构建乳腺癌新辅助化疗疗效分类模型的方法与流程
本发明涉及一种分类模型的构建方法及试剂盒,特别涉及利用血浆cfdna中tsss区域构建乳腺癌新辅助化疗疗效分类模型的方法。
背景技术:
新辅助化疗(neoadjuvantchemotherapy,nact)目前应用于局部晚期乳腺癌患者,或者有辅助化疗指征及强烈保乳需求的患者中。主要有以下4方面优势:首先,它可以使不可切除的肿瘤变为可切除;其次,增加保乳的机会;对于局部晚期并远处转移风险的患者,术前全身治疗被期望可以改善患者的生存时间;可以观察到肿瘤的缩小和成像变化,并且能够对临床疗效进行快速评价。因此新辅助化疗的使用可以明显减小手术前肿瘤的大小,降低临床分期,以及减少腋窝淋巴结的累及,避免乳房全切除,有助于提高患者的生活质量。
新辅助化疗的反应通常以病理完全反应(pcr)来评价,即在最终手术时,原发肿瘤床和局部淋巴结均无浸润性乳腺癌组织。pcr的结果与总体生存率和无病生存率的提高密切相关。新辅助化疗对于her2阳性和三阴性乳腺癌来说反应率较高,her2阳性中达到pcr的比例可以高达30%-60%以上,三阴性乳腺癌也可以高达20%-30%。对于er阳性的病人来说,luminala型的患者pcr率只有5%左右,而且pcr率与远期预后关系不大。对于luminalb型患者,在中国占乳腺癌患者大部分,在新辅助化疗后至少一半以上病人都可以达到肿瘤体积明显缩小,但仍有少数侵袭性细胞,且预后也明显改善,所以对于luminalb型的新辅助化疗可以用来缩瘤降期,而不以pcr做为新辅助治疗的目的。根据实体瘤疗效评价标准recist1.0可以分为四类:cr(所有病灶消失维持4周),pr(缩小30%,维持4周),sd(非pr/pd)和pd(增加20%,病灶增加前非cr/pr/sd)。
虽然新辅助化疗在过去的30多年取得了较大进展,但仍有20-30%的患者对新辅助化疗方案敏感性不佳,导致了病情延误,错过最佳治疗时机以及过度治疗。因此根据敏感性预测指标来制定乳腺癌个体化治疗方案是非常重要的,也可以避免治疗的盲目性。传统的辅助化疗疗效预测方案主要是针对er阳性、淋巴结未发生转移的早期乳腺癌患者的21基因复发风险评估,2019年nccn、asco和csco的乳腺癌指南中又新增加了70基因表达谱检测,主要针对er或者pr阳性,her-2阴性的乳腺癌患者,同时范围增加到对腋窝淋巴结有1-3个转移患者,这些检测对于早期乳腺癌患者的治疗决策,评估早期浸润性乳腺癌患者5年内的远处转移风险,使一部分乳腺癌患者在不影响长期疗效的基础上避免接受辅助化疗具有显著的意义。而目前针对预测乳腺癌新辅助化疗疗效尚无标准,可能的方案是应用新辅助化疗前的癌组织穿刺标本进行21基因复发风险评估或70基因表达谱检测,进而实现疗效预测及方案的确定。但由于穿刺的组织标本具有肿瘤异质性,样本量少等特点限制了其临床应用。
而在过去的几年中,液体活检越来越多应用于癌症的早期诊断,预后预测,复发风险评估,连续采样监测疾病进展和治疗反应等方向。相对于传统的组织活检,液态活检具有无创性、均质化及连续采样等优势。目前用于监测乳腺癌的标志物有肿瘤抗原15-3和27-29,但它们的敏感性估计仅为60-70%。除此之外患者血浆中循环肿瘤dna(ctdna),循环肿瘤细胞(ctcs)和外泌体(exosome)等的检出率和预后价值关系较大。其中ctdna中的fgfr、erbb2(her2)、erbb3(her3)、pi3k、brca1/2等基因突变为转移性乳腺癌的治疗策略提供参考。
技术实现要素:
本发明的目的在于克服现有技术的不足,提供一种构建乳腺癌新辅助化疗疗效分类模型的方法。
本发明所采取的技术方案是:
发明人研究发现,细胞游离dna(cfdna)主要来自于凋亡细胞,cfdna因被核小体保护而未被降解。基因组的核小体印记与基因转录水平密切相关,尤其在基因表达的转录起始位点(tsss)附近区域核小体的结合明显减少。因此本发明利用血浆中cfdna的全基因组测序,分析tsss区域覆盖度差异,构建一种用于预测乳腺癌新辅助化疗疗效的分类模型,为乳腺癌的治疗提供更为广阔和可靠的数据来源,具有更大的前景。
一种构建乳腺癌新辅助化疗疗效分类模型的方法,包括如下步骤:
cfdna的reads的获取:
获取已知化疗结果的不同乳腺癌分型病人的血浆cfdna测序数据,获得cfdna的reads数据;
tsss区域覆盖度分析:
提取基因转录起始位点tsss上下游区域的基因组位置信息,定义为原始启动子区域;
将cfdna的reads比对到人类参考基因组hg19上,去除比对数据中的重复序列,统计出所有蛋白编码基因的原始启动子区域的reads数后,对tsss区域覆盖度进行标准化处理;
覆盖度差异分析:
利用kruskal-wallis非参数单因子方差分析方法,筛选出各组中tsss区域覆盖度存在差异的基因启动子区域,对原始p值进行校正,筛选出fdr值小于0.1的基因启动子区域,最后确定差异tsss区域;
聚类分析:
对tsss区域覆盖度的数据进行样本间的标准化,采用等级聚类法根据样本间启动子覆盖度的相关性进行聚类分析;根据原始启动子区域的覆盖度差异和所述病人的已知疗效情况,将样本类型聚类为pcr组和npcr组或pr组和sd组,得到乳腺癌新辅助化疗疗效分类模型。
在一些方法的实例中,所述原始启动子区域为基因转录起始位点tsss上下游1kb的区域。
在一些方法的实例中,每个样本的所述cfdna测序数据量不少于6m。
在一些方法的实例中,所述乳腺癌分型包括luminalb型、her-2阳性和三阴性。
在一些方法的实例中,所述化疗使用的药物包括表柔比星、环磷酰胺、紫杉醇、卡铂、吉西他滨、卡培他滨中的至少一种。
在一些方法的实例中,对聚类分析数据进行可视化处理。
在一些方法的实例中,所述可视化处理的软件为treeview软件或者r语言heatmap函数。
在一些方法的实例中,tsss区域覆盖度分析时,所述标准化处理的方法选自rpkm标准化方法、fpkm标准化方法或按如下方法计算得到:
标准化数值=区域reads数目/总比对上的reads数目。
在一些方法的实例中,覆盖度差异分析时,使用holm法对原始p值进行校正。
在一些方法的实例中,使用z-score标准化方法对基因启动子覆盖度的数据进行样本间的标准化,其计算公式为:标准后数值
本发明的有益效果是:
本发明方法构建的模型,通过对待测病人的血浆cfdna序列可以实现对乳腺癌患者新辅助化疗进行疗效预测,确定乳腺癌的治疗策略,指导乳腺癌新辅助化疗。
本发明的模型,所需要的数据来源于外周血,属于无创检测范畴,克服了现有乳腺癌新辅助化疗疗效预测的材料需要有创性穿刺取样所导致的临床体验不佳、无法解决肿瘤异质性、穿刺禁忌证、造成肿瘤传播转移风险等缺点。
附图说明
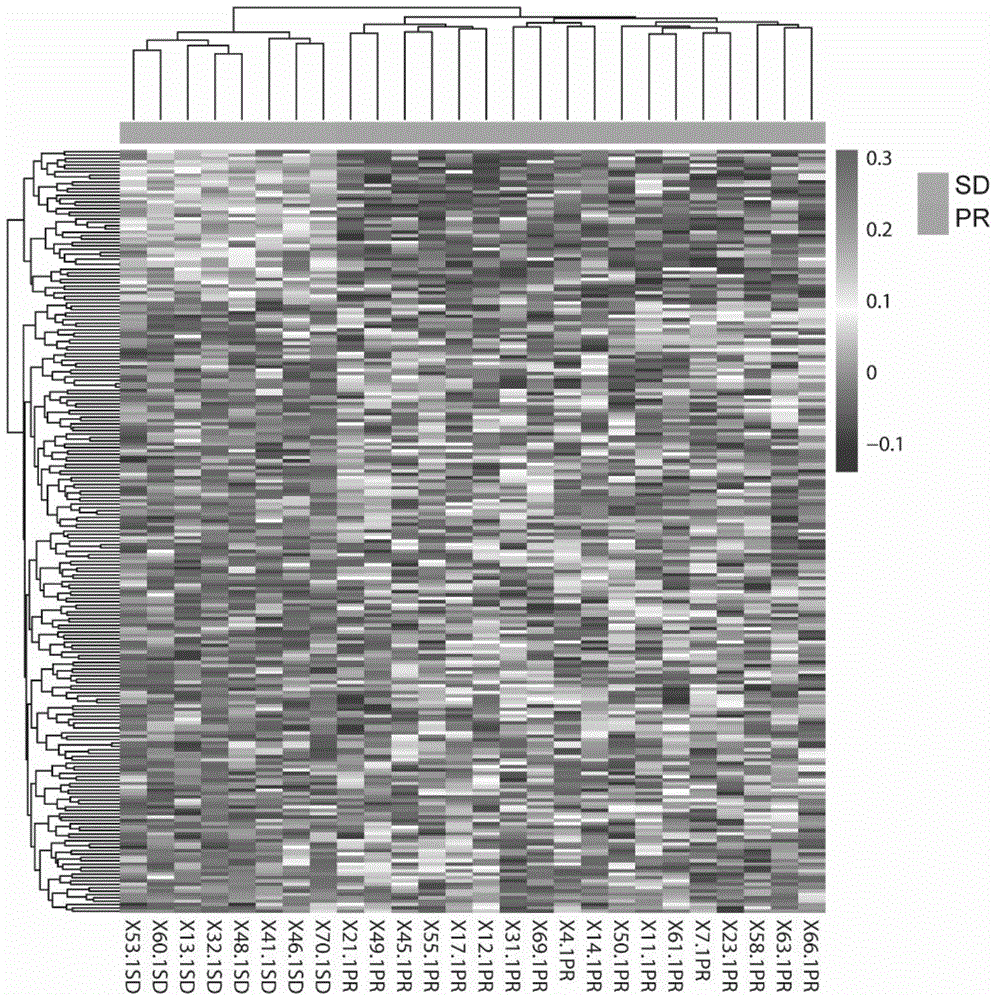
图1表柔比星、环磷酰胺、紫杉醇联合方案的乳腺癌pr组与sd组的化疗前血浆cfdna中tsss区域覆盖度差异的聚类结果。
图2表柔比星、环磷酰胺、紫杉醇联合方案的乳腺癌pcr组与npcr组的化疗前血浆cfdna中tsss区域覆盖度差异的聚类结果。
图3表柔比星、环磷酰胺、紫杉醇、卡铂联合方案的乳腺癌pcr组与npcr组的化疗前血浆cfdna中tsss区域覆盖度差异的聚类结果。
具体实施方式
一种构建乳腺癌新辅助化疗疗效分类模型的方法,包括如下步骤:
cfdna的reads的获取:
获取已知化疗结果的不同乳腺癌分型病人的血浆cfdna测序数据,获得cfdna的reads数据;
tsss区域覆盖度分析:
提取基因转录起始位点tsss上下游区域的基因组位置信息,定义为原始启动子区域;
将cfdna的reads比对到人类参考基因组hg19上,去除比对数据中的重复序列,统计出所有蛋白编码基因的原始启动子区域的reads数后,对tsss区域覆盖度进行标准化处理;
覆盖度差异分析:
利用kruskal-wallis非参数单因子方差分析方法,筛选出各组中tsss区域覆盖度存在差异的基因启动子区域,对原始p值进行校正,筛选出fdr值小于0.1的基因启动子区域,最后确定差异tsss区域;
聚类分析:
对tsss区域覆盖度的数据进行样本间的标准化,采用等级聚类法根据样本间启动子覆盖度的相关性进行聚类分析;根据原始启动子区域的覆盖度差异和所述病人的已知疗效情况,将样本类型聚类为pcr组和npcr组或pr组和sd组,得到乳腺癌新辅助化疗疗效分类模型。
基于该分类模型,通过对待测病人的血浆cfdna测序数据进行分析,即可准确确定其分类,便于制订相应的新辅助化疗策略,具有很好的指导效果。
在一些方法的实例中,所述原始启动子区域为基因转录起始位点tsss上下游1kb的区域。这样既可以避免遗漏,也可以提高数据的准确性。
在一些方法的实例中,每个样本的所述cfdna测序数据量不少于6m。这样有利于排除无效或可信度较差的数据。
在一些方法的实例中,所述乳腺癌分型包括luminalb型、her-2阳性和三阴性。
在一些方法的实例中,所述化疗使用的药物包括表柔比星、环磷酰胺、紫杉醇、卡铂、吉西他滨、卡培他滨中的至少一种。这些药物为乳腺癌新辅助化疗的常用药物。
在一些方法的实例中,对聚类分析数据进行可视化处理。这样可以更为直观地显示结果。
在一些方法的实例中,所述可视化处理的软件为treeview软件或者r语言heatmap函数等常见的可视化处理方案。
在一些方法的实例中,tsss区域覆盖度分析时,所述标准化处理的方法选自rpkm标准化方法、fpkm标准化方法或按如下方法计算得到:
标准化数值=区域reads数目/总比对上的reads数目。
通过对数据进行标准化处理,可以减少样本差异对结果准确度的影响。
在一些方法的实例中,覆盖度差异分析时,使用holm法对原始p值进行校正。。
在一些方法的实例中,使用z-score标准化方法对基因启动子覆盖度的数据进行样本间的标准化,其计算公式为:标准后数值
下面结合实施例,进一步说明本发明的技术方案。实施例用于进一步示例性说明本发明的技术方案,不应视为对本发明保护范围的局限。
以下实施例涉及的检测试剂包括血浆cfdna提取试剂、文库构建试剂、测序试剂、测序芯片等。其中文库构建试剂盒包括末端修复、接头连接、pcr扩增试剂,均为本领域常规试剂,均可通过市售获得。
实施例1乳腺癌新辅助化疗使用表柔比星、环磷酰胺、紫杉醇联合方案的疗效预测
步骤1:样本收集:收集乳腺癌患者采用表柔比星、环磷酰胺、紫杉醇联合方案新辅助化疗前的外周血26例,经8个周期化疗术后标本进行疗效评价,按照recist1.0疗效评价标准,其中达到pr效果18例,达到sd效果8例,按是否达到pcr标准达到pcr患者7例,未达到pcr(npcr)患者19例,具体如表1所示。
步骤2:血浆分离:收集的外周血经4℃,1600g离心10min,上清液4℃,16000g离心10min进行血浆分离,注意不要吸到白细胞。
步骤3:血浆cfdna提取:使用血浆游离dna提取试剂盒(magabio公司)提取血浆cfdna,严格按说明书进行操作,提取的dna使用qubit3.0(lifetechnologies)进行浓度测定。
步骤4:文库构建及定量:使用ionplusfragmentlibrarykit(lifetechnologies公司)试剂盒对dna进行末端修复,磁珠纯化后,两端添加上测序接头并扩增,得到测序文库,使用qubit3.0进行浓度测定。
步骤5:高通量测序:对10个文库进行等量混合,使用ionpitmhi-qtmot2200kit(lifetechnologies)对进行模板制备和模板富集。使用ionpitmhi-qtmsequencingkit(lifetechnologies)进行测序,iontorrentprotontm测序仪选择预先设置好的程序flow数为500,每个样本数据量大于6m)。
步骤6:生物信息学分析
(1)tsss区域覆盖度分析
从ucsc的refseq数据库下载人类蛋白编码基因的信息,我们根据数据库注释信息提取每个基因的转录起始位点tsss上下游1kb区域的基因组位置信息,使用bowtie软件将原始测序read比对到人类参考基因组hg19上,去除比对数据中的重复序列,统计出所有蛋白编码基因的原始启动子区域的reads数后,采用rpkm标准化方法对数据进行处理。
(2)覆盖度差异分析
通过kruskal-wallis非参数单因子方差分析,分别按照pr组和sd组,pcr组和npcr组进行分组,比较组间覆盖度数值差异,对原始p值利用holm法进行校正后得到fdr值,筛选得到fdr值小于0.1的tsss区域。
(3)聚类分析
利用cluster软件,对基因启动子覆盖度的数据进行样本间的标准化,采用等级聚类法根据样本间启动子覆盖度的相关性进行聚类分析,并利用r语言的pheatmap包对数据进行可视化展示。
结果如图1和图2表示。基于26例乳腺癌的采用表柔比星、环磷酰胺、紫杉醇联合方案的乳腺癌患者的血浆游离dna进行高通量检测,并进行tsss区域覆盖度的分析,以pr与sd评价标准,评估乳腺癌患者对表柔比星、环磷酰胺、紫杉醇联合方案的效果。我们发现pr组与sd组共存在227个基因的启动子覆盖度存在差异(表2)。其中45个基因的覆盖度在sd组中升高,182个基因的覆盖度在sd组间下降。通过差异的启动子覆盖度进行无监督聚类,结果发现基因启动子的覆盖度差异能够将治疗后pr组与sd组的患者分别聚成两组,两组间的覆盖度模式存在显著的差异(图1)。
若以pcr和npcr为指标,评估乳腺癌患者对表柔比星、环磷酰胺、紫杉醇联合方案的效果。我们发现pcr组与npcr组共存在398个基因的启动子覆盖度存在差异(表3)。其中73个基因的覆盖度在pcr组中升高,325个基因的覆盖度在pcr组间下降。通过差异的启动子覆盖度进行无监督聚类,结果发现基因启动子的覆盖度差异能够将pcr组与npcr组的患者分别聚成两组,两组间的覆盖度模式存在显著的差异(图2)。
实施例2乳腺癌新辅助化疗表柔比星、环磷酰胺、紫杉醇、卡铂联合方案的疗效预测
步骤1:收集乳腺癌患者采用表柔比星、环磷酰胺、紫杉醇、卡铂联合方案新辅助化疗前的外周血8例,经8个周期化疗术后标本进行疗效评价,其中达到pcr患者3例,未达到pcr(npcr)患者5例,具体如表1所示。
步骤2-6同实施例1的步骤2-6,分组方式只按照pcr与npcr的分组方式。
结果如图3所示,基于8例采用表柔比星、环磷酰胺、紫杉醇、卡铂联合方案的乳腺癌患者的血浆游离dna进行高通量检测,并进行tsss区域覆盖度的分析,发现pcr组与npcr组共存在555个基因的启动子覆盖度存在差异(表4)。其中228个基因的覆盖度在pcr组中升高,327个基因的覆盖度在pcr组间下降。通过差异的启动子覆盖度进行无监督聚类,结果发现基因启动子的覆盖度差异能够将pcr组与npcr组的患者分别聚成两组,两组间的覆盖度模式存在显著的差异(图3)。
表1、34例乳腺癌患者信息
表2、表柔比星、环磷酰胺、紫杉醇联合方案的乳腺癌pr组与sd组化疗前血浆cfdna差异tsss区域及相关基因列表
表3、表柔比星、环磷酰胺、紫杉醇联合方案的乳腺癌pcr组与npcr组化疗前血浆cfdna差异tsss区域及相关基因列表
表4、表柔比星、环磷酰胺、紫杉醇、卡铂联合方案的乳腺癌pcr组与npcr组化疗前血浆cfdna差异tsss区域及相关基因列表
- 还没有人留言评论。精彩留言会获得点赞!