药物组合物的制作方法
关联申请的参考本专利申请主张基于在2018年11月27日提出的日本申请特愿2018-221650号的优先权,该日本申请的全部公开内容通过引用而作为本发明的公开的一部分。本发明涉及药物组合物,更详细而言,涉及含有甲基巴多索隆或其药学上可接受的盐的药物组合物。
背景技术:
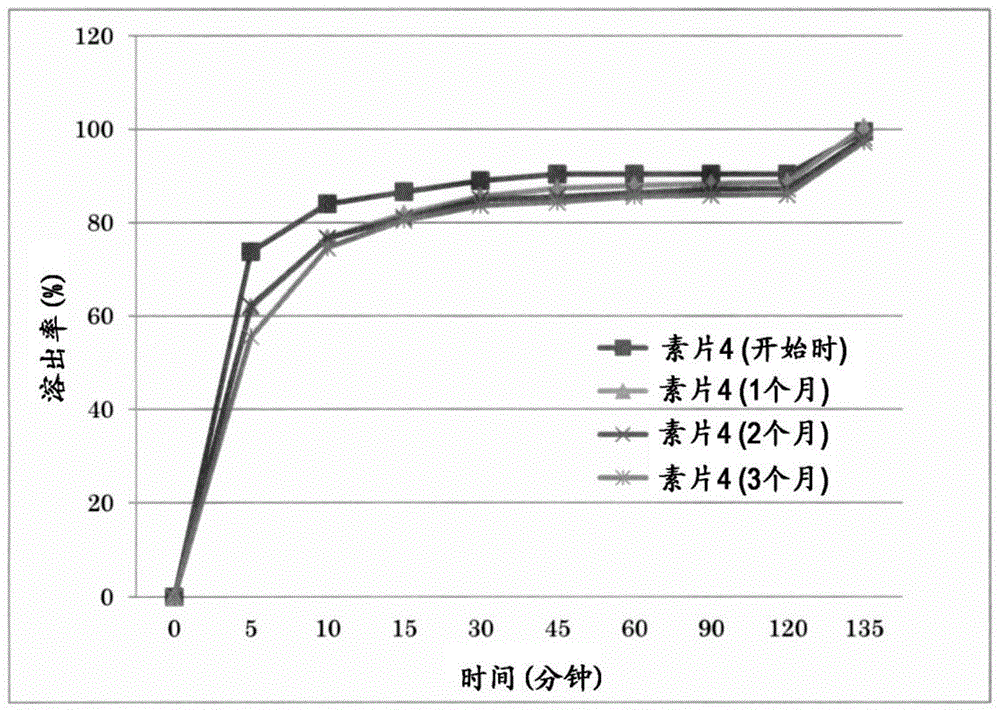
:作为合成萜类化合物,下式表示的甲基巴多索隆(2-氰基-3,12-二氧代齐墩果-1,9(11)-二烯-28-酸甲酯(methyl2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oate))(以下,有时也称为“化合物a”)是已知的(参见专利文献1),另外还已知该甲基巴多索隆具有多晶型形态(参见专利文献2)。[化学式1]已明确,甲基巴多索隆是将转录因子nrf2(其在体内的应激防御反应中发挥核心作用)激活的低分子化合物,具有广泛的抗氧化应激及抗炎症作用,在临床前研究及人类临床试验中,具有有效的抗炎症作用及抗肿瘤作用。还已知甲基巴多索隆尤其对进展期癌患者显示出显著的抗癌活性,此外具有改善由2型糖尿病导致的慢性肾病的罹患患者的肾脏功能、胰岛素抵抗性、血糖控制的测定值、及全身性循环器官疾病的能力(参见专利文献3)。另外,临床试验表明,通过甲基巴多索隆的施予而显著地改善了egfr值(肾功能的指标)(参见非专利文献1、非专利文献2、及非专利文献3)。虽然已进行了用于设计适合甲基巴多索隆的药物组合物的研究(参见专利文献4),但仍在寻求甲基巴多索隆的进一步稳定化。现有技术文献专利文献专利文献1:wo1999/65478号公报专利文献2:wo2009/023232号公报专利文献3:wo2009/089545号公报专利文献4:wo2010/093944号公报非专利文献非专利文献1:nengljmed,2013,369,p2492-2503非专利文献2:amjnephrol,2011,33,p469-476非专利文献3:nengljmed,2011,p327-336技术实现要素:发明要解决的课题本发明提供含有甲基巴多索隆或其药理学上可接受的盐、可接受作为医药品的稳定的药物组合物等。用于解决课题的手段本发明涉及以下的(1)~(26)。(1)药物组合物,其含有甲基巴多索隆或其药学上可接受的盐、崩解剂和粘合剂,所述药物组合物具有包衣被膜。(2)如(1)所述的药物组合物,其中,崩解剂为选自由交联聚维酮、交联羧甲基纤维素钠、低取代度羟丙基纤维素、羧甲基纤维素、羧甲基纤维素钙、羧甲基淀粉钠、部分α化淀粉、及淀粉组成的组中的1种以上。(3)如(2)所述的药物组合物,其中,崩解剂为选自由交联聚维酮、交联羧甲基纤维素钠、及低取代度羟丙基纤维素组成的组中的1种以上。(4)如(1)~(3)中任一项所述的药物组合物,其中,相对于药物组合物100重量份而言,包含0.1~20重量份的崩解剂。(5)如(1)~(4)中任一项所述的药物组合物,其中,粘合剂为选自由羟丙基纤维素、甲基纤维素、羟丙甲纤维素、羧甲基纤维素、羧甲基乙基纤维素、羟乙基纤维素、乙酸羟丙基甲基纤维素琥珀酸酯、羟丙基甲基纤维素邻苯二甲酸酯、羟丙基淀粉、羧基乙烯基聚合物、聚乙烯吡咯烷酮、聚乙烯吡咯烷酮乙酸乙烯酯共聚物、聚乙烯醇、甲基丙烯酸共聚物、聚乙二醇、淀粉、明胶、糊精、普鲁兰多糖、琼脂、及阿拉伯胶组成的组中的1种以上。(6)如(5)所述的药物组合物,其中,粘合剂为选自由羟丙基纤维素、羟丙甲纤维素、聚乙烯醇、及聚乙烯吡咯烷酮组成的组中的1种以上。(7)如(1)~(6)中任一项所述的药物组合物,其中,相对于药物组合物100重量份而言,包含0.1~30重量份的粘合剂。(8)如(1)~(7)中任一项所述的药物组合物,其还含有稳定剂。(9)如(8)所述的药物组合物,其中,稳定剂为有机酸。(10)如(9)所述的药物组合物,其中,有机酸为富马酸及/或苹果酸。(11)如(1)~(10)中任一项所述的药物组合物,其中,包衣被膜包含选自由水溶性聚合物、乳糖、白糖、甘露醇、氧化钛、滑石、碳酸钙、及三乙酸甘油酯组成的组中的1种以上的包衣剂。(12)如(11)所述的药物组合物,其中,水溶性聚合物为选自由聚乙二醇、聚乙烯醇聚乙二醇接枝共聚物、聚乙烯吡咯烷酮、羟丙甲纤维素、羟丙基纤维素、聚乙烯醇、及聚乙烯醇丙烯酸甲基丙烯酸甲酯共聚物组成的组中的1种以上。(13)如(11)或(12)所述的药物组合物,其中,包衣被膜还含有着色剂。(14)如(13)所述的药物组合物,其中,着色剂包含选自由黄色三氧化二铁、氧化铁、及氧化钛组成的组中的1种以上。(15)如(1)~(14)中任一项所述的药物组合物,其中,相对于包衣被膜100重量份而言,包含0.1~100重量份的包衣剂。(16)如(1)~(15)中任一项所述的药物组合物,其还含有光泽剂。(17)如(16)所述的药物组合物,其中,光泽剂为巴西棕榈蜡及/或硬脂酸镁。(18)如(1)~(17)中任一项所述的药物组合物,其中,相对于药物组合物100重量份而言,包含0.1~20重量份的甲基巴多索隆或其药学上可接受的盐,相对于药物组合物100重量份而言,包含3~20重量份的粘合剂,相对于药物组合物100重量份而言,包含0.1~15重量份的崩解剂,相对于包衣被膜100重量份而言,包含50~90重量份的包衣剂。(19)如(1)~(18)中任一项所述的药物组合物,其中,甲基巴多索隆为非晶质。(20)如(1)~(19)中任一项所述的药物组合物,其为固态制剂。(21)如(20)所述的药物组合物,其中,固态制剂为片剂。(22)泡罩包装品,其包含(1)~(21)中任一项所述的药物组合物、以及将聚合物层压而成的膜及铝箔。(23)如(22)所述的泡罩包装品,其中,将聚合物层压而成的膜为将选自聚丙烯、聚氯乙烯、聚偏二氯乙烯、及聚氯三氟乙烯中的1种以上的聚合物层压而成的膜。(24)药物包装品,其是包装体中封入有(22)或(23)所述的泡罩包装品而成的。(25)如(24)所述的药物包装品,其中,包装体为铝袋。(26)如(24)或(25)所述的药物包装品,其中,在包装体内还封入有脱氧剂及/或干燥剂。根据本发明,通过制成含有甲基巴多索隆或其药理学上可接受的盐、崩解剂和粘合剂、且具有包衣被膜的药物组合物,从而能够提高甲基巴多索隆或其药理学上可接受的盐的稳定性。附图说明[图1]为对素片4在保存前(开始时)、1个月后(1个月)、2个月后(2个月)、及3个月后(3个月)的经时溶出率(%)进行比较的图。纵轴表示溶出率(%),横轴表示时间(分钟)。[图2]为对素片5在保存前(开始时)、1个月后(1个月)、2个月后(2个月)、及3个月后(3个月)的经时溶出率(%)进行比较的图。纵轴表示溶出率(%),横轴表示时间(分钟)。[图3]为对素片6在保存前(开始时)、1个月后(1个月)、2个月后(2个月)、及3个月后(3个月)的经时溶出率(%)进行比较的图。纵轴表示溶出率(%),横轴表示时间(分钟)。[图4]为对素片7在保存前(开始时)、1个月后(1个月)、2个月后(2个月)、及3个月后(3个月)的经时溶出率(%)进行比较的图。纵轴表示溶出率(%),横轴表示时间(分钟)。具体实施方式本发明的药物组合物为含有甲基巴多索隆(以下,有时也称为“化合物a”)或其药学上可接受的盐、粘合剂和崩解剂、且具有包衣被膜的药物组合物。化合物a的化学结构如上文所述,可以通过国际公开第1999/65478号公报(专利文献1)中公开的方法、或遵照其的方法来制造。本发明中,所谓“药学上可接受”,是指对于制备通常安全、无毒、且在生物学及其他方面均无不良的药学组合物有用,并且不仅包括在针对人的药物用途中被接受,还包括在兽医学用途中被接受。本发明中,所谓“药学上可接受的盐”,表示上述所定义的药学上可接受且具有所期望的药理活性的盐。作为这样的盐,可举出与例如盐酸、氢溴酸、硫酸、硝酸、及磷酸等无机酸、或者例如马来酸、甲磺酸、草酸等有机酸形成的酸加成盐。作为药学上可接受的盐,也可举出存在的酸性质子能够与无机碱或有机碱反应时可形成的碱加成盐。作为药学上可接受的无机碱,可举出氢氧化钠、碳酸钠、氢氧化钾、氢氧化铝、及氢氧化钙。作为药学上可接受的有机碱,可举出乙醇胺、二乙醇胺、三乙醇胺、氨丁三醇、及n-甲基葡糖胺等。作为化合物a的药学上可接受的盐,可举出钠盐、钾盐等碱金属盐、钙盐、镁盐等碱土金属盐、胺盐等有机碱盐等。本发明的化合物a或其药学上可接受的盐包括其分子内盐、加成物、它们的溶剂合物、或水合物等中的任意物质。化合物a或其药学上可接受的盐中,也有可存在几何异构体、光学异构体等立体异构体、互变异构体等的情况,本发明包括包含上述在内的所有可能的异构体及它们的混合物。化合物a或其药学上可接受的盐中的各原子的一部分或全部可以被各自对应的同位素原子取代,本发明也包括这些经同位素原子取代的衍生物。本发明的化合物a或其药学上可接受的盐也包括化合物a的活性代谢物(作为活性代谢物,例如可举出各种结合物等)或其药学上可接受的盐。本发明的药物组合物中的化合物a或其药学上可接受的盐的含量没有特别限定,相对于药物组合物100重量份而言,优选为0.1~20重量份,更优选为1~20重量份,进一步优选为2~15重量份,特别优选为2~10重量份。化合物a可以使用晶体(形态a)、非晶质(形态b)或它们的混合物中的任一者,根据本发明的优选方式,化合物a主要为非晶质(形态b),相对于100重量份的化合物a而言,优选包含50重量份~100重量份的非晶质(形态b),更优选包含80重量份~99.9重量份,进一步优选包含95重量份~99重量份。根据本发明的更优选方式,化合物a为玻璃质基质中的非晶质的固体分散物,例如,为使化合物a与甲基丙烯酸共聚物的混合物的溶液或悬浮液喷雾干燥而得到的生成物。这样的固体分散物优选可举出将化合物a与甲基丙烯酸共聚物以4:6的重量比例混合而成的混合物,更优选可举出使化合物a与甲基丙烯酸共聚物的该混合物喷雾干燥而得到的生成物。为了得到非晶质化合物a的固体分散物,可以使用各种分取技术来生成。作为合适的非晶质化合物a的固体分散物的生成方法,可举出以往的各种热方法(例如,热熔挤出法)、溶剂法、及热/溶剂法(例如,粒剂的喷雾干燥或流动浸渍法)。关于固体分散体的制备方法,根据“水难溶性药物的经口制剂化技术最前线”(cmc出版,2016年)(参考文献1),从非晶质药物在固体分散体中的稳定性的观点考虑,固体分散体的玻璃化转变点越高越好。此外记载了,从制造上的观点考虑,优选喷雾干燥时的排气温度为固体分散体的玻璃化转变点以下(参见参考文献1的195页)。另外,作为基于喷雾干燥法的固体分散体中可使用的代表性溶剂,可举出水、甲醇、乙醇、丙酮、二氯甲烷等,根据药物和载体,从这些之中选择合适的溶剂。记载了:药物及载体以高浓度溶解时,能够提高制造效率,因此喷雾液的药物浓度优选为50mg/ml以上(参见参考文献1的196页)。本发明的药物组合物中,典型而言,包含治疗上有效的量的化合物a或其药学上可接受的盐。此处,所谓“治疗上有效的”量,是指向患者施予本发明的药物组合物时可获得所期望的药理效果的量。通常,治疗上有效的量可以参考患者的临床参数,按照经验来确定。本发明的药物组合物中含有的崩解剂只要是作为药物可使用的物质即可,没有特别限定,例如,可举出交联聚维酮等pvp系崩解剂;交联羧甲基纤维素钠、低取代度羟丙基纤维素、羧甲基纤维素、羧甲基纤维素钙等纤维素系崩解剂;羧甲基淀粉钠、部分α化淀粉、淀粉等淀粉系崩解剂,优选为选自由交联聚维酮、交联羧甲基纤维素钠、低取代度羟丙基纤维素、羧甲基纤维素、羧甲基纤维素钙、羧甲基淀粉钠、部分α化淀粉、及淀粉组成的组中的1种以上,更优选为选自由pvp系崩解剂及纤维素系崩解剂组成的组中的1种以上,进一步优选为选自由交联聚维酮、交联羧甲基纤维素钠、及低取代度羟丙基纤维素组成的组中的1种以上,进一步优选为交联羧甲基纤维素钠或低取代度羟丙基纤维素,特别优选为低取代度羟丙基纤维素。此处,低取代度羟丙基纤维素为纤维素的低取代度羟丙基醚,低取代度羟丙基纤维素干燥后进行定量时,含有5.0~16.0%的羟基丙氧基(-oc3h6oh:75.09)。本发明的药物组合物中的崩解剂的含量只要是作为药物可使用的量即可,没有特别限定,相对于药物组合物100重量份而言,优选包含0.1重量份~20重量份,更优选包含0.1重量份~18重量份,进一步优选包含0.1重量份~15重量份。本发明的药物组合物中含有的粘合剂只要是作为药物可使用的物质即可,没有特别限定,优选为选自由羟丙基纤维素、羟丙甲纤维素(羟丙基甲基纤维素)、甲基纤维素、羧甲基纤维素、羧甲基乙基纤维素、羟乙基纤维素、乙酸羟丙基甲基纤维素琥珀酸酯、羟丙基甲基纤维素邻苯二甲酸酯、羟丙基淀粉、羧基乙烯基聚合物、聚乙烯吡咯烷酮、聚乙烯吡咯烷酮乙酸乙烯酯共聚物、聚乙烯醇、甲基丙烯酸共聚物、聚乙二醇、淀粉(更优选为玉米淀粉、马铃薯淀粉)、明胶、糊精、普鲁兰多糖、琼脂、及阿拉伯胶组成的组中的1种以上,更优选为选自由羟丙基纤维素、羟丙甲纤维素、聚乙烯醇、及聚乙烯吡咯烷酮组成的组中的1种以上,进一步优选为羟丙基纤维素或羟丙甲纤维素,特别优选为羟丙甲纤维素。本发明的药物组合物中的粘合剂的含量只要是作为药物可使用的量即可,没有特别限定,相对于药物组合物100重量份而言,优选包含0.1重量份~30重量份,更优选包含0.5重量份~25重量份,进一步优选包含3重量份~20重量份。本发明的药物组合物除了包含化合物a、崩解剂、及粘合剂以外,还可以包含作为药物可使用的其他添加物,例如,可以包含药物制剂中使用的选自由稳定剂、赋型剂、润滑剂、着色剂、流动剂及光泽剂组成的组中的1种以上的添加物。本说明书中的稳定剂、赋型剂、润滑剂、着色剂、流动剂、及光泽剂不限于各自记载的用途(功能),也可用于其他用途(机能)(例如,将粘合剂用作赋型剂、将赋型剂用作粘合剂等)。本发明的药物组合物中含有的稳定剂只要是作为药物可使用的物质即可,没有特别限定,例如,可举出有机酸、碳酸钙等,优选为有机酸。作为本发明的药物组合物中含有的有机酸,例如,可举出富马酸、苹果酸、柠檬酸、柠檬酸水合物、柠檬酸酐、琥珀酸、己二酸、酒石酸、马来酸等,优选为富马酸及/或苹果酸,更优选为富马酸。通过使本发明的药物组合物含有富马酸及/或苹果酸,能够制成类似物质总量少、稳定性更高的良好制剂。本发明的药物组合物中的稳定剂(优选为有机酸)的含量只要是作为药物可使用的量即可,没有特别限定,相对于药物组合物100重量份而言,优选包含0.01重量份~30重量份的稳定剂,更优选包含0.05重量份~10重量份,进一步优选包含0.1重量份~5重量份。本发明的药物组合物中含有的赋型剂只要是作为药物可使用的物质即可,没有特别限定,例如,可举出糖、糖醇、结晶纤维素、硅酸处理结晶纤维素(硅酸化结晶纤维素)、无机盐等,优选为乳糖、白糖、麦芽糖、蔗糖、甘露醇、山梨糖醇、赤藓糖醇、麦芽糖醇、木糖醇、葡萄糖、结晶纤维素、硅酸处理结晶纤维素、磷酸一氢钙、磷酸二氢钙、磷酸二氢钠、磷酸钙,可以将2种以上的这些赋型剂组合使用,更优选为乳糖(优选为、乳糖水合物)、甘露醇、硅酸处理结晶纤维素,可以将2种以上的这些赋型剂组合使用。本发明的药物组合物中的赋型剂的含量只要是作为药物可使用的量即可,没有特别限定,相对于药物组合物100重量份而言,优选包含0.1重量份~99.9重量份的赋型剂,更优选包含1重量份~95重量份,进一步优选包含10重量份~90重量份。本发明的药物组合物中含有的润滑剂只要是作为药物可使用的物质即可,没有特别限定,例如,优选硬脂酸镁、硬脂酸钙、月桂基硫酸钠、滑石、单硬脂酸甘油酯、轻质硅酸酐、富马酸硬脂酸钠、蔗糖脂肪酸酯类(例如蔗糖硬脂酸酯、蔗糖棕榈酸酯、蔗糖油酸酯、蔗糖月桂酸酯等),可以将2种以上的这些润滑剂组合使用。本发明的药物组合物中的润滑剂的含量只要是作为药物可使用的量即可,没有特别限定,相对于药物组合物100重量份而言,优选包含0.05重量份~10重量份,更优选包含0.1重量份~5重量份,进一步优选包含0.5重量份~3重量份。本发明的药物组合物中含有的着色剂只要是作为药物可使用的物质即可,没有特别限定,优选包含选自由黄色三氧化二铁、氧化钛、滑石、三氧化二铁、黑氧化铁、氧化铁、铜叶绿素、铜叶绿酸钠、炭黑、药用炭、食用色素、甘草萃取物、绿茶末、核黄素、丁酸核黄素、核黄素磷酸钠、及肉豆蔻酸辛基十二烷基酯组成的组中的1种以上,更优选包含选自由黄色三氧化二铁、氧化铁、及氧化钛组成的组中的1种以上。本发明的药物组合物中的着色剂的含量只要是作为药物可使用的量即可,没有特别限定,相对于药物组合物100重量份而言,例如,可以包含0.01重量份~5重量份。本发明的药物组合物中含有的流动剂只要是作为药物可使用的物质即可,没有特别限定,例如,可举出含水二氧化硅、轻质硅酸酐、合成硅酸铝、合成水滑石、干燥氢氧化铝凝胶、高岭土、硅酸钙、硅铝酸镁、滑石等,可以将2种以上的这些流动剂组合使用。本发明的药物组合物中的流动剂的含量只要是作为药物可使用的量即可,没有特别限定,相对于药物组合物100重量份而言,例如,可以包含0.01重量份~5重量份。本发明的药物组合物中含有的光泽剂只要是作为药物可使用的物质即可,没有特别限定,例如,可举出巴西棕榈蜡、虫胶、蜂蜡、硬化油、硬脂酸镁等,可以将2种以上的这些光泽剂组合使用,优选为巴西棕榈蜡及/或硬脂酸镁。本发明的药物组合物中含有的光泽剂的含量只要是作为药物可使用的量即可,没有特别限定,相对于药物组合物100重量份而言,优选包含0.0001重量份~100重量份,更优选包含0.001重量份~10重量份,进一步优选包含0.01重量份~1重量份。本发明的药物组合物具有包衣被膜。该包衣被膜例如可通过对含有化合物a或其药学上可接受的盐、崩解剂和粘合剂的素制剂(例如,素片)进行包衣处理而赋予。该包衣处理例如可通过下述方式进行:利用喷雾包衣法等,将含有包衣剂的包衣液喷雾至含有化合物a的素制剂(例如,素片)。该包衣剂在包衣液中溶解、悬浮、分散等而使用,作为构成该包衣液的溶剂,例如,可举出水、甲醇、乙醇等醇类等,更优选水。本发明的药物组合物的包衣被膜没有特别限定,优选包含选自由水溶性聚合物、乳糖、白糖、甘露醇、氧化钛、滑石、碳酸钙、及三乙酸甘油酯组成的组中的1种以上的包衣剂,更优选包含三乙酸甘油酯。水溶性聚合物只要是作为药物可使用的物质即可,没有特别限定,优选为选自由聚乙二醇、聚乙烯醇聚乙二醇接枝共聚物、聚乙烯吡咯烷酮、羟丙甲纤维素、羟丙基纤维素、聚乙烯醇、及聚乙烯醇丙烯酸甲基丙烯酸甲酯共聚物组成的组中的1种以上。本发明的药物组合物的包衣被膜优选还含有着色剂,该着色剂更优选包含选自由黄色三氧化二铁、氧化铁、及氧化钛组成的组中的1种以上。包衣被膜中的包衣剂没有特别限定,相对于包衣被膜100重量份而言,优选包含0.1重量份~100重量份,更优选包含1重量份~95重量份,进一步优选包含50重量份~90重量份。作为该包衣处理中使用的包衣液的使用量,只要为能够对药物组合物赋予光稳定性等的量即可,没有特别限定,相对于素制剂100重量份而言,包衣被膜(剂皮)在干燥状态下优选为0.01重量份~50重量份,更优选为0.05重量份~30重量份,进一步优选为0.1重量份~20重量份。本发明的药物组合物优选还含有光泽剂,更优选含有巴西棕榈蜡及/或硬脂酸镁作为光泽剂。本发明的药物组合物优选为经口用制剂,可以进一步添加矫味剂等。本发明的药物组合物的形状没有特别限定,优选为固态制剂,更优选具有片剂、散剂、细粒剂、颗粒剂、胶囊剂或干糖浆的形状,进一步优选为片剂。通过使本发明的药物组合物的形状为片剂,具有下述优点:容易服用,具有充分的物理强度,与胶囊剂相比不易崩塌。作为本发明的药物组合物为片剂时的制造方法,将含有作为有效成分的甲基巴多索隆或其药学上可接受的盐、崩解剂和粘合剂的混合物进行造粒、压片,并经历薄膜包衣工序而制造。本发明的药物组合物的制造方法没有特别限定,例如,可以利用压缩成型等制剂学
技术领域:
中通常使用的方法来制造,例如,可以利用直接压缩法、干式颗粒压缩法(辊压缩成型法、碎渣压片法等)等来制造。在任意情况下均优选下述方法:例如,将化合物a或其药学上可接受的盐、及崩解剂、粘合剂等添加物混合,并根据需要进行造粒及整粒。接着,例如在制备片剂时,可举出使用压缩压片机将得到的干燥造粒物形成片剂。压片压强可以从例如300~3000kg/cm2的范围中适当选择。片剂尺寸没有特别限定,优选每1片的重量为20~3000mg、片剂的直径为5~15mm。对素片实施包衣处理时,可以用溶解/分散有包衣剂的溶液/分散液将得到的素片包衣,形成剂皮。作为溶解/分散该包衣剂的溶剂,例如,可举出水、乙醇、异丙醇、它们的混合溶剂等,这些之中,优选水。包衣例如使用以往型的盘型包衣机、通气式包衣机、流动层型包衣装置、转动流动型包衣装置等来进行。作为本发明的药物组合物的优选方式,为下述药物组合物(优选为片剂):相对于药物组合物100重量份而言,包含0.1~20重量份的化合物a或其药学上可接受的盐,相对于药物组合物100重量份而言,包含0.1~30重量份的粘合剂(优选为羟丙甲纤维素),相对于药物组合物100重量份而言,包含0.1~20重量份的崩解剂(优选为低取代度羟丙基纤维素),相对于包衣被膜100重量份而言,包含0.1~100重量份的包衣剂(优选在包衣被膜中包含选自由羟丙甲纤维素、乳糖、及三乙酸甘油酯组成的组中的一种以上的包衣剂)。作为本发明的药物组合物的另一优选方式,为下述药物组合物(优选为片剂):相对于药物组合物100重量份而言,包含0.1~20重量份的化合物a或其药学上可接受的盐,相对于药物组合物100重量份而言,包含0.1~30重量份的粘合剂(优选为羟丙甲纤维素),相对于药物组合物100重量份而言,包含0.1~20重量份的崩解剂(优选为低取代度羟丙基纤维素),相对于药物组合物100重量份而言,包含0.0001~10重量份的光泽剂(优选为巴西棕榈蜡及/或硬脂酸镁),相对于包衣被膜100重量份而言,包含0.1~100重量份的包衣剂(优选在包衣被膜中包含选自由羟丙甲纤维素、乳糖、及三乙酸甘油酯组成的组中的一种以上的包衣剂)。作为本发明的药物组合物的另一优选方式,为下述药物组合物(优选为片剂):相对于药物组合物100重量份而言,包含0.1~20重量份的化合物a或其药学上可接受的盐,相对于药物组合物100重量份而言,包含0.1~30重量份的粘合剂(优选为羟丙甲纤维素),相对于药物组合物100重量份而言,包含0.1~20重量份的崩解剂(优选为低取代度羟丙基纤维素),相对于药物组合物100重量份而言,包含0.01~30重量份的稳定剂(优选为有机酸,更优选为富马酸),相对于药物组合物100重量份而言,包含0.001~10重量份的光泽剂(优选为、巴西棕榈蜡及/或硬脂酸镁),相对于包衣被膜100重量份而言,包含0.1~100重量份的包衣剂(优选在包衣被膜中包含选自由羟丙甲纤维素、乳糖、及三乙酸甘油酯组成的组中的一种以上的包衣剂)。作为本发明的药物组合物的更优选的方式,为下述药物组合物(优选为片剂):相对于药物组合物100重量份而言,包含0.1~20重量份的化合物a或其药学上可接受的盐,相对于药物组合物100重量份而言,包含0.5~25重量份的粘合剂(优选为羟丙甲纤维素),相对于药物组合物100重量份而言,包含0.1~18重量份的崩解剂(优选为低取代度羟丙基纤维素),相对于包衣被膜100重量份而言,包含1~95重量份的包衣剂(优选在包衣被膜中包含选自由羟丙甲纤维素、乳糖、及三乙酸甘油酯组成的组中的一种以上的包衣剂)。作为本发明的药物组合物的另一更优选方式,为下述药物组合物(优选为片剂):相对于药物组合物100重量份而言,包含0.1~20重量份的化合物a或其药学上可接受的盐,相对于药物组合物100重量份而言,包含0.5~25重量份的粘合剂(优选为羟丙甲纤维素),相对于药物组合物100重量份而言,包含0.1~18重量份的崩解剂(优选为低取代度羟丙基纤维素),相对于药物组合物100重量份而言,包含0.001~10重量份的光泽剂(优选为巴西棕榈蜡及/或硬脂酸镁),相对于包衣被膜100重量份而言,包含1~95重量份的包衣剂(优选在包衣被膜中包含选自由羟丙甲纤维素、乳糖、及三乙酸甘油酯组成的组中的一种以上的包衣剂)。作为本发明的药物组合物的另一更优选方式,为下述药物组合物(优选为片剂):相对于药物组合物100重量份而言,包含0.1~20重量份的化合物a或其药学上可接受的盐,相对于药物组合物100重量份而言,包含0.5~25重量份的粘合剂(优选为羟丙甲纤维素),相对于药物组合物100重量份而言,包含0.1~18重量份的崩解剂(优选为低取代度羟丙基纤维素),相对于药物组合物100重量份而言,包含0.05~10重量份的稳定剂(优选为有机酸,更优选为富马酸),相对于药物组合物100重量份而言,包含0.001~10重量份的光泽剂(优选为巴西棕榈蜡及/或硬脂酸镁),相对于包衣被膜100重量份而言,包含1~95重量份的包衣剂(优选在包衣被膜中包含选自由羟丙甲纤维素、乳糖、及三乙酸甘油酯组成的组中的一种以上的包衣剂)。作为本发明的药物组合物的又一优选方式,为下述药物组合物(优选为片剂):相对于药物组合物100重量份而言,包含0.1~20重量份的化合物a或其药学上可接受的盐,相对于药物组合物100重量份而言,包含3~20重量份的粘合剂(优选为羟丙甲纤维素),相对于药物组合物100重量份而言,包含0.1~15重量份的崩解剂(优选为低取代度羟丙基纤维素),相对于包衣被膜100重量份而言,包含50~90重量份的包衣剂(优选在包衣被膜中包含选自由羟丙甲纤维素、乳糖、及三乙酸甘油酯组成的组中的一种以上的包衣剂)。作为本发明的药物组合物的另一更优选方式,为下述药物组合物(优选为片剂):相对于药物组合物100重量份而言,包含0.1~20重量份的化合物a或其药学上可接受的盐,相对于药物组合物100重量份而言,包含3~20重量份的粘合剂(优选为羟丙甲纤维素),相对于药物组合物100重量份而言,包含0.1~15重量份的崩解剂(优选为低取代度羟丙基纤维素),相对于药物组合物100重量份而言,包含0.01~1重量份的光泽剂(优选为巴西棕榈蜡及/或硬脂酸镁),相对于包衣被膜100重量份而言,包含50~90重量份的包衣剂(优选在包衣被膜中包含选自由羟丙甲纤维素、乳糖、及三乙酸甘油酯组成的组中的一种以上的包衣剂)。作为本发明的药物组合物的另一更优选方式,为下述药物组合物(优选为片剂):相对于药物组合物100重量份而言,包含0.1~20重量份的化合物a或其药学上可接受的盐,相对于药物组合物100重量份而言,包含3~20重量份的粘合剂(优选为羟丙甲纤维素),相对于药物组合物100重量份而言,包含0.1~15重量份的崩解剂(优选为低取代度羟丙基纤维素),相对于药物组合物100重量份而言,包含0.1~5重量份的稳定剂(优选为有机酸,更优选为富马酸),相对于药物组合物100重量份而言,包含0.01~1重量份的光泽剂(优选为巴西棕榈蜡及/或硬脂酸镁),相对于包衣被膜100重量份而言,包含50~90重量份的包衣剂(优选在包衣被膜中包含选自由羟丙甲纤维素、乳糖、及三乙酸甘油酯组成的组中的一种以上的包衣剂)。根据本发明的另一方式,提供提高甲基巴多索隆或其药学上可接受的盐的稳定性的方法,其特征在于,向甲基巴多索隆或其药学上可接受的盐中加入崩解剂及粘合剂,并进行包衣被膜处理。另外,根据本发明的另一优选方式,提供降低甲基巴多索隆或其药学上可接受的盐的类似物质总量的方法,其特征在于,向甲基巴多索隆或其药学上可接受的盐中加入崩解剂及粘合剂,并进行包衣被膜处理。这样的方法中,与上述的本发明的药物组合物同样地,可以加入除化合物a、崩解剂、及粘合剂以外的作为药物可使用的其他添加物,例如,可以加入药物制剂中使用的选自由稳定剂、赋型剂、润滑剂、着色剂、流动剂、及光泽剂组成的组中的1种以上的添加物。本发明的药物组合物可以收纳于瓶包装、泡罩包装、铝袋等气密容器中而提供。作为气密容器的原材料,只要能够抑制来自外部的水分的侵入即可,没有特别限定,可以使用医药品领域中出于不耐受水分的内容物的防湿等目的而使用的原材料,其中,特别优选泡罩包装品。本发明的泡罩包装品包含上述含有化合物a等的药物组合物、以及将聚合物层压而成的膜及铝箔。该将聚合物层压而成的膜只要为泡罩包装品中通常可使用的膜即可,没有特别限定,优选为将聚丙烯、聚氯乙烯、聚偏二氯乙烯、聚氯三氟乙烯(aclar(商标))等聚合物层压而成的膜等,更优选聚丙烯或硬质氯乙烯。作为该铝箔,只要为泡罩包装品中可使用的铝箔即可,没有特别限定,可以为通常使用的铝箔,优选为降低了粘接剂中的三聚氰胺树脂量的铝箔。本发明的泡罩包装品的制造方法没有特别限定,使用通常所用的泡罩包装机,在该将聚合物层压而成的膜中成型出泡孔(pocket),投入片剂,通过热等将铝箔密封,由此得到。本发明的药物包装品是将上述泡罩包装品封入包装体而成的。作为该包装体,只要为药物包装品中通常可使用的包装体即可,没有特别限定,优选为铝袋等。该药物包装品中,可以同时封入通常的封入药物包装品中的物质,优选将脱氧剂及/或干燥剂与上述泡罩包装品同时封入。本发明的药物包装品可以通过下述方式制造:将以上述方式制造的泡罩包装品等封入铝袋等包装体中,使用热密封机等进行密封。实施例以下,利用实施例,更具体地说明本发明,但本发明的技术范围不限于这些示例。薄膜包衣片的制造例配方例1:使用混合机(tbm-25,株式会社德寿工作所制),将含有40重量%化合物a(甲基巴多索隆)的固体分散体(利用喷雾干燥法制造,以下的“固体分散体”也同样地制造)480.8g、硅酸处理结晶纤维素(prosolv,jrspharma)1976.9g、乳糖水合物(日本药典)1715.4g、羟丙甲纤维素(tc-5e,信越化学工业株式会社制)250.0g、低取代度羟丙基纤维素(l-hpc,信越化学工业株式会社制)500.0g、轻质硅酸酐(adsolider101,freundcorporation株式会社制)38.5g进行混合。向该混合物中加入硬脂酸镁19.2g,进一步混合。使用辊压机(ccs-220,株式会社powrex制),对该混合物进行干式造粒及整粒。加入相对于得到的整粒品而言为0.38重量%的量的硬脂酸镁,进行混合,得到压片用混合品。使用旋转压片机(ht-ap15,畑铁工株式会社制)进行制片(重量:130mg,片剂的形状:圆形(直径7mm)),由此得到素片(配方例1)(参见表1)。将被膜剂混合品分散于水中,制备了固态成分浓度10重量%的包衣液(被膜剂混合品的组成示于下述表2)。针对素片,使用片剂包衣机(prc-7,株式会社powrex制),以相对于素片100重量份而言剂皮在干燥状态下成为5重量份的方式,喷洒包衣液来进行包衣。散布相对于素片而言成为0.03重量%的量的巴西棕榈蜡,进行上光,由此得到目标片剂。将得到的素片及片剂的组成示于以下的表1及2。[表1]表1:配方例1(素片)的配方[表2]表2:配方例1(素片)的薄膜包衣配方成分1片中(mg)含有率(重量%)羟丙甲纤维素3.9060.0氧化钛1.4622.4乳糖水合物0.6510.0三乙酸甘油酯0.396.0黄色三氧化二铁0.101.5氧化铁0.010.1合计6.50100.0配方例2使用混合机(tbm-25,株式会社德寿工作所制),将含有40重量%化合物a(甲基巴多索隆)的固体分散体(利用喷雾干燥法制造,以下的“固体分散体”也同样地制造)500.0g、硅酸处理结晶纤维素(prosolv,jrspharma)2068.0g、乳糖水合物(日本药典)1796.0g、羟丙甲纤维素(tc-5e,信越化学工业株式会社制)236.1g、低取代度羟丙基纤维素(l-hpc,信越化学工业株式会社制)520.0g、轻质硅酸酐(adsolider101,freundcorporation株式会社制)40.0g进行混合。向该混合物中加入硬脂酸镁20.0g,进一步混合。使用辊压机(ccs-220,株式会社powrex制),对该混合物进行干式造粒及整粒。加入相对于得到的整粒品而言成为0.38重量%的量的硬脂酸镁,进行混合,得到压片用混合品。使用旋转压片机(ht-ap15,畑铁工株式会社制)进行制片(重量:130mg,片剂的形状:圆形(直径7mm)),由此得到含有5mg化合物a的素片(配方例2)(参见表3)。将被膜剂混合品分散于水中,制备了固态成分浓度10重量%的包衣液(被膜剂混合品的组成示于下述表4)。针对素片,使用片剂包衣机(prc-7,株式会社powrex制),以相对于素片100重量份而言剂皮在干燥状态下成为5重量份的方式,喷洒包衣液来进行包衣。散布相对于素片而言成为0.03重量%的量的巴西棕榈蜡,进行上光,由此得到目标片剂。配方例3与5mg片配方例2同样地制备混合品,使用旋转压片机(ht-ap15,畑铁工株式会社制)进行制片(重量:260mg,片剂的形状:圆形(直径9mm)),由此得到含有10mg化合物a的素片(配方例3)(参见表3)。将被膜剂混合品分散于水中,制备了固态成分浓度15重量%的包衣液(被膜剂混合品的组成示于下述表4)。针对素片200g,使用片剂包衣机(drc-200,株式会社powrex制),以相对于素片100重量份而言剂皮在干燥状态下成为4重量份的方式,喷洒包衣液来进行包衣。散布相对于素片而言成为0.03重量%的量的巴西棕榈蜡,进行上光,由此得到目标片剂。配方例4与5mg片配方例2同样地制备混合品,使用旋转压片机(ht-ap15,畑铁工株式会社制),进行制片(重量:390mg,片剂的形状:椭圆形(长径13.5mm,短径7mm)),由此得到含有15mg化合物a的素片(配方例4)(参见表3)。将被膜剂混合品分散于水中,制备了固态成分浓度15重量%的包衣液(被膜剂混合品的组成示于下述表4)。使用片剂包衣机(prc-7,株式会社powrex制),以相对于素片100重量份而言剂皮在干燥状态下成为3.5重量份的方式,喷洒包衣液来进行包衣。散布相对于素片而言成为0.03重量%的量的巴西棕榈蜡,进行上光,由此得到目标片剂。将得到的素片及片剂的组成示于以下的表3及4。[表3]表3:配方例2~4(素片)的配方[表4]表4:用于对配方例2~4(素片)进行薄膜包衣、并且添加光泽剂的配方需要说明的是,上述表4中,使用硬脂酸镁代替巴西棕榈蜡作为光泽剂进行上光,也能够得到目标片剂。素片1~3的共通混合品的混合将含有40重量%化合物a的固体分散体336.5g、硅酸处理结晶纤维素(prosolv,jrspharma)807.7g、轻质硅酸酐13.5g在塑料袋内混合200次,得到含有化合物a的混合品(i)。另外,将硅酸处理结晶纤维素631.9g及轻质硅酸酐11.9g在塑料袋内混合200次,得到混合品(ii)。例1:素片1的制备方法将上述混合品(i)215.0g、乳糖水合物210.5g、羟丙基纤维素(hpc-ssl-sfp,日本曹达株式会社制)19.5g、交联羧甲基纤维素钠(ac-di-sol,fmcbiopolymer制)32.5g在塑料袋内混合200次,进而加入硬脂酸镁(partecklub,merck)2.5g,进一步混合50次。使用干式造粒机(tf-labo,freundcorporation株式会社制),对得到的混合物进行干式造粒。使用整粒机(comil,株式会社powrex制),对得到的干式造粒品进行整粒。将得到的整粒品443.1g、上述混合品(ii)124.6g及交联羧甲基纤维素钠(ac-di-sol,fmcbiopolymer制)30.0g在塑料袋内混合200次,进而加入硬脂酸镁2.3g,混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所)进行制片(重量130mg,片剂的形状:圆形(直径7mm)),由此得到素片1。例2:素片2的制备方法将上述混合品(i)148.9g、乳糖水合物145.7g、羟丙基纤维素(hpc-ssl-sfp,日本曹达株式会社制)13.5g、低取代度羟丙基纤维素(l-hpc,信越化学工业株式会社制)22.5g在塑料袋内混合200次,进而加入硬脂酸镁(partecklub,merck)1.7g,进一步混合50次。使用干式造粒机(tf-labo,freundcorporation株式会社制),对得到的混合物进行干式造粒。使用整粒机(comil,株式会社powrex),对得到的干式造粒品进行整粒。将得到的整粒品295.4g、上述混合品(ii)83.1g及低取代度羟丙基纤维素(l-hpc,信越化学工业株式会社制)20.0g在塑料袋内混合200次,进而加入硬脂酸镁1.5g,混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所)进行制片(重量130mg,片剂的形状:圆形(直径7mm)),由此得到素片2。例3:素片3的制备方法将上述混合品(i)148.9g、乳糖水合物145.7g、羟丙基纤维素(hpc-ssl-sfp、日本曹达株式会社制)13.5g、羟基乙酸淀粉钠(primojel,dfepharma)22.5g在塑料袋内混合200次,进而加入硬脂酸镁(partecklub,merck)1.7g,进一步混合50次。使用干式造粒机(tf-labo,freundcorporation株式会社制),对得到的混合物进行干式造粒。使用整粒机(comil,株式会社powrex制),对得到的干式造粒品进行整粒。将得到的整粒品295.4g、上述混合品(ii)83.1g及羟基乙酸淀粉钠(primojel,dfepharma)20.0g在塑料袋内混合200次,进而加入硬脂酸镁1.5g,混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所制)进行制片(重量130mg,片剂的形状:圆形(直径7mm)),由此得到素片3。试验例1:例1、例2、及例3中得到的素片1~3的稳定性的比较试验甲基巴多索隆的类似物质测定在以下的条件下利用液相色谱进行试验而测定。作为液相色谱用的柱,使用acquityuplchssc18、粒径1.8μm、2.1mm×50mm(waters制)或其同等品,将柱温维持为40℃。作为流动相a,为20mmol/l磷酸二氢钠·柠檬酸缓冲液(ph4.5),作为流动相b,为乙腈。试样溶液使用以甲基巴多索隆的浓度成为100μg/ml的方式用65重量%的乙腈进行稀释而成的溶液。在流速为0.6ml/min的条件下,使用紫外吸光光度计(测定波长:242nm)进行类似物质测定,求出相对于甲基巴多索隆的表示量而言的各类似物质总量(%)。稳定性试验按以下的条件进行。保存条件:30℃,75%rh,1个月保存形态:在打开了盖子的褐色瓶中放入素片1~3[表5]表5:素片1~3的稳定性的比较结果根据上述表5的结果可知,在添加任意崩解剂的情况下,均为类似物质总量少、在稳定性方面优异的制剂(另外,虽未显示数据,但在添加任意崩解剂的情况下,从开始时起的类似物质总量的增加量均少)。将上述三种素片进行比较可知,含有低取代度羟丙基纤维素作为崩解剂的素片为类似物质总量(及增加量)少、综合而言稳定性最优异的制剂。例4:素片4的制备方法将化合物a44.2g、硅酸处理结晶纤维素110.4g、轻质硅酸酐1.8g、乳糖水合物176.9g、羟丙甲纤维素(tc-5e,信越化学工业株式会社制)4.6g在塑料袋内混合200次,进而加入硬脂酸镁(partecklub,merck)1.8g,进一步混合50次。使用干式造粒机(tf-labo,freundcorporation株式会社制),对得到的混合物进行干式造粒。使用整粒机(comil,株式会社powrex制),对得到的干式造粒品进行整粒。将整粒品295.4g、硅酸处理结晶纤维素81.5g、轻质硅酸酐1.5g、及交联羧甲基纤维素钠(ac-di-sol,fmcbiopolymer制)20g在塑料袋内混合200次,进而加入硬脂酸镁1.5g,混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所)进行制片(重量130mg,片剂的形状:圆形(直径7mm)),由此得到素片4。例5:素片5的制备方法将化合物a44.2g、硅酸处理结晶纤维素90.9g、轻质硅酸酐1.8g、乳糖水合物159.6g、羟丙甲纤维素(tc-5e、信越化学工业株式会社制)41.4g在塑料袋内混合200次,进而加入硬脂酸镁(partecklub、merck)1.8g,进一步混合50次。使用干式造粒机(tf-labo,freundcorporation株式会社制),对得到的混合物进行干式造粒。使用整粒机(comil,株式会社powrex制),对得到的干式造粒品进行整粒。将整粒品295.4g、硅酸处理结晶纤维素81.5g、轻质硅酸酐1.5g、交联羧甲基纤维素钠(ac-di-sol、fmcbiopolymer制)20.0g在塑料袋内混合200次,进而加入硬脂酸镁1.5g,混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所)进行制片(重量130mg,片剂的形状:圆形(直径7mm)),由此得到素片5。例6:素片6的制备方法将化合物a44.2g、硅酸处理结晶纤维素98.0g、轻质硅酸酐1.8g、乳糖水合物166.3g、羟丙甲纤维素(tc-5e,信越化学工业株式会社制)4.6g在塑料袋内混合200次,进而加入硬脂酸镁(partecklub,merck)1.8g,进一步混合50次。使用干式造粒机(tf-labo,freundcorporation株式会社制),对得到的混合物进行干式造粒。使用整粒机(comil,株式会社powrex制),对得到的干式造粒品进行整粒。将整粒品275.4g、硅酸处理结晶纤维素81.5g、轻质硅酸酐1.5g、低取代度羟丙基纤维素(l-hpc,信越化学工业株式会社制)40.0g在塑料袋内混合200次,进而加入硬脂酸镁1.5g,混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所)进行制片(重量130mg,片剂的形状:圆形(直径7mm)),由此得到素片6。例7:素片7的制备方法将化合物a44.2g、硅酸处理结晶纤维素78.2g、轻质硅酸酐1.8g、乳糖水合物149.3g、羟丙甲纤维素(tc-5e,信越化学工业株式会社制)41.4g在塑料袋内混合200次,进而加入硬脂酸镁(partecklub,merck)1.8g,进一步混合50次。使用干式造粒机(tf-labo,freundcorporation株式会社制),对得到的混合物进行干式造粒。使用整粒机(comil,株式会社powrex),对得到的干式造粒品进行整粒。将整粒品275.4g、硅酸处理结晶纤维素81.5g、轻质硅酸酐1.5g、低取代度羟丙基纤维素(l-hpc,信越化学工业株式会社制)40.0g在塑料袋内混合200次,进而加入硬脂酸镁1.5g,混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所)进行制片(重量130mg,片剂的形状:圆形(直径7mm)),由此得到素片7。上述素片4~7中的崩解剂及粘合剂的组成如下所示。[表6]表6:素片4~7每1片(130mg)中的崩解剂及粘合剂的组成试验例2:例4、例5、例6、及例7中得到的素片4~7的溶出性的比较试验将得到的素片4~7放入褐色玻璃瓶中,在不加盖的状态下,在温度为30℃、相对湿度为75%的环境下,进行保存前(开始时)、1个月、2个月、及3个月保存。按照日本药典溶出试验第2法(桨法,50rpm)来实施溶出试验。试验液使用含有0.15重量%月桂基硫酸钠的日本药典溶出试验第2液900ml,利用液相色谱,对试验开始5、10、15、30、45、60、90、120、及135分钟时的化合物a的溶出率进行评价。作为柱,使用acquityuplchssc18、粒径1.8μm、2.1mm×50mm(waters制)或其同等品,维持于40℃。作为流动相a,使用20mmol/l磷酸二氢钠·柠檬酸缓冲液(ph4.5),作为流动相b,使用乙腈。在流速0.6ml/min的条件下,使用紫外吸光光度计(测定波长:242nm)进行测定。将保存前(开始时)、1个月后(1个月)、2个月后(2个月)、及3个月后(3个月)的素片4~7的经时溶出率(%)的结果示于图1~4。为根据素片的保存期间的不同而溶出率的偏差小的优异制剂,可知,对于化合物a,在粘合剂存在下,无论使用交联羧甲基纤维素钠及低取代度羟丙基纤维素(l-hpc)中的任一者作为崩解剂,溶出率的偏差均较小,任意素片(素片4~7)均可获得良好的溶出率。另外,若将该二种崩解剂进行比较,相较于交联羧甲基纤维素钠而言,低取代度羟丙基纤维素(l-hpc)的偏差更小,可知对于化合物a,低取代度羟丙基纤维素(l-hpc)是更优选的崩解剂。例8:片剂1的制备方法将含有40重量%化合物a的固体分散体144.2g、硅酸处理结晶纤维素(prosolv,jrspharma)346.2g、乳糖水合物(日本药典)564.2g及轻质硅酸酐5.8g进行混合,得到含有化合物a的混合品。将该混合品494.8g与羟丙基纤维素(hpc-ssl-sfp,日本曹达株式会社制)19.4g在塑料袋内旋转混合200次,进而将硬脂酸镁2.7g在塑料袋内旋转混合50次。使用辊压机(tf-labo,freundcorporation株式会社制),对该混合物进行干式造粒。使用整粒机(comil,株式会社powrex制),对得到的干式造粒品进行整粒。将得到的整粒品480g、硅酸处理结晶纤维素132.5g、轻质硅酸酐2.5g及交联羧甲基纤维素钠(ac-di-sol,fmcbiopolymer制)在塑料袋内进行混合。进而添加硬脂酸镁2.5g,在塑料袋内旋转混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所)进行制片(重量:130mg,片剂的形状:圆形(直径7mm)),由此得到素片。将被膜剂混合品各自分散于水中,制备了包衣液(被膜剂混合品的组成及包衣液的固态成分浓度示于下述表7)。针对素片200g,使用片剂包衣机(drc-200,株式会社powrex制),以相对于素片100重量份而言剂皮在干燥状态下成为5重量份的方式,喷洒各包衣液,进行包衣,由此得到目标片剂1。例9:片剂2的制备方法将含有40重量%化合物a的固体分散体144.2g、硅酸处理结晶纤维素(prosolv,jrspharma)346.2g、乳糖水合物(日本药典)564.2g及轻质硅酸酐5.8g进行混合,得到含有化合物a的混合品。将该混合品494.8g与羟丙基纤维素(hpc-ssl-sfp,日本曹达株式会社制)19.4g在塑料袋内旋转混合200次,进而将硬脂酸镁2.7g在塑料袋内旋转混合50次。使用辊压机(tf-labo,freundcorporation株式会社制),对该混合物进行干式造粒。使用整粒机(comil,株式会社powrex制),对得到的干式造粒品进行整粒。将得到的整粒品480g、硅酸处理结晶纤维素132.5g、轻质硅酸酐2.5g及交联羧甲基纤维素钠(ac-di-sol,fmcbiopolymer制)在塑料袋内进行混合。进而添加硬脂酸镁2.5g,在塑料袋内旋转混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所)进行制片(重量:130mg,片剂的形状:圆形(直径7mm)),由此得到素片。将被膜剂混合品各自分散于水中,制备了包衣液(被膜剂混合品的组成及包衣液的固态成分浓度示于下述表7)。针对素片200g,使用片剂包衣机(drc-200,株式会社powrex制),以相对于素片100重量份而言剂皮在干燥状态下成为5重量份的方式,喷洒各包衣液,进行包衣,由此得到目标片剂2。[表7]表7:薄膜包衣配方试验例3:例8及例9中得到的薄膜包衣片的稳定性的比较甲基巴多索隆的类似物质测定在以下的条件下利用液相色谱进行试验。作为柱,使用acquityuplchssc18、粒径1.8μm、2.1mm×50mm(waters制)或其同等品,维持于40℃。作为流动相a,使用20mmol/l磷酸二氢钠·柠檬酸缓冲液(ph4.5),作为流动相b,使用乙腈。试样溶液使用以化合物的浓度成为100μg/ml的方式用65重量%乙腈进行稀释而成的溶液。在流速为0.6ml/min的条件下,使用紫外吸光光度计(测定波长:242nm)进行类似物质测定,求出相对于甲基巴多索隆的表示量而言的各类似物质总量(%)。稳定性试验按以下的条件进行。保存条件:30℃,75%rh,1个月保存形态:在打开了盖子的褐色瓶中放入片剂1及2[表8]表8:例8及例9中得到的薄膜包衣片(片剂1及2)的稳定性的比较结果根据上述表8的结果可知,在使用任意薄膜包衣配方的情况下,只要为含有化合物a、崩解剂和粘合剂、且具有包衣被膜的片剂,则为类似物质总量少、稳定性高的良好制剂。可知特别是通过使薄膜包衣配方中含有三乙酸甘油酯,能够得到类似物质总量变少、稳定性高的良好制剂。例10:配方例2及素片8的制备方法将含有40重量%化合物a的固体分散体19.2g、硅酸处理结晶纤维素79.5g(prosolv,jrspharma)、乳糖水合物69.1g、羟丙甲纤维素(tc-5e,信越化学工业)9.1g、低取代度羟丙基纤维素(l-hpc,信越化学工业)20g、轻质硅酸酐(adsolider101,freundcorporation)1.5g在塑料袋内进行混合,进而加入硬脂酸镁0.8g(pertecklubmst,merck),进一步进行混合,得到与上述相同的配方例2的混合品。将含有40重量%化合物a的固体分散体19.2g、硅酸处理结晶纤维素78.5g(prosolv,jrspharma)、乳糖水合物68.2g、羟丙甲纤维素(tc-5e,信越化学工业)9.1g、低取代度羟丙基纤维素(l-hpc,信越化学工业)20.0g、轻质硅酸酐(adsolider101,freundcorporation)1.5g、有机酸(稳定剂)2.0g在塑料袋内进行混合,进而加入硬脂酸镁0.8g(pertecklubmst,merck),进一步进行混合,得到含有1重量%有机酸的混合品。使用干式造粒机(tf-labo,freundcorporation),对得到的混合物进行干式造粒。使用整粒机(comil,powrex),对得到的干式造粒品进行整粒。向得到的整粒品中,加入成为0.38重量%的量的硬脂酸镁(pertecklubmst,merck),在塑料袋内混合50次,得到压片用混合品。使用旋转压片机(virgo,菊水制作所)进行制片(重量130mg,片剂的形状:圆形(直径7mm)),由此得到素片8(含有有机酸)。将素片8的组成示于以下的表9。另外,作为参考,也示出配方例2的组成。[表9]表9:配方例2(素片)及素片8的组成试验例4:例10中得到的配方例2(素片)及素片8中是否添加有机酸而带来的稳定性的比较试验甲基巴多索隆的类似物质测定在以下的条件下利用液相色谱进行试验而测定。具体而言,作为柱,使用acquityuplchssc18、粒径1.8μm、2.1mm×50mm(waters制)或其同等品,维持于40℃。作为流动相a,使用10mmol/l磷酸缓冲液(ph2.5),作为流动相b,使用乙腈。试样溶液使用以化合物的浓度成为400μg/ml的方式用流动相a∶乙腈混合液(4∶6)进行稀释而成的溶液。在流速为0.3ml/min的条件下,使用紫外吸光光度计(测定波长:242nm)进行类似物质测定,求出相对于甲基巴多索隆的表示量而言的各类似物质总量(%)。稳定性试验按以下的条件进行。保存条件:40℃,75%rh,1个月或2个月保存形态:在打开了盖子的褐色瓶中放入配方例2(素片)及素片8(5种)[表10]表10:例10中得到的配方例2(素片)及素片8中的类似物质总量(%)根据上述表10的结果可知,与未添加有机酸的情况(配方例2)相比,添加了任一种有机酸时(素片8),类似物质总量的增加量均较少。另外可知,这些有机酸中,尤其是添加了富马酸及苹果酸的配方中类似物质总量及其增加量(1个月及2个月)均较少。铝袋包装品1~3的制造利用与配方例2同样的步骤,制作素片。具体而言,如下文所述。使用混合机(tbm-25,株式会社德寿工作所制),将含有40重量%化合物a(甲基巴多索隆)的固体分散体(利用喷雾干燥法制造,以下的“固体分散体”也同样地制造)500.0g、硅酸处理结晶纤维素(prosolv,jrspharma)2068.0g、乳糖水合物(日本药典)1796.0g、羟丙甲纤维素(tc-5e,信越化学工业株式会社制)236.0g、低取代度羟丙基纤维素(l-hpc,信越化学工业株式会社制)520.0g、轻质硅酸酐(adsolider101,freundcorporation株式会社制)40.0g进行混合。向该混合物中加入硬脂酸镁20.0g,进一步混合。使用辊压机(ccs-220,株式会社powrex制),对该混合物进行干式造粒及整粒。加入相对于得到的整粒品而言成为0.38重量%的量的硬脂酸镁,进行混合,得到压片用混合品。使用旋转压片机(ht-ap15,畑铁工株式会社制)进行制片(重量:130mg,片剂的形状:圆形(直径7mm)),由此得到含有5mg化合物a的素片x。针对得到的素片x,与配方例2同样地散布薄膜包衣及巴西棕榈蜡,实施上光,得到目标片剂x。将得到的素片x及片剂x的组成示于以下的表11及12。[表11]表11:素片x的配方[表12]表12:用于对素片x进行薄膜包衣、且添加光泽剂的配方针对基于上述的表11及12的配方制作的片剂x,使用aclar(商标)膜(sumilite(商标)fcl-1122,sumitomobakelite株式会社制)(以下,也称为“aclar”)或硬质氯乙烯膜(sumilite(商标)vss,sumitomobakelite株式会社制)(以下,也称为“硬质氯乙烯”)或聚丙烯膜(tas2230v,大成化工株式会社制)(以下,也称为“聚丙烯”)及铝箔(tokaitoyoaluminiumhanbai株式会社制或uacj制箔公司制),利用ptp包装机(pfd-100型,maruhohatsujyokogyo株式会社制),得到泡罩包装品1~3。将得到的泡罩包装品3片及干燥剂(ms-serum-w3g,东海化学工业所)放入铝袋中,使用热密封机(v-301-10w,fujiimpulse株式会社制)进行密封,得到目标铝袋包装品1~3。铝袋包装品4~6的制造与化合物a(甲基巴多索隆)一同添加有机酸12.0g(素片中包含0.2重量%的有机酸(富马酸)),将硅酸处理结晶纤维素减少至2060.0g、并将乳糖水合物减少至1792.0g,除此以外,利用与铝袋包装品1~3相同的步骤,得到素片y、片剂y、泡罩包装品4~6及铝袋包装品4~6。试验例5:铝袋包装品1~6的稳定性的比较试验针对铝袋包装品1~6(以下,也称为“包装品1~6”),在与上述的试验例4同样的保存条件(40℃,75%rh,1个月,敞开)下测定类似物质总量(%)(“开始时”及“40℃,75%rh,1个月,敞开”)。将结果示于以下的表13。甲基巴多索隆的类似物质测定在与试验例4同样的条件下利用液相色谱进行试验而测定。[表13]表13:铝袋包装品1~6的保存后的类似物质总量(%)根据上述表10、13及下述表14的结果可知,通过制成铝袋包装品,类似物质总量(%)的增加被抑制,稳定性提高。另外,根据上述表13的结果可知,在铝袋包装品中,通过添加相对于素片整体重量而言为0.2重量%的有机酸(富马酸),从而有下述倾向:类似物质总量(%)的增加被抑制,稳定性提高。此外可知,铝袋包装品中,与aclar(商标)膜(聚氯三氟乙烯)相比,使用了硬质氯乙烯膜或聚丙烯膜时,类似物质总量(%)的增加被进一步抑制,稳定性进一步提高。试验例6:铝袋包装品的稳定性的研究针对上述的铝袋包装品1~3的制造过程中得到的片剂x及泡罩包装品1(使用aclar)(未放入铝袋中),在与上述的试验例4及5同样的保存条件(40℃,75%rh,1个月,敞开)及保存状态(片剂x被放入打开了盖的褐色玻璃瓶中)下,测定类似物质总量(%)(“开始时”及“40℃,75%rh,1个月,敞开”)。将结果示于以下的表14。甲基巴多索隆的类似物质测定在与试验例4同样的条件下利用液相色谱进行试验而测定。[表14]表14:片剂x及泡罩包装品1的保存后的类似物质总量(%)根据上述表13及14的结果可知,与片剂x(薄膜包衣配方)相比,片剂x的泡罩包装品1及铝袋包装品1中类似物质总量及其增加量均较少。另外可知,与泡罩包装品1相比,片剂x的铝袋包装品1中若干类似物质总量及其增加量更少。因此可知,通过使本发明的薄膜包衣配方为泡罩包装品,能够提高稳定性,另外可知,通过使本发明的薄膜包衣配方为铝袋包装品,能够进一步提高稳定性。试验例7:薄膜包衣配方中的稳定性的比较试验以下的试验中使用:上述试验例5的比较试验中使用的片剂x;以使片剂y的素片中包含1重量%而非0.2重量%的富马酸的方式制作的片剂y-1;和,以代替片剂y中包含的富马酸而在素片中包含1重量%的苹果酸的方式制作的片剂z。片剂x、片剂y-1、及片剂z均为薄膜包衣配方。针对如此制作的片剂x、片剂y-1及片剂z,在与试验例5相同的保存条件及液相色谱的条件下,测定类似物质总量(%)(未显示数据)。结果可知,就添加有机酸(富马酸或苹果酸)而制作的片剂y-1(含有富马酸)及片剂z(含有苹果酸)而言,与未添加有机酸而制作的片剂x相比,类似物质总量(%)的增加被进一步抑制,稳定性进一步提高。另外可知,添加富马酸而制作的片剂y-1、与添加苹果酸而制作的片剂z在从开始时起的类似物质总量(%)的增加量方面基本没有区别。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1