用于结合亲和力预测的方法和系统以及生成候选蛋白-结合肽的方法与流程
背景技术:
本公开涉及结合剂分子与靶分子之间(例如肽与蛋白质之间、或蛋白质对之间)的结合亲和力的计算预测。
在许多生物学背景下,对分子结合的理解很重要。例如,在药物或生物疗法的开发中,重要的是要理解候选治疗分子与其预定的靶标如何结合以及结合强度如何,或者致病性肽与细胞表面蛋白如何结合以及结合强度如何。
在脊椎动物中,主要组织相容性复合体(mhc)分子已经进化为结合致病性肽或自身的肽以形成mhc-肽复合体,其随后通过细胞机制被转运到细胞表面。mhc分子通常被称为mhci类或mhcii类。尽管在功能上相似,但mhci类分子将内源性衍生的肽从细胞内递送到细胞表面,而mhcii类分子负责将外源性或细胞外的肽递送到细胞表面,随后发生cd8+(细胞毒性)或cd4+(辅助性)_t细胞对肽的识别。然后这种识别引发或传播免疫应答。在i类抗原呈递途径中有几个关键的相互依赖的步骤,其包括通过蛋白酶体和tap转运的抗原加工,和ii类途径包括外源性抗原的内化,在内体中的捕获和随后的蛋白酶介导的水解。然而,mhc结合步骤是内源性和外源性抗原加工途径的最重要的选择机制,并且对于致病性肽的成功呈递和对于癌症中突变的新抗原是必需的(尽管不够)。mhc结合作为适应性免疫应答的中心原则的重要性已经引起了在几个研究领域(包括传染病、疫苗开发、移植、自身免疫性疾病和癌症免疫疗法)中准确鉴定和测量mhc-肽结合亲和力的广泛研究。
在人类中,mhci类分子由i类人类白细胞抗原(hla)基因组区域中被称为hla-a、hla-b和hla-c的三个多态性基因编码。所有三个基因都是极其多态性的,具有超过10,000个表征的等位基因。与i类相似,mhcii类分子由被称为hla-dr、hla-dq和hla-dp的三种多态性基因编码。mhc分子中的高度多态性,以及抗原性肽序列中氨基酸的固有可变性,已经促使需要开发能够准确预测mhc-肽结合亲和力的计算工具,从而产生许多成功的方法。然而,需要大量实验数据的可用性来训练良好的预测模型。几年来,已经进行了大量的实验努力来为一些最常见的mhc等位基因积累mhc-肽结合亲和力测量。尽管如此,在目前可获得的mhc-肽结合亲和力测量的数据库中,仍未充分涵盖明显大多数的等位基因。对于这些等位基因中的几个,表现最佳的预测因子通常是“等位基因特异性”模型,这意味着它们仅在一个特定的mhc等位基因上进行训练,因此也只能预测一个特定的mhc等位基因。等位基因特异性模型通常需要大量的结合亲和力数据,使得它们仅适用于少量的充分研究的等位基因。几种算法方法已经利用了足够的训练数据的可用性来建立成功的等位基因特异性方法,范围从神经网络到支持向量机(svm),而且还利用了更机械地解释的方法,如位置特异性得分矩阵(pssm)或基于分子建模的方法。
已经进行了几种尝试来创建实施“泛等位基因”模型的计算工具,该模型可以生成跨不同mhc等位基因的结合亲和力的预测。这些主要是使用基于神经网络的方法来训练的,并且通常属于两个类别中的一个。“泛特异性”模型限于其已经训练的等位基因之间的预测,而“泛”模型基于mhc序列进行预测,因此可以对任意或从头mhc等位基因进行预测。
泛等位基因模型的首要目的是预测可应用于所有等位基因的一般结合模式,使得即使对于具有很少或没有训练数据的等位基因也能够进行预测。为了这样做,通常认为模型需要反映mhc分子与肽之间的物理相互作用(例如通过使用获得的mhc-肽复合体的晶体结构数据)。
从结构数据中已知mhc分子的肽相互作用组分由高度多态性结合裂隙组成,所述裂隙由形成两个几乎平行的螺旋的两个氨基酸链组成。对于mhci类,已经观察到形成mhci类分子的182个氨基酸中只有少量的氨基酸紧邻结合的肽氨基酸(例如,在结合的肽的任何氨基酸的4.0埃的距离内)。这些界定结合裂隙的肽近端mhc氨基酸可以被称为“伪序列”(参见,例如,nielsen等人,plosone2007,2:e796,其内容通过引用整体并入本文)。
肽与mhc蛋白结合的方式可以使用文献中被称为“接触点图”、“接触位置图”或简单地被称为“接触图”(其各自在下文可互换使用)来概括。通常,接触点图谱定义了结合剂分子(如肽)的氨基酸残基与与其结合的靶分子(如mhc蛋白)的相应氨基酸残基之间的映射(mapping),其中相应的氨基酸残基是靶分子的残基,其在距结合剂分子中的氨基酸残基的阈值距离内,使得它们与结合剂分子残基具有足够的物理相互作用,以有助于形成结合剂-靶复合体。例如,在粘合剂分子残基的4埃内的残基可以形成接触点图的一部分。
接触点图可以表示为表或矩阵,其中行代表靶分子伪序列的氨基酸,和列代表结合分子的氨基酸。如果结合剂分子氨基酸b在伪序列氨基酸t的预定距离内,则表的条目(t,b)等于1。mhci类分子的接触点图的具体实例可以发现于nielsen等人中。mhcii类分子的另一个实例可以发现于karosiene等人,immunogenetics2013,65:711-724中,其内容通过引用整体并入本文。
已经使用接触点图建立了所谓的基于“口袋”的肽-mhc结合模型,其中将肽的每个氨基酸(单体)作为单独的结合单位处理,所述结合单位根据接触点图与假定与单体接近的mhc伪序列的一个或多个氨基酸结合。mhc伪序列的近端氨基酸可以被称为“结合口袋”。这使得能够通过将结合口袋与训练数据中结构亚单位的类似出现进行匹配,然后将来自匹配亚单位的所有结合亲和力贡献加在一起,来预测很少或没有训练数据可用的等位基因的mhc结合模式。
理想的是提供预测结合亲和力的方法,其改进了上述方法,或者至少提供了有用的可替代方法。
概述
总的来说,本公开提出了预测结合亲和力的方法和系统,其中现有技术的口袋方法扩展到了单体使用之外。因此,在某些实施方案中,可以捕获肽和mhc分子两者中的上下文氨基酸n-mer长度残基(其中n为2至肽的长度)的影响。
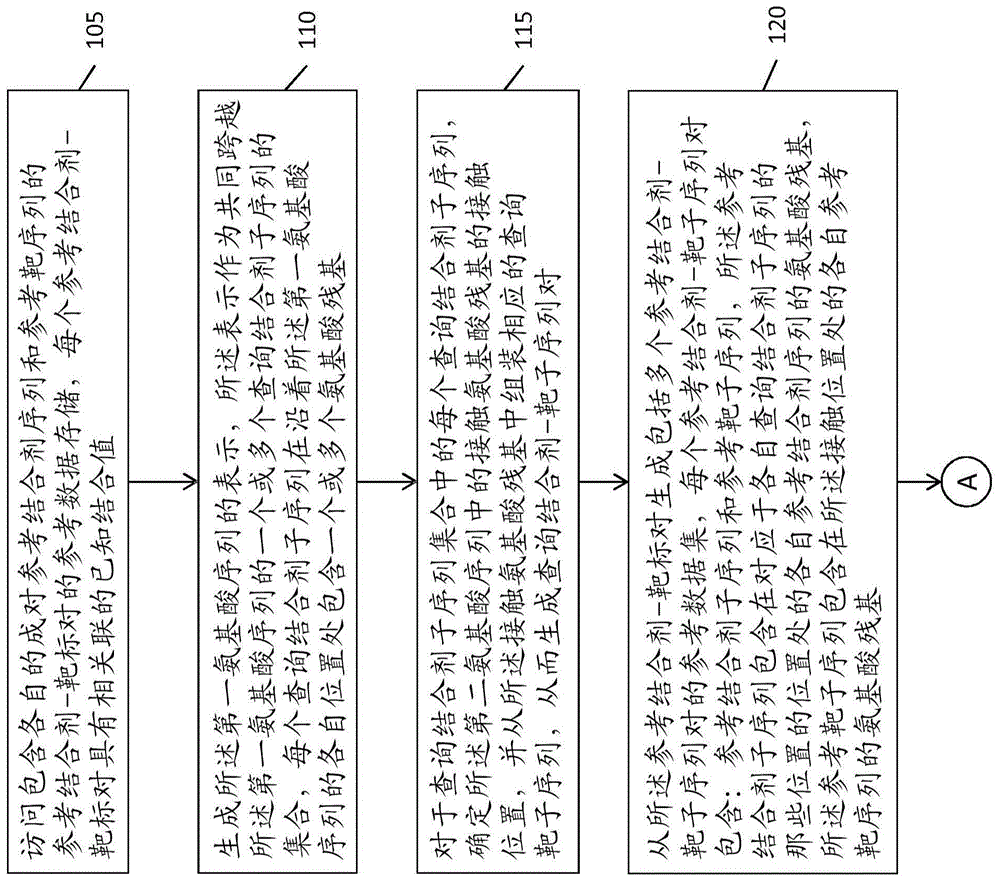
在本公开的第一方面,提供了预测查询结合剂分子与查询靶分子结合亲和力的计算机实施的方法,所述查询结合剂分子具有第一氨基酸序列,并且所述查询靶分子具有第二氨基酸序列,所述方法包括:用至少一个处理器,访问包含各自的成对参考结合剂序列和参考靶序列的参考结合剂-靶标对的参考数据存储,每个参考结合剂-靶标对具有相关联的已知结合值;用所述至少一个处理器,生成所述第一氨基酸序列的表示,所述表示作为共同跨越所述第一氨基酸序列的一个或多个查询结合剂子序列的集合,每个查询结合剂子序列在沿着所述第一氨基酸序列的各自位置处包含一个或多个氨基酸残基;对于查询结合剂子序列集中的每个查询结合剂子序列,确定所述第二氨基酸序列中的接触氨基酸残基的接触位置,并从所述接触氨基酸残基中组装相应的查询靶子序列,从而生成查询结合剂-靶子序列对;用所述至少一个处理器,从所述参考结合剂-靶标对生成包括多个参考结合剂-靶子序列对的参考数据集,每个参考结合剂-靶子序列对包含:参考结合剂子序列和参考靶子序列,所述参考结合剂子序列包含在对应于各自查询结合剂子序列的那些位置的位置处的各自参考结合剂序列的氨基酸残基,所述参考靶子序列包含在所述接触位置处的各自参考靶序列的氨基酸残基;其中每个参考结合剂-靶子序列对被分配基于从中产生参考结合剂-靶标对的已知结合值的参考结合值;用所述至少一个处理器,对所述各自的查询结合剂-靶子序列对和所述参考结合剂-靶子序列对进行至少一次相似性运算,以为每个查询结合剂-靶子序列对生成多个相似性得分;和用所述至少一个处理器,计算所述查询结合剂分子与所述查询靶分子的结合亲和力,作为所述参考结合剂-靶子序列对的所述参考结合值的加权组合,其中所述加权组合的权重基于相似性得分。
优选地,至少一个查询结合剂子序列可以包含至少两个氨基酸残基。
通过考虑mhc结合口袋的上下文关系,比本领域技术更可靠地确定预测的结合亲和力,并以更好地代表mhc-肽结合的生物复杂性的方式提高结合亲和力预测的准确性。与现有技术相比,该技术有效地降低了建模的组合复杂性。
相似性运算可以通过生成用于在查询结合剂子序列与参考结合剂子序列之间进行比较的第一相似性得分,以及用于在查询靶子序列与参考靶子序列之间进行比较的第二相似性得分,并且将所述第一相似性得分和所述第二相似性得分组合,来生成各自的相似性得分。
优选地,在准确匹配的情况下,所述第一相似性得分可以被赋予非零值,否则被赋予零值。
更优选地,在准确匹配的情况下,所述第二相似性得分可以被赋予非零值,否则被赋予零值。
在某些实施方案中,所述相似性运算可以包括查询结合剂子序列和参考结合剂子序列之间的序列比对,和/或查询靶子序列与参考靶子序列之间的序列比对。优选地,所述相似性运算使用blosum80矩阵。
在某些实施方案中,所述相似性运算可以包括:生成包括第一节点集和第二节点集的二分图,所述第一节点集仅包含结合剂子序列,并且所述第二节点集仅包含靶子序列,所述二分图的边缘权重等于所述相关联的已知结合剂值;和为所述第一节点集和/或所述第二节点集确定所述二分图的单分投影(monopartiteprojection),其中所述相似性得分为所述单分投影的边缘权重。
在这些实施方案中,可以通过确定第一集合的节点对的两个节点都与之连接的第二集合的公共节点集合以及计算所述二分图的对应边缘权重之间的线性相关性,来计算所述第一集合的节点对的相似性得分。可以通过确定所述第二集合的节点对的两个节点都与之连接的所述第一集合的公共节点集合以及计算所述二分图的对应边缘权重之间的线性相关性,来计算所述第二集合的节点对的相似性得分。
接触氨基酸残基的位置可以根据接触点图来确定。
确定接触位置的步骤还可以包括基于所述查询结合剂子序列来模拟在所述接触点图中使用的虚拟查询结合剂子序列,和/或模拟在所述接触点图中使用的接触点集,其中所述虚拟查询结合剂子序列具有与所述查询结合剂子序列不同的长度。所述接触点集中的接触点的量可以不同于所述查询结合剂子序列和/或参考结合剂子序列的长度。以这种方式,所述方法可以适用于不同的长度,例如,对于9-mer接触点图,其中k<9,或k>9。优选地,可以将所述接触点图限定为包含不同的长度信息。在某些实施方案中,所述方法可以包括减少或扩增更大或更小的查询结合子序列以适合所述接触点图或将多个氨基酸残基映射到接触点,使得所述接触点图的物理效应产生设定数量的口袋。
在某些实施方案中,查询结合剂子序列的最大长度可以是l(l>1),并且所述一个或多个查询结合剂子序列的集合可以包含长度为1至l的所述第一氨基酸序列的所有可能的子序列。
所述相似性运算可以包括根据各自的参考结合子序列的氨基酸位置对每个参考结合剂-靶子序列对进行加权。
优选地,所述第二氨基酸序列可以是mhc蛋白序列。更优选地,所述mhc蛋白序列可以是hla蛋白序列。
计算所述结合亲和力可以包括计算参考数据矩阵、查询数据转置矩阵和所述参考结合值的向量的乘积,其中所述参考数据矩阵的条目指示在各自的参考结合剂序列和/或参考靶序列中存在或不存在参考子序列,并且所述查询数据转置矩阵的条目指示在各自的查询结合剂序列和/或查询靶序列中存在或不存在参考子序列;并且其中根据所述相似性得分对所述参考数据矩阵的条目和所述查询数据矩阵的条目进行加权。在该实施方案中,可以经由稀疏矩阵计算技术来计算乘积。
本发明可应用于mhci类分子和mhcii类分子。
根据本发明的另一方面,可以提供生成至少一种候选蛋白结合肽的方法,所述方法包括:获取多种肽的氨基酸序列和蛋白质的氨基酸序列;通过本发明上述方面中任一方面的方法,为每种肽确定对所述蛋白质的预测的结合亲和力;和基于各自预测的结合亲和力选择所述多种肽中的一种或多种候选肽。
所述蛋白质的氨基酸序列可以通过以下方法之一获得:血清学抗体测试、寡核苷酸杂交方法、基于核酸扩增的方法(包括但不限于基于聚合酶链式反应的方法)、基于dna或rna测序的自动预测、从头肽测序、edman测序或质谱。
所述方法还可以包括合成所述一种或多种候选肽。
另外,所述方法还可以包括将所述候选肽编码入相应的dna或rna序列。此外,所述方法可以包括将所述序列掺入细菌或病毒递送系统的基因组中以产生疫苗。
因此,由于可以更有效地预测结合亲和力,特别是在针对等位基因的参考数据很少或没有参考数据的情况下,可以为个体患者更可靠地构建基于肽、dna或rna的疫苗。在交叉验证评估中,本发明已经证明了相对于本领域工具性能的竞争性能,并且对于在可获得的参考数据中几乎没有覆盖或没有覆盖的等位基因的任何子集,都具有优越的性能。
根据本发明的另一方面,可以提供用于预测查询结合剂分子与查询靶分子的结合亲和力的结合亲和力预测系统,所述查询结合剂分子具有第一氨基酸序列,并且所述查询靶分子具有第二氨基酸序列,所述系统包括与至少一个存储器装置通信的至少一个处理器,所述至少一个存储器装置具有存储在其上的指令,所述指令用于使所述至少一个处理器执行本发明上述方面中任一方面所述的方法。
附图简述
现在将参考附图仅以示例性的方式详细描述实施方案,其中:
图1a和图1b显示了用于预测结合剂与靶标的结合亲和力的方法的实施方案;
图2示意性地描绘了生成氨基酸序列作为子序列集的表示;
图3显示了在图1a和图1b的方法中使用的接触图的实例;
图4显示了在方法中使用的生成的靶(mhc)子序列的实例;
图5至图7描绘了在方法中使用的参考数据的生成阶段;和
图8至图10描绘了使与参考氨基酸序列不匹配的查询氨基酸序列适应的各种方法。
详述
根据某些实施方案的方法使得能够对查询结合剂分子(如肽)与查询靶分子(如蛋白质)的结合亲和力进行计算预测。查询结合剂分子和查询靶分子各自具有各自的氨基酸序列。基于包含参考结合剂-靶标对的参考数据进行预测,每对具有已知的(测量的)结合值,其可以是例如以nm测量的ic50值,或基于ic50的其它值。参考数据在本文中也可以被称为训练数据。
结合值不必是结合亲和力的直接量度,只要它反映结合剂与靶标之间的相对结合强度(即,相对于其它结合剂-靶标对)即可。通常,参考数据可以至少部分地从公共数据库获得,诸如免疫表位数据库(immuneepitopedatabase,iedb)(www.iedb.org)、gpcrdb(www.gpcrdb.org)或brenda(http://www.brenda-enzymes.org)。
参考图1,根据某些实施方案的方法100包括步骤105,其是访问参考结合剂-靶标对的参考数据存储。每个参考结合剂-靶标对包含参考结合剂氨基酸序列(如肽序列)和参考靶氨基酸序列(如mhc蛋白序列)。下面的讨论将集中于肽-mhc的结合,但是应当理解,下面所讨论的方法和系统可以容易地适用于其它数据集,其中成对的结合剂序列和靶序列以及相应的结合值是可获得的。
为了生成对不在参考数据中的查询肽-mhc对的预测,根据实施方案的方法首先生成(步骤110)查询肽序列的表示,所述表示作为共同跨越查询肽序列的一个或多个查询肽子序列的集合,这些子序列中的至少一个的长度为两个或更多个氨基酸。例如,如图2(b)中所示的,通过将肽序列分解成所有可能的连续的2-mer,产生一种可能的表示,其中2-mer以重叠的方式以1个残基间隔覆盖肽序列,并因此共同跨越肽序列。如图2(g)中所示的,另一种可能的表示是根据3-mer,其中至少一些3-mer子序列是不连续的。同样,9-mer肽的整个长度由3-mer共同跨越(即,每个肽残基包含在至少一个3-mer中)。
接下来,对于每个查询肽子序列,该方法确定(步骤115)查询mhc序列中的接触氨基酸残基的接触位置。通常,这是通过使用适当的接触图来完成的。例如,对于mhci类序列,可以使用nielsen等人的接触图,其在图3中被再现。然而,通常可以使用预测的或实验性的任何接触点图。使用nielsen等人的接触图,具有位置(1、2)的图2(b)的第一个2-mer在查询mhc序列中具有相应的接触位置c=(7、9、24、45、59、62、63、66、67、70、99、159、163、167、171)。
类似于现有技术的结合口袋术语,本实施方案的非单体结合单元可以被描述为“高阶口袋”或hop。在肽-mhc结合的上下文中,单独的查询肽子序列可以被称为查询肽hop,并且相应的查询mhc子序列可以被称为查询mhchop,所述相应的查询mhc子序列由在相应的接触位置(例如,如从接触图衍生的接触位置)处的mhc残基组装。因此,每个hop都是适当的肽或mhc序列的子序列。使用序列的一组或多组氨基酸位置,从肽或mhc序列生成hop的过程可以被称为hop分解。
一旦为每个查询肽hop确定了查询mhchop,就可以将hop对作为查询肽-mhc子序列(hop)对存储(例如,在ram中或数据库中)。
接下来,该方法使用参考结合剂-靶标(肽-mhc)对生成(步骤120)参考数据集。参考数据集可以包括多行,每行包含参考肽子序列、参考mhc子序列和参考结合值。参考肽子序列是参考肽hop,其使用与被用于生成查询肽hop的氨基酸位置相同的氨基酸位置生成。类似地,参考mhc子序列是mhc参考hop,其使用与被用于生成查询mhchop的接触位置相同的接触位置生成。参考肽-mhchop对与参考结合值相关联,所述参考结合值等于衍生hop的肽-mhc对的结合值。
现在将参考图4至图7描述hop分解和参考数据集的生成的具体实例。所示序列不是真正的mhc或肽序列,而是被简单地选择用于说明目的。如图5中所示的,与多种不同肽之一结合的等位基因hla-x和hla-y的结合值是可获得的,但在本实例中,期望预测新肽abydefghi和肽abcdefghi与等位基因hla-x的结合,所述新肽abydefghi在参考数据中未被表示,所述肽abcdefghi在参考数据中被表示,但是对于其与等位基因hla-z的结合是未知的。仅对肽的位置(1、2)和(1、3)说明了hop分解,但是应当理解,根据需要,对于肽的其它位置使用相同的程序,以上述所讨论的方式共同跨越整个肽序列。
未显示三个等位基因的完整序列,但是对于三个位置(1、2、3),可以通过使用如上所述的适当接触图来生成相应的mhchop。在图4所示的实例中,根据所使用的具体接触图,每个肽残基与3个mhc残基相邻。应当理解,通常,取决于接触图的确切形式或被用于确定接触位置的其他手段,不同的肽残基可能与比这个数更少的或更多的mhc残基接近。
对于查询肽abydefghi,位置(1、2)处的肽hop是通过确定沿着序列(即ab)的那些位置处的氨基酸而生成的。类似地,位置(1、3)处的肽hop是ay。等位基因hla-x的两组位置的相应mhchop通过图4中所示的hop的串接给出。因此,对于位置(1、2),hla-x的mhchop是abcdef,而对于位置(1、3),mhchop是abcabd。
如图5中所示的,可以对参考数据进行相同的处理。例如,如第一行所示的,肽abcdefghi在位置(1、2)处具有hop分解ab,并且在位置(1、3)处具有hop分解ac。不需要再生成hla-x的mhchop,因为在这种情况下参考mhc序列与查询mhc序列相同。
如图5中所示的,每个肽hop-mhchop对具有与生成它的肽-mhc对的结合值相等的相关联的结合值。因为不同序列对的hop分解不一定是唯一的,所以该方法可以包括例如通过取结合值的平均值或中值来计算重复的hop对的不同结合值的汇总的其它步骤。这在图5中被示出,其中,例如,配对abcdef-ab的结合值被计算为各个值(0.9、0.8、0.8、0.9、0.5)的平均值。
一旦对参考肽和mhc序列进行了所有的hop分解,并且对重复的hop对计算了适当的汇总,结果就是参考数据集,在这种情况下,所述数据集包括两个表:用于接触位置(1、2)的第一表510,和用于接触位置(1、3)的第二表520。应当理解,在跨越整个肽序列的全面接触位置集合的情况下,为了产生参考数据集,将生成参考数据的其它表。
回到图1b,在该方法的下一步骤125中,对各自的查询肽-mhchop对和参考肽-mhchop对进行至少一次序列相似性运算,以为每个查询肽-mhchop对生成多个相似性得分。
在一个实例中,在查询hop对与参考hop对之间的准确匹配的情况下,相似性得分可以被赋予值1,否则值为0。因此,例如,在肽的位置(1、2)处的查询hop对ab-abcdef将被分配相似性得分1,因为它与图5中的参考数据表的第一行中的ab-abcdef准确匹配,但是在位置(1、3)处的ay-abcabd将被分配得分0,因为在图5中的参考数据表中没有匹配。
在另一个实例中,相似性得分可以使用氨基酸替换矩阵(如图4中所示的一个矩阵)来计算。这使得能够从与查询序列相似(以生物学上有意义的方式)但不相同的序列中包含对结合亲和力预测的贡献。可以分别计算肽hop和mhchop的相似性得分。因此,例如,可以实施肽hop的准确匹配,但是对于mhchop,可以使用替换矩阵。
例如,查询mhc等位基因hla-z不在图5的参考数据中表示,但是已经被确定在位置(1、2)处具有mhchopabcdex,而在位置(1、3)处具有abcxyd。尽管abcdex在位置(1、2)的参考数据中不具有任何准确匹配,但是如果使用图4中的替换矩阵,则可以计算0.79的(归一化的)相似性得分,并被用于对abcdex对总体结合亲和力预测的贡献进行加权,这将在下面更详细地解释。
在另一个实例中,相似性得分可以使用网络邻域相似性运算来计算,其中从mhchop和肽hop生成二分网络,网络的边缘权重是各自肽-mhc对的结合值,并且进行网络的单分投影,这将在下面进一步详细解释。
回到图1b,该方法的下一个步骤130包括计算查询肽与查询mhc蛋白的结合亲和力,作为参考肽-mhchop对的结合值(来自参考数据)的加权组合,通常是总和。加权组合的权重基于相似性得分,并且可以包括来自多于一次的相似性运算的贡献。例如,可以应用序列相似性和网络邻域相似性。
实施方案将结合口袋的概念扩展到单体方法之外,并基于k-mer肽-mhc复合体(k≥n≥1)内所有可能的n-mer口袋进行推理,以改善mhc-肽结合预测。通过涵盖高阶的mhc-肽口袋,肽和mhc分子两者中的上下文氨基酸n-mer长度残基的影响被包括在内,从而以更好地代表mhc-肽结合的生物复杂性的方式提高了结合亲和力预测的准确性。
通过单独或联合引入序列和邻域相似性,可以对在可获得的参考数据中未很好表示(或根本没有表示)的mhc等位基因进行预测。本发明的方法提供了改进的物理上可解释的、高度可通用的mhc-肽结合预测子。
现在将更详细地描述上述实施方案的其它方面。
hop分解
在一个实例中,k-mer肽与mhc结合裂隙(例如9-mer)的结合可以模拟为8个2-mermhc-肽结合口袋的相互作用、7个3-mer结合口袋的相互作用等,作为(k-n+1)个数目的n-mer的相互作用。我们将仅使用连续的n-mer片段的这种方法表示为“连接的”方法。图2(b)和图2(c)说明了2-mer和3-mer肽hop的连接方法。
在另一个实例中,线性肽可以被认为是闭环的。对于从9-mer肽生成的3-merhop,这需要添加由肽位置(8、9、1)和(9、1、2)组成的“环连接的”3-mer。尽管此类实施方案通过以这种方式对mhc-肽相互作用建模而失去了物理可解释性的一些方面,但它们的优点在于它们在连接的方法中纠正了对中心肽残基的可能的偏倚。例如,在3-mer的情况下,连接方法中的末端肽氨基酸位置1和9将仅被3-mer(1、2、3)和(7、8、9)中的一个分别覆盖。残基位置2和8将被两个3-mer覆盖,并且其余位置被三个3-mer覆盖。使用闭环分解方法,将总是有9个从9-mer分解的n-mer。我们将这称为“环”方法。在图2(e)中示出了3-mer的环方法。
在其它实例中,我们可以考虑用于所讨论的mhc-肽结合口袋相互作用的所有可能的n-组合,其中n-mer的数目由二项式系数
对于与mhc分子结合的长度为k=9的肽,其9个结合口袋由接触点图给出,我们限定了以下n-mermhc-肽结合口袋的相互作用:
-接触位置由k个非重复的数字(对于九聚体,从1至9)组成,指示肽残基的位置,以及相应的mhc结合口袋(例如,根据接触图衍生的mhc结合口袋)。
-高阶口袋(hop)是在给定的接触位置实现mhc-肽结合复合体,即k个结合的肽残基和相应的mhc结合袋。
-肽hop是hop的肽组分。例如,接触位置(1、3、4)和肽“abcdefghi”产生肽hop“acd”。
-如在图1的上下文中所讨论的,mhchop是hop的mhc组分。
本发明方法的实施方案可以通过使用单个n值和单一方法(“全面的”、“环”或“连接的”),或者通过组合几个n值和方法来应用。已经发现“全面的”方法总体上给出了优异的性能。
为了以下讨论的目的,可以定义以下hop函数h:
h:(c,i,x)→(i,x)c,(1)
其中i是mhc分子,x是肽,和c∈c(n)是在所讨论的方法的接触位置c的集合中的单个接触位置和n值。(i,x)c表示在接触位置c处的i和x的hop实现,其中i是mhchop,和x是肽hop。我们用i和j表示mhc分子,用相应的小写字母i和j表示mhchop,用x和y表示完整的肽,用x和y相应地表示肽hop。应当注意,只有属于相同接触位置c的hop才可以被直接比较,并且如上所述的,不相等的mhc-肽对(i,x)可以分解成几个相同的hop(i,x)c,只要在mhc和肽氨基酸序列中存在适当的重叠。
考虑具有未知结合亲和力的查询mhc-肽对(i,x)和hop组分h(c,i,x)→(i,x)c。赋予具有相应的结合亲和力wjy的mhc-肽对(j,y)和hop组分h:(c,j,y)→(j,y)c的参考数据集,以下表达式可以被用于预测单个hop组分(i,x)c对i和x之间的结合亲和力的结合亲和力贡献:
其中,{(j,y)c}是具有c∈c(k)的所有唯一hop的集合。此外,sim(i,j;x,y)是提供任何两个hop(i,x)c与(j,y)c之间的定量相似性得分的通用相似性度量。权重可以由通用表达式表示:
其中,{h(c,j,y)=(j,y)c}是参考数据中mhc-肽对(j,y)分解为hop(j,y)c的所有条目的集合,并且wjy是与mhc-肽对(j,y)相关联的转换的结合亲和力值。相似性算子globalsim(i,j;x,y)是独立于hop的,即在框架内为“全局”,并且提供任何两个mhc-肽对(i,x)与(j,y)之间的定量相似性得分。在某些实施方案中,globalsim(i,j;x,y)≡1,这相当于使方程式(3)简单地为含有hop(j,y)c的所有mhc-肽对的结合亲和力值的平均值。通常,所有相似性度量被限定为0至1的范围内,其中1意味着完美的相似性,而0意味着没有相似性或最小相似性。
方程式(2)表示单一接触位置对mhc-肽对(i,x)的总结合亲和力预测的贡献。通过对所讨论的n-mer方法的所有接触位置求和,可以通过以下方程式计算查询mhc-肽对(i,x)的结合亲和力预测值:
其中a(c)是锚点值权重,使得在mhc-肽结合相互作用中某些位置比其它位置更重要。肽位置2和9是用于hla结合的此类位置的实例。
在某些实施方案中,可能期望包括具有多个n值的可能性,以及多个相关联的相似性度量选择。因此,在此类实施方案中,结合亲和力预测值可以通过如下方程式对来自n和sim的不同组合的贡献求和来获得:
其中n(n,sim)是所讨论的n和sim的唯一hop匹配的数目,即,方程式(4)中所有接触位置c上的方程式(2)中非零总和元素的数目。剩余量f(n,sim)是重要性-权重值,当例如通过使3-mer的重要性变为2-mer的两倍来组合几种单独的方法时,所述重要性-权重值使得能够具有灵活性。方程式(5)可以被认为是对多个方法进行整合的方式,这允许基于多种相似性度量和n值的几个模型对每个结合亲和力预测做出贡献。
根据上述方程式(2、4、5),显然本发明的方法提供了灵活性,因为hop相似性度量sim(i,j;x,y)的许多不同选择是可能的。
在某些实施方案中,对肽hop和mhchop的相似性运算分别进行。因此,通用相似性度量可以被表示如下:
sim(i,j;x,y)=mhcsim(i,j)·pepsim(x,y),(6)
其中,mhcsim(i,j)提供mhchopi与j之间的相似性得分,并且pepsim(x,y)提供肽hopx与y之间的相似性得分。
在某些实施方案中,查询肽的长度可以不同于参考数据和/或接触点图中肽的长度k。在这种情况下,查询肽可以被转换为一个或多个“虚拟的”k-mer。在图8至图10中示出了实施这一目的的不同方式。类似地,可以模拟该接触点的集合,以便产生“虚拟的”mhc结合裂隙或“虚拟的”口袋,这将从下面的实施例中变得清楚。
如图8(a)中所示的,例如,如果参考数据含有9-mer,并且查询肽是11-mer,在解决该问题的“连接”方法中,可以使2个氨基酸的凸出部分800穿过整个肽,以2个最外面的氨基酸悬挂在结合裂隙的任一端开始和结束。每个物理11-mer有10个此类虚拟肽(不是9个,因为在9-mer肽中有2个末端和8个“中间”)。如图8(b)中示出的另一种方法,被称为“全面的”方法,其允许长度为1的凸出部分和悬垂部分(802a、802b),因此产生更多的虚拟肽,总共为
“移动窗口”、“连接的”和“全面的”方法也适用于k<9,但是用间隙插入代替了凸起部分和悬挂部分。与k>9相比,虚拟肽的数量也更少,因为物理肽更小。
如图10(a)中所示的,对于k<9,我们还可以实施肽“伸展”框架,其中单个氨基酸1000可以占据多于一个的口袋,例如相邻的口袋对(1002、1004)或(1004、1006)。也就是说,可以插入双份的相邻氨基酸而不是插入间隙,以模拟单个氨基酸在口袋中伸展。
如图10(b)中所示的,对于k>9,我们可以另外实施肽“挤压”方法,其中复制一个或多个肽位置,将两个氨基酸挤入复制的单个mhc口袋中。例如,氨基酸1050和1052可以被“挤压”到口袋1060或口袋1062中等等。
在一些实施方案中,可以组合上述方法,例如通过允许组合所有k>9的方法,和使用优化或其它学习方法组合所有k<9的方法,以发现哪些虚拟肽/mhc构型在整个数据中最有意义。例如,可以将不同方法的各种组合应用于一个或多个训练数据集,并且可以选择当应用于测试数据集时给出最佳性能的组合作为最佳组合。因此,使用组合方法进行的结合亲和力预测可以包括对来自每个虚拟肽-mhc对的结合亲和力预测贡献的加权总和,产生每个物理肽-mhc对的一个结合亲和力预测值。
如已经注意到的,“虚拟的”k-mer或结合剂裂隙可以被认为是允许将多个长度查询与接触点图一起使用的模拟。在常规的接触点图中,线性固定长度的肽被映射到固定长度的mhc残基的集合。对于不同的长度,可以将肽分成k-mer的组合,并加工每个组合。
在所提出的模拟k-mer技术的实例中,k-mer查询肽的子集可以被映射到接触点的子集。通过将查询k-mer翻译(或转码)成可以被映射到接触点的不同序列,可以将多个肽氨基酸映射到任何数目的口袋中,并因此将残基映射到任何口袋中。
上述实例包括减少(或扩增)较大(或较小)的查询肽以适合接触点图或将多个氨基酸映射到接触点,使得物理效应产生设定数目的口袋。
以这种方式,接触点图技术可以应用于不同的肽长度,例如,对于9-mer接触点图,k<9或k>9。接触点图可以被限定为包含不同的长度信息。实际上,该技术可以被应用于与接触点图“设计”的长度不同的k-mer。提出了解决两个三维蛋白质/分子之间的复杂相互作用的方法,只要存在表达靶氨基酸子序列与结合剂氨基酸子序列(甚至对于许多到许多不连续的氨基酸子序列)之间的接触点的方法。
相似性度量sim(i,j;x,y)的实例
基本相似性
在一种形式中,方程式(6)中的相似性度量sim(i,j;x,y)的分量可以被表示为:
这相当于要求查询hop与参考数据集中的至少一个条目准确匹配,以便它有助于结合亲和力预测。通过应用这些度量,方程式(2)可以被写为:
其中n是在参考数据中发现的准确hop匹配的数目。
序列相似性
在一些实施方案中,相似性运算可以包括确定查询肽hop与参考肽hop之间的序列相似性,和/或查询mhchop与参考mhchop之间的序列相似性。
方程式(6)中的各个mhc相似性因子可以被写成:
并且对于肽序列相似性pepseqsim(x,y)是等价的。参数α是调谐参数,可以理解的是,可以应用该参数以提供软阈值,因为它调节了与弱相似性相比的强相似性的影响,而没有明确地去除数据(另一方面,硬阈值则会将低于阈值的相似性得分设置为零)。与在α=1的情况下相比,在训练过程中允许此类参数的调谐可能会产生更好的模型性能。可以使用真结合值与预测结合值之间的皮尔逊相关系数,以及通过将ic50值≤500nm的数据点视为结合类别中的数据点以及具有高于非结合类别中的阈值的值的那些数据点来生成接收子-运算子特性(thereceiver-operatorcharacteristic,roc)曲线下面积(auc)来评估模型性能。这些度量将被分别统称为pcc和auc。
在某些实施方案中,可以使用blosum比对来确定序列相似性。有利地,已经发现blosum80矩阵的使用在本发明发明人测试的blosum矩阵中产生最好的性能(同样,如通过pcc和auc所测量的)。然而,应当理解,也可以使用其他评分矩阵,无论是基于进化考虑、物理-化学考虑(如用于编码和非编码氨基酸的拓扑和结构信息的向量-vtsa,和/或疏水性、空间的和电子特性的主分量评分向量-vhse),还是两者。
二分网络邻域相似性
在一些实施方案中,相似性运算可以包括确定网络邻域相似性。在此类实施方案中,对于给定的接触位置c,可以生成二分网络(图),其中mhc和肽hop分量i和x分别是顶部和底部节点。在任何两个mhchop或任何两个肽hop之间没有直接连接或边缘(链接)。所有的直接边缘都在mhchop与肽hop之间,其中结合亲和力值作为相关联的边缘权重。使用跨越二分网络中的顶部或底部节点(mhc或肽hop)的这些边缘权重的协方差,可以对参考数据中不存在的hop进行预测。例如,在mhchop邻域相似性的情况下,通过用单一权重值代替任何mhchop对共享的所有肽hop来确定mhc-肽hop网络的加权单分投影,其指示mhchop对与共享肽hop的集合如何类似地相互作用。有多种方法来确定权重值,但是已经发现结合亲和力对之间的线性相关性在pcc和auc方面提供了最佳性能。然后,方程式(gensim)中的mhchop因子可以被表示为:
其中n是mhchop(顶部节点)i和j之间的共有相邻肽hop(底部节点)y的数目,并且w是所讨论的下标mhchop对的有效结合亲和力权重值(在广义框架中,方程式(3)也可以被应用于这些权重值)。对于肽邻域相似性pepnbrsim(x,y),使用等效表达式。
尽管邻域相似性运算不能实施从头hop分量的推理,但有利的是不需要测试数据来计算相似性得分。换句话说,可以在没有测试数据的任何先验知识的情况下训练模型。这可以允许更短的预测时间,这在至少一些情况下在短训练时间上可以是优选的。它还简化了其它技术(如自举)的实施。
在一些实施方案中,方程式(10)的替代是可能的。对于单分网络,加权拓扑重叠(wto)度量先前已经被成功地应用于各种生物系统。参见,例如,k.nowick、t.gernat、e.almaas和l.stubbs,“differencesinhumanandchimpanzeegeneexpressionpatternsdefiningaevolutioningnetworkoftranscriptionfactorinbrain”,proceedingsofthenationalacademyofsciences,vol.106,no.52,pp.22358–22363,2009,其全部内容通过引用并入本文。
节点/顶点i与j之间的wto可以通过以下方程式计算:
其中,aij是网络的加权邻接矩阵中的元素,并且ki是节点i的加权/未加权连通性(节点强度/度)。因此,该度量的二分版本可以被用作hop邻域相似度度量,即可以代替方程式(10)的右手侧。它可以被表示为:
其中,bwto代表“二分加权拓扑差异”,i、j是顶部节点,x是底部节点,并且ki是等价于wto表达式中的连通性(底部节点邻居的总和)的二分。如果分母是零,则bwto值也是零。可替代地,可以使用类似的“二分加权拓扑差异”(bwtd)度量来代替方程式(10b):
已经发现bwto和bwtd两者在一些情况中都表现良好,但是已经出现方程式(10)作为基于预测性能(pcc和auc)的总体最佳邻域相似性选择。
相似性参数
对于序列和邻域相似性以及其它可想象的相似性方法,应用如方程式(9)和(10)中所进行的调谐参数α,以便以此类方式调整相似性得分,即,当与不同的hop相比时,赋予相似的hop适当的结合亲和力贡献,这可能是有利的。在其中相似性值范围为1(相同的)至0(不同的)的实施方案中,我们可以应用α来调整相似性值之间的比率,同时保留它们的顺序。以最小相似性得分的形式或者以固定的最大数目m的相似得分的形式应用相似性截止也可能是有利的。在mhc序列相似性的情况下,例如,在mhcsim=mhcseqsim并且pepsim是基本的、二进制相似性度量的情况下,方程式(2)可以被写为:
其中,j至m的总和简单地意指我们在与mhchopi最相似的m个mhchopj上求总和。
参数优化
在通用框架中有几个自由参数,范围从相似性度量具体参数(如调谐参数和截止值)到更普遍适用的参数(如锚点位置权重和进近重要性权重f(n,sim))。可以使用各种优化策略来找到这些参数,包括局部和全局方法。
在一些实施方案中,可以使用下面详细描述的逐步参数优化方法。然而,应当理解,可以使用本领域技术人员已知的各种方法来进行许多其它参数优化过程。
1.已经发现锚点权重a(c)广泛地不受实验设置改变的影响,只要在泛等位基因设置中对它们进行评估。因此,它们是我们发现的第一个参数类别。几种优化方案将是合适的,但是我们选择应用差分进化(de),因为可以优化最佳auc值,其与de的全局性质一起被认为提供不太易于过度拟合的参数。可以选择1、2和3-mer的全面基本hop对作为该步骤的预测子。de算法针对几次训练/测试折叠运行几次,以确保参数通用,并将平均锚点值传递到下一步骤。应该注意的是,只有9个单体锚点权重被认为是变量,因为剩余的高阶锚点权重作为它们的单体贡献的平均值来计算。
2.该步骤对于每个相似性方法是特定的,由n值(该方法中口袋的数目)和相似性度量(即,方程式中的(n,sim))限定。在此,具有在先前步骤中发现的锚点参数的1、2、3-mer的全面基本hop对与由(n,sim)限定的特定(邻域或序列)相似性相结合,作为优化的预测子。使用与步骤1中类似的de优化算法,对于每个特定的优化,仅有2个变量要优化,即总和截止参数m和功率参数α。与局部方法(如最小二乘法)相比,de非常适合该步骤,因为m是整数。同样在此,针对几次训练/测试折叠运行几次优化,以确保参数通用,并且平均m和α值(对于每个特定的(n,sim)方法)被传递到下一步骤。
3.在步骤1和2之后,已经生成锚点权重a(c)以及各种相似性方法所需的所有m和α值。剩下的部分是调谐来自每种(n,sim)方法的预测贡献参数f,使得总的求和预测性能是最佳的。曲线拟合参数(y=ax+b中的a和b)也可以被添加到优化变量中,以便获得类似于训练数据中的实际ic50测量值的ic50预测值。由于可变空间是平滑的并且表现得非常好,所以可以使用最小二乘法优化,使真实值和预测值之间的总平方距离最小化。这几乎等同于对大的pcc值进行优化,这是回归方法所期望的特性。同样,针对几个训练/测试折叠,用不同的初始条件运行几次优化,以生成参数集列表(每一个都被发现对于它们的训练/测试集和初始条件是最优的)。通过对该列表应用一些统计方法,通过平均值(平均,中值)、最大总体相关性或其它方法来找到最终的参数集。就最大的整体相关性而言,我们意指与所有其它参数集具有最大平均相关性的参数集,其因此可以被称为“共有参数”。
在一个实例中,可以将来自iedb的数据分成两个子集,其中第一子集用于训练,第二子集用于测试。例如,第一和第二子集可以是(1)添加到iedb中直到2009的所有结合亲和力数据(本文中被称为kim09),和(2)添加在2010和2013之间的所有结合亲和力数据(本文中被称为kim13)。结合亲和力测量值(ic50/ec50)可以使用以下log转换转化:1-log(ic50)/log(5002),其中对数基数反映了mhc-肽对被认为结合的默认阈值(结合剂具有ic50≤500nm)。此外,可以省略一部分数据,以便在被称为数据盲化的程序中协助过拟合测试,例如,通过可重复地伪随机地省略上述结合数据的0.5%。
该方法的评估是经由几个数值实验进行的:(1)对kim09数据的训练和对kim13数据的测试,(2)使用kim09数据中的预定义的5倍来进行5倍交叉验证(其中如本文中所用的“倍”是指与任何其它子集不重叠的数据的子集),(3)对不包括在0.5%盲数据集中的所有数据的训练和对盲集的测试,和(4)将kim09和kim13数据集相结合,留出一个等位基因进行测试,同时对其余所有等位基因进行训练。四种实验类型在下面被称为:(1)kim09-kim13,(2)kim09-5倍,(3)盲,和(4)loao(留一等位基因缺失)。对于每个实验使用两个评估度量:真结合值和预测结合值之间的皮尔逊相关系数(pcc),以及使用ic50≤500nm的标准作为结合阈值(即,如果ic50小于或等于阈值,则将结合剂和靶标分类为结合的,否则为未结合的)的接收子-运算子特性(roc)曲线下面积(auc)。roc曲线可以通过本领域已知的任何方法生成。例如,python包numpy和scipy中可用的函数可以被用于生成roc曲线。
发现泛等位基因锚点定位置权重在随机子集中是高度稳定的,并且为了简单起见,选择固定作为kim0955倍交叉验证的最佳方案。针对序列相似性等位基因特异性预测框架实施等位基因特异性锚点,但在泛等位基因框架中未尝试。在20倍伪随机训练方案中使用差分进化发现相似性度量特定参数,其中5个等位基因和5%的数据被从训练中省去并用于测试。还尝试了更传统的5倍交叉验证,得到了除了loao实验以外的所有实验的良好性能的模型,这是可感测的,因为这些模型在训练过程中从未被“强制”预测未知的mhc等位基因。在足够的迭代次数之后,还发现相似性参数对于各种子集是稳定的,并且因此对于下面所讨论的结果中的所有泛方法也保持静态。为了确定进近重要性权重,我们选择了双重训练框架,其中:(1)使用与上述相同的20倍交叉验证方案训练用于loao实验的模型,而(2)使用标准交叉验证训练用于其它实验的模型,每倍1000个随机参数初始化,其中选择最终共有参数作为与所有其它数千个参数集具有最高平均相关性的集合。
结合亲和力预测值的计算
在一个实施方案中,方程式(2)、(4)和(5)中的结合亲和力预测值可以通过对参考数据进行适当的hop分解,将结果存储在一个或多个字典或散列表中,然后,对于每个查询hop,进行所存储的结果的查找来确定。可以通过循环不同的hop分解、相似性度量等来计算加权总和。
可替代地,一些实施方案可以使用稀疏矩阵编码来计算结合亲和力。例如,scipy的稀疏矩阵库可以与numpy功能一起使用,以实现内存效率和速度,远远不匹配更直接的迭代字典实施。
我们首先定义矩阵h,它是(#数据行×#hopid)的矩阵。hopid是唯一鉴定任何mhc-肽(结合剂-靶标)hop的整数。对于每个数据行,存在mhc序列(伪序列)和肽,其可以被分解成n个hop(如果仅包括单体hop,则n=9)。对于数据行p,每个hop具有相关联的hopidq。从基本框架开始,所有这些(p,q)对在矩阵中被赋予值1.0。作为参考,该基本矩阵中每行的总和为n。
矩阵有两种类型“训练”和“测试”,其中使用训练(参考)数据构建h训练,而使用测试(查询)数据构建h测试。以其最简单的形式(没有锚点,没有重复的hop平均),然后可以通过以下方程式找到基本预测值:
其中,y是包含与训练数据相关联的结合亲和力值的形状(#训练数据行×1)的列向量,
为了在重复的hop上求平均,我们沿着h训练的列进行求和,留下形状(1×#hopid)的行向量。将该总和向量沿列扩展到h训练的形状,我们可以用总和矩阵进行逐元素相除运算,用m表示:
类似地,如果我们想要应用锚点权重,我们简单地在具有与h相同的形状的锚点权重矩阵a与h测试或h训练之间进行逐元素相乘(符号:⊙)。作为参考,a具有n个唯一值的最大值,并且所有列由相同的元素组成(因为它是相同的hop)。逐一实施,这看起来不像它那么麻烦,因为我们可以利用a的这种可预测的结构,避免产生密集的h形矩阵。对于用m的上述运算也是如此。应用锚点:
h=h⊙a.(14)
到目前为止,我们还没有考虑相似性。将相似性添加到框架通常影响整个过程,包括锚点权重和归一化,并且必须在任何其他运算之前应用。如果使用邻域相似性或序列相似性或两者,则每行不再有n个hop。相反,每行的hop的数目受到n、sim、m和α的影响,并且通常对于每行是不相等的。然而,所有行的共同特征是每行总是只有n个元素等于1.0(对于非零α)。其它元素被赋予相似性值∈[0,1)。
用这些相似性元素填充矩阵的过程可以如下:
1.对于每个hop(从数据行p分解的hop),找到数据中存在的m个最相似的hop。
2.对于每个hop,找到与在1中找到的m个命中匹配的hopidq。
3.使元素(p,q)等于由sim和α赋予的原始hop与相似hop之间的相似性得分。
在上文中,hop可以是mhc-肽(完整的)hop、mhchop或肽hop,这取决于所选择的方法。这可以针对多种相似性来进行,使得
h=h基本的+hsima+hsimb+...,(15)
只要相似性之间没有重叠元素(如果重叠,则必须应用一些规则以确保元素为0至1)。
通常,序列相似性仅被应用于“测试”矩阵,因为查询序列确定我们需要计算预测的相似性。由于nbrsim局限于对已经存在于训练数据中的mhc和肽hop进行预测,所以通常仅将邻域相似性应用于“训练”矩阵。
序列和邻域相似性可以如下地被并入预测(自相似性从nbrsim和seqsim中被去除,因为它们已经被包含在基本矩阵中):
h训练=h训练基本的+h训练nbrsim.(16)
m以与上述所讨论的总和矩阵m类似的方式从h训练找到。然后,
此外,添加锚点:
h训练=h训练⊙a。
查询矩阵是:
h测试=h测试基本的+h测试seqsim
并且如前所述,通过以下方程式找到预测值:
如上所述,如果查询肽与参考肽具有不同的长度k,则可以使用以下这些步骤预测单个k-mer的结合亲和力:
-使用前述分解方法中的一种或组合将k-mer转换成虚拟肽-mhc对。
-预测每个虚拟肽-mhc对的结合亲和力值(下文被称为“虚拟预测值”)。
-k-mer的结合亲和力通过以下之一发现:
ο选择最佳(最低ic50值)虚拟预测的值。
ο计算所有虚拟预测的平均值(平均、中值等)。
ο对虚拟预测值(如前10%、或前3个最佳值)的子集进行平均。
ο如果可以获取虚拟肽-mhc对重要性权重的集合,则进行所有虚拟预测值的加权平均。
评估结果
基本泛等位基因法
应用基本泛等位基因方法(方程式(7)和(8)),使用在图2(h)中示意性描绘并被标记为“全面的1、2、3-mer”的hop分解。我们还允许分别在方程式(4)和(5)中的锚点和进近权重值a(c)和f(n)上进行优化。使用上面概述的验证实验,我们获得了表1中所示的1、2和3-mer基本泛等位基因方法的性能。本文中的每个表都用于9-mer来说明所描述的技术。对于kim09-kim13的实验,该方法提供的auc值为0.920,pcc值为0.752,与kim095倍交叉验证的值相当。这些值超过了方法上可比较的工具(如pickpocket、nebula和snebula)的报告值。因此,有利地,本发明的方法的基本泛等位基因版本能够实现与等位基因特异性方法竞争的预测,但不需要大量的训练数据。
loao预测具有auc=0.849和pcc=0.608的总加权平均值。在一些情况下,对与训练集中的mhc等位基因共享非常少的等位基因(hla-a-01-01和hla-a-29-02)或没有共享的等位基因(hla-b-46-01)的mhchop的预测值比平均值差。
具有序列相似性推理的泛等位基因法
通过使用方程式(9)添加序列相似性推理有效地增加了训练数据的大小,其中也添加了源自具有与查询hop足够相似的序列的mhc-肽对的数据点。如上所述的,序列相似性方法可以被应用于mhchop、肽hop或两者同时进行。为了生成以下结果,我们仅考虑单因子相似性度量(即mhc和肽序列相似性),而不考虑组合上述相似性因子两者的高阶序列相似性度量。
序列相似性方法的性能稍微超过基本方法的整体性能,其中例如对于kim09-kim13的实验,auc=0.926和pcc=0.765。如表2中所示的,loao实验的结果显示了对等位基因(如hla-a-01-01、hla-a-29-02和hla-b-46-01)的显著改进,这些等位基因在训练数据中具有很少或没有准确的mhchop匹配。有利地,通过允许该方法从具有与查询等位基因/肽相似的序列片段的mhc等位基因/肽获得数据,我们发现这些等位基因的性能有了大大的提高。对于hla-a-01-01,auc从0.551增加到0.795,对于hla-b-46-01,auc从0.5(即随机)增加到0.930,对于pcc值,也有相应的增加。序列相似性loao性能的总加权平均值产生auc=0.900和pcc=0.683,这考虑到了所有预测都是对其预测子的训练数据中不包含的等位基因进行的。这些结果表明,序列相似性方法可以很好地推广到训练数据中的从头等位基因或代表性不足的等位基因。
具有邻域相似数据插补的泛等位基因法
如上所述的,邻域相似性方法与序列相似性方法一样,是对基本方法的扩展,其中包括来自mhc和肽hop的贡献,所述mhc和肽hop在参考数据中共享结合特性。
泛等位基因网络邻域相似性方法将mhc和肽hop邻域相似性添加到基本泛等位基因1、2和3-mer方法,类似于序列相似性方法。表3中显示了邻域相似性方法的性能。观察到auc和pcc两者的得分均比基本方法适度增加。得分与序列相似性方法的得分几乎相同。kim09-kim13测试得到auc=0.928,pcc=0.766。这些跨等位基因实验展示了其中在训练数据中通常充分覆盖查询hop分量的情况下的邻域方法的强度。
loao实验遵循基本方法结果(表1)的总体趋势,其中平均auc=0.850和pcc=0.616,尽管对于一些单独的等位基因具有显著变化。
具有组合相似性策略的泛等位基因法
将序列相似性和邻域相似性方法组合通过利用训练数据插补和序列相似度推理相结合,允许本发明的方法兼顾了序列和网络邻域相似性策略的优点。对于组合策略,我们允许长度大于3的n-merhop,如果测试数据与训练数据非常相似,则这主要产生轻微的性能增加,否则由于过度拟合倾向而显著降低。因此,如前所述,loao实验限于1-、2-和3-mer。组合的loao结果与序列相似性方法的结果非常相似,尽管几乎所有等位基因都有轻微的至显著的改善,这产生了平均auc=0.921和pcc=0.731的结果。
如表4中所示的,对于所有实验,组合方法改进了先前的方法,其中对于kim09-kim13的测试,auc=0.930和pcc=0.770,如图5中所示的,对于其它实验,也有类似的增加。在盲数据集解除盲之后,模型不被改变或调整,这清楚地暗示了良好的普遍性,同时还提供了优于其它方法的性能。
从上面的讨论中可以理解,与现有技术的基于单体中心口袋的方法相比,本发明方法的实施方案在预测针对训练数据库中很少或没有覆盖的等位基因的mhc-肽结合亲和力方面提供了改进的性能。现有技术方法的至少一些局限性可以通过整合来自n-merhop二分网络邻域的相似性推理和/或序列比对来克服。整合的通用框架和网络推理的性能超过方法上可比较的泛等位基因方法,达到了接近用于标准iedb数据性能测试的最佳类内神经网络方法的总体性能。平均auc得分为0.93至0.95,pcc得分为0.77至0.81,证明了所述方法的优异分类和回归能力。在loao实验中可以看到最令人信服的结果,其证明本发明方法的实施方案的强有力的效用,这证明了auc=0.92和pcc=0.73的平均性能。因此,本发明方法能够对罕见的,很少研究的mhc等位基因进行有用的结合亲和力预测。
在本文中,我们提供了该方法在疫苗设计中的明确用途。然而,应当理解,本文所述的技术同样可应用于设计识别所鉴定的靶标的定制t细胞。类似地,该技术也可以被用于鉴定肿瘤中的新抗原负荷,并且其中所述新抗原负荷被用作生物标志物,即预测对疗法的应答。
现在转到图11,其显示了适于实施该方法的实施方案的系统的实例。系统1100包括与参考数据存贮器1120通信的至少一个服务器1110。服务器还可以例如通过通信网络1140与自动肽合成装置1130通信。
在某些实施方案中,服务器可以获得多个肽的氨基酸序列和蛋白质的氨基酸序列,并使用上述步骤为每个肽确定与蛋白质的预测的结合亲和力。基于各自预测的结合亲和力,服务器可以选择多种肽中的一种或多种候选肽。
候选肽可以被发送到自动肽合成装置1130以合成肽。自动肽合成装置1130合成地生成靶表位,即在本实施例中为靶肽。用于自动肽合成的技术是本领域熟知的,并且应当理解,可以使用任何已知的技术。通常,使用标准固相合成肽化学合成靶肽,并在配制成水性溶液之前使用反相高效液相色谱纯化。如果用于疫苗接种,则在施用之前,肽溶液通常在施用于患者之前与佐剂混合
肽合成技术已经存在20多年,但是近年来已经经历了快速的改进。为了简洁起见,我们没有详细描述此类机器,但是本领域技术人员将理解它们的操作,并且此类常规机器可以适于从服务器接收候选蛋白。
服务器可以包括上述功能,以预测查询结合剂分子与查询靶分子的结合亲和力。各自的结合亲和力可以被发送到进一步的处理模块,以基于适于产生疫苗的结合亲和力来鉴定靶表位。然而,服务器也可可操作为鉴定疫苗设计的靶表位。当然,应当理解,这些功能可以在计算机网络的不同处理实体和彼此通信的不同处理模块之间被细分。例如,服务器可以通过计算机网络接收一个或多个查询分子并返回合适的结合亲和力或候选表位的集合。该查询可以从计算机网络以电子方式接收或者输入到图形用户界面。
用于预测结合亲和力的技术和基于该结合亲和力的用于鉴定候选肽的技术可以整合到更广泛的生态系统中以用于定制的疫苗开发。示例性疫苗开发生态系统在本领域中是众所周知的,并且为了上下文而在高水平上进行了描述,但是为了简洁起见,我们没有详细描述生态系统。
在示例性生态系统中,第一个样品步骤可以是从肿瘤活组织检查和匹配的健康组织对照中分离dna。在第二个序列步骤中,对数据进行测序并鉴定变体(即突变)。在免疫剖析仪步骤中,相关的突变肽可在计算机上生成。
使用相关联的突变肽和本文所述的技术,可以预测和选择新抗原,并鉴定用于疫苗设计的靶表位。即,基于使用本文所描述的技术确定的预测的结合亲和力选择的候选肽序列。
然后使用如上所述的常规技术合成生成靶表位。在施用之前,肽溶液通常在施用于患者(接种疫苗)之前与佐剂混合。
通过本文所述的方法预测的合适的靶表位也可以被用于产生除基于肽的疫苗以外的其它类型的疫苗。例如,可以将肽靶标编码入相应的dna或rna序列中,并用于接种患者。注意,dna通常被插入到质粒构建体中。可替代地,可以将dna并入细菌或病毒递送系统(也可以是rna,这也取决于病毒递送系统)的基因组中,其可以被用于接种患者,因此在基因工程化的病毒或细菌中制造的疫苗在患者内(即体内)免疫后制造靶标。
图12中显示了合适的服务器1110的实例。在该实例中,服务器包括至少一个微处理器1200、存储器1201、任选的输入/输出装置1202(如键盘和/或显示器)以及外部接口1203,它们经由如图所示的总线1204互连。在该实例中,外部接口1203可以被用于将服务器1110与外围装置(如通信网络1140、参考数据存贮器1120、其它存储装置等)连接。尽管显示了单个外部接口1203,但是这仅是出于示例的目的,并且在实践中,可以提供使用各种方法(例如,以太网、串行、usb、无线等)的多个接口。
在使用中,微处理器1200以存储在存储器1201中的应用软件的形式执行指令,以允许进行所需的过程,其包括与参考数据存贮器1120通信,以便接收和处理输入数据,和/或与客户端装置通信,以便接收用于查询结合剂分子和查询靶分子的序列数据,并根据上述方法生成结合亲和力预测值。应用软件可以包括一个或多个软件模块,并且可以在适宜的执行环境(如运算系统环境等)中执行。
因此,可以理解,服务器1200可以由任何合适的处理系统形成,诸如适合编程的客户端装置、pc、网页服务器,网络服务器等。在一个具体实例中,服务器1200是标准处理系统(如基于英特尔架构的处理系统),其执行存储在非易失性(例如,硬盘)存储装置上的软件应用程序,但这不是必需的。然而,还将理解,处理系统可以是任何电子处理装置,诸如微处理器、微芯片处理器、逻辑门配置、可选地与诸如fpga(现场可编程门阵列)的实施逻辑相关联的固件,或任何其它电子装置、系统或布置。因此,当使用术语服务器时,这仅仅是为了示例的目的,而不是为了限制。
虽然服务器1200被显示为单个实体,但是应当理解,服务器1200可以分布在多个地理上分开的位置上,例如通过使用作为基于云的环境的一部分提供的处理系统和/或数据库1201。因此,上述布置不是必需的,也可以使用其它合适的配置。
表1:基本方法的结果
表2:序列相似性方法的结果
表3:邻域相似性方法的结果
表4:组合方法的结果
- 还没有人留言评论。精彩留言会获得点赞!