用于使用神经网络进行种系和体细胞变体调用的系统和方法与流程
相关申请的交叉引用
本申请要求2018年8月13日提交的美国临时申请号62/718,338和2019年5月31日提交的美国临时申请号62/855,541的优先权,这些临时申请各自借此以全文引用的方式并入。
通过引用并入
本说明书中提到的所有出版物和专利申请都以相同的程度通过引用并入本文,就好像每个单独的出版物或专利申请都被具体且单独地指出通过引用并入一样。
本发明的实施例总体上涉及用于变体调用的系统和方法,并且更具体地涉及使用机器学习算法进行变体调用。
背景技术:
使用测序数据鉴别遗传突变是生物信息学中的一项至关重要的任务,它对包括癌症在内的多种疾病的诊断、预后和治疗具有影响。这是一项不平凡的任务,尤其是当用于生成测序数据的测序技术具有较高的错误率(这是单分子测序技术所常见的)时或当突变以较低的频率(癌症突变尤其如此)或在复杂的基因组区域内发生时。测序样本中癌症突变的频率较低可能是由于样本污染(因为肿瘤样本中可能含有一些来自正常细胞的dna)或肿瘤异质性所致。
传统方法通过限制特定技术和待测突变类型来缩窄问题的范围。因此,存在用于鉴别种系和癌症突变的不同成套工具和框架,其根据突变是大还是小进一步分层。另外,用于低错误测序技术(例如,短读取测序)的方法不同于用于高错误测序技术(例如,单分子测序)的方法。差异主要体现在数据标志(datasignature)以及用于将错误与真实突变区分开的统计模型中。显式数据标志和统计模型的使用限制了传统方法对多种技术的通用性。此外,即使是传统方法,也需要调整参数来捕获样本的特定特征,这可能取决于多种因素,包括所使用的技术、测序方案、测序样本、样本纯度等。
深度学习是一种机器学习技术,只要有正确的训练数据,就可以使用多个数据层隐式地捕获数据标志和统计模型。就突变检测问题的通用性而言,这使得深度学习成为潜在的有吸引力的解决方案。已知几种用于对种系突变进行分类的深度学习解决方案。
poplin,r.等人(“creatingauniversalsnpandsmallindelvariantcallerwithdeepneuralnetworks”,biorxiv,092890(2016))提出了一种方法,其中首先通过基因组扫描鉴别候选变体定位,然后将在每个所鉴别的候选周围比对的读取的堆叠图像作为输入提供到卷积神经网络(cnn)中,然后该卷积神经网络预测候选变体的基因型。这种方法不是很实用,因为它需要复杂的神经网络架构和大量内存。
luo,r.等人(“clairvoyante:amulti-taskconvolutionaldeepneuralnetworkforvariantcallinginsinglemoleculesequencing.biorxiv,310458(2018))使用数据汇总方法,该方法包括扫描基因组以鉴别候选变体位点,并为每个候选位置准备多个输入矩阵,每种变体类型一个矩阵。chin,j.(“simpleconvolutionalneuralnetworkforgenomicvariantcallingwithtensorflow”,https://towardsdatascience.com/simple-convolution-neural-network-for-genomic-variant-calling-with-tensorflow-c085dbc2026f,2017年7月16日出版)中讨论了类似的技术。这些方法也不是最优的,因为它们不能清楚地表示整个窗口中的所有插入序列。例如,clairvoyante系统为插入事件分配了一个独立的输入矩阵,该矩阵仅捕获变体定位处的插入,而忽略窗口内其他定位处的真实/错误插入。有关整个窗口中插入序列精确定位的全图信息对于将真实插入变体与背景噪声区分开来至关重要。
torracinta,r.等人(“adaptivesomaticmutationscallswithdeeplearningandsemi-simulateddata.biorxiv,79087(2016))描述了将六层全连接神经网络应用于一组手动提取的特征。但是仅具有全连接层的系统不能利用卷积神经网络提供的功能,即,使用见于候选变体的局部基因组情境中的模式直接从原始序列数据中学习特征表示。此外,由于全连接的网络更加复杂,因此与使用cnn所能实现的相比,该方法的通用性和可扩展性较差。
技术实现要素:
本发明的总体上涉及用于变体调用的系统和方法,并且更具体地涉及使用机器学习算法进行变体调用。
在一些实施例中,提供了用于种系变体调用的方法。该方法可以包括获得参考序列、多条序列读取和候选变体在序列读取内的位置;通过将一个或多个空格插入一条或多条序列读取中而获得经扩充序列读取;通过将一个或多个空格插入参考序列中而获得经扩充参考序列;将候选变体周围的经扩充序列读取的片段转换为样本矩阵;将候选变体周围的经扩充参考序列的片段转换为参考矩阵;将样本矩阵和参考矩阵提供给受训神经网络;以及,在受训神经网络的输出端,获得关于多条序列读取内的变体的预测数据。
在一些实施例中,该方法进一步包括检测多条序列读取内的一个或多个插入碱基,其中扩充序列读取和参考序列包括:对于在任何序列读取中的任一条中检测到的每个插入碱基,在该插入碱基的位置处将空格插入参考样本中。
在一些实施例中,该方法进一步包括:对于在所述序列读取中的任一条中检测到的每个插入碱基,在该插入碱基的位置处将空格插入每一条在该插入碱基的位置处没有检测到插入的序列读取中。
在一些实施例中,样本矩阵包括至少四个表示四种类型的核苷酸碱基的行,每一行表示在经扩充序列读取的片段内不同位置处的相应核苷酸碱基类型的碱基数;和至少一个表示在经扩充序列读取的片段内不同位置处的插入的空格数的行。
在一些实施例中,参考矩阵具有与样本矩阵相同的维数,并且其中参考矩阵提供经扩充参考序列内不同核苷酸碱基和空格的具体定位的完全表示。
在一些实施例中,受训神经网络包括受训卷积神经网络。
在一些实施例中,该方法进一步包括将以下至少一项提供给受训神经网络:变体位置矩阵,其表示经扩充序列读取的片段内候选变体的位置;覆盖率矩阵,其表示经扩充序列读取的片段的覆盖率或深度;比对特征矩阵,其表示经扩充序列读取的比对特征;知识性碱基矩阵,其表示关于一个或多个变体的公开已知信息的信息。
在一些实施例中,关于变体的预测数据包括以下至少一项:变体的预测类型;变体的预测位置;变体的预测长度;以及变体的预测基因型。
在一些实施例中,关于变体的预测数据包括变体的预测类型,并且其中神经网络配置为产生用于变体的预测类型的多个值中的一个,该多个值包括:第一值,其指示变体为假阳性的概率;第二值,其指示变体为单核苷酸多态性变体的概率;第三值,其指示变体为缺失变体的概率;以及第四值,其指示变体为插入变体的概率。
在一些实施例中,提供了用于体细胞变体调用的方法。该方法可以包括获得多条正常序列读取和多条肿瘤序列读取;将正常序列读取的片段和肿瘤序列读取的片段分别转换为正常样本矩阵和肿瘤样本矩阵;将正常样本矩阵和肿瘤样本矩阵馈入受训卷积神经网络中;以及在受训卷积神经网络的输出端,获得多条肿瘤序列读取内的体细胞变体的预测类型。
在一些实施例中,多条肿瘤序列读取表示患者的肿瘤样本的遗传信息,并且多条正常序列读取表示患者的正常样本的遗传信息。
在一些实施例中,将正常序列读取转换为正常样本矩阵包括通过将一个或多个空格插入一条或多条正常序列读取中而扩充正常序列读取的片段;并且将肿瘤序列读取的片段转换为肿瘤样本矩阵包括通过将一个或多个空格插入一条或多条肿瘤序列读取中而扩充肿瘤序列读取的片段。
在一些实施例中,肿瘤样本矩阵包括至少一个用于每种核苷酸碱基类型的行,每一行表示在肿瘤序列读取的片段内每个位置处的相应核苷酸碱基类型的出现次数;和至少一个表示在肿瘤序列读取的片段内每个位置处的插入空格数的行。
在一些实施例中,该方法进一步包括向受训卷积神经网络提供一个或多个矩阵,该一个或多个矩阵表示从一个或多个其他变体调用者获得的特征,该调用者已经分析了多个肿瘤序列读取和/或多个正常序列读取。
在一些实施例中,该方法进一步包括获得参考序列;将参考序列转换为参考矩阵;以及将参考矩阵与正常样本矩阵和肿瘤样本矩阵一起馈入受训卷积矩阵中。
在一些实施例中,提供一种非暂时性计算机可读介质,其包含指令,该指令当由计算机系统的一个或多个处理器执行时,使计算机系统实施包括以下各项的操作:获得多条正常序列读取和多条肿瘤序列读取;将正常序列读取的片段和肿瘤序列读取的片段分别转换为正常样本矩阵和肿瘤样本矩阵;将正常样本矩阵和肿瘤样本矩阵馈入受训卷积神经网络中;以及在受训卷积神经网络的输出端,获得多条正常序列读取内的体细胞变体的预测类型。
在一些实施例中,提供一种计算机系统,其包含一个或多个处理器并且耦接至一个或多个存储有指令的非暂时性计算机可读存储器,该指令当由所述计算机系统执行时,使该计算机系统实施包括以下各项的操作:获得多条肿瘤序列读取;通过将一个或多个空格插入一条或多条肿瘤序列读取中获得经扩充肿瘤序列读取;将肿瘤序列读取的片段转换为肿瘤样本矩阵;将正常样本矩阵和肿瘤样本矩阵馈入受训神经网络中;以及在受训神经网络的输出端,获得多条肿瘤序列读取内的体细胞变体的预测类型。
在一些实施例中,提供了用于变体调用的方法。该方法可以包括:获得参考序列和多条序列读取;任选地对多条序列读取与参考序列实施第一比对,除非所获得的多条序列读取和参考序列是以已经比对的配置获得的;从经比对序列读取和参考序列中鉴别候选变体位置;在候选变体位置周围扩充序列读取和/或参考序列,以实现多条序列读取与参考序列的第二比对;从经扩充参考序列生成针对候选变体位置的参考矩阵,并从多条经扩充序列读取生成针对候选变体位置的样本矩阵;将参考矩阵和样本矩阵输入神经网络中;以及用该神经网络确定候选变体位置处是否存在变体类型。
在一些实施例中,扩充序列读取和/或参考序列的步骤包括将一个或多个空格引入序列读取和/或参考序列,以解释序列读取中的插入和/或缺失。
在一些实施例中,该方法进一步包括从训练数据集生成多个训练矩阵,其中训练矩阵具有与样本矩阵和参考矩阵相对应的结构,其中训练数据集包含序列数据,该序列数据包含多个突变,该多个突变包含单核苷酸变体、插入和缺失;以及使用多个训练矩阵训练神经网络。
在一些实施例中,训练数据集包含多个子集,其中每个子集包含从0%到100%范围内的肿瘤纯度水平,其中该子集中的至少两个子集各自具有不同的肿瘤纯度水平。
在一些实施例中,该子集中的至少三个子集各自具有不同的肿瘤纯度水平。
在一些实施例中,多个子集包括具有小于约30%的肿瘤纯度水平的第一子集、具有约30%和70%之间的肿瘤纯度水平的第二子集和具有至少约70%的第三肿瘤纯度水平的第三子集。
在一些实施例中,多个子集包括具有小于约40%的肿瘤纯度水平的第一子集、具有约40%和60%之间的肿瘤纯度水平的第二子集和具有至少约60%的肿瘤纯度水平的第三子集。
在一些实施例中,多个子集包括具有小于约10%的肿瘤纯度水平的子集。
在一些实施例中,多个子集包括具有小于约5%的肿瘤纯度水平的子集。
在一些实施例中,训练数据集包括合成数据。
在一些实施例中,合成数据包括人工生成的突变,其中人工生成的突变包含单核苷酸变体、插入和缺失。
在一些实施例中,训练数据集包括真实数据,其中真实数据包括真实突变,其中真实突变包括单核苷酸变体、插入和缺失。
在一些实施例中,训练数据集包含多个子集,其中每个子集包含从0%到100%范围内的变体等位基因频率,其中该子集中的至少两个子集各自具有不同的变体等位基因频率。
在一些实施例中,该子集中的至少三个子集各自具有不同的变体等位基因频率水平。
在一些实施例中,该子集中的至少一个子集具有至少2.5%的变体等位基因频率。
在一些实施例中,该子集中的至少一个子集具有至少5%的变体等位基因频率。
在一些实施例中,该子集中的至少一个子集具有至少10%的变体等位基因频率。
在一些实施例中,该方法进一步包括将来自至少一个突变调用算法的至少一个预测输入神经网络中。
在一些实施例中,至少一个预测包括来自至少三个独立的突变调用算法的至少三个预测。
在一些实施例中,至少一个预测包括来自至少五个独立的突变调用算法的至少五个预测。
在一些实施例中,训练数据集包括合成数据和真实数据的集合体。
在一些实施例中,训练数据集包括至少5%的合成数据。
在一些实施例中,训练数据集包括至少10%的合成数据。
在一些实施例中,训练数据集包括全基因组测序数据。
在一些实施例中,训练数据集包括全外显子组测序数据。
在一些实施例中,训练数据集包括靶向测序数据。
在一些实施例中,训练数据集包括从福尔马林固定石蜡包埋样本获得的数据。
在一些实施例中,训练数据集包括全基因组测序数据、全外显子组测序数据、靶向测序数据和从福尔马林固定石蜡包埋样本获得的数据中的至少两者。
在一些实施例中,训练数据集包括全基因组测序数据、全外显子组测序数据、靶向测序数据和从福尔马林固定石蜡包埋样本获得的数据中的至少三者。
在一些实施例中,训练数据集包括全基因组测序数据、全外显子组测序数据、靶向测序数据和从福尔马林固定石蜡包埋样本获得的数据。
在一些实施例中,提供了用于变体调用的方法。该方法可以包括获得参考序列、多条肿瘤序列读取和多条正常序列读取;任选地实施多条肿瘤序列读取和多条正常序列读取与参考序列的第一比对,除非所获得的多条肿瘤序列读取和多条正常序列读取及参考序列是以已经比对的配置获得的;从经比对肿瘤序列读取、正常序列读取和参考序列中鉴别候选变体位置;在候选变体位置周围扩充肿瘤序列读取和/或正常序列读取和/或参考序列以实现多条肿瘤序列读取和多条正常序列读取与参考序列的第二比对;从经扩充参考序列生成针对候选变体位置的参考矩阵,从多条经扩充肿瘤序列读取生成针对候选变体位置的肿瘤矩阵,并且从多条经扩充正常序列读取生成针对候选变体位置的正常矩阵;将参考矩阵、肿瘤矩阵和正常矩阵输入神经网络中;以及用该神经网络确定候选变体位置处是否存在变体类型。
在一些实施例中,该方法进一步包括从训练数据集生成多个训练矩阵,其中训练矩阵具有与肿瘤矩阵、正常矩阵和参考矩阵相对应的结构,其中训练数据集包括肿瘤序列数据和正常序列数据;以及使用多个训练矩阵训练神经网络。
在一些实施例中,肿瘤序列数据和正常序列数据两者均包括多个突变,该突变包括单核苷酸变体、插入和缺失。
在一些实施例中,正常序列数据包括最多5%的肿瘤序列数据。
在一些实施例中,正常序列数据包括最多10%的肿瘤序列数据。
在一些实施例中,肿瘤序列数据包括约10%至100%之间的肿瘤纯度水平。
在一些实施例中,训练数据集包括多个肿瘤序列数据子集,其中每个肿瘤序列数据子集包含从10%到100%范围内的肿瘤纯度水平,其中肿瘤序列数据子集中的至少两个子集各自具有不同的肿瘤纯度水平。
在一些实施例中,肿瘤序列数据子集中的至少三个子集各自具有不同的肿瘤纯度水平。
在一些实施例中,多个肿瘤序列数据子集包括具有小于约30%的肿瘤纯度水平的第一肿瘤序列数据子集、具有约30%和70%之间的肿瘤纯度水平的第二肿瘤序列数据子集和具有至少约70%的肿瘤纯度水平的第三肿瘤序列数据子集。
在一些实施例中,多个肿瘤序列数据子集包括具有小于约40%的肿瘤纯度水平的第一肿瘤序列数据子集、具有约40%和60%之间的肿瘤纯度水平的第二肿瘤序列数据子集和具有至少约60%的肿瘤纯度水平的第三肿瘤序列数据子集。
在一些实施例中,训练数据集包括合成数据。
在一些实施例中,合成数据包括人工生成的突变,其中人工生成的突变包含单核苷酸变体、插入和缺失。
在一些实施例中,训练数据集包括真实数据,其中真实数据包括真实突变,其中真实突变包括单核苷酸变体、插入和缺失。
在一些实施例中,训练数据集包括全基因组测序数据。
在一些实施例中,训练数据集包括全外显子组测序数据。
在一些实施例中,训练数据集包括靶向测序数据。
在一些实施例中,训练数据集包括从福尔马林固定石蜡包埋样本获得的数据。
在一些实施例中,提供了一种系统。该系统可以包括处理器,其配置成执行实施权利要求18至64中任一项中所述的步骤。
附图简要说明
本发明的新颖特征在所附的权利要求书中具体阐述。通过参考以下具体实施方式并结合附图,可以更好地理解本发明的特征和优点,具体实施方式阐述了其中利用本发明原理的说明性实施例,在附图中:
图1示出了根据一些实施例的包括测序装置和计算机系统的示例性系统;
图2a和图2b示出了根据一些实施例的用于种系变体调用的示例性方法;
图3示出了根据一些实施例的图2的示例性方法的扩充步骤;
图4示出了根据一些实施例的可以提供给神经网络和从神经网络获得的示例性数据;
图5示出了根据一些实施例的神经网络的示例性架构;
图6示出了根据一些实施例的用于体细胞变体调用的示例性方法;
图7示出了根据一些实施例的可作为图6的示例性方法的一部分实施的扩充步骤;
图8示出了根据一些实施例的可以提供给神经网络和从神经网络获得的数据的另一示例;
图9示出了根据一些实施例的神经网络的示例性架构;
图10a示出了针对给定的候选体细胞snv的输入矩阵的生成;图10b示出了输入矩阵和网络架构;图10c示出了针对给定的候选体细胞缺失的输入矩阵的生成;图10d示出了针对给定的候选体细胞插入的输入矩阵的生成;
图11a至图11c示出了不同检测方法在铂双样本混合物数据集上的性能;
图12a至图12c示出了不同检测方法在dream阶段3数据集上的性能;
图13a至图13c示出了不同检测方法在dream阶段4数据集上的性能;
图14a至图14c示出了两种不同检测方法在pacbio数据集上的性能;
图15a至图15b示出了序列覆盖率对于不同检测方法在全外显子组样本混合物数据集上的性能的影响;
图16a至图16d示出了不同检测方法在铂肿瘤掺入数据集上的性能;
图17a和图17c示出了不同的体细胞突变检测方法在全外显子组数据集上的性能;图17b和图17d示出了体细胞突变检测方法在靶向组数据集上的性能;
图18a示出了使用基于全基因组和全外显子组训练的模型对外显子组混合物数据集进行测试的性能分析;图18b示出了使用基于全基因组和全外显子组训练的模型对靶向组混合物数据集进行测试的性能分析;
图19示出了在dream阶段3、dream阶段4、铂双样本混合物、铂肿瘤掺入、pacbio和外显子组数据集中的基准真值(groundtruth)indel的尺寸分布;
图20a和图20b。图20a示出了针对各种数据集,各种检测方法基于预测的体细胞突变位置和类型对indel进行测试的性能分析;
图21示出了针对dream挑战阶段3数据集的交叉样本训练的性能分析;
图22a示出了不同体细胞突变检测方法在真实数据集colo-829上的性能;图22b示出了不同体细胞突变检测方法在真实数据集cll1上的性能;图22c示出了不同体细胞突变检测方法在真实数据集tcga-az-6601上的性能;
图23是用于microsoftazure实验的261个tcga癌症样本的列表;
图24a至图24m示出了所测试的不同网络架构;
图25示出了图24a至图24m所示的不同网络架构的性能分析;
图26a和图26b示出了不同体细胞突变检测算法的运行时比较;
图27示出了在由seqc-ii联合体分类为四个置信度水平(高、中、低和未分类)的针对hcc1395的调用结果超集合中,基准真值snv和indel体细胞突变的vaf分布;
图28a至图28e显示了neusomatic在seqc-ii数据集中的123个复本上的总体性能;
图29a至图29d显示了各种模型在21个wgs复本数据集上的性能比较。图29a示出了f1得分(%),图29b示出了跨不同体细胞突变调用者的精确率-召回率比较。在这里,seqc-wgs-gt50-掺入wgs10受训模型用于neusomatic和neusomatic-s。图29c显示了各种neusomatic受训模型的f1得分(%)比较。图29d显示了不同体细胞突变检测方法和基于wgs数据集的neusomatic网络受训模型的f1得分(%)性能;
图30a至图30d显示了肿瘤纯度数据集的性能比较。图30a显示了跨不同覆盖率(10×至300×)和肿瘤纯度(5%至100%)的不同体细胞突变调用者的snv和indelf1得分(%)比较。图30b显示了匹配的正常样本对肿瘤污染的鲁棒性:在80×覆盖率和5%至100%肿瘤纯度下,当匹配的正常样本被5%的肿瘤污染时,f1得分(%)与纯正常样本的f1得分(%)相比有所改变。对于图30a和图30b,seqc-wgs-gt50-掺入wgs10受训模型用于neusomatic和neusomatic-s。图30c显示了各种neusomatic受训模型的f1得分(%)比较。图30d显示了针对不同indel尺寸的性能分析。负indel尺寸反映缺失;
图30e和图30f显示了基于肿瘤纯度数据集的精确率-召回率分析。图30e显示了snv,并且图30f显示了跨不同覆盖率(10×至300×)和肿瘤纯度(5%至100%)的不同体细胞突变调用者的indel准确性比较;
图30g显示了基于肿瘤正常滴定数据集的各种neusomatic-s受训模型的精确率-召回率比较;
图30h显示了不同体细胞突变检测方法和基于肿瘤-正常滴定数据集的neusomatic网络受训模型的f1得分(%)性能;
图31a至图31f显示了文库制备、wes和ampliseq数据集的性能比较。图31a显示了跨不同文库-试剂盒和dna量的不同体细胞突变调用者的snv和indelf1得分(%)比较。图31b显示了针对不同文库-试剂盒和dna量的各种neusomatic受训模型的精确率-召回率比较。图31c和图31e显示了针对不同体细胞突变调用者的snvf1得分(%)比较。图31d和图31f显示了各种neusomatic受训模型的f1得分(%)比较。对于图31a、图31c和图31e,seqc-wgs-gt50-掺入wgs10受训模型用于neusomatic和neusomatic-s;
图31g显示了基于文库制备数据集的各种neusomatic-s受训模型的精确率-召回率比较;
图31h显示了不同体细胞突变检测方法和基于文库制备数据集的neusomatic网络受训模型的f1得分(%)性能;
图31i显示了不同体细胞突变检测方法和基于wes数据集的neusomatic网络受训模型的f1得分(%)性能;
图31j显示了不同体细胞突变检测方法和基于ampliseq数据集的neusomatic网络受训模型的f1得分(%)性能;
图32a至图32d显示了ffpe数据集的性能比较。图32a和图32b显示了基于16个ffpewgs复本的性能。图32c和图32d显示了基于14个ffpewgs复本的性能。图32a和图32c显示了跨ffpe和新鲜正常样本的不同体细胞突变调用者的f1得分(%)比较。图32b和图32d显示了各种neusomatic受训模型的f1得分(%)比较。图32b显示了使用ffpe和新鲜正常样本进行的不同体细胞突变调用者的16个ffpewgs复本的精确率-召回率图。对于图32a和图32c,seqc-wgs-gt50-掺入wgs10受训模型用于neusomatic和neusomatic-s;
图32e显示了不同体细胞突变检测方法和基于ffpewgs数据集的neusomatic网络受训模型的f1得分(%)性能;
图32f显示了不同体细胞突变检测方法和基于ffpewes数据集的neusomatic网络受训模型的f1得分(%)性能;
图33a和图33b是分析使用neusomatic(图33a)和neusomatic-s(图33b)针对来自不同数据集的九个复本进行训练对基于靶标样本训练的影响的图表。对于每个样本,使用两个样本特异性模型:一个仅基于靶标样本训练,另一个则使用来自seqc-wgs掺入模型的另外10%的训练数据进行训练;
图33c显示了seqc-wgs-掺入模型和两个样本特异性neusomatic网络受训模型的f1得分(%)性能;
图34a至图34d显示了不同indel尺寸、不同vaf分布以及在困难区域上的性能分析。图34a和图34b显示了使用不同调用者和不同训练方法对跨seqc-ii数据集的不同indel尺寸的性能分析。负indel尺寸反映缺失。图34c显示了使用不同调用者对跨seqc数据集的具有不同vaf范围的突变的性能分析。图34d显示了不同体细胞调用者在困难区域上的性能分析,该困难区域包括不同尺寸的串联重复序列(tr)和片段复制;
图35示出了使用不同调用者对跨seqc数据集的具有不同vaf范围的突变的性能分析。
图36a和图36b显示了使用不同的针对neusomatic(图36a)和neusomatic-s(图36b)的训练方法对跨seqc数据集的具有不同vaf范围的突变的性能分析;
图37a至图37c显示了使用不同的调用者(图37a)和针对neusomatic(图37b)和neusomatic-s(图37c)的训练方法(图37b和图37c)对肿瘤/正常滴定数据集的具有不同vaf范围的突变的性能分析;
图38显示了使用不同调用者在全基因组与困难基因组区域上的性能比较,该困难基因组区域包括不同尺寸的串联重复序列(tr)和片段复制;
图39a和图39b显示了不同的针对neusomatic(图39a)和neusomatic-s(图39b)的训练方法在困难区域上的性能分析,该困难区域包括不同尺寸的串联重复序列(tr)和片段复制;
图40a和图40b显示了使用不同的针对neusomatic(图40b)和neusomatic-s(图40c)的训练方法在全基因组和困难基因组区域上的性能比较,该困难区域包括不同尺寸的串联重复序列(tr)和片段复制;
图41a至图41c显示了使用不同的调用者(图41a)和针对neusomatic(图41b)和neusomatic-s(图41c)的训练方法(图41b和图41c)对肿瘤/正常滴定数据集上困难区域的性能分析,该困难区域包括不同尺寸的串联重复序列(tr)和片段复制;以及
图42显示了基于wgs数据集的私有fn调用结果的vaf分布的小提琴图比较。
具体实施方式
此外,本公开(除了描述其它以外)还描述一种用于种系变体调用的方法,该方法可以包括获得参考序列、多条序列读取和候选变体在序列读取内的位置;通过将一个或多个空格插入一条或多条序列读取中而获得经扩充序列读取;通过将一个或多个空格插入参考序列中而获得经扩充参考序列;将候选变体周围的经扩充序列读取的片段转换成样本矩阵;将候选变体周围的经扩充参考序列的片段转换成参考矩阵;将样本矩阵和参考矩阵提供给受训神经网络;以及,在受训神经网络的输出端,获得关于多条序列读取内的变体的预测数据。所公开的系统和方法允许直接从原始数据捕获重要的变体信号,并且对于不同的测序技术、样本纯度和测序策略(诸如全基因组与靶标富集)一致地实现高精确率。
图1阐述了说明性系统100,其包括通信耦接至计算机系统102的测序装置110。测序装置110可以直接(例如,通过一条或多条通信电缆)或通过网络130耦接至计算机系统102,网络130可以是互联网或广域网络、局域网络、有线网络和/或无线网络的任何其他组合。在一些实施例中,计算机系统102可以包括在或整合在测序装置110中。在一些实施例中,测序装置110可以对包含遗传物质的样本进行测序,并产生所得的测序数据。测序数据可以被发送到计算机系统102(例如,通过网络130),或存储在存储装置上并在以后的阶段被发送到计算机系统102(例如,通过网络130)。在一些实施例中,计算机系统102可以包括或可以不包括显示器108和一个或多个输入装置(未示出),用于从使用者或操作者(例如,技术人员或遗传学家)接收命令。在一些实施例中,计算机系统102和/或测序装置110可以由使用者或其他装置通过网络130远程访问。因此,在一些实施例中,本文讨论的各种方法可以在计算机系统102上远程运行。
计算机系统102可以包括一个计算机装置或任何类型的多个计算机装置的组合,诸如个人计算机、膝上型计算机、网络服务器(例如,本地服务器或公共/私有/混合云中包括的服务器)、移动装置等等,其中一些或所有装置可以互连。计算机系统102可以包括一个或多个处理器(未示出),每个处理器可以具有一个或多个核心。在一些实施例中,计算机系统102可以包括一个或多个通用处理器(例如,cpu)、专用处理器诸如图形处理器(gpu)、数字信号处理器、或这些和其他类型的处理器的任意组合。在一些实施例中,可以使用定制或可定制的电路诸如专用集成电路(asic)或现场可编程门阵列(fpga)来实现计算机系统中的一些或所有处理器。在一些实施例中,计算机系统102还可以检索并执行存储在整合到或以其他方式通信耦接至计算机系统102的一个或多个存储器或存储装置(未示出)中的非暂时性计算机可读指令。存储器/存储装置可以包括非暂时性计算机可读存储介质的任意组合,包括各种类型的半导体存储芯片(dram、sram、sdram、闪存、可编程只读存储器)等等。也可以使用磁盘和/或光盘。存储器/存储装置也可以包括可读取和/或可写入的可移动存储介质;此类介质的示例包括光盘(cd)、只读数字通用光盘(例如,dvd-rom、双层dvd-rom)、只读和可读blu-ray®磁盘、超密度光盘、闪存存储卡(例如,sd卡、小型sd卡、微型sd卡等)等等。在一些实施例中,数据和其他信息(例如测序数据)可以存储在一个或多个远程定位(例如,云存储)中,并与系统100的其他部件同步。
种系变体调用
图2a示出了根据一些实施例的用于种系变体调用的示例性方法200。方法200可以例如以软件的形式(即,存储在一个或多个非暂时性介质中的可由例如计算机系统102的一个或多个处理器访问和执行的一组指令)或以固件、硬件或其任意组合的形式来实现。
方法200可以在步骤210处开始,在步骤210处可以获得参考序列。参考序列可以例如从一个或多个私有或公共资源库获得,诸如参考序列(refseq,由美国国家生物技术信息中心(ncbi)建立的核苷酸碱基序列的开放获取、注释和管理集合)或ncbigenomesftp网站(其存储了一组不同生物的完整基因组)。应当理解,在一些实施例中,参考序列的特定副本可以本地存储(例如,存储在计算机系统102的存储器中),而在其他实施例中,参考序列可以从远程服务器获得,例如,通过网络130获得。此外,在一些实施例中,可以获得完整的参考序列,而在其他实施例中,可以获得参考序列的一个或多个区段,例如,仅与具体测定法相关联的一个或多个区段。因此,本文所用的“参考序列”通常是指参考序列的一个或多个区段,其可以包括或可以不包括完整的参考序列。
在步骤220,可以获得与由测序装置110测序的遗传样本(例如,包含患者的dna或rna物质的样本)相对应的多条序列读取。如上所述,可以直接从测序装置110或从通信耦接至计算机系统102的一个或多个本地或远程易失性或非易失性存储器、存储装置或数据库中获得序列读取。所获得的序列读取可以已经预处理的(例如,预先比对的),或者它们可以是“原始的”,在这种情况下,方法200还可以包括预处理(例如,预先比对)步骤(未示出)。而且,尽管在一些实施例中,可以获得完整的序列读取(由测序装置110生成),但在其他实施例中,可以仅获得序列读取的区段。因此,如本文所用,“获得序列读取”通常是指获得一条或多条(例如,相邻)序列读取的一个或多个区段。
在步骤230,可以获得序列读取内的多个候选变体位置。在一些实施例中,这包括扫描获得的多条序列读取,将它们与获得的参考序列进行比较,并确定该序列读取内的似乎包括一些类型的变体(例如,插入或缺失变体(indel)、单核苷酸变体(snv)或结构变体(sv))的一组一个或多个位置。查找候选变体位置可以包括例如检查参考中的所有位置,并确定至少一个序列读取区别于参考的位置。在确定候选位置时,也可以使用一些过滤器,例如,对于待视为候选变体位置的特定位置而言,针对必须不同于参考的读取数或百分比的过滤器。在其他实施例中,序列读取可能已经被扫描和分析(例如,通过独立的软件和/或硬件模块),在这种情况下,可以从耦接至计算机系统102的存储器或数据库中获得多个候选位置。在步骤240,该方法可以从多个鉴别出的候选变体位置前进到下一个候选变体位置。
在步骤250,在当前候选变体位置周围扩充序列读取和参考序列,以实现精确的多序列比对(msa)。结合图3可以更好地说明这一步骤,该图显示了多个示例性序列读取311、示例性参考序列310和示例性候选变体位置350。如图3所示,扩充参考序列310和序列读取311可包括将一个或多个空位或空格(例如,空格325)插入参考序列310内和/或一个或多个序列读取311中,从而产生经扩充参考序列320和与经扩充参考序列320精确比对的多条序列读取321。
具体地,如图3所示,在一些实施例中,扩充可包括:确定或检测具体序列读取中的插入碱基;在该插入碱基位置处将空格插入参考样本中;在相同位置处将空格插入每一条在该相同位置处没有检测到插入的序列读取中(在一些情况下,这对应于除检测到插入的具体序列读取以外的所有序列读取);以及对每个在每条序列读取中检测到的插入碱基重复该过程。应当理解,在检测到两个碱基或更长的潜在插入的位置中,可以分别插入两个或更多个空格。应当理解,可以使用其他合适的技术将空格插入序列读取和参考序列中,从而可以最终实现精确的多序列比对(msa),这意味着经扩充序列读取内的所有碱基(或至少在没有出现缺失或插入的那些定位)都可以彼此精确地比对,并与经扩充参考序列精确比对。
在一些实施例中,扩充还可以包括检测具体序列读取中的缺失碱基,并在潜在缺失变体位置处将空格插入该序列读取中。应当理解,在检测到缺失碱基的情况下,不需要扩充参考序列。
还应理解,“插入空格”可以包括或可以不包括对存储了序列读取和参考序列的存储器的实际修改,并且可以代之以其他操作的固有或隐含部分(例如,方法200的其他步骤)以实现相同的最终结果。例如,代替修改序列读取和参考序列的原始定位,可以以将会造成新副本将空格存储在所有正确位置处的方式将序列读取和参考序列选择性地从原始定位处复制到独立的存储器定位处。如本文所用,“存储空格”可以意味着存储除对应于四种类型的核苷酸碱基(a、c、t和g)的四种值以外的任何随机或预定值(例如,0)。作为另一个示例,如下所述,当将序列读取和参考序列转换成计数矩阵时,可能不是在任何步骤中物理地插入空格,而是可以在算法上考虑空格。
在一些实施例中,在候选变体位置周围扩充序列读取和参考序列可以意味着仅扩充包括候选变体位置的可变长度或预定长度的一部分。然而,在其他实施例中,可以一次性地扩充可用序列读取和参考序列的完整长度,在这种情况下,步骤250可以仅实施一次(例如,在步骤240之前或甚至在步骤230之前),而不是针对每一个候选变体位置重复步骤250。
在步骤260,可以将经扩充序列读取的片段(即,窗口)转换(或“总结”)成样本矩阵。该片段可以具有固定的长度,并且可以将候选变体位置放置在中间或可以不放置在中间。例如,在图3所示的示例中,扩充片段长16列(位置),并且包括候选变体位置350的左侧7列和右边8列,从而将候选变体位置350基本放置在扩充片段的中间。
在一些实施例中,样本矩阵可以是具有与正在被转换的片段相同的列数的计数矩阵。对于每种类型的核苷酸碱基(a、c、g和t),矩阵可以包括一个横行,每一横行表示相应核苷酸碱基在经扩充序列读取的每个位置(列)上出现的次数。参考图3的示例,样本矩阵331中的横行“a”包括值“6000000306520000”,指示碱基“a”在经扩充读取的片段内的第一位置出现6次;在第八位置出现3次;在第十位置出现6次;在第十一位置出现5次;在第十二位置出现两次;并且在片段内的任何其他位置出现0次(未出现)。
在一些实施例中,样本矩阵还可以包括一个横行,该横行表示空格(在图3中表示为“-”)在经扩充序列读取内的每个位置出现的次数。例如,在图3中,样本矩阵331的第一横行(标记为“-”)包括值“0000014000140000”,指示经扩充序列读取在片段内的位置6、7、11和12处分别包括1、4、1和4个空格。应当理解,尽管例如在图3的示例中将空格横行“-”显示为矩阵的第一横行,但是在其他实施例中,空格横行可以放置在矩阵内的其他地方(例如,最后)。还应当理解,横/列表示法是任意的,并且在其他实施例中,该表示法可以颠倒,这意味着样本矩阵可以具有5列(a、c、t、g、-),并且横行数对应于片段的长度。据此,通常可以将矩阵描述为具有至少五个“行”(横行或列),一个行表示每种核苷酸类型的计数,并且至少一个行表示经扩充序列读取内的空格计数。
在步骤270,可以类似地将经扩充参考序列的与序列读取的片段相对应的片段转换成具有与样本矩阵相同维数的参考矩阵。例如,在图3中,扩充参考320转换为参考矩阵330。类似于样本矩阵331,参考矩阵330可以具有四个横行,指示每种类型的核苷酸在经扩充参考序列内的每个位置出现的次数,以及空格横行,指示在扩充参考序列内的每个位置处的空格数目。然而,应当理解,在其他实施例中,参考矩阵330可以具有与图3所示不同的参考序列不同碱基的总结/表示法。
在图3所示的实施例中,参考矩阵320也被归一化以具有与样本矩阵331相同的值范围。如图3所示,归一化可以包括将每个计数乘以归一化因子,诸如序列读取的总数(在该示例中乘以6)。可以理解,在其他实施例中,代替对参考矩阵进行归一化,可以对样本矩阵或对两个矩阵实施归一化。在其他实施例中,矩阵可能根本不需要被归一化。还应当理解,步骤270不需要在步骤260之后实施,并且在一些实施例中可以在步骤260之前或与之并行实施。
在步骤280,可以将参考矩阵和样本矩阵作为输入提供给受训深度神经网络,并且在步骤290,可以获得受训深度神经网络的输出,其中,输出可以包括各种关于由样本矩阵表示的序列读取内包括的变体的预测,如下文更详细讨论。
本领域技术人员还应该理解,除非明确相反地指示,否则方法200中的步骤的数目和顺序不必限于如图2a所示的步骤的数目和顺序。因此,在不同的实施例中,方法200的一些步骤可以被重新排序或并行实施;某些步骤可以设为可选或省略;并且可以添加一些附加步骤而不脱离本公开的精神和范围。
例如,图2b示出了另一个实施例,其中在步骤210'处,获得参考序列和多条相对应的序列读取。可以以任何顺序(即顺序地或同时地)获得参考序列和多条相对应的序列读取。接下来,在步骤220'中,将参考序列和多条相对应的序列读取进行比对。在一些实施例中,可以以比对的格式获得参考序列和多条相对应的序列读取,这可以消除实施第一比对步骤的需要。在步骤230'处,从经比对序列读取和参考序列中鉴别候选变体位置。在步骤240'处,以与上述相同的方式(即,通过插入空位以解释插入和/或缺失)在候选变体位置周围扩充多条序列读取和/或参考序列,以实现多条序列读取与参考序列的第二比对。
在一些实施例中,可对任一候选变体位置上的约2至5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95或100个碱基的窗口进行扩充和比对。在一些实施例中,可对候选变体位置任一侧的至少约5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95或100个碱基的窗口进行扩充和比对。在一些实施例中,可对候选变体位置任一侧的不超过约5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95或100个碱基的窗口进行扩充和比对。
在步骤250'处,从经扩充参考序列生成针对候选变体位置的参考矩阵,并从多条经扩充序列读取生成针对候选变体位置的样本矩阵。如上所述,矩阵包括在每个序列位置的每个碱基或空位(即,a、c、g、t或空位)的频率。在步骤260'处,将参考矩阵和样本矩阵输入受训神经网络中。
在步骤270'处,神经网络确定在候选变体位置处是否存在变体类型。例如,神经网络可以包括多个卷积层,这些卷积层处理参考矩阵和样本矩阵,并生成由一个或多个分类器和回归器(即,突变类型的分类器和长度分类器,以及位置回归器)处理的输出,确定变体类型、尺寸和位置。在一些实施例中,神经网络可以包括多达约5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95或100层。在一些实施例中,卷积层可以被构造成块。在一些实施例中,块的数目是层的数目的大约一半。
如果参考序列和相对应的序列读取包括超过一个变体候选位置,则对于每个变体候选位置可以重复步骤230'至260'。另外,可以通过获得额外的参考序列和额外的相对应序列读取来重复步骤210'至260'。
尽管在图2a和图2b中示出的示例在针对种系变体调用的章节中进行了描述,但这些示例可以适用于体细胞变体调用。相反,在针对体细胞变体调用的章节中描述的实施例也可以适用于种系变体调用。例如,图2b中描述的多条序列读取可以细分为两个组:正常序列读取和肿瘤序列读取。正常序列读取可以从受试者的正常组织样本获得,而肿瘤序列读取可以从受试者的肿瘤组织样本获得。正常矩阵和肿瘤矩阵将会与参考矩阵一起生成,而不是生成单个样本矩阵。该方法的其余部分将以类似的方式进行。
图4示出了示例性实施例,其中参考矩阵330和样本矩阵331(连同下面讨论的其他可选输入)被馈入受训深度神经网络350中,然后,深度神经网络350输出关于样本矩阵331所表示的序列读取的一个或多个预测。
在一些实施例中,网络350可以是卷积神经网络(cnn),其说明性示例在图5中显示。如图5所示,网络350可以包括多个串联连接的卷积层(510a-510i),其最终馈入第一全连接层520a,其输出馈入四个独立的输出各种预测的全连接层520a-520c。在图5的示例中,不同的卷积层510可以具有不同的过滤器尺寸(例如1x3、3x3或5x5)并可以通过额外的处理层(未明确显示)互连,诸如整流线性单元(relu)、池化层、批归一化(bn)层等。此外,如图5所示,在一些实施例中,有时可以经由标识捷径连接515将输入到特定层中的输入连接到后续层的输出,以便帮助将信号保持在网络的更深层中。
在一些实施例中,网络350可以输出与经扩充序列读取的片段内的候选变体相关联的一个或多个预测。如图5的示例中所示,这样的预测之一可以在全连接层520b的输出端获得并且可以指示候选变体位置是否对应于变体,如果是,则指示该变体的类型。更具体地,在一些实施例中,预测可以包括与四种可能类型的变体相关联的至少四个概率值:none(无变体,即假阳性调用结果);snp/snv(单核苷酸多态性/变体);ins(插入变体);和del(缺失变体)。基于这些概率值,可以确定哪种类型的变异是最可能的,以及具体预测的置信度。
在一些实施例中,神经网络350还可以输出(例如,在全连接层520c的输出端)由样本矩阵331表示的经扩充序列读取的片段内的变体的预测位置。在其他实施例中,可以省略该输出,并且可以基于针对给定的候选变体而选择序列读取的片段的方式,假设变体的位置是已知的(例如,在片段的中心),如上所述。
在一些实施例中,神经网络350还可以输出(例如,在全连接层520d的输出端)变体的预测长度。例如,如果预测的变体是一个碱基长(例如,snp变体或一碱基del或ins变体),则可以输出“1”的长度;如果预测的变体是两碱基长的del或ins变体,则可以输出“2”的长度;以此类推。在一些实施例中,如果输出指示变体是2个碱基长或更长,则后处理步骤可以解析正被插入或缺失的确切序列。
在一些实施例中,神经网络350还可以输出(例如,在全连接层520e的输出端)与该变体相关联的预测基因型,例如,表示该变体的概率为(1)纯合参考(非变体);(2)杂合变体(只有母系或父系副本具有变体);(3)纯合变体(其中两个副本具有相同变体);或(4)其他(每个副本都有不同的变体)。应当理解,在其他实施例中,可以省略这些输出中的一些和/或可以添加额外的输出。
现在参考网络的输入,除了上面讨论的参考矩阵330和样本矩阵331之外,在一些实施例中,可以提供其他输入以进一步改善预测的准确性和/或扩展可以在网络的输出端获得的信息量。如图4的示例中所示,额外的输入可以包括,例如,变体位置矩阵340、覆盖率矩阵343和一个或多个比对特征矩阵344。
在一些实施例中,所有输入矩阵可以作为一个大的三维矩阵提供到网络350中。矩阵的维数可以是例如5xsxk,其中5对应于横行/列的数目(-、a、c、t、g);s对应于片段的长度(例如,16);并且k对应于上述的不同二维(例如,5xs)矩阵的数目。在一些实施例中,k可以高达30,或者甚至更高。
变体位置矩阵340可以包括参考矩阵和样本矩阵内的候选变体位置的二维表示。例如,矩阵可以具有五个横行(-、a、c、t、g),并且每一列可以表示片段内的位置。例如,变体位置矩阵340可以在对应于候选变体位置的列的所有横行中均包括一个值(例如,1),并且在所有其他列的所有横行中均包括另一个值(例如,0)。
覆盖率矩阵343可以表示序列读取的具体片段的覆盖率或深度。例如,在一些实施例中,覆盖率矩阵343可以在其所有元素中包括相同的值,该值表示片段内不同读取的覆盖率/深度(例如,平均覆盖率/深度)。在其他实施例中,覆盖率矩阵343可在不同列处包括不同的值,每个值表示每一列的相应覆盖率/深度。比对特征矩阵344可以表示各种与序列读取的质量及其比对相关联的度量。此类度量可以包括例如碱基质量、映射质量(mappingquality)、链偏向性、剪切信息等。这些比对特征可通过为序列读取提供更多情境来帮助改善预测准确性,例如,通过将序列碱基和/或其映射(mapping)的质量告知网络。网络350的额外输入(为简洁起见未示出)可以包括关于已知变体的各种数据。可以从例如dbsnp、cosmic、exac等公共和/或私有知识库中获取此类数据。
应当理解,图5仅显示了网络350的一种示例性配置,并且具有其他合适的配置/架构的神经网络可以用于分析经扩充序列读取(由样本矩阵表示)并预测变体的类型及其其他特征,而不脱离本公开的范围和精神。这样的替代配置可以包括更少的层、额外的层、具有不同参数的层、额外的输入或输出、更少的输入或输出等等。此外,在一些实施例中,网络350可以不是卷积神经网络(cnn),但可以是另一种类型的深度神经网络(dnn),即,在输入层与输出层之间具有多个层的另一种类型的人工神经网络(ann),而不脱离本公开的范围和精神。
体细胞变体调用
在一些实施例中,神经网络350也可以被训练以实施体细胞变体调用。图6示出了使用受训神经网络350进行体细胞变体调用的示例性方法600。
在步骤610处,方法600包括例如以上文结合方法200的步骤210所述者类似的方式获得参考序列。在步骤620处,方法600包括获得多条肿瘤序列读取和多条相对应的正常(非肿瘤)序列读取。例如,在一些实施例中,肿瘤序列读取可以包括患者的肿瘤(癌性)组织的测序结果,而正常序列读取可以包括取自相同患者的正常(非癌性)组织的测序结果。在其他实施例中,正常序列读取可以包括取自不同患者的正常组织的测序结果。可以例如以上文结合方法200的步骤220所述类似的方式获得两种类型的序列读取。
在步骤630处,可以通过从存储器或公共/私有数据库获得预定的变体位置即一组白名单候选位置,或通过实施在肿瘤序列读取与参考序列之间的比较(例如,以上文结合方法200的步骤230所述的方式),来获得多个候选体细胞变体位置。
在步骤640处,该方法进行到多个候选体细胞变体位置之一。在步骤660处,在候选体细胞变体位置周围将正常序列读取的片段转换为正常样本矩阵。类似地,在步骤665处,在候选体细胞变体位置周围将肿瘤序列读取的片段(在长度和定位上基本上对应于正常序列读取的片段)转化为肿瘤样本矩阵。在步骤670处,在候选体细胞变体位置周围将参考序列的片段(在长度和定位上基本上对应于正常序列读取的片段)转化为参考矩阵。
在一些实施例中,在将以上三个片段转换成它们的相应参考矩阵之前,可以使用上文结合方法200的步骤250描述的扩充技术来扩充它们中的每一个。在其他实施例中,可以在将片段转换成矩阵之前对它们实施不同类型的扩充技术;例如,可以使用k-mer塌缩方法,其中将长度l>k的均聚物塌缩成长度k,其中k为大于或等于1的任何数字。在其他实施例中,可以完全省略扩充步骤,并且可以将原始的、未处理的序列读取和参考序列直接转换为它们的相应矩阵。
在图7所示的示例中,实施类似于在方法200的步骤250实施的扩充。在该示例中,首先扩充参考序列310的片段、肿瘤序列读取311-a的片段和正常序列读取311-b的片段,以分别产生经扩充参考序列320、经扩充肿瘤序列读取321-a和经扩充正常序列读取321-b,然后分别转换为参考矩阵330、肿瘤样本矩阵331-a和正常样本矩阵331-b。
现在回到方法600,在步骤680处,将三个矩阵馈入(作为输入提供)受训神经网络中,并且在步骤690处,获得受训神经网络的输出,其中,输出至少包括对肿瘤序列读取(例如,在候选体细胞变体位置或其附近)中含有的体细胞变体类型的预测(概率估计)。
图8显示了用于体细胞变体调用的受训神经网络350的示例性图示。在该示例中,网络350接收参考矩阵330、肿瘤样本矩阵331-a和正常样本矩阵331-b作为其输入。
应当理解,尽管图8显示了输入至网络350中的所有三种类型的矩阵,但在一些实施例中,参考矩阵330和/或正常样本矩阵331-b可以被省略并且不提供给网络350,而且网络350仍然可以实施体细胞变体调用。在省略正常样本矩阵的实施例中,可以从方法600的步骤620中省略获得正常序列读取,并且可以完全省略将正常序列读取转换成矩阵的步骤660。
在一些实施例中,为了进一步改善预测的准确性和/或扩展可以在网络的输出端获得的信息量,网络还可以接收以下的额外矩阵作为输入:诸如表示候选体细胞变体位置的体细胞变体位置矩阵340;表示肿瘤序列读取的片段和正常序列读取的片段的相应覆盖率的肿瘤覆盖率矩阵343-a和正常覆盖率矩阵343-b;以及一个或多个比对特征矩阵344,该矩阵以类似于上文结合图4所述者类似的方式表示序列读取的特征。另外,尽管在图8中为了简洁起见未示出,但在一些实施例中,可以从诸如dbsnp、cosmic、exac等公共和/或私有知识库获得关于已知变体的数据,并将其作为输入提供给网络350。
在一些实施例中,为了进一步改善体细胞变体调用的准确性,还可以在其输入端向神经网络350提供一个或多个其他调用者的特征矩阵345。这些矩阵可以表示(例如,总结)从已经处理序列读取的相同片段的一个或多个其他(例如,第三方)变体调用应用程序或算法获得的一个或多个特征。此类特征可以包括由其他应用程序/算法预测的变体类型、长度和位置,以及质量得分、映射得分、变体显著性得分和其他比对特征。如上文结合图4所讨论的,在一些实施例中,馈入网络350的所有输入矩阵可以被“组合”并且提供为一个大的三维矩阵。
图9显示了示例性的受训神经网络350,其接收上述各种输入并输出预测,该预测至少指示在其输入端由体细胞样本矩阵331-a表示的体细胞序列读取的片段中包含的体细胞变体的变体类型、位置和长度。在一些实施例中,网络350还可以包括输出变体的基因型的全连接层520e(如图5所示),但是在体细胞变体调用的情况下,该基因型并不重要,因此可以从网络中省略。
基于图9和图5的示例,应当理解,在一些实施例中,具有相同或基本相同的架构的网络350可以用于预测种系变体和体细胞变体,取决于已经对其进行训练的数据类型(如下文所讨论的),并且取决于提供给它的输入类型。然而,在一些实施例中,可以针对种系变体调用或体细胞变体调用来修改和优化网络350的架构。
神经网络训练
本领域技术人员应当理解,在神经网络350可以开始实施准确的种系变体调用或体细胞变体调用之前,首先需要分别基于种系或体细胞训练数据(即,训练序列)对其进行训练。训练可以包括,例如,在大量训练序列上实施方法200或600的所有步骤,但随后还可以向网络提供“基准真值”数据(例如,实际已知的变体类型及其位置长度、基因型等),以便在每次运行之后能够通过被称为“反向传播”的过程来调整其可训练的过滤器和其他可训练的参数,从而使网络逐渐将其输出误差最小化。
在一些实施例中,网络350可以基于具有充分表征的基准真值变体的基因组诸如na12878基因组进行训练。在一些实施例中,代替真实基因组或除了真实基因组之外,可以使用各种基于模拟的策略来训练网络。例如,为了训练用于种系变体调用的网络,可以使用mu,j.c.等人(“varsim:ahigh-fidelitysimulationandvalidationframeworkforhigh-throughputgenomesequencingwithcancerapplications.”bioinformatics31,1469–1471(2015))中描述的方法模拟具有给定的一组变体的合成样本。作为另一个示例,为了训练用于体细胞变体调用的网络,可以例如使用eving,a.等人(“combiningtumorgenomesimulationwithcrowdsourcingtobenchmarksomaticsingle-nucleotide-variantdetection,”naturemethods(2015))中描述的方法,将其中已经掺入随机变体的正常样本馈入该网络中。替代地或额外地,可以将两个不同的正常种系样本以不同比例混合以生成合成的肿瘤/正常训练样本。作为另一个示例,可以通过在两个种系样本的变体定位之间切换读取来生成具有期望的等位基因频率分布的虚拟肿瘤/正常训练样本。
实例1
前言
体细胞突变是癌症发生、进展和治疗中的关键标志。由于肿瘤正常样本的交叉污染、肿瘤异质性、测序伪影和覆盖率,因此准确检测体细胞突变具有挑战性。通常,有效过滤由上述问题引入的假阳性调用结果并精确保留难以捕捉的真阳性调用结果(这些调用结果可能在等位基因频率(af)较低的情况下发生或在复杂性较低的区域发生),对于精确的体细胞突变检测算法至关重要。
迄今为止,已经开发了一系列工具来解决体细胞突变检测问题,包括mutect21、muse2、vardict3、varscan24、strelka25和somaticsniper6。这些工具采用不同的统计和算法方法,在为之设计这些工具的某些癌症或样本类型中表现良好;然而,它们在更广泛的样本类型和测序技术范围中的通用性受限,因此在这些场景中可能显示出次优的准确性7,8,9。在我们的早期工作,即somaticseq10中,我们使用通过整合算法正交方法来最大化灵敏度。它还使用机器学习来整合近百个特征以保持较高的精确率,从而导致所有单独方法的精确率都有所改善。不过,somaticseq中使用的机器学习主干依赖于突变定位的一组提取特征。结果,它无法在体细胞突变的基因组情境下完全捕获原始信息以进一步将真实的体细胞突变与背景错误区分开来,限制了其在挑战性情况(诸如低复杂度区域和低肿瘤纯度)下的性能。
这里,我们通过利用深度卷积神经网络(cnn)解决了肿瘤测序数据统计模型的通用性和复杂性的局限性。最近,cnn在不同结构域的分类问题中表现出了显著的性能,包括种系变体调用11、12、13和皮肤癌分类14。即便如此,仍未探索将cnn应用于体细胞突变检测这一具有挑战性的问题。之前基于深度学习的唯一尝试15是将六层全连接的神经网络应用于一组手动提取的特征。这种方法缺乏cnn架构提供的能力,cnn架构使用见于本地区域中的模式直接从原始数据中学习特征表示。此外,由于全连接网络的复杂性,它的通用性和可扩展性不如cnn。
我们引入了neusomatic,这是第一个基于cnn的体细胞突变检测方法,它可以有效地利用源自序列比对以及其他方法的信号来准确地鉴别体细胞突变。与其他专注于种系变体的基于深度学习的方法不同,neusomatic正在解决由于肿瘤样本的复杂性而在准确性方面尚未得到满足的更大需求。它可以直接从原始数据有效地捕获重要的突变信号,并且对于不同的测序技术、样本纯度和测序策略(诸如全基因组与靶标富集)一致地实现高精确率。
结果
neusomatic概述
neusomatic网络的输入是通过扫描肿瘤样本以及匹配的正常样本的序列比对而鉴别出的候选体细胞突变(图10a至图10d)。通过其他方法报告的体细胞突变也可以包括在此候选列表中。对于每个候选基因座,我们构造一个3维特征矩阵m(尺寸为k × 5× 32),由k个通道组成,每个通道的尺寸为5 × 32,以捕获来自以该基因座为中心的区域的信号。矩阵m的每个通道都有五个表示四种核苷酸碱基和空位碱基(“-”)的横行,以及32个表示候选定位周围的比对列的列。
图10a示出了用于给定的候选体细胞snv的输入矩阵制备的虚拟示例。提取候选体细胞突变周围七个碱基的窗口中的序列比对信息。然后通过添加空位以解释读取中的插入而扩充参考序列。然后将经扩充比对总结为参考矩阵、肿瘤计数矩阵和正常计数矩阵。计数矩阵在比对的每一列中记录a/c/g/t和空位(“-”)字符的数目,而参考矩阵在每一列中记录参考碱基。然后通过覆盖率对计数矩阵进行归一化,以反映每一列中的碱基频率。保留了独立的通道来记录肿瘤和正常覆盖率。
图10b示出了输入3维矩阵和所提出的neusomatic网络架构。输入矩阵由参考通道、肿瘤和正常频率通道、覆盖率和位置通道以及其后的几个总结比对特征的通道组成。在集成模式下使用时,neusomatic还包括用于其他各个方法特征的额外通道。neusomatic网络架构由九个卷积层组成,这些卷积层由四个具有捷径标识连接的块构成。我们在最后一层使用两个softmax分类器和一个回归器来预测突变类型、尺寸和位置。
图10c示出了用于给定的候选体细胞缺失的输入矩阵制备的虚拟示例。提取候选体细胞突变周围7个碱基的窗口中的序列比对信息。然后通过添加空位以解释读取中的插入而扩充参考序列。然后将经扩充比对总结为参考矩阵、肿瘤计数矩阵和正常计数矩阵。计数矩阵在比对的每一列中记录a/c/g/t和空位(“-”)字符的数目,而参考矩阵在每一列中记录参考碱基。然后通过覆盖率对计数矩阵进行归一化,以反映每一列中的碱基频率。保留了独立的通道来记录肿瘤和正常覆盖率。
图10d示出了用于给定的候选体细胞插入的输入矩阵制备的虚拟示例。提取候选体细胞突变周围7个碱基的窗口中的序列比对信息。然后通过添加空位以解释读取中的插入而扩充参考序列。然后将经扩充比对总结为参考矩阵、肿瘤计数矩阵和正常计数矩阵。计数矩阵在比对的每一列中记录a/c/g/t和空位(“-”)字符的数目,而参考矩阵在每一列中记录参考碱基。然后通过覆盖率对计数矩阵进行归一化,以反映每一列中的碱基频率。保留了独立的通道来记录肿瘤和正常覆盖率。
前三个通道分别是参考通道、肿瘤频率通道和正常频率通道,它们总结了候选基因座周围的参考碱基以及该区域中不同碱基的频率。为了捕获插入,我们在候选基因座周围使用与读取比对中的插入相对应的空位扩充了参考序列(图10a、图10c和图10d)。因此,肿瘤频率矩阵和正常频率矩阵的每一列分别代表肿瘤样本和正常样本的相对应多序列比对(msa)列中a/c/g/t/空位碱基的频率。其余的通道总结了针对支持不同碱基的读取的其他特征,诸如覆盖率、碱基质量、映射质量、链偏向性和剪切信息。如果在集成模式下使用neusomatic,则我们还将使用了用于由单独的体细胞突变检测方法报告的特征的额外通道。通过这种简洁而又全面的结构化表示,neusomatic可以利用肿瘤、正常和参考中的必要信息将具有低af的难以捕获的体细胞突变与种系变体以及测序错误区分开来。这种设计还可以在cnn中使用卷积过滤器来捕获矩阵子块中的情境模式。
为了与基因组问题中使用的其他cnn方法进行比较,deepvariant11使用读取堆积作为种系变体调用的输入。相反,我们使用每一列的碱基频率总结作为我们的网络的输入。这简化了cnn结构,从而允许实质上更有效的实现。例如,deepvariant花费约1000cpu核心小时来调用30×全基因组样本的种系变体16,而独立版本的neusomatic尽管处理了两个(肿瘤-正常)样本而不是一个样本并且在低于种系的50%或100%af的更低体细胞af的情况下寻找候选者,但它可以在约156cpu核心小时内从30×肿瘤-正常配对样本中检测体细胞突变。另一种种系变体调用方法clairvoyante12,使用三个通道来总结窗口中心的等位基因计数、缺失和插入的碱基计数。相反,我们使用早前所述的参考扩充方法将所有这些事件总结在单个碱基频率矩阵中,该方法可以清楚地表示整个窗口中的所有插入和缺失(indel)事件。
neusomatic采用新颖的cnn结构,该结构可在给定的特征矩阵m的情况下预测候选体细胞突变的类型和长度(图10b)。所提出的cnn由九个卷积层组成,该卷积层由四个具有捷径标识连接的块构成,其灵感来自resnet17,但具有不同的编队以适用于所提出的输入结构。我们在最后一层使用两个softmax分类器和一个回归器。第一个分类器鉴别候选者是否为非体细胞调用结果、snv、插入或缺失。第二个分类器通过四个类别预测体细胞突变的长度(0指示非体细胞,或者长度为1、2或大于2),并且回归器预测体细胞突变的定位。使用这些分类器的输出,我们鉴别了一组体细胞突变。如果预测indel的长度大于2,我们对与该位置重叠的读取实施简单的后处理步骤,以从读取的比对cigar字符串中解析indel序列。已经证明,这对于illumina测序器生成的数据表现良好。对于更高错误率的测序数据,执行更复杂的局部重排后处理以解析indel序列。
由于neusomatic可以在独立模式和集成模式下使用,因此我们使用neusomatic-s表示独立模式,而保留neusomatic来表示集成模式。我们将neusomatic和neusomatic-s与最新的体细胞突变检测方法(包括mutect21、muse2、somaticsniper6、strelka25、vardict3和varscan24)以及集成方法somaticseq10进行了比较。我们使用多个合成数据集和真实数据集进行比较并对比了性能。我们在下文报告,考虑到数据集中体细胞突变的af,合成数据集的体细胞突变检测难度依次递增。
基于铂样本混合物数据集的比较
对于第一个合成数据集,如先前的研究5,10一样,我们将两个正常铂基因组18样本,即na12877和na12878,以70:30、50:50和25:75的肿瘤纯度比率混合以创建三个肿瘤污染曲线,并且以5:95比率混合以创建污染的正常样本。我们还包括了对100%纯正常样本和50%纯肿瘤样本的测试。我们使用na12878中的种系变体(na12877中的参考调用结果)作为用于评估的真值集。neusomatic-s和neusomatic均显著优于所有其他方法(图11a、图11b和图11c)。neusomatic与其他方法相比的性能改善得益于更低、更具挑战性的肿瘤纯度(25:75混合物)。总而言之,neusomatic分别得到高达99.6%和97.2%的snv和indel的f1得分,相对于最低样本纯度下针对该数据集的最佳方法,其改善高达7.2%。对于肿瘤纯度为50%的样本,将正常样本纯度从100%降低至95%对neusomatic的性能影响不大(<0.3%),但它造成somaticseq准确性降低约3%。
图11a和图11b是用于铂双样本混合物数据集的性能分析的图表。在此数据集中,使用了四种肿瘤和正常样本纯度的情况(50%t:100%n、70%t:95%n、50%t:95%n和25%t:95%n)。图11a是用于精确率-召回率分析的图:置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在曲线上,并用实心圆标记。图11b是用于针对不同indel尺寸的indel准确性(f1得分)的性能分析的曲线图。图11c是显示不同体细胞突变检测方法在铂双样本混合数据集上的性能的表。对于每种方法,我们报告精确率-召回率曲线中达到最高f1得分的质量得分阈值的精确率、召回率和f1得分。(rc:召回率,pr:精确率,f1:f1得分)。
基于icgc-tcgadream挑战数据集的比较
对于第二个合成数据集,我们使用icgc-tcgadream挑战阶段3和阶段4数据集19,这些数据集是通过将突变计算地掺入到具有不同af的配对正常样本的健康基因组中而构建的(请参见“方法”)。我们将肿瘤和正常样本混合,以创建五种肿瘤/正常纯度情况。neusomatic-s用于阶段3和阶段4两个数据集的性能均优于所有其他独立方法,其中snv平均超过8%,而indel则平均超过22%(图12a、图12b、图12c、图13a、图13b、图13c)。随着肿瘤纯度的降低,性能的这种改善也增加。我们进一步观察到,即使neusomatic-s在更具挑战性的情况(诸如25:75混合物的snv和25:75和50:50混合物的indel)下仍然优于somaticseq,但neusomatic(集成模式)明显优于somaticseq和neusomatic-s。总而言之,neusomatic分别得到高达96.2%和93.5%的snv和indel的f1得分,相对于在最低样本纯度下的最佳方法,其改善高达34.6%。对于肿瘤纯度为50%的样本,将正常样本纯度从100%降低至95%对neusomatic的性能影响不大(平均约1.2%),而somaticseq和strelka2的f1得分降低>3%。
图12a和图12b是用于dream阶段3数据集的性能分析的曲线图。在此数据集中,使用了五种肿瘤和正常样本纯度的情况(100%t:100%n、50%t:100%n、70%t:95%n、50%t:95%n和25%t:95%n)。图12a是用于精确率-召回率分析的曲线图:置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在曲线上,并用实心圆标记。图12b是用于针对不同indel尺寸的indel准确性(f1得分)的性能分析的曲线图。图12c是显示不同体细胞突变检测方法在dream挑战阶段3数据集上的性能的表。对于每种方法,我们报告精确率-召回率曲线中达到最高f1得分的质量得分阈值的精确率、召回率和f1得分。(rc:召回率,pr:精确率,f1:f1得分)。
图13a和图13b是用于dream阶段4数据集的性能分析的曲线图。在此数据集中,使用了五种肿瘤和正常样本纯度的情况(100%t:100%n、50%t:100%n、70%t:95%n、50%t:95%n和25%t:95%n)。图13a是用于精确率-召回率分析的曲线图:置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在曲线上,并用实心圆标记。图13b是用于针对不同indel尺寸的indel准确性(f1得分)的性能分析的曲线图。图13c是显示不同体细胞突变检测方法在dream挑战阶段4数据集上的性能的表。对于每种方法,我们报告精确率-召回率曲线中达到最高f1得分的质量得分阈值的精确率、召回率和f1得分。(rc:召回率,pr:精确率,f1:f1得分)。
基于铂肿瘤掺入数据集的比较
对于第三个合成数据集,如以前的研究1,10一样,我们通过以从均值[0.05,0.1,0.2,0.3]的二项分布中采用的掺入频率,在na12878的变体位置处,将来自na12878的读取掺入到na12877中而构建肿瘤样本,并使用独立的na12877读取作为纯正常样本。请注意,与以固定比例混合样本得到固定af的体细胞突变的早期策略不同,这种混合方法以0.025到0.3范围内的不同af生成体细胞突变。neusomatic分别得到高达80.9%和66.7%的snv和indel的f1得分,相对于最佳方法,其改善高达4%(图16a至图16d)。对于低af体细胞突变,性能改善甚至更高(af = 0.025,改善11%;af=0.05,改善8%)(图16b)。图16a显示精确率-召回率分析,其中置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在曲线上,并用实心圆标记。图16b显示针对不同af的性能分析。图16c显示针对不同indel尺寸的indel准确性(f1得分)的性能分析。图16d显示,对于每种方法,精确率-召回率曲线中达到最高f1得分的质量得分阈值的精确率、召回率和f1得分。(rc:召回率,pr:精确率,f1:f1得分)。
基于全外显子组和靶向组的比较
为了评定neusomatic在不同靶标富集上的性能,我们使用了来自ashkenazijewishtrio20的全外显子组和靶向组数据集(图17a至图17d)。我们基于全外显子组数据集训练了neusomatic和somaticseq,并将受训模型应用于全外显子组和该组。对于全外显子组,neusomatic的snv和indel的f1得分分别高达99.3%和88.6%。对于靶向组,neusomatic和neusomatic-s的snvf1得分始终超过其他方法,为99.2%。
图17a示出了各种方法在外显子组样本混合物上的性能分析。在此数据集中,使用了四种肿瘤和正常样本纯度的情况(50%t:100%n、70%t:95%n、50%t:95%n和25%t:95%n)。置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在曲线上,并用实心圆标记。这里的训练是neusomatic、neusomatic-s和somaticseq基于外显子组数据的训练。
图17b示出了各种方法在靶标组样本混合物上的性能分析。在此数据集中,使用了四种肿瘤和正常样本纯度的情况(50%t:100%n、70%t:95%n、50%t:95%n和25%t:95%n)。置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在曲线上,并用实心圆标记。这里的训练是neusomatic、neusomatic-s和somaticseq基于外显子组数据的训练。
图17c示出了不同体细胞突变检测方法在全外显子组样本混合数据集上的性能。对于每种方法,我们报告精确率-召回率曲线中达到最高f1得分的质量得分阈值的精确率、召回率和f1得分。(rc:召回率,pr:精确率,f1:f1得分)。
图17d示出了不同体细胞突变检测方法在靶向组数据集上的性能。对于每种方法,我们报告精确率-召回率曲线中达到最高f1得分的质量得分阈值的精确率、召回率和f1得分。(rc:召回率,pr:精确率,f1:f1得分)。
将基于全基因组铂混合物数据训练的模型应用于两个靶标富集集,得到相似的性能,这证实了neusomatic的鲁棒性(图18a和图18b)。与其他数据集类似,对于肿瘤纯度为50%的样本,将正常样本纯度从100%降低至95%,将会最小限度地减少基于全外显子组数据集的neusomaticf1得分(平均约0.3%),而somaticseq和strelka2的f1得分降低>5%。
图18a示出了使用基于全基因组(铂数据,基因组混合物)和全外显子组(hg003-hg004外显子组混合物)训练的模型对外显子组混合物数据集进行测试的性能分析。置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分指示在图例中,并用实心圆标记在曲线上。
图18b示出了使用基于全基因组(铂数据,基因组混合物)和全外显子组(hg003-hg004外显子组混合物)训练的模型对靶标组混合物数据集进行测试的性能分析。置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分指示在图例中,并用实心圆标记在曲线上。
基于pacbio数据集的比较
我们进一步评估了neusomatic在错误率较高的读取上的性能,特别是在来自长读取测序平台的读取上的性能。我们使用基于原始pacbio读取模拟的具有20%、30%和50%af体细胞突变的肿瘤-正常配对样本(图14a、图14b和图14c)。neusomatic鉴别出的体细胞snv和indel的f1得分分别高达98.1%和86.2%,其性能优于vardict3,针对snv,胜出高达34.4%,而针对indel,胜出高达53.2%。该分析证实了neusocomic在检测体细胞突变方面的能力,即使序列读取的错误率如同pacbio长原始读取一样高。
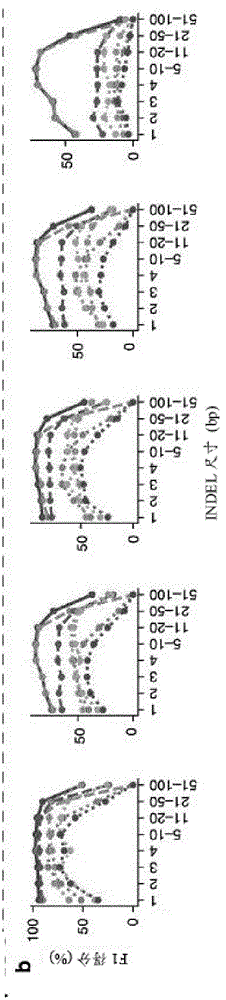
针对不同indel尺寸的比较
值得注意的是,对于不同数据集中的各种indel尺寸,neusomatic始终优于其他方法(图11b、图12b、图13b、图14b、图16c和图19)。对于大(大于50个碱基)的indel,由于大多数具有体细胞indel的短读取都是软跳过的,因此indel信息会在堆积计数矩阵中丢失。对于此类情况,neusomatic受益于其他方法的预测,因为诸如vardict和mutect2之类的一些方法使用本地程序集进行预测。
图14a和图14b是不同体细胞突变检测方法在pacbio数据集上的性能分析的曲线图。在此数据集中,使用了三种肿瘤和正常样本纯度的情况(70%t:95%n、50%t:95%n和25%t:95%n)。图14a是用于精确率-召回率分析的曲线图:置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在曲线上,并用实心圆标记。图14b是用于针对不同indel尺寸的indel准确性(f1得分)的性能分析的曲线图。图14c显示,对于每种方法,精确率-召回率曲线中达到最高f1得分的质量得分阈值的精确率、召回率和f1得分。(rc:召回率,pr:精确率,f1:f1得分)。图19示出了在dream阶段3、dream阶段4、铂双样本混合物、铂肿瘤掺入、pacbio和外显子组数据集中的基准真值(groundtruth)indel的尺寸分布。负尺寸对应于缺失。
indel类型和位置准确性
对于所有讨论的数据集,我们还评定了通过不同体细胞突变检测方法进行indel调用的性能,使用更为宽松的标准,即简单地正确预测体细胞indel的位置(并忽略确切的indel序列)。再次,我们观察到neusomatic相对于其他方案的相似优势,指示主要的改善是由所提出的cnn结构而不是后处理indel解析步骤贡献的(图20a和图20b)。
图20a示出了在三种肿瘤纯度(50%、30%和20%)以及95%正常纯度的情况下,基于针对pacbio数据集的所预测的体细胞突变的位置和类型(而忽略了确切的所预测indel序列的准确性)的indel的性能分析。置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在曲线上,并用实心圆标记。
图20b示出了针对dream阶段3、dream阶段4、铂双样本混合物、全外显子组和铂肿瘤掺入数据集的基于所预测的体细胞突变的位置和类型的indel的性能分析(而忽略了确切的所预测indel序列的准确性)。对于前四个数据集,使用了三种肿瘤纯度方案(70%、50%和25%),而正常样本的纯度为95%。置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在曲线上,并用实心圆标记。
读取覆盖率分析
为了评估序列覆盖率对不同技术的影响,我们对全外显子组数据集进行了下采样,以获得序列覆盖率在20×和100×范围内的样本(图15a和图15b)。对于不同的覆盖率,neusomatic始终优于其他技术。对于低覆盖率样本,随着问题更具挑战性,改善也有所增加。此外,将覆盖范围从100×减少到50×对neusomatic的影响很小(对于snv约为1.5%,对于indel约为5%),而somaticseq的snv和indel的f1得分均下降约20%,并且strelka2的snv的f1得分下降了约13%且indel的f1得分下降了约15%。该分析显示了neusomatic对覆盖率扰动的鲁棒性以及在挑战性场景中的优势,这可以在较低覆盖率下看到。
图15a是用于序列覆盖率对全外显子组样本混合物数据集的影响的性能分析的曲线图。在该示例中,肿瘤的纯度为50%,正常样本的纯度为95%。y轴示出了每种算法在20×到100×范围内进行的样本比对所能获得的最高f1得分。
图15b是用于序列覆盖率对全外显子组样本混合物数据集的影响的性能分析的曲线图。在该示例中,肿瘤的纯度为50%,正常样本的纯度为95%。肿瘤和正常比对的覆盖范围是20x至100x。置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分指示在图例中,并用实心圆标记在曲线上。
训练鲁棒性
我们通过基于dream挑战阶段3数据集针对不同纯度进行训练和测试,评定neusomatic针对特定纯度进行训练的鲁棒性。图21显示,即使当我们在非常不同的肿瘤纯度上进行训练和测试时,其性能也仅会轻微降低。我们还观察到,使用从多个肿瘤纯度汇总的数据进行的训练与对在靶标肿瘤纯度上进行的训练一样好(图21)。这表明并入了多个肿瘤纯度的训练集足以获得对肿瘤纯度变化具有鲁棒性的模型。
图21示出了针对dream挑战阶段3数据集的交叉样本训练的性能分析。我们使用基于不同纯度训练的neusomatic模型以及基于来自所有不同纯度的集合输入训练的模型,对肿瘤纯度分别为70%、50%和25%的每个样本进行了测试。置信度或质量得分用于推导出精确率-召回率曲线。通过每种算法获得的最高f1得分打印在图例中,并用实心圆标记在曲线上。
基于真实数据的比较
在不存在针对体细胞突变21的高质量的、全面的基准真值数据集比如针对种系变体的“瓶中基因组金集”(genome-in-a-bottlegoldset)22的情况下,我们将不可能在合成数据之外计算f1准确性。幸运的是,我们可以采用具有经过验证的体细胞突变的现有数据集来评估neusomatic在真实数据上的准确性性能(有关如何在真实数据上外推f1得分的更多详细信息,请参见“方法”)。我们使用了两个数据集:cll123是具有961个经过验证的体细胞snv的慢性淋巴细胞白血病患者全基因组数据,而colo-82924,25是具有454个经过验证的体细胞snv的永存转移性恶性黑色素瘤细胞系衍生全基因组数据集。为了用这两个真实wgs样本评估neusomatic,我们使用了基于dream挑战阶段3训练的模型。如图22a和图22b中所示,neusomatic分别针对colo-829恶性黑色素瘤样本和cll1慢性淋巴细胞性白血病样本获得了99.7%和93.2%的最高外推f1得分。我们还用结直肠腺癌的tcga26,27全外显子组测序(wes)样本(tcga-az-6601)评估了neusomatic,获得了超过99.6%的最高外推f1得分(图22c)。
图22a显示了不同体细胞突变检测方法在真实数据集colo-829上的性能。图22b显示了不同体细胞突变检测方法在真实数据集cll1上的性能。图22c显示了不同体细胞突变检测方法在真实数据集tcga-az-6601上的性能。
为了证明neusomatic在云上的可扩展性和经济效益,我们还使用集成模式和独立模式在microsoftazure云平台上处理了来自tcga的261个全外显子组测序癌症样本(图23)。这些样本是从多种癌症类型中采集的,包括结肠直肠腺癌、卵巢浆液性腺癌、宫颈鳞状细胞癌和宫颈内腺癌。尽管云平台使我们能够按需自动启动计算实例(computeinstances),但集成模式和独立模式分别平均需要2.42小时和0.72小时的时间来处理每个样本。使用azure的可抢占型计算实例(使用标准的h16实例类型,每个实例具有16个核心),每个样本的处理成本较低,对于集成模式和独立模式分别为0.77usd和0.23usd。我们还通过与这些样本中的44,270个经过验证的snp进行比较,评定了neusomatic在这些样本上的准确性,这为我们提供了分别为98.9%和97.2%的整体模式召回率和独立模式召回率。因此,neusomatic不仅可以用于不同的测序技术或测序策略,而且可以在各种计算平台(包括本地hpc集群和弹性云计算架构)上运行。
图23是用于microsoftazure实验的261个tcga癌症样本的列表。样本取自三种癌症类型:结肠直肠腺癌(coad)、卵巢浆液性腺癌(ov)以及宫颈鳞状细胞癌和宫颈内腺癌(cesc)。
讨论
neusomatic是第一个基于深度学习的体细胞突变检测框架,它是高性能且通用的。当使用相同的cnn架构时,它可以针对改变的肿瘤纯度跨从合成到真实的多个数据集、从全基因组到靶向的多种测序策略以及从短读取测序到高错误率的长读取测序的多种测序技术范围而实现最佳的准确性。具体而言,对于低肿瘤纯度和低等位基因频率,neusomatic明显优于其他最新的体细胞突变检测方法,从而证明了其解决疑难问题的能力。neusomatic利用卷积神经网络的有效实现来快速、准确地解决体细胞突变检测问题。它使用肿瘤/正常比对信息的新颖总结作为一组输入矩阵,可以有效地捕获基因组情境中的主要信号。基于这些矩阵训练所提出的cnn架构,可以直接从原始数据中学习特征表示。从观察到的训练数据中学到的深层特征可以准确地鉴别重要的突变标志,这些标志可以将真实的调用结果与由测序错误、交叉污染或覆盖率偏差所引入的伪影区分开来。我们相信,neusomatic通过为体细胞突变检测提供非常广泛适用的方法,可以极大地改进最新技术。
方法
icgc-tcgadream挑战数据
阶段3数据由正常样本和肿瘤样本组成,这些样本是通过将7903个snv和7604个indel突变计算地掺入到具有三个不同af(50%、33%和20%)的相同正常样本的健康基因组中而构建的,以创建合成但真实的肿瘤正常样本对。阶段4的数据具有相似的构造,但是在30%和15%af的两个亚克隆中具有16,268个snv和14,194个indel。然后,我们通过将95%正常读取与5%肿瘤读取混合而构建不纯的正常读取。我们还通过分别以100:0、70:30、50:50和25:75的比率混合肿瘤和正常读取来构建四种肿瘤混合物。因此,跨这四个肿瘤混合比率的体细胞突变,对于阶段3数据,af的范围为5%至50%,而对于阶段4数据集,af的范围为3.75%至30%。
铂合成肿瘤数据
我们下载了200×铂基因组样本na12878和na12877及其真值种系变体(v2017-1.0)18,以构建虚拟肿瘤和正常对(ena登录号prjeb3246)。对于正常样本,我们将na12877下采样为50×。对于肿瘤样本,我们通过分别以15×、25×和37.5×独立地下采样na12877,并将每一个与35×、25×和12.5×的经下采样na12878混合,从而构建了三个肿瘤纯度分别为70%、50%和25%的50×计算机模拟混合物样本。我们使用na12878中的杂合子变体和纯合子变体(它们是na12877中的参考调用结果,与na12877变体相距至少五个碱基并且彼此相距300个碱基)作为训练和评估步骤的真值集(1,103,285个snv和174,754个indel)。因此,取决于na12878中种系变体的接合性,跨这三种肿瘤混合物比率的体细胞突变的af范围为12.5%至70%。
我们还通过在na12878中的杂合子变体定位和纯合子变体定位(它们是na12877中的参考调用结果)处,将来自经下采样(至50×覆盖率)na12878的读取随机掺入到经下采样(至50×覆盖率)na12877数据中,从而生成了另一个50×虚拟肿瘤样本。对于每个变体,我们用均值[0.05、0.1、0.2、0.3]随机分配来自二项式分布的掺入读取的频率。因此,取决于变体的接合性,均值体细胞突变af的范围为2.5%至30%。为了避免在真值集中产生歧义,我们仅使用相关配对末端读取不与任何其他变体重叠的变体(316,050个snv和46,978个indel)。这产生了具有来自na12878的读取的被污染肿瘤样本。我们还使用na12877的另一个独立的经下采样(至50×)数据用作纯正常样本。
对于这两个实验,均下载了fastq文件和真值种系变体,并先后与bwa-mem(v0.7.15)28和picardmarkduplicates(v2.10.10)(https://broadinstitute.github.io/picard)以及gatkindelrealignerandbasequalityscorerecalibration(v3.7)29比对。
真实的肿瘤-正常对数据
我们使用cll1慢性淋巴细胞白血病数据集23(登录:https://www.ebi.ac.uk/ega/datasets/egad00001000023)和colo-829永存转移性恶性黑色素瘤细胞系数据集24,25(登录:https://www.ebi.ac.uk/ega/studies/egas00000000052),基于真实肿瘤-正常配对数据以及已发布的经过验证的体细胞突变清单,评定我们的方法。
colo-829数据集由80×全基因组测序肿瘤样本及其匹配的60×正常血液colo-829bl样本组成,并具有454个经过验证的体细胞snv。cll1具有覆盖率分别为53×和42×的全基因组测序肿瘤样本和匹配的正常样本,并具有961个已公开的体细胞snv。
tcga-az-660126,27数据集是来自tcga的结肠腺癌肿瘤样本及其匹配的正常组织的全外显子组测序。肿瘤和正常样本分别以145×和165×的深度测序。我们使用tcga30和cosmic31数据库中的952个经过验证的snv作为该样本的基准真值体细胞突变。
对于真实数据,我们将外推精确率计算为所预测的体细胞突变的百分比,该体细胞突变已经被至少两种独立方法调用,或者已经在cosmic数据库中报告为至少两个相同癌症类型样本中已证实的体细胞突变。然后,我们基于召回率的调和平均数和该外推精确率来计算外推的f1得分。
全外显子组和靶向组数据
为了用不同靶标富集实验评定neusomatic,我们使用了来自ashkenazijewishtrio20的全外显子组数据集。我们下载了hg003和hg004的深度测序(200×覆盖率)全外显子组比对文件(ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/),以及高置信度的种系变体(瓶中基因组v3.3.2版)。然后,我们使用hg004的随机70×、50×和25×下采样以及hg003的30×、50×和75×下采样的混合物,分别构建了70%、50%和25%的纯肿瘤样本。通过将95×hg003下采样比对和5×hg004下采样比对混合,我们还构建了95%的纯正常样本。我们使用agilentsureselecthumanallexonv5bed文件进行我们的分析。与铂合成肿瘤数据(11,720个snv,878个indel)相似,鉴别了基准真值体细胞突变。取决于hg004中种系变体的接合性,跨这三种肿瘤混合物比率的体细胞突变的af范围为12.5%至70%。
为了验证在靶标组上的性能,我们将上述比对和真值数据限制为illumina的trusight遗传疾病组bed文件(216个snv,5个indel)。由于靶标组区域中真实indel的数量有限,我们仅评估在snv上的性能。
pacbio数据
对于长读取分析,我们下载了hg002样本的高置信度种系变体(瓶中基因组v3.3.2版)(ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/)20。我们使用chm1数据集32(sra登录号srx533609)建立了长读取误差分布。然后,我们使用varsim模拟框33与longislnd计算机模拟长读取测序仪模拟器34的组合,模拟了100×纯正常样本。使用一组随机体细胞突变,我们还模拟了具有相同误差分布的100×纯肿瘤样本。我们使用ngmlr(v0.2.6)35来比对序列。然后,我们将纯正常比对的47.5×下采样与纯肿瘤比对的2.5×下采样混合以形成纯度为95%的50×正常对,并将正常比对的40×、35×和25×独立下采样分别与纯肿瘤比对的10×、15×和25×下采样混合以构建纯度为20%、30%和50%的50×肿瘤混合物。我们将训练集限制在1号染色体上的120兆碱基区域(带有39,818个真值体细胞snv和38,804个真值体细胞indel),并将测试集限制在整个22号染色体(带有12,201个真值体细胞snv和12,185个真值体细胞indel)。跨三种肿瘤混合物比率的体细胞突变的af范围为20%至50%。
候选突变制备
第一步,我们扫描肿瘤读取比对以找到具有突变证据的候选定位。由于基因组区域的复杂性或测序伪影,这些位置中的许多位置要么具有种系变体,要么被错误调用。我们将一组自由过滤器应用于一组候选定位,以确保此类定位的数量合理。通常,对于snv,我们要求af≥0.03或超过两个读取支持snv并且phred缩放的碱基质量得分大于19(对于真实wes数据集,大于14)作为最低要求。对于1个碱基的indel,我们要求af≥0.02或超过一个读取支持。对于大于1个碱基的indel,我们要求af≥0.03。对于集成方法,我们还包括通过其他体细胞突变检测方法检测到的任何体细胞突变作为输入候选。对于pacbio数据集,对于大于1个碱基的snv和indel,我们使用af≥0.1,对于1个碱基的indel,af≥0.15。
对于dream挑战数据集,我们从输入候选中排除了dbsnp36中存在的变体。为了公平比较,我们还过滤了所有其他体细胞突变检测工具的dbsnp调用结果。
输入突变矩阵
对于每个候选位置,我们制备了一个3维矩阵m,其具有尺寸为5 × 32的k个通道(图10a、图10c和图10d)。每个通道中的五个横行对应于四种dna碱基a、c、g和t以及空位字符(“-”)。矩阵的32列中的每一列代表比对的一列。
对于每个候选定位,我们提取了肿瘤和正常读取比对。如图10a所示,然后,我们将肿瘤和正常样本与参考的读取比对作为msa。为此,当读取中存在插入时,我们通过在参考序列上添加空位而扩充参考序列。必须注意,此过程不需要对输入bam文件的原始读取比对进行任何进一步的重新比对,而只需通过在发生插入的位置处分配其他列,即可将比对重构为msa格式。如果在特定位置之后的多次读取中存在多个不同插入,我们将其视为经左比对序列并将它们放在同一组列中(例如,参见图10a中虚拟示例的第九列中的a碱基插入和c碱基插入)。使用此读取表示法,我们在每一列中找到a/c/g/t/-字符的频率,并为肿瘤和正常样本(矩阵m中的通道c2和c3)记录单独的矩阵。在通道c1中,我们在每一列中记录参考碱基(或空位)。通道ci(4 ≤ i ≤ k)记录肿瘤和正常样本中的其他比对信号,诸如覆盖率、碱基质量、映射质量、链、剪切信息、编辑距离、比对得分和配对末端信息。例如,对于碱基质量通道,每个样本都有一个尺寸为5 × 32的矩阵,该矩阵记录在每列中具有给定碱基(对于给定的横行)的读取的平均碱基质量。另举一例,对于编辑距离通道,每个样本都有一个尺寸为5 × 32的矩阵,该矩阵记录在每列中具有给定碱基(对于给定的横行)的读取的平均编辑距离。矩阵m的一个通道专用于指定候选者所在的列。在当前实现中,我们在独立模式的neusomatic-s方法中总共使用了26个通道。
对于neusomatic的集成扩展,我们还包括额外的通道来捕获由所使用的六个单独方法中的每个方法报告的特征。在该实现中,我们使用了93个额外通道来表示从其他方法中提取的特征以及由somaticseq报告的比对。因此,neusomatic的集成模式具有针对每个候选矩阵的119个输入通道。
对于每个候选定位,我们在候选者周围七个碱基的窗口中报告比对信息。我们保留32列,以考虑到具有插入的经扩充比对。在少数情况下,如果插入量较大,则32列可能不足以表示比对。对于此类情况,我们将截短插入,以便可以记录候选者附近的至少三个碱基。
cnn架构
所提出的cnn(图10b)由如下构造的九个卷积层组成。输入矩阵被馈入具有64个输出通道、1 × 3核尺寸和relu激活的第一卷积层中,之后被馈入到批归一化和最大池化层中。然后,这一层的输出被馈入一组四个块中,这些块具有类似于resnet结构的捷径标识连接。这些块由一个具有3 × 3个核的卷积层和之后的批归一化和一个具有5× 5个核的卷积层组成。在这些捷径块之间,我们使用批归一化层和最大池化层。最终块的输出被馈入尺寸为240的全连接层中。然后将所得的特征向量馈入两个softmax分类器和一个回归器中。第一个分类器是4向分类器,可从非体细胞、snv、插入和缺失这四类中预测突变类型。第二个分类器根据0、1、2和≥3这四个类别来预测所预测突变的长度。非体细胞调用结果标注为零尺寸突变,snv和1个碱基的indel标注为尺寸1,而2个碱基的且尺寸≥3的indel分别标注为2和≥3尺寸的突变。回归器预测矩阵中突变的列,以确保预测靶向正确的位置,并使用平滑的l1损失函数对其进行优化。
cnn的参数少于900k,这使我们能够通过使用大的批次规模来实现高效的实现。在配备8台teslak80nvidiagpu的机器上,全基因组训练过程耗时约8小时。
cnn训练
对于dream挑战数据集、铂数据集和靶标富集数据集,我们将基因组区域随机分为50%训练集和50%测试集。对于pacbio数据集,我们基于1号染色体上的120兆碱基区域训练neusomatic,并在22号染色体上对其进行测试。
对于每个数据集,我们合并了来自所有不同肿瘤/正常纯度场景的所生成的训练输入矩阵,并使用合并的集合来训练网络。然后,我们将该统一受训模型应用于每个独立的肿瘤/正常纯度设置下的测试。
dream挑战数据集具有阶段3的15,507个体细胞突变和阶段4的30,462个体细胞突变。为了更好地进行网络训练,我们使用bamsurgeon19将af分布类似于原始dream数据的约95k的更多snv和约95k的更多indel掺入到阶段3和阶段4的肿瘤样本中。
我们使用1000个的批次规模和sgd优化器以0.01的学习速率和0.9的动量对网络进行训练,并且每400代训练就将学习速率乘以0.1。
一般而言,由于输入候选定位的非体细胞(参考或种系)调用结果比真实体细胞突变多得多,因此在每一代训练中,我们都使用训练集中的所有真实体细胞突变并随机选择的非体细胞候选者,该非体细胞候选者的数目是真实体细胞突变数的两倍。我们使用加权的softmax分类损失函数来平衡每个类别中候选者的数量。对于dream挑战数据,由于我们在训练集中添加了更多的合成突变,因此我们增加了非体细胞类别的权重,以在测试集中实现更高的精确率。
为了评定合成靶标富集数据集,我们使用了全外显子组和全基因组数据作为训练集。
为了测试真实wgs样本cll1和colo-829,我们使用了基于dream挑战阶段3训练的somaticseq和neusomatic模型。对于真实wes样本tcga-az-6601,我们使用来自另一个tcgawes数据集tcga-az-431530的数据制备了训练集。我们将来自该数据集的肿瘤比对和正常比对混合,并将混合物分成两个相等的比对。然后,我们使用一个比对作为纯正常样本,并使用bamsurgeon将约91k的随机snv和约9k的随机indel掺入到另一个比对中,以生成用于训练的合成肿瘤样本。我们使用基于该合成肿瘤-正常wes数据集训练的模型在真实wes数据集tcga-az-6601上测试neusomatic和somaticseq。对于261个真实tcga样本的实验,我们使用了类似的方法来使用12个tcga样本制备训练集。基于该合成数据集训练的模型用于在261个tcga样本上进行测试。
超参数调优
对于超参数调优,我们在dream挑战阶段3实验中使用了10%的基因组,并在所有其他实验中使用了推导出的参数。
我们进一步探索了不同的网络架构,诸如具有4至16个resnet块的预激活resnet架构(包括resnet-18和resnet-34架构)(图24a至图24e),以及所提出的残差neusomatic架构的一些变体(图24f至图24m)。为了评估这些网络,我们将dream阶段3数据集中的训练数据分为两个相等的部分,使用其中的一个来训练不同架构,并使用另一个以独立模式评估这些不同架构。图25在准确性、网络参数数量、存储器使用和速度方面比较了这些架构。通常,与传统的体细胞突变检测方法相比,所有这些网络都可以获得相对较高的准确性。这一发现揭示了所提出的数据总结方法的重要性,该方法在候选者的基因组情境中捕获主要信号并有助于卷积网络在体细胞突变检测问题上的有效实现。与具有所提出的残差块的那些(图24f至图24m)相比,具有两个3 × 3卷积过滤器(图24a至图24e)的缺省resnet架构具有较低的平均准确性。另外,具有跨步卷积的网络(图24a至图24g)具有更多的网络参数和运行时要求。总而言之,尽管每种网络架构在一些比较方面均显示出优势,但我们还是选择了所提出的neusomatic网络架构(图10b;图24k)作为缺省网络架构以兼顾所有这些因素,而其他网络则可以轻松实现根据用户的使用情况和时间/计算约束对其进行调整。
图24a至图24m示出了所测试的不同网络架构。图24a至图24e示出了具有不同数量的预激活残差块的具有缺省3x3卷积层的resnet架构。在这里,跨步卷积与通道扩展一起使用。图24f和图24g示出了具有3x3和5x5的卷积层以及一些扩张卷积的多个定制的残差块。在这里,跨步卷积与通道扩展一起使用。图24h示出了具有3x3和5x5的卷积层以及一些扩张卷积的四个定制的残差块。这里,不使用跨步卷积。图24i至图24m示出了具有不同残差块和全连接尺寸的neusomatic残差架构。
图25示出了图24a至图24m所示的不同网络架构的性能分析。在这里,所有网络都在经过了600代训练之后以1000个的批次规模进行评定。
其他体细胞突变检测算法
在我们的分析中,我们使用了缺省设置的strelka2(v2.8.4)、mutect2(v4.0.0.0)、somaticsniper(v1.0.5.0)、muse(v1.0rc)、vardict(v1.5.1)、varscan2(v2.3.7)和somaticseq(v2.7.0)体细胞突变检测算法。
我们使用vardict作为基于pacbio数据的neusomatic的替代方法。为了能够在高错误率的长读取上检测体细胞突变,我们使用了具有“−m10000−q1−q5−x1−c1−s2−e3−g4−k0”参数设置的vardict。此外,与neusomatic一样,对于snv,使用af≥0.1,对于indel,使用af≥0.15。
为了训练somaticseq,我们还遵循了与neusomatic所用相同的50%训练/测试区域划分。此外,与neusomatic一样,对于每个数据集,我们将来自所有不同肿瘤/正常纯度场景的训练数据组合,以训练somaticseqsnv分类器和indel分类器。然后使用这些统一的分类器来预测每个单独的肿瘤/正常纯度设置。
对于精确率-召回率分析,基于每种工具分配的置信度或质量得分对体细胞突变进行分类。对于muse,我们使用层分配作为排序标准。对于vardict、varscan2、mutect2、strelka2和somaticsniper,我们分别使用了vcf文件中报告的ssf值、ssc值、tlod值、somaticevs值和ssc值进行排序。对于somaticseq和neusomatic,我们使用了qual领域中的体细胞突变质量得分。neusomatic基于cnn预测的概率报告所预测的体细胞突变的质量得分。
为了分析真实样本的性能,我们使用了来自不同方法的pass体细胞调用结果(对于vardict,我们仅限于具有strongsomatic状态的调用结果)。对于neusomatic,我们将wgs的质量得分阈值设为0.97,将wes的质量得分阈值设为0.6。
计算复杂性
对于全基因组数据,在双14核intelxeoncpue5-2680v42.40ghz机器上,扫描30×肿瘤和正常比对以找到候选者、提取特征并制备输入矩阵可能耗费约3.9小时。在配备8台teslak80nvidiagpu的机器上,全基因组训练过程可能耗费约8小时(规模为580,000的每一代训练约90秒)。取决于候选体细胞突变的截止值af,使用8台teslak80nvidiagpu计算对于30×全基因组数据上候选突变的网络预测可能耗费约35分钟(af截止值为0.05,3.9m个候选者)至约100分钟(af截止值为0.03,11.5m个候选者)。对于125×全外显子数据,网络预测的整个扫描、制备和计算可能耗费约30分钟。将使用neusomatic集成方法和独立方法(仅cpu模式)在125×全外显子组数据集和30×全基因组数据集上预测体细胞突变的端到端运行时与图26a和图26b中的其他体细胞突变检测技术进行比较。
图26a示出了不同体细胞突变检测算法的运行时比较。显示了cpu核心-小时,以预测125x全外显子组测序数据集上的体细胞突变。图26b示出了不同体细胞突变检测算法的运行时比较。显示了cpu核心-小时,以预测30x全基因组测序数据集上的体细胞突变。
代码可用性
neusomatic用python和c++编写。它的深度学习框架是使用pytorch0.3.1实现的,以使gpu应用程序能够进行训练/测试。根据知识共享署名非商业共享(creativecommonsattribution-noncommercial-sharealike)4.0国际许可,源代码位于https://github.com/bioinform/neusomatic。本文的结果是基于neusomaticv0.1.3的。
报告总结
有关实验设计的更多信息,请参见与本文链接的《自然研究报告汇编》(natureresearchreportingsummary)。
资料可用性
这项研究的序列数据是从各种来源收集的,即,欧洲核苷酸档案库(登录号:prjeb3246;https://www.ebi.ac.uk/ena)、序列读取档案库(登录号:srx1026041;https://www.ncbi.nlm.nih.gov/sra)、国际癌症基因组联合体(项目:icgc-tcgadream突变调用挑战,受控访问:https://icgc.org/)、癌症基因组图集(登录号:tcga-az-6601、tcga-az-4315;受控访问:https://gdc.cancer.gov/)、欧洲基因组-现象档案库(登录号:egas00000000052、egad00001000023;受控访问:https://www.ebi.ac.uk/ega/)和瓶中基因组(登录号:hg002、hg003、hg004;ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/)。使用https://github.com/bioinform/neusomatic/blob/paper/etc/data_scripts.zip的脚本从上述数据集中生成了合成数据。所有其他相关数据可应要求提供。
参考文献
1.cibulskis,k.等人,sensitivedetectionofsomaticpointmutationsinimpureandheterogeneouscancersamples.nat.biotechnol.31,213(2013)。
2.fan,y.等人,muse:accountingfortumorheterogeneityusingasample-specificerrormodelimprovessensitivityandspecificityinmutationcallingfromsequencingdata.genomebiol.17,178(2016)。
3.lai,z.等人,vardict:anovelandversatilevariantcallerfornext-generationsequencingincancerresearch.nucleicacidsres.44,e108–e108(2016)。
4.koboldt,d.c.等人,varscan2:somaticmutationandcopynumberalterationdiscoveryincancerbyexomesequencing.genomeres.22,568–576(2012)。
5.kim,s.等人,strelka2:fastandaccuratecallingofgermlineandsomaticvariants.nat.methods15,591–594(2018)。
6.larson,d.e.等人,somaticsniper:identificationofsomaticpointmutationsinwholegenomesequencingdata.bioinformatics28,311–317(2011)。
7.wang,q.等人,detectingsomaticpointmutationsincancergenomesequencingdata:acomparisonofmutationcallers.genomemed.5,91(2013)。
8.alioto,t.s.等人,acomprehensiveassessmentofsomaticmutationdetectionincancerusingwhole-genomesequencing.nat.commun.6,10001(2015)。
9.roberts,n.d.等人,acomparativeanalysisofalgorithmsforsomaticsnvdetectionincancer.bioinformatics29,2223-2230(2013)。
10.fang,l.t.等人,anensembleapproachtoaccuratelydetectsomaticmutationsusingsomaticseq.genomebiol.16,197(2015)。
11.poplin,r.等人,auniversalsnpandsmall-indelvariantcallerusingdeepneuralnetworks.nat.biotechnol.36,983(2018)。
12.luo,r.、sedlazeck,f.j.、lam,t.-w.和schatz,m.,clairvoyante:amulti-taskconvolutionaldeepneuralnetworkforvariantcallinginsinglemoleculesequencing.https://www.biorxiv.org/content/early/2018/04/28/310458(2018)。
13.simpleconvolutionalneuralnetworkforgenomicvariantcallingwithtensorflow.https://towardsdatascience.com/simple-convolution-neural-network-for-genomic-variant-calling-with-tensorflow-c085dbc2026f(2017)。
14.esteva,a.等人,dermatologist-levelclassificationofskincancerwithdeepneuralnetworks.nature542,115–118(2017)。
15.torracinta,r.等人,adaptivesomaticmutationscallswithdeeplearningandsemi-simulateddata.https://www.biorxiv.org/content/early/2016/10/04/079087(2016)。
16.runningdeepvariant.https://cloud.google.com/genomics/docs/tutorials/deepvariant(2018)。
17.he,k.、zhang,x.、ren,s.和sun,j.,deepresiduallearningforimagerecognition.proc.ieeeconf.comput.vis.patternrecognit.770–778(2016)http://openaccess.thecvf.com/content_cvpr_2016/html/he_deep_residual_learning_cvpr_2016_paper.html。
18.eberle,m.a.等人,areferencedatasetof5.4millionphasedhumanvariantsvalidatedbygeneticinheritancefromsequencingathree-generation17-memberpedigree.genomeres.27,157-164(2017)。
19.ewing,a.d.等人,combiningtumorgenomesimulationwithcrowdsourcingtobenchmarksomaticsingle-nucleotide-variantdetection.nat.methods12,623(2015)。
20.zook,j.m.等人,extensivesequencingofsevenhumangenomestocharacterizebenchmarkreferencematerials.sci.data3,160025(2016)。
21.xu,c.,areviewofsomaticsinglenucleotidevariantcallingalgorithmsfornext-generationsequencingdata.comput.struct.biotechnol.j.16,15–24(2018)。
22.zook,j.m.等人,integratinghumansequencedatasetsprovidesaresourceofbenchmarksnpandindelgenotypecalls.nat.biotechnol.32,246(2014)。
23.puente,x.s.等人,whole-genomesequencingidentifiesrecurrentmutationsinchroniclymphocyticleukaemia.nature475,101(2011)。
24.morse,h.g.和moore,g.e.,cytogenetichomogeneityineightindependentsitesinacaseofmalignantmelanoma.cancergenet.cytogenet.69,108–112(1993)。
25.pleasance,e.d.等人,acomprehensivecatalogueofsomaticmutationsfromahumancancergenome.nature463,191(2010)。
26.network,c.g.a.等人,comprehensivemolecularcharacterizationofhumancolonandrectalcancer.nature487,330(2012)。
27.grasso,c.s.等人,geneticmechanismsofimmuneevasionincolorectalcancer.cancerdiscov.8,730-749(2018)。
28.li,h.,aligningsequencereads,clonesequencesandassemblycontigswithbwa-mem.https://arxiv.org/abs/1303.3997(2013)。
29.vanderauwera,g.a.等人,fromfastqdatatohigh-confidencevariantcalls:thegenomeanalysistoolkitbestpracticespipeline.curr.protoc.bioinforma.43,10-11(2013)。
30.grossman,r.l.等人,towardasharedvisionforcancergenomicdata.n.engl.j.med.375,1109–1112(2016)。
31.forbes,s.a.等人,cosmic:somaticcancergeneticsathigh-resolution.nucleicacidsres.45,d777–d783(2016)。
32.chaisson,m.j.等人,resolvingthecomplexityofthehumangenomeusingsingle-moleculesequencing.nature517,608(2015)。
33.mu,j.c.等人,varsim:ahigh-fidelitysimulationandvalidationframeworkforhigh-throughputgenomesequencingwithcancerapplications.bioinformatics31,1469-1471(2014)。
34.lau,b.等人,longislnd:insilicosequencingoflengthyandnoisydatatypes.bioinformatics32,3829-3832(2016)。
35.sedlazeck,f.j.等人,accuratedetectionofcomplexstructuralvariationsusingsingle-moleculesequencing.nat.methods15,461-468(2018)。
36.sherry,s.t.等人,dbsnp:thencbidatabaseofgeneticvariation.nucleicacidsres.29,308–311(2001)。
实例2
体细胞突变的准确检测具有挑战性,但对于了解癌症的形成、进展和治疗至关重要。我们最近提出了neusomatic,这是第一个基于深度卷积神经网络的体细胞突变检测方法,并展示了在计算机模拟数据上的性能优势。在这项研究中,我们使用了seqc-ii联合体的首批全面且充分表征的体细胞参考样本,以探究在癌症突变检测中利用深度学习框架的最佳实践。利用该联合体为这些参考样本建立的高置信度体细胞突变,我们鉴别了用于在源自表示真实场景的样本的多个数据集上建立鲁棒模型的策略。所提出的策略在用于新鲜和ffpedna输入、不同的肿瘤/正常纯度和不同的覆盖率(范围从10×至2000×)的多种测序技术诸如wgs、wes、ampliseq靶标测序中实现了高鲁棒性。neusomatic在一般情况下以及在诸如低覆盖率、低突变频率、dna损伤和困难基因组区域等挑战性情况下,都大大优于传统检测方法。
前言
体细胞突变是关键的癌症驱动因素。准确的体细胞突变检测可对癌症患者进行精确的诊断、预后和治疗1。已经开发了几种工具来鉴别来自下一代测序技术的体细胞突变2-11。一般而言,传统技术通过各种统计学/算法建模方法,采用从肿瘤-正常配对测序数据2-10中提取的一组手工测序特征,从背景噪声、种系变体和/或交叉污染中准确地指出体细胞突变。这些人工设计的方法不易推广到特定癌症类型、样本类型或为其开发的测序策略之外。
最近,已经提出了一种基于深度学习的体细胞突变检测方法,称为neusomatic,该方法使用卷积神经网络(cnn)直接从原始数据中学习特征表示法11。neusomatic使用肿瘤/正常比对信息的新颖总结作为一组输入矩阵,可以采用这些输入矩阵来有效地训练模型,以学习如何有效地将真实的体细胞突变与伪影区分开来。由neusomatic训练的网络模型可以直接从读取比对和基因组情境中捕获重要的突变信号,而无需人工干预。因此,neusomatic提供了一个框架,通过基于适当的数据进行训练,可轻易地应用于不同问题的陈述,包括各种测序技术、癌症类型、肿瘤和正常纯度以及突变等位基因频率。此外,它可以实现为独立的体细胞突变检测方法,或与现有方法结合以实现最高的准确性。已经基于计算机模拟数据集证明,neusomatic明显优于传统技术。鉴于缺乏具有已知“基准真值”体细胞突变的彻底表征的基准样本,对真实样本的性能评估仅限于对少数经过验证的体细胞变体进行部分敏感性分析。因此,尽管看到了通过基于计算机模拟数据实现neusomatic的基于cnn的框架所具有的优势,但迄今为止,尚未全面评估neusomatic在真实癌症样本上的准确性和再现性。
最近,由fda领导的测序质量控制ii期(seqc-ii)联合体的体细胞突变工作组开发了参考匹配的肿瘤-正常样本:人三阴性乳腺癌细胞系(hcc1395)和匹配的b淋巴细胞来源的正常细胞系(hcc1395bl)12,13。seqc-ii联合体使用正交测序技术、多个测序复本和多个生物信息学分析渠道,已经为hcc1395开发了定义明确的体细胞单核苷酸变体(snv)和插入/缺失(indel)的“金集”。
作为第一个全面且特征明确的经配对肿瘤-正常参考癌症样本,该数据集以及基于多个位点和技术制备的伴随测序数据为实现两个重要目的提供了独特的资源。首先,使用联合体针对这些参考样本开发的高置信度体细胞突变调用结果集,与传统方案相比,我们在真实癌症样本中对基于深度学习的体细胞突变检测实施了深入分析。其次,我们探索了使用seqc-ii数据训练neusomatic中的cnn的各种模型构建策略,并鉴别了基于源自表示真实场景的样本的多个数据集的有效训练方法。我们评估了所提出的基于全基因组测序(wgs)、全外显子组测序(wes)和ampliseq靶向测序数据集的策略,覆盖率范围为10×至2000×。wgs和wes数据源自福尔马林固定石蜡包埋(ffpe)和新鲜dna,使用三种具有不同输入量的文库制备方案,并且来自多个平台/测序位点。使用肿瘤和正常滴定法,我们针对5%至100%肿瘤纯度,5%的污染匹配正常样本以及10×至300×wgs覆盖率,评估了不同方法。所提出的用以训练和实现neusomatic中的深度学习框架的策略在所有上述真实场景中均实现了高鲁棒性,并且显著优于传统的经配对肿瘤-正常体细胞突变检测方法。
我们对seqc-ii参考癌症样本的分析表明,在neusomatic中实现的深度学习方案有助于克服体细胞突变检测中的主要挑战,这些挑战使用传统技术难以解决。因此,源自我们的研究的深度学习模型和策略可以为研究团体提供可行的最佳实践建议,以进行可靠的癌症突变检测。
结果
参考样本和数据集
为了对体细胞突变检测问题进行全谱分析,我们使用了由seqc-ii联合体的体细胞突变工作组开发的首个全面的全基因组特征性参考肿瘤-正常配对乳腺癌细胞系(hcc1395和hcc1395bl)12,13。我们利用该联合体推导的高置信度体细胞突变(39,536个snv和2,020个indel)作为我们的基准真值集,如图27所示,其示出了在由seqc-ii联合体分类为四个置信度级别(高、中、低和未分类)的hcc1395超级调用结果集中,基准真值snv和indel体细胞突变的vaf分布。高置信度和中置信度的调用结果被归为一组体细胞突变的“真值”。为了广泛评定预测的一致性和再现性,我们使用了来自各种数据集的123个复本,这些数据集表示真实的癌症检测应用,包括具有不同覆盖率、肿瘤纯度和文库制备的真实wgs、wes和ampliseq靶标测序,这些癌症检测应用于在六个中心的多个平台上测序的ffpe和新鲜dna(请参见“方法”)。
图27示出了在由seqc-ii联合体分类为四个置信度水平(高、中、低和未分类)的针对hcc1395的调用结果超集合中,基准真值snv和indel体细胞突变的vaf分布。高置信度和中置信度的调用结果被归为一组体细胞突变的“真值”。
分析概述
我们使用各种受训网络模型评估了neusomatic的基于深度学习的体细胞突变检测方法,并将其与七个广泛使用的传统体细胞突变调用算法进行了比较,该传统算法包括mutect22、muse3、somaticsniper4、strelka25、vardict6、tnscope7和lancet8。我们以其独立模式(显示为neusomatic-s)和其集成模式对neusomatic进行了评定,其中除原始数据外,还包括由mutect2、somaticsniper、vardict、muse和strelka2报告的预测作为输入通道。
图28a至图28e显示了neusomatic在seqc-ii数据集中的123个复本上的总体性能。图28a显示了当应用于seqc-ii数据时,基于dream挑战阶段3数据集训练的neusomatic模型优于其他技术。图28b显示了使用seqc-ii参考样本训练不同的neusomatic模型导致平均f1得分的额外4%至5%的绝对改善。图28c显示,使用基于seqc-ii数据训练的模型,neusomatic跨wgs、wes、ffpe、ampliseq和不同的文库制备数据集中不同纯度/覆盖率的各种复本集实现了与其他技术相比的一致优势。在该子图中,对于每个复本,最佳f1得分是通过不同方法计算得出的。热图示出了体细胞突变检测方法中的任一种方法的f1得分与最佳f1得分之间的绝对差异。在每张图中,显示了每种方法跨123个复本的平均f1得分。图28d比较了不同技术跨snv和indel的六个数据集中123个复本的性能。对于每个复本,最佳f1得分是通过不同方法计算得出的。热图示出了体细胞突变检测方法中的任一种方法的f1得分与最佳f1得分之间的绝对差异。显示了每种方法跨123个复本的平均f1得分。
在我们的分析中,我们使用了几种不同的训练模型。首先,我们使用了最近发布的已经可用的模型11,该模型是使用来自dreamchallengestage3数据集13的计算机模拟掺入训练的。尽管用于训练dream3模型的样本类型、测序平台、覆盖率、掺入突变频率和样本的异质性之间存在巨大差异,但该模型在各种特征的真实癌症数据集上的性能优于其他传统技术,对于snv和indel两者,该模型跨不同样本的平均f1得分平均值比其他传统技术高了约4%(图28a)。尽管这种优越性支持neusomatic对所指定变体的鲁棒性,但它也表明,通过学习来自真实癌症样本的测序特征和突变标志,neusomatic中使用的深度学习框架甚至可以表现得更好,特别是用于预测indel和用于覆盖率更高的经pcr富集数据集比如ampliseq。
为了鉴别使用真实癌症样本建立网络模型的最有效策略,我们使用seqc-ii参考样本评估了neusomatic的另外十种训练方法(图28e)。
第一模型(seqc-wgs-掺入)基于一组计算机模拟肿瘤-正常复本的wgs对进行训练,其中通过将突变掺入到不同的正常复本品来设计计算机模拟肿瘤。第二模型(seqc-wgs-gt-50)使用hcc1395中的基准真值体细胞突变基于50%基因组上的一组真实wgs肿瘤-正常复本对进行训练。第三模型(seqc-wgs-gt50-掺入wgs10)通过将来自第一模型的训练数据的10%添加到用于第二模型的训练数据中来制备,以利用大量的掺入突变和真实的体细胞突变。这三个模型都基于所有数据集上进行了测试。对于诸如ffpe和wes之类的特定数据集,我们还使用一组合成的计算机模拟肿瘤和正常复本的合成对准备了另外六个专门模型。对于所有模型,我们评估了在基因组50%保留区域(未用于seqc-wgs-gt-50模型)上的性能。我们还使用类似于seqc-wgs-gt-50模型的所有基准真值突变但基于全基因组训练了一个模型(seqc-wgs-gt-all)。seqc-wgs-gt-all并非直接适用于已经看到所有真实突变的wgs数据集,它对于在hcc1395以外的其他数据集或样本上的性能分析将是有益的。
使用基于seqc-ii样本训练的模型,我们通过将平均f1得分额外改善约4%至5%而提高了dream3模型的平均性能(图28b)。所提出的模型构建策略跨各种样本类型和测序策略中始终是最佳的,就snv和indel的平均f1得分而言,其性能分别比传统方法高出5.7%和7.8%以上。类似地,在所有样本中,我们都观察到了比其他传统技术改善了5.6%以上的中位数f1得分(图28c和图28d)。
wgs数据集
我们在使用hiseqx10、hiseq4000和novaseqs6000平台在六个测序中心测序的21个wgs复本上评估了上述体细胞调用技术和网络模型的性能(图29a至图29d)。
neusomaticseqc-wgs-gt50-掺入wgs10模型的性能始终优于其他方案,但在复本之间存在细微差异,表明了鲁棒性和再现性(图29a)。neusomatic的snv和indel的平均f1得分分别为94.6%和87.9%,分别比其他传统体细胞突变检测方案的snv和indel的平均f1得分高出5.6%和10.2%。精确率-召回率分析表明,与其他技术相比,neusomatic的高精确率推动了这种优势(图29b)。比较不同的模型训练策略还发现,neusomatic-sindel调用从使用基准真值体细胞突变的训练中受益更多,而一般而言,与dream3模型相比,我们使用seqc-ii参考样本观察到了高达11%的显著改善(图29c)。
肿瘤纯度和受污染的正常样本
由于肿瘤纯度和匹配的正常样本中的肿瘤细胞的污染会极大地影响突变检测的准确性,因此我们通过wgs样本的肿瘤-正常滴定探究了在不同测序深度和样本纯度下的模型鲁棒性。我们首先针对10×至300×覆盖率研究了与纯正常样本配对的纯度为5%至100%的肿瘤样本(图30a至图30h)。通常,尽管肿瘤纯度和覆盖率有所变化,neusomatic仍保持优于其他方案的优势,这反映了其鲁棒性(图30a、图30e和图30f)。在更具挑战性的情况比如较低的覆盖率下,它比传统方案具有最大优势(例如,对于具有10×覆盖率和100%纯度的样本,f1得分为约20%)。
我们进一步在80x覆盖率下,针对10%至100%范围内的肿瘤样本纯度,分析了正常样本对于5%肿瘤污染的鲁棒性(图30b)。neusomatic对肿瘤-正常的交叉污染具有高鲁棒性,f1得分的中位数绝对变化少于5%。在纯正常样本中具有高f1得分的其他技术中,strelka2也显示出对肿瘤污染的高鲁棒性(f1得分的中位数变化为8.4%)。mutect2、muse、lancet和tnscope尽管在纯正常样本场景中具有高f1得分,但在使用受污染的正常样本时,f1得分却下降了约50%。
基于基准真值训练的模型比dream3模型具有最高的优势,通常主要是由于更高的精确率所致(图30c和图30g)。与基于基准真值训练的模型相比,基于计算机模拟肿瘤训练的seqc-wgs-掺入模型也显示出较低的精确率。通常,indel、低纯度和低覆盖率样本从基于seqc-ii数据训练中受益最大。
分析跨各种覆盖率和纯度设置的不同indel尺寸的f1得分,表明neusomatic对于插入和缺失两者的尺寸变化的鲁棒性(图30d)。
文库制备和dna输入
为了衡量文库制备对预测鲁棒性的影响,我们使用我们的模型来测试使用truseq-nano和nexteraflex方案制备的六个复本以及三个dna输入量:1ng、10ng和100ng(图31a至图31f和图31h)。在不同的文库制备方法中,neusomatic始终优于其他技术。对于1ngtruseq-nano文库,由于去除多余读取后的有效覆盖率受限(约7x),所有方法的性能均较差。平均而言,与传统的体细胞突变检测技术相比,neusomatic将snv的f1得分改善了8.4%。对于indel,lancet的基于程序集的indel调用者比neusomaticseqc-wgs-gt50-掺入wgs10模型的性能高出约4%(图31a)。相反,neusomatic的seqc-wgs-gt50模型在indel方面的性能与lancet相似(图31b和图31g)。与dream3模型相比,基于seqc-ii掺入或基准真值数据的训练导致snv的f1得分总体改善了约8.4%。在更具挑战性的情况下(包括输入为1ng的truseq-nano文库),优势更加明显。我们还观察到,neusomatic-s从这些模型中受益更多。
经捕获(wes)组和靶向(ampliseq)组
我们使用在六个测序位点进行测序的16个wes复本以及ampliseq数据集中的三个复本,对我们的模型进行了测试(图31c至图31f、图31i和图31j)。尽管基于seqc-iiwgs样本训练的模型具有不同的覆盖率和平台偏差,但neusomatic在2000x覆盖率下在wes和ampliseq数据集上均表现良好。对于wes,neusomatic的snvf1平均得分达到了95.4%,比替代方案的f1平均得分改善了2.6%。基于wes和wgs数据训练的模型在wes数据集上的表现类似,f1得分为约95%。在ampliseq数据集上,neusomaticseqc-wg-gt50和seqc-wes-掺入模型的平均f1得分大于90%,与其他模型/方案以及strelka2相比是最高的。
ffpe处理的效果
为了衡量neusomatic对经ffpe处理样本的预测的鲁棒性,我们使用了8个wgs和7个wesffpe复本,分别用1小时、2小时、6小时和24小时的四个不同的甲醛固定时间进行了制备。我们用ffpe正常匹配样本和新鲜的正常匹配样本评估了每个ffpe复本。尽管存在ffpe伪像,neusomatic仍然始终具有比其他技术优越的性能,并且其在不同固定时间和所使用的匹配正常样本上的表现在很大程度上保持不变(图32a、图32e和图32f)。基于wgsffpe数据,neusomatic得到的snv和indel的平均f1得分分别为86.1%和76.9%,比替代技术的平均f1得分改善了4%和6%。类似地,对于ffpewes数据,neusomatic的平均f1得分为78.9%,比传统方案的平均f1得分高出4%以上(图32c)。
通常,当我们利用seqc-ii样本进行训练时,我们观察到了相对于dream3模型的显著提升(图32b和图32d)。仅对于使用neusomatic进行indel预测而言,基于ffpe样本训练的模型似乎比基于新鲜样本训练的模型有所改善,但对于snv,基于新鲜样本训练的模型更为优越。
样本特异性模型
尽管基于seqc-ii训练的通用模型已表现出比其他传统体细胞突变检测方案始终更好的性能,但在这里我们使用跨不同seqc-ii数据集的九个复本,探讨了经样本特异性训练的模型是否可以给我们带来额外的准确性提升。对于每个样本,我们使用为该样本制备的计算机模拟肿瘤-正常复本训练了neusomatic和neusomatic-s模型。我们还通过将用于seqc-wgs-掺入模型的10%的训练候选者与针对每个样本推导出的训练数据组合而训练了一个截然独特的模型。我们将这两个样本特异性模型与通用seqc-wgs-掺入模型进行了比较(图33a至图33c)。平均而言,与用于neusomatic的seqc-wgs-掺入模型相比,样本特异性模型的snv和indelf1得分分别改善了约0.5%和约5%。文库制备和ffpe样本从样本特异性训练中受益最大。例如,用nexteraflex方案制备的具有1ngdna量的文库制备样本的snv和indel绝对f1得分分别获得了1.6%和19.4%的改善。同样,对于snv和indel,具有匹配的新鲜正常样本的24小时持续时间ffpewgs样本的f1得分分别改善了1.8和14.8个百分点。对于neusomatic-s,只有indel似乎从样本特异性训练中受益。
indel性能
我们进一步评估了跨多个数据集检测不同尺寸的indel的准确性(图34a和图34b)。尽管neusomatic没有明确地将本地程序集用于indel检测,但是在大范围的indel尺寸、覆盖率和肿瘤纯度下,它仍然始终优于其他方法,包括基于程序集的技术,例如lancet(图34a和图30d)。总体而言,对于2个以上碱基对(bps)的插入和缺失而言,neusomatic得到的f1得分比lancet分别高24.4%和6.5%,而lancet是最好的替代传统技术。比较跨不同neusomatic训练方法的indel准确性,dream3模型主要受到插入检测准确性低的困扰,而seqc-ii受训模型则可靠地鉴别了不同尺寸的插入和缺失两者(图34b)。
不同变体等位基因频率(vaf)的性能分析
我们分析了跨体细胞突变vaf的不同方法的准确性(图34c和图35至图37c)。在不同的数据集上,我们观察到neusomatic在vaf小于20%的情况下显示出优于其他方案的优势(图34c和图35)。我们还观察到,neusomatic对于vaf的变化具有高鲁棒性,对于低至5%的vaf具有一致的预测。对于具有5%至20%vaf的突变,seqc-ii受训模型显然比dream3模型表现更好(图36a和图36b)。对于所有覆盖率和肿瘤纯度设置,观察到相似的鲁棒性(图37a至图37c)。
在困难基因组区域上的性能
为了说明对于基因组情境复杂性的鲁棒性,我们评估了不同体细胞调用方案在困难基因组区域上的性能,该困难基因组区域包括不同长度的串联重复序列(tr)和片段复制(图34d和图38至图41c)。尽管许多其他方案在困难基因组区域上的性能显著下降(图38),但neusomatic仍然具有高鲁棒性,与全基因组分析相比,其与其他方案相比的改善甚至更为显著(图34d和图38)。平均而言,对于这些困难基因组区域,neusomatic的snv和indel两者的f1得分比最好的替代传统体细胞调用者高出15%以上。用seqc-ii样本训练neusomatic可以大大改善在这些困难基因组区域上的准确性,其中最大的优势是使用基于基准真值突变训练的模型的串联重复区域(图39a至图40b)。在不同的覆盖率和肿瘤纯度下,观察到了在困难基因组区域上的类似性能(图41a至图41c)。
讨论
我们探索了neusomatic的深度学习框架在跨各种实验设置的集合而检测体细胞突变中的鲁棒性,这些实验设置的集合是在具有不同覆盖率、样本纯度和文库制备的真实wfp、wes和ampliseq样本中见到的,这些检测在使用多个平台在六个中心测序的ffpe和新鲜dna上进行。实验证实,neusomatic具有从原始数据中捕获真实的体细胞突变标志以将真实调用结果与测序伪影区分开来的潜力。基于seqc-ii参考样本训练的neusomatic模型,都使用掺入突变和seqc-ii研究推导出的一组基准真值体细胞突变两者,可以提高性能以实现最高的准确性。该分析重点介绍了可用于各种场景中的构建模型的最佳实践策略。与基线dream3网络模型相比,从seqc-ii参考样本推导出的所提出模型显示可通过学习正确的突变信号而减少snv和indel两者的假阳性,改善插入调用准确性,并增强对困难基因组区域的体细胞检测。
尽管neusomatic保持对匹配正常样本中肿瘤污染的鲁棒性,但许多体细胞调用方法如mutect2、lancet、muse和tnscope受到了严重影响,因为它们拒绝了正常样本中具有相对应读取的真实体细胞突变。因此,他们的召回率显著下降。基于wgs训练的neusomatic模型在靶向测序数据上同样表现良好。另一方面,tnscope采用机器学习(随机森林)模型,而lancet使用局部彩色的debruijn图方法来检测突变。两种工具均设计为用于全基因组的体细胞突变检测,因此不适用于靶向测序(通常覆盖少于1mb的基因组)。
分析跨包括突变类型、indel长度、vaf和基因组区域在内的不同规范的预测准确性,结果表明,neusomatic在挑战性(诸如覆盖率低、vaf小于20%、困难基因组区域如串联重复序列、长度超过2bp的indel、具有dna损伤的ffpe或污染的匹配正常样本)情况下比其他方案具有最大的改善。作为将测序或样本特异性伪影与真实体细胞突变相混淆的结果,传统方案具有许多假阳性(因此精确率较低),这些假阳性通常见于低复杂性基因组区域中(图34d和图38)。但是,通过基于来自多个平台和多个中心的wgs样本进行训练,neusomatic学习了易于出错的基因组情境,从而在不同条件下(包括难以调用的低复杂性基因组情境)不断提高了准确性。类似地,通过不同方法对遗漏的调用结果的分析表明,由例如strelka2之类的传统方案产生的私有假阴性调用结果(这些调用结果被neusomatic正确地预测到)具有低vaf(图42)。这表明了其他方案在检测低vaf突变方面的劣势。另一方面,通过从原始数据中学习正确的标志,neusomatic可以更准确地区别挑战性的低vaf调用结果与伪影。
图42显示了基于wgs数据集的私有fn调用结果的vaf分布的小提琴图比较。在每个子图中,我们将一种传统的体细胞突变检测方案与neusomatic进行了比较。对于x-vs-y子图,我们鉴别了xfn,这是在21个wgs复本的至少11个中被算法x(x中的fn)遗漏的基准真值snv集。同样,我们鉴别了yfn,这是在21个wgs复本的至少11个中被算法y(y中的fn)遗漏的基准真值snv集。然后,该图显示了x和y的私有fn调用结果的vaf分布。换言之,小提琴图以蓝色显示了xfn/yfn集合中调用结果的vaf分布,并且以红色显示了yfn/xfn集合中调用结果的vaf分布。对于绝大多数传统方案例如strelka2,由neusomatic正确预测的私有fn具有较低的vaf,这表明传统方案在检测低vaf突变方面的劣势。在每个x-vs-y子图中,在顶部报告x和y的私有fn数量。
方法
seqc-ii肿瘤-正常测序数据和基准真值
在我们的分析中,我们使用了seqc-ii参考匹配的样本、人三阴性乳腺癌细胞系(hcc1395)和匹配的b淋巴细胞来源的正常细胞系(hcc1395bl)。详细的样本信息可在seqc-ii参考样本手稿12、13中找到。seqc-ii体细胞突变工作组已经为这些样本建立了体细胞突变的金标准真值集13(图27)。使用来自不同测序中心的多个肿瘤-正常测序复本和正交突变检测生物信息学渠道定义了真值集。为了给突变调用结果分配置信度得分并最小化平台、中心和渠道的特定偏差,实现了somaticseq10机器学习框架以基于计算机模拟肿瘤-正常复本对训练一组分类器,该计算机模拟肿瘤-正常复本对通过将合成突变掺入到hcc1395bl比对文件中而生成,并且每个复本对都与另一个hcc1395bl匹配。使用这些分类器,基于交叉比对器和交叉测序中心的再现性,将突变调用结果分为四个置信度级别(高置信度、中置信度、低置信度和未分类)。高置信度和中置信度调用结果归为一组体细胞突变的“真值集”(v1.0),其包含总计39,536个snv和2,020个indel。hcc1395中体细胞突变的真值集可在ncbi的ftp网站(ftp://ftp-trace.ncbi.nlm.nih.gov/seqc/ftp/somatic_mutation_wg/)上获得而公用。
该研究中使用的所有测序数据均可通过ncbi的sra数据库(srp162370)公开获得。对于所有样本,首先使用trimmomatic15修剪fastq文件,然后与bwa-mem(v0.7.15)16比对,之后与picardmarkduplicates(https://broadinstitute.github.io/picard)比对。
训练数据集和模型
使用不同的训练数据集来推导出有效的neusomaticcnn模型,既使用了用合成的掺入突变制备的计算机模拟肿瘤复本,也使用了具有已知的高置信度体细胞突变真值集的真实肿瘤复本(图28e):
dream3模型
作为基线wgs模型,我们采用了dream3模型,该模型是最近通过基于icgc-tcgadream挑战阶段3数据训练14而开发的11。阶段3数据集由正常样本和肿瘤样本组成,这些样本是通过将7903个snv和7604个indel突变计算地掺入到具有三个不同af(50%、33%和20%)的相同正常样本的健康基因组中而构建的,以创建合成但真实的肿瘤正常样本对。使用具有与原始dream数据相似的af分布的bamsurgeon17,将额外的约95ksnv和约95kindel掺入到肿瘤样本中11,以进行更好的网络训练。通过将来自五个不同肿瘤正常纯度设置(100t:100n、50t:100n、70t:95n、50t:95n和25t:95n11)的训练数据(在基因组的50%中)组合而训练该模型。基于在这五个复本对中鉴别出的约29.2m个候选突变训练neusomatic和neusomatic-s,这些候选突变包括约450k个具有体细胞snv/indel标记的候选者和标记为非体细胞的约28.7m个候选者。
tcga模型
作为基线wes模型,我们使用了最近通过基于12个tcga样本集18训练而开发的tcga模型11。将这些样本中每一个的肿瘤比对和正常比对混合并分成两个相等的比对。将一个比对作为正常复本处理,并且将突变掺入到另一个比对中以构建肿瘤复本。对于每个样本,将约88k个snv和约44k个indel掺入以生成用于训练的合成肿瘤样本。基于在这12个复本对中鉴别出的约5.9m个候选突变训练neusomatic和neusomatic-s,这些候选突变包括约1.5m个具有体细胞snv/indel标记的候选者和标记为非体细胞的约4.4m个候选者。
seqc-wgs-掺入模型
为了制备用于构建该模型的训练数据,我们采用bamsurgeon通过将计算机模拟突变掺入到一个hcc1395bl复本中并将其与独特的hcc1395bl复本配对作为正常匹配来构建合成肿瘤。使用这种方法,我们制备了十个计算机模拟肿瘤-正常样本对。十个复本对中的八个是从wgs复本中选择的,这些复本在四个不同位点进行了测序,平均覆盖率为40×至95×。通过合并来自illumina的多个novaseq测序复本而获得其他两个复本对,从而获得约220×肿瘤样本覆盖率和约170×正常匹配样本覆盖率的计算机模拟。在每一个计算机模拟肿瘤中,我们掺入了约92k个snv和约22k个indel。从β分布(形状参数α=2且β=5)中随机选择掺入突变的af。对于十个重复对中的每一个,我们还通过混合95%正常读取和5%肿瘤读取来构建不纯正常样本。因此,我们总共使用了20个计算机模拟肿瘤-正常配对来训练seqc-wgs-掺入模型。然后,我们基于在这20个复本对中鉴别出的约272m个候选突变训练neusomatic和neusomatic-s,这些候选突变包括约2m个具有体细胞snv/indel标记的候选者和标记为非体细胞的约270m个候选者。
seqc-wgs-gt-50模型
使用真实wgs肿瘤正常复本以及seqc-ii高置信度真值集作为基准真值体细胞突变来构建该模型。我们使用八个wgs肿瘤-正常复本作为基础样本来训练该模型。前七个wgs复本对来自hiseq和novaseq平台上的六个不同测序中心,平均覆盖率为40×至95×,最后一个是通过将illumina的9个novaseq测序复本组合而构建的,得到具有约390×覆盖率的复本对。对于这八个复本对中的每一个,我们构建了另外两个纯度变异,一个通过将95%正常读取和5%肿瘤读取混合而获得95%正常纯度,另一个通过将10%肿瘤读取和90%正常读取混合而获得10%肿瘤纯度。因此,对于每个复本对,我们有一个具有100%肿瘤和正常的版本、一个100%纯肿瘤与95%纯正常匹配的版本和一个10%纯肿瘤与100%纯正常匹配的版本。因此,我们总共使用了24个肿瘤-正常复本来训练seqc-wgs-gt-50模型。为了进行公正的评估,我们仅使用了50%的基因组来训练该模型,而其余50%则用于评估。为了制备训练和评估区域,我们将基因组分为尺寸约92kbps的小区域,并随机选择一半的断裂区域进行训练,另一半进行评估。我们将每个灰色区突变周围的5个碱基的填充区域排除在训练区域之外,所排除区域包括在seqc-ii调用结果的超集合中具有低置信度标签的调用结果,以及在seqc-ii调用结果的超集合中那些具有超过30%的vaf的具有未分类标签的调用结果。然后,我们基于在这24个复本对中鉴别出的约137m个候选突变训练neusomatic和neusomatic-s,这些候选突变包括约416k个具有体细胞snv/indel标记的候选者和标记为非体细胞的约136.5m个候选者。
seqc-wgs-gt-all模型
该模型的制备类似于seqc-wgs-gt-50模型,但是使用了全基因组上的所有高置信度基准真值体细胞突变。因此,它不能用来评估用于训练目的的wgs样本,并且鉴于所有样本都已用于训练,因此它可能会偏向于真值集突变。该模型对于其他数据集或非seqc-ii样本的性能分析仍然是有益的。我们基于在用于seqc-wgs-gt-50模型的这24个复本对中鉴别出的约272m个候选突变来训练用于neusomatic和neusomatic-s的模型,这些候选突变包括约839k个具有体细胞snv/indel标记的候选者和标记为非体细胞的约271m个候选者。
seqc-wgs-gt50-掺入wgs10模型
该模型的训练数据是通过将seqc-wgs-掺入模型中使用的10%的训练候选者与seqc-wgs-gt-50中的所有候选者合并而成的。这种组合利用了大量的掺入突变和真实肿瘤-正常数据中所见的真实体细胞突变特征。我们基于164m个候选突变的组合集训练了neusomatic和neusomatic-s,这些候选突变包括具有体细胞snv/indel标签的约574k个候选者和标记为非体细胞的约163.5m个候选者。
seqc-wes-掺入模型
与seqc-wgs-掺入模型类似,我们使用bamsurgeon构建了7个计算机模拟肿瘤-正常wes复本,以训练该模型。复本对是从wes数据集中选择的,这些复本在四个不同位点进行了测序,平均覆盖率为60×至550×。在每一个计算机模拟肿瘤中,我们掺入了约97k个snv和约19k个indel。从β分布(形状参数α=2且β=5)中随机选择掺入突变的af。然后,我们基于在这7个复本对中鉴别出的约3.7m个候选突变训练neusomatic和neusomatic-s,这些候选突变包括约755k个具有体细胞snv/indel标记的候选者和标记为非体细胞的约3m个候选者。
seqc-ffpe-掺入模型
与seqc-wgs-掺入模型类似,我们使用bamsurgeon构建了8个计算机模拟肿瘤-正常wgsffpe复本,以训练该模型。复本对是从ffpe数据集中选择的,这些复本在两个不同位点进行了测序,具有四种不同制备时间。在每一个计算机模拟肿瘤中,我们掺入了约92k个snv和约22k个indel。从β分布(形状参数α=2且β=5)中随机选择掺入突变的af。在我们的训练中,我们还将每个计算机模拟肿瘤与一个新鲜的wgs复本进行匹配,以包括ffpe肿瘤与新鲜的正常场景。因此,我们总共使用了16个计算机模拟肿瘤-正常配对来训练seqc-ffpe-掺入模型。我们基于在这7个复本对中鉴别出的约191m个候选突变训练neusomatic和neusomatic-s,这些候选突变包括约1.7m个具有体细胞snv/indel标记的候选者和标记为非体细胞的约190m个候选者。
seqc-ffpe-wes-掺入模型
与其他掺入模型类似,我们使用bamsurgeon构建了7个计算机模拟肿瘤-正常wesffpe复本,以训练该模型。复本对是从在两个不同位点测序的wesffpe数据集中选择的,并在四个不同的时间间隔内制备。由于我们没有在该数据集中包括两个具有相同ffpe制备时间和测序位点的正常复本,因此我们将每个复本对的肿瘤比对和正常比对混合在一起,并将混合物分成两个相等的比对。然后,我们将一个比对作为正常复本处理,而将突变掺入到另一个比对中以构建肿瘤复本。在每一个计算机模拟肿瘤中,我们掺入了约90k个snv和约19k个indel。从β分布(形状参数α=2且β=5)中随机选择掺入突变的af。在我们的训练中,我们还将每个计算机模拟肿瘤与一个新鲜的wes复本进行匹配,以包括ffpe肿瘤与新鲜的正常场景。因此,我们总共使用了14个计算机模拟肿瘤-正常配对来训练seqc-ffpe-wes-掺入模型。我们基于在这7个复本对中鉴别出的约9.6m个候选突变训练neusomatic和neusomatic-s,这些候选突变包括约1.4m个具有体细胞snv/indel标记的候选者和标记为非体细胞的约8.2m个候选者。
seqc-wes-掺入wgs10、seqc-ffpe-掺入wgs10、seqc-ffpe-wes-掺入wgs10模型
这些模型的训练数据是通过将10%的用于seqc-wgs-掺入模型的训练候选者与分别来自seqc-wes-掺入、seqc-ffpe-掺入和seqc-ffpe-wes-掺入的所有候选者组合而制备的。这种组合既利用了大量的掺入wgs突变,又利用了wes和ffpe样本的样本偏差。
样本特异性模型
除了上述通用模型外,对于跨多种数据类型的一组九个样本,我们还推导出了样本特异性模型。选择的样本包括通过novaseq仪器测序的wgs样本、wes样本、使用nexteraflex文库制备试剂盒制备的dna量为1ng的样本、肿瘤纯度为50%的30×wgs样本、肿瘤纯度为10%的100×wgs样本、以及各自用福尔马林处理24小时并与新鲜/ffpe正常样本匹配的两个wgs和两个wesffpe样本。对于每个样本,我们使用随机掺入制备了计算机模拟肿瘤。对于10%肿瘤样本,从β分布(形状参数α=2且β=18)中随机选择掺入突变的af。对于其他样本,使用β分布(形状参数α=2且β=5)选择af。然后,对于每个样本,我们使用计算机模拟肿瘤-正常复本训练了neusomatic和neusomatic-s模型。此外,对于每个样本,我们还通过将用于seqc-wgs-掺入模型的10%的训练候选者与针对该样本推导出的训练数据组合而训练了一个独特的模型。
体细胞突变检测算法
除了neusomatic以外,我们使用了七种体细胞突变调用者:mutect2(4.beta.6)2、somaticsniper(1.0.5.0)4、lancet(1.0.7)8、strelka2(2.8.4)5、tnscope(201711.03)7、muse(v1.0rc)3和vardict(v1.5.1)6,并且使用缺省参数或用户手册推荐的参数来运行它们中的每一个。对于somaticsniper,我们使用了“-q1-q15-s1e-05”。对于tnscope(201711.03),我们使用了在sevenbridges’scgc中实现的版本,命令如下:“sentieondriver-i$tumor_bam-i$normal_bam-r$ref--algotnscope--tumor_sample$tumor_sample_name--normal_sample$normal_sample_name-d$dbsnp$output_vcf”。对于lancet,我们使用“--cov-thr10--cov-ratio0.005--max-indel-len50-e0.005”。在最终的vcf文件中标记为“pass”的高置信度输出将用于我们的比较分析。对于vardict,我们还限制了具有“somatic”状态的调用结果。来自每个调用者的用于比较的结果全部是候选突变体,否则用户会将其视为此调用者检测到的“真实”突变。
我们以集成和独立两种模式使用了neusomatic(0.1.4)。对于集成模式,除了通过neusomatic的“比对扫描”步骤鉴别的候选变体之外,我们还合并了来自五个单独的调用者(mutect2、somaticsniper、strelka2、muse和vardict)的调用结果,并将它们的出现表示为用于每个候选变体的额外输入通道。对于neusomatic和neusomatic-s,我们在预处理步骤中使用了“--scan_maf0.01--min_mapq10--snp_min_af0.03--snp_min_bq15--snp_min_ao3--ins_min_af0.02--del_min_af0.02--num_threads28--scan_window_size500--max_dp100000”。对于训练,我们使用了“--coverage_thr300--batch_size8000”。
困难基因组区域
我们使用了“瓶中基因组”联合体推导出的一组困难基因组区域19。这些区域包括串联重复序列(分为小于50bp和大于50bp的两个不同类别的重复序列)以及片段复制区域。我们在这些区域上评估了不同的技术,以示出对于基因组情境复杂性的鲁棒性。
假阴性调用结果分析
为了更好地理解不同技术的性能差异,我们对neusomatic的假阴性snv的vaf分布与其他方案基于wgs数据集进行了成对比较(图42)。在每个x-vs-y分析中,我们鉴别了xfn,这是在21个wgs复本的至少11个中被算法x(x中的假阴性)遗漏的基准真值snv集。同样,我们鉴别了yfn,这是在21个wgs复本的至少11个中被算法y(y中的假阴性)遗漏的基准真值snv集。然后,我们计算x的私有假阴性,定义为xfn/yfn,以及y的私有假阴性,定义为yfn/xfn。然后,对于那些集合中每个集合中的体细胞突变,我们计算突变的vaf的分布。
评估过程
为了公平地比较,我们在50%保留基因组区域(未用于训练seqc-wgs-gt-50模型的区域)上评估了所有模型和体细胞突变算法。这个约1.4gb的区域包含来自用于hcc1395的seqc-ii真值集的约21ksnv和约1.3kindel,除用于seqc-wgs-gt-all模型外,这些nuv未用于训练任何neusomatic模型。
seqc-ii联合体将针对hcc1395标记为高置信度和低置信度的调用结果归为一组,作为本文使用的体细胞突变的“真值集”。我们采用此真值集来计算所有渠道在hcc1395的所有复本中的真阳性和假阴性。根据seqc-ii联合体的建议,我们还将低置信度调用结果列入了黑名单,因为它们的验证率较低。由于该联合体提供的真值集的vaf检测极限为5%并且深度检测极限为50×,因此对于覆盖率数据较高的调用结果或vaf较低的调用结果,我们无法确定其真实的体细胞状态。因此,对于不属于真值集的任何渠道报告的私有调用结果,我们排除了在评估中被认为模棱两可的调用结果。总而言之,对于手头的平均覆盖率为c且肿瘤纯度为p的肿瘤复本,如果渠道报告该复本有一个为d个读取所支持的私有体细胞突变(该体细胞突变不在真值集中),则仅当其在100%纯度和50x覆盖率下的支持读取推断数量≥3时,我们才将此调用结果标记为假阳性。换句话说,如果d≥3cp/50,则此调用结果为假阳性;否则,我们将从评估中排除此调用结果。
对于wes和ampliseq数据,由于评估区域中真正indel的数量非常有限,因此我们仅报告了snv评估。
参考文献
1.xu,c.,areviewofsomaticsinglenucleotidevariantcallingalgorithmsfornext-generationsequencingdata.comput.struct.biotechnol.j.16,15–24(2018)。
2.cibulskis,k.等人,sensitivedetectionofsomaticpointmutationsinimpureandheterogeneouscancersamples.nat.biotechnol.31,213(2013)。
3.fan,y.等人,muse:accountingfortumorheterogeneityusingasample-specificerrormodelimprovessensitivityandspecificityinmutationcallingfromsequencingdata.genomebiol.17,178(2016)。
4.larson,d.e.等人,somaticsniper:identificationofsomaticpointmutationsinwholegenomesequencingdata.bioinformatics28,311–317(2011)。
5.kim,s.等人,strelka2:fastandaccuratecallingofgermlineandsomaticvariants.nat.methods15,591–594(2018)。
6.lai,z.等人,vardict:anovelandversatilevariantcallerfornext-generationsequencingincancerresearch.nucleicacidsres.44,e108–e108(2016)。
7.freed,d.、pan,r.和aldana,r.tnscope:accuratedetectionofsomaticmutationswithhaplotype-basedvariantcandidatedetectionandmachinelearningfiltering.biorxiv250647
8.narzisi,g.等人,lancet:genome-widesomaticvariantcallingusinglocalizedcoloreddebruijngraphs.communicationsbiology1,20(2018)。
9.koboldt,d.c.等人,varscan2:somaticmutationandcopynumberalterationdiscoveryincancerbyexomesequencing.genomeres.22,568–576(2012)。
10.fang,l.t.等人,anensembleapproachtoaccuratelydetectsomaticmutationsusingsomaticseq.genomebiol.16,197(2015)。
11.sahraeian,s.m.e.、liu,r.、lau,b.、podesta,k.、mohiyuddin,m.和lam,h.y.,deepconvolutionalneuralnetworksforaccuratesomaticmutationdetection.naturecommunications.10,1041(2019)。
12.thesomaticmutationworkinggroupofseqc-iiconsortium.achievingreproducibilityandaccuracyincancermutationdetectionwithwhole-genomeandwhole-exomesequencing.https://doi.org/10.1101/626440(2019)。
13.fang,l.t.等人,establishingreferencesamplesfordetectionofsomaticmutationsandgermlinevariantswithngstechnologies,https://doi.org/10.1101/625624(2019)。
14.ewing,a.d.等人,combiningtumorgenomesimulationwithcrowdsourcingtobenchmarksomaticsingle-nucleotide-variantdetection.nat.methods12,623(2015)。
15.bolger,a.m.、lohse,m.和usadel,b.,trimmomatic:aflexibletrimmerforilluminasequencedata.bioinformatics30,2114-2120(2014)。
16.li,h.,aligningsequencereads,clonesequencesandassemblycontigswithbwa-mem.https://arxiv.org/abs/1303.3997(2013)。
17.ewing,a.d.等人,combiningtumorgenomesimulationwithcrowdsourcingtobenchmarksomaticsingle-nucleotide-variantdetection.nat.methods12,623(2015)。
18.grossman,r.l.等人,towardasharedvisionforcancergenomicdata.n.engl.j.med.375,1109–1112(2016)。
19.krusche,peter、lentrigg、paulc.boutros、christophere.mason、m.francisco、benjaminl.moore、margonzalez-porta等人,“bestpracticesforbenchmarkinggermlinesmall-variantcallsinhumangenomes.”naturebiotechnology1(2019)。
尽管已经参照一些说明性实施例描述了本公开,但应当理解,本领域技术人员可以在本公开原则的精神和范围内设计出许多其他的修改和实施例。更具体地,在不违背本公开的精神的情况下,在上述公开、附图和所附权利要求的范围内,所述主题组合排列的组成部分和/或布置可以进行合理的变化和修改。除了在所述组成部分和/或布置中的变化和修改外,对于本领域技术人员来说,替代用途也将是显而易见的。
本说明书中提到的和/或应用数据表中列出的所有美国专利、美国专利申请公开、美国专利申请、国外专利、国外专利以及非专利公开均通过引用整体并入本文。如果需要采用各种专利、申请和出版物的概念来提供其它实施例,则可以修改实施例的各方面。
当特征或元件在本文中被称为在另一特征或元件“上”时,它可以直接位于另一特征或元件上,或者也可以存在中间特征和/或元件。相反,当特征或元件被称为“直接位于”另一特征或元件“上”时,则不存在中间的特征或元件。还应理解,当特征或元件被称为“连接”、“附接”或“耦接”至另一特征或元件时,它可以直接连接、附接或耦接至另一特征或元件,或可能存在中间特征或元件。相反,当特征或元件被称为“直接连接”、“直接附接”或“直接耦接”到另一特征或元件时,则不存在中间的特征或元件。尽管关于一个实施例进行了描述或显示,但是如此描述或显示的特征和元件可以应用于其他实施例。本领域的技术人员还将意识到,提及与另一特征“相邻”设置的结构或特征可具有与相邻特征重叠或位于其下的部分。
本文使用的术语只是为了描述特定实施例的目的,并非旨在限制本发明。例如,如本文所用,单数形式“一”、“一个”、“该”和“所述”也旨在包括复数形式,除非上下文另外明确指出。将进一步理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指定了所述特征、步骤、操作、元件和/或组件的存在,但并不排除存在或添加一个或多个其他特征、步骤、操作、元件、组件和/或它们的组。如本文所用,术语“和/或”包括一个或多个相关联列出的项目的任意和所有组合,并且可以缩写为“/”。
本文可使用空间相对术语诸如“下方”、“在……下”、“下面”、“上方”、“上面”等来描述如图中所示出的一个元件或特征与另一元件或特征的关系。应理解的是,除了图中所描绘的取向之外,空间相对术语还意图涵盖装置在使用或操作中的不同方位。例如,如果附图中的装置被翻转,则被描述为在其他元件或特征“下方”或“在……下方”的元件将定向为在其他元件或特征“上方”。因此,示例性术语“下方”可涵盖上方和下方两种取向。可以其他方式定向装置(转动90°或以其他取向),并且相应地解释本文所使用的空间相对描述语。类似地,除非另外具体指出,否则术语“向上”、“向下”、“竖直”、“水平”等在本文中仅用于解释的目的。
尽管本文可以使用术语“第一”和“第二”来描述各种特征/元件(包括步骤),但是除非上下文另外指出,否则这些特征/元件不应受这些术语的限制。这些术语可以用于将一个特征/元件与另一特征/元件区分开来。因此,在不脱离本发明教导的情况下,下面讨论的第一特征/元件可以被称为第二特征/元件,并且类似地,下面讨论的第二特征/元件可以被称为第一特征/元件。
在整个本说明书和随后的权利要求中,除非上下文另外要求,否则词语“包含”以及诸如“包括”和“含有”的变体意味着可以在方法和物品中共同使用各种组分(例如,组合物以及包括设备和方法的设施)。例如,术语“包含”将被理解为暗示包括任何陈述的元件或步骤,但是不排除任何其他元件或步骤。
如本文在说明书和权利要求书中所用,包括在实施例中所用,并且除非另有明确说明,否则所有数字可以被读作与以词语“约”或“近似地”开头一样,即使该术语没有明确出现。当描述幅度和/或位置时,可以使用短语“约”或“大约”来指示所描述的值和/或位置在值和/或位置的合理预期范围内。例如,数字值的值可以是规定值(或值的范围)的+/-0.1%、规定值(或值的范围)的+/-1%、规定值(或值的范围)的+/-2%、规定值(或值的范围)的+/-5%、规定值(或值的范围)的+/-10%等。此处给出的任何数值应除非上下文另外指出,否则也应理解为包括约或大约该值。例如,如果公开值“10”,则还公开“约10”。本文叙述的任何数值范围旨在包括其中包含的所有子范围。还应理解,如本领域技术人员适当理解的那样,当公开的值“小于或等于”该值时,还公开了“大于或等于该值”以及值之间的可能范围。例如,如果公开了值“x”,则还公开了“小于或等于x”以及“大于或等于x”(例如,其中x是数值)。还应理解,在整个申请中,以多种不同格式提供数据,并且该数据表示端点和起点以及数据点的任意组合的范围。例如,如果公开了特定数据点“10”和特定数据点“15”,则应理解,大于、大于或等于、小于、小于或等于以及等于10和15视为已公开,且介于10和15之间也视为已公开。还应理解,还公开了两个特定单位之间的每个单位。例如,如果公开了10和15,则还公开了11、12、13和14。
尽管上面描述了各种说明性实施例,但是在不脱离如权利要求所描述的本发明的范围的情况下,可以对各种实施例进行多种改变中的任何一种。例如,在替代实施例中,可以经常改变所描述的各种方法步骤的顺序,而在其他替代实施例中,可以完全跳过一个或多个方法步骤。在一些实施例中可以包括各种装置和系统实施例的任选特征,而在其他实施例中可以不包括。因此,前面的描述主要是为了示例性目的而提供,并且不应解释为限制本发明的范围,因为本发明的范围在权利要求中所阐述。
本文包括的实例和示例性说明通过示例性说明而非限制的方式示出了可以实践本主题的特定实施例。如所提及的,可以利用其他实施例并从中得出其他实施例,从而可以在不脱离本公开的范围的情况下进行结构和逻辑上的替换和改变。仅出于方便起见,如果事实上公开了多个发明或发明构思,本文中可以单独地或共同地用术语“发明”来指代本发明主题的这些实施例,而无意将本申请的范围限制为任何单个发明或发明构思。因此,尽管本文中已经示例性说明和描述了特定实施例,但是为实现相同目的而计算的任何布置都可以代替所示的特定实施例。本公开意图覆盖各种实施例的任何和所有修改或变化。通过阅读以上描述,以上实施例的组合以及本文中未具体描述的其他实施例对于本领域技术人员将是显而易见的。
- 还没有人留言评论。精彩留言会获得点赞!