包含组蛋白脱乙酰酶6抑制剂的药物组合物的制作方法
本发明涉及用于预防或治疗cmt病的药物组合物,其包含组蛋白脱乙酰酶6(hdac6)抑制剂作为活性成分;使用组蛋白脱乙酰酶6(hdac6)抑制剂的治疗方法;以及组蛋白脱乙酰酶6(hdac6)抑制剂在制备用于预防或治疗cmt病的药物中的用途。
背景技术:
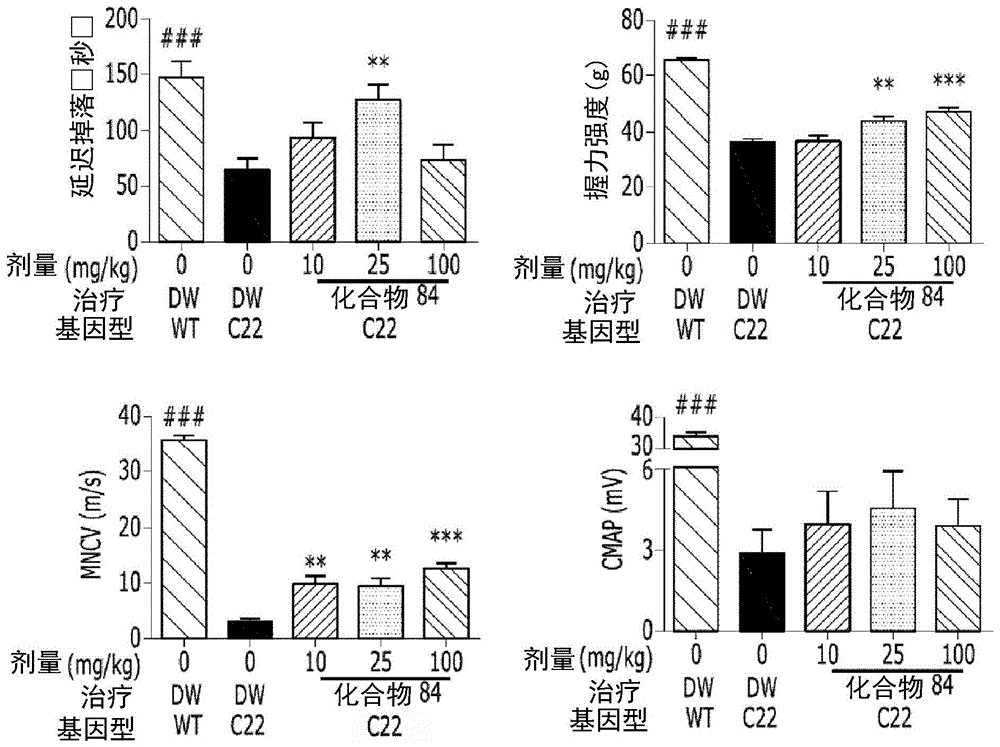
:夏科-马里-图思(cmt,遗传性运动和感觉神经病:hmsn)病是最常见的遗传性周围神经疾病类型,其由构成神经的蛋白质突变引起。迄今为止,已经从大约90个基因中鉴别出多于1,000种突变(timmerman等人,(2014)genes5:13-32)。随着夏科-马里-图思(cmt)病的发展,周围神经的进行性变性导致受神经分布影响的肌肉萎缩,由此患者呈现出手和足肌肉逐渐萎缩,以及出现畸形的手和足症状。cmt在遗传和临床上非常多样且复杂,并且已知其症状根据突变类型从接近正常状态至束缚在轮椅上的状态变化。cmt主要出现在青少年时期,每2,500人中就有1例发生(krajewski等人(2000)brain123:1516)。cmt属于罕见疾病,是一种遗传性周围神经疾病。但是,其人群中的患病率是1/2,500。韩国大约有20,000名患者,且全球大约有2,800,000名患者。迄今为止,cmt的治疗仅限于康复、辅助工具、疼痛控制、手术治疗等,但是尚未开发出成功的治疗剂。因此,迫切需要开发用于cmt的治疗剂。例如,关于cmt(即遗传性运动和感觉神经病中最常见的类型),已对抗坏血酸进行了大规模的临床试验,通过一起培养神经膜细胞与背根神经节细胞的实验已证明其对于在周围神经系统髓鞘形成是必不可少的物质,但这种试验未能证明有效性(pareyson等人(2011)10(4):3205)。[现有技术参考]韩国专利注册号1697518krajewski等人,(2000)brain123:1516pareyson等人,(2011)10(4):3205发明公开技术问题本发明的目的是提供用于预防或治疗夏科-马里-图思(cmt)病的药物组合物,其包含本发明的化合物、其异构体或其药学上可接受的盐。本发明的另一个目的是提供本发明的化合物、其异构体或其药学上可接受的盐在制备用于预防或治疗夏科-马里-图思(cmt)病的药物中的用途。本发明的另一目的是提供用于预防或治疗夏科-马里-图思(cmt)病的方法,其包括向个体给药治疗有效量的本发明的化合物、其异构体或其药学上可接受的盐的步骤。本发明的另一个目的是提供本发明的化合物、其异构体或其药学上可接受的盐在预防或治疗夏科-马里-图思(cmt)病中的用途。【技术方案】下面对此进行详细描述。同时,本发明中公开的每个描述和实施方案也可以分别应用于本发明的其它描述和实施方案。换句话说,本发明公开的各种元件的所有组合都落入本发明的范围内。而且,不能看出本发明的范围限于以下描述的具体描述。本发明提供用于预防或治疗夏科-马里-图思(cmt)病的药物组合物,其包含组蛋白脱乙酰酶6抑制剂。在本发明中,所述组蛋白脱乙酰酶6抑制剂可为由下式i表示的化合物、其异构体或其药学上可接受的盐:[式i]其中r1为氢或ch3,r2为氢或ch3,其中当r1为氢时,r2为-ch3,并且当r1为-ch3时,r2为氢,l为-(c4-c5烷基)-;-(c1-c3烷基)-l1-;-c(=o)-l1-或-s(=o)2-l1-,其中-(c4-c5烷基)-和-(c1-c3烷基)-可以是未取代的或者被-ch3取代,l1为-(c3-c6)环烷基-;a1和a2各自独立地为-n-或-cr3-,其中a1和a2二者不可以同时为-n-,r3为氢;-f、-cl、-br、-i或-oh,并且a3为-nh-或-o-,q选自-(c1-c6)烷基-;-(c2-c6)烯基-;-c(=o)-;-c(=s)-;-s(=o)2-和其中-(c1-c6)烷基-和-(c2-c6)烯基-可以是未取代的或者各自独立地被1至3个-ch3基团或卤素原子取代,q1为氢;-f、-c1、-br或-i,n为0、1或2的整数,其中当q为时,n为0,当q为-c(=o)-;-c(=s)-或-s(=o)2-时,n为1,并且当q为-(c1-c6)烷基-或-(c2-c6)烯基-时,n为1或2,并且x可选自-c1-c6烷基;-c3-c6环烷基;-c2-c6烯基;-c3-c6环烯基;-(c0-c2烷基)ar;-oar;-(c0-c2烷基)het;萘基和以下基团:其中r4为h或-c1-c4烷基,-c0-c2烷基、-c2-c6烯基和-c1-c6烷基可以是未取代的或者被1至2个-ch3基团;1至3个-f基团;或二者取代,ar为c6单环芳族化合物,其可以是未取代的或者被一个或多个卤素原子;-oh;-nh2;-c1-c6烷基;-o(c1-c6)烷基;-c3-c6环烯基;-nh(c1-c6烷基);-n(c1-c3烷基)2;-ch2n(c1-c3烷基)2;-s(=o)2-(c1-c3烷基)或苯基基团取代,其中-c1-c3烷基;-c1-c6烷基和-c3-c6环烯基可各自独立地被1至5个-f或-ch3基团取代,并且het为4至6元杂芳族或非芳族环化合物,其包含1至3个选自n、o和s的元素同时具有0至3个双键,并且可以是未取代的或者被一个或多个卤素原子;-c1-c6烷基;-c(=o)(c1-c3烷基);-s(=o)2(c1-c3烷基)或苄基基团取代,其中-c1-c3烷基和-c1-c6烷基可各自独立地被-oh;1至5个-f或-ch3基团取代。如本文中所使用,“c0烷基”意指没有碳,并因此表示键。例如,“-(c0烷基)ar”意指-ar。本发明的由式i表示的化合物可为由下式ii或式iii表示的化合物:[式ii][式iii]其中l为-(c5烷基)-;-(c1-c2烷基)-l1-;-c(=o)-l1-或-s(=o)2-l1-,其中-(c5烷基)-和-(c1-c2烷基)-为直链并且可以是未取代的或者被-ch3取代,l1为-(c3-c6)环烷基-;a1和a2各自独立地为-n-或-cr3-,其中a1和a2二者不可以同时为-n-,r3为氢;-f或-oh,并且a3为-nh-或-o-,q选自-(c1-c3)烷基-;-c(=o)-;-c(=s)-;-s(=o)2-和其中-(c1-c3)烷基-可以是未取代的或者被1至3个-ch3基团或卤素原子取代,q1为氢;-f或-cl,n为0或1的整数,其中当q为时,n为0,并且当q为-c(=o)-、-c(=s)-、-s(=o)2-或-(c1-c3)烷基-时,n为1,并且x可选自-c1-c6烷基;-c3-c6环烷基;-c2-c6烯基;-c3-c6环烯基;-(c0-c2烷基)ar;-oar;-(c0-c2烷基)het;萘基;以及以下基团:其中r4为h或-c1-c4烷基,-c0-c2烷基;-c2-c6烯基和-c1-c6烷基可以是未取代的或者被1或2个-ch3基团或1至3个-f基团取代,ar为c6单环芳族化合物,其可以是未取代的或者被一个或多个卤素原子;-oh;-nh2;-c1-c6烷基;-o(c1-c6)烷基;-c3-c6环烯基;-nh(c1-c6烷基);-n(c1-c3烷基)2;-ch2n(c1-c3烷基)2;-s(=o)2-(c1-c3烷基)或苯基基团取代,其中-c1-c3烷基;-c1-c6烷基和-c3-c6环烯基可各自独立地被1至5个-f或-ch3基团取代,并且het为4至6元杂芳族或非芳族环化合物,其包含1至3个选自n、o和s的元素同时具有0至3个双键,并且可以是未取代的或者被一个或多个卤素原子;-c1-c6烷基;-c(=o)(c1-c3烷基);-s(=o)2(c1-c3烷基)或苄基基团取代,其中-c1-c3烷基和-c1-c6烷基可各自独立地被-oh;或1至5个-f或-ch3基团取代。在本发明的优选的实施方案中,式i、ii和iii中的l、q和x可如下定义:l为-ch2-l1-,其中l1为a1和a2各自独立地为-n-或-cr3-,其中a1和a2二者不可以同时为-n-,r3为氢;-f或-oh,并且a3为-nh-或-o-,q为-ch2-、-c(=o)-或-s(=o)2-,并且x可选自-c1-c6烷基;-(c0-c2烷基)ar;-(c0-c2烷基)het;-oar;以及以下基团:其中r4为h或-c1-c4烷基,-c0-c2烷基和-c1-c6烷基可以是未取代的或者被1或2个-ch3基团和/或1至3个-f基团取代,并且ar和het各自独立地如在式i至式iii中所定义。在本发明的实施方案中,上文作为式i至式iii中的x或取代基提及的杂环化合物(het)可具有选自以下基团的结构:其中r5各自独立地为氢;-f;-cl;-c1-c6烷基;-c(=o)(c1-c3烷基);-s(=o)2(c1-c3烷基)或苄基,其中-c1-c3烷基和-c1-c6烷基可各自独立地被-oh;1至5个-f或-ch3基团取代,m为0、1、2或3的整数,并且当m为0时,het是未取代的,并且当m为1、2或3时,het可以被独立的r5取代。本发明的由式i表示的化合物可为由下式i-1表示的化合物:[式i-1]其中a为-c(=o)-;-ch2-;-nh(c=o)-;-c(=s)-或-s(=o)2-,并且r1为直链或支链c1-c4烷基;直链或支链c1-c4烯基;c3-c5环烷基;-oc6c5;-(c0-c2烷基)cf3;苯基(其可以是未取代的或者被c1-c3烷基;吡咯;-och3;-cf3;-f;-cl或-oh取代);吡啶基;呋喃基;噻吩基;苄基(其可以是未取代的或者被一个或多个c1-c3烷基;吡咯;-och3;-cf3;-f;-cl或-oh基团取代);苯乙基;萘基;噁唑基;嘧啶基;吡唑基或吡嗪基。由式i-1表示的化合物优选为如本文中公开的化合物82、83、84、98、99、100、120、121、122、123、125、126、127、128、145、146、147、148、149、159、160、161、177、184、188、204、211、212、213、214、222、223、224、225、232、255、265、266、267、270、272、275、290、291、292、293、294、295、296、297、354、355、403、404和405。所述由式i表示的化合物可为由下式i-2表示的化合物:[式i-2]其中r2、r3或r4可各自独立地为氢或选自以下的基团中的任意一种:所述由式i-2表示的化合物优选为如本文中公开的化合物309、327、328、329、330、331、332、342、343、344、345、346和347。本发明的由式i表示的化合物可为由下式i-3表示的化合物:[式i-3]其中r5可选自直链或支链c1-c4烷基;-coch3;-ch2cf3和-so2ch3。所述由式i-3表示的化合物优选为如本文中公开的化合物481、482、483、484和485。本发明的由式i表示的化合物可为由式i-4表示的化合物:[式i-4]其中n为0、1或2的整数,并且r6、r7或r8可各自独立地为氢或者选自以下基团中的任意一种:所述由式i-4表示的化合物优选为如本文中公开的化合物234、242、243、244、245、246、247、283、284、285、286、288、326和340。本发明的由式i表示的化合物可为由式i-5表示的化合物:[式i-5]其中r9可选自吗啉;哌啶;吡咯烷;氮杂环丁烷;哌嗪;-c1-c2伯或仲烷基胺;其中吡咯烷可以是未取代的或者被一个或多个-f或-cl基团取代,氮杂环丁烷可以是未取代的或者被一个或多个-f或-cl基团取代,并且哌嗪可以是未取代的或者被一个或多个直链或支链c1-c5烷基;苄基;-coch3;-ch2cf3或-so2ch3基团取代。所述由式i-5表示的化合物优选为如本文中公开的化合物356、376、382、383、384、385、411、412、413、426、427、428、429、430、431、452、453、454、455、456、457、466、467、468和486。本发明的由式i表示的化合物可为由下式i-6a或式i-6b表示的化合物:[式i-6a][式i-6b]其中r10可选自吡咯烷;哌啶;c1-c2伯或仲烷基胺;哌嗪;吗啉;其中吡咯烷可以是未取代的或者被一个或多个-f或-cl基团取代,并且哌嗪可以是未取代的或者被一个或多个直链或支链c1-c5烷基;-coch3;-ch2cf3或-so2ch3基团取代。所述由式i-6a表示的化合物优选为如本文中公开的化合物423、424、425、432、433、434、435、439、440、441、442、443、444、458、459、460、461、462和463。此外,由式i-6b表示的化合物优选为如本文中公开的化合物446。本发明的由式i表示的化合物可为由下式i-7表示的化合物:[式i-7]其中x可选自-oh;-f;-cl或-br。所述由式i-7表示的化合物优选为如本文中公开的化合物386和387。本发明的由式i表示的化合物可为由下式i-8表示的化合物:[式i-8]其中b可选自其中w为氢;-f;-c1或-oh。所述由式i-8表示的化合物优选为如本文中公开的化合物154、171、172、173、194、218、219、520、571、574、652、812、813、814、818、820、822、823和824。本发明的由式i表示的化合物可为由下式i-9表示的化合物:[式i-9]其中a为-c(=o)-;-ch2-;-nh(c=o)-;-c(=s)-或-s(=o)2-,并且r11可选自呋喃基;苄基;吡咯取代的苯基或吡咯取代的苯胺基。所述由式i-9表示的化合物优选为如本文中公开的化合物321、322和323。本发明的由式i表示的化合物可为由下式i-10表示的化合物:[式i-10]由式i-10表示的化合物优选为如本文中公开的化合物472。本发明的由式i表示的化合物可为由下式i-11表示的化合物:[式i-11]由式i-11表示的化合物优选为如本文中公开的化合物402。本发明的由式i表示的化合物可为由下式i-12表示的化合物:[式i-12]由式i-12表示的化合物优选为如本文中公开的化合物380和388。本发明的由式i表示的化合物可为由下式i-13表示的化合物:[式i-13]其中a为-c(=o)-;-ch2-;-ch(ch3)-;-nh(c=o)-;-nch3(c=o)-;-c(=s)-或-s(=o)2-,并且r12可选自直链或支链c1-c4烷基;苯基(其可以是未取代的或者被一个或多个直链或支链c1-c3烷基;-f;-cl;cf3;-och3;-c1-c2伯或仲烷基胺;-so2ch3;噻吩基;吡咯;吡唑基;呋喃基;三唑基或咪唑基取代);吡啶基;噻吩基;呋喃基;苄基(其可以是未取代的或者被-f、-cl或-och3取代);吲哚(其可以是未取代的或者被一个或多个直链或支链c1-c3烷基基团取代);二氢苯并呋喃基;苯胺(其可以是未取代的或者被直链或支链c1-c3烷基取代);二氢吲哚或萘基。由式i-13表示的化合物优选为如本文中公开的化合物80、81、103、104、105、106、107、108、109、110、111、112、113、114、115、118、162、163、164、165、166、167、168、183、185、186、187、189、196、197、215、220、230、231、233、256、268、271、273、274、298、299、300、301、302、303、304和305。本发明的由式i表示的化合物可为由下式i-14表示的化合物:[式i-14]其中r2、r3和r4可各自独立地为氢或选自以下的基团中的任意一种:由式i-14表示的化合物优选为如本文中公开的化合物348、349、350、351、352、396、400和401。本发明的由式i表示的化合物可为由下式i-15表示的化合物:[式i-15]其中n为0、1或2的整数,并且r6、r7和r8可各自独立地为氢或选自以下的基团中的任意一种:由式i-15表示的化合物优选为如本文中公开的化合物250、251、252、253、257、258、259、260、261、262、263、276、277、278、279和280。本发明的由式i表示的化合物可为由下式i-16表示的化合物:[式i-16]其中b可选自其中w可被-f或-cl取代。所述由式i-16表示的化合物优选为如本文中公开的化合物174、175、176和195。本发明的由式i表示的化合物可为由下式i-17表示的化合物:[式i-17]其中a为-c(=o)-;-ch2-;-nh(c=o)-;-c(=s)-或-s(=o)2-,并且b可选自此外,r13可选自吡咯烷和-c1-c2伯或仲烷基胺。所述由式i-17表示的化合物优选为如本文中公开的化合物475、476、478、479、480和487。本发明的由式i表示的化合物可为由下式i-18表示的化合物:[式i-18]所述由式i-18表示的化合物优选为如本文中公开的化合物477。本发明的由式i表示的化合物可为由下式i-19表示的化合物:[式i-19]其中r14可选自所述由式i-19表示的化合物优选为如本文中公开的化合物119和193。本发明的由式i表示的化合物可为由下式i-20表示的化合物:[式i-20]所述由式i-20表示的化合物优选为如本文中公开的化合物198。本发明的由式i表示的化合物可为由下式i-21表示的化合物:[式i-21]其中r15可选自所述由式i-21表示的化合物优选为如本文中公开的化合物248和249。本发明的由式i表示的化合物可为由下式i-22表示的化合物:[式i-22]所述由式i-22表示的化合物优选为如本文中公开的化合物569和573。本发明的由式i表示的化合物可为由下式i-23表示的化合物:[式i-23]其中b可选自其中w为氢;-f;-cl或-oh。所述由式i-23表示的化合物优选为如本文中公开的化合物609、653和696。所述由式i、式ii、式iii和式i-1至i-23表示的化合物如下所示。优选地,本发明的由式i表示的化合物或其药学上可接受的盐可选自化合物080、081、082、083、084、098、099、100、106、107、108、109、110、112、120、121、122、123、125、126、127、128、145、146、147、148、149、159、160、161、171、173、174、175、177、186、188、193、194、195、196、198、211、214、219、248、249、250、251、252、255、265、266、267、272、283、284、285、286、292、295、297、305、326、328、329、330、332、342、343、344、345、346、349、354、356、376、380、382、383、384、385、386、387、388、403、411、413、430、431、432、433、434、435、439、440、441、442、443、444、452、453、454、455、456、467、468、481、482、483、484、485、486、569、696、813和823。更优选地,本发明的由式i表示的化合物或其药学上可接受的盐可选自化合物082、083、084、098、100、120、121、122、123、125、126、127、128、145、146、148、149、159、160、161、171、174、177、194、211、249、255、283、305、326、328、329、330、332、342、343、344、345、346、349、354、356、376、382、383、387、388、411、413、431、439、441、444、452、453、454、467、468、481、482、483、484、485、696和813。如本文所使用的,术语“药学上可接受的盐”是指通常用于药学领域的任何盐。药学上可接受的盐的实例包括但不限于与无机离子诸如钙、钾、钠或镁离子的盐,与无机酸诸如盐酸、硝酸、磷酸、溴酸、碘酸、高氯酸、酒石酸或硫酸的盐,与有机酸诸如乙酸、三氟乙酸、柠檬酸、马来酸、琥珀酸、草酸、苯甲酸、酒石酸、富马酸、苦杏仁酸、丙酸、柠檬酸、乳酸、乙醇酸、葡糖酸、半乳糖醛酸、谷氨酸、戊二酸、葡糖醛酸、天冬氨酸、抗坏血酸、碳酸、香草酸、氢碘酸等的盐,与磺酸诸如甲磺酸、乙磺酸、苯磺酸、对甲苯磺酸或萘磺酸的盐,与氨基酸诸如甘氨酸、精氨酸或赖氨酸的盐,以及与胺诸如三甲胺、三乙胺、氨、吡啶或甲基吡啶的盐。在本发明中,优选的盐包括与盐酸、三氟乙酸、柠檬酸、溴酸、马来酸、磷酸、硫酸、酒石酸等的盐,并且这样的化合物的优选实例包括如本文中公开的化合物230、245、250、251、253、266、270、271、273、274、275、290、291、292、293、294、295、296、297、478和486。由式i表示的化合物可以含有一个或多个不对称碳原子,并因此可以以外消旋体、外消旋混合物、单一对映异构体(光学异构体)、非对映异构体混合物和单独的非对映异构体的形式存在。式i化合物可以通过本领域已知的方法(例如柱色谱法或hplc)分离成这样的异构体。或者,可以通过使用已知构型的光学纯原料和/或试剂来进行立体定向合成而合成式i化合物的立体异构体。特别地,所述异构体可为光学异构体。在本发明中,由式i表示的化合物可以通过韩国专利注册号1697518中公开的方法来制备,但不限于此。包含本发明的组蛋白脱乙酰酶6抑制剂的药物组合物用于预防或治疗cmt病。夏科-马里-图思(cmt)病(遗传性运动和感觉神经病:hmsn)是遗传性周围神经疾病的最常见类型,已知主要由轴突转运缺陷引起。在本发明中,cmt病可以是选自以下的至少一种类型:1型cmt、2型cmt、4型cmt、cmtx、dsn、ch(先天性髓鞘形成减少)、hnpp(具有压迫性麻痹易感的遗传性神经病)和gan(巨轴突神经病),但不限于此。在本发明中,可以通过给药根据本发明的药物组合物来预防或治疗cmt病。例如,本发明的药物组合物可以通过包含具有组蛋白脱乙酰酶6(hdac6)抑制活性的新型二甲基哌嗪衍生化合物并因此通过改善轴突转运来预防或治疗与组蛋白脱乙酰酶6活性相关的cmt病。在本发明的一个实施方案中,由式i表示的化合物对组蛋白脱乙酰酶6(hdac6)选择性地具有抑制活性,因此可以减轻运动能力的下降并改善电生理指标,可以治疗或减轻由cmt病引起的症状,例如轴突萎缩和髓鞘神经损失,以及可抑制或延迟这种症状的表现。特别地,本发明的由式i表示的化合物可有效地治疗或缓解在cmt病中表现的症状,如在运动神经传导速度的降低、复合肌肉动作电位降低、进行性神经元变性、肌无力、异常感觉、施万细胞增生、髓鞘神经损失和轴突萎缩,以及可以抑制或延迟这种症状的表现。对于给药,除了式i化合物、其异构体或其药学上可接受的盐之外,本发明的药物组合物还可含有至少一种药学上可接受的载体。用于本发明中的药学上可接受的载体可以是以下中的至少一种:生理盐水、无菌水、林格氏溶液、缓冲盐水、右旋糖溶液、麦芽糖糊精溶液、甘油、乙醇,及其中一种或多种的混合物。如果必要,所述组合物可以含有其它常规添加剂,如抗氧化剂、缓冲剂或抑菌剂。另外,可以使用稀释剂、分散剂、表面活性剂、粘合剂和润滑剂将所述组合物配制成注射制剂(如溶液剂、混悬剂、浑浊的液体等)、丸剂、胶囊剂、颗粒剂和片剂。因此,本发明的组合物可以是贴剂、液体剂、丸剂、胶囊剂、颗粒剂、片剂、栓剂等的形式。这些制剂可以通过本领域中用于制剂的常规方法,或通过在remington’spharmaceuticalscience(最新版),mackpublishing公司,eastonpa中公开的方法来制备。此外,本发明的组合物可以依据疾病或组分制备为多种制剂。作为使用本发明药物组合物制备口服给药的制剂的非限制性实例,可以是片剂、锭剂、糖锭剂、水溶性混悬剂、油混悬剂、制备的散剂、颗粒剂、乳剂、硬胶囊剂、软胶囊剂、糖浆剂、酏剂等。为了将本发明的药物组合物配制成口服给药制剂,可以使用以下物质:粘合剂,例如乳糖、蔗糖、山梨糖醇、甘露醇、淀粉、支链淀粉、纤维素、明胶等;赋形剂,例如磷酸二钙等;崩解剂,例如玉米淀粉、甘薯淀粉等;润滑剂,例如硬脂酸镁、硬脂酸钙、硬脂酰富马酸钠、聚乙二醇蜡等;其中也可以使用甜味剂、调味剂、糖浆等。此外,在胶囊剂的情况下,除了上述材料以外,还可以使用如脂肪油等液体载体。作为使用本发明药物组合物的肠胃外制剂的非限制性实例,可以是可注射的溶液剂、栓剂、用于呼吸吸入的粉末、用于喷雾的气雾剂、软膏剂、用于涂敷的粉末、油、霜剂等。将本发明的药物组合物制成肠胃外给药的制剂,可以使用以下物质:灭菌水溶液、非水溶剂、悬浮液,乳剂、冻干制剂、外用制剂等。作为非水溶剂和混悬剂,可以使用以下物质,但不限于此:丙二醇、聚乙二醇、如橄榄油的植物油、如油酸乙酯的可注射酯等。根据预期用途,本发明的组合物可以口服或肠胃外给药(例如经静脉内、皮下、腹膜内或局部给药)。所述药物组合物的剂量根据患者的体重、年龄、性别、健康状况、饮食、给药时间、给药方式、排泄率、疾病的严重程度等而变化。本发明的药物组合物的药物有效量和有效剂量可以根据药物组合物的配制方法、给药方式、给药时间和/或给药途径等而不同,并且可以根据各种因素而多样化,所述因素包括通过给药所述药物组合物要实现的反应类型和程度,待给药的个体的类型(例如个体的年龄、体重、一般健康状况、疾病症状或严重程度、性别、饮食和排泄),在同一时间或不同时间用于相应个体的其他药物组合物的成分,以及在制药领域熟知的其他类似因素,并且本领域技术人员可以容易地确定及开出有效剂量以用于预期治疗。本发明的药物组合物可以每天给药一次或每天分成数次给药。本发明的药物组合物可以作为单独的治疗剂或与其他治疗剂组合给药,并且可以与常规治疗剂顺序或同时给药。考虑到以上所有因素,本发明的药物组合物可以以最小量实现最大效力而没有副作用的量给药,并且这种量可以由本发明所属领域技术人员容易地确定。除了由式i表示的化合物、其异构体或其药学上可接受的盐之外,本发明的药物组合物还可进一步包含至少一种有效成分,该有效成分显示与其相同或相似的药用效果。本发明的药物组合物即使单独使用也可以显示优异的效果,但是为了提高治疗效率,可以进一步与诸如激素治疗、药物治疗等各种方法联合使用。本发明还提供预防或治疗cmt病的方法,其包括向需要其的个体给药治疗有效量的由式i表示的化合物、其异构体或其药学上可接受的盐或者包含其的药物组合物的步骤。如本文所用,术语“治疗有效量”是指由式i表示的化合物、其异构体或其药学上可接受的盐有效预防或治疗cmt病的量。在本发明的治疗方法中,由式i表示的化合物、其异构体或其药学上可接受的盐的合适的每日总量可以由负责医生在正确医学决定范围内确定,并且可以是例如在约0.1-10,000mg/kg范围内、在约1-8,000mg/kg范围内、在约5-6,000mg/kg范围内或在约10-4,000mg/kg范围内,优选地,在约50-2,000mg/kg范围内的量可以每天给药一次或每天分成数次给药。然而,出于本发明的目的,优选根据各种因素应将特定的治疗有效剂量不同地应用于每个特定患者,所述因素包括要由此实现的反应的类型和程度,包括在一些情况中存在使用的其他制剂的特定组合物,患者的年龄、体重、一般健康状况、性别和饮食,给药时间,给药途径,所述组合物的分泌率,治疗疗程及与特定组合物一起或同时使用的药物,以及在制药领域熟知的其他类似因素。本发明的用于预防或治疗cmt病的方法包括不仅在疾病表现其症状之前进行处置,而且还包括通过给药由式i表示的化合物、其异构体或其药学上可接受的盐来抑制或避免这种症状。在疾病管理中,某些活性成分的预防或治疗剂量可以根据疾病或病症的性质和严重程度以及给药所述活性成分的途径而变化。剂量及其频率可以根据个体患者的年龄、体重和反应而变化。自然地鉴于这些因素,本领域技术人员可以容易地选择合适的剂量和用法。另外,本发明的用于预防或治疗cmt病的方法可以进一步包括与由式i表示的化合物、其异构体或其药学上可接受的盐一起给药治疗有效量的有助于治疗所述疾病的另外的活性剂,所述另外的活性剂可与由式i表示的化合物一起呈现出协同效应或累加效应。本发明还提供由式i表示的化合物、其异构体或其药学上可接受的盐在制备用于预防或治疗cmt病的药物中的用途。为了制备药物,可以将由式i表示的化合物、其异构体或其药学上可接受的盐与可接受的辅剂、稀释剂、载体等组合,并且可以与其他活性剂一起制备成复合制剂,并因此具有活性成分的协同作用。本发明还提供由式i表示的化合物、其异构体或其药学上可接受的盐在预防或治疗夏科-马里-图思(cmt)病中的用途。如果彼此不矛盾,本发明的用途、组合物和治疗方法中提及的事项是等同应用的。本发明的有益效果包含本发明的由式i表示的化合物、其异构体或其药学上可接受的盐的药物组合物具有选择性地组蛋白脱乙酰酶6(hdac6)抑制活性,并因此有效预防或治疗cmt病。附图简要描述图1显示根据本发明的一个实施方案在将化合物84向2.5周龄的cmt1a小鼠(c22)重复给药两周之后的行为和电生理改善程度的评价结果(n=10只/组。正常组:给药dw的wt,对照组:给药dw的cmt同窝出生小鼠(cw22)。n=10),(a)延迟掉落的时间,(b)握力强度(在给药后两周内测量),(c)mncv,和(d)cmap(在给药后三周内测量)。数据以平均值±sem表示。使用dunnett检验的单因素方差分析用于比较tg组和药物治疗组。*p<0.05,**p<0.01,***p<0.0001。使用t检验比较wt和tg。#p<0.05,###p<0.001。图2显示根据本发明的一个实施方案在将化合物84向2.5周龄的cmt1a小鼠(c22)重复给药两周之后评估组织病理学改善程度的结果(n=4只/组。正常组:给药蒸馏水(dw)的野生型小鼠(wt),对照组:给药蒸馏水(dw)的cmt同窝出生小鼠,n=3),(a&b)有髓鞘神经纤维和无髓鞘神经纤维的大小分布直方图,(c)有髓鞘的轴突和无髓鞘的轴突的数目,(d)轴突的总数目,以及(e)每组甲苯胺蓝染色的坐骨神经横切面的代表性组织学图像。比例尺:50μm(x200)。向wt小鼠给药dw,并且向c22小鼠给药dw或以10mg/kg、25mg/kg或100mg/kg给药化合物84,持续两周。数据以平均值±sem表示。使用dunnett检验的单因素方差分析用于比较对照组和药物治疗组。*p<0.05,**p<0.01,***p<0.0001。使用t检验比较正常组(wt)和对照组(tg)。#p<0.05,###p<0.001。发明方式下文将通过制备实施例和实施方案对本发明进行更详细的描述。然而,这些制备实施例和实施方案仅仅是为举例说明本发明的目的而提供,因此本发明并不限于此。制备实施例1:化合物84(4-(((3r,5s)-4-苄基-3,5-二甲基哌嗪-1-基)甲基)-n-羟基苯甲酰胺)的合成步骤1:4-(((3r,5s)-3,5-二甲基哌嗪-1-基)甲基)苯甲酸甲酯的合成在0℃下,将(2s,6r)-2,6-二甲基哌嗪(50.000g,437.867mmol)和cs2co3(171.199g,525.440mmol)在乙腈(200ml)中溶解,并将4-(溴甲基)苯甲酸甲酯(式1-1,80.242g,350.293mmol)加入至所述溶液,随后在室温下搅拌5小时。将反应混合物通过玻璃滤器过滤以移除固体,并将滤液在减压下浓缩以移除溶剂。将水加入至浓缩物,随后用乙酸乙酯萃取。将有机层用氯化钠的饱和水溶液洗涤,用无水硫酸镁干燥,过滤,并在减压下浓缩。将己烷(100ml)加入至浓缩物并搅拌,并将沉淀的固体过滤,用己烷洗涤,并干燥以得到白色固体形式的期望化合物(85.200g,74.2%)。步骤2:4-(((3r,5s)-4-苄基-3,5-二甲基哌嗪-1-基)甲基)苯甲酸甲酯的合成在室温下,将4-(((3r,5s)-3,5-二甲基哌嗪-1-基)甲基)苯甲酸甲酯(式1-2,30.000g,114.351mmol)、溴化苄(14.961ml,125.786mmol)和k2co3(23.707g,171.527mmol)在乙腈(150ml)中溶解,并将溶液在相同温度下搅拌17小时。将反应混合物通过玻璃滤器过滤以移除固体,并将滤液在减压下浓缩以移除溶剂。将水加入至浓缩物,随后用乙酸乙酯萃取。将有机层用氯化钠的饱和水溶液洗涤,用无水硫酸镁干燥,过滤,并在减压下浓缩。将浓缩物通过柱色谱法(二氧化硅;120g筒;乙酸乙酯/己烷=由0%至30%)纯化,并浓缩以得到白色固体形式的期望化合物(22.400g,55.6%)。步骤3:化合物84的合成在0℃下,将4-(((3r,5s)-4-苄基-3,5-二甲基哌嗪-1-基)甲基)苯甲酸甲酯(式1-3,15.000g,42.557mmol)、羟胺(52.061ml,851.136mmol,50.00%水溶液)和氢氧化钾(23.879g,425.568mmol)在甲醇(300ml)中溶解,并将溶液在相同温度下搅拌1小时。将反应混合物在减压下浓缩以移除溶剂,并将碳酸氢钠的饱和水溶液加入至浓缩物,随后用二氯甲烷萃取。将有机层用氯化钠的饱和水溶液洗涤,用无水硫酸镁干燥,过滤,并在减压下浓缩。将浓缩物通过柱色谱法(二氧化硅;120g筒;甲醇/二氯甲烷=由0%至20%)纯化并浓缩,然后将所得的物质由乙醚(200ml)和二氯甲烷(50ml)中在25℃下结晶并过滤。将所得的固体用乙醚洗涤并干燥以得到白色固体形式的化合物84(12.580g,83.6%)1hnmr(400mhz,dmso-d6)611.18(brs,1h),9.03(brs,1h),7.70(d,2h,j=8.2hz),7.36-7.33(m,4h),7.28(dd,2h,j=7.5,7.5hz),7.18(dd,1h,j=7.2,7.2hz),3.73(s,2h),3.44(s,2h),2.64(d,2h,j=10.6hz),2.61-2.53(m,2h),1.81(t,2h,j=10.5hz),0.90(d,6h,j=6.1hz);lrms(es)m/z354.2(m++1).制备实施例2:化合物382(4-(((3r,5s)-3,5-二甲基-4-(3-(哌啶-1-基甲基)苄基)哌嗪-1-基)甲基)-n-羟基苯甲酰胺)的合成步骤1:4-(((3r,5s)-3,5-二甲基-4-(3-(哌啶-1-基甲基)苄基)哌嗪-1-基)甲基)苯甲酸甲酯的合成将4-(((3r,5s)-4-(3-甲酰基苄基)-3,5-二甲基哌嗪-1-基)甲基)苯甲酸甲酯(式5-1,0.150g,0.394mmol)和哌啶(0.038ml,0.434mmol)在二氯甲烷(10ml)中溶解,并将溶液在室温下搅拌30分钟。将na(oac)3bh(0.125g,0.591mmol)加入至反应溶液,随后在相同温度下搅拌17小时。然后将碳酸氢钠的饱和水溶液加入至反应混合物,随后用二氯甲烷萃取。将萃取物通过塑料滤器过滤以移除固体残渣和水层,然后在减压下浓缩。将浓缩物通过柱色谱法(二氧化硅;4g筒;甲醇/二氯甲烷=由0%至10%)纯化并浓缩,以得到淡黄色油形式的期望化合物(0.095g,53.6%)。步骤2:化合物382的合成在室温下,将4-(((3r,5s)-3,5-二甲基-4-(3-(哌啶-1-基甲基)苄基)哌嗪-1-基)甲基)苯甲酸甲酯(式5-2,0.044g,0.098mmol)、羟胺(0.060ml,0.979mmol,50.00%水溶液)和氢氧化钾(0.127g,1.957mmol)在甲醇(3ml)中溶解,并将溶液在相同温度下搅拌30分钟。然后将反应混合物在减压下浓缩以移除溶剂,并将碳酸氢钠的饱和水溶液加入至浓缩物,随后用二氯甲烷萃取。将萃取物通过塑料滤器过滤以移除固体残渣和水层,然后在减压下浓缩以得到白色固体形式的化合物382(0.037g,83.9%)。1hnmr(400mhz,dmso-d6)δ7.68(d,2h,j=8.2hz),7.30-7.28(m,3h),7.22-7.20(m,2h),7.08(d,1h,j=6.2hz),3.72(s,2h),3.41(s,2h),3.40(s,2h),2.65-2.62(m,2h),2.58-2.53(m,2h),2.29(s,4h),1.80(t,2h,j=10.7hz),1.49-1.46(m,4h),1.40-1.38(m,2h),0.90(d,6h,j=6.1hz);lrms(es)m/z451.2(m++1).制备实施例3:化合物454(n-羟基-4-(((3r,5s)-4-(3-((4-异戊基哌嗪-1-基)甲基)苄基)-3,5-二甲基哌嗪-1-基)甲基)苯甲酰胺)的合成步骤1:4-异戊基哌嗪-1-甲酸叔丁酯的合成在室温下,将哌嗪-1-甲酸叔丁酯(2.000g,10.738mmol)、1-溴-3-甲基丁烷(1.352ml,11.275mmol)和cs2co3(4.198g,12.886mmol)在乙腈(150ml)中溶解,并将溶液在相同温度下搅拌17小时。将反应混合物通过玻璃滤器过滤以移除固体,并将滤液在减压下浓缩以移除溶剂。将浓缩物通过柱色谱法(二氧化硅;12g筒;乙酸乙酯/己烷=由0%至10%)纯化并浓缩,以得到无色油形式的期望化合物(1.220g,44.3%)。步骤2:1-异戊基哌嗪三氟乙酸盐的合成在室温下,将4-异戊基哌嗪-1-甲酸叔丁酯(1.000g,3.900mmol)在二氯甲烷(10ml)/三氟乙酸(5ml)中溶解,并将溶液在相同温度下搅拌17小时。将反应混合物在减压下浓缩以移除溶剂,并将浓缩物由乙酸乙酯(20ml)在室温下结晶并过滤,并将所得的固体用乙酸乙酯洗涤并干燥以得到白色固体形式的期望化合物(0.929g,94.0%)。步骤3:4-(((3r,5s)-4-(3-((4-异戊基哌嗪-1-基)甲基)苄基)-3,5-二甲基哌嗪-1-基)甲基)苯甲酸甲酯的合成将4-(((3r,5s)-4-(3-甲酰基苄基)-3,5-二甲基哌嗪-1-基)甲基)苯甲酸甲酯(式5-1,0.220g,0.578mmol)和1-异戊基哌嗪三氟乙酸盐(0.220g,0.867mmol)在二氯甲烷(5ml)中溶解,并将溶液在室温下搅拌1小时。将na(oac)3bh(0.245g,1.156mmol)加入至反应溶液,随后在相同温度下进一步搅拌17小时。然后将碳酸氢钠的饱和水溶液加入至反应混合物,随后用二氯甲烷萃取。将萃取物通过塑料滤器过滤以移除固体残渣和水层,然后在减压下浓缩。将浓缩物通过柱色谱法(二氧化硅;4g筒;甲醇/二氯甲烷=由0%至10%)纯化并浓缩,以得到淡黄色油形式的期望化合物(0.232g,77.0%)。步骤4:化合物454的合成在室温下,将4-(((3r,5s)-4-(3-((4-异戊基哌嗪-1-基)甲基)苄基)-3,5-二甲基哌嗪-1-基)甲基)苯甲酸甲酯(式5-2,0.232g,0.446mmol)、羟胺(0.545ml,8.910mmol,50.00%水溶液)和氢氧化钾(0.250g,4.455mmol)在甲醇(5ml)中溶解,并将溶液在相同温度下搅拌30分钟。将反应混合物在减压下浓缩以移除溶剂,并将碳酸氢钠的饱和水溶液加入至所得的浓缩物,随后用二氯甲烷萃取。将萃取物通过塑料滤器过滤以移除固体残渣和水层,然后在减压下浓缩以得到白色固体形式的化合物454(0.067g,29.6%)。1h-nmr(400mhz,dmso-d6)δ7.69(d,2h,j=8.2hz),7.30(d,2h,j=8.1hz),7.27(s,1h),7.23-7.21(m,2h),7.09-7.08(m,1h),3.72(s,2h),3.42(s,2h),3.41(s,2h),2.63(d,2h,j=10.2hz),2.58-2.53(m,2h),2.34-2.27(m,8h),2.24(t,2h,j=7.6hz),1.80(t,2h,j=10.5hz),1.57-1.53(m,1h),1.31-1.25(m,2h),0.89(d,6h,j=6.1hz),0.85(d,6h,j=6.6hz).<实施例1>hdac6特异性抑制剂对线粒体轴突转运速度的影响分析(体外)分析hdac6特异性抑制剂对线粒体轴突转运的影响,以鉴定本发明的由式i表示的化合物是否选择性抑制hdac6活性,并因此增加微管蛋白(即hdac6的关键底物)的乙酰化,以表现改善线粒体的转运速度(其已通过在神经元轴突内用β淀粉样蛋白处理而降低)的效应。为此,在授精的第17至18天(e17-18),将来自sprague-dawley(sd)大鼠胎儿的海马神经元在已用胞外基质包被并用1m浓度的β淀粉样蛋白片段处理的培养容器中培养7天以用于成像。24小时后,在体外培养的第8天,将这种神经元用化合物处理。3小时后,将其用mitotrackerredcmxros(lifetechnologies,ny,usa)处理最后五分钟,以染色线粒体。染色的神经元线粒体的轴突转运图像用共聚焦显微镜(leicasp8;leicamicrosystems,uk)以1秒钟的间隔拍摄一分钟,使用imaris分析程序(bitplane,zurich,瑞士)测量每秒每个线粒体的转运速度,并设置一个节段,其中与媒介物相比,β淀粉样蛋白处理组显示线粒体的转运速度显著降低。之后,用媒介物设置为100%并且用β淀粉样蛋白处理设置为0%的标准化结果在下表1中描述。[表1]用化合物处理后,速度分布表示为:*,0%-50%;**,50%-100%;***,>100%。从以上结果可见,鉴定了本发明的由式i表示的化合物显示出改善线粒体的轴突转运速度的优异效果,如上表1所示。<实施例2>hdac6/1酶抑制测定(体外)以现有的开发的材料作为对照组进行对比实验,以通过对hdac1和hdac6酶活性的抑制实验鉴定本发明的由式i表示的化合物对hdac6的选择性。使用enzolifescience,inc.的hdac荧光药物发现试剂盒(bml-[1424]ak511,516)测量hdac酶的活性。为了测试hdac1酶活性,将人重组hdac1(bml-se456)用作酶源,fluorde“sirt1(bnl-ki177)”用作底物。将化合物的5倍稀释液分入96孔平板中,然后将0.3μg的酶和10μm的底物插入每个孔中,在30℃下反应60分钟,由此将fluordedeveloperii(bmlki176)插入其中,并反应30分钟且完成。之后,用多板酶标仪(flexstation3,moleculardevice)测量荧光值(ex360,em460)。通过使用calbiocheminc.的人重组hdac6(382180),按照与hdac1酶活性测试方法相同的方案对hdac6酶进行实验。使用graphpadprism4.0程序计算各ics0值,最终结果值在下表2中描述。[表2]化合物hdac1(μm)hdac6(μm)hdac6选择性(倍数)8411.90.023517382>100.083>120454nd0.044>237如上表2所示,鉴定了由本发明的由式i表示的化合物显示出优异的选择性hdac6抑制活性。<实施例3>hdac6的抑制效果和对其他hdac选择性的分析在酶水平上鉴定由式i表示的化合物的hdac6抑制作用和选择性。实验由reactionbiologycorp.(malvern,pa,usa)要求,并根据组织内部制定的检测方法进行。特别地,将化合物84的系列稀释液分入平板中,然后将底物(即rhk-k(ac)-amc)和酶一起插入50mmtris-hcl缓冲液(ph8.0,137mmnacl,2.7mmkcl,1mmmgcl2、1mg/mlbsa)中以诱导反应。之后,将含有2mm烟酰胺和16mg/ml胰蛋白酶的50mmtris-hcl缓冲液(ph8.0,137mmnacl,2.7mmkcl,1mmmgcl2)插入其中,然后进行反应,之后在ex.360nm/em.460nm测量荧光信号以测量酶活性,其结果示于下表3。使用pan-hdac抑制剂(即lbh589)作为对照组。[表3]如上表3所示,鉴定了化合物84的hdac6的ic50为72.2nm,hdac8的ic50为5170nm,而其他hdac同种型完全没有被抑制。换句话说,发现化合物84具有优异的hdac6抑制能力,并且相对于其他hdac同种型具有非常高的选择性。<实施例4>药物作用评价为了鉴定化合物84对cmt病的治疗效果和作用机理,将cmt1a小鼠(c22)用化合物84处理,以鉴定动物的行为症状以及组织学改善和蛋白质表达变化。cmt1a在由构成髓磷脂的pmp22基因重复引起的无髓鞘cmt病组中以最高频率发生。已知测试中使用的cmt1a小鼠(c22)中插入了七个人pmp22基因,其比内源性小鼠pmp22mrna过表达多大约2.7倍。在2.5周龄之前,小鼠出现诸如运动功能下降、感觉异常等症状,并且由于周围神经变性而从中观察到肌肉无力、蹄和尾巴拖拉。在组织学上,观察到髓鞘减少、施万细胞过度生长、洋葱头样结构(onionbulb)、轴突变性/损失以及肌肉萎缩。特别地,将化合物84每天两次以10、25和100mg/kg向2.5周龄的cmt1a小鼠(c22)重复给药,持续两周。在最后一天进行行为评价和电生理分析,之后立即处死动物以进行组织病理学分析和评价。所有结果均以平均值±标准误表示,通过阴性对照组与各测试材料组之间的统计学显著性确定药物效果的有效性。对于统计学分析,使用anova(单次测量为单因素方差分析,重复测量为双因素方差分析)鉴定离散同质性。dunnett或bofferroni事后检验的结果是,如果p值小于0.05,则确定为具有统计学意义。<实施例4-1>动物行为的评价每天进行一次小鼠转棒实验、平衡木和握力强度测试,持续2天。此后,基于转棒实验、握力强度和体重的结果,通过z-阵列方法分配动物。将c22小鼠分为四组,其由dw处理组和三个化合物84处理组(10、25、100mg/kg)组成,每组10只小鼠。每天两次重复口服给药测试品,且在给药测试品后30分钟进行行为测试。转棒实验进行转棒实验(rotarod,le8205,panlab)以评价运动协调功能/运动功能。在测试前三天,将所有测试动物置于8rpm转棒上,每天进行五次适应性训练。从转棒上掉落的时间达到150-180秒的动物用于测试(包括大约80%的动物)。在分组和本实验中,在8rpm固定速度下测量动物从转棒上掉落的时间,持续3分钟。在一个实验中,转棒实验总共进行了3次。在其三个结果中,最大值用作评估值。其结果示于图1(a)。在图1中,dwwt(同窝出生)是指给药蒸馏水的小鼠的正常组,而dwc22组(同窝出生)是指给药蒸馏水的cmt小鼠(对照组)。作为将化合物84向2.5周龄的cmt1a小鼠(c22)重复给药持续两周的结果,可以看出与对照组(给药dw的同窝出生小鼠(dwc22))相比,延迟掉落的时间以剂量依赖方式增加,从而减轻cmt病,如图1(a)所示。握力强度测试cmt(遗传性运动和感觉神经病)显示出一个关键症状,其中由于神经变性导致控制肌肉的神经分布减少而导致肌肉萎缩。在实际的临床测试中,通过直腿屈踝检查和握力测试来评估肌肉萎缩的程度。gst(握力强度测试,bio-gs3,bioseb)主要用于动物实验。对于gst,用网格评估四爪的强度。所有实验均由一个人进行。当小鼠尝试抓握时,在此处施加一点点拉力以使小鼠保持抓握,然后以大约15°的坡度牵拉小鼠略高于水平线以测量最大拉力。在连续测量五次的值中,最大值用权重校准,然后将其用作评估结果。在给药之前和给药期间,以与上述相同的方式每周一次进行测试,持续两周。其结果示于图1(b)。作为将化合物84向2.5周龄的cmt1a小鼠(c22)重复给药持续两周的结果,可以看出与对照组(给药dw的同窝出生小鼠(dwc22))相比,握力强度以剂量依赖方式增加,如图1(b)所示。神经传导研究(ncs)如下所述进行神经传导研究(nsc)的评估。将超大刺激施加于近端周围神经,然后记录由神经控制的远端肌肉产生的复合肌肉动作电位(cmap),以测量潜伏期、振幅、神经传导速度(ncv)、持续时间、形状、面积等,然后将其与正常值进行比较并进行评价。对于神经传导研究(nsc),对用异氟烷麻醉的小鼠的坐骨神经给与电刺激,然后用vikingquest(natusmedicalinc.)测量对腓肠神经的电刺激。其结果示于图1(c)和(d)。作为将化合物84向2.5周龄cmt1a小鼠(c22)重复给药持续两周的结果,可以看出与对照组(给药dw的同窝出生小鼠(dwc22))相比,运动神经传导速度(mncv)和复合肌肉动作电位(cmap)以剂量依赖方式增加,如图1(c)和(d)所示。因此,化合物84改善了在c22小鼠中缺陷的运动能力和电生理指数。因此,发现化合物84具有作为cmt病的治疗剂的药用效果。<实施例4-2>组织病理学分析如下所述进行组织病理学分析。在最后一次给药化合物84之后0.5小时,通过颈脱位法使小鼠安乐死,然后由其收集坐骨神经。为了进行组织病理学检查,将收集的坐骨神经固定在2.5%的戊二醛溶液中。通过组织处理方法将固定的组织制备成塑料包埋块,然后以500nm的半薄厚度进行显微切片,以制备坐骨神经组织片段,然后用甲苯胺蓝染色。用光学显微镜以400×放大倍数对染色的神经组织进行拍照。在拍摄的整个区域中,使用图像j程序测量全部有髓鞘纤维和全部无髓鞘纤维的内部轴突直径。通过使用全部有髓鞘纤维的数目和全部无髓鞘纤维的数目以及各个神经纤维的直径的大小分布图来分析其结果。其结果示于图2。在图2中,dwwt(同窝出生)或wt-veh(同窝出生)是指给药蒸馏水的小鼠的正常组,dwc22(同窝出生)或c22-veh(同窝出生)是指给药蒸馏水的cmt小鼠。如图2(e)所示,在坐骨神经的组织病理学评价中发现,与小鼠正常组(wt,dw)相比,小鼠对照组(c22,dw)显示出中等程度的髓鞘神经纤维损失、轴突萎缩和施万细胞增生,而给药化合物84的动物显示在小鼠对照组中已经出现的形态学变化(髓鞘神经纤维的损失、轴突萎缩和施万细胞增生)得到显著改善。特别地,通过量化有髓鞘神经和无髓鞘神经的比率,可以确定与对照组小鼠相比,给药化合物84的组的所述量化比率显著增加(图2(c)和(d))。作为大小分布的分析结果,可以看出与正常组(wt-veh)相比,对照组(c22-veh)显示髓鞘神经的直径大小降低,而与对照组相比,给药化合物84的组显示髓鞘神经直径的大小增加(图2(a))。因此,可以看出,化合物84减轻已经在cmt小鼠(c22)中观察到的轴突萎缩和髓鞘神经的损失。化合物84对cmt病的治疗作用可从本测试中鉴定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1