(例如,pam序列),则罚分越高。
ο插入和缺失具有额外的罚分应用
·包括针对编辑化合物的经识别的区域的可替代的、较不优选部分(例如,用于单个rna指导编辑化合物的次级或可替代性的pam)的位点将被罚分。
实例1
选择总共12个近交系作为用于waxy(蜡质基因)基因组编辑的靶标系。(关于waxy编辑的靶标系的细节,参见公开号pct/us17/14903,将其通过引用并入本文中)。专有的等位基因模型序列资源库包括总共582个玉蜀黍近交种的下一代测序(ngs)序列,所述近交种中的38个具有相对深的覆盖范围(30x),其余具有平均3x的覆盖范围。使用bowtie2(langmead等人.2012),将所有序列与b73参考基因组比对。snp基因座是从具有相对深的覆盖范围的近交种定义的。为定义为snp,基因座必须满足以下标准:
1.至少一个近交种表现出与参考不同的纯合基因型。
2.仅允许4个近交种(约38个中的10%)具有缺失数据。
3.仅6%的具有观察数据的近交种可以携带杂合基因型。(在所有38个近交种显示观察数据的情况下,该标准将允许2个近交种是杂合的)。
4.对于跨所有近交种的基因座,仅观察到两个纯合等位基因。
“纯合的”基因型被定义为其中至少98%的观察到的读段包含相同等位基因的情况。
目的基因组区域包含66个snp基因座,将这些基因座用于鉴定哪些近交种在wx基因区域内是相同状态。582个近交种的66个基因座基因型产生了38,412个可能的基因型得分的矩阵,其中9,411个未观察到。为促进高通量管道中的单倍型构建,通过最近邻方法推算了这些未观察到的基因型。给定目的近交种和未观察到得分的基因座,将围绕所述基因座的300个snp基因座的基因型与数据集中彼此近交的基因型进行比较。最近邻近交种被定义为相对于在300个snp窗口内snp基因座的目的近交种而言,具有最低错配得分的近交种。一对近交种的错配得分由基因组窗口中每个snp基因座的错配得分之和组成(类似于roberts等人.2007)。两个纯合基因型之间的错配被记录得分为2,并且具有缺失数据的位点被记录得分为1。一个近交种是纯合子而另一个是杂合子的错配也被记录得分为1。如果希望更保守的估算,则可以修改缺失数据或杂合基因座的错配得分。
基于跨300个基因座观察到的和经估算的snp基因型的相似性,将近交种分为具有相同状态的单倍型的集。通过在每个基因座选择两个纯合等位基因之一来分配所有近交种的基因型,以用作任意参考等位基因。与参考等位基因不匹配的基因型被重编码为0,而与参考等位基因匹配的基因型被编码为1。缺失的基因型被编码为0.5。将基因型编码为数值时,两个近交种之间的距离d根据它们的基因型计算如下:
其中a和b是每个近交种的重编码基因型的向量,并且n是目的区域中snp基因型的数量。该距离度量通常被称为“曼哈顿(manhattan)”距离。然后基于这些距离,以分层、聚合的方式,使用完全连锁对所述近交种聚簇,这是针对聚簇问题的标准方法(james等人.2013)。在初始迭代中,所有近交种都被放置在自己的聚簇中。在连续的迭代中,比较了所有成对的聚簇,并将它们之间具有最小距离的聚簇连接。使用完全连锁方法,两个聚簇a和b之间的距离d被定义为:
其中d(a,b)如在公式1中定义。选择阈值t作为可以连接两个聚簇的最大可允许距离。通过以下条件来定义单倍型组,其中所有聚簇对具有高于阈值t的距离:
使用曼哈顿距离来定义基因型距离并使用完全连锁来定义聚簇距离,这允许将单倍型组解释为由基因型距离均小于阈值t的近交种的集组成。相关值s被定义为:
可以被认为是“相似性截止值”,它设置了单倍型组内允许的最小基因型相似性。
对具有相似性截止值s=0.98的582个近交种执行单倍型组分配的前述程序,对于wx目的区域产生至少3个近交种的10个相同状态的组。
实例2
该实例证实了根据本发明的方法设计的核酸靶向序列的用途,以在产生靶向基因组编辑的同时使无意脱靶编辑最小化。
当评估waxy1(wx1:grmzm2g024993)的指导rna情形时,在等位基因模型序列中鉴定候选cas9靶位点,随后是研究人员从候选池中选择靶位点,并然后针对b73参考基因组和编辑的基因型或脱靶位点的等位基因模型检查所选择的靶标。
探索了用于指导rna设计的许多情形。在一个实施例中,可以将个体等位基因模型序列提供给实施这些方法的网络或命令行界面,并且可以生成对每个输入等位基因模型特定的输出。可以选择过滤偏好,例如使一个或多个参考基因组中发现的脱靶命中最小化,并将结果进行比较以鉴定保守的核酸靶向序列。
其他实施例包括检查排序最前的等位基因模型序列的共有序列。在此类实施例中,可以部署任何可接受的多序列比对(msa)工具(例如,www.ebi.ac.uk/tools/msa),以经由编辑位点候选者鉴定部分中描述的方法生成共有序列输入序列用于检查。可以使用对本领域技术人员可用的clustalw(2)、mafft、muscle、kalign或可替代性程序,以产生有效的多序列比对和所得共有序列组装。诸如sequencher、alignx或其他dna/rna/蛋白质序列软件套件的程序通常包含嵌入式clustalw或其他msa工具,并可以按多种格式(诸如fasta)输出共有序列。可以使用默认值或自定义参数生成共有序列文件,所述默认值或自定义参数控制共有序列如何产生(同一性/多个),以及对于多态核苷酸,核苷酸或残基多态性可以如何使用iupac代码显示。在优选的实施例中,将通过对多于两个等位基因模型组进行比对而产生的共有序列文件提交至封装上述方法的命令行或网络工具,以搜索合适的位点,所述位点在被选择用于设计指导rna时能够使得cas9编辑化合物在相同的编辑化合物情况下对waxy1等位基因模型中的所有主要单倍型组做出编辑。将共有序列和单倍型的多个比对用于鉴定具有高度序列相似性的waxy1等位基因模型的合适的子区域,以便于多个单倍型可以被相同的编辑化合物进行有效地靶向。另外,将针对靶向区域的共有序列和单倍型的比对用于鉴定位置,如果所述位置被能够靶向所述位点的编辑化合物靶向则指导其仅修饰某些单倍型或单倍型的分组,所述单倍型之间共享可靶向的序列保守性,但与在所述位点处的其他单倍型存在实质性的差异。当搜索位点外(off-site)命中时,通过实施编辑位点候选者鉴定和编辑位点候选者中的选择部分中所述的方法的网站和命令行工具,将任何iupac取代残基转换为任何碱基代码n。
在优选的实施例中,经由msa工具产生的共有序列文件可以经受对于基因组编辑从业者已知的众多生物信息学重复掩蔽算法中的任何一种,所述算法基于与已知或发现对于任何基因组或使用本领域公认的多种方法重新鉴定的散布重复序列重复的序列的相似性关系来滤除序列重复残基。在优选的实施例中,可以提交从任何msa工具导出的共有序列等位基因模型序列(具有或不具有针对多态性残基的iupac取代),以将产生输出文件的掩蔽算法进行重复,所述算法用模糊占位符(例如,x或n)掩蔽重复残基。
实例(双链的)重复掩蔽的waxy1(启动子)共有序列等位基因模型序列,表明保守的指导rna靶向cr10和cr4。
实例3
重复掩蔽的waxy1共有序列等位基因模型序列通过pam位点扫描运行,以鉴定所有pam位点,并然后过滤到具有在参考基因组序列中不超过单个拷贝的完全匹配的靶序列的那些候选序列。将bowtie(“bowtie-a-v0”)用于搜索玉蜀黍参考基因组中靶序列的完全匹配命中。总共鉴定了109个靶pam位点,其具有完全靶序列的至多一个拷贝,并且其中存在68个靶pam位点(具有种子序列的至多一个拷贝),所述靶pam位点成为候选位点。
接下来,通过基于参考的脱靶扫描运行每个候选pam位点的靶序列,以使用bwa(“bwaaln-n4”)来鉴定具有高达4的编辑距离的所有可能的脱靶。在参考基因组中发现的与靶序列不完全相同但非常相似的脱靶,然后用于进一步将候选列表滤至没有编辑距离为1的脱靶的那些序列。例如,对于cr4andcr10,列出了在玉蜀黍b73参考基因组中编辑距离为0至4的脱靶数目。对于两个位点,存在具有编辑距离大于2的脱靶,但总数足够低以确认所述两个位点都对waxy序列具有特异性。
最后,通过无参考脱靶扫描运行每个靶序列,以鉴定具有在三个玉蜀黍近交系的ngs短读段中编辑距离高达4的所有可能的脱靶,其中使用illuminahi-seq对每个近交系按75x+深度进行测序。然后,在ngs读段中发现的脱靶进一步确认,除了靶序列以外,在这些近交种中均未发现完全匹配的命中。例如,对于cr4和cr10,将近交种中编辑距离为0至4的脱靶数目如下列出。发现两个重叠群在cr10的近交种2_ngs中具有完全匹配,但随后通过使用cap3的另一轮组装(其中同一性截止值放宽至95%)将两个重叠群确认为来自相同来源。相同的情况适用于在cr4的近交种1_ngs中具有完全匹配的两个重叠群。在每个编辑距离处的脱靶数目仍然很低,足以确认其对waxy序列的特异性。
表1:
实例4
关于包含在582个近交种群组内的典型杂种优势组(例如硬柄合成(stiffstalksynthetic(sss))、非硬柄(non-stiffstalk(nss))、flint或其他杂种优势组分类),可以检查单倍型组的总体分布。在waxy1基因(wx1,grmzm2g024993)的情况下,可以将10个相同状态的组进一步解析为在sss和nss杂种优势池中主要的基于pilon组装的等位基因模型组(参见图5)。
表2:
在该wx1实例中,前10个独特的等位基因模型代表在n=582个近交种集中所有系的96%。针对wx1的crispr-cas实验的设计可以集中在对应于特定靶向的近交种基因型的个体等位基因模型上;或集中在等位基因模型分布中观察到的主要等位基因上;或集中在来自等位基因模型分布的罕见等位基因上;或集中在通过对来自等位基因模型分布的两个或更多个序列进行比较而生成的共有序列文件上。在seqidno.1、wx1_pro_cr10和wx1_pro_cr4中所述的指导rna作为实例跨所有主要单倍型是100%保守的,具有通过以上报道的位点鉴定和选择方法的我们基于网络和基于命令行的一种或多种实施检测的最小位点外靶标,并且有望在相关种质中跨所有主要iis单倍型在切割dna方面具有作为cas9试剂的活性。
- 还没有人留言评论。精彩留言会获得点赞!
- 肿瘤靶向核磁共振/荧光双模态成像造影剂及其制备和应用的制造方法与工艺
- pH氧化还原双敏感的PAMAM靶向纳米给药载体及其制备方法与制造工艺
- 具有卵巢肿瘤靶向D构型多肽及其基因递释系统的制造方法与工艺
- 靶向survivin纳米抗体及其制备方法和应用与制造工艺
- 用于PSMA靶向的成像和放射疗法的金属/放射性金属标记的PSMA抑制物的制造方法与工艺
- 一种线粒体靶向的粘度荧光探针及其制备方法和应用与制造工艺
- 一种兼具荧光可视、磁靶向和pH敏多功能的复合空心微球药物载体及其制备方法与制造工艺
- 一种用于修饰微泡的多肽以及靶向GBM的药物制剂的制造方法与工艺
- 白果内酯作为脑靶向增效剂在制备防治脑肿瘤药物的应用的制造方法与工艺
- 机器人辅助精确靶向穿刺软组织目标柔性针夹紧固定装置的制造方法