含有PLK1活化抑制剂作为活性成分的用于预防或治疗癌症的药物组合物的制作方法
本发明涉及一种用于预防、治疗或减轻癌症的组合物,其包含作为活性成分的polo样激酶1(plk1)抑制剂及其药学上可接受的盐,所述polo样激酶抑制剂通过结合到plk1的polo盒结构域(pbd)来抑制蛋白质的活性。
背景技术:
有丝分裂是指其中所有细胞的成分被分成两个新细胞的分裂。当有丝分裂开始时,染色体凝聚、纺锤极体分离并迁移到两个极,中间的染色体列队,最后所有细胞成分都发生分离。当细胞开始分裂时,染色体应形成特定的结构以进行有效的双向分离,而此类有丝分裂特异性的染色体结构通常取决于三种多蛋白复合体、两种缩素蛋白复合体和一种黏连蛋白复合体。黏连蛋白复合体与其姐妹染色单体结合,而凝聚素复合体使染色体内部变粗和变短。每个缩素蛋白复合体由两个atpase亚基异二聚体、染色体的结构维持(smc2和smc4)和三个非smc调节亚基组成。这三个调节组分的独特集合将定义每个缩素蛋白复合体,并且例如,ncap-d2、ncap-g和ncap-h是缩素蛋白复合体i的组成元素,而ncap-d3、ncap-g2和ncap-h2是缩素蛋白复合体ii的组成元素。smc2和4亚基异二聚体是利用其atp酶活性进行有丝分裂dna凝聚的交联剂。ncap-h和ncap-h2是连接smc亚基异二聚体和其他两个调节亚基的kleisin蛋白,并且ncapg、ncapg2、ncad2和ncapd3是包含对应于可变框架的heat重复结构域的每个缩素蛋白复合体的调节亚基。缩素蛋白复合体i在间期过程中位于胞质溶胶中,在核膜塌陷后立即通过极光激酶b掺入染色体中,并保留在染色体臂中直至胞质分裂过程。相反,缩素蛋白复合体ii导致染色体在细胞分裂过程中被浓缩,同时即使在间期中也保留在细胞核中,并且缩素蛋白复合体ii通过蛋白磷酸酶2a(pp2a)催化活性非依赖性功能被掺入染色体中。包括染色体脱模、染色质重塑和复杂体i凝聚在内的各种其他作用使染色体凝聚得以维持直至胞质分裂。此外,存在于酵母菌种中的缩素蛋白i是用于真核染色体凝聚的经典缩素蛋白复合体。缩素蛋白ii不仅调节染色体刚性,而且还调节各种细胞活动,例如染色体分离、dna修复、细胞凋亡、姐妹染色单体解析、基因表达调节和组蛋白调节。有意思的是,所有线虫缩素蛋白复合体ii组分的突变型纯合子均表现出异常大小或异质核分布。在人细胞中,缩素蛋白复合体ii的任何组分的缺乏均导致染色体列队或分离的缺陷。关于染色体分离作用,最近的报告报道了ncapd3有助于plk1向染色体癌的迁移。
染色体分离是向每个子细胞传递保守的遗传信息的最重要过程。染色体分离的第一步是微管与动粒的附着。动粒是一种蛋白质复合装配体,其对应于姐妹染色单体所结合的染色体的着丝粒。微管-动力结合需要通过各种蛋白质的精细调节以用于进行精确的双向相互作用。这些过程通过调节适当的时间并通过由激酶和磷酸酶(例如aurorab和/或pp2a磷酸酶)的精细磷酸化梯度对此类激酶/磷酸酶底物激活进行定位来执行。
在此类过程中,已知作为一种丝氨酸/苏氨酸激酶的polo样激酶1(plk1)对于染色体分离和染色体完整性是必不可少的。plk1介导微管附着到动粒的初始阶段。其根据有丝分裂期间微管的迁移而不同地位于染色体、动粒和中体中,并且从前中期到中期位于动粒中,直到完成中期板中的染色体列队。此外,当每个动粒不正确地附着到微管时,位于动粒上的plk1使bubr1磷酸化,以等待后期的开始。即,plk1在细胞增殖中起关键作用,作用于有丝分裂和dna损伤修复的各种过程。
从结构上讲,plk1是一种磷酸化酶,并由具有磷酸化活性的激酶位点和识别底物的polo-盒结构域(pbd)组成,与其他磷酸化酶不同。激酶位点和pbd位点形成如下结构,其中当底物彼此不竞争时磷酸化酶活性受到干扰,并且当底物与pbd结合时,在打开结构时激酶位点和pbd位点具有磷酸化活性。因此,已知大多数底物与pbd结合并被磷酸化,但是当产生抑制pbd或kd的一种功能的突变体时,似乎仍保留了细胞的plk1功能,因此已知即使底物与pbd结合,也存在与kd功能无关的底物和功能。据报道,在许多癌中,在细胞分裂过程中起多种作用的plk1的表达增加,特别是由于该表达对癌细胞是致命的,因此已知抑制plk1的活性通过维持对细胞异常的单轴纺锤丝状态来诱导细胞凋亡。因此,在各种研究中进行了靶向plk1的抗癌药物开发研究。在最初的研究阶段,plk1抑制剂作为一种抑制plk1的磷酸化酶活性的atp竞争性抑制剂来开发,并且目前在临床实践中作为plk1抑制剂的大多数药物是此类n端atp结合位点抑制剂。然而,由这些抑制剂靶向以抑制磷酸化活性的激酶位点示出与其他plk家族或其他磷酸化酶的相似性,这使得其难以选择性靶向plk1,并且即使在各种恶性肿瘤中均示出治疗效果,但由于药效学问题,其临床应用也受到限制。
因此,本发明人从先前的研究中证实,缩素蛋白复合体ii的亚基ncapg2通过结合到plk1的pbd位点影响动粒中plk1的定位和底物磷酸化活性,并通过实际研究ncapg2的pbd结合位点并基于此,肽被鉴定为plk1抑制剂。然而,该肽具有局限性,例如对自溶作用的不稳定性和低细胞内渗透性。
因此,利用肽和plk1pbd的结合结构进行分子建模的设计以及通过筛选低分子量化合物来发现对plk1具有高结合强度的有效、低毒性和低分子量化合物,结果具有抑制癌细胞生长的能力已经成为主要挑战,并且对此进行了研究(韩国公开专利10-2016-0045957),但是研究仍然不充分。
技术实现要素:
[技术问题]
为了解决如上所述的本发明的问题,本发明人筛选了340,000种化合物的文库,以便通过根据ncapg2衍生肽与plk1的pbd的结合结构设计分子模型来发现对plk1的pbd具有高结合亲和力和低毒性的低分子量化合物,从而鉴定出有效的plk1抑制化合物。
另外,本发明人发现,该化合物在细胞水平上有效地阻碍了各种癌细胞系的生长,从而基于此完成了本发明。
因此,本发明的目的是提供用于预防、减轻或治疗癌症的组合物,其包含由以下化学式1或2表示的化合物或其药学上可接受的盐作为活性成分。
[化学式1]
[化学式2]
然而,本发明要解决的技术问题不限于上述问题,并且本领域技术人员从以下描述中可以清楚地理解未提及的其他问题。
[技术方案]
为了实现该目的,本发明提供了一种用于预防或治疗癌症的药物组合物,其包含由以下化学式1或2表示的化合物或其药学上可接受的盐作为活性成分。
[化学式1]
[化学式2]
(在化学式1或2中,r1为h、烷基或-cnh2ncooh(n为1至4的整数),r2为h、烷基、-cmh2mcn、-cmh2mor5或-cph2p(ch(oh))qr6,r5为被一个或多个c1-3烷基取代的苯基,r6为h、烷基或-oph2o3,m为2至4的整数,p为1至3的整数,并且q为2至4的整数,r3为h、卤素、-nh2、烷基或-ch=o,并且r4为h、烷基、-cooh或-cx3,并且x为卤素)
此外,本发明提供了一种用于缓解癌症的保健功能食品组合物,其包含由以下化学式1或2表示的化合物或其药学上可接受的盐作为活性成分。
作为本发明的示例性实施方案,在化学式1或2中,
r1可以为h、-ch3或-ch2cooh,
r2可以为h、-ch3、-c2h4cn、-ch2(ch(oh))3ch2oh、-ch2(ch(oh))3oph2o3,或
r3可以为h、cl、-nh2、-ch3或-ch=o,并且
r4可以为h、-ch3、-cooh或-cf3。
作为本发明的另一示例性实施方案,由化学式1或2表示的化合物可以选自以下化合物:
2,4-二氧-1,2,3,4-四氢苯并[g]蝶啶-7-羧酸;
10-甲基-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
8-氯-1h,2h,3h,4h-苯并[g]蝶啶-2,4-二酮;
10-甲基-7-(三氟甲基)-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
8-氨基-1,3-二甲基-1h,2h,3h,4h-苯并[g]蝶啶-2,4-二酮;
8-氨基-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
7,8,10-三甲基-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
7,10-二甲基-2,4-二氧-2h,3h,4h,10h-苯并[g]蝶啶-8-甲醛;
4,10-二氢-7,8,10-三甲基-2,4-二氧苯并[g]蝶啶-3(2h)-乙酸;
3-{7,8-二甲基-2,4-二氧-2h,3h,4h,10h-苯并[g]蝶啶-10-基}丙腈;
10-[2-(3-甲基苯氧基)乙基]-7-(三氟甲基)-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
7,8-二甲基-10-[(2s,3s,4r)-2,3,4,5-四羟基戊基]苯并[g]蝶啶-2,4-二酮;以及
[(2r,3s,4s)-5-(7,8-二甲基-2,4-二氧苯并[g]蝶啶-10-基)-2,3,4-三羟基戊基]二氢磷酸酯。
作为本发明的又一示例性实施方案,所述癌症可以是选自下列中的一种或多种肝癌、乳腺癌、血液癌、子宫颈癌和前列腺癌。
作为本发明的又一个示例性实施方案,所述化合物可以结合于polo样激酶1(plk1)的polo盒结构域(pbd)。
作为本发明的又一个示例性实施方案,所述组合物可抑制癌细胞的生长。
作为本发明的又一个示例性实施方案,所述组合物可诱导癌细胞的细胞凋亡。
此外,本发明提供了一种用于预防或治疗癌症的方法,该方法包括:将药物组合物施用于个体,所述药物组合物包含由化学式1或2表示的化合物或其药学上可接受的盐作为活性成分。
此外,本发明提供了药物组合物用于预防或治疗癌症的用途,所述药物组合物包含由化学式1或2表示的化合物或其药学上可接受的盐作为活性成分。
[有利效果]
通过对化合物进行文库筛选以发现具有低毒性同时对plk1的pbd具有高结合亲和力的低分子量化合物,本发明人鉴定出由化学式1或化学式2表示的有效化合物,并且证实了该化合物以低浓度有效结合到plk1的pbd,并显着抑制肝癌、乳腺癌、血液癌、宫颈癌和前列腺癌细胞的生长。
因此,与相关领域中靶向激酶结构域的atp结合位点抑制剂相比,根据本发明的化合物通过选择性结合到plk1的pbd而具有对plk1的高选择性和结合亲和力以及低毒性的优点。
因此,根据本发明的plk1抑制剂化合物可通过抑制各种癌细胞的生长而有效地用作抗癌剂,并且可预期除了其单独给药之外,还通过共同给药表现出与现有开发的抗癌剂的协同作用。
附图说明
图1示出了用于根据本发明的示例性实施方案发现选择性结合到plk1的pbd以抑制plk1的活性的低子量化合物的fp分析方法(荧光偏振竞争测定)的原理。
图2是示出根据本发明的示例性实施方案的化合物的fp测定分析结果和ic50的一组图。
图3是示出根据本发明的示例性实施方案的化合物的fp测定分析结果和ic50的图。
图4是示出根据本发明的示例性实施方案的化合物的fp测定分析结果和ic50的图。
图5a和5b是根据本发明的实施例3,用于测量化合物2(m2)抑制癌细胞生长的能力的图。
图5c是根据本发明的实施例3,用于测量m2和m3变体抑制jimt1细胞的癌细胞生长的能力的一组图。
图6是根据本发明的实施例3,用于测量化合物2(m2)、化合物3(m4)、化合物4(m21)和索拉非尼抑制肝癌细胞生长的能力的一组图。
图7是根据本发明的实施例3,用于测量化合物3(m4)抑制癌细胞生长的能力的一组图。
图8是根据本发明的实施例3,用于测量化合物3(m4)抑制癌细胞生长的能力的一组图。
图9a是根据本发明的实施例3,用于测量化合物5(m23)和化合物6(m25)抑制肝癌细胞生长的能力的一组图。
图9b是根据本发明的实施例3,用于测量m2和m3变体抑制hepg2细胞中癌细胞生长的能力的一组图。
图9c是根据本发明的实施例3,用于测量m2和m3变体抑制snu449细胞中的癌细胞生长的能力的一组图。
图10是根据本发明的实施例3,用于测量仅化合物2(m2)以及化合物2(m2)和bi2536的混合治疗抑制肝癌细胞系的生长的能力。
图11确认了根据本发明的实施例4,在根据本发明的示例性实施方案的化合物治疗期间,位于中心体的r微管蛋白、plk1和染色体(dapi)之间的相互位置关系。
图12是示出根据本发明的实施例4的在染色体臂和中心体中的ncapg2的染色程度的照片和图。
图13是根据本发明的实施例5,在用化合物2(m2)处理的情况下对细胞循环的影响的一组图。
图14a示出了在用m2或bi2536处理之后,hepg2细胞的相对细胞面积。
图14b示出了通过在用m2或bi2536处理的hepg2细胞中进行核染色观察到的图像。
图15a示出了在分别用m2和bi2536处理hepg2细胞之后进行流式细胞术的结果。
图15b是示出在分别增加m2和bi2536的剂量同时处理hepg2细胞后细胞凋亡的结果的图。
图16是根据本发明的实施例8,分别向小鼠腹膜内注射化合物4和dmso之后去除并对肺、心脏、肝脏、肾脏、脾脏和皮肤进行组织病理学分析的一组照片。
图17是分别示出根据本发明的实施例8的肿瘤大小和小鼠体重的变化的一组图。
图18是示出根据本发明的实施例8,去除的癌发生组织的外观的一组照片。
图19是示出根据本发明的实施例8对plk1进行免疫组织化学染色以比较plk1在去除的组织中的表达的照片,plk1本身的表达差异不明显,但是细胞数在有丝分裂期中减少。
图20a示出根据本发明的实施例9,在用m2治疗小鼠之后移植的肿瘤的宏观形态和mri图像。
图20b是使用图20a的mri图像示出移植的肿瘤的体积变化和m2的肿瘤生长减少效果的图。
图20c示出了在根据本发明的实施例9的移植的肿瘤组织中,细胞数在有丝分裂期中减少。
图20d是示出使用图20c中所示的组织病理学观察结果计算的每个治疗组的有丝分裂指数的图。
图21a由mri图像示出了根据本发明的实施例9的用m2治疗小鼠后移植的肿瘤大小的变化。
图21b示出了根据本发明实施例9的用m2治疗小鼠后移植的肿瘤的最终重量。
图21c示出了根据本发明实施例9的用m2治疗小鼠后移植的肿瘤的体积变化。
图22a示出了根据本发明的实施例10的在对照中移植到小鼠中的肿瘤的mri图像。
图22b示出了根据本发明的实施例10的用m2治疗的组中移植到小鼠中的肿瘤的mri图像。
图22c示出了根据本发明的实施例10的用bi2536治疗的组中移植到小鼠中的肿瘤的mri图像。
图22d是比较了根据本发明的实施例10的用m2或bi2536治疗的组中的肿瘤体积、肿瘤重量和体重的变化一组图。
具体实施方式
因为本发明可被修改成各种形式并且包括各种示例性实施方案,因此特定的示例性实施方案将在附图中示出并且在具体实施方式中进行详细描述。然而,该描述并不旨在将本发明限制于特定的示例性实施方案,并且应当理解,属于本发明的精神和技术范围的所有改变、等同物和替代物均包括在本发明中。当确定在描述本发明时相关公知技术的详细描述可能使本发明的主旨模糊不清时,将省略其详细描述。
发明涉及plk1抑制剂及其用途,更具体地讲,涉及对plk1的pbd具有高结合亲和力的低毒性化合物以及用于预防、减轻或治疗癌症的组合物,所述组合物包含所述化合物作为活性成分。在下文中,将详细描述本发明。
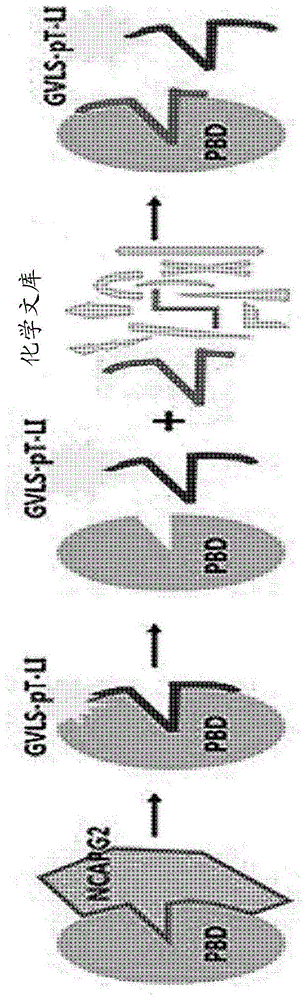
通过先前的研究,本发明人发现以定位于ncapg2的1010位的磷酸化苏氨酸为中心的gvlsptli肽与polo-盒结构域(pbd)结合,该polo-盒结构域是丝氨酸/苏氨酸-蛋白激酶1(plk1)的底物结合位点,并且这种结合触发将纺锤丝定位到染色体中,这对于plk1的有丝分裂期作用而言非常重要。然而,由于存在这样的问题,即在开发肽作为抗癌剂时需要克服诸如肽的不稳定性和低细胞内渗透性之类的局限性,因此在本发明中已尝试模拟该肽的pbd结合结构并且发现了一种低分子量化合物,其能够基于肽和plk1pbd结合位点的晶体结构而竞争性结合到pbd。
因此,在本发明的示例性实施方案中,通过计算机分析对340,000种化合物的文库进行初步筛选,得到了700种候选化合物,并且通过对化合物进行fp分析方法发现了有效抑制肽与plk1之间结合的有效化合物,即,plk1抑制剂(参见实施例1和2)。
在本发明的另一个示例性实施方案中,为了研究通过该示例性实施方案最终发现的化合物是否能够实际上抑制各种癌细胞系的生长,在细胞水平用该化合物处理肝癌、乳腺癌、血液癌、子宫颈癌和前列腺癌细胞系后测量细胞。证实了该化合物与治疗浓度成比例地有效抑制肝癌、乳腺癌、血液癌、子宫颈癌和前列腺癌细胞,并且可以证实对正常细胞的相对生长的抑制作用相对较小(参见实施例3)。
在本发明的又一个示例性实施方案中,已证实该化合物作为癌细胞中正常细胞分裂过程所涉及的plk1抑制剂与磷酸化活性的作用不同,并且证实靶向pbd的命中(hit)物质阻碍了plk1本身在细胞中的正常位置以使其确切配偶体的位置不合适,从而显示出在有丝分裂期之前的阶段抑制细胞生长进程的效果(参见实施例4和5)。
在本发明的另一个示例性实施方案中,证实了用m2处理hepg2细胞的结果,出现了细胞的抗增殖作用(参见实施例6)。
在本发明的另一个示例性实施方案中,证实了用m2处理hepg2细胞的结果,凋亡群体增加了(参见实施例7)。
在本发明的另一个示例性实施方案中,证实了化合物的毒性试验和化合物在肝癌异种移植模型中抑制癌症生长的能力(参见实施例8)。
在本发明的另一个示例性实施方案中,证实了化合物在肝癌原位异种移植模型中抑制癌症生长的能力(参见实施例9和10)。
通过该结果,可将根据本发明由以下化学式1或2表示的化合物或其药学上可接受的盐用作各种癌的治疗剂,具体地讲肝癌、乳腺癌、血液癌、子宫颈癌和前列腺癌。
因此,本发明提供了一种用于预防或治疗癌症的药物组合物,其包含由以下化学式1或2表示的化合物或其药学上可接受的盐作为活性成分。
[化学式1]
[化学式2]
在化学式1或2中,
r1为h、烷基或-cnh2ncooh(n为1至4的整数),
r2为h、烷基、-cmh2mcn、-cmh2mor5或-cph2p(ch(oh))qr6,r5为被一个或多个c1-3烷基取代的苯基,r6为h、烷基或-oph2o3,m为2至4的整数,p为1至3的整数,并且q为2至4的整数,
r3为h、卤素、-nh2、烷基或-ch=o,并且
r4为h、烷基、-cooh或-cx3,并且x为卤素。
优选地,在化学式1或2中,
r1可以为h、-ch3或-ch2cooh,
r2可以为h、-ch3、-c2h4cn、-ch2(ch(oh))3ch2oh、-ch2(ch(oh))3oph2o3,或
r3可以为h、cl、-nh2、-ch3或-ch=o,并且
r4可以为h、-ch3、-cooh或-cf3。
此外,更优选地,由化学式1或2表示的化合物可选自以下化合物:
2,4-二氧-1,2,3,4-四氢苯并[g]蝶啶-7-羧酸;
10-甲基-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
8-氯-1h,2h,3h,4h-苯并[g]蝶啶-2,4-二酮;
10-甲基-7-(三氟甲基)-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
8-氨基-1,3-二甲基-1h,2h,3h,4h-苯并[g]蝶啶-2,4-二酮;
8-氨基-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
7,8,10-三甲基-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
7,10-二甲基-2,4-二氧-2h,3h,4h,10h-苯并[g]蝶啶-8-甲醛;
4,10-二氢-7,8,10-三甲基-2,4-二氧苯并[g]蝶啶-3(2h)-乙酸;
3-{7,8-二甲基-2,4-二氧-2h,3h,4h,10h-苯并[g]蝶啶-10-基}丙腈;
10-[2-(3-甲基苯氧基)乙基]-7-(三氟甲基)-2h,3h,4h,10h-苯并[g]蝶啶-2,4-二酮;
7,8-二甲基-10-[(2s,3s,4r)-2,3,4,5-四羟基戊基]苯并[g]蝶啶-2,4-二酮;以及
[(2r,3s,4s)-5-(7,8-二甲基-2,4-二氧苯并[g]蝶啶-10-基)-2,3,4-三羟基戊基]二氢磷酸酯。
在下文中,将概述根据本发明的实施例1和2发现的化合物2,4-二氧-1,2,3,4-四氢苯并[g]蝶啶-7-羧酸及其衍生物。
[表1]
“癌症”是通过本发明的药物组合物预防或治疗的疾病,统称为由具有以下特性的细胞引起的疾病:其中细胞忽略正常的生长极限并分裂和生长的侵袭特性、浸润周围组织的浸润特征和扩散到体内其他部位的转移特性。在本发明中,癌症可以是选自下列中的一种或多种:肝癌、乳腺癌、血液癌、前列腺癌、卵巢癌、胰腺癌、胃癌、结肠直肠癌、脑癌、甲状腺癌、膀胱癌、食道癌、子宫癌和肺癌,并且可以更优选为肝癌、乳腺癌、血液癌、子宫颈癌或前列腺癌,但不限于此。
除非另有说明,否则本文中使用的所有技术和科学术语具有与本发明所属领域的技术人员通常理解的含义相同的含义。因此,例如,术语“烷基”是指通过去除单个原子而衍生自直链或支链饱和烃的一价基团,其具有1至8个碳原子,优选地1至6个碳原子。
“卤素”是指氟、氯、溴和碘。
如本文所用,术语“预防”是指通过施用根据本发明的药物组合物来抑制或延迟癌症发作的所有作用。
如本文所用,术语“治疗”是指通过施用根据本发明的药物组合物来减轻或有利地改变由癌症引起的症状的所有作用。
在本发明中,由药学上可接受的游离酸形成的酸加成盐可用作盐。该酸加成盐得自无机酸,诸如盐酸、硝酸、磷酸、硫酸、氢溴酸、氢碘酸、亚硝酸或亚磷酸,以及无毒有机酸,诸如脂族一元和二元羧酸盐、苯基-取代的链烷酸酯、羟基链烷酸酯和链烷二酸酯、芳族酸、脂族和芳族磺酸。这些药学上无毒的盐包括硫酸盐、焦硫酸盐、硫酸氢盐、亚硫酸盐、亚硫酸氢盐、硝酸盐、磷酸盐、磷酸一氢盐、磷酸二氢盐、偏磷酸盐、焦磷酸盐氯化物、溴化物、碘化物、氟化物、乙酸盐、丙酸盐、癸酸盐、辛酸盐、丙烯酸盐、甲酸盐、异丁酸盐、癸酸盐、庚酸盐、丙炔酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、富马酸盐、马来酸盐、1,4-丁炔二酸盐、1,6-己二酸酯、苯甲酸盐、氯苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、羟基苯甲酸盐、甲氧基苯甲酸盐、邻苯二甲酸盐、对苯二甲酸盐、苯磺酸盐、甲苯磺酸盐、氯苯磺酸盐、二甲苯磺酸盐、苯乙酸盐、苯丙酸盐、苯基丁酸盐、柠檬酸盐、乳酸盐、β-羟基丁酸盐、乙醇酸盐、苹果酸盐、酒石酸盐、甲磺酸盐、丙磺酸盐、萘-1-磺酸盐、萘-2-磺酸盐或扁桃酸盐。
根据本发明的酸加成盐可以通过典型的方法制备,例如,将由化学式1或2表示的化合物溶解在过量的酸水溶液中,并使用与水混溶的有机溶剂(例如,甲醇、乙醇、丙酮或乙腈)使该盐沉淀。此外,酸加成盐还可通过使溶剂或过量的酸从该混合物中蒸发,然后干燥混合物或吸滤沉淀的盐来制备。
另外,可使用碱制备药学上可接受的金属盐。通过例如将化合物溶解在过量的碱金属氢氧化物或碱土金属氢氧化物溶液中,过滤不溶性化合物盐,蒸发滤液,并干燥所得产物来获得碱金属或碱土金属盐。在这种情况下,制备钠盐、钾盐或钙盐作为金属盐是药学上合适的。通过使碱金属或碱土金属盐与合适的银盐(例如,硝酸银)反应获得对应于此的银盐。
根据本发明的药物组合物包含由化学式1或2表示的化合物或其药学上可接受的盐作为活性成分,并且还可包含药学上可接受的载体。药学上可接受的载体通常用于制剂中,并且包括盐水、无菌水、林格氏溶液、缓冲盐水、环糊精、葡萄糖溶液、麦芽糖糊精溶液、甘油、乙醇、脂质体等,但不限于此,并且如果需要,还可包括其他典型的添加剂,例如抗氧化剂和缓冲液。此外,可通过另外添加稀释剂、分散剂、表面活性剂、粘合剂、润滑剂等将组合物配制成可注射制剂,诸如水溶液、悬浮液、乳液、滴丸剂、胶囊剂、颗粒剂或片剂等。关于合适的药学上可接受的载体和制剂,可通过使用remington文献中所公开的方法优选地根据每种成分来配制组合物。本发明的药物组合物在制剂方面没有特别限制,但是可配制成注射剂、吸入剂、皮肤外用剂、口服药物等。
本发明的药物组合物可以根据目标方法口服给药或可胃肠外给药(例如静脉内、皮下、通过皮肤、鼻腔或呼吸道给药),并且给药剂量可根据患者的病症和体重、疾病的严重程度、药物形式以及给药途径和时间而变化,但是可由本领域技术人员适当地选择。
本发明的组合物以药学上有效的量施用。在本发明中,“药学上有效的量”意指足以以适用于医疗的合理的受益/风险比来治疗疾病的量,并且有效剂量水平可根据包括下列的因素来确定:患者的疾病类型、疾病严重程度、药物活性、对药物的敏感性、给药时间、给药途径、排泄率、治疗时间和同时使用的药物、以及医学领域中的其他众所周知的因素。根据本发明的组合物可以单独的治疗剂形式或与其他治疗剂组合来施用,可以与相关技术中的治疗剂顺序或同时地施用,并且可以单剂量或多剂量施用。考虑到所有上述因素,重要的是以可获得最大效果但不具有任何副作用的最小量施用组合物,并且该量可以由本领域技术人员容易地确定。
具体地,本发明的组合物的有效量可根据患者的年龄、性别和体重而变化,并且通常,每天或每1mg组合物0.001至150mg,优选0.01至100mg组合物。每天或每隔一天,可施用0.001至150mg组合物/1kg体重,并且优选地0.01至100mg组合物/1kg体重,或者每天一至三次地施用0.001至150mg组合物/1kg体重,并且优选地0.01至100mg组合物/1kg体重。然而,由于有效量可根据给药途径、肥胖的严重程度、性别、体重、年龄等而增加或减少,因此该剂量不旨在以任何方式限制本发明的范围。
作为本发明的另一方面,本发明提供了一种用于缓解癌症的保健功能食品组合物,其包含由化学式1或2表示的化合物或其药学上可接受的盐作为活性成分。
本发明中使用的术语“缓解”是指至少减小与待治疗的病症相关的参数,例如症状的程度的所有作用。
根据本发明的食品组合物可通过将活性成分直接添加到食品中来使用,或者可与其他食品或食品成分一起使用,但是可以通过典型方法适当地使用。活性成分的混合量可根据其使用目的(用于预防或减轻)来适当地确定。通常,当制备食品或饮料时,基于原料计,本发明的组合物的添加量为15重量%或更少,优选为地10重量%或更少。然而,出于保健和卫生目的或为了健康管理目的而长期摄入时,该量可以低于上述范围。
除了本发明的保健功能食品组合物以指定比率包含活性成分作为必要成分之外,其他成分没有特别限制,并且本发明的食品组合物可包含各种调味剂、天然碳水化合物等作为典型饮料中的附加成分。上述天然碳水化合物的示例包括典型的糖,诸如单糖,例如葡萄糖、果糖等;二糖,例如麦芽糖、蔗糖等;和多糖,例如糊精、环糊精等,以及糖醇诸如木糖醇、山梨糖醇和赤藓糖醇。作为除了上述调味剂以外的调味剂,可有利地使用天然调味剂(甜蛋白、甜叶菊提取物(例如莱鲍迪甙a、甘草甜素等)和合成调味剂(糖精、阿斯巴甜等)。天然碳水化合物的比例可通过本领域技术人员的选择适当地确定。
除了添加剂之外,本发明的保健功能食品组合物还可包含各种营养素、维生素、矿物质(电解质)、调味剂诸如合成调味剂和天然调味剂、着色剂和填充剂(奶酪、巧克力等)、果胶酸及其盐、藻酸及其盐、有机酸、保护性胶体增稠剂、ph调节剂、稳定剂、防腐剂、甘油、醇、碳酸化饮料中使用的碳酸化剂等。这些成分可以单独使用或以其组合使用。这些添加剂的比率也可以由本领域技术人员适当地选择。
在下文中,将提出有助于理解本发明的优选实施例。然而,提供以下实施例仅为了更容易理解本发明,并且本发明的内容不受以下实施例的限制。
实施例1.使用荧光偏振(fp)方法进行化合物筛选
通过先前的研究,本发明人发现以位于ncapg2的1010位的磷酸化苏氨酸为中心的gvlsptli肽与polo-盒结构域(pbd)结合并溶解该结合的晶体结构,该polo-盒结构域是丝氨酸/苏氨酸蛋白激酶1(plk1)的底物结合位点,并且该结合触发纺锤丝的结合位点到染色体中,这对于plk1的有丝分裂期作用而言是非常重要的。基于这些研究结果,本发明人模拟了肽的pbd-结合结构,并试图发现能够竞争性结合到pbd的低分子量化合物。
因此,韩国化学研究所通过计算机分析对340,000种化合物的文库进行了初步筛选,并对从中获得的700种候选化合物进行了实验。
出于该目的,通过将在溶液中纯化的plk1的pbd位点和结合fitc荧光的肽(fitc标记的1010pt(gvlsptli-nh2))的缀合物与待筛选的低分子量化合物混合来进行荧光偏振竞争测定。分析方法的原理在图1中示出,并且如图1所示,测量原理是,在荧光缀合的肽结合到plk1的pbd结构域的状态下,当能够竞争性结合到相同结合位点的低分子量化合物结合到结合位点时,肽从plk1分离时荧光减少的程度来测量低分子量化合物与plk1的结合强度。
更具体地讲,通过在每种浓度下制备4μmplk1-pbd蛋白、10nm肽(fitc标记的1010pt(gvlsptli-nh2))和20μm候选化合物在室温下将反应进行30分钟后,将各个组分放入黑色的96孔板中进行混合,使用infinitef200pro(tecangroupltd,switzerland)测量荧光偏振(mp)值。通过相同的方法进行三次该实验,求出平均值,并将激发波长和发光波长分别设定为485nm和535nm。
通过上述方法筛选候选化合物的结果是,发现示出荧光偏振值为180【该值显着低于在未添加化合物的情况下(当仅添加fitc标记的1010pt肽和plk1-pbd蛋白时)测量为约300的荧光偏振值】的化合物,即2,4-二氧-1,2,3,4-四氢苯并[g]蝶啶-7-羧酸,该化合物被确定为命中化合物,并且进行以下实验。
实施例2.命中化合物和衍生物化合物的ic50测量
通过对该化合物和通过一级和二级化合物筛选发现的命中化合物及其各种衍生物进行实施例1中所示的fp测定法分析该化合物的ic50。出于该目的,使用gst树脂分离与gst-标签结合的靶蛋白,并通过最终进行凝胶过滤来获得15mg/ml的与gst-标签结合的纯靶蛋白。将靶蛋白用反应缓冲液稀释,并分别以12um、3um和1.5um的浓度制备,并将储存在棕色试管中的fitc结合的肽(fitc标记的1010pt(gvls-pt-li-nh2))用反应缓冲液稀释,并以30nm的浓度制备。
此外,将浓度为100mm的化合物用反应缓冲液稀释,并分别以160.0um、80.0um、40.0um、20.0um、10.0um、5.0um、2.5um、1.25um、0.625um、0.3125um、0.15625um和0.0um制备。接着,将三种浓度的靶蛋白在96孔黑色平板的12孔中等分,即3行各12孔,并通过在每个孔中等分将结合肽与靶蛋白混合,在每个孔中等分靶蛋白。然后,将各浓度的化合物等分到靶蛋白质和结合肽在其中混合的每个孔中,在室温下反应30分钟。当反应完成时,在将激发波长和发射波长分别设置为485nm和535nm并将g-因子设定为1.077之后,使用infinitef200pro(tecangroupltd,switzerland)测量荧光偏振值。在这种情况下,由于g因子根据肽的特性而略有不同,因此在实验开始之前仅对肽进行了采样以固定使用前的值。使用graphpadprism(graphpadsoftware,sandiego,ca,usa)分析结合曲线。
作为实验的结果,获得了根据化合物浓度的fp测定分析结果,并在图2至4中示出,并且基于该结果计算出化合物的ic50。结果,测得2,4-二氧-1,2,3,4-四氢苯并[g]蝶啶-7-羧酸的ic50值为约25μm,并如图2至4所示,测得该化合物的衍生物的ic50值为0.45至27μm。
同时,在化合物2(m2)、化合物4(m21)、化合物5(m23)和化合物6(m25)的情况下,测量fitc标记的1010pt(fitc-gvlsptli-nh2)、cdc25cpt(fitc-llcsptpn-nh2)和pbip肽(fitc-lhspta-nh2)的ic50值,并在图3中示出。
实施例3.命中化合物和衍生物化合物抑制各种癌细胞生长的能力的分析
旨在研究通过实施例1和2发现的特异性结合至plk1的pbd结构域的化合物在癌细胞分裂期间是否实际上与plk1结合以抑制细胞分裂并抑制细胞生长。
出于该目的,使用肝癌、乳腺癌、血液癌、子宫颈癌和前列腺癌细胞系进行实验,将鼠肝癌细胞系hepa1-6和乳腺癌细胞系mda-mb-468在补充有10%胎牛血清(fbs)和1%青霉素/链霉素的dmem培养基中进行培养,并将其他细胞系在补充有相同添加剂的rpmi1640培养基中进行培养,并且将细胞系用于实验中。
为检查该化合物抑制乳腺癌细胞系生长的能力,在细胞附着后第1天和第3天以每种指定的μm浓度处理化合物,并用0.1%溶剂(dmso)处理对照。再过两天后,将附着在培养板上并生长的细胞系用1xpbs冲洗,并在室温下用4%多聚甲醛处理10分钟以固定细胞。然后,在将细胞用pbs冲洗两次后,将固定的细胞用0.5%tritonx-100溶液处理并在室温下反应15分钟,再用pbs冲洗三次,然后用0.5μg/ml的dapi试剂处理,并在37℃下反应10分钟以染色细胞核。在用pbs另外漂洗细胞一次之后,通过cytation3对用dapi染色的细胞进行拍照,并用gen5软件(biotek,usa)分析所得图像。同时,未附着在培养板的情况下处于漂浮方式的细胞用4%多聚甲醛溶液处理,在室温下反应10分钟以固定细胞,然后通过cytation3在明场中拍照,并通过gen5软件((biotek,usa)对所得图像进行分析。
将每孔2x103mda-mb-468细胞等分在96孔板中,以与上述相同的方式培养,并用化合物2(m2)和化合物3(m4)处理,然后分析抑制细胞生长的能力。结果,如图5a、5b和7所示,证实了细胞数与化合物的处理浓度成比例地显著减少。
此外,为了确定m2变体中乳腺癌细胞的反应性,使用jimt1人乳腺癌细胞在96孔板上等分每孔2x103个细胞,并按照上述相同的方式进行培养,并作为另外进行实验的结果,如图5c所示,证实了m2、m202和m203均以剂量依赖性方式显著减少癌细胞的数量。
此外,为了研究化合物2(m2)和3(m4)抑制血液癌细胞系生长的能力,在96孔板中等分每孔1x103个血液癌细胞系hl-60和u937,并且以与上述相同的方式进行实验。
结果,如图5a、5b和7所示,该化合物在两种细胞系中示出非常高的抑制细胞生长的能力。
另外,为了分析化合物2(m2)和3(m4)抑制子宫颈癌和前列腺癌细胞系生长的能力,将子宫颈癌细胞系hela和pc-3细胞(其为前列腺癌细胞系)等分在96孔板中,并以与上述相同的方式用各种浓度的化合物进行处理,然后测量细胞数。
结果,如图5a、5b和7所示,证实了用化合物处理抑制子宫颈癌中的细胞生长的能力,并且如图5a、5b和8所示,证实了取决于化合物的变体,在前列腺癌细胞中的作用存在差异。
为分析抑制肝癌细胞系生长的能力,将每孔6.6x103hepg2细胞,每孔各1x103hep3b、snu-475和snu-449细胞和每孔2x103snu-387细胞等分在96孔板中,以与上述相同的方式培养,用化合物2(m2)、化合物3(m4)、化合物4(m21)、化合物5(m23)和化合物6(m25)进行处理,然后分析抑制细胞生长的能力。
结果,如图5a、5b、6、8和9a所示,确认了细胞数量与化合物的处理浓度成比例地显著减少。
相反,如图5a所示,可以看出当用该化合物处理正常细胞系hdf时,该处理直到20um都不会显著影响细胞凋亡。
作为分析的结果,如5a至9a所示,证实了根据本发明的命中化合物的变体对细胞活力的影响不同。其中,已确认了化合物2(m2)有效且始终抑制癌细胞出现在相对不同细胞中的能力。
另外,作为以与hepg2相同的方式,关于抑制肝癌细胞系生长的能力,对hepg2人肝癌细胞中的m2变体的反应性进行实验的结果,如图9b所示,在m2和m202的情况下,肝癌细胞的面积以剂量依赖性方式显著减少,但是在m217的情况下显示相对较低的反应性,并且作为对snu449人肝癌细胞的m2变体的反应性进行实验的结果,如图9c所示,在m2、m202和m203的情况下,癌细胞数以剂量依赖性方式显著减少,然而在m206、m209、m217和m218的情况下,不呈现相对显著的癌细胞减少作用。
此外,当用低浓度的化合物2(m2)和作为plk1激酶抑制剂的bi2536的混合物处理癌细胞时,如图10所示,连同化合物2(m2)的反应性,可观察到bi2536抑制癌细胞的协同能力。
实施例4.确认由于命中化合物和衍生物化合物而导致的plk1位置变化,以及染色体臂和动粒内的ncapg2染色程度的比较
为了确认在用该化合物处理细胞后,细胞内plk1位置是否存在变化,确认了位于中心体和染色体(dapi)中的r-微管蛋白和plk1之间的相互位置关系。
如图11所示,可看到在细胞分裂的中间阶段,plk1清楚地仅确切地位于中心体和染色体的中心动粒中(图11中的对照)。然而,可看到,在用化合物2(m2)处理的情况下,位于中心体的r-微管蛋白也被弱染色,并且plk1也难以位于正常位置,从而使得难以确认一个清晰的位置(图11中的m2)。
相反,bi2536的处理似乎在plk1或r-微管蛋白本身的位置方面不产生相对较大的差异,但是由于其活性异常而观察到异常的染色体分离(图11中的bi2536)。
进一步地,如图12所示,与对照(图12中的对照)相比,在以50um的浓度用化合物2(m2)处理并持续24小时的hek293细胞系中,ncapg2(其为plk1的动粒中的pbd结合蛋白)在染色体臂和动粒中的染色程度降低。
实施例5.确认命中化合物和衍生物化合物对细胞周期的影响
在用化合物2(m2)处理的情况下,使用流式细胞仪确认对细胞周期的影响。在1天和3天后,用浓度分别为20、40和80μm的化合物2(m2)处理snu-449(肝癌细胞系中的一种),并在另外2天后采集。
此外,旨在使用能够仅特异性地对停在细胞分裂中期的细胞进行染色的磷酸-组蛋白h3(ser10)抗体,更加特异性地对停止在细胞分裂中期的细胞群染色。作为阳性对照,将细胞用20nm的被称为plk1激酶抑制剂的bi2536处理,并用于观察处于细胞分裂中期中的细胞和在g2/m期中的增加。
作为实验的结果,如图13所示,当用化合物2(m2)处理细胞时,与ct相比,磷酸-组蛋白h3阳性细胞的比例降低,这是与用bi2536处理细胞时的比例增加相反的结果。
此外,关于细胞周期,还可看到,当用化合物2(m2)处理细胞时,在所有处理浓度下,具有多倍体染色体数目的细胞的比例并未增加,然而当用bi2536处理时,多倍体细胞的比例显著增加(图13)。由此可见,化合物2(m2)以不同于bi2536的方式影响细胞的生长和死亡,并且通过多倍体细胞比例没有增加的事实,可以看出在进入细胞分裂周期之前,细胞的生长被抑制,并且多倍体细胞的比例不增加。
通过该结果,确认了通过实施例1和2发现的与plk1的pbd结构域结合的化合物实际上有效地抑制了肝癌、乳腺癌、血液癌、子宫颈癌和前列腺癌细胞的生长,并且可确认的是,对正常细胞相对生长的抑制作用相对较小。
此外,确认了该化合物作为参与癌细胞中正常细胞分裂过程的plk1抑制剂与磷酸化活性的作用不同,并且确认了靶向pbd的命中物质阻碍了plk1本身在细胞中的正常定位,使得其确切的配偶体的位置不合适,从而显示出在有丝分裂期之前的阶段抑制细胞生长进程的效果。
实施例6.用m2处理后细胞活性变化的确认
将hepg2细胞(一种肝癌细胞系)在每种命中化合物浓度下在补充有培养基的96孔微量滴定板中接种,4至6个重复。根据实验设计,第二天添加溶于dmso中的命中化合物,并通过在作为细胞对照处理的方案的最后一天达到80%的细胞密度来确定接种细胞的数量。细胞接种24小时后,用各种浓度的命中化合物(m2和bi2536)处理细胞,并且在一次处理后48小时,吸出培养基,然后进行二次处理。在48小时后,通过在37℃下2.5μmhoechst33342染色30分钟使细胞核可视化,然后吸出培养基并用新鲜培养基洗涤。使用cytationtm3(biotek,usa)读取板,分析细胞活性,并且结果表示为与对照处理相比,用命中化合物处理后的存活细胞的相对百分比。
结果,如图14a和14b所示,证实了m2和bi2536的处理浓度越高,细胞活性变得相对越小。
实施例7.用m2处理后由于细胞凋亡引起的细胞死亡
为了分析通过暴露于m2引起的凋亡模式,将hepg2细胞(一种肝癌细胞系)分别用20μm或100μmm2和20nm或100nmbi2536处理并持续3天。通过坏死和凋亡细胞的膜联蛋白v-异硫氰酸荧光素(膜联蛋白v-fitc)和碘化丙啶(pi)染色来检测细胞凋亡。首先,收获细胞并用pbs洗涤一次。然后将细胞重悬于含有4μlannexinv(bd,51-65874x)和pi(bd,51-66211e)的100μl结合缓冲液中。将细胞在黑暗中于37℃染色15分钟,然后使用facsan(bd,sanjose,ca)进行分析。使用cellquest软件(bd)分析数据。
结果,如图15a所示,在经m2和bi2536处理组中凋亡细胞均增加,并且如图15b所示,在经m2和bi2536治疗组中,细胞凋亡均以剂量依赖性方式增加。
实施例8.在肝癌异种移植模型中分析命中化合物衍生化合物抑制癌症生长的能力(化合物4的毒性试验)
将化合物4(m21)在300μlpbs中稀释,并分别以按小鼠体重计,1mg/kg、5mg/kg和10mg/kg每周3次进行腹膜内注射,并且在对照中,将稀释于300μlpbs的dmso以3%进行腹膜内注射。2周后,通过处死小鼠移除肺、心脏、肝脏、肾脏、脾脏和皮肤,并固定在福尔马林溶液中。在固定组织的组织病理学分析中未观察到单独的急性毒性引起的变化(图16)。
同时,通过向免疫缺陷小鼠(balb/c-nu)的皮下脂肪层中注射5×106hepg2细胞来制备异种移植模型。3周后,将化合物4(m21)和化合物2(m2)分别稀释在300μlpbs中,分别以5mg/kg和10mg/kg每周五次进行腹膜内注射,并且在对照中,将稀释于300μlpbs的dmso以3%进行腹膜内注射。
每周三次测量肿瘤大小和小鼠体重,并且结果示于图15中。在施用物质12天后(施用:总共10次),处死小鼠。移除肿瘤,称重,固定在福尔马林溶液中,并冷冻。
如图17和18所示,可以观察到,与对照相比,在用化合物处理的情况下,移除的产生癌的组织的重量减小。
同时,如图19所示,可以看出组织中plk1自身的表达差异不明显,但有丝分裂期的细胞数减少。
实施例9.在肝癌原位异种移植模型(hepg2细胞系)中抑制癌症生长的能力的分析
将5x106hepg2细胞(一种肝癌细胞系)注射到balb/c裸鼠的背部皮肤中,在大约3周后形成足够的癌组织时,取出该组织,均匀切成1mm3,并且通过在1cm内切开腹部将其移植到肝的右中叶。
基于本发明人先前对hcc细胞的体外实验,选择9.1mg/kgm2和1mg/kgbi2536的剂量。在细胞注射后7天开始施用,对于每个实验对于最高浓度的药物使用等量的dmso作为溶剂对照。每种药物每周注射5次,共注射19次。
结果,如图20a所示,m2和bi2536抑制了肿瘤细胞的生长,并且如图20b所示,m2和bi2536均抑制了通过生长抑制指数计算的hcc异种移植的进程。此外,证实组织学染色显示,与对照相比(如图20c所示),经m2处理的小鼠中的有丝分裂指数降低。图20d中的降低的有丝分裂指数与m2处理后的细胞周期分析一致,并且m2在体外和体内具有不同于bi2536的作用机制。
通过改变实验方法,对同一肝癌细胞系进行了另一实验。将5x106hepg2细胞注射到balb/c裸鼠的背部,大约3周后形成足够的癌组织时,将该组织取出,均匀切成1mm3,并通过在1cm内切开腹部移植到肝的右中叶。
通过上述方法在将肝癌组织移植到20只balb/c裸鼠中后约10天,在mri图像上确认有小斑点,因此,将充分构建肝癌的啮齿动物分为3组(约50%至60%)(参见图21a),每两天一次将对照和命中(m2)物质以5mg/kg和20mg/kg注入腹腔,其使用少于1.5%的dmso作为溶剂(载体),并且每周一次,使用mri图像以选择(跟踪)有恒定组织生长的啮齿动物。然后,在快速生长的癌组织为1cm或更小的状态下,在处死小鼠后观察癌组织(10次处理并持续3周)。
结果,如图21b和21c所示,随着m2剂量的增加,癌组织的重量和体积显著减少,从而可确认m2的优异抗癌作用。
实施例10.在使用人肝癌pdx的原位异种移植模型中抑制癌症生长的能力分析
将从人肝癌中分离出的癌组织移植到balb/c裸鼠的皮肤组织中,并使该皮肤组织长成癌组织,从而使用作为pdx模型建立的组织,均匀切成1mm3,然后通过在1cm内切开腹部移植到肝的右中叶。通过上述方法在将肝癌组织移植到20只balb/c裸鼠中后约10天,在mri图像上确认有小斑点,因此,将充分构建肝癌的啮齿动物分为3组(参见图22a、22b、以及22c),每两天一次将溶剂对照(dmso)、命中(m2)物质(40mg/kg)和bi2536(4mg/kg)注入腹腔,其使用少于1.5%的dmso作为溶剂(载体),并且每周一次,使用mri图像以选择(跟踪)有恒定组织生长的啮齿动物。然后,在快速生长的癌组织为1cm或更小时,在处死小鼠后观察癌组织(11次处理并持续3周)。
结果,如图22b至22d所示,根据m2的施用,癌组织的重量和体积显著减少,从而可证实m2的优异的抗癌作用。
提供本发明的上述描述是出于说明性目的,并且本发明所属领域的技术人员将理解,在不改变本发明的技术精神或基本特征的情况下,可容易地将本发明修改为其他特定形式。因此,应当理解,上述实施方案在所有方面仅是示例性的,而不是限制性的。
[工业实用性]
与相关技术中靶向激酶结构域的atp结合位点抑制剂相比,本发明的化合物通过与plk1的pbd选择性结合而具有高选择性和结合亲和力以及低毒性的优点。因此,本发明的化合物可有效地用作抑制各种癌细胞生长的抗癌剂,并且除了单次给药外,还可预期通过与其他现有抗癌剂共同给药的协同作用,从而化合物不仅可广泛用于制药工业,而且还可广泛用于保健食品行业。
- 还没有人留言评论。精彩留言会获得点赞!