用于蛋白质鉴定的解码方法与流程
交叉引用本申请要求2017年12月29日提交的第62/611,979号美国临时专利申请和2018年10月20日提交的第pct/us2018/056807号国际申请的权益,其中每一个均通过引用整体并入本文。
背景技术:
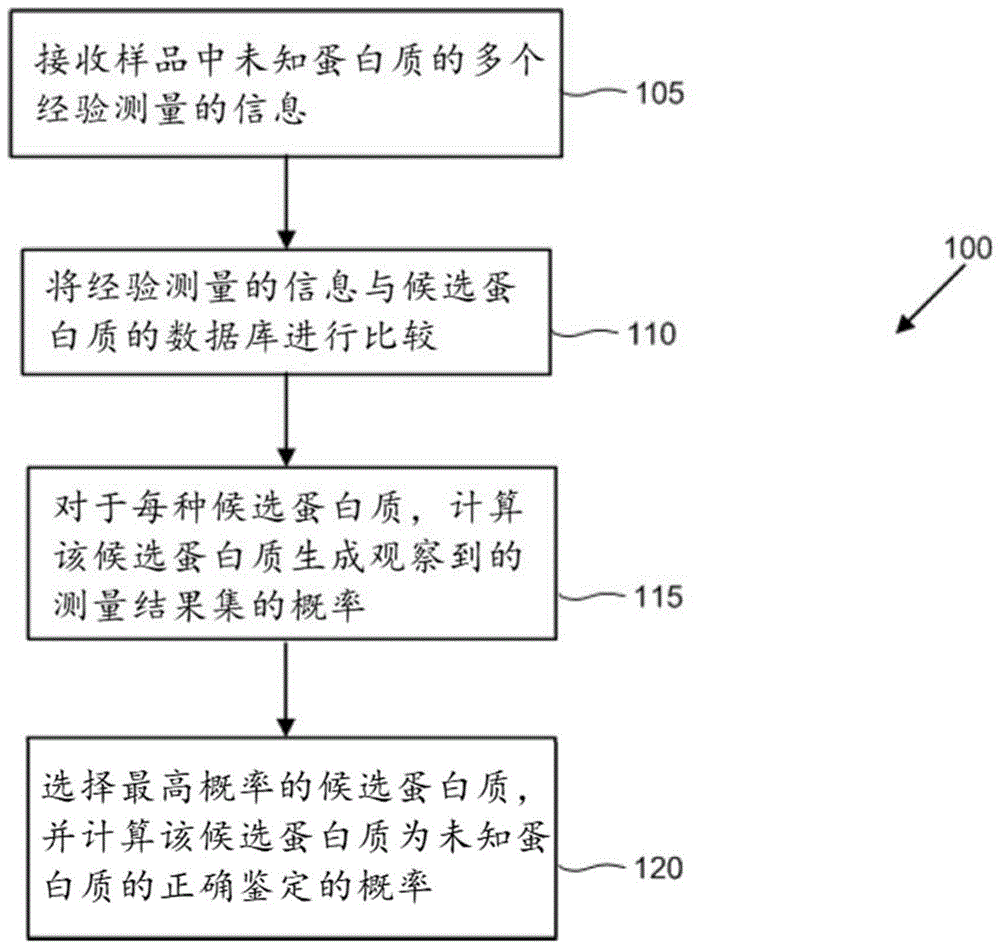
:当前用于蛋白质鉴定的技术通常依赖于高度特异性和灵敏性亲和试剂(如抗体)的结合和随后读出,或者依赖于来自质谱仪的肽读取数据(长度通常为大约12-30个氨基酸)。可以将这类技术应用于样品中的未知蛋白质,以基于对高度特异性和敏感性亲和试剂与目的蛋白质的结合测量值的分析,确定候选蛋白质的存在、不存在或量。技术实现要素:本文认识到需要改进未知蛋白质样品中蛋白质的鉴定和定量。本文提供的方法和系统可以显著减少或消除鉴定样品中的蛋白质的错误,从而改善所述蛋白质的定量。这类方法和系统可以实现未知蛋白质样品内候选蛋白质的准确和有效鉴定。这样的鉴定可以基于使用信息的计算,该信息例如是被配置为选择性地与一种或多种候选蛋白质结合的亲和试剂探针的结合测量、蛋白质长度、蛋白质疏水性和等电点。在一些实施方案中,未知蛋白质的样品可以暴露于单独的亲和试剂探针、合并的亲和试剂探针或单独的亲和试剂探针和合并的亲和试剂探针的组合。所述鉴定可以包括估计所述样品中存在一种或多种候选蛋白质中的每一种的置信水平。本文提供的方法和系统可以包括用于基于对完全完整的蛋白质或蛋白质片段进行的一系列实验来鉴定蛋白质的算法。每个实验可以是对蛋白质进行的经验测量,并且可以提供可用于鉴定该蛋白质的信息。实验的实例包括对亲和试剂(例如,抗体或适体)的结合、蛋白质长度、蛋白质疏水性和等电点的测量。关于实验结果的信息可用来计算蛋白质候选物的概率或可能性,和/或用来通过从蛋白质候选物列表中选择使观察到的实验结果的可能性最大化的蛋白质来推断蛋白质身份。本文提供的方法和系统还可包括蛋白质候选物的集合,以及用来计算实验结果来自这些蛋白质候选物中的每一种的概率的算法。在一个方面,本公开提供了一种鉴定未知蛋白质样品中的蛋白质的计算机实现的方法,该方法包括:(a)通过所述计算机接收对所述样品中的所述未知蛋白质进行的多个经验测量的信息;(b)通过所述计算机,将所述多个所述经验测量的所述信息的至少一部分与包含多个蛋白质序列的数据库进行比较,每个蛋白质序列对应于多种候选蛋白质中的候选蛋白质;以及(c)对于所述多种候选蛋白质中的一种或多种候选蛋白质中的每一种,通过所述计算机,基于所述多个所述经验测量的所述信息的所述至少一部分与包含所述多个蛋白质序列的所述数据库的所述比较,生成以下一项或多项:(i)所述候选蛋白质生成所述多个经验测量的所述信息的概率,(ii)假定所述样品中存在所述候选蛋白质,未观察到所述多个经验测量的概率,以及(iii)所述样品中存在所述候选蛋白质的概率。在一些实施方案中,所述多个经验测量中的两个或更多个选自:(i)一个或多个亲和试剂探针中的每一个与所述样品中所述未知蛋白质的结合测量,每个亲和试剂探针被配置为选择性地与所述多种候选蛋白质中的一种或多种候选蛋白质结合;(ii)所述样品中的一种或多种所述未知蛋白质的长度;(iii)所述样品中的一种或多种所述未知蛋白质的疏水性;以及(iv)所述样品中的一种或多种所述未知蛋白质的等电点。在一些实施方案中,生成所述多个概率进一步包括接收多个附加亲和试剂探针中的每一个的结合测量的附加信息,每个附加亲和试剂探针被配置为选择性地与所述多种候选蛋白质中的一种或多种候选蛋白质结合。在一些实施方案中,所述方法进一步包括针对所述一种或多种候选蛋白质中的每一种,生成所述候选蛋白质与所述样品中的所述未知蛋白质之一相匹配的置信水平。在一些实施方案中,所述多个亲和试剂探针包含不超过50个亲和试剂探针。在一些实施方案中,所述多个亲和试剂探针包含不超过100个亲和试剂探针。在一些实施方案中,所述多个亲和试剂探针包含不超过200个亲和试剂探针。在一些实施方案中,所述多个亲和试剂探针包含不超过300个亲和试剂探针。在一些实施方案中,所述多个亲和试剂探针包含不超过500个亲和试剂探针。在一些实施方案中,所述多个亲和试剂探针包含超过500个亲和试剂探针。在一些实施方案中,所述方法进一步包括生成鉴定所述样品中的所述蛋白质的纸质或电子报告。在一些实施方案中,所述样品包括生物样品。在一些实施方案中,所述生物样品从受试者获得。在一些实施方案中,所述方法进一步包括至少基于所述多个概率来确定所述受试者中的疾病状态。在一些实施方案中,(c)包括对于所述多种候选蛋白质中的一种或多种候选蛋白质中的每一种,通过所述计算机生成(i)所述候选蛋白质生成所述多个经验测量的所述信息的所述概率。在一些实施方案中,(c)包括对于所述多种候选蛋白质中的一种或多种候选蛋白质中的每一种,通过所述计算机生成(ii)假定所述样品中存在所述候选蛋白质,未观察到所述多个经验测量的所述概率。在一些实施方案中,(c)包括对于所述多种候选蛋白质中的一种或多种候选蛋白质中的每一种,通过所述计算机生成(iii)所述样品中存在所述候选蛋白质的所述概率。在一些实施方案中,所述测量结果包含亲和试剂探针的结合。在一些实施方案中,所述测量结果包含亲和试剂探针的非特异性结合。在一些实施方案中,所述测量结果包含亲和试剂探针的结合。在一些实施方案中,所述测量结果包含亲和试剂探针的非特异性结合。在一些实施方案中,所述经验测量包含亲和试剂探针的结合。在一些实施方案中,所述经验测量包含亲和试剂探针的非特异性结合。在一些实施方案中,所述方法进一步包括生成具有预定阈值的蛋白质鉴定的灵敏度。在一些实施方案中,所述预定阈值为小于1%不正确。在一些实施方案中,所述样品中的所述蛋白质是截短的或降解的。在一些实施方案中,所述样品中的所述蛋白质不是源自蛋白质末端。在一些实施方案中,所述经验测量包括所述样品中的一种或多种所述未知蛋白质的长度。在一些实施方案中,所述经验测量包括所述样品中的一种或多种所述未知蛋白质的疏水性。在一些实施方案中,所述经验测量包括所述样品中的一种或多种所述未知蛋白质的等电点。在一些实施方案中,所述经验测量包括对抗体混合物进行的测量。在一些实施方案中,所述经验测量包括对从多个物种获得的样品进行的测量。在一些实施方案中,所述经验测量包括在由非同义单核苷酸多态性(snp)引起的单氨基酸变异(sav)的存在下对样品进行的测量。基于仅示出并描述了本公开的说明性实施方案的以下详细描述,本公开的其他方面和优点对本领域技术人员而言将变得显而易见。应当认识到,本公开能够具有其他不同的实施方案,并且其若干细节能够在各个明显的方面进行修改,所有这些都不脱离本公开内容。因此,附图和说明书在本质上将被视为说明性的,而非限制性的。援引并入本说明书中所提及的所有出版物、专利和专利申请均通过引用并入本文,其程度犹如具体地且单独地指出每个单独的出版物、专利或专利申请均通过引用而并入。如果通过引用而并入的出版物和专利或专利申请与本说明书中包含的公开内容存在矛盾,则本说明书旨在取代和/或优先于任何这样的矛盾材料。附图说明本发明的新颖特征在所附权利要求书中具体阐述。通过参考以下对利用本发明原理的说明性实施方案加以阐述的详细描述以及附图(在本文中也称为“图”),将会对本发明的特征和优点获得更好的理解,在这些附图中:图1示出了根据所公开的实施方案,生物样品中未知蛋白质的蛋白质鉴定的示例流程图。图2示出了根据所公开的实施方案,对于三种不同的实验情况(使用50、100和200个探针,分别用灰色、黑色和白色圆圈表示),相对于亲和试剂探针中的探针识别位点(例如,三聚体结合表位)数目(范围最多100个探针识别位点或三聚体结合表位)绘制的亲和试剂探针的灵敏度(例如,在小于1%的错误检测率(fdr)下鉴定的底物的百分比)。图3示出了根据所公开的实施方案,对于三种不同的实验情况(使用50、100和200个探针,分别用灰色、黑色和白色圆圈表示),相对于亲和试剂探针中的探针识别位点(例如,三聚体结合表位)数目(范围最多700个探针识别位点或三聚体结合表位)绘制的亲和试剂探针的灵敏度(例如,在小于1%的错误检测率(fdr)下鉴定的底物的百分比)。图4示出的图示显示了根据所公开的实施方案,对于使用100个(左)、200个(中心)或300个探针(右)的实验,蛋白质鉴定的灵敏度。图5示出的图示显示了对于使用各种蛋白质片段化方法的实验,蛋白质鉴定的灵敏度。在顶行和底行的每一行中,根据所公开的实施方案,以50、100、200和300个亲和试剂测量(在从左至右的4幅图中)显示了蛋白质鉴定性能,其中最大片段长度值为50、100、200、300、400和500(分别以六边形、朝下的三角形、朝上的三角形、菱形、矩形和圆形表示)。图6示出的图示显示了根据所公开的实施方案,对于使用测量类型的各种组合的实验,人类蛋白质鉴定的灵敏度(在小于1%的fdr下鉴定的底物的百分比)。图7示出的图示显示了根据所公开的实施方案,对于使用50、100、200或300个针对来自大肠杆菌、酵母或人的未知蛋白质的亲和试剂探针(分别由圆圈、三角形和正方形表示)的实验,蛋白质鉴定的灵敏度。图8示出的图示显示了根据所公开的实施方案,相对于迭代(x轴)的结合概率(y轴,左)和蛋白质鉴定的灵敏度(y轴,右)。图9显示了根据所公开的实施方案,对于模拟的200个探针的实验,估计的错误鉴定率与实际错误鉴定率的比较证明了准确的错误鉴定率估计。图10示出了被编程或以其他方式配置为实现本文提供的方法的计算机控制系统。图11示出了删截的(censored)蛋白质鉴定与未删截的(uncensored)蛋白质鉴定方法的性能。图12示出了删截的蛋白质鉴定和未删截的蛋白质鉴定方法对随机“假阴性”结合结果的容忍度。图13示出了删截的蛋白质鉴定和未删截的蛋白质鉴定方法对随机“假阳性”结合结果的容忍度。图14示出了采用被高估或被低估的亲和试剂结合概率,删截的蛋白质鉴定和未删截的蛋白质鉴定方法的性能。图15示出了使用具有未知结合表位的亲和试剂,删截的蛋白质鉴定和未删截的蛋白质鉴定方法的性能。图16示出了使用具有遗漏的结合表位的亲和试剂,删截的蛋白质鉴定和未删截的蛋白质鉴定方法的性能。图17示出了使用针对蛋白质组中的前300个最丰富的三聚体、蛋白质组中的300个随机选择的三聚体或蛋白质组中的300个最不丰富的三聚体的亲和试剂,删截的蛋白质鉴定和未删截的蛋白质鉴定方法的性能。图18示出了使用具有随机或生物类似脱靶位点的亲和试剂,删截的蛋白质鉴定和未删截的蛋白质鉴定方法的性能。图19示出了使用一组最佳亲和试剂(探针),删截的蛋白质鉴定和未删截的蛋白质鉴定方法的性能。图20示出了使用未混合的候选亲和试剂和候选亲和试剂的混合物,删截的蛋白质鉴定和未删截的蛋白质鉴定方法的性能。图21示出了根据一些实施方案,两个杂交步骤对亲和试剂与蛋白质之间的结合的增强。图22示出了根据一些实施方案,使用用于选择性修饰和检测4种氨基酸(k、d、c和w)的试剂集合的蛋白质鉴定性能。图23示出了根据一些实施方案,使用用于选择性修饰和检测20种氨基酸(r、h、k、d、e、s、t、n、q、c、g、p、a、v、i、l、m、f、y和w)的试剂集合的蛋白质鉴定性能。图24示出了根据一些实施方案,使用氨基酸顺序的测量的蛋白质鉴定的性能,其中以x轴上示出的检测概率(等于反应效率)测量所有氨基酸,并且y轴表示在低于1%的错误发现率下鉴定的样品中蛋白质的百分比。具体实施方式尽管已经在本文中显示并描述了本发明的多个实施方案,但是对本领域技术人员显而易见的是这些实施方案仅作为实例提供。在不偏离本发明的情况下,本领域技术人员可以想到许多改变、变化和替换。应当理解,可以使用本文所述发明的实施方案的各种替代方案。如本文所用的术语“样品”通常是指生物样品(例如,含有蛋白质的样品)。样品可取自组织或细胞,或者取自组织或细胞的环境。在一些实例中,样品可以包含或来源于组织活检物、血液、血浆、细胞外液、干燥的血液斑点、培养的细胞、培养基、废弃组织、植物物质、合成蛋白质、细菌样品和/或病毒样品、真菌组织、古菌或原生动物。在采集之前,样品可能已从来源中分离。样品可包含法医证据。非限制性实例包括在采集之前从主要来源中分离的指纹、唾液、尿液、血液、粪便、精液或其他体液。在一些实例中,蛋白质在样品制备过程中从其主要来源(细胞、组织、体液如血液、环境样品等)中分离。样品可以来源于灭绝的物种,包括但不限于来源于化石的样品。该蛋白质可以从或者可以不从其主要来源纯化或以其他方式富集。在一些情况下,在进一步加工之前将主要来源均质化。在一些情况下,使用缓冲液如ripa缓冲液裂解细胞。在此阶段也可使用变性缓冲液。可以对样品进行过滤或离心以去除脂质和颗粒物质。样品也可以被纯化以去除核酸,或者可以用rna酶和dna酶处理。样品可含有完整蛋白质、变性蛋白质、蛋白质片段或部分降解的蛋白质。样品可取自患有疾病或病症的受试者。所述疾病或病症可以是传染病、免疫病症或疾病、癌症、遗传病、退行性疾病、生活方式疾病、损伤、罕见疾病或年龄相关性疾病。该传染病可由细菌、病毒、真菌和/或寄生虫引起。癌症的非限制性实例包括膀胱癌、肺癌、脑癌、黑素瘤、乳腺癌、非霍奇金淋巴瘤、宫颈癌、卵巢癌、结直肠癌、胰腺癌、食管癌、前列腺癌、肾癌、皮肤癌、白血病、甲状腺癌、肝癌和子宫癌。遗传疾病或病症的一些实例包括但不限于多发性硬化(ms)、囊性纤维化、charcot–marie–tooth病、亨廷顿病(huntington'sdisease)、peutz-jeghers综合征、唐氏综合症、类风湿性关节炎和tay–sachs病。生活方式疾病的非限制性实例包括肥胖症、糖尿病、动脉硬化、心脏病、中风、高血压、肝硬化、肾炎、癌症、慢性阻塞性肺病(copd)、听力问题和慢性背痛。损伤的一些实例包括但不限于擦伤、脑损伤、瘀伤、烧伤、脑震荡、充血性心力衰竭、建筑损伤、脱位、连枷胸、骨折、血胸、椎间盘突出、髋骨隆凸挫伤、低体温、撕裂、神经挟捏、气胸、肋骨骨折、坐骨神经痛、脊髓损伤、肌腱韧带筋膜损伤、创伤性脑损伤和鞭伤。可在治疗患有疾病或病症的受试者之前和/或之后取得样品。可在治疗之前和/或之后取得样品。可在治疗或治疗方案期间取得样品。可从受试者取得多个样品以监测治疗随时间的效果。可从已知或疑似患有没有可用诊断性抗体的传染病的受试者取得样品。样品可取自疑似患有疾病或病症的受试者。样品可取自经历不明原因的症状如疲劳、恶心、体重减轻、酸痛和疼痛、虚弱或记忆丧失的受试者。样品可取自具有明确原因的症状的受试者。样品可取自由于诸如家族史、年龄、环境暴露等因素、生活方式风险因素或存在其他已知风险因素而具有发生疾病或病症的风险的受试者。样品可取自胚胎、胎儿或孕妇。在一些实例中,样品可包含从母亲血浆中分离的蛋白质。在一些实例中,蛋白质从母亲血液中的循环胎儿细胞分离。样品可以取自健康个体。在一些情况下,样品可以纵向地取自同一个体。在一些情况下,可以对纵向获取的样品进行分析,目的是监测个体的健康状况并早期检测健康问题。在一些实施方案中,可以在家庭环境或照护点环境下采集样品,随后在分析之前通过邮递、快递或其他运输方法来运输该样品。例如,家庭用户可以通过手指点刺采集血斑样品,该血斑样品可以被干燥并且随后在分析之前通过邮递来运输。在一些情况下,可以使用纵向获取的样品来监测对预期会影响健康、运动表现或认知表现的刺激的反应。非限制性实例包括对药物、节食或运动方案的反应。可对样品的蛋白质进行处理以去除可能干扰表位结合的修饰。例如,可对蛋白质进行酶处理。例如,可对蛋白质进行糖苷酶处理以去除翻译后糖基化。可用还原剂处理蛋白质以减少该蛋白质内的二硫键。可用磷酸酶处理蛋白质以去除磷酸基团。可以去除的翻译后修饰的其他非限制性实例包括乙酸基团、酰胺基团、甲基、脂质、遍在蛋白、豆蔻酰化、棕榈酰化、异戊二烯化或异戊烯化(例如法尼醇和香叶基香叶醇)、法尼基化、香叶基香叶酰化、糖基磷脂酰肌醇化、脂化、黄素部分附接、磷酸泛酰巯基乙胺化和亚视黄基席夫碱形成。样品的蛋白质可以通过修饰一个或多个残基以使其更易于被亲和试剂结合或检测来处理。在一些情况下,可以处理样品的蛋白质以保留可促进或增强表位结合的翻译后蛋白质修饰。在一些实例中,可将磷酸酶抑制剂添加到样品中。在一些实例中,可添加氧化剂以保护二硫键。可以使样品的蛋白质完全或部分变性。在一些实施方案中,可以使蛋白质完全变性。可通过施加外部应激如去污剂、强酸或强碱、浓无机盐、有机溶剂(例如,醇或氯仿)、辐射或热而使蛋白质变性。可通过添加变性缓冲液而使蛋白质变性。也可将蛋白质沉淀、冻干和悬浮在变性缓冲液中。蛋白质可通过加热而变性。不太可能对蛋白质造成化学修饰的变性方法可能是优选的。在缀合之前或之后,可对样品的蛋白质进行处理以产生更短的多肽。剩余的蛋白质可以用酶如蛋白酶k部分消化以生成片段,或者可以保持其完整。在进一步的实例中,蛋白质可暴露于蛋白酶如胰蛋白酶。蛋白酶的另外的实例可包括丝氨酸蛋白酶、半胱氨酸蛋白酶、苏氨酸蛋白酶、天冬氨酸蛋白酶、谷氨酸蛋白酶、金属蛋白酶和天冬酰胺肽裂合酶。在一些情况下,去除极大的和小的蛋白质(例如肌联蛋白)可能是有用的,例如,此类蛋白质可以通过过滤或其他适当的方法去除。在一些实例中,极大蛋白质可包括至少约400千道尔顿(kd)、450kd、500kd、600kd、650kd、700kd、750kd、800kd或850kd的蛋白质。在一些实例中,极大蛋白质可包括至少约8,000个氨基酸、约8,500个氨基酸、约9,000个氨基酸、约9,500个氨基酸、约10,000个氨基酸、约10,500个氨基酸、约11,000个氨基酸或约15,000个氨基酸的蛋白质。在一些实例中,小蛋白质可包括小于约10kd、9kd、8kd、7kd、6kd、5kd、4kd、3kd、2kd或1kd的蛋白质。在一些实例中,小蛋白质可包括少于约50个氨基酸、45个氨基酸、40个氨基酸、35个氨基酸或约30个氨基酸的蛋白质。可以通过大小排阻色谱法去除极大或小蛋白质。极大蛋白质可通过大小排阻色谱法分离,用蛋白酶处理以产生中等大小的多肽,并与样品的中等大小的蛋白质重新组合。例如,可以用可辨识的标签标记样品的蛋白质,以允许样品的多路化。可辨识的标签的一些非限制性实例包括:荧光团、荧光纳米颗粒、量子点、磁性纳米颗粒或dna条形码化的碱基连接体。所使用的荧光团可包括荧光蛋白,如gfp、yfp、rfp、egfp、mcherry、tdtomato、fitc、alexafluor350、alexafluor405、alexafluor488、alexafluor532、alexafluor546、alexafluor555、alexafluor568、alexafluor594、alexafluor647、alexafluor680、alexafluor750、pacificblue、香豆素、bodipyfl、pacificgreen、oregongreen、cy3、cy5、pacificorange、tritc、texasred、藻红蛋白和别藻蓝蛋白。可以对任何数目的蛋白质样品进行多路化。例如,多路化的反应可含有来自2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、约20、约25、约30、约35、约40、约45、约50、约55、约60、约65、约70、约75、约80、约85、约90、约95、约100个或多于约100个初始样品的蛋白质。可辨识的标签可提供探询每种蛋白质的来源样品的方式,或者可指导来自不同样品的蛋白质隔离到固体支持物上的不同区域。在一些实施方案中,随后将蛋白质施加至官能化的基底上,从而以化学方式将蛋白质附接至基底。任何数目的蛋白质样品可以在分析之前混合,而不进行标记或多路化。例如,多路化的反应可含有来自2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、约20、约25、约30、约35、约40、约45、约50、约55、约60、约65、约70、约75、约80、约85、约90、约95、约100个或多于约100个初始样品的蛋白质。例如,可以对合并的样品进行罕见病况的诊断。然后可以只对该诊断检测呈阳性的样品池中的样品进行单个样品的分析。可以使用组合池化设计对样品进行多路化而无需标记,在该设计中,以某种方式将样品混合到池中,该方式允许使用计算多路分解从分析的池中解析出来自各个样品的信号。如本文所用的术语“基底”通常是指能够形成固体支持物的基底。基底或固体基底可指蛋白质可以共价或非共价附接至其上的任何固体表面。固体基底的非限制性实例包括颗粒、珠子、载玻片、装置元件的表面、膜、流动池、孔、腔室、宏观流体腔室、微流体腔室、通道、微流体通道或其他任何表面。基底表面可以是平的或弯曲的,或者可以具有其他形状,并且可以是光滑的或有纹理的。基底表面可含有微孔。在一些实施方案中,基底可以由玻璃、碳水化合物如葡聚糖、塑料如聚苯乙烯或聚丙烯、聚丙烯酰胺、胶乳、硅、金属如金,或纤维素组成,并且可被进一步修饰以允许或增强蛋白质的共价或非共价附接。例如,基底表面可通过用特定官能团如马来酸或琥珀酸部分修饰进行官能化,或者通过用化学反应性基团如氨基、巯基或丙烯酸基团修饰(例如通过硅烷化)进行衍生化。合适的硅烷试剂包括氨基丙基三甲氧基硅烷、氨基丙基三乙氧基硅烷和4-氨基丁基三乙氧基硅烷。基底可用n-羟基琥珀酰亚胺(nhs)官能团进行官能化。玻璃表面还可以使用例如环氧硅烷、丙烯酸硅烷或丙烯酰胺硅烷,通过诸如丙烯酸或环氧基等其他反应性基团进行衍生化。供蛋白质附接的基底和方法优选地对于反复的结合、洗涤、成像和洗脱步骤是稳定的。在一些实例中,基底可以是载玻片、流动池或者微尺度或纳米尺度结构(例如,有序结构,如微孔、微柱、单分子阵列、纳米球、纳米柱或纳米线)。基底上官能团的间隔可以是有序的或随机的。可通过例如光刻法、蘸笔(dip-pen)纳米刻蚀法、纳米压印刻蚀法、纳米球刻蚀法(nanospherelithography)、纳米球刻蚀法(nanoballlithography)、纳米柱阵列、纳米线刻蚀法、扫描探针刻蚀法、热化学刻蚀法、热扫描探针刻蚀法、局部氧化纳米刻蚀法、分子自组装、模版刻蚀法或电子束刻蚀法来创建官能团的有序阵列。有序阵列中的官能团可被定位成使得每个官能团与其他任何官能团相距小于200纳米(nm),或约200nm、约225nm、约250nm、约275nm、约300nm、约325nm、约350nm、约375nm、约400nm、约425nm、约450nm、约475nm、约500nm、约525nm、约550nm、约575nm、约600nm、约625nm、约650nm、约675nm、约700nm、约725nm、约750nm、约775nm、约800nm、约825nm、约850nm、约875nm、约900nm、约925nm、约950nm、约975nm、约1000nm、约1025nm、约1050nm、约1075nm、约1100nm、约1125nm、约1150nm、约1175nm、约1200nm、约1225nm、约1250nm、约1275nm、约1300nm、约1325nm、约1350nm、约1375nm、约1400nm、约1425nm、约1450nm、约1475nm、约1500nm、约1525nm、约1550nm、约1575nm、约1600nm、约1625nm、约1650nm、约1675nm、约1700nm、约1725nm、约1750nm、约1775nm、约1800nm、约1825nm、约1850nm、约1875nm、约1900nm、约1925nm、约1950nm、约1975nm、约2000nm或超过2000nm。可以以一定的浓度提供随机间隔的官能团,使得官能团与其他任何官能团平均相距至少约50nm、约100nm、约150nm、约200nm、约250nm、约300nm、约350nm、约400nm、约450nm、约500nm、约550nm、约600nm、约650nm、约700nm、约750nm、约800nm、约850nm、约900nm、约950nm、约1000nm或超过100nm。基底可被间接地官能化。例如,可对基底进行聚乙二醇化,并且可将官能团施加至全部或一组peg分子。可使用适合于微尺度或纳米尺度结构(例如,有序结构,如微孔、微柱、单分子阵列、纳米球、纳米柱或纳米线)的技术对基底进行官能化。基底可包含任何材料,包括金属、玻璃、塑料、陶瓷或其组合。在一些优选的实施方案中,固体基底可以是流动池。流动池可以由单层或多层组成。例如,流动池可包含基层(例如,硼硅酸盐玻璃层)、覆盖在基层上的通道层(例如,蚀刻的硅层)以及覆盖层或顶层。当这些层组装在一起时,可以形成封闭的通道,在任一端具有穿过覆盖层的入口/出口。每层的厚度可以变化,但优选小于约1700μm。这些层可由诸如光敏玻璃、硼硅酸盐玻璃、熔融硅酸盐、pdms或硅等合适的材料组成。不同的层可由相同的材料或不同的材料组成。在一些实施方案中,流动池可在流动池底部上包含通道开口。流动池可在可被离散地可视化的位置上包含数百万个附接的靶标缀合位点。在一些实施方案中,与本发明的实施方案一起使用的各种流动池可包含不同数目的通道(例如,1个通道、2个或更多个通道、3个或更多个通道、4个或更多个通道、6个或更多个通道、8个或更多个通道、10个或更多个通道、12个或更多个通道、16个或更多个通道,或超过16个通道)。各种流动池可包含不同深度或宽度的通道,深度或宽度可在单个流动池内的通道之间不同,或在不同流动池的通道之间不同。单个通道的深度和/或宽度也可以变化。例如,在通道内的一个或多个点处,通道可以是小于约50μm深、约50μm深、小于约100μm深、约100μm深、约100μm至约500μm深、约500μm深或超过约500μm深。通道可具有任何横截面形状,包括但不限于圆形、半圆形、矩形、梯形、三角形或卵形的横截面。可将蛋白质点样、滴加、移液、流动、洗涤或以其他方式施加至基底。在基底已经用诸如nhs酯的部分进行官能化的情况下,不需要对蛋白质进行修饰。在基底已经用替代部分(例如巯基、胺或连接体核酸)进行官能化的情况下,可以使用交联试剂(例如辛二酸二琥珀酰亚胺酯、nhs、磺酰胺)。在基底已经用连接体核酸进行官能化的情况下,可以用互补核酸标签修饰样品的蛋白质。可以使用可光活化的交联剂来引导样品与基底上的特定区域的交联。可以使用可光活化的交联剂通过将每个样品附接在基底的已知区域中来允许蛋白质样品的多路化。可光活化的交联剂可以例如通过在蛋白质交联之前检测荧光标签来允许已经成功标记的蛋白质的特异性附接。可光活化的交联剂的实例包括但不限于n-5-叠氮基-2-硝基苯甲酰基氧基琥珀酰亚胺、6-(4'-叠氮基-2'-硝基苯基氨基)己酸磺基琥珀酰亚胺酯、4,4'-氮杂戊酸琥珀酰亚胺酯、4,4'-氮杂戊酸磺基琥珀酰亚胺酯、6-(4,4'-氮杂戊酰胺基)己酸琥珀酰亚胺酯、6-(4,4'-氮杂戊酰胺基)己酸磺基琥珀酰亚胺酯、2-((4,4'-氮杂戊酰胺基)乙基)-1,3'-二硫代丙酸琥珀酰亚胺酯和2-((4,4'-氮杂戊酰胺基)乙基)-1,3'-二硫代丙酸磺基琥珀酰亚胺酯。多肽可通过一个或多个残基附接至基底。在一些实例中,多肽可经由n末端、c末端、两个末端或经由内部残基附接。除了永久性交联剂之外,使用可光切割的连接体对于一些应用也可能是合适的,并且这样做使得能够在分析后从基底中选择性地提取蛋白质。在一些情况下,可光切割的交联剂可用于几种不同的多路化样品。在一些情况下,可光切割的交联剂可用于多路化反应中的一个或多个样品。在一些情况下,多路化反应可包含经由永久性交联剂交联至基底的对照样品和经由可光切割的交联剂交联至基底的实验样品。每个缀合的蛋白质可在空间上彼此分开,使得每个缀合的蛋白质是光学可辨析的。因此,蛋白质可以用独特空间地址单独标记。在一些实施方案中,这可以通过使用低浓度蛋白质和基底上的低密度附接位点进行缀合以使得每个蛋白质分子在空间上彼此分开来实现。在使用可光活化的交联剂的实例中,可以使用光图案,使得蛋白质附着至预定的位置。在一些实施方案中,每个蛋白质可与独特空间地址相关联。例如,一旦蛋白质在空间上分离的位置处附接至基底,则每个蛋白质可以例如通过坐标被分配索引化的地址。在一些实例中,预先分配的独特空间地址的网格可以预先确定。在一些实施方案中,基底可含有易于辨识的固定的标志,使得可以相对于该基底的固定的标志确定每个蛋白质的放置。在一些实例中,基底可具有永久地标记在表面上的网格线和/或“原点”或其他基准点。在一些实例中,可永久地或半永久地标记基底的表面,以提供用以定位所交联的蛋白质的参考。图案化的形状本身,如缀合多肽的外部边界,也可以用作基准点,以供确定每个斑点的独特位置。基底还可以含有缀合的蛋白质标准品和对照。缀合的蛋白质标准品和对照可以是已缀合在已知位置上的已知序列的肽或蛋白质。在一些实例中,缀合的蛋白质标准品和对照可充当测定中的内部对照。蛋白质可以从纯化的蛋白质储备物施加至基底,或者可以通过诸如核酸可编程蛋白质阵列(nucleicacid-programmableproteinarray,nappa)等过程在基底上合成。在一些实例中,基底可包含荧光标准品。这些荧光标准品可用来校准测定之间的荧光信号强度。这些荧光标准品也可用来将荧光信号强度与区域中存在的荧光团的数目相关联。荧光标准品可包含在测定中使用的一些或所有不同类型的荧光团。一旦基底与来自样品的蛋白质缀合,就可进行多亲和试剂测量。本文所述的测量过程可以采用各种亲和试剂。在一些实施方案中,可以将多种亲和试剂混合在一起,并且可以对该亲和试剂混合物与蛋白质-基底缀合物的结合进行测量。在一些情况下,对亲和试剂混合物的结合进行的测量可以在不同的溶剂条件和/或蛋白质折叠条件方面发生变化;因此,可以在这类变化的溶剂条件和/或蛋白质折叠条件下,对相同的亲和试剂或一组亲和试剂进行重复测量,以获得不同组的结合测量。在一些情况下,可以通过对样品进行重复测量来获得不同组的结合测量,在该样品中蛋白质已经受到酶处理(例如,用糖苷酶、磷酸化酶或磷酸酶处理)或未经酶处理。如本文所用的,术语“亲和试剂”通常是指以可再现的特异性结合蛋白质或肽的试剂。例如,亲和试剂可以是抗体、抗体片段、适体、微蛋白质结合物或肽。在一些实施方案中,微蛋白质结合物可以包括长度可以在30-210个氨基酸之间的蛋白质结合物。在一些实施方案中,可以设计微蛋白质结合物。例如,蛋白结合物可以包括肽大环(例如,如[hosseinzadeh等人,“comprehensivecomputationaldesignoforderedpeptidemacrocycles,”science,2017年12月15日;358(6369):1461-1466]中所述,其通过引用整体并入本文)。在一些实施方案中,单克隆抗体可能是优选的。在一些实施方案中,抗体片段如fab片段可能是优选的。在一些实施方案中,亲和试剂可以是可商购获得的亲和试剂,如可商购获得的抗体。在一些实施方案中,可通过筛选可商购获得的亲和试剂以鉴定具有有用特性的亲和试剂来选择所需的亲和试剂。亲和试剂可具有高、中或低特异性。在一些实例中,亲和试剂可识别若干不同的表位。在一些实例中,亲和试剂可识别存在于两种或更多种不同蛋白质中的表位。在一些实例中,亲和试剂可识别存在于多种不同蛋白质中的表位。在一些情况下,在本公开的方法中使用的亲和试剂可以对单个表位是高度特异性的。在一些情况下,在本公开的方法中使用的亲和试剂可以对含有翻译后修饰的单个表位是高度特异性的。在一些情况下,亲和试剂可具有高度相似的表位特异性。在一些情况下,可以专门设计具有高度相似的表位特异性的亲和试剂,以解析高度相似的蛋白质候选物序列(例如,具有单氨基酸变体或同种型的候选物)。在一些情况下,亲和试剂可具有高度多样的表位特异性,以使蛋白质序列的覆盖最大化。在一些实施方案中,由于探针与蛋白质-基底结合的随机性质,可以用相同的亲和探针重复进行实验,预期是结果可以不同,从而为蛋白质鉴定提供额外的信息。在一些情况下,被亲和试剂识别的一个或多个特定表位可能不是完全已知的。例如,可以针对与一种或多种完整蛋白质、蛋白质复合物或蛋白质片段的特异性结合来设计或选择亲和试剂,而无需知道特定结合表位。通过鉴定过程,可能已经详细了解了该试剂的结合谱。即使特定结合表位是未知的,使用所述亲和试剂的结合测量也可以用来确定蛋白质身份。例如,针对与蛋白质靶标结合而设计的可商购获得的抗体或适体可以用作亲和试剂。在测定条件(例如,完全折叠、部分变性或完全变性)下鉴定后,该亲和试剂与未知蛋白质的结合可提供关于未知蛋白质的身份的信息。在一些情况下,在知道或不知道它们所靶向的特定表位的情况下,可以使用蛋白质特异性亲和试剂(例如,可商购获得的抗体或适体)的集合来生成蛋白质鉴定。在一些情况下,蛋白质特异性亲和试剂的集合可包含约50、100、200、300、400、500、600、700、800、900、1000、2000、3000、4000、5000、10000、20000种或超过20000种亲和试剂。在一些情况下,亲和试剂的集合可包含证明在特定生物体中具有靶标反应性的所有可商购获得的亲和试剂。例如,可以连续地对蛋白质特异性亲和试剂的集合进行测定,并单独地对每种亲和试剂进行结合测量。在一些情况下,可以在结合测量之前混合蛋白质特异性亲和试剂的子集。例如,对于每个结合测量运行,可以选择亲和试剂的新混合物,该新混合物包含从完整组中随机选择的亲和试剂的子集。例如,每种后续混合物可以以相同的随机方式生成,期望许多亲和试剂将存在于多于一种混合物中。在一些情况下,可以使用蛋白质特异性亲和试剂的混合物更快速地生成蛋白质鉴定。在一些情况下,蛋白质特异性亲和试剂的此类混合物可增大亲和试剂在任何单独的运行中结合的未知蛋白质的百分比。亲和试剂的混合物可以包含所有可用亲和试剂的约1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%或更多。在单个实验中评估的亲和试剂的混合物可能会或可能不会共享单独的亲和试剂。在一些情况下,在集合内可能有多种与相同蛋白质结合的不同的亲和试剂。在一些情况下,该集合中的每种亲和试剂都可以与不同的蛋白质结合。在对相同蛋白质具有亲和力的多种亲和试剂与单个未知蛋白质结合的情况下,未知蛋白质的身份是所述亲和试剂的共同靶标的置信度可能增加。在一些情况下,在多种亲和试剂结合相同蛋白质上的不同表位的情况下,使用靶向相同蛋白质的多种蛋白质亲和试剂可能会提供冗余,并且仅靶向该蛋白质的亲和试剂子集的结合可能会受到翻译后修饰或结合表位的其他空间位阻的干扰。在一些情况下,结合表位未知的亲和试剂的结合可与结合表位已知的亲和试剂的结合测量一起使用,以生成蛋白质鉴定。在一些实例中,可以选择一种或多种亲和试剂来结合给定长度如2、3、4、5、6、7、8、9、10或多于10个氨基酸的氨基酸基序。在一些实例中,可以选择一种或多种亲和试剂来结合具有2个氨基酸至40个氨基酸的一系列不同长度的氨基酸基序。在一些情况下,亲和试剂可以用核酸条形码进行标记。在一些实例中,核酸条形码可用来纯化使用后的亲和试剂。在一些实例中,核酸条形码可用来分选亲和试剂以供重复使用。在一些情况下,亲和试剂可以用荧光团进行标记,该荧光团可用来分选使用后的亲和试剂。亲和试剂家族可包含一种或多种类型的亲和试剂。例如,本公开的方法可以使用亲和试剂家族,其包含抗体、抗体片段、fab片段、适体、肽和蛋白质中的一种或多种。可以修饰亲和试剂。修饰的实例包括但不限于检测部分的附接。检测部分可以直接或间接附接。例如,检测部分可以直接共价附接至亲和试剂,或者可以通过连接体附接,或者可以通过亲和反应附接,如互补核酸标签或生物素链霉亲和素对。能够经受亲和试剂的温和洗涤和洗脱的附接方法可能是优选的。亲和试剂可以用例如可辨识的标签进行标记,以允许结合事件的鉴定或定量(例如,采用结合事件的荧光检测)。可辨识的标签的一些非限制性实例包括:荧光团、磁性纳米颗粒或核酸条形码化的碱基连接体。所使用的荧光团可包括荧光蛋白,如gfp、yfp、rfp、egfp、mcherry、tdtomato、fitc、alexafluor350、alexafluor405、alexafluor488、alexafluor532、alexafluor546、alexafluor555、alexafluor568、alexafluor594、alexafluor647、alexafluor680、alexafluor750、pacificblue、香豆素、bodipyfl、pacificgreen、oregongreen、cy3、cy5、pacificorange、tritc、texasred、藻红蛋白和别藻蓝蛋白。或者,亲和试剂可以未标记,例如当直接检测结合事件时,例如采用结合事件的表面等离子体共振(spr)检测。检测部分的实例包括但不限于荧光团、生物发光蛋白质、包含恒定区和条形码区的核酸区段,或用于与纳米颗粒如磁性颗粒连接的化学系链(tether)。例如,亲和试剂可以用dna条形码标记,然后可以在其位置处进行明确测序。作为另一个示例,可以通过荧光共振能量转移(fret)检测方法,使用不同荧光团的组作为检测部分。检测部分可包括具有不同激发或发射模式的几种不同的荧光团。检测部分可以是可从亲和试剂上切下的。这可以允许从不再感兴趣的亲和试剂上去除检测部分以减少信号污染的步骤。在一些情况下,亲和试剂是未修饰的。例如,如果亲和试剂是抗体,则可以通过原子力显微术检测抗体的存在。亲和试剂可以是未修饰的,并且可以例如通过对一种或多种亲和试剂具有特异性的抗体来检测。例如,如果亲和试剂是小鼠抗体,则可以通过使用抗小鼠第二抗体来检测该小鼠抗体。或者,亲和试剂可以是适体,该适体由对该适体具有特异性的抗体来检测。可以用如上所述的检测部分修饰第二抗体。在一些情况下,可以通过原子力显微术检测第二抗体的存在。在一些实例中,亲和试剂可包含相同的修饰,例如缀合的绿色荧光蛋白,或者可包含两种或更多种不同类型的修饰。例如,每种亲和试剂可与各自具有不同激发或发射波长的几种不同荧光部分之一缀合。这可以允许亲和试剂的多路化,因为可以组合和/或区分几种不同的亲和试剂。在一个实例中,第一亲和试剂可与绿色荧光蛋白缀合,第二亲和试剂可与黄色荧光蛋白缀合,而第三亲和试剂可与红色荧光蛋白缀合,因此这三种亲和试剂可以是多路化的并通过它们的荧光进行鉴定。在另一个实例中,第一、第四和第七亲和试剂可与绿色荧光蛋白缀合,第二、第五和第八亲和试剂可与黄色荧光蛋白缀合,而第三、第六和第九亲和试剂可与红色荧光蛋白缀合;在这种情况下,第一、第二和第三亲和试剂可以一起多路化,而第二、第四和第七亲和试剂以及第三、第六和第九亲和试剂形成两个进一步的多路化反应。可以一起多路化的亲和试剂的数目可取决于用来区分它们的检测部分。例如,用荧光团标记的亲和试剂的多路化可能受到可用的独特荧光团数目的限制。对于进一步的实例,用核酸标签标记的亲和试剂的多路化可由核酸条形码的长度决定。核酸可以是脱氧核糖核酸(dna)或核糖核酸(rna)。可以在用于测定之前确定每种亲和试剂的特异性。可以在使用已知蛋白质的对照实验中确定亲和试剂的结合特异性。可以使用任何合适的实验方法来确定亲和试剂的特异性。在一个实例中,基底可以在已知位置处负载已知的蛋白质标准品并用于评估多种亲和试剂的特异性。在另一个实例中,基底可包含实验样品以及对照和标准品的小组,使得每种亲和试剂的特异性可从与对照和标准品的结合来计算,然后用于鉴定实验样品。在一些情况下,可以包括具有未知特异性的亲和试剂以及已知特异性的亲和试剂,来自已知特异性的亲和试剂的数据可用来鉴定蛋白质,而未知特异性的亲和试剂与所鉴定的蛋白质的结合模式可用来确定其结合特异性。还可以通过使用其他亲和试剂的已知结合数据来重新确认任何单独的亲和试剂的特异性,以评估该单独的亲和试剂结合哪些蛋白质。在一些情况下,亲和试剂与缀合至基底上的每种已知蛋白质结合的频率可以用来得出与基底上的任何蛋白质结合的概率。在一些情况下,与包含表位(例如氨基酸序列或翻译后修饰)的已知蛋白质结合的频率可以用来确定亲和试剂与特定表位结合的概率。因此,通过亲和试剂组的多次使用,亲和试剂的特异性可以随着每次迭代而逐渐改善。虽然可以使用对特定蛋白质具有独特特异性的亲和试剂,但是本文所述的方法可能不需要它们。另外,方法可能对一系列特异性有效。在一些实例中,当亲和试剂对任何特定蛋白质都不具有特异性,但对氨基酸基序(例如三肽aaa)具有特异性时,本文所述的方法可能特别有效。在一些实例中,可以选择具有高、中或低结合亲和力的亲和试剂。在一些情况下,具有低或中结合亲和力的亲和试剂可能是优选的。在一些情况下,亲和试剂可具有约10-3m、10-4m、10-5m、10-6m、10-7m、10-8m、10-9m、10-10m或低于约10-10m的解离常数。在一些情况下,亲和试剂可具有大于约10-10m、10-9m、10-8m、10-7m、10-6m、10-5m、10-4m、10-3m、10-2m或高于10-2m的解离常数。在一些情况下,具有低或中koff速率或者中或高kon速率的亲和试剂可能是优选的。可以选择一些亲和试剂来结合修饰的氨基酸序列,如磷酸化的或遍在蛋白化的氨基酸序列。在一些实例中,可以选择对可由一种或多种蛋白质包含的表位家族具有广泛特异性的一种或多种亲和试剂。在一些实例中,一种或多种亲和试剂可以结合两种或更多种不同的蛋白质。在一些实例中,一种或多种亲和试剂可以与其一种或多种靶标弱结合。例如,亲和试剂可以与其一种或多种靶标以低于10%、低于10%、低于15%、低于20%、低于25%、低于30%或低于35%结合。在一些实例中,一种或多种亲和试剂可以与其一种或多种靶标中等或强烈地结合。例如,亲和试剂可以与其一种或多种靶标以超过35%、超过40%、超过45%、超过60%、超过65%、超过70%、超过75%、超过80%、超过85%、超过90%、超过91%、超过92%、超过93%、超过94%、超过95%、超过96%、超过97%、超过98%或超过99%结合。为了补偿弱结合,可将过量的亲和试剂施加至基底。亲和试剂可以相对于样品蛋白质以约1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1或10:1过量施加。亲和试剂可以相对于样品蛋白质中表位的预期出现率以约1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1或10:1过量施加。为了补偿高亲和试剂解离速率,可以将连接体部分附接至每种亲和试剂,并用来将结合的亲和试剂可逆地连接至其结合的基底或未知蛋白质。例如,dna标签可以附接至每种亲和试剂的末端,而不同的dna标签附接至基底或每种未知蛋白质上。在亲和试剂与未知蛋白质杂交后,可以在芯片上洗涤在一端与亲和试剂相关dna标签互补而在另一端与基底相关标签互补的连接体dna,以使亲和试剂与基底结合,并防止亲和试剂在测量前解离。结合后,可以通过在破坏dna连接键的热或高盐浓度的存在下洗涤来释放连接的亲和试剂。图21示出了根据一些实施方案,两个杂交步骤对亲和试剂与蛋白质之间的结合的增强。特别是,图21的步骤1示出了亲和试剂杂交。如步骤1中所见,亲和试剂2110与蛋白质2130杂交。蛋白质2130与载玻片2105结合。如步骤1中所见,亲和试剂2110附接有dna标签2120。在一些实施方案中,亲和试剂可以附接有超过一个dna标签。在一些实施方案中,亲和试剂可以附接有1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20个或超过20个dna标签。dna标签2120包括具有识别序列2125的单链dna(ssdna)标签。另外,蛋白质2130包含两个dna标签2140。在一些实施方案中,可以使用与蛋白质中的半胱氨酸反应的化学法添加dna标签。在一些实施方案中,蛋白质可以附接有超过一个dna标签。在一些实施方案中,蛋白质可以附接有1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95、100个或超过100个dna标签。每个dna标签2140包括具有识别序列2145的ssdna标签。如步骤2中所见,dna连接体2150与分别附接至亲和试剂2110和蛋白质2130的dna标签2120和2140杂交。dna连接体2150包含具有分别与识别序列2125和2145互补的序列的ssdna。此外,识别序列2125和2145位于dna连接体2150上,以允许dna连接体2150同时与dna标签2120和2140两者结合,如步骤2中所示。特别是,dna连接体2150的第一区域2152选择性地与识别序列2125杂交,而dna连接体2150的第二区域2154选择性地与识别序列2145杂交。在一些实施方案中,第一区域2152和第二区域2154可以在dna连接体上彼此间隔开。特别是,在一些实施方案中,dna连接体的第一区域和dna连接体的第二区域可以在第一区域与第二区域之间用非杂交间隔序列间隔开。此外,在一些实施方案中,识别序列的序列可以小于与dna连接体完全互补,并且仍可以与dna连接体序列结合。在一些实施方案中,识别序列的长度可以小于5个核苷酸、5个核苷酸、6个核苷酸、7个核苷酸、8个核苷酸、9个核苷酸、10个核苷酸、11个核苷酸、12个核苷酸、13个核苷酸、14个核苷酸、15个核苷酸、16个核苷酸、17个核苷酸、18个核苷酸、19个核苷酸、20个核苷酸、21个核苷酸、22个核苷酸、23个核苷酸、24个核苷酸、25个核苷酸、26个核苷酸、27个核苷酸、28个核苷酸、29个核苷酸或30个核苷酸或超过30个核苷酸。在一些实施方案中,识别序列可以与互补dna标签序列具有一个或多个错配。在一些实施方案中,识别序列的大约十分之一的核苷酸可以与互补dna标签序列错配,并且仍然可以与互补dna标签序列杂交。在一些实施方案中,识别序列的不到十分之一的核苷酸可以与互补dna标签序列错配,并且仍然可以与互补dna标签序列杂交。在一些实施方案中,识别序列的大约十分之二的核苷酸可以与互补dna标签序列错配,并且仍然可以与互补dna标签序列杂交。在一些实施方案中,识别序列的超过十分之二的核苷酸可以与互补dna标签序列错配,并且仍然可以与互补dna标签序列杂交。亲和试剂还可包含磁性组分。该磁性组分可用于将一些或所有结合的亲和试剂操纵到同一成像平面或z堆叠(stack)中。将一些或所有亲和试剂操纵到同一成像平面中可以改善成像数据的质量并降低系统中的噪声。如本文所用的,术语“检测器”通常是指能够检测信号的装置,该信号包括指示亲和试剂与蛋白质的结合事件存在与否的信号。该信号可以是指示结合事件存在与否的直接信号,如表面等离子体共振(spr)信号。该信号可以是指示结合事件存在与否的间接信号,如荧光信号。在一些情况下,检测器可以包括可以检测信号的光学和/或电子组件。术语“检测器”可以在检测方法中使用。检测方法的非限制性实例包括光学检测、光谱检测、静电检测、电化学检测、磁性检测、荧光检测、表面等离子体共振(spr)等。光学检测方法的实例包括但不限于荧光测定法和紫外线-可见光吸收。光谱检测方法的实例包括但不限于质谱法、核磁共振(nmr)光谱法和红外光谱法。静电检测方法的实例包括但不限于基于凝胶的技术,如凝胶电泳。电化学检测方法的实例包括但不限于在高效液相色谱法分离扩增产物后对扩增产物的电化学检测。样品中的蛋白质鉴定蛋白质是活生物体的细胞和组织的重要结构单元。给定的生物体产生一大组不同的蛋白质,通常被称为蛋白质组。蛋白质组可以随时间而变化,并且随细胞或生物体经历的各个阶段(例如,细胞周期阶段或疾病状态)而变化。对蛋白质组的大规模研究或测量(例如,实验分析)可被称为蛋白质组学。在蛋白质组学中,存在多种鉴定蛋白质的方法,包括免疫测定(例如酶联免疫吸附测定(elisa)和western印迹法)、基于质谱学的方法(例如,基质辅助激光解吸/电离(maldi)和电喷雾电离(esi))、混合方法(例如,质谱免疫测定(msia))和蛋白质微阵列。例如,单分子蛋白质组学方法可以尝试通过多种方法来推断样品中蛋白质分子的身份,这些方法的范围从氨基酸的直接官能化到使用亲和试剂。从此类方法收集的信息或测量值通常通过合适的算法进行分析,以鉴定样品中存在的蛋白质。由于缺乏灵敏度、缺乏特异性和检测器噪声,蛋白质的准确定量也可能会遇到挑战。特别是,由于检测器信号水平的随机和不可预测的系统变异,样品中蛋白质的准确定量可能会遇到挑战,这可能会导致蛋白质鉴定和定量错误。在一些情况下,可以通过监测仪器诊断学和共模行为来校准和去除仪器和检测系统学。然而,蛋白质(例如,通过亲和试剂探针)的结合本质上是一个概率过程,其结合灵敏度和特异性可能均不理想。本公开提供了用于精确且有效鉴定蛋白质的方法和系统。本文提供的方法和系统可以显著减少或消除鉴定样品中的蛋白质的错误。这类方法和系统可以实现未知蛋白质样品内候选蛋白质的准确和有效鉴定。该蛋白质鉴定可以基于使用样品中未知蛋白质的经验测量的信息的计算。例如,经验测量可以包括被配置为选择性地与一种或多种候选蛋白质结合的亲和试剂探针的结合信息、蛋白质长度、蛋白质疏水性和/或等电点。可以将蛋白质鉴定优化为可在最小内存占用下计算。该蛋白质鉴定可以包括估计样品中存在一种或多种候选蛋白质中的每一种的置信水平。在一个方面,本文公开了一种鉴定未知蛋白质样品内蛋白质的计算机实现的方法100(例如,如图1所示)。该方法可以独立地应用于样品中的每种未知蛋白质,以生成样品中鉴定的蛋白质的集合。蛋白质的量可以通过对每种候选蛋白质的鉴定数进行计数来计算。鉴定蛋白质的方法可以包括通过计算机接收样品中未知蛋白质的多个经验测量的信息(例如,步骤105)。经验测量可以包括(i)一个或多个亲和试剂探针中的每一个与样品中的一种或多种未知蛋白质的结合测量,(ii)一种或多种未知蛋白质的长度;(iii)一种或多种未知蛋白质的疏水性;和/或(iv)一种或多种未知蛋白质的等电点。在一些实施方案中,多个亲和试剂探针可包含多个单独亲和试剂探针的池。例如,亲和试剂探针池可包含2、3、4、5、6、7、8、9、10种或超过10种类型的亲和试剂探针。在一些实施方案中,亲和试剂探针池可包含2种类型的亲和试剂探针,它们组合构成该亲和试剂探针池中亲和试剂探针的大部分组成。在一些实施方案中,亲和试剂探针池可包含3种类型的亲和试剂探针,它们组合构成该亲和试剂探针池中亲和试剂探针的大部分组成。在一些实施方案中,亲和试剂探针池可包含4种类型的亲和试剂探针,它们组合构成该亲和试剂探针池中亲和试剂探针的大部分组成。在一些实施方案中,亲和试剂探针池可包含5种类型的亲和试剂探针,它们组合构成该亲和试剂探针池中亲和试剂探针的大部分组成。在一些实施方案中,亲和试剂探针池可包含超过5种类型的亲和试剂探针,它们组合构成该亲和试剂探针池中亲和试剂探针的大部分组成。每个亲和试剂探针可以被配置为选择性地与多种候选蛋白质中的一种或多种候选蛋白质结合。所述亲和试剂探针可以是k-聚体亲和试剂探针。在一些实施方案中,每个k-聚体亲和试剂探针被配置为选择性地与多种候选蛋白质中的一种或多种候选蛋白质结合。经验测量的信息可以包含被认为已经与未知蛋白质结合的一组探针的结合测量。接下来,可以通过计算机将未知蛋白质的经验测量信息的至少一部分与包含多个蛋白质序列的数据库进行比较(例如,步骤110)。每个蛋白质序列可以对应于多种候选蛋白质中的候选蛋白质。所述多种候选蛋白质可包含至少10种、至少20种、至少30种、至少40种、至少50种、至少60种、至少70种、至少80种、至少90种、至少100种、至少150种、至少200种、至少250种、至少300种、至少350种、至少400种、至少450种、至少500种、至少600种、至少700种、至少800种、至少900种、至少1000种或超过1000种不同的候选蛋白质。接下来,对于多种候选蛋白质中的一种或多种候选蛋白质中的每一种,可以通过计算机来计算或生成对候选蛋白质的经验测量将会生成观察到的测量结果的概率(例如,在步骤115中)。如本文所用的,术语“测量结果”是指在进行测量时观察到的信息。例如,亲和试剂结合实验的测量结果可以是阳性或阴性结果,如试剂的结合或不结合。作为另一个示例,测量蛋白质长度的实验的测量结果可以是417个氨基酸。另外或备选地,对于多种候选蛋白质中的一种或多种候选蛋白质中的每一种,可以通过计算机来计算或生成对候选蛋白质的经验测量将不会生成观察到的测量结果的概率。另外或备选地,可以通过计算机来计算或生成对候选蛋白质的经验测量将会生成未观察到的测量结果的概率。另外或备选地,可以通过计算机来计算或生成对候选蛋白质的一系列经验测量将会生成结果集的概率。如本文所用的,“结果集”是指蛋白质的多个独立的测量结果。例如,可以对未知蛋白质进行一系列经验亲和试剂结合测量。每种单独的亲和试剂的结合测量包括测量结果,并且所有测量结果的集合是结果集。在一些情况下,结果集可以是所有观察到的结果的子集。在一些情况下,结果集可以由未凭经验观察到的测量结果组成。另外或备选地,对于多种候选蛋白质中的一种或多种候选蛋白质中的每一种,可以通过计算机来计算或生成未知蛋白质是候选蛋白质的概率。步骤115和/或120的计算或生成可以迭代地或非迭代地进行。可以基于未知蛋白质的经验测量结果与包含针对所有候选蛋白质的多个蛋白质序列的数据库的比较来生成步骤115中的概率。因此,算法的输入可包括候选蛋白质序列的数据库和未知蛋白质的一组经验测量值(例如,被认为已经与未知蛋白质结合的探针、未知蛋白质的长度、未知蛋白质的疏水性和/或未知蛋白质的等电点)。在一些情况下,算法的输入可包括与估计任何亲和试剂针对任何候选蛋白质生成任何结合测量值的概率(例如,每种亲和试剂的三聚体水平结合概率)有关的参数。算法的输出可包括(i)在给出假设的候选蛋白质身份的情况下,观察到测量结果或结果集的概率,(ii)针对未知蛋白质,从这组候选蛋白质中选出的最可能的身份,以及在给出测量结果或结果集的情况下该鉴定为正确的概率(例如,在步骤120中),和/或(iii)一组高概率候选蛋白质身份以及未知蛋白质是该组蛋白质之一的相关概率。在假定候选蛋白质是被测量的蛋白质的情况下观察到测量结果的概率可以被表示为:p(测量结果|蛋白质)。在一些实施方案中,p(测量结果|蛋白质)是完全通过计算机计算的。在一些实施方案中,p(测量结果|蛋白质)是基于蛋白质的氨基酸序列的特征来计算的,或来源于蛋白质的氨基酸序列的特征。在一些实施方案中,p(测量结果|蛋白质)不依赖于对蛋白质的氨基酸序列的了解来计算。例如,p(测量结果|蛋白质)可以通过以下方式凭经验确定:在针对候选蛋白质分离物的重复实验中获取测量值,并根据以下频率计算p(测量结果|蛋白质):(具有结果的测量数/测量的总数)。在一些实施方案中,p(测量结果|蛋白质)来源于过去对蛋白质的测量的数据库。在一些实施方案中,p(测量结果|蛋白质)是通过以下方式计算的:从测量结果被删截的未知蛋白质的集合生成一组确信蛋白质鉴定,然后计算确信地被鉴定为候选蛋白质的一组未知蛋白质之间测量结果的频率。在一些实施方案中,可以使用p(测量结果|蛋白质)的种子值来鉴定未知蛋白质的集合,并且基于确信地与候选蛋白质匹配的未知蛋白质之间的测量结果的频率来精炼该种子值。在一些实施方案中,重复该过程,其中基于更新的测量结果概率生成新的鉴定,然后从更新的确信鉴定集合生成新的测量结果概率。假定候选蛋白质是被测量的蛋白质,则未观察到测量结果的概率可以被表示为:p(未测量结果|蛋白质)=1–p(测量结果|蛋白质)。假定候选蛋白质是被测量的蛋白质,则观察到由n个单独测量结果组成的测量结果集的概率可以被表示为每个单独测量结果的概率的乘积:p(结果集|蛋白质)=p(测量结果1|蛋白质)*p(测量结果2|蛋白质)*…*p(测量结果m|蛋白质)未知蛋白质为候选蛋白质(蛋白质i)的概率可以基于每种可能的候选蛋白质的结果集的概率来计算。在一些实施方案中,测量结果集包含亲和试剂探针的结合。在一些实施方案中,测量结果集包含亲和试剂探针的非特异性结合。在一些实施方案中,样品中的蛋白质是截短的或降解的。在一些实施方案中,样品中的蛋白质不含原始蛋白质的c末端。在一些实施方案中,样品中的蛋白质不含原始蛋白质的n末端。在一些实施方案中,样品中的蛋白质不含原始蛋白质的n末端且不含原始蛋白质的c末端。在一些实施方案中,所述经验测量包括对抗体混合物进行的测量。在一些实施方案中,所述经验测量包括对来自多个物种的含有蛋白质的样品进行的测量。在一些实施方案中,所述经验测量包括对来源于人类的样品进行的测量。在一些实施方案中,所述经验测量包括对来源于不同于人类的物种的样品进行的测量。在一些实施方案中,所述经验测量包括在由非同义单核苷酸多态性(snp)引起的单氨基酸变异(sav)的存在下对样品进行的测量。在一些实施方案中,所述经验测量包括在基因组结构变异如影响样品中蛋白质的序列的插入、缺失、易位、倒位、区段重复或拷贝数变异(cnv)的存在下对样品的测量。在一些实施方案中,所述方法进一步包括将该方法应用于样品中测量的所有未知蛋白质。在一些实施方案中,所述方法进一步包括针对所述一种或多种候选蛋白质中的每一种,生成候选蛋白质与样品中所测量的未知蛋白质相匹配的置信水平。该置信水平可以包括概率值。或者,该置信水平可以包括具有误差的概率值。或者,该置信水平可以包括概率值的范围,任选地具有置信度(,例如,约90%、约95%、约96%、约97%、约98%、约99%、约99.9%、约99.99%、约99.999%、约99.9999%、约99.99999%、约99.999999%、约99.9999999%、约99.99999999%、约99.999999999%、约99.9999999999%、约99.99999999999%、约99.999999999999%、约99.9999999999999%置信度或高于99.9999999999999%置信度)。在一些实施方案中,所述方法进一步包括生成样品中存在候选蛋白质的概率。在一些实施方案中,所述方法进一步包括独立于样品中的每种未知蛋白质生成蛋白质鉴定和相关概率,并生成在样品中鉴定出的所有独特蛋白质的列表。在一些实施方案中,该方法进一步包括对为每种独特候选蛋白质生成的鉴定数进行计数,以确定样品中每种候选蛋白质的量。在一些实施方案中,可以过滤蛋白质鉴定和相关概率的集合,以仅包含高评分、高置信度和/或低错误发现率的鉴定。在一些实施方案中,可以生成亲和试剂与全长候选蛋白质的结合概率。在一些实施方案中,可以生成亲和试剂与蛋白质片段(例如,完整蛋白质序列的子序列)的结合概率。例如,如果以某种方式处理未知蛋白质并将其与基底缀合,使得每种未知蛋白质仅前100个氨基酸被缀合,则可以为每种蛋白质候选物生成结合概率,使得将前100个氨基酸以外的表位结合的所有结合概率都被设置为零,或者设置为表示错误率的极低概率。如果每种蛋白质的前10、20、50、100、150、200、300、400个或超过400个氨基酸与基底缀合,则可以使用类似的方法。如果最后10、20、50、100、150、200、300、400个或超过400个氨基酸与基底缀合,则可以使用类似的方法。在一些实施方案中,在不能将单个蛋白质候选物匹配分配给未知蛋白质的情况下,可以将一组潜在的蛋白质候选物匹配分配给未知蛋白质。可以将置信水平分配给未知蛋白质,该未知蛋白质是该组中任何蛋白质候选物之一。该置信水平可以包括概率值。或者,该置信水平可以包括具有误差的概率值。或者,该置信水平可以包括概率值的范围,任选地具有置信度(,例如,约90%、约95%、约96%、约97%、约98%、约99%、约99.9%、约99.99%、约99.999%、约99.9999%、约99.99999%、约99.999999%、约99.9999999%、约99.99999999%、约99.999999999%、约99.9999999999%、约99.99999999999%、约99.999999999999%、约99.9999999999999%置信度或高于99.9999999999999%置信度)。例如,未知蛋白质可能与两种蛋白质候选物强烈匹配。这两种蛋白质候选物可能彼此具有高度的序列相似性(例如,两种蛋白质同种型,如与规范序列相比具有单个氨基酸变体的蛋白质)。在这些情况下,可能没有单独的蛋白质候选物被分配有高置信度,但是高置信度可归因于与包含这两种强烈匹配蛋白质候选物的“蛋白质的组”的单个但未知的成员匹配的未知蛋白质。在一些实施方案中,可以努力检测其中未知蛋白质未被光学拆分的情况。例如,在极少数情况下,两个或更多个蛋白质可能会结合在基底的同一“孔”或位置中,尽管尽力避免这种情况发生。在一些情况下,可以用非特异性染料处理缀合的蛋白质,并测量来自该染料的信号。如果两个或更多个蛋白质没有被光学拆分,则由染料产生的信号可高于包含单个蛋白质的位置,并且可用来标示出具有多个结合蛋白质的位置。在一些实施方案中,通过对从中获得或衍生出未知蛋白质样品的人类或生物体的dna或rna进行测序或分析来生成或修饰多种候选蛋白质。在一些实施方案中,所述方法进一步包括获得关于未知蛋白质的翻译后修饰的信息。关于翻译后修饰的信息可包括翻译后修饰的存在,而无需了解具体修饰的性质。该数据库可以被认为是ptm的指数产物。例如,一旦已经将蛋白质候选物序列分配给未知蛋白质,就可以将针对所测定的蛋白质的亲和试剂结合的模式与包含来自先前实验的亲和试剂与相同候选物的结合测量值的数据库进行比较。例如,结合测量值的数据库可衍生自与在已知位置含有已知序列的未修饰蛋白质的核酸可编程蛋白质阵列(nucleicacid-programmableproteinarray,nappa)的结合。另外或备选地,可以从先前的实验获得结合测量值的数据库,在所述实验中,蛋白质候选物序列被确信地分配给未知蛋白质。被测蛋白质与现有测量值数据库之间的结合测量值差异可提供关于翻译后修饰可能性的信息。例如,如果亲和剂与数据库中的候选蛋白质具有高结合频率,但不与测定的蛋白质结合,则该蛋白质上某处存在翻译后修饰的可能性较高。如果存在结合差异的亲和试剂的结合表位已知,则翻译后修饰的位置可以定位在亲和试剂的结合表位处或附近。在一些实施方案中,关于特定翻译后修饰的信息可以通过在用特异性去除特定翻译后修饰的酶处理蛋白质-基底缀合物之前和之后进行重复亲和试剂测量而得出。例如,可以在用磷酸酶处理基底之前获取一系列亲和试剂的结合测量值,然后在用磷酸酶处理后重复测量。在磷酸酶处理之前结合未知蛋白质但在磷酸酶处理之后不结合(差异结合)的亲和试剂可提供磷酸化的证据。如果被差异结合亲和试剂识别的表位已知,则磷酸化可以位于该亲和试剂的结合表位处或附近。在一些情况下,可以使用针对特定翻译后修饰的亲和试剂的结合测量值来确定特定翻译后修饰的计数。例如,可以使用识别磷酸化事件的抗体作为亲和试剂。该试剂的结合可以指示未知蛋白质上存在至少一个磷酸化。在一些情况下,可通过对针对特定翻译后修饰特异性亲和试剂所测量的结合事件的数目进行计数来确定未知蛋白质上特定类型的离散翻译后修饰的数目。例如,磷酸化特异性抗体可以与荧光报道分子缀合。在这种情况下,荧光信号的强度可用来确定与未知蛋白质结合的磷酸化特异性亲和试剂的数量。与未知蛋白质结合的磷酸化特异性亲和试剂的数量继而可用来确定未知蛋白质上的磷酸化位点的数目。在一些实施方案中,可以将来自亲和试剂结合实验的证据与可能被翻译后修饰的氨基酸序列基序或特定蛋白质位置的已有知识(例如,来自dbptm、phosphositeplus或uniprot)相组合,以得出翻译后修饰的更准确的计数、鉴定或定位。例如,如果不能仅从亲和力测量值准确地确定翻译后修饰的位置,则可能支持包含经常与目的翻译后修饰相关的氨基酸序列基序的位置。在一些实施方案中,迭代地生成概率,直到满足预定条件。在一些实施方案中,预定条件包括以至少50%、至少55%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.9%、至少99.99%、至少99.999%、至少99.9999%、至少99.99999%、至少99.999999%、至少99.9999999%、至少99.99999999%、至少99.999999999%、至少99.9999999999%、至少99.99999999999%、至少99.999999999999%、至少99.9999999999999%的置信度或高于99.9999999999999%的置信度生成多个概率中的每一个。在一些实施方案中,所述方法进一步包括生成鉴定样品中的一种或多种未知蛋白质的纸质或电子报告。该纸质或电子报告可以进一步针对每种候选蛋白质指示样品中存在候选蛋白质的置信水平。该置信水平可以包括概率值。或者,该置信水平可以包括具有误差的概率值。或者,该置信水平可以包括概率值的范围,任选地具有置信度(,例如,约90%、约95%、约96%、约97%、约98%、约99%、约99.9%、约99.99%、约99.999%、约99.9999%、约99.99999%、约99.999999%、约99.9999999%、约99.99999999%、约99.999999999%、约99.9999999999%、约99.99999999999%、约99.999999999999%、约99.9999999999999%置信度或高于99.9999999999999%置信度)。该纸质或电子报告可以进一步指示在预期的错误发现率阈值以下(例如,低于10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.4%、0.3%、0.2%或0.1%的错误发现率)鉴定的蛋白质候选物列表。错误发现率可以如下估计:首先以置信度的降序对蛋白质鉴定进行排序。然后,可以将排序列表中任意点的估计错误发现率计算为1-avg_c_prob,其中avg_c_prob是该列表中当前点之处或之前(例如,比其更高的置信度)的所有蛋白质的平均候选物概率。然后,可以通过返回排序列表中错误发现率高于阈值的最早点之前的所有蛋白质鉴定,来生成低于所需错误发现率阈值的蛋白质鉴定的列表。或者,可以通过返回排序列表中错误发现率低于或等于所需阈值的最后点之前(含)的所有蛋白质,来生成低于所需错误发现率阈值的蛋白质鉴定的列表。在一些实施方案中,样品包括生物样品。该生物样品可以从受试者获得。在一些实施方案中,所述方法进一步包括至少基于所述多个概率来确定受试者中的疾病状态或病症。在一些实施方案中,所述方法进一步包括通过计数对每种蛋白质候选物进行的鉴定的次数来量化蛋白质。例如,样品中存在的蛋白质的绝对量(例如,蛋白质分子数)可以通过对从该蛋白质候选物生成的确信鉴定的数目进行计数来计算。在一些实施方案中,该量可以计算为所测定的未知蛋白质总数的百分比。在一些实施方案中,可以对原始鉴定计数进行校准,以从仪器和检测系统去除系统误差。在一些实施方案中,可以对该量进行校准,以消除由蛋白质候选物的可检测性变化引起的量偏差。蛋白质的可检测性可以通过经验测量或计算机模拟来评估。所述疾病或病症可以是传染病、免疫病症或疾病、癌症、遗传病、退行性疾病、生活方式疾病、损伤、罕见疾病或年龄相关性疾病。该传染病可由细菌、病毒、真菌和/或寄生虫引起。癌症的非限制性实例包括膀胱癌、肺癌、脑癌、黑素瘤、乳腺癌、非霍奇金淋巴瘤、宫颈癌、卵巢癌、结直肠癌、胰腺癌、食管癌、前列腺癌、肾癌、皮肤癌、白血病、甲状腺癌、肝癌和子宫癌。遗传疾病或病症的一些实例包括但不限于多发性硬化(ms)、囊性纤维化、charcot–marie–tooth病、亨廷顿病(huntington'sdisease)、peutz-jeghers综合征、唐氏综合症、类风湿性关节炎和tay–sachs病。生活方式疾病的非限制性实例包括肥胖症、糖尿病、动脉硬化、心脏病、中风、高血压、肝硬化、肾炎、癌症、慢性阻塞性肺病(copd)、听力问题和慢性背痛。损伤的一些实例包括但不限于擦伤、脑损伤、瘀伤、烧伤、脑震荡、充血性心力衰竭、建筑损伤、脱位、连枷胸、骨折、血胸、椎间盘突出、髋骨隆凸挫伤、低体温、撕裂、神经挟捏、气胸、肋骨骨折、坐骨神经痛、脊髓损伤、肌腱韧带筋膜损伤、创伤性脑损伤和鞭伤。在一些实施方案中,所述方法包括鉴定和量化小分子(例如,代谢物)或聚糖,而不是蛋白质,或除蛋白质以外还鉴定和量化小分子(例如,代谢物)或聚糖。例如,可以使用亲和试剂,例如以不同倾向结合糖或糖组合的凝集素或抗体来鉴定聚糖。亲和试剂结合各种糖或糖组合的倾向可以通过分析与可商购获得的聚糖阵列的结合来表征。例如,未知的聚糖可以使用羟基反应性化学与官能化的基底缀合,并且可以使用聚糖结合亲和试剂获得结合测量值。亲和试剂与基底上未知聚糖的结合测量值可直接用来量化具有特定糖或糖组合的聚糖的数目。或者,可以使用本文所述的方法将一个或多个结合测量值与从候选聚糖结构数据库预测的结合测量值进行比较,以鉴定每种未知聚糖的结构。在一些实施方案中,将蛋白质与基底结合,并用聚糖亲和试剂进行结合测量,以鉴定附接至蛋白质的聚糖。此外,可以在单个实验中使用聚糖和蛋白质亲和试剂两者进行结合测量,以生成蛋白质骨架序列和缀合的聚糖鉴定。作为另一个实例,可以使用针对代谢物中常见的偶联基团如巯基、羰基、胺或活性氢的化学,将代谢物缀合至官能化的基底。可以使用对特定官能团、结构基序或代谢物具有不同倾向的亲和试剂进行结合测量。可以将所得结合测量值与候选小分子数据库的预测结合测量值进行比较,并且可以用本文所述的方法鉴定基底上每个位置处的代谢物。实施例1:通过亲和试剂结合的蛋白质鉴定本文描述的方法可以与亲和结合试剂(例如,适体或抗体)结合测量组合使用,以分析和/或鉴定样品中的蛋白质。在这种情况下,待计算的测量结果概率是亲和结合试剂(例如,亲和试剂或亲和探针)与候选蛋白质的结合或不结合事件的概率。可以将结合概率建模为以表位的存在为条件,该表位被蛋白质序列中存在的亲和结合试剂所识别。例如,表位可以是“三聚体”(三个氨基酸的序列)。可将亲和试剂设计为靶向特定表位(例如,gav)。亲和试剂的脱靶结合(例如,亲和试剂与不同于其靶表位的表位的结合)可以通过包括与其他表位结合的非零概率来建模。例如,亲和试剂可以被设计为结合gav三聚体,但是可以具有与三个另外的识别位点的脱靶结合:cld、tyl和iad。对于该亲和试剂,结合概率可以被建模为:p(亲和探针结合|蛋白质)={如果gav、cld、tyl或iad存在于蛋白质序列中则为0.25;否则为0}。也可以有小概率的亲和试剂与蛋白质的非特异性结合,其可以被表示为:p(亲和探针结合|蛋白质)={如果gav、cld、tyl或iad存在于蛋白质序列中则为0.25;否则为0.00001}。在此,概率测量抗体结合的检测结果。作为一个示例,考虑分析来自人源样品的蛋白质的情况。假定样品中的蛋白质在人类“参考”蛋白质组中被代表(例如,在uniprot规范蛋白质序列和功能信息数据库中发现)。也就是说,蛋白质候选物列表是uniprot数据库中约21000种蛋白质和相关序列的集合。从样品中衍生出未知蛋白质的集合,并且在一系列亲和试剂结合实验中探测每种未知蛋白质,测量并记录结果(结合或不结合)。例如,这类实验可以包括顺序添加不同的亲和试剂并观察亲和试剂与未知蛋白质的结合。亲和试剂或“探针”被选择为靶向蛋白质候选物列表中最常观察到的三聚体(在约800种可能的三聚体中)。在靶向的三聚体之外,每个探针都具有与随机选择的许多其他三聚体的脱靶结合。探针与蛋白质序列结合的概率可以被表示为:p(亲和探针结合|蛋白质)=1–[p(无非特异性结合)*p(无特异性结合)]。假定:n=蛋白质候选物的序列长度;q=识别位点的长度(例如3);s=非特异性三聚体结合概率(例如10-5);p=特异性结合概率(例如0.25);则p(无非特异性结合)和p(无特异性结合)项可以被表示为:p(无非特异性结合)=(1–s)n–q+1=(1–10-5)n–3+1和p(无特异性结合)=∏对于每个识别位点(1-p)位点在蛋白质中出现的次数。最后,探针不与蛋白质结合的概率可以被表示为:p(亲和探针不结合|蛋白质)=1–p(亲和探针结合|蛋白质)。图2示出了对于三种不同的实验情况(使用50、100和200个探针,分别用灰色、黑色和白色圆圈表示),相对于亲和试剂探针中的探针识别位点(例如,三聚体结合表位)数目(范围最多100个探针识别位点或三聚体结合表位)绘制的亲和试剂探针的灵敏度(例如,在小于1%的错误检测率(fdr)下鉴定的底物的百分比)。如图2所示,所用探针的数目对正确鉴定蛋白质的能力具有重要影响。在y轴上绘出的是灵敏度,灵敏度是正确鉴定的未知蛋白质的百分比,其阈值(例如上限)为小于1%鉴定不正确。例如,如果每个探针包含5个识别位点或三聚体结合表位(1个靶向位点和4个脱靶位点),则当使用50个探针时,蛋白质鉴定的灵敏度小于10%,当使用100个探针时,该灵敏度约为60%,而当使用200个探针时,该灵敏度约为90%。实际上,当使用300个探针时,灵敏度超过95%(结果未在图中显示)。这种蛋白质鉴定方法支持具有许多脱靶结合位点的探针。即使具有60个识别位点或三聚体结合表位(1个靶向位点和59个脱靶位点),在100个探针的实验中,鉴定灵敏度约为55%,而在200个探针的实验中,鉴定灵敏度约为90%。然而,如图3所示,当探针具有超过100个结合位点或三聚体结合表位时,鉴定蛋白质的能力迅速下降。图3示出了对于三种不同的实验情况(使用50、100和200个探针,分别用灰色、黑色和白色圆圈表示),相对于亲和试剂探针中的探针识别位点(例如,三聚体结合表位)数目(范围最多700个探针识别位点或三聚体结合表位)绘制的亲和试剂探针的灵敏度(例如,在小于1%的错误检测率(fdr)下鉴定的底物的百分比)。例如,如果每个探针包含100个识别位点或三聚体结合表位(1个靶向位点,99个脱靶位点),则当使用50个探针时,蛋白质鉴定的灵敏度约为1%,当使用100个探针时,该灵敏度约为30%,而当使用200个探针时,该灵敏度约为70%。然而,如果每个探针包含200个识别位点或三聚体结合表位(1个靶向位点,199个脱靶位点),则当使用50个探针时,蛋白质鉴定的灵敏度小于1%,当使用100个探针时,该灵敏度小于20%,而当使用200个探针时,该灵敏度小于40%。实施例2:蛋白质亲和试剂与已被截短或降解的蛋白质的结合本文描述的方法可以应用于分析和/或鉴定样品中已被截短的蛋白质。在这类实验中,亲和探针与蛋白质结合的概率计算被修改为仅考虑与截短的蛋白质序列的结合,而不是与完整蛋白质序列的结合。例如,图4示出的图示显示了对于使用100个(左)、200个(中心)或300个探针(右)的实验,蛋白质鉴定的灵敏度。在每幅图中,针对测量4种底物长度的实验,确定了亲和试剂探针的灵敏度(例如,在小于1%的错误检测率(fdr)下鉴定的底物的百分比):(1)完整(完全)的蛋白质,(2)蛋白质的50长度的n或c末端片段,(3)蛋白质的100长度的n或c末端片段,和(4)蛋白质的200长度的n或c末端片段。n和c末端片段分别用实心条和条纹条表示。每个探针均与靶向的三聚体和4个其他随机脱靶三聚体结合。如图4所示,例如,即使当蛋白质被截短为仅包含100个氨基酸的片段并且进行200个探针的实验时,也可以鉴定出相当大比例的蛋白质(约40%)。如果使用300个探针,则在蛋白质被截短为仅包含100个氨基酸的片段的情况下,可以鉴定出约70-75%的蛋白质。图4还显示了含有n末端片段的截短蛋白质比含有c末端片段的片段略微更易于鉴定(例如,具有较高的蛋白质鉴定灵敏度)。实施例3:既不包含衍生出该片段的完整蛋白质的c末端也不包含其n末端的蛋白质片段本文描述的方法可以应用于分析和/或鉴定样品中的蛋白质片段,该蛋白质片段不包含衍生出该片段的完整蛋白质的原始2个末端中的任一个。在这样的实验中,亲和探针与蛋白质结合的概率计算被修改为仅考虑与截短的蛋白质序列而不是与完整蛋白质序列的结合。图5示出的图示显示了对于使用各种蛋白质片段化方法的实验,蛋白质鉴定的灵敏度。在顶行和底行的每一行中,以50、100、200和300个亲和试剂测量(在从左至右的4幅图中)显示了蛋白质鉴定性能,其中最大片段长度值为50、100、200、300、400和500(分别以六边形、朝下的三角形、朝上的三角形、菱形、矩形和圆形表示)。参见图5的顶行,当使用由片段起始位置和片段长度限定的特定片段产生方法时,每个子图上的每个点代表灵敏度(蛋白质鉴定率)。片段是在每种蛋白质上的特定起始位置处生成的,该起始位置按照氨基酸中距n末端的距离(例如,距其氨基酸(aa)数)来索引(如在x轴上绘出的)。每个蛋白质片段的末端被选择为生成长度为50、100、200、300、400或500个氨基酸的片段(最大片段长度或最大片段长度值),分别由六边形、朝下的三角形、朝上的三角形、菱形、矩形和圆形表示。如果由于蛋白质太短而无法生成给定指定长度的片段,则保留比所要求的包含c末端的长度更短的片段。例如,当使用50种亲和试剂进行实验时,只能鉴定出很小百分比的蛋白质(如在y轴上绘出的)。然而,当使用最大长度为200个氨基酸的片段,以200个亲和试剂探针进行实验时,取决于片段的起始位点(如在x轴上绘出的),可以鉴定出约50%至约85%的蛋白质(如在y轴上绘出的)。随着片段起始位点进一步远离n末端,蛋白质鉴定灵敏度存在普遍下降趋势。这种趋势可以用以下事实来解释:随着片段起点远离n末端移动,生成包括c末端且小于最大片段长度的更多片段。参见图5的底行,此处的4个子图显示出与顶行相似的结果,只是在灵敏度和错误发现率计算之前,从分析中丢弃不匹配最大片段长度的任何片段(例如,不包含c末端的片段)。仅在可能已生成有效片段的那些蛋白质中计算蛋白质鉴定的灵敏度。图5的底行显示,在没有固定片段长度的情况下,在最大片段长度处,相对于片段起始位点的位置,蛋白质鉴定灵敏度没有统计学上显著的变化。片段长度是蛋白质鉴定率的主要决定因素,而不是蛋白质序列内的片段位置。实施例4:通过长度、疏水性和/或等电点测量进行的蛋白质鉴定本文描述的方法可以应用于使用来自蛋白质测量的信息来分析和/或鉴定样品中的蛋白质,该信息包括长度、疏水性和/或等电点(pi)。测量蛋白质查询候选物的特定长度的概率可以被表示为:其中σ=|cv*预期结果值|u=(测得的结果值–预期结果值)/σ在这种情况下,测量结果是测得的未知蛋白质的长度,而预期结果值是蛋白质查询候选物的长度。该模型还使用变异系数(cv)值,该值描述了测量方法的预期精度。使用相同的公式计算测量蛋白质的特定疏水性的概率,并将预期结果值设置为根据蛋白质候选物序列计算的亲疏水性总平均值(gravy)得分。例如,可以使用用于计算分子生物学的biopython工具执行kyte-doolittle计算方法(例如,如[kyte等人,“asimplemethodfordisplayingthehydropathiccharacterofaprotein,”j.mol.biol.,1982年5月5日;157(1):105-32]所述,其通过引用整体并入本文),来计算这样的gravy得分。类似地,按照[tabb,davidl.,“analgorithmforisoelectricpointestimation,”<http://fields.scripps.edu/dtaselect/20010710-pi-algorithm.pdf>,2003年6月28日,其通过引用整体并入本文]描述的方法,以使用用来实现bjellqvist的方法(例如,如[audain等人,“accurateestimationofisoelectricpointofproteinandpeptidebasedonaminoacidsequences,”bioinformatics,2015年11月14日;32(6):821-27]所述,其通过引用整体并入本文)的biopython从蛋白质候选物序列计算的预期pi值对等电点(pi)建模。在所有情况下,实验测量精度均设置为cv值为0.1。图6示出的图示显示了对于使用测量类型的各种组合的实验,人类蛋白质鉴定的灵敏度(在小于1%的fdr下鉴定的底物的百分比)。仅使用蛋白质长度、疏水性或pi测量,实际上无法鉴定出蛋白质(例如,灵敏度<1%)。组合所有三种类型的测量(长度+疏水性+pi),实际上仍然无法得到鉴定。然而,蛋白质长度、疏水性或pi测量可以用来加强从亲和试剂探针结合实验的测量。例如,可以基于这些特性中的任一种来分级分离蛋白质,并且将每个级分缀合至基底上的不同空间位置。在该分级分离和缀合之后,可以进行亲和试剂结合测量,并且可以根据蛋白质的空间地址来确定疏水性、蛋白质长度或pi的测量值。可以基于凝胶过滤(sds-page)或大小排阻色谱法,按分子量来分级分离变性的蛋白质。可以通过用分子量除以氨基酸的平均质量(111da)来估算蛋白质的长度。可以使用疏水相互作用色谱法根据疏水性来分级分离蛋白质。可以使用离子交换色谱法根据pi来分级分离蛋白质。例如,通过以0.1的cv值分级分离来进行蛋白质长度的附加测量将使用100个探针(1个靶向三聚体,每个探针4个额外的脱靶位点)的实验的鉴定灵敏度从约55%(不使用蛋白质长度测量)提高至约65%(使用蛋白质长度测量)。类似地,通过以0.1的cv值进行蛋白质长度的附加测量将使用200个探针(1个靶向三聚体,每个探针4个额外的脱靶位点)的实验的鉴定灵敏度从约90%(不使用蛋白质长度测量)提高至约95%(使用蛋白质长度测量)。实施例5:通过用抗体混合物测量进行的蛋白质鉴定本文描述的方法可以应用于使用来自实验的信息来分析和/或鉴定样品中的蛋白质,其中在每个结合实验中测量亲和试剂的混合物。与公开的实施方案一`致,通过使用来自santacruzbiotechnology,inc.的市售抗体池获得结合测量值来对1,000种未知人类蛋白质的鉴定进行基准化。从包含约21,005种蛋白质的uniprot蛋白质数据库中随机选择了1,000种蛋白质。可从santacruzbiotechnology目录获得的对人类蛋白质具有反应性的单克隆抗体的列表从在线抗体登录中心下载。该列表包含22,301种抗体,并被过滤为与uniprot人类蛋白质数据库中的蛋白质相匹配的14,566种抗体的列表。在该实验中建模的抗体的完整集合包含这14,566种抗体。抗体混合物与1,000种未知蛋白质候选物结合的实验评估如下所述进行。首先,模建了50种抗体混合物。为了产生任何单一混合物,从抗体的总集合中随机选择了5,000种抗体。接下来,对于每种混合物,确定该混合物与任何未知蛋白质的结合概率。注意到尽管从目的是推断它们的身份的意义上来说这些蛋白质是“未知的”,但是该算法知道每种“未知蛋白质”的真实身份。如果混合物中含有针对未知蛋白质的抗体,则结合概率指定为0.99。如果混合物不包含针对未知蛋白质的抗体,则将结合概率指定为0.0488。换言之,抗体混合物的结合结果的概率被建模为:p(结合结果|蛋白质)={如果混合物包含针对蛋白质的抗体则为0.99;否则为0.0488}。0.0488的值表示该混合物针对蛋白质发生非特异性(脱靶)结合事件的概率。基于任何单个抗体结合除其靶标以外的蛋白质的预期概率以及混合物中蛋白质的数目,对混合物的非特异性结合概率进行建模。抗体混合物的非特异性结合事件的概率是该混合物中任何单个抗体非特异性结合的概率。该概率是基于混合物中的抗体数(n)和任何单个抗体的非特异性结合的概率(p)而计算的,并且可以由以下方程式表示:混合物的非特异性结合概率=1–(1–p)n在这种情况下,假设单个抗体结合除其靶蛋白以外的其他蛋白质的非特异性结合事件的概率为0.00001(10-5)。因此,任何单一抗体的非特异性结合概率(p)为10-5,得出:混合物的非特异性结合概率=1–(1–10-5)5000=0.0488。另外,与蛋白质不结合的结果的概率被计算为:p(不结合结果|蛋白质)=1–p(结合结果|蛋白质)。对于每种未知蛋白质,基于混合物与未知蛋白质的结合概率,评估所测量的每种抗体混合物的结合。对最小值为0且最大值为1的均匀分布进行随机采样,并且如果所得到的数字小于抗体混合物与未知蛋白质的结合概率,则该实验导致该混合物的结合事件。否则,该实验导致该混合物的非结合事件。评估所有结合事件后,如下进行蛋白质推断:对于每种未知蛋白质,针对uniprot数据库中21,005种蛋白质候选物中的每一种,评价了所评估的结合事件(总计50个,每种混合物1个)的顺序。更具体地,针对每种候选物计算观察到结合事件顺序的概率。通过将测量的所有50种混合物中每个单独混合物结合/非结合事件的概率相乘,计算出概率。以与上述相同的方式计算结合概率,并且非结合的概率为1减去结合概率。具有最高结合概率的蛋白质查询候选物是未知蛋白质的推断身份。通过将头部单独候选物的概率除以所有候选物的总概率,计算出针对该单个蛋白质正确的鉴定概率。采用对1,000种未知蛋白质中的每一种推断出的身份,未知蛋白质按照其鉴定概率的降序进行排序。选择鉴定概率截断值,以使列表中所有先前鉴定中不正确鉴定的百分比为1%。总体而言,在1,000种未知蛋白质中,有551种得到鉴定,不正确鉴定率为1%。因此,蛋白质鉴定的灵敏度为55.1%。实施例6:在许多物种中的蛋白质鉴定本文描述的方法可以应用于分析和/或鉴定从许多不同物种获得的样品中的蛋白质。例如,亲和试剂结合实验的序列的结果可以用来鉴定大肠杆菌(e.coli)、酿酒酵母(saccharomycescerevisiae)(酵母)或智人(homosapiens)(人)中的蛋白质,分别用圆圈、三角形和正方形表示。为了适应每种物种的分析方法,必须从物种特异性序列数据库(例如从uniprot下载的物种的参考蛋白质组)生成蛋白质候选物列表。图7示出的图示显示了对于使用50、100、200或300个针对来自大肠杆菌、酵母或人的未知蛋白质的亲和试剂探针(分别由圆圈、三角形和正方形表示)的实验,蛋白质鉴定的灵敏度。每个探针都与一个靶向的三聚体和另外四个脱靶位点结合,概率为0.25。对于所测试的三个物种中的每一个,使用200个探针进行的实验的灵敏度(在小于1%的错误鉴定率下鉴定的未知蛋白质的百分比)约为90%。实施例7:在snp的存在下的蛋白质鉴定本文描述的方法可以应用于在由非同义单核苷酸多态性(snp)引起的单氨基酸变异(sav)的存在下分析和/或鉴定样品中的蛋白质。除少数单氨基酸变异(sav)外具有相同序列的蛋白质可能难以区分。例如,在使用一系列亲和试剂测量的实验中,除非在实验中包括对蛋白质的多态性区域具有高度选择性的亲和试剂,否则几乎不可能将蛋白质的规范形式与其变异形式区分开。在通过任何亲和试剂测量都无法区分多态性区域的情况下,对于规范和变异蛋白质查询候选物,任一种蛋白质形式的测量都将返回相似的概率(可能性)(例如,l(规范蛋白质|证据)=0.8且l(变异蛋白质|证据)=0.8)。在这样的情况下,任何一种单独的蛋白质候选物可能都不会返回高于0.5的概率,例如,如下对于规范蛋白质所表示的(其中cprot=规范蛋白质,vprot=变异蛋白质):其中l其他是除规范蛋白质和变异蛋白质外,所有蛋白质查询候选物的可能性总和,并且是大于或等于零的数字。在这种情况下,可对未知蛋白质返回成组的潜在蛋白质鉴定。例如,前两种最可能的蛋白质查询候选物的概率可以被表示为:使用这种方法,可以从未知蛋白质获得确信的鉴定,尽管其无法解析规范蛋白质和变异蛋白质。特别是,其中l其他接近零的情况可能会导致确信的鉴定。实施例8:从经验结果对概率模型的迭代改进在使用期望最大值或相关方法的蛋白质鉴定的计算期间,可以使用经验测量来迭代地改进在本文所述的一种或多种方法中使用的概率模型。在此针对亲和试剂结合实验描述了一种这样的方法。首先,用估计值初始化每个亲和试剂探针的结合概率。例如,200个探针的集合可各自靶向单个三聚体,并且估计的结合概率为0.5。使用本文其他地方(例如,参见实施例1)公开的方法鉴定蛋白质。接下来,如以下步骤所概述的,基于经验测量值迭代地精炼每个探针的结合概率:(1)使用以估计的错误发现率<0.01鉴定的未知蛋白质的集合来更新结合概率:对于每个探针,使用该集合中包含被探针识别的结合位点(三聚体)的蛋白质的比例来计算更新的结合概率:更新“集合中结合位点>20的蛋白质的数目”的探针概率:如果更新的概率<10-5,则将其设置为10-5(以避免指定0的概率)。(2)使用更新的结合概率进行另一项蛋白质鉴定。重复第1步和第2步以进行多次迭代(例如,总共1、2、3、4、5、6、7、8、9、10次或超过10次迭代)。使用采用200个探针的实验测试这种迭代方法,每个探针以0.25的结合概率识别单个三聚体。200个探针的结合测量针对2000种未知蛋白质进行建模,其中探针结合概率的初始估计值被设置为0.5。在执行该迭代算法5次迭代后,更新的探针结合概率变得更加准确(接近0.25),并且蛋白质鉴定灵敏度增加。图8示出的图示显示了相对于迭代(x轴)的结合概率(y轴,左)和蛋白质鉴定的灵敏度(y轴,右)。如图8所示,细线显示每个单独探针的探针结合概率,细线之间的黑线是探针结合概率的中值,而粗线显示每次迭代的蛋白质鉴定灵敏度。实施例9:从蛋白质候选物匹配概率估计鉴定错误发现率在本文所述的一种或多种方法中使用的用于蛋白质推断或鉴定的概率模型产生针对每种未知蛋白质的蛋白质序列匹配列表以及该序列匹配为正确的相关概率作为直接结果。在许多情况下,只有一部分蛋白质鉴定可能是正确的。因此,下面描述了一种可用于估计并控制一组蛋白质的错误鉴定率的方法。首先,完整的蛋白质鉴定集按照蛋白质鉴定概率的降序排序,如下所示(其中prot=蛋白质):prot1概率(p1):0.99prot2概率(p2):0.97prot3概率(p3):0.92prot4概率(p4):0.9prot5概率(p5):0.8prot6概率(p6):0.75prot7概率(p7):0.6prot8概率(p8):0.5接下来,将该列表中每个点的预期错误发现率计算为其中是该列表中给定点和更早点的所有概率的平均值(如下所示):如图9所示,对于模拟的200个探针的实验,估计的错误鉴定率与实际错误鉴定率的比较证明了准确的错误鉴定率估计。参见图9的上图,将鉴定灵敏度与实际错误鉴定率和估计的错误鉴定率进行比较。参见图9的下图,将估计的错误鉴定率相对于实际错误鉴定率(如实线所示)作图,而虚线表示理想的完美准确的错误鉴定率估计。估计的错误鉴定(id)率可用来根据错误鉴定的容忍度来确定蛋白质鉴定列表的阈值。实施例10:错误发现率估计方法的推导考虑蛋白质鉴定的列表,每个蛋白质鉴定包括对于未知蛋白质最可能的蛋白质匹配,以及该匹配为正确的相关概率(p(蛋白质|证据))。例如:prot1–macd2,p1=0.99prot2–kcnu1,p2=0.97prot3–rgl2,p3=0.92prot4–mtlr,p4=0.9该列表中预期的错误发现数目为1-该列表中所有蛋白质的平均匹配概率。在这种情况下:该方法的基本原理如下。考虑n个蛋白质鉴定的列表,并且每个蛋白质鉴定proti是随机变量,其中如果鉴定正确,则proti=1,如果鉴定不正确,则proti=0。在这种情况下,任何列表中正确鉴定(correctids)的数目是这些随机变量的总和:每个单独蛋白质鉴定的期望值等于正确鉴定的概率:e(proti)=1*pi+0*(1-pi)=pi通过期望值的线性,可以得出:预期的正确发现率(正确鉴定的数目/鉴定数目)是平均候选物概率:错误发现率为1-正确发现率,或:实施例11:使用结合测量结果的蛋白质鉴定本文所述的方法可以应用于与亲和试剂与未鉴定的蛋白质的结合和/或非结合相关的数据的不同子集。在一些实施方案中,本文所述的方法可以应用于其中不考虑所测量的结合结果的特定子集(例如,非结合测量结果)的实验。这些不考虑所测量的结合结果的子集的方法在本文中可以被称为“删截的”推断方法(例如,如实施例1中所述)。在图10描述的结果中,由删截的推断方法得到的蛋白质鉴定是基于评估与特定未鉴定的蛋白质相关的结合事件的发生率。因此,删截的推断方法在确定未知蛋白质的身份时不考虑非结合结果。这种类型的删截的推断方法与“未删截的”方法不同,在“未删截的”方法中,所有获得的结合结果都被考虑(例如,与特定未鉴定的蛋白质相关的结合测量结果和非结合测量结果)。在一些实施方案中,在预期特定结合测量或结合测量结果更容易发生错误或可能偏离蛋白质的预期结合测量结果(例如,结合测量结果由该蛋白质生成的概率)的情况下,可采用删截的方法。例如,在亲和试剂结合实验中,可以基于与主要具有线性结构的变性蛋白质的结合来计算结合测量结果和非结合测量结果的概率。在这些条件下,表位可能容易被亲和试剂接近。然而,在一些实施方案中,可以在非变性或部分变性条件下收集对测定的蛋白质样品的结合测量,在这些条件下,蛋白质以具有明显三维结构的“折叠”状态存在,这在许多情况下会导致蛋白质上在线性形式下可及的亲和试剂结合表位由于折叠状态中的空间位阻而成为不可及的。例如,如果亲和试剂识别蛋白质的表位在折叠蛋白质的结构上可及的区域中,则可以预期在未知样品上获得的经验结合测量值将与从线性化蛋白质得出的计算结合概率一致。然而,例如,如果被亲和试剂识别的表位在结构上不可及,则可以预期非结合结果将多于从线性化蛋白质得出的计算结合概率所预期的结果。此外,基于蛋白质周围的特定条件,可以以多种不同的可能构型来配置三维结构,并且基于所需亲和试剂的可及性程度,不同的可能构型中的每一个可以具有结合特定亲和试剂的独特预期。因此,可以预期非结合结果与针对每种蛋白质计算出的结合概率有所偏差,并且仅考虑结合结果的删截的推断方法可能是合适的。在图10中提供的“删截的”推断方法中,仅考虑测量的结合结果(换句话说,要么不测量非结合结果,要么不考虑测量的非结合结果),使得结合结果集的概率仅考虑导致结合测量值的m个测量的结合结果,它是既包含结合测量结果又包含非结合测量结果的n个总测量结合结果的子集。这可以用以下表达式描述:p(结果集|蛋白质)=p(结合事件1|蛋白质)*p(结合事件2|蛋白质)*…*p(结合事件m|蛋白质)当采用删截的方法时,将比例因子应用于p(结合结果集|蛋白质)以校正偏差可能是适当的。例如,更长的蛋白质通常具有更高的生成潜在结合结果的概率(例如,因为它们包含更多的潜在结合位点)。为了纠正这种偏差,可以通过将p(结合结果集|蛋白质)除以可以基于蛋白质上的潜在结合位点数从蛋白质生成的m个结合位点的独特组合的数目,来计算每种候选蛋白质的比例似然sl。对于具有三聚体识别位点的长度为l的蛋白质,可能存在l-2个潜在结合位点(例如,完整蛋白质序列的每个可能的长度为l的子序列),以使:在给定结果集的情况下,从q种可能的候选蛋白质的集合中选择出任何候选蛋白质的概率可以由下式给出:在图10中绘出了删截的蛋白质推断与未删截的蛋白质推断方法的实施方案的性能。在图10中绘出的数据在表1中提供。表1在图10所示的比较中,将蛋白质鉴定灵敏度(例如,鉴定出的独特蛋白质的百分比)相对于针对在线性化蛋白质基底上使用的删截推断和未删截推断所测量的亲和试剂循环数作图。所使用的亲和试剂针对蛋白质组中排名前列的最丰富的三聚体,并且每种亲和试剂对另外四个随机三聚体具有脱靶亲和力。当使用100个亲和试剂循环时,未删截的方法要比删截的方法表现好十倍以上。当使用更多循环时,未删截的推断胜过删截的推断的程度降低。实施例12:蛋白质鉴定对随机假阴性和假阳性亲和试剂结合的容忍度在一些情况下,亲和试剂结合的假阴性结合测量结果可能具有高发生率。“假阴性”结合结果表现为亲和试剂结合测量的发生频率低于预期。例如,由于结合检测方法、结合条件(例如,温度、缓冲液组成等)、蛋白质样品的破坏或亲和试剂储备液的破坏等问题,可能会出现这样的“假阴性”结果。为了确定假阴性测量值对删截的蛋白质鉴定和未删截的蛋白质鉴定方法的影响,通过在计算机中将1/10、1/100、1/1,000、1/10,000或1/100,000的随机观察到的结合事件切换为非结合事件,有意破坏亲和试剂测量循环的子集。以这种方式破坏了总共300个亲和试剂循环中的0、1、50、100、200或300个。如图11中绘出的结果所示,删截的蛋白质鉴定方法和未删截的蛋白质鉴定方法均能容忍这种类型的随机假阴性结合。在图11中绘出的数据在表2中提供。表2类似地,“假阳性”结合结果表现为亲和试剂结合测量的发生频率高于预期。通过将结合结果的子集从非结合结果切换到结合结果来估计对“假阳性”结合结果的容忍度。该评估的结果在表3中提供。表3这些结果在图12中绘出,图12表明,随着随机假阳性测量值的发生率逐渐增加,删截的蛋白质鉴定方法的性能比未删截的蛋白质鉴定方法衰减得更快。然而,这两种方法都可以容忍每个亲和试剂循环中1/1000的假阳性率,或亲和试剂循环的子集中1/100的假阳性率。实施例13:采用被高估或低估的亲和试剂结合概率的蛋白质推断的性能采用正确估计的亲和试剂与三聚体的结合概率,并采用被高估或低估的亲和试剂结合概率,使用蛋白质鉴定来评估蛋白质鉴定灵敏度。真实结合概率为0.25。被低估的结合概率为:0.05、0.1和0.2。被高估的结合概率为0.30、0.50、0.75和0.90。总共获取了300个循环的亲和试剂测量。没有(0)、所有300种或一部分(1、50、100、200种)亲和试剂应用了被高估或低估的结合概率。其他所有蛋白质都在蛋白质鉴定中使用了正确的结合概率(0.25)。该分析的结果在表4中提供。表4这些结果在图13中绘出,显示在可能无法准确估计结合概率的一些情况下,删截的蛋白质鉴定可能是优选的方法。实施例14:使用具有未知结合表位的亲和试剂进行的蛋白质推断方法的性能在一些情况下,亲和试剂可具有许多未知的结合位点(例如,表位)。使用输入蛋白质鉴定算法中的以概率0.25各自结合五个三聚体位点(例如,靶向的三聚体和四个随机脱靶位点)的亲和试剂,比较了采用亲和试剂结合测量的删截的蛋白鉴定和未删截的蛋白鉴定方法的灵敏度。亲和试剂的子集(0/300、1/300、50/300、100/300、200/300或300/300个)具有另外1个、4个或40个额外的结合位点,每个额外的结合位点针对一个随机三聚体,结合概率为0.05、0.1或0.25。该分析的结果在表5中示出。表5这些结果在图14中绘出,显示未删截的推断更能容忍包含另外的隐藏结合位点,并且当300种亲和试剂中的50种含有40个另外的结合位点时,这两种推断方法的性能都显著受损。实施例15:使用具有遗漏的结合表位的亲和试剂进行的蛋白质推断方法的性能在一些情况下,可能有表征不当的亲和试剂具有许多不存在的带注释的结合表位(例如,额外的预期结合位点)。即,用于生成亲和试剂的预期结合概率的模型包含不存在的额外预期位点。使用输入蛋白质鉴定算法中的以概率0.25各自结合随机三聚体位点(例如,靶向的三聚体和四个随机脱靶位点)的亲和试剂,比较了采用亲和试剂结合测量的删截的蛋白鉴定和未删截的蛋白鉴定方法的灵敏度。亲和试剂的子集(0/300、1/300、50/300、100/300、200/300或300/300个)具有1个、4个或40个额外的结合位点,每个额外的结合位点针对一个随机三聚体,结合概率为0.05、0.1或0.25,添加到蛋白质推断算法使用的亲和试剂的模型中。该分析的结果在表6中示出。表6这些结果在图15中绘出,显示未删截的推断更能容忍在亲和试剂结合模型中包含额外的预期结合位点,并且当大多数亲和试剂含有40个额外的预期结合位点时,这两种蛋白质推断方法的性能都一定程度地受损。实施例16:采用替代缩放策略,对亲和试剂结合分析的删截的推断可以使用亲和试剂结合测量值与各种概率缩放策略的组合,将本文所述的方法应用于推断蛋白质身份(例如,鉴定未知蛋白质)。实施例11中描述的删截的推断方法基于蛋白质上潜在的结合位点数(蛋白质长度-2)和观察到的结合结果数(m)来缩放观察到的蛋白质结果的概率:本文所述的方法可以与用于计算缩放的可能性的替代方法一起应用。该实施例应用了替代的归一化方法,该方法对从用于测量蛋白质的亲和试剂集中为长度为k的蛋白质生成n个结合事件的概率进行建模,并基于该概率进行缩放。首先,对于每个探针,计算探针结合样品中身份未知的三聚体的概率:其中p(三聚体j)是相对于蛋白质组中所有8,000个三聚体的总计数的三聚体发生频率。对于长度为k的任何蛋白质,探针i结合该蛋白质的概率可以由下式给出:p(蛋白质结合|探针i,k)=1-(1-p(三聚体结合|探针i))k-2对长度为k的蛋白质观察到的成功结合事件的数目可以遵循n次试验的poisson-binomial分布,其中n为对该蛋白质进行的探针结合测量的次数,分布的参数p探针,k表示每次试验的成功概率:p探针,k=[p(结合|探针1,k),p(结合|探针2,k),p(结合|探针3,k)…p(结合|探针n,k)]。用一组特定的探针从长度为k的蛋白质生成n个结合事件的概率可以由用p参数化的poisson二项式分布(pmfpoibin)的概率质量函数给出,其在n处评价:p(n结合事件|探针,k)=pmfpoibin(n,p探针,k)基于以下概率计算特定结果集的缩放概率:实施例17:使用随机选择的亲和试剂本文所述的方法可以应用于任何组亲和试剂。例如,蛋白质鉴定方法可以应用于针对蛋白质组中最丰富的三聚体或针对随机三聚体的一组亲和试剂。表7a-7c中分别示出了来自使用亲和试剂的人类蛋白质推断分析的结果,所述亲和试剂针对蛋白质组中的前300个最不丰富的三聚体、蛋白质组中的300个随机选择的三聚体或蛋白质组中的300个最丰富的三聚体。表7a-c表7a-针对蛋白质组中最不丰富的三聚体的300种亲和试剂表7b-针对蛋白质组中的随机三聚体的300种亲和试剂表7c-针对蛋白质组中最丰富的三聚体的300种亲和试剂这些结果在图16中绘出。在所有情况下,每种亲和试剂与目标三聚体的结合概率均为0.25,与其他随机选择的三聚体的结合概率为0.25至4。基于灵敏度(例如,鉴定出的蛋白质的百分比)来测量每个亲和试剂组的性能。每个亲和试剂组进行5次重复评估,每次重复的性能以点绘出,用垂直线连接来自同一组亲和试剂的重复测量。由前300种最丰富的亲和试剂组成的亲和试剂组的结果为蓝色,后300种为绿色。生成并评估了针对随机三聚体的总共100个包含300种亲和试剂的不同组。这些组中的每一个都由用垂直灰线连接的一组5个灰点(每次重复有一个)表示。根据该分析中使用的未删截的推断,与靶向随机三聚体相比,靶向更丰富的三聚体可提高鉴定性能。实施例18:具有生物类似脱靶位点的亲和试剂本文所述的方法可以应用于采用具有不同类型的脱靶结合位点(表位)的亲和试剂的亲和试剂结合实验。在该实施例中,比较采用两类亲和试剂的性能:随机的,和“生物类似的”亲和试剂。来自这些评估的结果在表8a-8d中示出。表8a-d表8a-采用亲和试剂的删截推断的性能,所述亲和试剂具有生物类似的脱靶位点并且针对蛋白质组中的300个最丰富的三聚体删截的循环数探针类型灵敏度真100生物类似0.00634真200生物类似31.97667真300生物类似68.73336表8b-采用亲和试剂的未删截推断的性能,所述亲和试剂具有生物类似的脱靶位点并且针对蛋白质组中的300个最丰富的三聚体表8c-采用亲和试剂的删截推断的性能,所述亲和试剂具有随机的脱靶位点并且针对蛋白质组中的300个最丰富的三聚体删截的循环数探针类型灵敏度真100随机0.082414真200随机74.68619真300随机93.13427表8d-采用亲和试剂的未删截推断的性能,所述亲和试剂具有随机的脱靶位点并且针对蛋白质组中的300个最丰富的三聚体删截的循环数探针类型灵敏度假100随机60.02916假200随机95.47356假300随机98.51021与随机亲和试剂不同,生物类似的亲和试剂具有在生物化学上与靶表位相似的脱靶结合位点。随机亲和试剂和生物类似亲和试剂都以0.25的结合概率识别其靶表位(例如,三聚体)。每种随机类别的亲和试剂均具有4个随机选择的脱靶三聚体结合位点,结合概率为0.25。相反,“生物类似的”亲和试剂的4个脱靶结合位点是与亲和试剂所靶向的三聚体最相似的四个三聚体,它们的结合概率为0.25。对于这些生物类似的亲和试剂,三聚体序列之间的相似性是通过对每个序列位置处氨基酸对的blosum62系数进行求和而计算出的。随机亲和试剂和生物类似的亲和试剂都靶向人类蛋白质组中最丰富的前300个三聚体,其中丰度是通过包含该三聚体的一个或多个实例的独特蛋白质的数目来衡量的。图17显示了当使用具有随机(蓝色)或生物类似(橙色)脱靶位点的亲和试剂时,删截的(虚线)和未删截的(实线)蛋白质推断方法在人类样品中鉴定出的蛋白质百分比方面的性能。在该比较中,未删截的推断优于删截的推断,在生物类似的亲和试剂的情况下,未删截的推断有更好的性能,在随机亲和试剂的情况下,删截的推断有更好的性能。或者,不是使用针对蛋白质组中最丰富的三聚体的亲和试剂,而是可以基于可以测量的候选蛋白质(例如,人类蛋白质组)、所进行的蛋白质推断的类型(删截或未删截的)以及使用的亲和试剂的类型(随机的或生物类似的),为特定方法选择最佳的一组三聚体靶标。如下所述,可以使用“贪婪(greedy)”算法来选择一组最佳亲和试剂:1)初始化所选亲和试剂(ar)的空列表。2)初始化一组候选ar(例如,8,000种ar的集合,每种ar针对具有随机脱靶位点的独特三聚体)。3)选择一组蛋白质序列,以针对(例如,uniprot参考蛋白质组中的所有人类蛋白质)进行优化。4)重复以下操作,直到选择了所需数目的ar:a.对于每种候选ar:i.模拟候选ar与蛋白质集的结合。ii.使用来自候选ar的模拟结合测量值和来自所有先前选择的ar的模拟结合测量值,对每种蛋白质进行蛋白质推断。iii.通过将通过蛋白质推断确定每种蛋白质的正确蛋白质鉴定的概率进行加和,计算候选ar的评分。b.将评分最高的ar添加到所选ar的集合中,并将其从候选ar列表中删除。贪婪方法用来从针对人类蛋白质组中最丰富的前4,000个三聚体的随机亲和试剂或生物类似亲和试剂集合中选择300种最佳亲和试剂。针对删截的蛋白质推断和未删截的蛋白质推断均进行了优化。来自这些优化的结果在表9a-9d中提供。表9a-d表9a-采用亲和试剂的删截推断的性能,所述亲和试剂具有生物类似的脱靶位点并且针对蛋白质组中的300个最佳三聚体删截的循环数探针类型灵敏度真100生物类似25.58007真200生物类似87.82173真300生物类似95.15025表9b-采用亲和试剂的未删截推断的性能,所述亲和试剂具有生物类似的脱靶位点并且针对蛋白质组中的300个最佳三聚体删截的循环数探针类型灵敏度假100生物类似76.76556假200生物类似97.2106假300生物类似99.03005表9c-采用亲和试剂的删截推断的性能,所述亲和试剂具有随机的脱靶位点并且针对蛋白质组中的300个最佳三聚体删截的循环数探针类型灵敏度真100随机24.93343真200随机88.06263真300随机95.8476表9d-采用亲和试剂的未删截推断的性能,所述亲和试剂具有随机的脱靶位点并且针对蛋白质组中的300个最佳三聚体删截的循环数探针类型灵敏度假100随机65.72841假200随机96.38012假300随机98.56092图18中绘出了用于删截的蛋白质推断和未删截的蛋白质推断的优化探针集的性能。使用通过贪婪优化算法选择的一组亲和试剂,可以改善在使用删截的蛋白推断和未删截的蛋白推断方法时,随机和生物类似亲和试剂集的性能。另外,当使用贪婪方法选择亲和试剂时,随机亲和试剂集的性能几乎与生物类似亲和试剂集相同。实施例19:使用亲和试剂混合物的结合的蛋白质推断本文所述的方法可以应用于分析和/或鉴定已经使用亲和试剂混合物测量的蛋白质。当通过亲和试剂混合物进行测定时,特定蛋白质生成结合结果的概率可以如下计算:1)计算即混合物中每种亲和试剂的非特异性表位结合的平均概率。2)基于蛋白质的长度(l)和亲和试剂表位的长度(k)计算蛋白质上的结合位点数:num结合位点=l–k+1。不发生非特异性结合事件的概率为3)对于混合物中的每种亲和试剂,计算不发生表位特异性结合事件的概率:4)混合物生成蛋白质的非结合结果的概率为:5)混合物生成结合结果的概率为:p(结合|蛋白质)=1-p(无结合|蛋白质)用于计算来自蛋白质混合物的结合或非结合结果的概率的该方法与本文所述的方法结合使用,以分析亲和试剂混合物用于蛋白质鉴定的性能。该分析中的每种单独的亲和试剂以0.25的概率结合其靶向的三聚体表位,并且以0.25的概率结合与该表位靶标结合的4个最相似的三聚体。对于这些亲和试剂,通过针对所比较的三聚体中每个序列位置处的氨基酸,将来自blosum62替换矩阵的系数相加来计算三聚体相似性。另外,每种亲和试剂结合20个额外的脱靶位点,结合概率根据脱靶位点与使用blosum62替换矩阵计算的靶向三聚体之间的序列相似性来缩放。这些额外的脱靶位点的概率为:其中sot是脱靶位点与靶位点之间的blosum62相似性,而sself是靶序列与其本身之间的blosum62相似性。将结合概率低于2.45x108的任何脱靶位点调整为具有2.45x108的结合概率。在该实例中,非特异性表位结合概率为2.45x108。使用贪婪方法,针对删截的和未删截的蛋白质推断生成了一组最佳的300种亲和试剂混合物:1)初始化所选亲和试剂(ar)混合物的空列表。2)初始化候选亲和试剂列表(在该实施例中,由使用实施例18中详述的贪婪方法计算出的300种最优试剂组成)。3)选择一组蛋白质序列以针对(例如,uniprot参考蛋白质组中的所有人类蛋白质)进行优化。4)重复以下步骤,直到生成所需数目的ar混合物:a.初始化空混合物。b.对于每种候选ar:i.使用添加了候选ar的当前混合物模拟结合结果。ii.使用来自i的模拟结合测量值和来自先前生成的混合物的模拟结合测量值,对每种蛋白质进行蛋白质推断。iii.通过将通过蛋白质推断确定每种蛋白质的正确蛋白质鉴定的概率进行加和,计算具有该候选ar的混合物的评分。c.将评分最高的候选ar添加到该混合物中。d.对于尚未在该混合物中的每种候选ar,如i-iii中所述,对添加了ar的混合物进行评分,并且如果评分最高的候选物的评分高于添加到该混合物中的先前候选物,则将其添加到该混合物中并重复此步骤。当评分最佳的候选ar相对于先前添加的候选ar降低了混合物的评分时,或者当所有候选ar已添加到混合物中时,该混合物即完成。图19示出了当未混合的候选亲和试剂与删截的蛋白质推断和未删截的蛋白质推断一起使用时,以及当使用混合物时,蛋白质鉴定的灵敏度。在图19中绘出的数据在表10a-10b中示出。表10a-b表10a-在对单个探针结合(未混合)或探针混合物(混合)进行测量时,删截的推断的性能删截的混合类型循环数探针类型灵敏度真混合100生物类似2.244199真未混合100生物类似1.363002真混合200生物类似72.16939真未混合200生物类似76.51198真混合300生物类似86.91518真未混合300生物类似91.5684表10b-在对单个探针结合(未混合)或探针混合物(混合)进行测量时,未删截的推断的性能删截的混合类型循环数探针类型灵敏度假混合100生物类似65.76011假未混合100生物类似50.79244假混合200生物类似97.81286假未混合200生物类似96.30404假混合300生物类似99.14416假未混合300生物类似98.56726混合物的使用在采用未删截的推断时改善了性能,但是如果采用删截的推断,则可能会负面影响其性能。实施例20–采用包含7种候选聚糖的数据库的聚糖鉴定考虑数据库含有7种候选聚糖的情况:id结构19galb1-4glcnacb1-6(galb1-4glcnacb1-3)galnac52glcnacb1-2mana1-6(glcnacb1-2mana1-3)manb1-4glcnacb1-4glcnac344glcnaca1-4galb1-3galnac378neu5aca2-3galb1-4(fuca1-3)glcnacb1-3galnac430fuca1-3glcnacb1-6(galb1-4glcnacb1-3)galb1-4glc519galnaca1-3(fuca1-2)galb1-4glcnacb1-6galnac534neu5aca2-3galb1-4(fuca1-3)glcnacb1-2man另外,使用4种亲和试剂(ar)进行该实验,每种亲和试剂具有25%的结合给定二糖的可能性。这些试剂所结合的其他二糖在该数据库中的任何聚糖中均未发现。对于针对数据库中的每个序列的亲和试剂,构建命中表(行=亲和试剂#1至#4,列=seqid)需要注意的是,该信息以渐进的方式达到,因此可以迭代地进行计算。从命中表中,评价p(聚糖_i|ar_j)以生成概率矩阵,如下所示。请注意,对于给定的条目,如果命中表≥1,则使用p_降落_ar_n=真实降落率=0.25;否则,如果命中表=0,则使用p(检测器错误)=0.00001注意,许多单元具有0.00001的概率。这种小概率说明了可能的检测器错误。聚糖的初始、未归一化的概率被计算为每种候选聚糖的概率的乘积:19523443784305195342.5e-162.5e-161e-201.5625e-076.25e-122.5e-160.00390625接下来,计算大小归一化,这是指一定数目的亲和试剂可以降落在给定聚糖上的方式的数目,它是该聚糖的潜在结合位点数的函数。大小归一化由choose(位点_i,n)项给出。例如,候选id52具有6个二糖位点,大小归一化为[6select4],即15。如果结合事件多于可用二糖位点的数目,则大小归一化因数被设置为1。通过除以大小归一化,对每种聚糖的未归一化概率进行归一化,以考虑该大小校正,其得出:接下来,对概率进行归一化,以使整个数据库中的整个概率集总计为一个。这是通过将大小归一化的概率相加为0.00390641并将每个大小归一化的概率除以该归一化以获得最终平衡概率来实现的:实施例21:删截的蛋白质鉴定在含有蛋白质同种型的样品中的性能本文描述的蛋白质鉴定方法可以应用于含有蛋白质同种型的样品。规范蛋白质的同种型可指通过与规范蛋白质相同的基因或与规范蛋白质相同的基因家族中的另一个基因的可变剪接形成的规范蛋白质的变体。蛋白质同种型可以在结构上类似于规范蛋白质,通常与规范蛋白质共有大部分序列。蛋白质样品和亲和试剂为了确定同种型序列的存在对蛋白质鉴定的影响,对由20,374种独特的规范人类蛋白质和这些规范蛋白质的21,987种独特同种型组成的蛋白质集合进行亲和试剂结合分析。规范蛋白质和同种型蛋白质是在作为uniprot数据库的一部分可获得的参考人类蛋白质组中列出的蛋白质。该分析中仅包括带有“swiss-prot”名称的蛋白质,该名称用来指定已手动注释并审查的蛋白质。对于每种单独的规范蛋白质包括的同种型的数目为0至36个同种型。该集合中规范蛋白质的同种型的平均数为1.08。使用384个亲和试剂循环分析样品,每个循环测量独特亲和试剂与样品中每种蛋白质的结合结果。每种亲和试剂以0.25的概率结合靶向的三聚体,并以0.25的概率结合与靶向的三聚体最相似的四个三聚体。其他脱靶三聚体以2.45x10-8和0.25*1.5-x中数值较大的概率结合,其中x是靶向的三聚体与其本身的相似度减去脱靶三聚体与三聚体靶标的相似度。三聚体序列之间的相似度可以通过,例如,对于三个序列位置中每一个处的氨基酸对,将blosum62系数求和来计算。如实施例18所述,使用贪婪方法选择亲和试剂三聚体靶标,以针对人类蛋白质组进行优化。使用未知同种型序列的蛋白质鉴定性能使用仅包含蛋白质样品中20,374种规范蛋白质的序列的数据库,对样品的结合结果进行删截的蛋白质推断。由于用于蛋白质推断的数据库缺少样品中21,987种蛋白质同种型的序列,因此当样品中潜在蛋白质同种型的序列未知时,该分析的结果指示性能。通过以这种方式进行蛋白质推断,对样品中83.9%的蛋白质鉴定了正确的蛋白质家族,错误发现率为1%。如本文所用的,术语“蛋白质家族”通常是指包括规范蛋白质序列和该规范蛋白质序列的所有同种型的一组序列。如果推断的蛋白质身份与所分析的蛋白质在同一蛋白质家族内,则可以鉴定该蛋白质的正确蛋白质家族。使用已知同种型序列的蛋白质鉴定性能当使用由样品中所有蛋白质序列(规范蛋白质序列和同种型蛋白质序列)组成的序列数据库进行蛋白质推断时,对于样品中60.9%的蛋白质鉴定出正确的蛋白质序列,错误发现率为1%。如果鉴定出蛋白质的确切序列,则鉴定了该蛋白质的正确蛋白质序列。此外,对于样品中89.8%的蛋白质,鉴定出正确的蛋白质家族。蛋白质家族和确切蛋白质序列的鉴定率之间的差异可能是由于难以解析具有相似序列的多种同种型候选物之间蛋白质的身份而引起的。使用先验定义的蛋白质家族的蛋白质鉴定性能当规范蛋白质序列和同种型蛋白质序列分组为蛋白质家族先验地已知时,可以通过直接计算蛋白质家族概率来提高蛋白质家族的鉴定率。对于所测量的单个蛋白质,可以通过将组成该家族的单个蛋白质序列的每个概率相加来计算该蛋白质是该蛋白质家族的成员的概率。将对于所分析的蛋白质具有最高概率的蛋白质家族指定为蛋白质家族鉴定。当以这种方式计算蛋白质家族概率时,以1%的错误发现率为样品中97.2%的蛋白质鉴定正确的蛋白质家族。相比之下,当不直接计算蛋白质家族概率时,以1%的错误发现率为样品中89.8%的蛋白质鉴定正确的蛋白质家族。实施例22:删截的蛋白质鉴定在包含具有单氨基酸变异(sav)的蛋白质的样品中的性能本文描述的蛋白质鉴定方法可以应用于含有具有单氨基酸变异的蛋白质的样品。如本文所用的,规范蛋白质的单氨基酸变异(sav)通常是指规范蛋白质的变体,其区别在于单个氨基酸。单氨基酸变异蛋白质通常可由编码该蛋白质的基因中的错义单核苷酸多态性(snp)引起。蛋白质样品和亲和试剂为了确定sav蛋白质的存在对蛋白质鉴定的影响,对由20,374种独特的规范人类蛋白质和这些规范蛋白质的12,827种独特sav组成的蛋白质集合进行亲和试剂结合分析。规范蛋白质是在作为uniprot数据库的一部分可获得的参考人类蛋白质组中列出的蛋白质。对于每种规范蛋白质,如果sav数据库中存在该蛋白质的一个或多个sav,则样品中包含随机选择的sav。使用的sav数据库是uniprot人类多态性和疾病突变索引。该分析中仅包括带有“swiss-prot”名称的蛋白质,该名称用来指定已手动注释并审查的蛋白质。使用384个亲和试剂循环分析样品,每个循环测量独特亲和试剂与样品中每种蛋白质的结合结果。每种亲和试剂以0.25的概率结合靶向的三聚体,并以0.25的概率结合与靶向的三聚体最相似的四个三聚体。其他脱靶三聚体以2.45x10-8和0.25*1.5-x中数值较大的概率结合,其中x是靶向的三聚体与其本身的相似度减去脱靶三聚体与三聚体靶标的相似度。三聚体序列之间的相似度可以通过,例如,对于三个序列位置中每一个处的氨基酸对,将blosum62系数求和来计算。如实施例18所述,使用贪婪方法选择亲和试剂三聚体靶标,以针对人类蛋白质组进行优化。使用已知sav序列的蛋白质鉴定性能使用仅包含蛋白质样品中20,374种规范蛋白质的序列的数据库,对样品的结合结果进行删截的蛋白质推断。由于用于蛋白质推断的数据库缺少样品中12,827种sav蛋白质的序列,因此当样品中所有潜在sav的序列未知时,该分析的结果指示性能。通过以这种方式进行蛋白质推断,对样品中96.0%的蛋白质鉴定了正确的sav蛋白质家族,错误发现率为1%。如本文所用的,术语“sav蛋白质家族”通常是指包括规范蛋白质序列和该规范蛋白质序列的所有sav的一组序列。如果推断的蛋白质身份与所分析的蛋白质在同一sav蛋白质家族内,则可以鉴定该蛋白质的正确sav蛋白质家族。使用已知sav序列的蛋白质鉴定性能当使用由样品中所有蛋白质序列(规范蛋白质序列和sav蛋白质序列)组成的序列数据库进行蛋白质推断时,对于样品中27.1%的蛋白质鉴定出正确的蛋白质序列,错误发现率为1%。如果鉴定出蛋白质的确切序列,则鉴定了该蛋白质的正确蛋白质序列。此外,对于样品中96.1%的蛋白质,鉴定出正确的sav蛋白质家族。sav蛋白质家族和确切蛋白质序列的鉴定率之间的差异可能是由于难以解析规范蛋白质序列和极为相似的sav序列的身份而引起的。使用先验定义的sav蛋白质家族的蛋白质鉴定性能通过直接计算sav蛋白质家族的概率可以提高sav蛋白质家族的鉴定率。对于所测量的单个蛋白质,该蛋白质是sav蛋白质家族的成员的概率可以通过将组成该家族的各个蛋白质序列的每个概率相加来计算。将对于所分析的蛋白质具有最高概率的sav蛋白质家族指定为sav蛋白质家族鉴定。当以这种方式计算sav蛋白质家族概率时,以1%的错误发现率为样品中96.5%的蛋白质鉴定正确的sav蛋白质家族。相比之下,当不直接计算蛋白质家族概率时,以1%的错误发现率为样品中96.1%的蛋白质鉴定正确的sav蛋白质家族。实施例23:删截的蛋白质推断对来自物种混合物的包含蛋白质的样品的性能在一些情况下,蛋白质样品可以包含来自多个物种中的每一种的蛋白质。蛋白质样品可以包含源自诸如化石等外部来源的蛋白质。在一些实施方案中,蛋白质样品可以包含合成的、修饰的或工程化的蛋白质,如重组蛋白,或通过体外转录和翻译合成的蛋白质。在一些实施方案中,合成的、修饰的或工程化的蛋白质可以包含非天然序列(例如,源自crispr-cas9修饰或其他人工基因构建体)。例如,每个物种可以是动物,如哺乳动物(例如,人、小鼠、大鼠、灵长类动物或猿猴)、农场动物(生产牛、奶牛、家禽、马、猪等)、竞技动物、伴侣动物(例如,宠物或支持动物);植物、原生生物、细菌、病毒或古生物。在该实施例中,来自小鼠肿瘤异种移植模型的样品可以包含大量的小鼠和人类来源的蛋白质。为了确定蛋白质推断对具有来自进行蛋白质推断的物种混合物的蛋白质的样品的性能,对由2,000种独特的小鼠蛋白质和2,000种独特的人类蛋白质组成的蛋白质集合进行了亲和试剂结合分析。人类蛋白质和小鼠蛋白质均从各自物种的uniprot参考蛋白质组中的规范swiss-prot序列条目的集合中随机选择。使用384个亲和试剂循环分析该样品,每个循环测量独特亲和试剂与样品中每种蛋白质的结合结果。每种亲和试剂以0.25的概率结合靶向的三聚体,并以0.25的概率结合与靶向的三聚体最相似的四个三聚体。其他脱靶三聚体以2.45x10-8和0.25*1.5-x中数值较大的概率结合,其中x是靶向的三聚体与其本身的相似度减去脱靶三聚体与三聚体靶标的相似度。三聚体序列之间的相似度可以通过,例如,对于三个序列位置中每一个处的氨基酸对,将blosum62系数求和来计算。如实施例18所述,使用贪婪方法选择亲和试剂三聚体靶标,以针对人类蛋白质组进行优化。当使用仅包含来自人类蛋白质组的候选蛋白质的序列(uniprot人类参考蛋白质组中的规范swiss-prot序列条目)的数据库对混合物样品进行蛋白质推断时,结果显示没有鉴定出样品中的蛋白质(例如,鉴定率为0%),错误发现率阈值低于1%。相比之下,当使用包含来自人类蛋白质组和小鼠蛋白质组的候选蛋白质的序列的数据库对混合物样品进行蛋白质推断时,以低于1%的错误发现率阈值鉴定出样品中85.3%的蛋白质。这种性能差异表明,对于包含来自多个物种的蛋白质的样品(例如,混合物样品),当使用包含来自混合物样品中代表的所有物种的候选蛋白质的序列的数据库进行蛋白质推断分析时,蛋白质鉴定性能得到显著改善。实施例24:针对靶向的一组蛋白质的亲和试剂集的设计可以设计为鉴定样品中蛋白质的特定子集而优化的一组亲和试剂。例如,与使用为鉴定整个蛋白质组而优化的集合相比,可以使用亲和试剂的最佳集合以较少的亲和试剂结合循环来鉴定特定一组靶蛋白质。在该实施例中,为了最佳地鉴定25种人类蛋白质生成了一组亲和试剂,这些蛋白质是对癌症免疫疗法治疗的临床反应的潜在生物标志物。表11中列出了靶向组中的蛋白质。表11:针对癌症免疫疗法的反应的靶向组中包括的蛋白质为了生成为鉴定完整蛋白质组而优化的一组亲和试剂,应用如实施例18所述的贪婪选择方法。这组亲和试剂可以被称为“蛋白质组优化的”亲和试剂集。为了生成为鉴定表11中的蛋白质而优化的一组亲和试剂,进行实施例18中步骤4)i)的修改形式,其中,不是通过对通过蛋白质推断确定的每种蛋白质的正确蛋白质鉴定的每个概率进行求和来计算候选亲和试剂的得分,而是通过仅对靶向组中的蛋白质的正确蛋白质鉴定的每个概率进行求和来计算候选亲和试剂的得分。该亲和试剂集可以被称为“组优化的”亲和试剂集。在包含来自uniprot的swiss-prot人类参蛋白质组中每种独特的规范蛋白质(20,374种蛋白质)的人类蛋白质组样品上测试蛋白质组优化的和组优化的亲和试剂组的性能。该样品包括靶组中的所有25种蛋白质。这两个亲和试剂集均用于分析蛋白质样品,并使用删截的推断对样品中的每种蛋白质生成蛋白质鉴定。表12中示出了通过蛋白质组优化的和组优化的亲和试剂集鉴定的靶向组蛋白质的数目。为了将靶向组蛋白质计数为成功的鉴定,它必须以低于1%的错误发现率存在于样品中鉴定的所有蛋白质的列表中。用不同数目的亲和试剂循环进行鉴定。例如,150个亲和试剂循环表明蛋白质推断是在数据集上进行的,该数据集包含用来自蛋白质组优化的或组优化的集合的前150种亲和试剂进行的分析,每种亲和试剂均在单独的循环中进行分析。表12:针对25种靶蛋白质的靶组的蛋白质鉴定性能表12中显示的结果表明,应用组优化的亲和试剂成功地提高了靶向组蛋白质的鉴定率。表13示出了对于组优化的和蛋白质组优化的亲和试剂集,以低于1%的错误发现率鉴定的所有蛋白质的百分比。表13:对于样品中的所有蛋白质的蛋白质鉴定性能表13中显示的结果表明,可以生成组优化的亲和试剂集,以改善鉴定特定靶向组中的一组蛋白质的性能。然而,可能会遇到折衷,其中所得的组优化的亲和试剂集对于鉴定靶向组之外的蛋白质而言可能是次优的,如通过表13中组优化的试剂的总蛋白质鉴定率降低所表明的。实施例25:使用单独氨基酸的存在、计数或顺序的检测,蛋白质推断的性能本文描述的蛋白质推断方法可以应用于蛋白质和肽中的特定氨基酸的测量。例如,可以对蛋白质进行测量,其指示蛋白质或肽中是否存在氨基酸(二进制)、蛋白质或肽中的氨基酸计数(计数)或蛋白质中的氨基酸顺序(顺序)。在该实施例中,通过一系列反应修饰蛋白质,每个反应选择性地修饰特定的氨基酸。这一系列反应中的每个反应的反应效率在0到1之间,表明该反应成功地修饰蛋白质中任何单个氨基酸底物的概率。对蛋白质样品进行此类修饰反应后,可以检测是否存在选择性修饰的氨基酸,可以检测选择性修饰的氨基酸的计数,并且/或者可以检测蛋白质内特定的一组选择性修饰的氨基酸的顺序。从氨基酸的存在与否测量进行的检测为了从指示是否存在氨基酸的一系列二进制测量生成蛋白质鉴定,可以将概率pr(检测到氨基酸存在|蛋白质)表示为1-(1–raa)caa,其中raa是该氨基酸的反应效率,而caa是该氨基酸在该蛋白质中出现的次数的计数。概率pr(未检测到氨基酸存在|蛋白质)可以被表示为1–pr(检测到氨基酸存在|蛋白质)。如果进行一系列多个氨基酸检测测量,则可以将概率相乘以确定给定候选蛋白质的完整一组n个测量的概率,其被表示为:pr(结果集|蛋白质)=pr(氨基酸1的测量结果|蛋白质)*pr(氨基酸2的测量结果|蛋白质)*...pr(氨基酸n的测量结果|蛋白质)。对于所测量的蛋白质,特定候选蛋白质为正确鉴定的概率可以被表示为其中(结果集|蛋白质i)是由p种蛋白质组成的蛋白质序列数据库中每种可能的蛋白质的结果集的概率之和。从氨基酸的计数测量进行的检测为了从氨基酸的一系列计数测量生成蛋白质鉴定,概率pr(氨基酸计数测量|蛋白质)可以被表示为其中raa是该氨基酸的反应效率,caa是该氨基酸在该蛋白质中出现的次数的计数,且m是对该蛋白质中的该氨基酸测得的计数。如果m>caa,则返回概率0。如果进行一系列多个氨基酸计数测量,则可以将概率相乘以确定给定候选蛋白质的完整一组n个测量的概率,其被表示为:pr(结果集|蛋白质)=pr(氨基酸1的测量结果|蛋白质)*pr(氨基酸2的测量结果|蛋白质)*...pr(氨基酸n的测量结果|蛋白质)。对于所测量的蛋白质,特定候选蛋白质为正确鉴定的概率可以被表示为其中(结果集|蛋白质i)是由p种蛋白质组成的蛋白质序列数据库中每种可能的蛋白质的结果集的概率之和。从氨基酸的顺序测量进行的检测在一些实施方案中,可以测量蛋白质中选择性修饰的氨基酸的顺序。例如,如果对氨基酸i和n进行修饰和测量,则具有序列tinyprtein的蛋白质可生成测量结果inin。类似地,在氨基酸修饰和/或测量的子集不成功的情况下,同一蛋白质可生成测量结果inn或iin。概率pr(测量结果|蛋白质)可以表示为pr(氨基酸计数|蛋白质)*numorder。其中raai是氨基酸i的反应效率,mi是测得氨基酸i的次数(例如,在inn的测量结果中,n测得2次),caai是氨基酸i在候选蛋白质序列中出现的次数,且氨基酸1至l都是在蛋白质中测得的独特的氨基酸(例如,i和n,对于测量结果inin)。如果针对任何特定氨基酸测得的计数均大于该氨基酸在该蛋白质候选物序列中出现的次数,则将概率pr(氨基酸计数|蛋白质)设置为零。numorder是可以从蛋白质序列生成特定结果的方式的数目。例如,可以通过以下方式从蛋白质tinyprtein生成in的测量结果:{tinyprtein,tinyprtein,tinyprtein},因此对于该特定结果和蛋白质序列,numorder为3。请注意,在不可能从蛋白质生成特定结果(例如,无法从蛋白质tinyprtein生成inni的测量结果)的情况下,numorder的值为零。对于所测量的蛋白质,特定候选蛋白质为正确鉴定的概率可以被表示为其中(测量结果|蛋白质i)是由p种蛋白质组成的蛋白质序列数据库中每种可能的蛋白质的测量结果的概率之和。在(测量结果|蛋白质i)等于零的情况下,将候选蛋白质的概率设置为零。使用用于选择性修饰和检测氨基酸k、d、c和w的试剂集合进行的蛋白质鉴定的性能在图22和表14中示出。如x轴所示,反应以不同的效率进行。每个条的阴影指示检测方式(“二进制”、“计数”或“顺序”分别指示检测氨基酸的存在与否、氨基酸计数或氨基酸顺序)。每个条的高度指示在低于1%的错误发现率下鉴定出的样品中蛋白质的百分比。所测样品是含有1,000种蛋白质的人类蛋白质样品。结果表明,可使用氨基酸顺序测量以0.9或更高的反应效率鉴定出大量蛋白质。如果使用氨基酸计数的测量,则可以以0.99或更高的反应效率鉴定出大量蛋白质。没有一个所测试的场景中,对氨基酸的存在与否的测量足以生成蛋白质检测。表14:使用4种氨基酸(k、d、c和w)的选择性修饰和检测,蛋白质鉴定的性能实验名称实验类型灵敏度反应效率kdwc二进制0.5二进制00.5kdwc计数0.5计数10.5kdwc顺序0.5顺序58.10.5kdwc二进制0.9二进制00.9kdwc计数0.9计数10.10.9kdwc顺序0.9顺序94.90.9kdwc二进制0.99二进制00.99kdwc计数0.99计数76.40.99kdwc顺序0.99顺序95.40.99kdwc二进制0.999二进制00.999kdwc计数0.999计数92.20.999kdwc顺序0.999顺序95.20.999如图23所示,扩大了用于选择性修饰和检测氨基酸的试剂的集合,以包括r、h、k、d、e、s、t、n、q、c、g、p、a、v、i、l、m、f、y和w这20种氨基酸。检测方式由线条的阴影表示,而反应效率在x轴上表示。y轴表示在低于1%的错误发现率下在样品中鉴定的蛋白质的百分比。图23和表15中示出的结果表明,如果反应效率大于约0.6并且使用氨基酸计数的测量,则这样的试剂集合对于蛋白质鉴定非常有效。然而,如果使用氨基酸存在或不存在的测量代替氨基酸计数的测量,则只能鉴定出很小百分比的蛋白质。表15:使用20种氨基酸(r、h、k、d、e、s、t、n、q、c、g、p、a、v、i、l、m、f、y和w)的选择性修饰和检测,蛋白质鉴定的性能图24示出了使用氨基酸顺序的测量的蛋白质鉴定的性能,其中以x轴上示出的检测概率(等于反应效率)测量氨基酸。y轴表示在低于1%的错误发现率下鉴定出的样品中蛋白质的百分比。采用在每种蛋白质的n末端25、50、100或200个氨基酸处测得的氨基酸顺序的测量来进行实验,并且候选蛋白质序列数据库分别由uniprot参考人类蛋白质数据库中每个规范蛋白质序列的前25、50、100或200个氨基酸组成。图24和表16中示出的性能表明,对于约0.3的检测概率,对每种蛋白质的至少前100个氨基酸进行测序是最佳的。对于高于约0.6的检测概率,对前25个或更多个氨基酸进行测序似乎已足够。表16:使用氨基酸顺序的测量,蛋白质鉴定的性能图25示出了各种方法对由1,000种独特人类蛋白质组成的样品的胰蛋白酶消化物的性能。该样品包含长度大于12的所有完全胰蛋白酶消化的肽,没有丢失因这些蛋白质引起的切割。黑线表示使用所有氨基酸的顺序的测量进行蛋白质鉴定时的性能,这些氨基酸以不同的检测概率(相当于反应效率)进行测量。浅线表示当在不同的检测概率(相当于反应效率)下仅测量氨基酸k、d、w和c的顺序时的性能。用于推断的序列数据库包含长度大于12的每个完全胰蛋白酶消化的肽的序列,没有丢失因这些蛋白质引起的切割,这些序列来源于从uniprot下载的人类参考蛋白质组数据库中的每个规范蛋白质序列。实线表示在低于1%的错误发现率下鉴定的样品中的肽的百分比。虚线表示在低于1%的错误发现率下鉴定的样品中的蛋白质的百分比。如果在低于1%的错误发现率下鉴定出具有对该蛋白质独特的序列的肽,则该蛋白质得到鉴定。这些结果表明,仅测量氨基酸k、d、w和c的顺序可能不足以从胰蛋白酶消化样品中检测蛋白质。此外,以等于或高于约0.5的检测概率(等于反应效率)测量所有氨基酸的顺序足以鉴定胰蛋白酶消化物中的大多数蛋白质。计算机控制系统本公开提供了计算机控制系统,其被编程用于实现本公开的方法。图10示出了计算机系统1001,其被编程或以其他方式配置为:接收样品中未知蛋白质的经验测量的信息,将经验测量的信息与包含对应于候选蛋白质的多个蛋白质序列的数据库进行比较,生成候选蛋白质生成观察到的测量结果集的概率,以及/或者生成在样品中正确鉴定候选蛋白质的概率。计算机系统1001可以调节本公开的方法和系统的各个方面,例如,接收样品中未知蛋白质的经验测量的信息,将经验测量的信息与包含对应于候选蛋白质的多个蛋白质序列的数据库进行比较,生成候选蛋白质生成观察到的测量结果集的概率,以及/或者生成在样品中正确鉴定候选蛋白质的概率。计算机系统1001可以是用户的电子设备,或者是相对于该电子设备位于远程的计算机系统。该电子设备可以是移动电子设备。计算机系统1001包括中央处理单元(cpu,本文中也称为“处理器”和“计算机处理器”)1005,中央处理单元1405可以是单核或多核处理器,或者用于并行处理的多个处理器。计算机系统1001还包括存储器或存储器位置1010(例如,随机存取存储器、只读存储器、闪速存储器)、电子存储单元1015(例如,硬盘)、用于与一个或多个其他系统通信的通信接口1020(例如,网络适配器)和外围设备1025,如高速缓冲存储器、其他存储器、数据存储和/或电子显示适配器。存储器1010、存储单元1015、接口1020和外围设备1025通过诸如主板的通信总线(实线)与cpu1005通信。存储单元1015可以是用于存储数据的数据存储单元(或数据存储库)。计算机系统1001可以借助于通信接口1020可操作地耦合至计算机网络(“网络”)1030。网络1030可以是因特网、互联网和/或外联网,或者与因特网通信的内联网和/或外联网。网络1030在一些情况下是电信和/或数据网络。网络1030可以包括能够实现分布式计算如云计算的一个或多个计算机服务器。例如,一个或多个计算机服务器可使得能够进行经网络1030的云计算(“云”),以执行本公开的分析、计算和生成的各个方面,例如,接收样品中未知蛋白质的经验测量的信息,将经验测量的信息与包含对应于候选蛋白质的多个蛋白质序列的数据库进行比较,生成候选蛋白质生成观察到的测量结果集的概率,以及/或者生成在样品中正确鉴定候选蛋白质的概率。这样的云计算可以由诸如amazonwebservices(aws)、microsoftazure、googlecloudplatform和ibm云等云计算平台提供。在一些情况下,网络1030借助于计算机系统1001可以实现对等网络,这可以使得耦合至计算机系统1001的设备能够起到客户端或服务器的作用。cpu1005可以执行一系列可以在程序或软件中体现的机器可读指令。所述指令可以存储在诸如存储器1010的存储器位置中。所述指令可被导向cpu1005,其随后可对cpu1005进行编程或以其他方式进行配置,以实现本公开的方法。由cpu1005执行的操作的实例可以包括获取、解码、执行和写回。cpu1005可以是电路如集成电路的一部分。系统1001中的一个或多个其他组件可被包括在该电路中。在一些情况下,该电路是专用集成电路(asic)。存储单元1015可以存储文件,如驱动程序、文库和保存的程序。存储单元1015可以存储用户数据,例如,用户偏好和用户程序。在一些情况下,计算机系统1001可以包括位于计算机系统1001外部(诸如位于通过内联网或因特网与计算机系统1001通信的远程服务器上)的一个或多个附加数据存储单元。计算机系统1001可以通过网络1030与一个或多个远程计算机系统通信。例如,计算机系统1001可与用户的远程计算机系统进行通信。远程计算机系统的实例包括个人计算机(例如,便携式pc)、平板或平板pc(例如,ipad、galaxytab)、电话、智能电话(例如,iphone、支持android的设备、)或个人数字助理。用户可以通过网络1030访问计算机系统1001。如本文所述的方法可通过存储在计算机系统1001的电子存储位置上(例如存储器1010或电子存储单元1015上)的机器(例如,计算机处理器)可执行代码来实现。该机器可执行代码或机器可读代码可以以软件的形式提供。在使用期间,该代码可以由处理器1005执行。在一些情况下,该代码可从存储单元1015中检索并存储在存储器1010上,以备处理器1005访问。在一些情况下,可以不包括电子存储单元1015,而将机器可执行指令存储在存储器1010上。可将该代码预编译并配置用于与具有适于执行该代码的处理器的机器一起使用,或者可以在运行过程中对其进行编译。该代码可以以编程语言的形式提供,该编程语言可以被选择为使得该代码能够以预编译或实时编译的方式执行。本文提供的系统和方法的各方面,如计算机系统1001,可以在编程中体现。该技术的各个方面可以被认为是“产品”或“制品”,其一般为在某种类型的机器可读介质中携带或体现的机器(或处理器)可执行代码和/或关联数据的形式。机器可执行代码可以存储在电子存储单元如存储器(例如,只读存储器、随机存取存储器、闪速存储器)或硬盘上。“存储”型介质可以包括计算机、处理器等的任何或全部有形存储器,或其相关模块,如各种半导体存储器、磁带驱动器、磁盘驱动器等,其可以随时为软件编程提供非暂时性存储。软件的全部或部分可以不时地通过因特网或各种其他电信网络进行通信。例如,这样的通信可以使软件能够从一台计算机或处理器加载到另一台计算机或处理器,例如,从管理服务器或主机加载到应用服务器的计算机平台。因此,可以承载软件元件的另一类型的介质包括光波、电波和电磁波,诸如跨越本地设备之间的物理接口、通过有线和光学陆线网络以及经由各种空中链路所使用的。携带这类波的物理元件,如有线或无线链路、光学链路等,也可以被认为是承载软件的介质。除非局限于非暂时性有形“存储”介质,否则如本文所用的诸如计算机或机器“可读介质”等术语是指参与向处理器提供指令以供执行的任何介质。因此,机器可读介质,如计算机可执行代码,可以采取许多形式,包括但不限于有形存储介质、载波介质或物理传输介质。非易失性存储介质包括,例如,光盘或磁盘,如任何计算机中的任何存储设备等,例如可用来实现附图中所示的数据库等。易失性存储介质包括动态存储器,如这样的计算机平台的主存储器。有形传输介质包括同轴电缆、铜线和光纤,包括构成计算机系统内的总线的导线。载波传输介质可以采取电信号或电磁信号或者声波或光波的形式,诸如在射频(rf)和红外线(ir)数据通信期间生成的那些信号或波。因此,计算机可读介质的常见形式包括,例如:软盘、柔性盘、硬盘、磁带、其他任何磁性介质、cd-rom、dvd或dvd-rom、其他任何光学介质、穿孔卡片纸带、其他任何具有孔洞图案的物理存储介质、ram、rom、prom和eprom、flash-eprom、其他任何存储器芯片或匣盒、传输数据或指令的载波、传输这类载波的线缆或链路,或者计算机可以从中读取编程代码和/或数据的其他任何介质。这些计算机可读介质形式中的许多形式可以参与将一个或多个指令的一个或多个序列运载到处理器以供执行。计算机系统1001可以包括电子显示器1035或与电子显示器235通信,电子显示器235包括用于提供例如算法、结合测量数据、候选蛋白质和数据库的用户选择的用户界面(ui)1040。ui的实例包括但不限于图形用户界面(gui)和基于网络的用户界面。本公开的方法和系统可以通过一个或多个算法来实现。算法可以通过软件在由中央处理单元1005执行时实现。例如,该算法可以接收样品中未知蛋白质的经验测量的信息,将经验测量的信息与包含对应于候选蛋白质的多个蛋白质序列的数据库进行比较,生成候选蛋白质生成观察到的测量结果集的概率,以及/或者生成在样品中正确鉴定候选蛋白质的概率。虽然本文已经显示并描述了本发明的优选实施方案,但是对于本领域技术人员显而易见的是,这些实施方案仅以示例的方式提供。并非打算用本说明书中提供的具体实例来限制本发明。尽管已经参照上述说明书对本发明进行了描述,但并不意味着对本文实施方案的描述和说明以限制性的意义来解释。在不脱离本发明的情况下,本领域技术人员现将想到许多变化、改变和替换。此外,应当理解,本发明的所有方面均不限于本文所阐述的具体描述、配置或相对比例,其取决于多种条件和变量。应当理解,在实施本发明的过程中可以采用本文所述的本发明实施方案的各种替代方案。因此可以预期,本发明还应涵盖任何这类替代、改变、变化或等同物。旨在以所附权利要求书限定本发明的范围,由此涵盖在这些权利要求范围内的方法和结构及其等同物。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1