分析实时扩增数据的方法
本发明涉及用于多维分析实时扩增数据的方法、系统、计算机程序和计算机可读介质。
背景
从一开始,实时聚合酶链反应(qpcr)就已成为分子生物学中用于检测和定量核酸的常规技术。这主要是由于其较大的动态范围(7到8个数量级),理想的灵敏度(5到10个分子)和可重复的定量结果。改进qpcr数据分析的新方法在许多分析领域都具有不可估量的价值,包括环境监测和临床诊断。尽管近年来研究已经饱和,但在实时pcr中使用标准曲线对核酸进行绝对定量无疑是重要的,并且在各个生物医学领域中都意义重大。
当前对于特定靶序列进行绝对定量的“金标准”是循环阈值(ct)法。ct值是扩增曲线的特征,该特征被定义为存在可检测的荧光增加的指数区域中的循环数。自从提出该方法以来,已经开发了几种替代方法,以期在准确性、精度和鲁棒性方面改进绝对定量。现有研究的重点基于单一特征的计算,例如与初始浓度线性相关的cy和-log10(f0)。这为绝对定量提供了一种简单的方法,但是,基于这样的单一特征的数据分析是受限的。因此,对使用标准曲线进行核酸绝对定量的改进方法的研究已经趋于平稳,并且都是非常渐进式的改进。
rutledge等人2004提出了基于三个动力学参数(fc、fmax和f0)的s型曲线拟合(scf)进行定量。sisti等人2010开发了“基于形状的离群点检测”方法,该方法不基于扩增效率,而是使用对参数化pcr扩增谱的非线性拟合。基于形状的离群点检测方法采用多维方法来定义扩增曲线之间的相似性度量,但是依赖于使用特定的扩增模型,即5参数s型模型,而这不是通用方法。此外,基于形状的离群点检测方法通常作为附件使用,并且仅使用多维方法来进行离群点检测,从而仅使用一维方法考虑定量。guescini等人2013提出了cy0法,该方法类似于ct方法,但考虑了扩增曲线的动力学参数,并可能补偿所比较样本之间的微小变化。bar等人2013提出了基于扩增效率计算的方法(kod),以用于早期非最佳测定条件检测。
本发明旨在至少部分地克服现有技术中固有的问题。
技术实现要素:
本发明由所附权利要求限定。本文的支持性公开内容提出了一个框架,该框架显示,在多维环境中观察时,标准曲线的益处延伸到绝对定量之外。现有研究的重点一直放在与靶标浓度线性相关的单个值(在本文中称为“特征”)的计算上,因此,在利用多种特征方面,现有方法存在差距。现在已经认识到,结合线性特征的益处是重要的。先前的方法已被局限于常规标准曲线例如金标准循环阈值(ct)法的简单性。这种新的方法能够增强核酸的定量、单通道多路复用、离群点检测、多维空间中与扩增动力学相关的特性模式以及增加的样本鉴定和定量的鲁棒性。
关于机器学习领域,当前公开的方法采取多维视图,结合多个特征(例如线性特征),以便利用现有方法背后的信息和原理并在其基础上改进来分析实时扩增数据。公开的方法涉及两个新概念:多维标准曲线及其“原点”(特征空间)。它们共同扩展了标准曲线的功能,允许同时进行绝对定量、离群点检测并提供对扩增动力学的洞察力。本发明描述了一种通用方法,该方法首次展示了多维标准曲线,从而增加了数据分析的自由度,并且从而能够揭示现有qpcr仪器(例如来自rochelifescience的lightcycler96system)获得的实时扩增数据中的趋势和模式。可以相信,本发明重新定义了分析实时核酸扩增数据的基础,并在核酸研究领域中实现了新的应用。
在本发明的第一方面,提供了一种定量包含靶核酸的样本的方法,该方法包括:为多个靶标浓度中的每个获得第一实时扩增数据集;从第一数据集中提取多组n个特征,其中每个特征使第一数据集与靶标的浓度相关;通过特征对在n维空间中定义的多个点拟合成线,每个点与多个靶标浓度中的一个相关,其中该线定义了核酸靶标特有的多维标准曲线,所述多维标准曲线可用于定量靶标浓度。
可选地,该方法还包括:获得与未知样本相关的第二实时扩增数据;从第二数据中提取相应的多组n个特征;和通过相应的多组n个特征,计算n维空间中的线与n维空间中定义的点的距离度量。可选地,该方法还包括:根据距离度量来计算扩增曲线之间的相似性度量,该相似性度量可以可选地用于识别离群点或对靶标进行分类。
可选地,每个特征不同于其他特征中的每个,并且可选地其中每个特征与靶标的浓度线性相关,并且可选地其中特征中的一个或多个包括ct、cy和-log10(f0)中的一个。
可选地,该方法进一步包括将n维空间中的线映射到与靶标浓度相关的一维函数m0,并且可选地其中一维函数与靶标浓度线性相关,和/或可选地其中一维函数定义了用于定量靶标浓度的标准曲线。可选地,映射使用降维技术来执行,并且可选地,其中降维技术包括以下中的至少一种:主成分分析;随机样本一致性;偏最小二乘回归;和投影到单一特征上。可选地,映射包括对特征中的每个应用各自的标量特征权重,并且可选地,其中各自的特征权重由优化目标函数的优化算法确定,并且可选地,其中目标函数被布置以用于优化量化性能。
可选地,计算距离度量包括将n维空间中的点投影到与n维空间中的线垂直的平面上,并且可选地,其中计算距离度量还包括基于投影点计算欧氏距离(euclideandistance)和/或马氏距离(mahalanobisdistance)。可选地,该方法还包括基于距离度量来计算相似性度量,并且可选地,其中,计算相似性度量包括将阈值应用于相似性度量。可选地,该方法还包括基于相似性度量来确定n维空间中的点是在群点(inlier)还是离群点。可选地,该方法还包括:如果n维空间中的点被确定为离群点,则从将在n维空间中定义的多个点拟合成线的步骤所基于的训练数据中排除该点,并且如果n维空间中的点未被确定为离群点,则另外基于n维空间中的点在n维空间中重新拟合成线。
可选地,该方法还包括:基于多维标准曲线,并且可选地进一步基于距离度量,以及可选地在从属于权利要求4时基于定义标准曲线的所述一维函数,确定靶标浓度。可选地,该方法还包括在显示器上显示靶标浓度。
可选地,该方法还包括以下步骤:将第一数据集拟合成曲线,其中特征提取基于曲线拟合的第一数据,并且可选地,其中曲线拟合使用5参数s型模型、指数模型和线性插值中的一种或多种来执行。可选地,预处理与熔解温度有关的第一数据集,并且在处理后的第一数据集上进行曲线拟合,并且可选地,其中预处理包括以下中的一种或多种:减去基线;和归一化。
可选地,与熔解温度有关的数据来自对应样本温度而采得的一个或多个物理度量,并且可选地其中一个或多个物理度量包括荧光读数。
在第二方面,提供了一种系统,其包括至少一个处理器和/或至少一个集成电路,该系统被布置为执行根据第一方面的方法。
在第三方面,提供了一种计算机程序,其包括指令,所述指令在由一个或多个处理器执行时使所述一个或多个处理器执行根据第一方面的方法。
在第四方面,提供了一种存储指令的计算机可读介质,所述指令在由至少一个处理器执行时使所述至少一个处理器执行根据第一方面的方法。
在第五方面,提供了根据第一方面的方法,其用于检测基因组材料,并且可选地,其中基因组材料包括一种或多种病原体,并且可选地,其中所述病原体包括一种或多种产碳青霉烯酶肠杆菌,并且可选地,其中病原体包含一个或多个碳青霉烯酶基因,所述碳青霉烯酶基因来自包含blaoxa-48、blavim、blandm和blakpc的集合。
在第六方面,提供了一种通过根据第一方面的方法检测一种或多种病原体来诊断感染的方法,并且可选地,其中所述病原体包括一种或多种产碳青霉烯酶肠杆菌,并且可选地,其中病原体包含一个或多个碳青霉烯酶基因,所述碳青霉烯酶基因来自包含blaoxa-48、blavim、blandm和blakpc的集合。
在第七方面,提供了一种通过根据第一方面的方法检测一种或多种病原体来即时(point-of-care)诊断传染病的方法,并且可选地,其中所述病原体包括一种或多种产碳青霉烯酶肠杆菌,并且可选地,其中病原体包含一个或多个碳青霉烯酶基因,所述碳青霉烯酶基因来自包含blaoxa-48、blavim、blandm和blakpc的集合。
如果本文公开的方法用于诊断,则可以在体外或离体进行。实施例可以用于单通道多路复用而无需pcr后操作。
根据本发明,将认识到,本文描述的某些方面和/或实施例的某些特征可以有利地与其他方面和/或实施例的那些特征组合。因此,特定实施例的以下描述不应被解释为指示所有描述的步骤和/或特征是必要的。相反,将理解,某些步骤和/或特征由于它们的功能或目的是可选的,即使在那些步骤或特征没有明确地被描述为可选的情况下也是如此。因此,上述方面并非旨在限制本发明,而是由所附权利要求书来限定本发明。
附图说明
为了可以理解本发明,下面参考附图通过示例描述优选的实施例。在附图中,相似的特征设有相似的附图标记。附图不一定按比例绘制。
图1是与所提出的多维框架相比,现有一维方法中的培训和测试的表示。
图2a至2c示出了使用本文所述的多维方法进行训练的过程。
图2d至2f示出了使用本文所述的多维方法进行测试的过程。
图3是用于优化特征权重的算法的表示。
图4a是多维标准曲线的表示。
图4b是通过主成分回归进行降维后获得的产生的定量曲线的示意图。
图5显示了特征空间中离群点的均值,以及离群点的均值在标准曲线上的正交投影。
图6a是通过投影到垂直于标准曲线的平面上,特征空间沿着多维标准曲线的轴线的视图的表示。
图6b是根据图6a的所得投影点的表示。
图6c是图6b的特征空间的正交视图在欧氏距离等效于原始空间中的马氏距离的情况下变换为新空间的表示。
图7显示了整个训练集叠加有2个自由度的χ2分布的马氏距离平方的直方图。
图8a显示了与温度相关的多维模式。
图8b显示了与引物混合物浓度相关的多维模式。
图8c显示了针对低浓度核酸的训练数据点沿多维标准曲线轴线的变化。
图9是实验工作流程的图示,以及实时一维对比多维标准曲线的比较。
图10显示了使用ct、cy和-log10(f0),使用针对四种靶基因修正的单一引物混合物(通过多重实时pcr)而构建的多维标准曲线。
图11显示了训练样本的实时扩增数据和熔解曲线分析(出于验证目的)。
图12显示了四个多维标准曲线中的每个的马氏空间。
图13是其中可以实现本发明的实施例的示例联网计算机系统的表示。
图14是诸如图13所示的示例计算设备的表示。
图15a-15d显示了根据一个示例,针对训练数据(15a)、离群点(15b)、引物浓度实验(15c)和温度变化实验(15d)的熔解曲线分析。
图16显示了在示例中从标准点到样本测试的平均马氏距离。其可以用于仅基于通过多重pcr测定获得的实时扩增曲线而将样本分类为blaoxa-48、blandm、blavim和blakpc基因。
具体实施方式
本发明的结构如下。为了理解所提出的框架,以相同的语言总体了解在常规方法中所做的工作非常有用。首先,介绍了常规方法,然后介绍了所提出的多维框架。为了更容易理解,将解释和讨论所公开方法的理论和优点。此外,通过举例的方式,给出了该新方法的示例实例,其中使用了以λdna为模板的实时数据的集合,并探讨了所公开方法的具体应用。
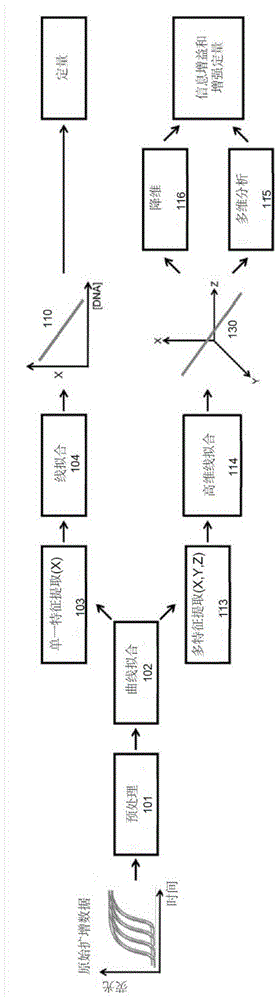
图1是显示了所公开的多维方法(底部分支)与基于对已知靶标进行连续稀释的靶标绝对定量的常规方法(顶部分支)相比的框图。
常规方法
在常规方法中,通常会预处理几种已知浓度靶标的原始扩增数据,并用适当的曲线拟合所述数据。从每个曲线中提取单一特征,例如循环阈值ct。以特征相对浓度来拟合成线,以便可以推断未知的样本浓度。这里,这两个术语,即训练和测试(正如在机器学习领域中使用的),分别用于描述标准曲线110的构建和定量未知样本。在传统的定量方法中,使用与具有已知特性的样本的熔融温度有关的第一集数据进行训练通过四个阶段来实现:预处理101、曲线拟合102、单一线性特征提取103和线拟合104,如图1所示的上部分支。
可以可选地执行预处理101以减少诸如背景噪声的因素,从而可以实现样本之间的更准确的比较。
曲线拟合102(例如,使用5参数s型模型、指数模型和/或线性插值)是可选的,并且鉴于扩增曲线在时间/温度上是离散的,且大多数技术需要在给定的时间/温度情况下未明确测量的荧光读数,因此曲线拟合102是有益的。
特征提取103涉及选择和确定靶标数据的特征(或“特性”,例如ct、cy、-log10(f0)、fdm、sdm)。
线(或曲线)拟合104涉及将所确定的特征数据相对于靶标浓度拟合成线(或曲线)110。
预处理101的示例包括基线减除和归一化。曲线拟合102的示例包括使用5参数s形模型、指数模型和线性插值。在特征提取103步骤中提取的特征的示例包括ct、cy或-log10(f0)。线拟合104技术的示例包括主成分分析和随机样本一致性(ransac)。
通过使用相同的前3个框(预处理101、曲线拟合102,线性特征提取103)作为训练,并在训练过程中使用从最终线拟合104步骤生成的线110,完成未知样本的测试(即,基于与未知样本中包含的靶标的熔解温度相关的第二数据,对未知样本中的靶标浓度进行定量),以便对样本进行定量。
提出的方法
提出的方法建立在上一段所述的常规技术的基础上,通过增加标准曲线的维数(在测试阶段中数据与该标准曲线对比)来一起探索、研究和充分利用多个特征。这个新框架在图1的下部分支中给出。
为了进行训练,在该示例实施例中,共有6个阶段:预处理101,曲线拟合102,多特征提取113,高维线拟合114,多维分析115和降维116。测试遵循类似的过程:预处理101,曲线拟合102,多特征提取113,多维分析115和降维116。对于常规方法,预处理101和曲线拟合102是可选的,并且利用合适的多维分析技术,降维的明确步骤也可以是可选的。
同样,预处理101的示例包括基线减除和归一化,曲线拟合102的示例包括使用5参数s型模型、指数模型和线性插值。在多特征提取113步骤中提取的特征的示例包括ct、cy、-log10(f0)、fdm、sdm。高维线拟合114技术的示例包括主成分分析和随机样本一致性(ransac)。多维分析115技术的示例包括计算欧氏距离、计算置信界限、使用标量αi对特征加权,如下文进一步所述。降维116技术的示例包括主成分回归、计算局部最小二乘和投影到原始特征上,如下文进一步所述。
图2a-2c示出了训练过程,图2d-2f显示了使用多维方法的测试。从训练开始,图2a显示了通过将已知核酸靶标连续稀释至已知浓度而对从常规qpcr仪器获得的实时核酸扩增曲线进行处理和曲线拟合。与常规训练相反,从处理后的扩增曲线中提取使用虚拟标记x、y和z表示的多个特征,而非提取单一线性特征。因此,每个扩增曲线已减少为多个3个值(例如x1、y1和z1)的集合,因此可以看作是在3维空间中对彼此绘制的多个点,如图2b所示。重点强调的是,尽管这是3d示例(为了使过程可视化),但是可选地,可以选择任意数量的特征。鉴于本示例中的所有特征均已被选择而使得它们与初始浓度线性相关,训练数据在3d空间中形成1d线,然后使用高维线拟合114对该线进行近似,以生成所谓的多维标准曲线130。尽管数据形成线,但重要的是要理解,数据点不一定精确地位于线上。因此,存在探索该多维空间(称为特征空间)的巨大空间,这将在本文中进行讨论。尽管在该示例中,仅考虑了线性特征(即与靶标浓度线性相关的特征),但是通过进行适当的改变,可以将所公开的方法应用于非线性特征。
出于定量目的,将多维标准曲线映射到单维m0,该函数与靶标的初始浓度线性相关。为了将由这样的函数所描述的曲线与常规标准曲线区分开,此处将其称为定量曲线150。如图2c所示,这是使用降维技术(drt)实现的。在数学上,这意味着drt是以下形式的多元函数:
一旦训练完成,就可以通过如下的测试对至少另一个(例如未知的)样本进行分析(例如定量和/或分类)。与训练相似,显示了处理后的扩增数据(图2d)及其在特征空间中的各自的相应的点(图2e)。鉴于测试点可能位于特征空间中的任何位置,有必要将它们投影到训练中生成的多维标准曲线130上。使用在训练中产生的drt函数
鉴于先前没有公开过这个更高维度的空间,因此有效地突出了通过常规透镜观察定量过程时在该新框架中并不存在的自由度。产生了以下优势:
优势1.每个提取特征的权重可以通过标量α1、…αn来控制。关于该自由度有两个主要观察结果。第一个观察结果是,可以通过将关联的α设置为小值来抑制定量性能差的特征。这引入了非常有用的被称为分离原理的框架属性。分离原理意味着,如果适当选择α,则包括增强多维分析的特征不会对定量性能产生负面影响。优化算法可用于基于目标函数设置α。因此,对于给定的目标,使用所提出的框架的定量性能以最佳单一特征性能为下限。第二个观察结果是,使用数个缩放特征的性能没有上限。因此,有可能胜过本报告中所示的单一特征。
优势2.这种多维思维方式的通用性意味着存在多种降维方法,例如:主成分回归、偏最小二乘回归、甚至投影到单一特征上(例如,使用常规方法中使用的标准曲线110)。鉴于drt可以是非线性的并且可以利用多种特征,因此可以改善预测性能。
优势3.训练和测试数据点不一定像传统技术那样完全位于直线上。该属性是为何在更高维度上拥有更多信息的支柱。例如,特征空间中的两个点越近,它们的扩增曲线就越可能相似(类似于“再生核希尔伯特空间(reproducingkernelhilbertspaces)”)。因此,特征空间中的距离度量可以提供一种计算扩增曲线之间相似性度量的方法。重要的是要理解,距离度量不一定并且实际上不太可能与相似性度量线性相关。例如,离多维标准曲线两倍远的点以两倍可能性出现不一定为真。可以使用训练数据本身来近似这种关系。在训练的情况下,相似性度量可用于识别和消除可能使定量性能产生偏差的离群点。对于测试,相似性度量可以给出未知数据是标准曲线的离群点(即非特异性的或由于qpcr伪像导致的)的可能性,而无需进行pcr后分析(例如熔解曲线或琼脂糖凝胶)。
优势4.反应条件变化的影响,例如退火温度或引物混合物浓度,可以通过特征空间中的模式来捕获。揭示这些趋势和模式对于理解数据是非常有洞察力的。在常规情况下这也是可能的,例如ct如何随温度变化,但是,由于反应条件对不同特征的影响不同,因此在所提出的多维技术中,例如如果在多维空间中观察图案,可以得出置信度较高的结论。例如,考虑到以下因素:温度变化δt会导致不同特征的不同变化,例如δx、δy和δz。因此,如果(如常规技术中)仅使用单一特征x并观察到变化δx,则不太可能以高置信度捕获变化的来源,即δt。而考虑多个特征(如提出的多维技术)并同时观察δx、δy和δz,则可以更加确定该来源是由于δt。
优势4的扩展与靶标浓度变化的影响有关。显然,使靶标浓度变化的模式是已知的:沿着多维标准曲线130的轴线。因此,数据本身足以提示某个具体样本的浓度是否与另一个样本不同。这具有重要意义,因为它允许识别并可能补偿重复之间的差异(这可能是由于诸如稀释和混合之类的实验误差)。这对于低浓度尤其重要,在低浓度情况下,这样的误差通常更为明显。有趣的是观察到,如果使用了多个特征,并且选择了drt以便将多维曲线投影到单个特征(例如ct)上,则定量性能与常规过程相似(例如所提出的框架的仅使用单一特征的特殊情况),但仍保留了由于采用多维空间而获得的机会和洞察力。
示例方法
已经确定可以使用几种不同的技术来实现所提出的方法中的每个步骤(如图1的下部分支所示),以图中作为示例。用于每个框的特定技术可以取决于应用,但是这里描述了特定示例方法以说明该方法的能力和通用性。然而,将理解的是,所描述的方法不限于那些特定示例。
预处理101
在该示例中执行的唯一预处理101是背景减除。这是使用基线减除来实现的:从每条扩增曲线中去除前5个荧光读数的均值。然而,在其他实施例中,可以省略预处理,或者可以执行其他或额外的预处理步骤(例如归一化),并且可以可选地执行更高级的预处理步骤,从而提高性能和/或准确性。
曲线拟合102
曲线拟合的示例模型是5参数s型模型(richards曲线),由下式给出:
其中x是循环数,f(x)是循环x处的荧光,fb是背景荧光,fmax是最大荧光,c是拐点的部分循环,b与曲线的斜率有关,d允许不对称形状(richard’s系数)。
用于对数据拟合成曲线的示例优化算法是信赖域法(trust-regionmethod),基于内部反射牛顿法。在此,由于可以选择5个参数的界限以促成独特而现实的解决方案,因此选择信赖域法要优于莱文贝格-马夸特(levenberg-marquardt)算法。5个参数[fb、fmax、c、b、d]的示例上下限分别为:[-0.5、-0.5、0、0、0.7]和[0.5、0.5、50、100、10]。
多特征提取113
可以提取的特征数n是任意的,但是在此示例中选择了3个特征,以增强框架每个步骤的可视化:ct、cy和-log10(f0),以便于说明。作为结果,在该示例中,特征空间中的每个点都是3维空间中的向量,
例如p=[ct,cy,-log10(f0)]t
其中[·]t表示转置运算符。
请注意,按照惯例,向量是列,并且是粗体小写字母。矩阵是粗体大写字母。这些特征的细节不是本发明的重点,因此将不进一步描述,并假定读者熟悉所述细节。
高维线拟合114
当构建多维标准曲线时,必须在n维空间中拟合一条线。这可以通过多种方式实现,例如使用主成分分析(pca)中的第一主成分,或者使用在有足够数据的情况下对离群点具有鲁棒性的技术(例如随机样本一致性(ransac))来实现。本示例使用前者(pca),因为使用了相对少的训练点来构建标准曲线。
距离和相似性度量(多维分析115)
在本发明中给出了两种距离度量作为示例:欧氏距离和马氏距离,尽管可以理解,还可以使用其他距离度量。
可以通过将一个点正交投影到多维标准曲线130上,然后使用简单几何学计算欧氏距离e,来计算点p与多维标准曲线之间的欧氏距离:
e=|(p-q1)-(q1+p·(q2-q1))|(3)
其中φ计算点p∈rn在多维标准曲线上的投影,点q1、q2∈rn是位于标准曲线上的任意两个不同的点,|·|表示绝对值运算符。
马氏距离定义为多维空间中的点p与分布d之间的距离。类似于欧氏距离,首先将点投影到多维标准曲线130上,并使用以下公式计算马氏距离d:
其中p、p、q1和q2在方程(2)中给出,并且σ是用于对分布d近似的训练数据的协方差矩阵。
为了将距离量度转换为相似度量度,可以显示,如果数据近似正态分布,则马氏距离平方即d2遵循χ2分布。因此,可以使用χ2分布表将特定的p值转换为距离阈值。例如,对于具有2个自由度的χ2分布,p值0.05和0.01分别对应于5.991和9.210的马氏距离平方。
特征权重
如前所述,可以为每个特征分配不同的权重α。为了达到这个目的,可以实现一种简单的优化算法。等效地,可以将误差度量最小化。图3是如何针对所公开的方法使用优化算法来找到最佳参数α的图示。在这个示例中,最小化的误差度量是以下小节中描述的品质因数(figureofmerit)。举例来说,合适的优化算法是权重初始化至统一的nelder-mead单纯型算法,即从不假设用于定量的特征有多好开始。这是基本算法,仅使用20次迭代即可找到权重,因此几乎没有计算开销。
降维116
在该示例中,使用了主成分回归,例如由方程(2)得出的m0=p,并将其与将标准曲线投影到所有三个维度即ct、cy和-log10(f0)上进行比较。
评估标准曲线
与有关评估标准曲线的现有文献相一致,例如,可以分别使用相对误差(re)和平均变异系数(cv)来测量准确性和精度。可以在将标准曲线归一化之后计算出每种浓度的cv,从而可以在整个标准曲线实现合理的比较。两种度量的公式由以下给出:
其中n是训练点数,i是给定训练点的索引,xi是第i个训练数据的真实浓度,x^i是使用标准曲线对xi的估值。
其中,m是浓度数,j是给定浓度的指数,x^j是给定浓度下由j索引的估计浓度的向量。函数std(·)和mean(·)分别执行其向量辐角(vectorargument)的标准差和均值。
参考统计领域,该示例还使用“留一交叉验证”(loocv)误差作为稳定性和总体预测性能的量度。稳定性是指去除训练点时的预测性能。计算loocv的方程为:
其中n是训练点数,i是给定训练点的索引,zi是除第i个训练点之外所有训练点的真实浓度的矢量,z^i是在没有第i个训练点的情况下由标准曲线生成的zi的估值。
为了用于计算α的优化算法以同时最小化三个前述度量,可以方便地引入品质因数q来捕获所有所需的属性。因此,q被定义为所有三个误差之间的乘积,并且可用于启发式地比较遍及定量方法的性能。
q=re×cv×loocv(8)
示例荧光数据集
举例来说,使用几个dna靶标进行qpcr扩增:
(i)使用含有噬菌体λdna序列的合成双链dna(gblocks片段基因,integrateddnatechnologies)来构建和评估标准曲线(每个反应的dna浓度为102-108个拷贝)。参见附录a。
(ii)使用从以下的纯培养物中分离的基因组dna进行离群点检测实验:抗碳青霉烯的(a)携带blaoxa-48的肺炎克雷伯菌(klebsiellapneumoniae),(b)携带blandm的大肠埃希氏菌(escherichiacoli)和(c)携带blakpc的肺炎克雷伯菌。参见附录b。
(iii)使用噬菌体λdna(newenglandbiolabs,目录号n3011s)进行引物变化实验(最终引物浓度范围为每种25nm至每种850nm)和温度变化实验(退火温度为52℃至72℃)。
该示例中使用的所有寡核苷酸均由idt(德国integrateddnatechnologies)合成,并如表1所示。使用primer3(http://biotools.umassmed.edu/bioapps/primer3_www.cgi)内部设计用于λ噬菌体的特异性pcr引物,而用于碳青霉烯抗性基因的特异性检测的引物对则获取自monteiro等人2012。根据制造商的说明,使用faststartessentialdnagreenmaster(roche)进行实时pcr扩增,引物浓度可变,5μl最终反应体积中dna的量可变。使用lightcycler96(roche)进行热循环,首先在95℃下孵育10分钟,然后进行40个如下的循环:95℃下持续20秒;62℃(对于λ)或68℃(对于碳青霉烯抗性基因)下持续45秒;在72℃下持续30秒,在每个循环结束时获取单个荧光读数。每个反应组合,起始dna和特定pcr扩增混合物均一式八份地进行。所有操作均通过熔解曲线分析完成,以确认扩增的特异性并且没有引物二聚体。使用qubit3.0荧光计(lifetechnologies)确定所有dna溶液的浓度。每个实验中包括适当的阴性对照。
表1本示例中使用的特异性pcr引物
结果
以下示例结果说明了使用上述方法的示例实例,所提出的框架的前述优势。鉴于定量性能与特征空间洞察力之间存在分离原理,本节分为两部分:定量性能和多维分析。第一部分显示了由于优势1和2中引入的两个自由度而产生的结果,而后者则针对多维空间中有趣的观察探索了优势3和4。
图4显示了多维标准曲线130和使用来自所有特征的信息进行定量。在图4a中,针对浓度值为102至108(右上至左下)的λdna,使用ct、cy和-log10(f0)构建了多维标准曲线130。每个浓度重复8次。使用主成分分析来实现线拟合。在图4b中,通过使用主成分回归对多维标准曲线进行降维来获得定量曲线150。
定量性能
在该示例中,使用合成的双链dna来构建多维标准曲线130,并评估其相对于单一特征方法的定量性能。在图4a中使所得多维标准曲线130可视化,该多维标准曲线130使用特征ct、cy和-log10(f0)而构建。附录c中给出了按从102至108的范围的浓度分组的每个扩增曲线的计算特征和曲线拟合参数。图4b显示了通过主成分回归进行降维116后获得的一维定量曲线150。为了进行比较,通过将多维标准曲线投影到每个特征上来计算常规示例的标准曲线,如附录d所示。
在该示例中,优化算法经过20次迭代后,用于控制每个特征对定量的贡献的最佳特征权重α收敛到α=[1.6807,1.0474,0.0134],其中权重分别对应于ct、cy和-log10(f0)。该结果易于解释,提示在该三个特征中,-log10(f0)表现出最差的定量性能;这与现有知识一致。再次重点强调的是,尽管-log10(f0)的权重相对于改善定量的其他特征受到抑制,因为它可以揭示多维空间的趋势,因此对其进行保留仍然有很多价值:这将在后文中变得明显。
表2给出了针对所提出的框架的这一特定实例与传统实例的性能度量和品质因数q。附录d中提供了按浓度分组的每个计算误差的分解。可以观察到ct提供最小的re,即准确性,而m0在cv和loocv(即精度和总体预测)上胜过其他方法。就品质因数而言,结合所有误差,与ct、cy和-log10(f0)相比,框架的任意实现分别使定量提高了6.8%、25.6%和99.3%。
表2本示例中使用的定量方法的性能度量以及启发式品质因数q。
re=相对误差,cv=变异系数,loocv=留一交叉验证。
多维分析
鉴于特征空间是新概念,因此存在探索可以实现的目标的空间。在本节中,将探讨特征空间中距离的概念,并通过离群点检测的示例进行证明。此外,显示了在该示例中,当改变反应条件时。模式存在于特征空间中。
图5显示了特征空间中的离群点,特别是λdna的多维标准曲线130以及三个碳青霉烯酶离群点:blaoxa、blandm和blakpc。图5的右侧显示了特征空间区域的放大视图,其中包含重复项的均值以及离群点在标准曲线上的投影。
在该示例中,使用携带碳青霉烯酶基因(即blaoxa、blandm和blakpc)的基因组dna作为多维标准曲线130的故意离群点(deliberateoutlier)。图5显示了特征空间中离群点的均值。附录e中显示了该示例中离群点扩增曲线的计算特征和曲线拟合参数,并使用附录f和图15a至15d中提供的熔解曲线,分析确认了离群点的特异性。鉴于离群测试点不完全位于多维标准曲线130上,图5还显示了离群点均值在多维标准曲线130上的正交投影;如所提出的框架中所述。
为了完全捕获离群点在特征空间中的位置,沿着多维标准曲线130的轴线查看特征空间是方便的。这可以通过将特征空间中的数据点投影到垂直于多维标准曲线130的平面上(如图6a所示)来实现。所得的投影点如图6b所示。
图6显示了使用特征空间进行聚类和检测离群点的多维分析。特别地,图6a显示了针对浓度值为102至108(右上至左下)的λdna使用ct、cy和-log10(f0)的多维标准曲线130。正交于标准曲线的任意超平面以灰色显示。图6b示出了当所有数据点都已经投影到前述超平面上时的特征空间的视图。数据点由对应于blaoxa、blandm和blakpc的训练标准点和离群点组成。由eoxa=1.16、endm=0.77和ekpc=1.41,给出与从多维标准曲线到离群点均值的欧氏距离e对应的误差。对应于p值为0.001的99.9%的置信度以黑色实线显示。图6c在正交视图中显示了变换的空间,其中欧氏距离d等效于马氏距离。黑色圆圈对应于为0.001的p值。
可以观察到,所有三个离群点601、602、603都可以聚类,并且可以与训练数据610清楚地区分开。此外,在该示例中,由eoxa=1.16,endm=0.77和ekpc=1.41,给出从多维标准曲线130到离群点均值的欧氏距离e。鉴于在该示例中,就欧氏距离而言,分别给出距多维标准曲线130最远的训练点为0.22:eoxa、endm、ekpc与0.22之比,为5.27、3.5、6.41。因此,该比率可以用作相似性度量,并且三个集群可以分类为离群点。但是,这种相似性度量有两个隐含的假设:(i)数据遵循统一的概率分布。也就是说,距离为两倍的点成为离群点的可能性也是两倍。通常在没有足够的信息来推断分布的情况下做出该假设。(ii)在不同方向(例如,沿不同轴线)的距离的可能性相同。在特征空间中,这在直觉上是不正确的,因为沿一个方向(例如ct)的变化对扩增曲线的影响不如沿另一个方向(例如-log10(f0))的变化大。重要的是强调特征空间中的方向含有有关扩增动力学变化多少的信息,因此,应沿着相同方向进行扩增反应之间的直接比较。在前述的先前(一维)数据分析中没有捕获该信息。
为了解决前述两个假设,可以使用马氏距离d。显然,通过观察图6b,数据主要在给定方向上变化。马氏距离可以直接使用方程(4)计算。为了可视化马氏距离,可以将特征空间的正交视图(图6b)变换为新的空间(图6c中的“变换的空间”),其中欧氏距离e等效于原始空间中的马氏距离d(即图6b中所示的空间)。从图6c可以看出,在所有方向上的数据都是等概率的,即训练数据610形成了圆形分布。由doxa=12.65,dndm=18.87和dkpc=19.36,给出从多维标准曲线130到离群点601、602、603的均值的马氏距离d。与欧氏距离相比,发现当考虑数据分布时,离群点的位置显著改变。例如,基于欧氏距离,blandm601是最接近的离群点,而使用马氏距离,则提示为blaoxa603。
马氏距离的有用属性是,如果数据近似正态分布,则其平方值遵循χ2分布。因此,可以将距离转换为概率,以捕获不均匀分布。图7显示了将整个训练集叠加有2个自由度的χ2分布的马氏距离d平方的直方图。在该示例中,基于χ2分布表,任何超过约3.717的点都有99.9%的可能性(p值<0.01)是离群点。因此,图7从所有训练数据点的马氏距离平方的直方图的角度显示了数据分布,该所有训练数据点用于构建叠加有2个自由度的x2分布的多维标准曲线。由于所有离群点的马氏距离均显著大于约3.717,因此它们可以被检测为离群点。可以选择其他距离(更大或更小)作为针对马氏距离进行测试的标准,这取决于点为在群点或离群点所需的置信水平。已经显示了距离为3.717,因为它对应于99%的概率,但是还可以选择对应于诸如80%、95%、99.9%的其他概率的距离。
第二示例多维分析(如图8所示)与相对于反应条件的观察模式有关。图8显示了与改变反应条件有关的模式。在所有图中,针对浓度值为102至108个拷贝/反应(右上至左下)的λdna,多维标准曲线均使用ct、cy和-log10(f0)。在图8a中,放大的图像显示了在5×106个拷贝/反应下,将λdna的反应温度从52℃改变为72℃的效果。在图8b中,放大的图像显示了在5×106个拷贝/反应下,对于λdna的每种引物,将引物混合物浓度从25nm改变为850nm的效果。在图8c中,放大的图像显示了针对给定低浓度(102个拷贝/反应),在特征空间中单个训练样本的位置。
在所示的示例中,选择了退火温度和引物混合物浓度来说明这一想法。如熔解曲线分析所示,qpcr的特异性不受影响(参见附录f和图15a至15d)。图8a显示了退火温度对标准曲线的影响。52.0℃至69.9℃的温度仅影响-log10(f0),而69.9℃至72.0℃的变化主要影响ct和cy(参见附录g)。类似地,图8b显示了与引物混合物浓度相关的模式:主要沿-log10(f0)方向观察每种引物从25nm到850nm的变化(参见附录h)。两项实验均表明,ct和cy对退火温度和引物混合物浓度的变化更稳定,这对定量性能有利。此外,在主要由于-log10(f0)的特征空间中观察模式。
基于此发现,先前的(一维)进行方式将表明在后续实验中使用ct或cy。但是,已经认识到,这意味着-log10(f0)生成的模式中包含的信息会丢失。因此,所提出的多维方法结合了对定量性能和模式识别有利的特征:在不损害定量性能的情况下保留所有信息。
最后,另一个有趣的观察结果是,对于低浓度的核酸,训练数据点沿多维标准曲线130的轴线变化,如图8c所示。因此,可以假设这种变化是由于浓度的波动而不是反应动力学的变化导致的。该假设有两个含义:(i)所有点都是在群点,并因此可能是特异的,不需要消耗大量资源的pcr后分析。如附录f所示,使用熔解曲线分析确定特异性。(ii)绝对定量的结果基于3个特征,而不是单一特征,这意味着估计的靶标浓度的置信度增加。
尽管已将所公开的框架描述为考虑了与初始靶标浓度线性相关的特征,但是选择了示例设计选择以降低分析的复杂性,但是可以可选地使用诸如非线性相关特征的其他特征。
此外,将要注意的是,如果两个不相关的pcr反应表现出完全对称的s型扩增曲线,则它们各自的标准曲线可能会重叠,因此会出现一个问题,即是否可以在扩增曲线之间捕获足够的信息,以便在特征空间中区分它们。但是,从分子的角度来看,可以通过调节化学反应来减轻这种影响,以便在不影响反应性能(例如速度、灵敏度、特异性等)的情况下充分改变扩增曲线。
结论
总之,本发明提出了通用的方法、多维标准曲线和特征空间,其实现了先前无法实现的技术和优点。已经说明,使用多个特征的优点是提高了定量的可靠性。此外,除了信任单一特征(例如ct)以外,其他特征(例如cy和-log10(f0))也可以用于检查定量结果是否相似。先前的一维思维方式未能考虑多个自由度以及最终能够由本文公开的通用框架实现的优势。因此,所公开的方法实现了四个主要功能:
(i)选择多个特征并基于定量性能对其加权的能力。
(ii)选择将多个特征映射到代表靶标浓度的单个值的最佳数学方法的灵活性。前两种能力导致了框架的定量性能的下限为最佳单一特征的分离原理,但是仍然保留了来自多个特征的洞察力和多维分析。有趣的是,对于该提出的方法中使用的示例数据集,金标准ct方法胜过其他单一特征。这是说明为什么对使用其他特征存在技术偏见的示例,因为结果是取决于数据的。所公开的框架提供了一种绝对定量的方法,而无需选择具有保证的定量性能的特定特征。本发明表明,与仅使用单一特征相比,通过使用多个特征实际上可以提高定量性能。
(iii)实现应用,例如通过特征空间的元素(例如距离度量、方向、数据分布)所捕获的信息增益来进行离群点检测,这些元素通常在先前的一维方法中没有意义或没有被考虑。
(iv)以特征空间中的特性模式观察反应条件下特定扰动的能力。
公开的方法的示例应用
核酸的绝对定量和多路复用检测单个反应中的几个靶标在生物医学相关领域,尤其是在即时医疗应用中,都有自己的重要和广泛的用途。对于先前的方法,使用qpcr检测多个靶标的能力与靶标数量成线性比例,因此是一项昂贵且耗时的作业。在本发明中,基于多维标准曲线而提出了方法,该方法扩展了通过普通qpcr仪器获得的实时pcr数据的用途。通过应用本文公开的方法,仅使用实时扩增数据即可实现对单个孔中的多个靶标(即在单个反应中使用来自临床样本中的细菌分离物,而无需pcr后操作,例如荧光探针、琼脂糖凝胶、熔解曲线分析或测序分析)同时进行单通道多路复用和鲁棒定量。鉴于应对抗菌耐药性挑战的重要性和需求,本示例中显示了所提出的方法,该方法同时对四种不同的碳青霉烯酶基因进行定量和多路复用:blaoxa-48、blandm、blavim和blakpc,它们占英国报道的产碳青霉烯酶肠杆菌科(carbapenemase-producingenterobacteriaceae)的97%。
核酸(dna和rna)的定量检测在生物医学领域中有许多应用,包括基因表达分析、遗传疾病易感性、突变检测和临床诊断。一种这样的应用是筛选细菌中的抗生素抗性基因:产碳青霉烯酶肠杆菌(cpe)的出现和扩散代表着全球范围内对公共卫生的迫在眉睫的威胁之一。抗碳青霉烯酶菌株的侵袭性感染与高死亡率(高达40-50%)关联,代表了全世界关注的的主要公共卫生问题。快速准确地筛选产碳青霉烯酶肠杆菌科(cpe)的携带,对于成功的感染预防和控制策略以及病床管理至关重要。然而,基于碳青霉烯易感性的常规cpe实验室检测是具有挑战性的:i)基于培养的方法由于易于获得且成本低廉而是方便的,但是其有限的灵敏度和较长的周转时间对于感染控制实践来说可能并不总是最佳的;(ii)与基于培养的方法相比,诸如qpcr的核酸扩增技术(naat)提供了快速的结果和增加的灵敏度和特异性。但是,这些方法通常过于昂贵,并且需要使用精细复杂的设备作为医疗保健系统中的筛选工具;和(iii)多路复用naat具有显著的灵敏度、成本和周转时间优势,从而增加了结果的通量和可靠性,但是生物技术行业一直在努力使用可用技术满足对高水平多路复用不断增长的需求。因此,对于可在现有医疗保健机构中被成功采用的新分子工具的临床需求尚未得到满足。
目前,qpcr是快速检测cpe和其他细菌感染的金标准。该技术以基于荧光的数据检测为基础,从而能实时监控pcr扩增的动力学。使用不同的方法来分析qpcr数据,循环阈值(ct)法是确定特定靶序列绝对浓度的优选方法。ct法假设所比较的样本具有相似的pcr效率,并将其定义为存在可检测的明显的荧光增加的扩增的对数线性区域中的循环数。已经开发出替代方法来定量模板核酸,包括标准曲线法、线性回归和非线性回归模型,但是它们都不允许同时进行靶标区分。多路复用分析系统允许在一种测定中检测多个核酸靶标,并可以提供所需的样本表征速度,同时仍节省成本和资源。但是,在实际情况下,多重定量实时pcr(qpcr)会受到实时定量热循环仪的检测通道数量的限制,并且通常依赖于熔解曲线分析、琼脂糖凝胶或测序来进行靶标确认。这些pcr后过程增加了诊断时间,限制了高通量应用,并导致扩增子受到实验室环境的污染。因此,迫切需要开发灵敏、准确且低成本的简化分子工具。
所公开的方法允许现有技术获得多重pcr的益处作为回报,同时降低了cpe筛选的复杂性;到账成本降低。这是由于以下事实:所提出的方法(i)使用单个荧光通道实现多参数成像;(ii)与未修饰的寡核苷酸相容;以及(iii)不需要pcr后处理。这可以通过使用多维标准曲线来实现,在本示例中,该多维标准曲线使用从扩增曲线中提取的ct、cy和-log10(f0)特征而构建。在该示例中,我们表明所描述的方法可以成功地应用于cpe筛选。这提供了一个概念证明,可以仅使用实时扩增数据在单个通道中对几个核酸靶标进行多路复用。然而,将理解的是,所公开的方法可以应用于任何核酸的检测以及任何病原性或非病原性基因组材料的检测。
如参考图9至图12和图16所描述的,所公开的方法的该示例应用描述了本文所公开的方法,该方法被应用于生成多维标准曲线(msc)以便仅使用扩增形状来同时进行dna定量、多重靶标区分和离群点检测。本文中,我们提出了msc以用于同时进行核酸定量、离群点检测和单通道多路复用,而无需熔解曲线分析或任何其他pcr后操作。本文公开的方法结合了与靶标浓度呈线性关系的扩增曲线的多个特征(例如ct、f0和cy0),以针对每个扩增曲线生成特性指纹。然后,将指纹绘制在多维空间中以生成多变量标准曲线,该多变量标准曲线可提供足够的信息增益,以便同时进行定量、多路复用和离群点检测。该方法已经被验证用于快速筛选四个临床上最相关的碳青霉烯酶基因(blakpc、blavim、blandm和blaoxa-48),并且显示与目前的方法相比增强定量。因此,所提出的方法具有提供更全面和可操作的诊断的潜力,从而改善了患者护理并降低了医疗保健费用。
图9是使用一维和多维分析方法进行单通道多重定量pcr的示例实验流程的说明。在本示例中,针对靶标1、2和3,通过多重qpcr扩增了未知dna样本。从扩增曲线中提取出诸如α、β和γ的特征。重点强调的是,可以选择任何数量的靶标和特征。
在图9(a)所示的示例常规一维分析中,通过使用单一特征对已知靶标进行系列稀释生成了三个常规标准曲线。鉴于无法基于这些标准曲线鉴定靶标,需要进行pcr后分析以进行靶标鉴定和定量。例如,将阈值ct相对于参考靶标1的log10浓度绘图,并生成拟合数据的回归线,从而构建standard1(std1)。从一维标准推断出未知样本中靶标丰度的相对值。但是,在单通道qpcr多路复用测定中,由于无法将单一特征外推到特定的标准曲线,因此多个标准曲线的存在会妨碍未知样本中靶标的鉴定和定量。因此,需要pcr后分析(例如琼脂糖凝胶、熔解曲线或测序)来进行靶标鉴定和定量。
在本文公开的多维分析(b)中,多维标准曲线和特征空间用于仅基于扩增曲线来同时定量和区分所关注靶标:消除了对昂贵耗时的pcr后操作的需要。与常规标准曲线相似,在统一的实验条件下,通过使用浓度已知的标准溶液来生成多维标准曲线。在该示例中,从每个扩增曲线中提取出多个特征α、β和γ并针对彼此进行绘制。因为每个扩增曲线已减小为三个值,所以它可以表示为3d空间中的单个点(在实施例中可以使用更多或更少的维度)。在该示例中,针对给定靶标,来自每种浓度的扩增曲线将生成三维集群,这些集群可以通过高维线拟合而连接,以生成靶标特异的多维标准曲线130。包含所有数据点的多维空间称为特征空间,可以将这些数据点投影到正交于标准曲线的任意超平面,以进行靶标分类和离群点检测。可以通过使用聚类技术对未知样本进行可靠分类,并且可以通过将所有特征组合成称为m0的统一特征中来实现增强的定量。重点强调的是,可以已经选择了任何数量的靶标和特征,在该示例中已经选择了三重测定和三个特征以全面地说明该概念。
实施例引物和扩增反应条件
所有寡核苷酸均由integrateddnatechnologies(荷兰)合成,无需另外纯化。引物名称和序列如表3所示。每次扩增反应均以5μl最终体积进行,其中含有2.5μlfaststartessentialdnagreenmaster2x浓缩液(德国rochediagnostics)、1μlpcr级水、0.5μl含有四种引物集的10x多重pcr引物混合物(每种引物5μm)和1μl不同浓度的合成dna或细菌基因组dna。pcr扩增由以下组成:在95℃下进行10分钟,然后进行45个如下的循环:在95℃下进行20秒,在68℃下进行45秒和72℃下进行30秒。为了验证产物的特异性,一个熔解循环如下进行:在95℃下进行10秒钟,在65℃下进行60秒钟,在97℃下进行1秒钟(从65℃到97℃连续读数)。使用lightcycler96实时pcr系统(德国rochediagnostics),将反应加载到lightcycler480多孔板96(德国,rochediagnostics)中,每个实验条件运行5至8次。
表3用于cpe多重qpcr测定的引物。
序列从5'到3'方向给出。大小表示pcr扩增产物。
合成dna样本和基因组dna样本
四种
表4该示例中使用的样本。
公开方法的示例
使用本文先前描述的方法实现了用于同时定量和多路复用的数据分析。因此,数据分析有以下几个阶段:预处理101、曲线拟合102、多维特征提取113、高维线拟合114、相似性度量(多维分析)115和降维116。
预处理101:(可选)在该示例中,通过基线校正进行背景减除。这通过从每条原始扩增曲线中去除前5个荧光读数的均值来实现。
曲线拟合102:(可选)在该示例中,拟合了5参数s型模型(richard’s曲线),以对扩增曲线进行建模:
其中x是循环数,f(x)是循环x处的荧光,fb是背景荧光,fmax是最大荧光,c是拐点的部分循环,b与曲线的斜率有关,d允许不对称形状(richard’s系数)。在该示例中,用于将数据拟合成曲线的优化算法是信赖域法且基于内部反射牛顿法。5个参数[fb、fmax、c、b、d]的上下限分别为:[-0.5、-0.5、0、0、0.7]和[0.5、0.5、50、100、10]。
特征提取113:在本示例中选择了三个特征以构建多维标准曲线:ct、cy和-log10(f0)。这些特征的细节不是本发明的重点。将意识到,在其他示例中可以使用更少或更多数量的特征。
线拟合114:在该示例中,使用最小二乘法进行线拟合,即主成分分析(pca)中的第一主成分。
相似性度量(多维分析)115:该示例中使用的相似性度量为马氏距离d:
其中p、p、q1和q2在方程(2)中给出,并且σ是用于对分布d近似的训练数据的协方差矩阵。
特征权重:为了使定量性能最大化,可以为每个特征分配不同的权重α。为了达到这个目的,可以实现简单的优化算法。等效地,可以将误差度量最小化。在这个示例中,最小化的误差度量是以下小节中描述的品质因数(figureofmerit)。优化算法是权重初始化至统一的nelder-mead单纯型算法(32,33),即以不假设用于定量的特征有多好开始。这是基本算法,仅使用20次迭代即可找到权重,因此几乎没有计算开销。
降维116:为了比较它们的性能,使用了三种降维技术。前三个是对每个单独特征(即ct、cy和-log10(f0))进行简单投影。最终的方法使用主成分回归来使用向量计算被称为m0的特征
p=[ct,cy,-log10(f0)]t
其中[·]t表示转置运算符。
如方程(2)所示,用于针对任意数量特征计算m0的一般形式为:
其中φ计算点p∈rn在多维标准曲线130上的投影。点q1、q2∈rn是位于标准曲线上的任意两个不同的点。
如以上总体公开中所述,执行标准曲线的评估。
结果
在该示例中,显示通过使用多维标准曲线分析qpcr中的荧光扩增曲线,可以实现对细菌分离物中的blaoxa-48、blandm,blavim和blakpc内酰胺酶基因同时进行鲁棒定量和多路复用检测。本节分为两部分:多路复用和鲁棒定量。首先,证明了可以实现单通道多路复用,这是有意义并且非常有利的。
使用多维分析的靶标区分
图11显示了针对blaoxa、blandm、blavim和blakpc基因的四条扩增曲线及其各自的衍生熔解曲线。四条曲线已被选择为具有相似的ct(19.40.5),因此每个反应具有不同的靶标dna浓度。仅使用该信息,即在常规技术中,将需要pcr后处理(例如熔解曲线分析)来区分靶标。相同的论点也适用仅观察cy和f0时。
本文公开的多维方法显示,考虑多个特征会提供足够的信息增益,以便使用多维标准曲线130区分离群点与特定靶标。利用该属性,可以建立几条多维标准曲线以区分多个特定靶标。图10显示了使用ct、cy和-log10(f0),使用针对四种靶基因修正的单一引物而构建的多维标准曲线1301、1302、1303、1304。直观地观察到,这四个标准在多维空间中足够远以区分训练样本。也就是说,仅使用从单个通道的扩增曲线中提取的特征就可能将未知的dna样本分类为多个特定靶标(或离群点)中的一个。
为了证明这一点,针对多维标准1301、1302、1303、1304测试了表4中给出的11个样本。用于对未知样本进行分类的相似性度量是马氏距离,使用0.01的p值作为阈值。为了完全捕获离群点在特征空间中的位置,沿着多维标准曲线1301、1302、1303、1304的轴线查看特征空间是方便的。图11中提供了熔解曲线,以证明实时扩增曲线属于不同的qpcr产物。在开发该方法之前,不可能使用单通道将扩增曲线与特定的测定相关联。因此,使用熔解曲线作为确认方法。
图12显示了该示例中针对四个标准的马氏空间。通过将所有数据点投影到与每个标准曲线正交的任意超平面上来构建该可视化,如上文公开的一般方法中所述。第一个观察结果是,来自每个标准的训练点(合成dna)都一起聚类在各自的马氏空间中,其p值<0.01。这证实了以下事实:在3个选定的特征中有足够的信息来区分4条捕获扩增反应动力学的标准曲线。
图12使用所公开的多维分析,该多维分析使用特征空间对未知样本进行聚类和分类。如前所述,对于该示例,已经使用了与每个多维标准曲线正交的任意超平面来投影所有数据点,包括四个多维标准(训练标准点)和八个未知样本(测试点)的各自的浓度的重复。圆形标注被放大以使样本相对于每个所关注标准的位置可视化。每个放大的圆形标注中的黑色圆形点表示所关注标准(每个浓度5至8个重复),默认情况下(0,0)放置在马氏空间的中心;深灰色星号代表其他标准;浅灰色星号代表测试点(每个样本3个重复);菱形显示每个样本的均值。每个黑色圆圈对应于为0.001的p值。
第二个观察结果是,具有单一抗性的测试样本(细菌分离株)的均值(样本1-8)落在正确的训练点集群(p值<0.01)之内。如附录中所述,使用熔解曲线分析来验证结果。可以在条形图中简便地捕获来自测试的结果,如图16所示。然而,重要的是使数据可视化以确认马氏距离是合适的相似性度量。当特征空间中的训练数据点近似正态分布时,则马氏空间中训练数据点的分布近似为圆形,如图6c所示。图16显示了在该示例中从标准点到样本测试的平均马氏距离。样本测试点和标准测试点的分布之间的平均距离已用于鉴定未知样本中碳青霉烯酶基因的存在。当数据近似正态分布时,马氏距离可以转换为概率。可以将相对于所关注标准的平均距离小于约3.717的样本测试点分类在该集群内(p值<约0.01)。样本1、2和5分类在blaoxa-48集群内,样本4和6分类在blandm集群内,样本3和7分类在blavim集群内,样本8分类在blakpc集群内。样本9不属于任何集群(p值>=约0.01)。dna扩增后,还进行了样本的熔解曲线分析,以确定多重qpcr产物的特异性。熔解曲线分析与基于马氏距离的样本分类非常吻合。
可以观察到,在每个变换的空间中使用适当的聚类技术,可以区分一个点是否属于靶标。此外,如果将概率分配给每个数据点,则可以将样本可靠地分类为给定标准,并同时对其进行定量。鉴于训练数据近似服从多元正态分布,马氏距离平方可以提供概率的度量。
鲁棒定量
如果已经建立了多路复用,则可以使用任何常规方法(例如金标准循环阈值ct)获得定量。但是,如本文公开的一般方法所示,可以使用特征m0来实现增强的定量,该特征m0组合了所有用于最佳绝对定量的特征。在本研究中,最优性的度量是将准确性、精度、鲁棒性和整体预测能力相结合的品质因数,如方程x所示。表5显示该本示例中使用的3个选定特征(ct、cy和-log10(f0))和m0的品质因数。还显示了改进百分比。可以看出,与最佳单一特征相比,定量总是得到改善的。对于blaoxa-48、blandm、blavim和blakpc,分别改善了30.69%、14.39%、2.12%和35.00%。这是多维框架的结果。进一步关注的是,观察到在常规方法中,没有单独的方法能对所有靶标均表现最佳。因此,就始终将m0视为最佳执行方法的意义而言,m0是最具鲁棒性的方法。
表5针对绝对定量将常规特征与m0进行比较的品质因数。
%imp.=m0相对下一种最佳方法的改善百分比(均为粗体)
*品质因数值使用-log10(f0)来计算
附录a
合成双链dna的核苷酸序列订购自integrateddnatechnologies,其中包含λ噬菌体dna靶标。
粗体表示正向λpcr引物,斜体表示反向λ引物。
附录b
从细菌分离物中制备模板以用于实时pcr测定。
将来自纯培养物的一个菌落环悬浮在50μl消化缓冲液(tris-hcl10mmol/l,edta1mmol/l,ph8.0,含5u/μl溶菌酶)中,并在37℃下干浴孵育30分钟。随后添加0.75μl的20μg/μl蛋白酶k(sigma),并将溶液在56℃孵育30分钟。煮沸10分钟后,将样本以10000×g离心5分钟,并将上清液转移至新试管中,并在使用前储存于-80℃。
附录c
λdna标准构建的实验值。
使用242bp的双链dnaλ噬菌体由标准曲线构建含有所需靶标序列的分子(gblock基因片段,idt)。每个条件均一式八份地运行。
附录d
附录e
离群点检测实验的实验值
基因组dna提取自纯细菌培养物。所有靶标为1.00e+05gdna拷贝/反应。
每个条件均一式八份地运行。
附录f
λdna标准实验的熔解曲线分析如图15a所示:该图显示了使用内部λ引物、使用242bp双链dna分子(订购自idt的gblock基因片段)的合成λdna标准实验的平均熔解曲线峰。该实验中使用从108到101个拷贝/反应的十倍稀释液,每个测试浓度8次反应。所有阳性反应的平均熔解曲线峰为80.49℃(sd=0.08℃),在其他退火温度下未观察到二次熔解事件。
离群点检测实验的熔解曲线分析,如图15b所示:该图显示blaoxa48的平均熔解曲线峰为80.66℃(sd=0.07℃),blandm的平均熔解曲线峰为83.97℃(sd=0.10℃),blakpc的平均熔解曲线峰为90.76℃(sd=0.10℃)。每个gdna样本进行一式八份的反应,106个基因组拷贝/反应。在其他退火温度下未观察到二次熔解事件。特异性引物组选自monteiro等人2012。
引物浓度变化实验的熔解曲线分析,如图15c所示:该图显示了使用噬菌体λdna和内部λ引物进行引物浓度实验的平均熔解曲线峰。对于测试的引物浓度,观察到的平均熔解曲线峰为:25nm时为80.18℃(sd=0.09℃);100nm时为80.10℃(sd=0.09℃);175nm时为80.18℃(sd=0.04℃);250nm时为80.13℃(sd=0.11℃);325nm时为80.21℃(sd=0.21℃);400nm时为80.34℃(sd=0.06℃);475nm时为80.46℃(sd=0.08℃);550nm时为80.50℃(sd=0.09℃);625nm时为80.63℃(sd=0.09℃);700nm时为80.66℃(sd=0.07℃);775nm时为80.73℃(sd=0.06℃);850nm时为80.87℃(sd=0.07℃)。每种引物浓度进行一式八份的反应。在其他退火温度下未观察到二次熔解事件。
温度变化实验的熔解曲线分析,如图15d所示:该图显示了使用噬菌体λdna和内部引物进行温度变化实验的平均熔解曲线峰。对于测试的温度,观察到的平均熔解曲线峰为:52.0℃时为80.53℃(sd=0.10℃);53.0℃时为80.52℃(sd=0.13℃);54.9℃时为80.48℃(sd=0.03℃);57.3℃时为80.53℃(sd=0.07℃);59.9℃时为80.53℃(sd=0.06℃);62.7℃时为80.43℃(sd=0.17℃);65.4℃时为80.51℃(sd=0.09℃);67.8℃时为80.51℃(sd=0.09℃);69.9℃时为80.47℃(sd=0.13℃);71.3℃时为80.35℃(sd=0.09℃);71.9℃时为80.35℃(sd=0.08℃);72.0℃时为80.36℃(sd=0.08℃)。每个测试温度进行进行一式八份的反应。在其他退火温度下未观察到二次熔解事件。
附录g
温度变化实验的实验值
λdna作为靶标(neb,目录号n3011s),106个基因组拷贝/反应。温度以摄氏度为单位。每个实验条件均一式八份地运行。
附录h
引物浓度变化实验的实验值
λdna作为靶标(neb,目录号n3011s),106个基因组拷贝/反应。引物浓度以纳摩尔(nm)为单位,每种引物的范围为25nm至850nm。每个实验条件均一式八份地运行。
根据前文的描述和附图,包括以上提到的那些方面和实施例的优点和技术效果对于本领域技术人员将是显而易见的。
将会理解,所描述的方法可以由一台或多台计算机在布置成执行所述方法的一个或多个计算机程序的控制下执行,所述计算机程序被存储在一个或多个存储器和/或其他种类的计算机可读介质中。
图13显示了可用于实现本文描述的方法的计算机系统1300的示例,所述计算机系统1300包括一个或多个服务器1310、一个或多个数据库1320以及一个或多个计算设备1330,所述服务器1310、数据库1320和计算设备1330通过计算机网络1340彼此通信连接。网络1340可以包括适合于数据传输或通信的任何种类的计算机网络中的一种或多种,例如局域网、广域网、城域网、互联网、无线通信网络1350、电缆网络、数字广播网络、卫星通信网络、电话网络等。计算设备1330可以是移动设备、个人计算机或其他服务器计算机。数据也可以通过物理计算机可读介质(例如记忆棒、cd、dvd、蓝光光盘等)进行通信,在这种情况下,可以省略全部或部分网络。
一个或多个服务器1310和/或计算设备1330中的每一个可以在一个或多个计算机程序的控制下操作,所述一个或多个计算机程序被布置为执行参考任何实施例描述的方法步骤的全部或子集,从而与一个或多个服务器1310和/或计算设备1330中的另一个进行交互,以便与一个或多个数据库1320一起共同执行所描述的方法步骤。
参照图14,图13中的一个或多个服务器1310和/或计算设备1330中的每一个可以包括如在此作为示例所示的特征。所示的计算机系统1400包括处理器1410、存储器1420、计算机可读存储介质1430、输出接口1440、输入接口1450和网络接口1460,它们可以借助于一个或多个数据总线1470彼此通信。应当理解,取决于所述系统的所需功能,可以省略这些特征中的一个或多个,并受制于实现所述方法/系统所需的功能,可以使用具有较少组件或额外/替代的其他计算机系统代替。
计算机可读存储介质可以是任何形式的非易失性和/或非暂时性数据存储设备,例如磁盘(例如硬盘驱动器或软盘)或光盘(例如cd-rom、dvd-rom或蓝光光盘)或存储设备(例如rom、ram、eeprom、eprom、闪存或便携式/可移动存储设备)等,并且可以存储数据、根据本文公开的一个或多个实施例的应用程序指令和/或操作系统。该存储介质对处理器可以是本地的,或者可以通过计算机网络或总线来访问。
处理器可以是能够执行根据实施例的方法步骤的任何装置,并且可以例如包括单个数据处理单元或并行或彼此协作操作的多个数据处理单元,或者可以被实现为可编程逻辑阵列、图形处理器或数字信号处理器或其组合。
输入接口被布置为从用户接收输入并将其提供给处理器,并且可以包括例如鼠标(或其他定点设备)、键盘和/或触摸屏设备。
输出接口可选地在处理器的控制下向系统用户提供视觉、触觉和/或听觉输出。
最后,网络接口为计算机提供了通过一个或多个数据通信网络发送/接收数据的功能。
实施例可以在任何合适的计算或数据处理设备上执行,例如服务器计算机、个人计算机、移动智能电话、机顶盒、智能电视等。这样的计算设备可以包含合适的操作系统,例如,unix、windows(rtm)或linux。
应当理解,可以在不影响方法和系统的功能或其优点/技术效果的情况下改变上述的功能划分。为了能够理解本发明,上述功能划分是作为示例给出的,因此是概念性的而非限制性的,本发明由所附权利要求书限定。本领域技术人员还将认识到,所描述的方法步骤可以以不同的顺序组合或执行,而不影响如权利要求中限定的本发明所产生的优点和技术效果。
还将意识到,所描述的功能可以作为硬件(例如使用现场可编程门阵列、asic或其他硬件逻辑)、固件和/或软件模块实现,或者作为这些模块的混合实现。还应当理解,承载了被布置为实现本发明的一个或多个方面的计算机程序的计算机可读存储介质和/或传输介质(例如,通信信号、数据广播、两个或多个计算机之间的通信链路等)可以体现本发明的方面。如本文所用,术语“计算机程序”是指设计用于在计算机系统上执行的一系列指令,并且可以包括源代码或目标代码、一个或多个功能、模块、可执行应用程序、小程序、小服务程序、库和/或其他可由计算机处理器执行的指令。
还将认识到,可以通过上述联网的计算机系统组件来获得第一数据(训练数据)集和第二数据(未知样本数据)集,诸如通过从存储中检索,由用户通过输入设备输入来获得。诸如在群点/离群点确定以及确定的样本浓度的结果数据也可以使用上述存储元件存储,和/或输出到显示器或其他输出设备。也可以使用这样的存储元件来存储多维标准曲线130和/或由一维函数定义的标准曲线。如本文所述,前述处理器可以处理这样的存储和输入的数据,并相应地存储/输出结果。
如本领域技术人员将理解的,可以改变以上实施例的细节而不脱离由所附权利要求限定的本发明的范围。对以上实施例的特征的许多组合、修改或变更对于技术人员来说将是显而易见的,并且旨在形成本发明的一部分。鉴于以上公开内容,可以通过做出对技术人员显而易见的适当改变而在任何其他实施例中使用与一个实施例或示例具体相关的所描述的任何特征。
- 还没有人留言评论。精彩留言会获得点赞!