使用(R)-2-(4-异丙基苯基)-N-(1-(5-(2,2,2-三氟乙氧基)吡啶-2-基)乙基)乙酰胺治疗原发性震颤的制作方法
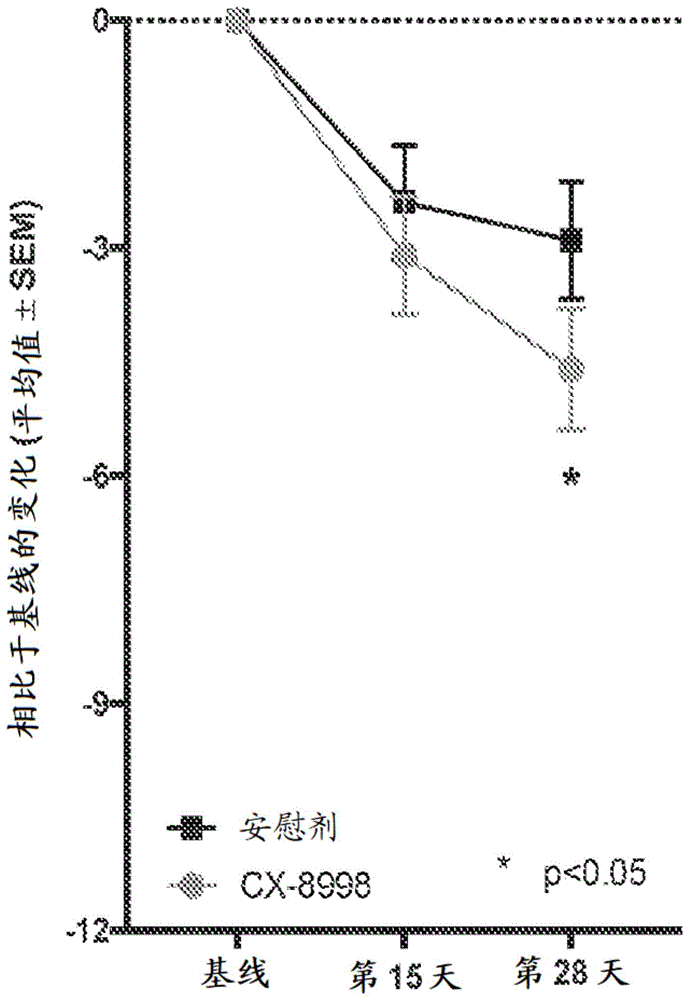
相关申请的交叉引用本申请要求于2018年10月3日提交的美国专利申请第62/740,755号的权益,并且要求于2018年12月14日提交的美国专利申请第62/780,049号的权益。先前申请的公开内容被视为本申请的公开内容的部分(并且通过引用并入其中)。本发明涉及用于治疗患有或处于患上一种或多种运动障碍(例如原发性震颤、癫痫和/或帕金森病(parkinson'sdisease))的风险下的哺乳动物的方法和材料。特别是,本文件涉及包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物,以及将所述组合物施用于患有或处于患上一种或多种运动障碍(例如原发性震颤、癫痫和/或帕金森病)的风险下的哺乳动物以治疗所述哺乳动物。
背景技术:
:原发性震颤是成人的所有运动障碍中最盛行者。在2010荟萃分析中,louis等人(1998movementdisorders.13(1):5-10)估计汇总的盛行率(所有年龄段)为0.9%,各研究之间具有在统计上显著的异质性(i2=99%,p<0.001)。据估计,≥65岁成人中的盛行率为4.6%(louis和ferreira,2010movdisord.25(5):534-541)。尽管et不会缩短预期寿命,但其对患者在家中和工作场所实施日常生活活动(adl,例如书写和进食)的能力的影响会给生活质量、社交互动和精神状态带来负面影响(lorenz等人,2006movdisord.21(8):1114-1118;louis等人,2015parkinsonismrelatdisord.21(7):729-735;george等人,1994psychosomatics.35(6):520-523;以及zesiewicz等人,2011neurology.77(19):1752-1755)。越来越多的人认识到et并非一种单症状性病症(bermejo-pareja,2011naturereviewsneurology.7(5):273-282)。对认知功能的影响是异质的,并且包括注意力、执行功能、言语流畅性、视觉空间功能、记忆和工作记忆的损害(bermejo-pareja等人,2012“v.cognitivefeaturesofessentialtremor:areviewoftheclinicalaspectsandpossiblemechanisticunderpinnings,”tremorandotherhyperkineticmovements(louis编辑)2012中的第2:02-74-541-1页)。睡眠障碍和疲劳在患有et的患者中比在年龄匹配的对照中也更为常见(chandran等人,2012actaneurolscand.125:332-7)。原发性震颤通常随着时间的推移而恶化,并且在一些人中可能是严重的。它是一种显著的失能,会影响许多日常生活活动并且可能是社交尴尬、恐惧症、抑郁症和焦虑症的来源。t型钙通道是电压活化钙通道(cav)家族的成员,其各自由具有不同生理性质的独特成孔α亚基定义,所述生理性质可通过与各种辅助亚基结合来进一步调节(zamponi等人,2015pharmacolrev.67:821-70)。t型钙通道的独特且有识别力的性质是其能够在相对低电压下在膜的较小去极化时活化,从而允许大量钙进入兴奋性细胞,从而导致进一步膜去极化,活化额外离子通道亚型,并启动动作电位。这类通道的另一个重要特征是其相对较快的失活(“t型”中的“t”是瞬态的)和从这种失活状态相对较慢的功能恢复。总之,这些性质能够使cav3通道瞬时响应于感觉输入的细微变化,并且然后迅速复位,从而使其在静息膜电位的设置中发挥关键作用,并且因此在细胞的总体兴奋性和振荡活动中发挥关键作用(iftinca等人,2009trendspharmacolsci.30:32-40)。在20世纪90年代早期发现并克隆了t型钙通道cav3、其同种型(cav3.1、cav3.2和cav3.3)及其基因cacna1g、cacna1h和cacna1i,并且阐明了其作为低阈值、电压闸控钙通道的功能(perez-reyes,1998bioenergbiomembr.30:313-8;cribbs等人,1998circres.83:103-9;lee等人,1999jneurosci.19(6):1912-21)。cav3的同种型在整个中枢神经系统(cns)和周围神经系统中表达,包括丘脑皮层途径,其中cav3.1是最常见的同种型(ertel等人,2000neuron.25:533-5)。已注意到深小脑核、黑质、外苍白球、内苍白球、底丘脑核(stn)在健康宿主中具有振荡,并且在具有神经系统病理状况的动物和人类中具有过度节律。已发现,cav3是在震颤、神经病性疼痛、癫痫和帕金森病中发现的病理生理状态下亚阈值振荡和过度节律的介体(llinás,2003anracadnacmed(madr).120:267-90;handforth等人,2005epilepsia.46:1860-70;llinás等人,2007procnatlacadsci.104:17819-24;park等人,2013frontneuralcircuits.7:172)。下橄榄体(io)似乎起到震颤发生器的作用,并且动物模型表明io起到固有心律调节器的作用(long等人,2002jneurosci.22:10898-905)。原发性震颤(et)可能是由于io中神经元群体的过度节律性同步放电引起,其影响小脑的功能(elbe等人,1996movdisord.11:70-78)。cav3在io和小脑中高度表达。cav3.1是在io中表达的主要cav3同种型。在小脑系统内,它也在purkinje细胞体、深小脑核、星状、篮状、树突状和高尔基体细胞上发现(molineux等人,2006procnatlacadsciusa.103:5555-60)。在这些位置中,cav3起到震颤发生器和持续节律心律调节器的作用(park等人,2010procnatlacadsciusa.107:10731-6)。技术实现要素:本文件提供了用于治疗患有或处于患上运动障碍(例如原发性震颤、癫痫和/或帕金森病)的风险下的哺乳动物的方法和材料。例如,可向有需要的哺乳动物(例如患有或处于患上运动障碍的风险下的哺乳动物)施用包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物以治疗所述哺乳动物。cx-8998是高度选择性电压活化的cav拮抗剂。如本文所表明,包含cx-8998的组合物的口服剂型可被配制以包括被设计用于延迟和/或持续cx-8998释放的第一组分和被设计用于立即cx-8998释放的第二组分,使得在经口施用于哺乳动物(例如具有运动障碍的哺乳动物)时,剂型可持续约12小时(例如对于每日两次投药)或约24小时(例如对于每日一次投药)为哺乳动物提供在治疗有益范围内的cx-8998的血浆水平。当前,普萘洛尔(propranolol)是美国唯一批准用于治疗原发性震颤的药物。2011年神经病学研究院(academyofneurology)关于原发性震颤治疗的循证指南更新缺乏任何新的积极建议(与2005年指南相比),证明目前的药物发现方法收效不佳(zesiewicz等人,2011neurology.77(19):1752-1755)。因此,以治疗量递送一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的能力为治疗患有原发性震颤的人类提供独特而未实现的机会。此外,以非侵袭性方式递送一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998),同时维持治疗量持续一段时间,可使白天的功效最大化,限制cmax有关的不良事件,并且限制夜间睡眠障碍(以及潜在激素效应),同时减少治疗益处所需的投药频率。一般来说,本文件的一个方面特征在于包含cav3拮抗剂或其药学上可接受的盐的口服剂型,其中所述剂型包括包含cav3拮抗剂的受控释放组分,并且其中所述剂型任选地含有包含cav3拮抗剂的立即释放组分;其中所述口服剂型在施用于约35岁或以上的人时可有效地提供:a)cav3拮抗剂在稳态下的cmax不大于24小时内平均血浆浓度的两倍;以及b)cav3拮抗剂在稳态下的血浆浓度为约400nm至约1000nm达至少18小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂在稳态下的cmax为24小时内平均血浆浓度的1至2倍。所述口服剂型可每日一次施用于人。所述cav3拮抗剂可具有以下结构:cav3拮抗剂可包括盐酸盐。口服剂型可为胶囊、丸剂、片剂或悬浮液。受控释放组分可包括粒子、片剂、微型片剂、溶液、悬浮液、胶囊或它们的混合物。粒子可为颗粒、丸粒、珠粒、微粒或纳米粒子或它们的混合物。口服剂型可包括胶囊。胶囊可为明胶胶囊或羟丙基甲基纤维素胶囊。受控释放组分中包含cav3拮抗剂的多个粒子中的至少一个粒子可包括包含ph敏感性肠溶聚合物的包衣。在一些情况下,ph敏感性肠溶聚合物包衣的至少一部分可在约6的ph下溶解。ph敏感性肠溶聚合物包衣可在约6的ph下溶解,ph敏感性肠溶聚合物可包括以重量计约6.25%的甲基丙烯酸共聚物l、约6.25%的甲基丙烯酸共聚物s、约1.25%的柠檬酸三乙酯和约6.25%的滑石。在一些情况下,ph敏感性肠溶聚合物包衣的至少一部分可在约7的ph下溶解。ph敏感性肠溶聚合物包衣可在约7的ph下溶解,ph敏感性肠溶聚合物可包括以重量计约17.4%的fs30d和约2.6%的t20。受控释放组分可包括以重量计约5.0%的cav3拮抗剂、约57.7%的乳糖一水合物、约25.0%的交聚维酮、约8.0%的无水柠檬酸、约2.0%的月桂基硫酸钠(sls)、约2.0%的羟丙基纤维素(hpc)、约0.3%的丁基化羟基苯甲醚(bha)和约0.1%的丁基化羟基甲苯(bht)。口服剂型还可包括立即释放组分,其中所述立即释放组分包括多个包含cav3拮抗剂的粒子。立即释放组分中的一个或多个粒子可包括以重量计约5.0%的cav3拮抗剂、约57.7%的乳糖一水合物、约25.0%的交聚维酮、约8.0%的无水柠檬酸、约2.0%的sls、约2.0%的hpc、约0.3%的bha和约0.1%的bht。在口服剂型施用于人时,立即释放组分中的一个或多个粒子可在施用45分钟内释放立即释放组分中存在的cav3拮抗剂的至少80%。口服剂型可包括立即释放组分中约30%的cav3拮抗剂和受控释放组分中约70%的cav3拮抗剂。口服剂型在施用于人时可在30分钟内有效达到cav3拮抗剂的cmax的至少25%。cav3拮抗剂可降低t型钙通道的活性。在另一方面,本文件提供了包含cav3拮抗剂或其药学上可接受的盐的口服剂型,其中所述剂型包括包含cav3拮抗剂的受控释放组分,并且其中所述剂型任选地含有包含cav3拮抗剂的立即释放组分;其中所述口服剂型在施用于小于约35岁的人时可有效地提供:a)cav3拮抗剂在稳态下的cmax不大于24小时内平均血浆浓度的2.5倍;以及b)cav3拮抗剂在稳态下的血浆浓度为约400nm至约1000nm达至少15小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂在稳态下的cmax为24小时内平均血浆浓度的1至2.5倍。所述口服剂型可每日一次施用于人。所述cav3拮抗剂可具有以下结构:cav3拮抗剂可包括盐酸盐。口服剂型可为胶囊、丸剂、片剂或悬浮液。受控释放组分可包括粒子、片剂、微型片剂、溶液、悬浮液、胶囊或它们的混合物。粒子可为颗粒、丸粒、珠粒、微粒或纳米粒子或它们的混合物。口服剂型可包括胶囊。胶囊可为明胶胶囊或羟丙基甲基纤维素胶囊。受控释放组分中包含cav3拮抗剂的多个粒子中的至少一个粒子可包括包含ph敏感性肠溶聚合物的包衣。在一些情况下,ph敏感性肠溶聚合物包衣的至少一部分可在约6的ph下溶解。ph敏感性肠溶聚合物包衣可在约6的ph下溶解,ph敏感性肠溶聚合物可包括以重量计约6.25%的甲基丙烯酸共聚物l、约6.25%的甲基丙烯酸共聚物s、约1.25%的柠檬酸三乙酯和约6.25%的滑石。在一些情况下,ph敏感性肠溶聚合物包衣的至少一部分可在约7的ph下溶解。ph敏感性肠溶聚合物包衣可在约7的ph下溶解,ph敏感性肠溶聚合物可包括以重量计约17.4%的fs30d和约2.6%的t20。受控释放组分可包括以重量计约5.0%的cav3拮抗剂、约57.7%的乳糖一水合物、约25.0%的交聚维酮、约8.0%的无水柠檬酸、约2.0%的sls、约2.0%的hpc、约0.3%的bha和约0.1%的bht。口服剂型还可包括立即释放组分,其中立即释放组分包括多个包含cav3拮抗剂的粒子。立即释放组分中的一个或多个粒子可包括以重量计约5.0%的cav3拮抗剂、约57.7%的乳糖一水合物、约25.0%的交聚维酮、约8.0%的无水柠檬酸、约2.0%的sls、约2.0%的hpc、约0.3%的bha和约0.1%的bht。在口服剂型施用于人时,立即释放组分中的一个或多个粒子可在施用45分钟内释放立即释放组分中存在的cav3拮抗剂的至少80%。口服剂型可包括立即释放组分中约30%的cav3拮抗剂和受控释放组分中约70%的cav3拮抗剂。口服剂型在施用于人时可在30分钟内有效达到cav3拮抗剂的cmax的至少25%。cav3拮抗剂可降低t型钙通道的活性。在另一方面,本文件提供了包含cav3拮抗剂或其药学上可接受的盐的口服剂型,其中所述剂型可包括胶囊,所述胶囊包括:a)包括一个或多个包含cav3拮抗剂的颗粒的立即释放组分,以及b)包括一个或多个包含cav3拮抗剂的颗粒的受控释放组分;其中所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少12小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少18小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达约12小时至约24小时。所述cav3拮抗剂可具有以下结构:cav3拮抗剂可包括盐酸盐。cav3拮抗剂可降低t型钙通道的活性。在另一方面,本文件提供了包含cav3拮抗剂或其药学上可接受的盐的口服剂型,其中所述剂型可包括片剂,所述片剂包括:a)包括一个或多个包含cav3拮抗剂的颗粒的立即释放组分,以及b)包括一个或多个包含cav3拮抗剂的颗粒的受控释放组分;其中所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少12小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少18小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达约12小时至约24小时。所述cav3拮抗剂可具有以下结构:cav3拮抗剂可包括盐酸盐。cav3拮抗剂可降低t型钙通道的活性。在另一方面,本文件提供了包含cav3拮抗剂或其药学上可接受的盐的口服剂型,其中所述剂型包括胶囊,所述胶囊包括:a)包含cav3拮抗剂的立即释放组分,以及b)包括一个或多个包含cav3拮抗剂的片剂的受控释放组分;其中所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少12小时。立即释放组分可包括多个颗粒。立即释放组分可包括多个珠粒和/或丸粒。一个或多个片剂可包括一个或多个微型片剂。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少18小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达约12小时至约24小时。所述cav3拮抗剂可具有以下结构:cav3拮抗剂可包括盐酸盐。cav3拮抗剂可降低t型钙通道的活性。在另一方面,本文件提供了包含cav3拮抗剂或其药学上可接受的盐的口服剂型,其中所述剂型可包括胶囊,所述胶囊包括:a)包含cav3拮抗剂的立即释放组分,其中立即释放组分可为液体,以及b)包含cav3拮抗剂的受控释放组分,其中受控释放组分包括一个或多个固体组分,其可为颗粒、珠粒、丸粒和/或微型片剂;其中所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少12小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少18小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达约12小时至约24小时。所述cav3拮抗剂可具有以下结构:cav3拮抗剂可包括盐酸盐。cav3拮抗剂可降低t型钙通道的活性。在另一方面,本文件提供了包含cav3拮抗剂或其药学上可接受的盐的口服剂型,其中所述剂型包括含有内部胶囊的胶囊,其中内部胶囊装配在口服剂量胶囊内,使得在内部胶囊的外表面与口服剂量胶囊的内表面之间存在空间;其中内部胶囊的外表面与口服剂量胶囊的内表面之间存在的空间可含有包含cav3拮抗剂的立即释放组分,其中所述立即释放组分可为液体;其中内部胶囊含有包含cav3拮抗剂的受控释放组分;并且其中所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少12小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少18小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达约12小时至约24小时。所述cav3拮抗剂可具有以下结构:cav3拮抗剂可包括盐酸盐。cav3拮抗剂可降低t型钙通道的活性。在另一方面,本文件提供了包含cav3拮抗剂或其药学上可接受的盐的口服剂型,其中所述剂型包括悬浮液,所述悬浮液包括:a)包含cav3拮抗剂的立即释放组分,其中立即释放组分是液体,以及b)包含cav3拮抗剂的受控释放组分,其中受控释放组分包括一个或多个固体组分,其可为颗粒、珠粒、丸粒和/或微型片剂;其中一个或多个固体组分悬浮于液体内;并且其中所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少12小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少18小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达约12小时至约24小时。所述cav3拮抗剂可具有以下结构:cav3拮抗剂可包括盐酸盐。cav3拮抗剂可降低t型钙通道的活性。在另一方面,本文件提供了包含cav3拮抗剂或其药学上可接受的盐的口服剂型,其中所述剂型包括悬浮液,所述悬浮液包括:a)液相、b)包含cav3拮抗剂的立即释放组分,其中立即释放组分包括一个或多个固体组分,其可为颗粒、珠粒、丸粒和/或微型片剂;其中一个或多个固体组分悬浮于液体内,以及c)包含cav3拮抗剂的受控释放组分,其中受控释放组分包括一个或多个固体组分,其可为颗粒、珠粒、丸粒和/或微型片剂;其中一个或多个固体组分悬浮于液体内;并且其中所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少12小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达至少18小时。所述口服剂型在施用于人时可有效维持cav3拮抗剂的平均血浆浓度为约400nm至约1000nm达约12小时至约24小时。所述cav3拮抗剂可具有以下结构:cav3拮抗剂可包括盐酸盐。cav3拮抗剂可降低t型钙通道的活性。在另一方面,本文件提供了治疗患有运动障碍的患者的方法。所述方法可包括以下,或基本上由以下组成:向有需要的患者(例如患有运动障碍的患者)施用本文提供的口服剂型。患者可为35岁或以上的成年人。患者可为小于约35岁的成年人。运动障碍可为原发性震颤、伴有失神型癫痫发作的特发性全身性癫痫或与帕金森病相关的震颤。口服剂型可在6am与中午之间施用(例如可每日一次施用)于患者。口服剂型可在睡醒后4小时内施用(例如可每日一次施用)于患者。受控释放组分可包括包含cav3拮抗剂的多个粒子,其中受控释放组分中包含cav3拮抗剂的多个粒子中的至少一个粒子包括包含ph敏感性肠溶聚合物的包衣,并且其中ph敏感性肠溶聚合物包衣的至少一部分在肠内ph下溶解。在施用口服剂型之前,患者可禁食至少4小时。cav3拮抗剂可有效减少或消除与运动障碍相关的震颤。与仅施用cav3拮抗剂的立即释放组分的人相比,患者可经历减少的不良事件。不良事件可为眩晕、头痛、亢奋、注意力障碍、感觉异常、幻觉、失眠、口腔干燥、味觉障碍、感觉迟钝、嗜眠、嗜睡、睡眠障碍、恶心、呕吐、静坐不能、意识水平下降、晕厥、记忆障碍、焦虑、不安定、疲劳、易怒、便秘、耳鸣、厌食、情绪障碍、性无能、复视、眼球震颤、困倦、麻疹样皮疹、粒细胞减少、粒细胞缺乏、红细胞发育不全、不发育或其任何组合。除非另有定义,否则本文所用的所有技术和科学术语都具有与本发明所属领域的技术人员通常所理解的含义相同的含义。举例来说,除非另有明确阐述,否则术语“例如”和“诸如”及其语法等效形式、短语“并且不限于”应理解为如下。举例来说,除非上下文另外明确指明,否则单数形式“一个”、“一种”和“所述”包括复数个指示物。举例来说,术语“约”是指“大约”(例如加上或减去所指示值的大约10%)。尽管可使用类似或等效于本文所述的方法和材料的方法和材料来实践本发明,但下文阐述合适的方法和材料。应了解,为清楚起见而阐述于单独实施方案上下文中的本发明的某些特征也可在单一实施方案中组合提供。相反,为简便起见在单一实施方案上下文中阐述的本发明的各种特征也可单独或以任何合适的子组合提供。本文所提及的所有出版物、专利申请、专利和其他参考文献都通过引用整体并入本文。倘若出现冲突,则以本说明书(包括定义)为准。另外,材料、方法和实施例仅为阐释性而无意具有限制性。在附图和以下说明中陈述本发明的一个或多个实施方案的细节。根据本说明和附图以及权利要求书,本发明的其他特征、目标和优点将变得显而易见。附图说明图1是显示如由研究者评定的震颤研究组(trg)原发性震颤等级评估量表(tetras)–表现子量表(ps)中从基线至第28天的变化的图。图2是显示tetras-日常生活活动(adl)从基线至第15天和第28天的变化的图。图3是显示tetras总评分从基线至第15天和第28天的变化的图。图4是显示tetras-ps螺线任务从基线至第28天的变化的图。图5是显示通过数字螺线描记法测量的功能性震颤频率从基线至第15天和第28天的变化的图。图6是显示在第28天临床整体改善印象(cgi-i)相比于安慰剂的差异的图。图7含有显示在第15天和第28天的患者整体变化印象(pgic)相比于安慰剂的差异的图。图8是显示目标达成量表(gas)相比于安慰剂的差异的图。图9是显示在第15天和第28天对抗震颤药物感到满意的个体的百分比相对于安慰剂增加的图(quest子项目)。图10是显示在第15天和第28天产生临床功效的cx-8998血浆浓度的图。图11a-11d含有显示在每日两次(bid)投用cx-8998后cx-8998代谢物暴露的图。图11a显示cx-8998代谢物m01。图11b显示cx-8998代谢物m02。图11c显示cx-8998代谢物m03。图11d显示cx-8998代谢物m04。图12是显示cx-8998血浆浓度与反应之间的关系的图。基线和安慰剂反应由阴影区表示。图13含有显示利用剂量滴定对不良事件的耐受性的图。图14含有显示安全性分析集中的心电图离群值的概述的图。图15含有显示安全性分析集中的cx-8998与安慰剂之间无差异的图。图16是医师评定的全分析集中的tetras表现子量表总评分的亚组分析的数据集和森林图。图17是全分析集中的tetras日常生活活动子量表的亚组分析的数据集和森林图。图18含有显示代谢物m01、m02、m03和m04的部分分离和次最佳峰宽度的cx-8998的色谱图。图19含有显示产生差的峰形的不期望次要相互作用的cx-8998的色谱图。图20含有显示cx-8998及其代谢物m01、m02、m03和m04的近基线分离的cx-8998的色谱图。图21含有显示在碱性条件下利用乙酸酐的乙酰化似乎对于m04具有选择性并且显示高反应产率的色谱图。图22是显示在个别分析物反应对混合分析物反应中形成的m04-ac在50℃下的反应时程的图。图23是显示人血浆提取物中在50℃进行30分钟衍生反应期间乙酸酐或吡啶增加后剩余的m04百分比的图。图24是显示基于峰面积的含有个别分析物的样品中在50℃下的反应时程的图。图25是人血浆样品中cx-8998、m01、m02、m03和m04的代表性色谱图。图26是显示从立即释放(ir)珠粒释放cx-8998的图。图27含有显示单一日剂量的cx-8998的立即释放和日剂量的稳态的曲线的图。图28含有显示老年男性中单一剂量和年轻男性中7个日剂量的cx-8998的释放曲线的图。图29是显示年龄对cx-8998释放的影响的图。图30是显示年龄对药代动力学(pk)的影响的图。图31含有老年患者中单一早晨剂量后cx-8998的血浆浓度的图。图32含有显示输出相关性的图。图33含有年轻患者中单一早晨剂量后cx-8998的血浆浓度的图。图34含有显示输出相关性的图。图35含有年轻患者中多个剂量后cx-8998的血浆浓度的图。图36含有显示爆发服用约6mg并接着在12小时内以几乎恒定速率服用约12mg的年轻成年男性的释放曲线的图。图37含有显示爆发服用约7mg并接着在12小时内以稍微减小速率服用约12mg的老年成年男性的释放曲线的图。图38含有显示从渗透泵片剂的零级释放的图。图39含有显示从胶囊中具有包衣珠粒的基质片剂的一级释放的图。图40含有显示从具有ir释放包衣的渗透泵片剂的爆发和零级释放的图。图41含有显示从具有ir包衣的基质片剂中ir珠粒和缓慢释放珠粒的混合物的爆发和一级释放的图。图42含有显示从具有肠溶包衣和ir包衣的ir片剂的ir珠粒和ph6肠溶包衣珠粒的混合物延迟3小时的双重爆发的图。图43含有显示从具有肠溶包衣和ir包衣的ir片剂的ir珠粒和ph7肠溶包衣珠粒的混合物延迟6小时的双重爆发的图。图44含有显示tetras和整体反应者中cx-8998的目标范围暴露的图。图45含有显示剂量下限的图。图46是显示在施用cx-899810mgbid后血浆和tam中总cx-8998和未结合cx-8998的目标暴露水平和pk曲线的图。图47a-47b显示示例性t-calm设计。图47a含有示例性t-calm设计的示意图。图47b含有示例性t-calm设计的流程图。图48a-48d含有显示研究者评定的tetras-ps相比于基线的变化的图。图49a-49d含有显示tetras-adl和总tetras相比于基线的变化的图。图50a-50b含有显示第28天的临床整体改善印象的图。图51含有显示tetras表现子评分的差异的图。上图显示通过项目的差异评分,其中负的评分指示由独立视频评定者(cr)的评分比研究者(pr)低。上图显示所有访视间的差异评分。图52含有显示cx-8998游离碱(fb)和cx-8998hcl制剂的固有溶解的图。图53含有显示从ir珠粒的释放的图。图54a-54b含有显示从具有第一制剂(图54a)和第二制剂(图54b)的ir珠粒的溶解速率的图。图55a-55b含有显示在酸性条件(图55a)和ph中性条件(图55b)中cx-8998制剂的溶解速率的图。图56含有显示ir珠粒、mr1珠粒和mr2珠粒的cx-8998释放曲线的图。图57a-57c含有显示在储存后ir珠粒(图57a)、mr1珠粒(图57b)和mr2珠粒(图57c)的cx-8998释放曲线的图。图58含有显示老年人中各种比率的ir和mr1珠粒的模拟溶解速率的图。图59含有显示老年人中各种比率的ir和mr2珠粒的模拟溶解速率的图。图60含有显示非老年人中各种比率的ir和mr1珠粒的模拟溶解速率的图。图61含有显示非老年人中各种比率的ir和mr2珠粒的模拟溶解速率的图。图62a-62c含有显示各种剂型25mg(图62a)、15mg(图62b)和8mg(图62c)的具有40%ir、25%mr1(ph6)和35%mr2(ph7)的制剂的模拟溶解速率的图。图63a-63b含有显示在单一剂量施用cx-8998(1×8mg)后cx-8998的cmax(图63a)和auc0-72h值(图63b)的示例性分布的图。图64a-64d含有显示在单一剂量施用cx-8998(1×8mg)后不良事件的起始时间(图64a)、消退时间(图64b)、持续时间(图64c)和起始集中度(图64d)的图。图65含有显示与cns和精神病学不良事件集中度相比的单一剂量施用ircx-8998(1×8mg)的pk曲线的图。阴影区域表示第5-第95百分位数。图66含有显示与cns和精神病学不良事件集中度相比单一剂量施用mr1cx-8998(1×8mg)的pk曲线的图。阴影区域表示第5-第95百分位数。图67含有显示与cns和精神病学不良事件集中度相比单一剂量施用mr2cx-8998(1×8mg)的pk曲线的图。阴影区域表示第5-第95百分位数。图68含有显示在治疗期期间的不良事件的图。图69含有显示来自批次l364-01036的ircx-8998珠粒的示例性溶解曲线的图。图70含有显示来自批次l364-01037的cr(ph6)cx-8998珠粒的示例性溶解曲线的图。图71含有显示来自批次l364-01038的cr(ph7)cx-8998珠粒的示例性溶解曲线的图。图72a-72c含有显示在单一剂量施用ircx-8998(图72a)、mr1cx-8998(图72b)或mr2cx-8998(图72c)后cx-8998、代谢物m01和代谢物m02的血浆浓度的图。图73a-73b含有显示在禁食对进食患者中平均(图73a)和对于个别患者(图73b)的cx-8998血浆浓度随时间(以小时计)的图,其中菱形表示禁食状态样品并且三角形表示进食状态样品。图74含有显示mr2cx-8998的示例性每日一次投用和ircx-8998的每日两次投用的图。在各个附图中,相同的参考符号指示相同的元件。具体实施方式本文件提供了用于治疗患有或处于患上运动障碍(例如原发性震颤、癫痫和/或帕金森病)风险下的哺乳动物(例如人类患者)的方法和材料。在一些情况下,本文件提供了包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物,以及所述组合物在制造用于治疗运动障碍(例如原发性震颤、癫痫和/或帕金森病)的药物中的用途。在一些情况下,包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物可为包括组合物的一种或多种组分的受控释放组合物,其中每一组分可被配制以在施用于人类患者时调节一种或多种t型钙通道拮抗剂从组合物的释放速率(例如以达到特异性靶标药代动力学结果)。例如,包含cx-8998的组合物可为具有被设计用于延迟和/或持续cx-8998释放的第一组分和任选地被设计用于立即cx-8998释放的第二组分的口服剂型,使得所述剂型在经口施用于哺乳动物(例如患有运动障碍的人类患者)时可为哺乳动物提供治疗有效血浆水平的cx-8998达约12小时或约24小时。在一些情况下,包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物可被设计以在施用于人类患者时提供特异性靶标药代动力学结果。例如,由于当人类患者中的血浆浓度过高时,t型钙通道拮抗剂可引起不期望的症状(例如不良作用或副作用),并且由于当血浆浓度过低时,t型钙通道拮抗剂可具有极小作用或无作用(例如极小或无治疗作用),因此期望所述剂型在施用于人类患者后可提供高于有效最小浓度的t型钙通道拮抗剂的血浆浓度,同时最小程度地超过最大浓度,高于所述最大浓度,某些不期望症状可变得更常见,并且可持续延长时间段。例如,包括一种或多种t型钙通道拮抗剂的组合物在作为单一口服剂量施用时可在人类患者中产生平均不超过1200nm的最大血浆浓度,同时维持血浆浓度平均高于400nm,并且可维持所述血浆浓度达至少12小时、至少15小时或至少18小时。在一些情况下,本文件提供了使用包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物的方法。例如,可将包括一种或多种t型钙通道拮抗剂的组合物(例如具有被设计用于延迟和/或持续cx-8998释放的第一组分和任选地被设计用于立即cx-8998释放的第二组分的口服剂型,使得所述剂型在经口施用于患有运动障碍的患者时可为患者提供治疗有效血浆水平的cx-8998达约12小时)施用于患有或处于患上运动障碍风险下的患者以治疗所述患者。本文提供的方法和材料可用于在长时间段(例如约24小时)内为有需要的患者提供持续cx-8998作用。例如,当包含cx-8998的组合物的口服剂型包括被设计用于延迟和/或持续cx-8998释放的第一组分和被设计用于立即cx-8998释放的第二组分时,立即释放组分可有效地在患者内快速达到cx-8998的治疗有效血浆水平,并且延迟和/或持续释放组分可有效地在患者内维持cx-8998的治疗有效血浆水平。组合物本文件提供了包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)。在一些情况下,t型钙通道拮抗剂可抑制(例如可降低或消除)t型钙通道的活性。在一些情况下,本文件提供了包含cx-8998的组合物。cx-8998(也称为mk-8998)是可抑制cav3(例如可以纳摩尔浓度(nm)效能抑制cav3的)高度选择性电压活化的钙通道(cav)拮抗剂,并且对于cav3具有高度选择性(例如针对其他离子通道靶标具有>100倍选择性)。cx-8998可用于剂量依赖性震颤减轻、减少和/或消除癫痫发作和/或减少和/或消除疼痛。如本文所用,术语“cx-8998”也可指cx-8998结构类似物,条件是cx-8998结构类似物维持如本文所述的cx-8998的药物功能(例如剂量依赖性震颤减轻、减少和/或消除癫痫发作和/或减少和/或消除疼痛)。cx-8998的化学名称包括(但不限于)(r)-2-(4-异丙基苯基)-n-(1-(5-(2,2,2-三氟乙氧基)吡啶-2-基)乙基)乙酰胺和2-(4-异丙基苯基)-n-{1r)-1-(5-(2,2,2-三氟乙氧基)吡啶-2-基)乙基}乙酰胺盐酸盐。cx-8998的化学结构如下文所示。cx-8998可呈cx-8998的任何适当形式。在一些情况下,cx-8998可呈碱形式(例如化合物的游离碱形式)。在一些情况下,cx-8998可呈盐形式(例如化合物的盐形式)。在cx-8998是盐的情况下,cx-8998盐可为任何适当盐。cx-8998盐可包括与任何适当酸(例如盐酸、柠檬酸、氢溴酸、马来酸、磷酸、硫酸、富马酸和酒石酸)形成的盐。例如,cx-8998可为cx-8998盐酸盐(例如cx-8998-hcl)。在一些情况下,cx-8998可被氘化。在一些情况下,cx-8998可呈cx-8998多形体形式。在一些情况下,cx-8998可呈cx-8998的结构异构体(例如cx-8998互变异构体)的形式。在一些情况下,本文提供的组合物(例如药学上可接受的组合物)(例如包括一种或多种t型钙通道拮抗剂如cx-8998的组合物)的一种或多种组分可跨越血脑屏障。例如,当组合物包括cx-8998时,cx-8998可跨越血脑屏障。例如,在包括cx-8998的组合物也包括一种或多种额外组分时,cx-8998可从组合物释放并且可跨越血脑屏障。例如,在包括cx-8998的组合物也包括一种或多种额外组分时,cx-8998可从组合物释放并且可跨越血脑屏障,同时一种或多种额外组分不跨越血脑屏障。在一些情况下,本文提供的组合物(例如药学上可接受的组合物)(例如包括一种或多种t型钙通道拮抗剂如cx-8998的组合物)的一种或多种组分不跨越血脑屏障。例如,在包括cx-8998的组合物也包括一种或多种额外组分时,cx-8998可从组合物释放并且可跨越血脑屏障,同时一种或多种额外组分不跨越血脑屏障。除了t型钙通道拮抗剂之外或代替t型钙通道拮抗剂,包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)可包括t型钙通道拮抗剂的代谢物。在一些实现方式中,本文所述组合物中存在的t型钙通道拮抗剂可代谢成(例如在向哺乳动物施用组合物后由哺乳动物代谢)t型钙通道拮抗剂的一种或多种代谢物。在一些情况下,t型钙通道拮抗剂的代谢物(例如cx-8998的代谢物)可为t型钙通道拮抗剂(例如cav3拮抗剂)。例如,当组合物包括cx-8998时,组合物可包括一种或多种cx-8998代谢物,和/或cx-8998可代谢成一种或多种cx-8998代谢物。cx-8998代谢物可为任何适当代谢物。在一些情况下,代谢物可结合至蛋白质(例如血浆蛋白质)。在一些情况下,代谢物可为游离的(例如不结合至任何蛋白质)。在一些情况下,代谢物可跨越血脑屏障(例如可存在于脑脊髓液(csf)和/或cns中)。cx-8998代谢物的实例包括(但不限于)代谢物01(m01)、m02、m03和m04。在一些情况下,本文所述组合物可包括和/或代谢成m01和m02。示例性cx-8998代谢物的化学结构如下文所示。在一些情况下,包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)中的t型钙通道拮抗剂可对所有3个cav3同种型具有选择性(例如可选择性结合至所有3个cav3同种型)。“选择性”在此上下文中是指与其他电压活化的钙通道(例如高电压活化的通道,例如l型心脏通道(cav1))相比和/或与其他类型的离子通道(例如氯离子通道、钾通道和钠通道)相比,cav3拮抗剂在拮抗cav3通道方面更强效。选择性可使用任何适当方法测定。例如,选择性可通过比较cav3拮抗剂在抑制第一类型的离子通道(例如cav3通道)中的ic50与其在抑制第二类型的离子通道(例如钠通道)中的ic50来测定。如果抑制第一类型的通道的ic50低于抑制第二类型的通道的ic50,则cav3拮抗剂可被视为对第一类型的通道具有选择性。ic50比率为0.1(或更低)表示10倍(或更大)选择性。ic50比率为0.01(或更低)表示100倍(或更大)选择性。ic50比率为0.001(或更低)表示1000倍(或更大)选择性。在一些情况下,与其他类型的离子通道相比,本文所述组合物中的t型钙通道拮抗剂对cav3的选择性可为10倍或更大、100倍或更大、或1000倍或更大。例如,在本文所述组合物中的t型钙通道拮抗剂是cx-8998时,相对于其他离子通道,组合物可具有大于100倍选择性。在一些情况下,本文所述组合物中的t型钙通道拮抗剂可选择性靶向一个或多个cav3同种型(例如cav3.1、cav3.2和/或cav3.3)。在一些情况下,本文所述组合物可选择性靶向所有三个cav3同种型(例如cav3.1、cav3.2和cav3.3)。在一些情况下,本文所述组合物(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)中的t型钙通道拮抗剂可以状态依赖性方式对cav3具有选择性。在一些情况下,本文所述组合物中的t型钙通道拮抗剂对cav3的选择性在超极化条件下与去极化条件相比高约29倍至约45倍。例如,在本文所述组合物中的t型钙通道拮抗剂是cx-8998时,cx-8998可对cav3(例如cav3.3)具有选择性,在去极化条件下可具有约3.6nm的ic50,并且在超极化条件下可具有约161nm的ic50。例如,在本文所述组合物中的t型钙通道拮抗剂是cx-8998时,cx-8998在去极化条件下可具有约84nm的拮抗cav3.3的ic50,并且在超极化条件下可具有约2.4μm的拮抗cav3.3的ic50。例如,在本文所述组合物中的t型钙通道拮抗剂是cx-8998时,cx-8998可对cav3.1具有选择性,在去极化条件下可具有约69nm的ic50,并且在超极化条件下可具有约2.4μm的ic50。在一些情况下,包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)中的t型钙通道拮抗剂可为强效cav3拮抗剂。例如,在本文所述组合物中的t型钙通道拮抗剂是cx-8998时,cx-8998在拮抗cav3方面可比其他离子通道更强效。例如,cx-8998可具有nm功效(例如cx-8998的nm值足以拮抗cav3)。在一些情况下,本文所述组合物中的t型钙通道拮抗剂可强效地抑制一个或多个cav3同种型(例如cav3.1、cav3.2和/或cav3.3)。在一些情况下,本文所述组合物中的t型钙通道拮抗剂可强效地抑制所有三个cav3同种型(例如cav3.1、cav3.2和cav3.3)。包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)可包括一种或多种cav3拮抗剂作为唯一活性成分。包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)可包括一种或多种t型钙通道拮抗剂以及一种或多种额外活性成分。例如,组合物可包括一种或多种t型钙通道拮抗剂以及用于治疗一种或多种运动障碍(例如原发性震颤、癫痫和/或帕金森病)的一种或多种额外药剂。用于治疗一种或多种运动障碍的药剂的实例包括(但不限于)β阻断剂(例如普萘洛尔(propranolol))、抗癫痫发作药物(例如扑米酮(primidone)、加巴喷丁(gabapentin)、托吡酯(topiramate)、唑尼沙胺(zonisamide)、乙琥胺(ethosuximide)、普瑞巴林(pregabalin)、丙戊酸盐、苯妥英(phenytoin)和米贝地尔(mibefradil))、安定药(例如阿普唑仑(alprazolam)和可那氮平(clonazepam)),施用一种或多种多巴胺激动剂(例如卡比多巴-左旋多巴(carbidopa-levodopa)、普拉克索(pramipexole)、罗匹尼罗(ropinirole)、罗替戈汀(rotigotine)和阿朴吗啡(apomorphine))、单胺氧化酶b(maob)抑制剂(例如司来吉兰(selegiline)、雷沙吉兰(rasagiline)和沙非酰胺(safinamide))、儿茶酚o-甲基转移酶(comt)抑制剂(例如恩卡朋(entacapon)和托卡朋(tolcapone))和抗副交感神经药(例如苯托品(benztropine)和三己芬迪(trihexyphenidyl))。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可包括一种或多种额外组分(例如一种或多种无活性成分)。例如,可将治疗有效量的cx-8998配制到包括一种或多种额外组分的组合物中并且施用于患有或处于患上一种或多种运动障碍(例如原发性震颤、癫痫和/或帕金森病)风险下的哺乳动物以治疗所述哺乳动物。用于配制本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂)的任何额外组分(例如药学上可接受的载体)可具有高纯度并且可基本上不含潜在有害的污染物(例如至少国家配方(nf)级和/或美国药典(usp)级)。例如,当打算施用于人类时,本文所述组合物可在如美国食品药品管理局(u.s.foodanddrugadministration)和/或其他国家的类似监管机构的适用规则中所定义的良好制造实践标准下制造或配制。例如,本文所述组合物可为无菌的和/或基本上等渗的和/或完全符合美国食品药品管理局的所有(例如当前)良好制造实践规则。本文所述组合物(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可配制成药学上可接受的组合物。例如,可将治疗有效量的cx-8998配制到药学上可接受的组合物中并且施用于患有或处于患上一种或多种运动障碍(例如原发性震颤、癫痫和/或帕金森病)风险下的哺乳动物以治疗所述哺乳动物。本文所用,术语“药学上可接受”是指在合理医学判断范围内适于与人类和动物的组织接触使用而无过度毒性、刺激性、过敏反应或其他问题或并发症并且与合理益处/风险比相称的化合物、材料、组合物和/或剂型。例如,可将治疗有效量的cx-8998与一种或多种药学上可接受的载体(例如添加剂、稀释剂和/或赋形剂)配制在一起。在一些情况下,药学上可接受的载体可为本文所述组合物的活性组分。在药学上可接受的载体是本文所述组合物的活性组分时,药学上可接受的载体在组合物中可具有一种或多种功能。例如,药学上可接受的载体可为本文所述组合物中的填充剂、稀释剂、增量剂、粘合剂、崩解剂、芯吸剂、基质形成聚合物、包衣剂、ph敏感性聚合物、孔形成剂、助流剂、润滑剂、渗透剂、保湿剂、抗氧化剂、抗微生物剂、溶解增强剂、渗透促进剂、结晶抑制剂和/或溶剂。在一些情况下,药学上可接受的载体可为本文所述组合物的无活性组分。药学上可接受的载体可为固体、半固体或液体材料。药学上可接受的载体可为用作一种或多种t型钙通道拮抗剂的媒介物、载体或介质的任何药学上可接受的化合物。可用于本文所述药学上可接受的组合物中的药学上可接受的载体的实例包括(但不限于)乳糖一水合物、交聚维酮、柠檬酸、月桂基硫酸钠、离子交换剂、氧化铝、硬脂酸铝、卵磷脂、血清蛋白质(例如人类血清白蛋白);缓冲物质,例如磷酸盐、甘氨酸、山梨酸、山梨酸钾;饱和植物脂肪酸的偏甘油酯混合物、水、盐或电解质,例如硫酸鱼精蛋白、磷酸氢二钠、磷酸氢钾、氯化钠、锌盐、胶状二氧化硅、三硅酸镁、聚乙烯吡咯烷酮、纤维素、基于纤维素的物质、羧甲基纤维素钠、聚丙烯酸酯、蜡、聚乙烯-聚氧基丙烯嵌段聚合物、聚乙二醇(peg;含有peg的分子,例如维生素epeg琥珀酸盐和peg-200)、tween如tween-80、环糊精、二甲亚砜(dmso)、羊毛脂、乳糖、右旋糖、蔗糖、山梨糖醇、甘露糖醇、淀粉、阿拉伯树胶、磷酸钙、海藻酸盐、黄蓍胶、明胶、硅酸钙、微晶纤维素、聚乙烯基吡咯烷酮、糖浆和甲基纤维素。本文所述组合物还可包括例如填充剂(例如乳糖、蔗糖、甘露糖醇、改性或未改性淀粉;纤维素衍生物,例如微晶纤维素、羟丙基纤维素和交联羧甲基纤维素钠;和矿物质盐,例如磷酸钙)、崩解剂(例如交联羧甲纤维素、淀粉羟乙酸钠和交聚维酮)、粘合剂(例如亲水聚合物,例如纤维素衍生物(例如羟丙基甲基纤维素)、卡波普(carbopol)、聚维酮和改性淀粉)、表面活性剂(例如磺酸酰基酯、聚山梨醇酯如聚山梨醇酯-80、脂肪酸及其衍生物、泊洛沙姆(poloxamer)、特吉特(tergitol)、和其他两亲性分子)、润滑剂(例如滑石、硬脂酸镁和矿物油)润湿剂、乳化和悬浮剂、防腐剂(例如苯甲酸甲基酯和苯甲酸羟丙基酯)、甜味剂和/或调味剂。在一些情况下,包括本文所述一种或多种t型钙通道拮抗剂的药学上可接受的组合物可包括乳糖一水合物、交聚维酮、柠檬酸和月桂基硫酸钠。例如,在本文所述组合物包括cx-8998时,组合物可包括以重量计约5%的cx-8998、约60%的乳糖一水合物、约25%的交聚维酮、约8%的柠檬酸和约2%的月桂基硫酸钠。应理解,某些额外组分(例如药学上可接受的载体)的使用可导致从一种或多种t型钙通道拮抗剂形成药用盐(例如cx-8998的药学上可接受的盐,例如cx-8998-hcl盐)。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可配制成单位剂型。术语“单位剂型”是指适于作为用于人类受试者和其他哺乳动物的单位剂量的物理离散单位,每个单位含有经计算以产生期望治疗作用的预定量的活性成分与合适的药物赋形剂的组合。在一些情况下,单位剂量可每天施用一次(例如每日一次剂量)。在一些情况下,单位剂量可每天施用一次以上(例如bid或每天三次(tid))。例如,单位剂量可含有约0.5mg至约1,000mg(1g)的一种或多种t型钙通道拮抗剂/天(例如约0.5mg至约900mg、约0.5mg至约800mg、约0.5mg至约700mg、约0.5mg至约600mg、约0.5mg至约500mg、约0.5mg至约400mg、约0.5mg至约300mg、约0.5mg至约200mg、约0.5mg至约100mg、约0.5mg至约50mg、约0.5mg至约30mg、约0.5mg至约20mg、约5mg至约1,000mg、约10mg至约1,000mg、约25mg至约1,000mg、约100mg至约1,000mg、约200mg至约1,000mg、约300mg至约1,000mg、约400mg至约1,000mg、约500mg至约1,000mg、约600mg至约1,000mg、约750mg至约1,000mg、约5mg至约750mg、约10mg至约500mg、约15mg至约400mg、约20mg至约300mg、约25mg至约250mg、约30mg至约200mg、约3mg至约30mg、约5mg至约25mg、约8mg至约16mg、约40mg至约150mg或约50mg至约100mgcx-8998/天)。在一些情况下,每个剂量含有约8mgcx-8998。在一些情况下,每个剂量含有约10mgcx-8998。在一些情况下,每个剂量含有约16mgcx-8998。在一些情况下,每个剂量含有约20mgcx-8998。在一些情况下,每个剂量含有约24mgcx-8998。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可包括任何适当量的一种或多种t型钙通道拮抗剂(例如cx-8998)。本文所述组合物中一种或多种t型钙通道拮抗剂的量(例如比例或浓度)可根据多种因素而变,所述因素包括剂量、化学特征(例如疏水性)和施用途径。例如,当组合物包括cx-8998时,组合物可包括以重量计约0.5%至约60%cx-8998(例如约1%至约60%、约3%至约60%、约5%至约60%、约8%至约60%、约10%至约60%、约12%至约60%、约15%至约60%、约20%至约60%、约25%至约60%、约30%至约60%、约40%至约60%、约50%至约60%、约0.5%至约50%、约0.5%至约40%、约0.5%至约30%、约0.5%至约20%、约0.5%至约15%、约0.5%至约10%、约0.5%至约7%、约0.5%至约5%、约0.5%至约2%、约1%至约50%、约5%至约30%、约10%至约20%、约1%至约5%、约5%至约10%、约10%至约15%、约15%至约20%、约20%至约30%、约30%至约40%、或约40%至约50%cx-8998)。在一些情况下,组合物可包括以重量计约5%cx-8998。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可被配制以任何适当形式施用。本文所述组合物可呈固体形式(例如片剂或胶囊)、液体形式(例如溶液或悬浮液)、或半固体形式(例如凝胶、洗剂、乳膏剂、软膏剂、软凝胶或胶粘物)。本文所述组合物可配制成的形式的实例包括(但不限于)溶液、悬浮液、片剂、丸粒、珠粒、丸剂、粉末(例如冻干粉末)、颗粒、糖锭、小药囊、扁囊剂、酏剂、乳液、糖浆、气雾剂(例如固体气雾剂或液体气雾剂)、洗剂、乳膏剂和软膏剂。在一些情况下,本文所述组合物的剂型还可包括一种或多种额外组分(例如化妆品和/或调味剂组分)。在组合物呈固体形式时,固体组合物可包括一个或多个粒子(例如一个或多个颗粒、一个或多个丸粒、一个或多个珠粒、一个或多个微粒和/或一个或多个纳米粒子;或颗粒、丸粒、珠粒、微粒和纳米粒子的混合物,及其子组合)。在组合物包括一个或多个粒子时,粒子可为任何适当大小。例如,粒子可具有约0.2mm至约2.0mm(例如约0.2mm至约1.8mm、约0.2mm至约1.5mm、约0.2mm至约1.2mm、约0.2mm至约1.0mm、约0.2mm至约0.8mm、约0.2mm至约0.6mm、约0.5mm至约2.0mm、约0.7mm至约2.0mm、约1.0mm至约2.0mm、约1.2mm至约2.0mm、约1.5mm至约2.0mm、约1.8mm至约2.0mm、约0.3mm至约1.8mm、约0.5mm至约1.5mm、约0.5mm至约1.0mm或约1.0mm至约1.5mm)的最长尺寸(例如直径)。在一些情况下,粒子可具有约0.5mm至约1mm的最长尺寸。在组合物包括一个或多个粒子时,粒子可为任何适当形状。例如,粒子可为圆形(例如球形)。例如,剂型可包括一个或多个非功能包衣(例如一个或多个不包括一种或多种t型钙通道拮抗剂并且不改变本文所述组合物中一种或多种t型钙通道拮抗剂的释放的包衣)。在一些情况下,一种或多种形式的本文所述组合物(例如口服剂型)可包含于胶囊(例如软明胶胶囊、硬明胶胶囊或硬羟丙基甲基纤维素胶囊)中。例如,本文所述组合物可包含于硬明胶胶囊或硬羟丙基甲基纤维素胶囊中。胶囊可为任何形状(例如椭圆形)。在本文所述组合物包含于胶囊中时,胶囊可为任何适当大小(例如4号、3号、2号、1号、0号、00号、000号、0el号或00el号胶囊)。在一些情况下,含有一种或多种t型钙通道拮抗剂的药学上可接受的组合物可为无菌组合物。例如,含有一种或多种t型钙通道拮抗剂的药学上可接受的组合物可包括水性和非水性无水注射溶液,其可含有抗氧化剂(例如丁基化羟基苯甲醚(bha)和/或丁基化羟基甲苯(bht))、稳定剂、缓冲剂、抑菌剂和使得组合物与预期接受者的血液等渗的溶质;以及水性和非水性无菌悬浮液,其可包括悬浮剂和增稠剂。在一些情况下,包括本文所述一种或多种t型钙通道拮抗剂的药学上可接受的组合物可包括含有多个含有cav3拮抗剂的丸粒和/或含有cav3拮抗剂的珠粒的胶囊。例如,在本文所述组合物包括cx-8998时,组合物可包括含有多个含有cx-8998的丸粒和/或含有cx-8998的珠粒的胶囊。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可被配制以任何适当时间施用。在一些情况下,含有cx-8998的药学上可接受的组合物可为立即释放组合物。如本文所用的术语“立即释放”是指组合物被配制以在施用组合物后的约30分钟(例如约35分钟、约40分钟、约45分钟、约50分钟或约60分钟)内释放全部(例如100%)或基本上全部(例如至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少97%、至少98%或至少99%)活性成分(例如一种或多种t型钙通道拮抗剂如cx-8998)。例如,包括cx-8998的立即释放组合物可被配制以在约45分钟内释放至少75%的cx-8998。在一些情况下,在立即释放组合物中,未刻意努力调节活性成分的释放速率。在一些情况下,含有cx-8998的药学上可接受的组合物可为受控释放组合物(例如调节释放组合物),例如延迟释放组合物、持续释放组合物或延长释放组合物。如本文所用的术语“受控释放”是指组合物被配制以可优化活性成分的血浆浓度曲线和/或对于患者更方便(例如允许较不频繁地施用)的方式释放活性成分(例如一种或多种t型钙通道拮抗剂如cx-8998)。在一些情况下,受控释放组合物可含有单一组分。在一些情况下,受控释放组合物可包括多个(例如两个、三个或更多个)组分,其各自被配制以特定方式释放活性成分。例如,受控释放组合物可包括被配制用于立即释放的一种或多种组分和被配制用于延迟和/或持续释放的一种或多种组分。如本文所用的术语“延迟释放”是指组合物被配制以延迟活性成分(例如一种或多种t型钙通道拮抗剂如cx-8998)的释放。例如,在组合物是口服剂型时,延迟释放组合物可被配制以延迟活性成分的释放,直到组合物通过胃(例如,以防止药物被例如胃液破坏或不活化,或者其可刺激胃粘膜)。在一些情况下,延迟释放组合物可被配制以延迟释放,直到剂型或剂型的一种或多种组分进一步向下通过肠道。例如,包括cx-8998的组合物可用延迟释放包衣包覆以获得延迟释放组合物。在一些情况下,延迟释放包衣可从一种或多种缓慢腐蚀的聚合物形成。在一些情况下,延迟释放包衣可从一种或多种ph独立性聚合物形成。在一些情况下,延迟释放包衣可从一种或多种ph敏感性聚合物形成。例如,ph敏感性肠溶聚合物在低ph下不可溶,但在较高ph下可溶,使得肠溶聚合物可形成在胃的低ph中(例如在约1至约3的ph下)形成不渗透层,但可溶胀并且然后一旦暴露于肠的更中性ph(例如在约5至约8的ph下)、例如如小肠中大于约6的ph或如结肠中大于约7的ph就会腐蚀。例如,ph敏感性卡波姆(carbomer)在低ph下(例如在约1至约5的ph下)基本上不溶,但在中性和碱性ph值下(例如在大于约6的ph下)溶胀以形成稠凝胶。在一些情况下,含有cx-8998的药学上可接受的组合物可为持续释放组合物。在组合物(例如包括一个或多个粒子(例如一个或多个颗粒、一个或多个丸粒、一个或多个珠粒、一个或多个微粒和/或一个或多个纳米粒子)的固体组合物)含有包衣时,包衣可为任何适当厚度。例如,包衣的厚度可为约50μm至约100μm(例如约50μm至约90μm、约50μm至约80μm、约50μm至约70μm、约50μm至约60μm、约60μm至约100μm、约70μm至约100μm、约80μm至约100μm、约90μm至约100μm、约60μm至约90μm、约70μm至约80μm、约60μm至约70μm、约70μm至约80μm或约80μm至约90μm)。例如,包衣的厚度可有效地将包衣粒子的重量(例如与无包衣粒子相比)增加约20%。如本文所用的术语“持续释放”(sr)是指组合物被配制以可维持基本上恒定药物浓度(例如最小有效浓度)达延长时间段的速率释放活性成分(例如一种或多种t型钙通道拮抗剂如cx-8998)。在一些情况下,持续释放组合物可维持基本上恒定药物浓度(例如最小有效浓度)达特定时间段。例如,持续释放组合物可包括一种或多种亲水聚合物,其在接触水时可溶胀并且可阻碍活性剂的释放。例如,持续释放组合物可包括一个或多个包含不溶性聚合物(例如乙基纤维素)和可溶性成孔剂的包衣。例如,在持续释放组合物配制为珠粒或丸粒时,持续释放组合物在制剂中可包括一种或多种聚合物(例如ph不敏感性聚合物和/或ph敏感性聚合物)。在一些情况下,药学上可接受的含有cx-8998的组合物可为受控释放组合物。在一些情况下,受控释放组合物在所谓“爆发释放”中快速释放活性成分(例如一种或多种t型钙通道拮抗剂如cx-8998)的至少一部分。在一些情况下,除组合物的立即释放组分外,也可使用从受控释放组合物或从受控释放组合物的组分的爆发释放。在一些情况下,从受控释放组合物或从受控释放组合物的组分的爆发释放可用作组合物的立即释放组分的替代。在一些情况下,包括本文所述一种或多种t型钙通道拮抗剂的药学上可接受的组合物可包括包含延迟释放和/或持续释放组合物的至少一种组分(例如第一组分)和任选地包括立即释放组合物的组分(例如第二组分)。例如,在本文所述组合物包括cx-8998时,在一些实施方案中,组合物可至少包括被配制用于延迟和/或持续释放cx-8998的第一组分和任选地被配制用于立即释放cx-8998的第二组分。在药学上可接受的组合物包括本文所述一种或多种t型钙通道拮抗剂、至少包括被配制用于延迟和/或持续释放cx-8998的第一组分和任选地被配制用于立即释放cx-8998的第二组分的一些情况下,组合物可包括任何适当比率的组分。在包括本文所述一种或多种t型钙通道拮抗剂的药学上可接受的组合物包括被配制用于延迟和/或持续释放cx-8998的第一组分和被配制用于立即释放cx-8998的第二组分时,组合物可包括任何适当量(例如约10%、约15%、约20%、约25%、约30%、约35%、约40%、约45%、约50%、约55%、约60%、约65%、约70%、约75%、约80%、约85%、约90%或约95%)的第一组分和任何适当量(例如约10%、约15%、约20%、约25%、约30%、约35%、约40%、约45%、约50%、约55%、约60%、约65%、约70%、约75%、约80%、约85%、约90%或约95%)的第二组分。例如,组合物可包括约40%的被配制用于延迟和/或持续释放cx-8998的第一组分和约60%的被配制用于立即释放cx-8998的第二组分。例如,组合物可包括约25%的被配制用于延迟和/或持续释放cx-8998的第一组分和约35%的被配制用于延迟和/或持续释放cx-8998的第二组分(例如被配制用于延迟和/或持续释放cx-8998并且与第一组分不同(例如其组成和/或其释放曲线不同)的第二组分),以及约40%的被配制用于立即释放cx-8998的第三组分。在组合物被设计以在两个或更多个不同时间释放活性成分(例如一种或多种t型钙通道拮抗剂如cx-8998)的一些情况下,第一释放(例如延迟和/或持续释放)和随后释放(例如立即释放)可重叠。在组合物被设计以在两个或更多个不同时间释放活性成分(例如一种或多种t型钙通道拮抗剂如cx-8998)的一些情况下,第一释放(例如延迟和/或持续释放)和随后释放(例如立即释放)可为连续的(例如不重叠)。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)的释放速率可使用任何适当方法测定。例如,可使用利用高效液相色谱(hplc)和/或分光光度法的溶解测试(例如单一阶段或两阶段溶解测试)以测定一种或多种t型钙通道拮抗剂(例如cx-8998)从含有一种或多种t型钙通道拮抗剂的组合物释放的速率。在一些情况下,组合物的释放速率可如别处所述测定(参见例如美国药典(usp)第117章)。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)中的t型钙通道拮抗剂可具有任何适当溶解速率。在一些情况下,本文所述组合物中的t型钙通道拮抗剂可具有与大体上纯t型钙通道拮抗剂(例如组合物中不存在的t型钙通道拮抗剂)基本上相同的溶解速率(例如溶解速率为约90%至约110%相同)。例如,在本文所述组合物中的t型钙通道拮抗剂是cx-8998时,组合物中的cx-8998可具有与大体上纯cx-8998基本上相同的溶解速率(例如在溶解介质的表面积、搅拌速度、ph和/或离子强度保持恒定时)。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可被设计以任何适当类型施用(例如口服、局部、肠胃外或吸入施用)。在一些情况下,组合物可被设计用于口服施用(例如作为口服剂型)。例如,当组合物包括cx-8998时,组合物可被设计为口服cx-8998剂型。在通过口服施用来施用时,含有一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的药物组合物可呈例如丸剂、片剂、胶囊、溶液或悬浮液的形式。在一些情况下,包括本文所述一种或多种t型钙通道拮抗剂的药学上可接受的组合物可包括包含多个含有cav3拮抗剂的丸粒和/或含有cav3拮抗剂的珠粒的胶囊。在一些情况下,胶囊可包括包含延迟释放和/或持续释放组合物的组分(例如第一组分)和任选地包括立即释放组合物的组分(例如第二组分)。例如,当组合物包括cx-8998时,组合物可包括包含多个含有cx-8998的丸粒和/或含有cx-8998的珠粒的胶囊,其一部分被配制用于立即释放并且一部分被配制用于延迟和/或持续释放。在一些情况下,包括本文所述一种或多种t型钙通道拮抗剂的药学上可接受的组合物可包括包含多个含有cav3拮抗剂的颗粒的胶囊。例如,当组合物包括cx-8998时,组合物可包括包含多个含有cx-8998的颗粒的胶囊,其一部分被配制用于立即释放并且一部分被配制用于延迟和/或持续释放。在一些情况下,包括本文所述一种或多种t型钙通道拮抗剂的药学上可接受的组合物可包括包含两个或更多个(例如,两个、三个、四个或更多个)层的片剂。在一些情况下,片剂可包括包含延迟释放和/或持续释放组合物的组分(例如第一层)和任选地包括立即释放组合物的组分(例如第二层)。例如,当组合物包括cx-8998时,组合物可包括含有被配制用于立即释放的cx-8998层和被配制用于延迟和/或持续释放的cx-8998层的片剂。在一些情况下,包括本文所述一种或多种t型钙通道拮抗剂的药学上可接受的组合物可包括包含核和一个或多个(例如,一个、两个、三个或更多个)包衣的片剂。例如,在片剂包括核和包衣的一些情况下,片剂核可含有被配制用于延迟和/或持续释放的cx-8998,并且片剂核可用含有被配制用于立即释放的cx-8998的包衣包覆。例如,在片剂包括核和包衣的一些情况下,含有cx-8998的片剂核可用包衣包覆,所述包衣延迟和/或持续cx-8998从核的释放,并且片剂核可进一步被配制用于立即释放的cx-8998包覆。在片剂包括核和包衣的一些情况下,片剂可为渗透泵片剂(例如,片剂具有渗透压驱动的递送系统)。例如,渗透泵片剂可包括核和包衣,其中片剂核可包括cx-8998和渗透剂,并且片剂核可用含有具有一个或多个孔(例如至少一个精确钻孔)的半透膜的包衣包覆,使得水跨越半透膜扩散可产生渗透压,其迫使呈溶液和/或悬浮液形式的cx-8998经由孔离开片剂核。在一些情况下,渗透泵片剂可用一个或多个含有cx-8998(例如被配制用于立即释放的cx-8998)的包衣包覆。在一些情况下,胶囊可包括包含立即释放组合物的组分(例如一个或多个粒子,例如片剂、珠粒、丸粒、颗粒和/或粉末、或它们的混合物)和包括被配制用于延迟释放和/或持续释放的cx-8998的组分(例如片剂或包衣)。例如,当组合物包括cx-8998时,组合物可包括含有被配制用于延迟和/或持续释放的cx-8998的第一片剂和任选地含有被配制用于立即释放的cx-8998的第二片剂。例如,当组合物包括cx-8998时,组合物可包括多个被配制用于立即释放的含有cx-8998的珠粒、丸粒、颗粒和/或粉末和含有被配制用于延迟和/或持续释放的cx-8998的片剂。在一些情况下,组合物包含一种或多种用于立即释放的组分和一种或多种用于延迟和/或持续释放的组分。在通过局部施用来施用时,含有一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的药物组合物可透皮、经表皮、经眼和/或至粘膜(例如鼻内、经阴道和经直肠)施用。在通过局部施用来施用时,含有一种或多种cav3拮抗剂的药物组合物可呈例如透皮贴剂、软膏剂、洗剂、乳膏剂、凝胶、滴剂、栓剂、喷雾、液体和粉末(例如冻干粉末)的形式。在通过肠胃外施用来施用时,含有一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的药物组合物可通过静脉内、动脉内、皮下、腹膜内、肌内、颅内(例如鞘内或室内)注射或输注施用。在通过肠胃外施用来施用时,含有一种或多种t型钙通道拮抗剂的药物组合物可呈例如液体、凝胶、滴剂、栓剂、喷雾、粉末(例如冻干粉末)、乳液、悬浮液、微粒、原位凝胶和含药物的植入物的形式。在通过肠胃外施用来施用时,含有一种或多种t型钙通道拮抗剂的药物组合物可以一个或多个团注剂量施用,或可通过连续灌注(例如通过输注泵或通过提供逐渐释放的可植入装置)来施用。在通过吸入施用(例如吸入)来施用时,含有一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的药物组合物可经肺(例如可施用于鼻腔、上呼吸道或下呼吸道)。例如,含有一种或多种t型钙通道拮抗剂的药物组合物的施用可包括吸入或吹入液体(例如气雾剂)和/或固体(例如粉末)。在通过吸入施用来施用时,含有一种或多种t型钙通道拮抗剂的药物组合物可呈例如液体(例如溶液和悬浮液)、凝胶和粉末(例如冻干粉末)的形式。在通过吸入施用来施用时,含有一种或多种t型钙通道拮抗剂的药物组合物可通过例如鼻喷雾泵、雾化器、计量剂量吸入器或干粉吸入器来施用。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可局部或全身施用。在一些情况下,含有一种或多种t型钙通道拮抗剂的组合物可通过口服施用于哺乳动物(例如人类)来全身施用。例如,当组合物包括cx-8998时,组合物可配制为口服剂型,例如胶囊(例如含有多个含有cx-8998的丸粒和/或含有cx-8998的珠粒的胶囊,其一部分被配制用于立即释放并且一部分被配制用于延迟和/或持续释放)。可使用任何适当方法以配制本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)。可用于配制本文所述固体组合物的方法的实例包括(但不限于)干式共混、湿法粒化、喷雾干燥、热熔融挤出、辊压、压缩、挤出、球形化、流化床干燥、盘干燥、流化床涂布、盘式涂布和囊封。可用于配制本文所述液体和半固体组合物的方法的实例包括(但不限于)叶片混合、高剪切混合、均质化、微流化、介质研磨、过滤、冻干和高压釜处理。在本文所述组合物包括丸粒和/或珠粒的情况下,丸粒和/或珠粒可通过挤出和球形化湿物质来产生,所述湿物质含有活性成分(例如一种或多种t型钙通道拮抗剂如cx-8998)和任选地额外组分,例如所选赋形剂(例如乳糖一水合物、交聚维酮(例如polyplasdonexl-10)、柠檬酸、月桂基硫酸钠(sls)、羟丙基纤维素(hpc)如hpc-sl、微晶纤维素、交联羧甲基纤维素钠、硬脂酸镁、羟丙基甲基纤维素、聚维酮和滑石)。例如,cx-8998、乳糖一水合物、交聚维酮、柠檬酸和月桂基硫酸钠的湿物质的挤出和球形化可用于产生含有以重量计约5%cx-8998、约60%乳糖一水合物、约25%交聚维酮、约8%柠檬酸和约2%sls的丸粒和/或珠粒(例如立即释放丸粒和/或珠粒)。例如,cx-8998、乳糖一水合物、交聚维酮、柠檬酸和月桂基硫酸钠的湿物质的挤出和球形化可用于产生含有以重量计约5%cx-8998、约60%乳糖一水合物、约25%交聚维酮、约8%柠檬酸和约2%sls的丸粒和/或珠粒(例如立即释放丸粒和/或珠粒)。在一些情况下,丸粒和/或珠粒可通过共混干燥成分、在行星式混合器中搅拌的同时缓慢添加水、挤出所得湿物质、使挤出物球形化以及然后在流化床干燥器中干燥所得丸粒和/或珠粒来制备。这些工艺可由本领域技术人员所了解。在本文所述组合物包括颗粒的情况下,颗粒可通过湿物质的湿法粒化来产生,所述湿物质含有活性成分(例如一种或多种t型钙通道拮抗剂如cx-8998)和任选地额外组分,例如所选赋形剂(例如乳糖一水合物、交聚维酮(例如polyplasdonexl-10)、柠檬酸、sls、hpc如hpc-sl、微晶纤维素、交联羧甲基纤维素钠、硬脂酸镁和tpgs)。例如,cx-8998、乳糖一水合物、微晶纤维素、交联羧甲基纤维素钠、hpc、硬脂酸镁和tpgs的湿物质的湿法粒化可用于产生以重量计约2%cx-8998、约20%乳糖一水合物、约63%微晶纤维素、约3%交联羧甲基纤维素钠、约3%hpc、约0.5%硬脂酸镁和约10%tpgs的颗粒(例如立即释放颗粒)。在一些情况下,颗粒可通过以下制备:在v形共混器中或在行星式混合器或高剪切粒化器的碗中共混,在行星式混合器或高剪切混合器中利用添加水或粘合剂溶液制粒,研磨或筛选,在流化床干燥器或盘干燥器中干燥,任选地研磨干燥的颗粒,以及然后将其与润滑剂和/或助流剂共混。这些工艺将由本领域技术人员所了解。在本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)包括延迟释放丸粒和/或珠粒和/或持续释放丸粒和/或珠粒的情况下,延迟释放丸粒和/或珠粒和/或持续释放丸粒和/或珠粒可通过用持续释放包衣和/或延迟释放包衣包覆丸粒和/或珠粒来产生。例如,丸粒和/或珠粒(例如立即释放丸粒和/或珠粒)可使用具有wurster插入物的流化床涂布器进行持续释放包衣和/或延迟释放包衣包覆。例如,惰性可食用球(例如糖球(小糖珠)或微晶纤维素球(苏丽芯(suglets))(例如含有一种或多种t型钙通道拮抗剂(例如用其包覆)的可食用球)可使用具有wurster插入物的流化床涂布器进行持续释放包衣和/或延迟释放包衣包覆。在一些情况下,包衣可使用膜涂布方法(例如使用流化床喷涂方法)施加(例如,至球、丸粒和/或珠粒)。涂布球、丸粒和/或珠粒的其他方法为本领域技术人员所了解。在一些情况下,持续释放珠粒/丸粒和/或延迟释放丸粒/珠粒可通过涂布立即释放丸粒/珠粒来产生。例如,延迟释放丸粒/珠粒和/或持续释放珠粒/丸粒可通过用持续释放包衣和/或延迟释放包衣包覆立即释放丸粒/珠粒来产生。在一些情况下,延迟释放丸粒/珠粒和/或持续释放珠粒/丸粒可独立于任何立即释放丸粒/珠粒而产生。例如,延迟释放丸粒/珠粒和/或持续释放珠粒/丸粒可通过配制丸粒/珠粒(例如无包衣的丸粒/珠粒)以提供活性成分(例如cx-8998)的逐渐释放来产生。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可使用任何适当方法来灭菌。在一些情况下,本文所述组合物可通过常规灭菌技术来灭菌。在一些情况下,本文所述组合物可经过滤灭菌。本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可具有任何适当ph。例如,含有一种或多种t型钙通道拮抗剂的药学上可接受的组合物的ph可为约3至约11。在一些情况下,含有一种或多种t型钙通道拮抗剂的药学上可接受的组合物的ph可为5至约9。在一些情况下,含有一种或多种t型钙通道拮抗剂的药学上可接受的组合物的ph可为约7至约8。应理解,某些额外组分(例如药学上可接受的载体)的使用可改变(例如,可用于改变)本文所述组合物的ph。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可包括一个或多个可检测标记。在一些情况下,可检测标记可适于在哺乳动物(例如人类)内体内使用。可检测标记的实例包括(但不限于)放射性同位素(例如14c)、非放射性同位素(例如氘和13c)、自旋标记、荧光标签和酶。例如,当组合物包括cx-8998时,cx-8998可为14c标记的cx-8998。例如,可对可检测标记进行检测以容许在施用于哺乳动物后使组合物成像。成像可使得医师或业内的另一专业人员能够确定哺乳动物内所施用组合物在空间和/或时间上的存在。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可包括一种或多种t型钙通道拮抗剂、甲基纤维素、聚乙二醇(peg)400、peg-200、tween-80和/或环糊精。例如,当组合物包括cx-8998时,组合物可包括cx-8998和约0.5至约1%甲基纤维素。例如,当组合物包括cx-8998时,组合物可包括cx-8998和约90%peg400。例如,当组合物包括cx-8998时,组合物可包括cx-8998和约80%peg-200。例如,当组合物包括cx-8998时,组合物可包括cx-8998和约10%tween-80。例如,当组合物包括cx-8998时,组合物可包括cx-8998和约30%环糊精。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可包括一种或多种t型钙通道拮抗剂、甲基纤维素和/或聚山梨醇酯-80。例如,当组合物包括cx-8998时,组合物可包括cx-8998、约0.5%甲基纤维素和约10%聚山梨醇酯-80。例如,当组合物包括cx-8998时,组合物可包括cx-8998、约0.5%甲基纤维素和约0.1%聚山梨醇酯-80。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可包括约2毫克一种或多种t型钙通道拮抗剂,其呈一起湿法制粒的组分与硬脂酸镁的共混物形式。例如,当组合物包括cx-8998时,组合物可包括约2毫克cx-8998(例如游离碱当量的cx-8998),其呈与一起湿法制粒的微晶纤维素、乳糖、交联羧甲基纤维素钠、维生素epeg琥珀酸盐和羟丙基纤维素与硬脂酸镁的共混物形式。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可包括立即释放组分中的约10mg一种或多种t型钙通道拮抗剂和延迟释放组分中的约10mg一种或多种t型钙通道拮抗剂。例如,当组合物包括cx-8998时,组合物可包括立即释放组分中的约10mgcx-8998(例如游离碱当量的cx-8998)和延迟释放组分中的约10mgcx-8998(例如游离碱当量的cx-8998)。在一些情况下,立即释放组分可包括通过挤出和球形化形成的立即释放珠粒,并且延迟释放组分可包括通过挤出和球形化形成并且用具有约ph5.5至约ph7(例如ph6、ph6.5或ph7)的溶解ph的一种或多种ph敏感性肠溶聚合物包覆的珠粒。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可包括立即释放组分中的约7毫克一种或多种t型钙通道拮抗剂和延迟释放组分中的约13毫克一种或多种t型钙通道拮抗剂。例如,当组合物包括cx-8998时,组合物可包括立即释放组分中的约7毫克cx-8998(例如游离碱当量的cx-8998)和延迟释放组分中的约13毫克cx-8998(例如游离碱当量的cx-8998)。在一些情况下,立即释放组分可包括通过挤出和球形化形成的立即释放珠粒,并且延迟释放组分可包括通过挤出和球形化形成并且用具有约ph5.5至约ph7(例如ph6、ph6.5或ph7)的溶解ph的一种或多种ph敏感性肠溶聚合物包覆的珠粒。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可为固体剂型(例如丸剂、胶囊和片剂)中的受控释放组合物,其包括具有被配制用于延迟和/或持续释放cx-8998的含有cx-8998的颗粒的第一组分和具有被配制用于立即释放cx-8998的含有cx-8998的颗粒的第二组分。例如,受控释放胶囊可包括具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的颗粒的第一组分,并且任选地还可包括具有含有cx-8998并且被配制用于立即释放cx-8998的颗粒的第二组分。例如,受控释放片剂可包括具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的颗粒的第一组分,并且任选地还可包括具有含有cx-8998并且被配制用于立即释放cx-8998的颗粒的第二组分。在一些情况下,具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的颗粒的第一组分和任选地具有含有cx-8998并且被配制用于立即释放cx-8998的颗粒的第二组分可压缩成片剂(例如单块片剂和具有两个或更多个层的片剂)。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可为包括两个或更多个(例如,两个、三个、四个或更多个)固体剂型(例如丸剂、胶囊和片剂)的受控释放组合物,其中至少一个固体剂型含有被配制用于立即释放cx-8998的cx-8998并且至少一个固体剂型含有被配制用于延迟和/或持续释放cx-8998的cx-8998。例如,受控释放组合物包括两个或更多个固体剂型,其中至少一个固体剂型含有被配制用于立即释放cx-8998的cx-8998并且至少一个固体剂型含有被配制用于延迟和/或持续释放cx-8998的cx-8998,两个或更多个固体剂型可构成单一剂量。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可为具有胶囊的受控释放组合物,所述胶囊具有第一组分,所述第一组分具有一个或多个(例如,一个、两个、三个、四个或更多个含有被配制用于延迟和/或持续释放cx-8998的cx-8998的片剂,和/或一个或多个(例如,一个、两个、三个、四个或更多个)含有被配制用于延迟和/或持续释放cx-8998的cx-8998的微型片剂;和任选地第二组分,所述第二组分含有被配制用于立即释放cx-8998的cx-8998。例如,受控释放胶囊可包括第一组分,所述第一组分具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的一个或多个片剂和/或一个或多个微型片剂;并且任选地还可包括第二组分,所述第二组分含有cx-8998并且被配制用于立即释放cx-8998。在一些情况下,具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的一个或多个片剂和/或一个或多个微型片剂的第一组分和任选地具有含有cx-8998并且被配制用于立即释放cx-8998的一个或多个片剂和/或一个或多个微型片剂的第二组分可在胶囊内。在一些情况下,具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的一个或多个片剂和/或一个或多个微型片剂的第一组分和任选地具有含有cx-8998并且被配制用于立即释放cx-8998的粉末的第二组分可在胶囊内。在一些情况下,具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的一个或多个片剂和/或一个或多个微型片剂的第一组分和任选地具有含有cx-8998并且被配制用于立即释放cx-8998的颗粒的第二组分可在胶囊内。在一些情况下,具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的一个或多个片剂和/或一个或多个微型片剂的第一组分和任选地具有含有cx-8998并且被配制用于立即释放cx-8998的珠粒和/或丸粒的第二组分可在胶囊内。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可为具有胶囊的受控释放组合物,所述胶囊包含含有被配制用于延迟和/或持续释放cx-8998的cx-8998的第一固体组分(例如颗粒、珠粒、丸粒和片剂)和任选地含有被配制用于立即释放cx-8998的cx-8998的第二液体组分(例如溶液或悬浮液)。例如,受控释放胶囊可包括具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的颗粒的第一固体组分并且可包括含有被配制用于立即释放cx-8998的cx-8998的第二液体组分。在一些情况下,具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的颗粒的第一组分和任选地含有被配制用于立即释放cx-8998的cx-8998的第二液体组分可在胶囊内。在一些情况下,具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的珠粒和/或丸粒的第一组分和任选地含有被配制用于立即释放cx-8998的cx-8998的第二液体组分可在胶囊内。在一些情况下,具有含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的一个或多个(例如,一个、两个、三个、四个或更多个)片剂的第一组分和任选地含有被配制用于立即释放cx-8998的cx-8998的第二液体组分可在含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的胶囊和/或一个或多个(例如,一个、两个、三个、四个或更多个)微型片剂内。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可为受控释放组合物,其具有第一(例如外部)胶囊和第二(例如内部)胶囊(例如经定尺寸装配在第一胶囊内的第二胶囊,使得在第二胶囊的外表面与第一胶囊的内表面之间存在空间)并且含有包含cx-8998并且被配制用于延迟和/或持续释放cx-8998的第一组分和任选地含有cx-8998并且被配制用于立即释放的第二组分。例如,包括在受控释放胶囊内装配的内部胶囊的受控释放胶囊可包括含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的第一组分,并且任选地可包括具有含有cx-8998的颗粒并且被配制用于立即释放cx-8998的第二组分,其包含于内部胶囊内和/或内部胶囊与外部胶囊之间的空间中。在一些情况下,外部胶囊可含有内部胶囊,所述内部胶囊含有作为含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的固体组分(例如颗粒、珠粒、丸粒和片剂,例如微型片剂)的第一组分,和任选地作为含有cx-8998并且被配制用于立即释放cx-8998的液体组分(例如溶液或悬浮液)的第二组分,其中含有cx-8998并且被配制用于立即释放cx-8998的液体组分在存在时存于内部胶囊的外表面与外部胶囊的内表面之间的空间中。在一些情况下,外部胶囊可含有具有含有cx-8998并且被配制用于立即释放cx-8998的第一组分的内部胶囊,并且内部胶囊可包括可延迟含有cx-8998并且被配制用于立即释放cx-8998的第一组分的释放的包衣。在一些情况下,本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)可为受控释放组合物,其为包括液相(例如溶液)和多个悬浮于液相中的固体组分(例如颗粒、珠粒、丸粒和片剂,例如微型片剂)的悬浮液。例如,包括液相和多个固体组分的受控释放悬浮液可包括液相中的一种或多种t型钙通道拮抗剂(例如cx-8998),可包括悬浮于液相中的固体组分中的一种或多种t型钙通道拮抗剂,或者可包括液相和悬浮于液相中的固体组分两者中的一种或多种t型钙通道拮抗剂。在一些情况下,含有cx-8998并且被配制用于立即释放cx-8998的多个固体组分和含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的第二多个固体组分可悬浮于受控释放悬浮液的液相中。在一些情况下,受控释放悬浮液可包括含有cx-8998并且被配制用于立即释放cx-8998的液相,并且可包括含有cx-8998并且被配制用于延迟和/或持续释放cx-8998的悬浮于液相中的多个固体组分。治疗方法本文件也提供了治疗患有或处于患上一种或多种运动障碍风险下的哺乳动物(例如人类)的方法。例如,可向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用本文所述组合物(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)以治疗哺乳动物。术语“治疗(treating或treatment)”是指以下中的一者或多者:(1)预防运动障碍;例如,预防可易患运动障碍但尚未经历或展现运动障碍的病理学或症状学的个体的运动障碍;(2)抑制运动障碍;例如,抑制正经历或展现运动障碍的病理学或症状学的个体的运动障碍(即,停止病理学和/或症状学的进一步发展);以及(3)改善运动障碍;例如,改善正经历或展现运动障碍的病理学或症状学的个体的运动障碍(即,逆转病理学和/或症状学),例如降低运动障碍的严重程度或减轻或缓和运动障碍的一种或多种症状。例如,在向患有或处于患上运动障碍风险下的哺乳动物施用包括cx-8998的组合物时,包括cx-8998的组合物可有效减少或消除运动障碍的一种或多种症状。运动障碍的症状的实例包括(但不限于)震颤(例如节律性震颤)、不稳定步态(例如共济失调)、张力障碍性运动、冷冻(运动徐缓)、抽搐、痉挛、其他运动障碍性运动、癫痫发作、疼痛、感觉问题、精神病学症状、认知损害、听力损害和情绪变化。当症状是震颤时,震颤可为任何适当类型的震颤(例如家族性震颤、动作性震颤、姿势性震颤、运动性震颤和/或静止性震颤)。在一些情况下,可向患有或处于患上运动障碍风险下的哺乳动物施用包括cx-8998的组合物,以减少或消除哺乳动物的震颤。当症状是震颤时,震颤可影响哺乳动物的任何适当部分(例如,手、头部、声音、臂、手指、腿、下巴和哺乳动物身体的其他部分)。在一些情况下,可向患有或处于患上运动障碍风险下的哺乳动物施用包括cx-8998的组合物,以减少或消除哺乳动物的手的震颤。例如,可向如本文所述有需要的哺乳动物(例如患有或处于患上运动障碍风险下的哺乳动物)施用一种或多种t型钙通道拮抗剂,以将哺乳动物的运动障碍的一种或多种症状的严重程度降低例如10、20、30、40、50、60、70、80、90、95或更大百分比。可使用任何适当方法来评价运动障碍的严重程度和/或运动障碍的症状。在一些情况下,运动障碍的严重程度和/或运动障碍的症状可包括评价一个或多个整体功能量度。在一些情况下,运动障碍的严重程度和/或运动障碍的症状可包括评价一个或多个具体功能量度。可用于评价运动障碍的严重程度和/或运动障碍的症状的方法的实例包括(但不限于)测量日常生活活动(例如,如使用tetras所测量)、临床医师整体改善印象(例如,如使用cgi-i所测量)、患者整体变化印象(例如,如使用pgic所测量)、震颤特异性目标达成(例如,如使用gas所测量)、生活质量(例如,如使用quest震颤药物满足子项目所测量)和阿基米德螺线(archimedesspiral)(例如,如使用笔和纸如在tetras-ps子项目中所测量和/或使用平板电脑和记录针如在imotor中数字测量)。在一些情况下,测量运动障碍的严重程度和/或运动障碍的症状的方法可阐述于实施例部分中。例如,当使用tetras测量日常生活活动时,评分越高,则震颤越严重,使得评分减小是震颤改善。在一些情况下,可用于评价运动障碍的严重程度和/或运动障碍的症状的方法可如别处阐述(参见例如elble等人,2013movementdisorders,281793;fahn等人,“clinicalratingscalefortremor,”第225-34页:jankovikj和tolosae.parkinson'sdiseaseandmovementdisorders.1988baltimore-milnich:urban&schwarzenberg;和haubenberger等人,2016movementdisorders,31,第9期;fahn等人,“membersoftheupdrsdevelopmentcommittee,”第153-163页和第293-304页:fahn等人(编辑)recentdevelopmentsinparkinson'sdisease,第2卷,1987florhampark,nj.macmillanhealthcareinformation;美国神经病学会关于原发性震颤的治疗指南,例如可在网站aan.com/guidelines/home/guidelinedetail/492获得的那些;以及美国神经病学会关于帕金森病的治疗指南,例如可在网站movementdisorders.org/mds-files1/resources/pdfs/treatmentsformotorsymptomsofpd-2018.pdf获得的那些。在一些情况下,在哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)处于禁食状态时,可向所述哺乳动物施用包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)。例如,在施用包括一种或多种t型钙通道拮抗剂的组合物之前,哺乳动物可已禁食(或已被指示禁食)至少4小时(例如,约4小时、约5小时、约6小时、约7小时、约8小时、约9小时、约10小时、约11小时或约12小时)。在一些情况下,当哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)呈进食(例如,非禁食)状态时,可向所述哺乳动物施用包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)。可如本文所述治疗患有或处于患上运动障碍(例如原发性震颤、癫痫和/或帕金森病)风险下的任何适当哺乳动物。可如本文所述治疗(例如通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)的哺乳动物的实例包括(但不限于)人类、非人类灵长类动物(例如猴)、狗、猫、马、牛、猪、绵羊、小鼠和大鼠。在一些情况下,可用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物治疗患有或处于患上运动障碍风险下的人类,以减少或消除运动障碍的一个或多个症状。当如本文所述治疗患有一种或多种运动障碍的哺乳动物(例如通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)时,哺乳动物可为任何适当年龄。在一些情况下,进行治疗的适当人类是成人(例如年轻成人、中年人和老年人)。例如,成年人可为18岁或以上(例如20岁或以上、30岁或以上、40岁或以上、50岁或以上、60岁或以上、65岁或以上、70岁或以上、或75岁或以上)。在一些情况下,成人可为约18岁至约100岁(例如,约18岁至约90岁、约18岁至约80岁、约18岁至约75岁、约18岁至约70岁、约18岁至约65岁、约18岁至约60岁、约18岁至约55岁、约18岁至约50岁、约18岁至约45岁、约18岁至约40岁、约20岁至约100岁、约30岁至约100岁、约40岁至约100岁、约50岁至约100岁、约60岁至约100岁、约65岁至约100岁、约70岁至约100岁、约75岁至约100岁、约80岁至约100岁、约20岁至约55岁、约20岁至约40岁、约22岁至约39岁、约40岁至约80岁、约40岁至约66岁、约40岁至约60岁、约50岁至约80岁、约55岁至约75岁、约57岁至约75岁、或约57岁至约70岁)。在一些情况下,进行治疗的适当人是青少年(例如不超过18岁的人)。例如,青少年人可为约1岁至约18岁(例如,约1至约17岁、约1至约16岁、约1至约15岁、约1至约14岁、约1至约13岁、约1至约12岁、约1至约11岁、约1至约10岁、约1至约8岁、约1至约5岁、约1至约3岁、约5至约18岁、约10至约18岁、约12至约18岁、约15至约18岁、约16至约18岁、约2至约16岁、约5至约15岁、约8至约12岁、约2至约8岁、或约5至约15岁)。例如,青少年可为约16岁。当如本文所述治疗患有一种或多种运动障碍的哺乳动物(例如通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)时,哺乳动物可为雌性或雄性。当如本文所述治疗患有一种或多种运动障碍的哺乳动物(例如通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)时,含有一种或多种t型钙通道拮抗剂如cx-8998的组合物可在任何适当时间施用。在一些情况下,可在早晨向哺乳动物施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物。例如,可在约6am至约中午(例如,约6am至约11am、约6am至约10am、约6am至约9am、约6am至约8am、约6am至约7am、约7am至约中午、约8am至约中午、约9am至约中午、约10am至约中午、约11am至约中午、约7am至约11am、约8am至约10am、约7am至约9am、约8am至约10am、或约9am至约11am)之间向哺乳动物施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物。例如,可在约4小时内(例如睡醒后约4小时、睡醒后约3小时、睡醒后约2小时、睡醒后约1小时、睡醒后约30分钟内或睡醒后立即)向哺乳动物施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物。当如本文所述治疗患有一种或多种运动障碍的哺乳动物(例如通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)时,哺乳动物可患有任何适当的运动障碍。在一些情况下,运动障碍可包括震颤。例如,本文提供的方法和材料可用于治疗患有或处于患上震颤风险下的哺乳动物。可通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物治疗的运动障碍的实例包括(但不限于)原发性震颤、帕金森病(例如与帕金森病相关的震颤)、共济失调(例如脊髓小脑性共济失调和弗里德赖希氏共济失调(friedreich'sataxia))、癫痫(例如伴有失神型癫痫发作的特发性全身性癫痫和儿科癫痫)、肌张力障碍(例如泛发性肌张力障碍、局灶肌张力障碍、泛酸盐激酶相关的神经退化(pkan)和哈勒沃登-施帕茨病(hallervorden-spatzdisease))、偏侧颤搐、小脑病症、手足徐动症、痉挛状态、运动徐缓、迟发性运动障碍、亨廷顿氏病(huntington'sdisease)、肌阵挛、妥瑞氏综合征(tourette'ssyndrome)、舞蹈症、抽搐、进行性核上性麻痹、多系统萎缩、皮质基底神经节变性(corticobasoganlionicdegeneration)、可塑性、不宁腿/臂综合征、直立震颤、僵人综合征、多动型运动障碍、瑞特综合征(rettsyndrome)和威尔逊氏病(wilson'sdisease)。在一些情况下,可通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物治疗患有或处于患上原发性震颤风险下的哺乳动物。在一些情况下,可通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物治疗患有或处于患上帕金森病风险下的哺乳动物。在一些情况下,可向患有或处于患上其他疾患和/或病症(例如但不限于神经病性疼痛、精神异常、自闭症谱系障碍和强迫症)的哺乳动物施用包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)。当如本文所述治疗患有一种或多种运动障碍的哺乳动物(例如通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)时,可向哺乳动物施用任何适当量的本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)。在一些情况下,可向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用治疗有效量的含有一种或多种t型钙通道拮抗剂如cx-8998的组合物。术语“治疗有效量”是指活性化合物(例如一种或多种t型钙通道拮抗剂如cx-8998)在组织、系统、动物、个体或人中引发由研究者、兽医师、医生或其他临床医师所寻求的生物或医学反应的量。本文所述一种或多种t型钙通道拮抗剂的有效量可根据以下变化:运动障碍和/或与运动障碍相关的一种或多种症状/并发症的严重程度、施用途径、哺乳动物的年龄和一般健康状况、赋形剂使用、与其他治疗性治疗共使用的可能性(例如使用其他药剂)和主治医师的判断。例如,组合物(例如用于兽医和/或研究动物如狗或大鼠中的组合物)可包括可为以下的一种或多种t型钙通道拮抗剂的剂量:约1mg/kg至约1000mg/kg体重/天(例如,约5mg/kg至约1000mg/kg、约10mg/kg至约1000mg/kg、约20mg/kg至约1000mg/kg、约25mg/kg至约1000mg/kg、约30mg/kg至约1000mg/kg、约50mg/kg至约1000mg/kg、约75mg/kg至约1000mg/kg、约100mg/kg至约1000mg/kg、约250mg/kg至约1000mg/kg、约500mg/kg至约1000mg/kg、约750mg/kg至约1000mg/kg、约1mg/kg至约750mg/kg、约1mg/kg至约500mg/kg、约1mg/kg至约300mg/kg、约1mg/kg至约250mg/kg、约1mg/kg至约200mg/kg、约1mg/kg至约150mg/kg、约1mg/kg至约125mg/kg、约1mg/kg至约100mg/kg、约1mg/kg至约75mg/kg、约1mg/kg至约50mg/kg、约1mg/kg至约30mg/kg、约5mg/kg至约500mg/kg、约10mg/kg至约300mg/kg、约15mg/kg至约200mg/kg、约20mg/kg至约100mg/kg、约25mg/kg至约75mg/kg、约10mg/kg至约30mg/kg、约20mg/kg至约50mg/kg、约30mg/kg至约60mg/kg、或约50mg/kg至约75mg/kg体重/天)。例如,当组合物包括cx-8998时,cx-8998的剂量可为约10mg/kg体重/天。例如,当组合物包括cx-8998时,cx-8998的剂量可为约30mg/kg体重/天。例如,当组合物包括cx-8998时,cx-8998的剂量可为约60mg/kg体重/天。例如,组合物(例如用于人类中的组合物)可包括可为以下的一种或多种t型钙通道拮抗剂的剂量:约10μg/kg至约1000μg/kg体重/天(例如约10μg/kg至约900μg/kg、约10μg/kg至约800μg/kg、约10μg/kg至约700μg/kg、约10μg/kg至约600μg/kg、约10μg/kg至约500μg/kg、约10μg/kg至约400μg/kg、约10μg/kg至约300μg/kg、约10μg/kg至约200μg/kg、约10μg/kg至约100μg/kg、约10μg/kg至约50μg/kg、约50μg/kg至约1000μg/kg、约100μg/kg至约1000μg/kg、约200μg/kg至约1000μg/kg、约300μg/kg至约1000μg/kg、约400μg/kg至约1000μg/kg、约500μg/kg至约1000μg/kg、约600μg/kg至约1000μg/kg、约700μg/kg至约1000μg/kg、约800μg/kg至约1000μg/kg、约900μg/kg至约1000μg/kg、约50μg/kg至约900μg/kg、约100μg/kg至约800μg/kg、约200μg/kg至约700μg/kg、约300μg/kg至约600μg/kg、约400μg/kg至约500μg/kg、约100μg/kg至约200μg/kg、约200μg/kg至约300μg/kg、约300μg/kg至约400μg/kg、约400μg/kg至约500μg/kg、约500μg/kg至约600μg/kg、约600μg/kg至约700μg/kg、约700μg/kg至约800μg/kg、或约800μg/kg至约900μg/kg体重/天)。例如,当组合物包括cx-8998时,cx-8998的剂量可为约100μg/kg体重/天。例如,当组合物包括cx-8998时,cx-8998的剂量可为约300μg/kg体重/天。例如,当组合物包括cx-8998时,cx-8998的剂量可为约600μg/kg体重/天。在一些情况下,所述剂量可取决于例如以下变量:疾病或病症的类型和进展程度、特定患者的总体健康状态、所选化合物的相对生物功效、赋形剂的配制及其施用途径。在一些情况下,剂量可随时间增加。例如,可在一段时间(例如1周、2周、3周或4周)内将剂量滴定至最终剂量。在一些情况下,剂量可随时间减少。例如,在一种或多种症状减轻或消除后,可将剂量减少至维持剂量。剂量可例如每天一次、每天两次、每天三次或每天四次施用。在一些情况下,剂量可每日施用一次。在一些情况下,剂量可每日施用两次。在一些情况下,可从源自体外或体内模型测试系统的剂量-反应曲线外推本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)中的一种或多种t型钙通道拮抗剂的有效量。有效量(例如治疗有效量)可保持恒定或者可根据哺乳动物对治疗的反应调节为浮动标度或可变剂量。各种因子可影响用于特定应用的实际有效量。例如,食物效应(例如施用组合物的哺乳动物是处于进食状态或禁食状态)、施用频率、治疗持续时间、多种治疗剂的使用、施用途径和运动障碍(例如原发性震颤、癫痫和/或帕金森病)的严重程度)可能需要增加或减少所施用实际有效量。施用频率可为减轻运动障碍(例如原发性震颤、癫痫和/或帕金森病)的严重程度而不对哺乳动物产生显著毒性的任何频率。例如,施用频率可为约每周一次至约每天三次、约每月两次至约每天六次、或约每周两次至约每天一次。施用频率可保持恒定或可在治疗的持续时间内可变。利用本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)的疗程可包括静息时段。例如,可在两周时段内每日施用含有治疗有效量的一种或多种t型钙通道拮抗剂的药学上可接受的组合物,接着是两周静息时段,并且这种方案可重复多次。至于有效量,各种因子可影响用于特定应用的实际施用频率。例如,有效量、治疗持续时间、多种治疗剂的使用、施用途径和运动障碍(例如原发性震颤、癫痫和/或帕金森病)的严重程度可能需要增加或减小施用频率。施用本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)的有效持续时间可为减轻运动障碍(例如原发性震颤、癫痫和/或帕金森病)的严重程度而不会对哺乳动物产生显著毒性的任何持续时间。例如,有效持续时间可从若干天至若干周、月或年变化。在一些情况下,治疗运动障碍(例如原发性震颤、癫痫和/或帕金森病)的有效持续时间可在约1个月至约10年的持续时间范围内。在一些情况下,治疗运动障碍(例如原发性震颤、癫痫和/或帕金森病)的有效持续时间可为无限的(例如所治疗哺乳动物的寿命)。例如,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括一种或多种t型钙通道拮抗剂的组合物达约1周至约9周(例如约1周至约8周、约1周至约7周、约1周至约6周、约1周至约5周、约1周至约4周、约1周至约3周、约1周至约2周、约2周至约9周、约3周至约9周、约4周至约9周、约5周至约9周、约6周至约9周、约7周至约9周、约8周至约9周、约2周至约8周、约3周至约7周、约4周至约6周、约2周至约4周、约3周至约5周、约4周至约6周、约5周至约7周、或约6周至约8周)。在一些情况下,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括cx-8998的组合物达约2周(例如约15天)。在一些情况下,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括cx-8998的组合物达约4周(例如约28天)。例如,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括一种或多种t型钙通道拮抗剂的组合物达约8周或更长时间。例如,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括一种或多种t型钙通道拮抗剂的组合物达约12周或更长时间。例如,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括一种或多种t型钙通道拮抗剂的组合物达约2个月或更长时间。例如,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括一种或多种t型钙通道拮抗剂的组合物达约4个月或更长时间。例如,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括一种或多种t型钙通道拮抗剂的组合物达约6个月或更长时间。例如,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括一种或多种t型钙通道拮抗剂的组合物达约12个月或更长时间。例如,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括一种或多种t型钙通道拮抗剂的组合物达约24个月或更长时间。多种因子可影响用于特定治疗的实际有效持续时间。例如,有效持续时间可随着施用频率、有效量、多种治疗剂的使用、施用途径和所治疗疾患的严重程度而变。在一些情况下,当向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括cx-8998的组合物时,组合物可有效地达到cx-8998的最小有效浓度(mec)(例如哺乳动物的血浆中产生本文所述治疗作用所需的cx-8998的最小浓度)。例如,cx-8998的mec可为约100nm至约1800nm(例如约200nm至约1800nm、约300nm至约1800nm、约400nm至约1800nm、约500nm至约1800nm、约700nm至约1800nm、约1000nm至约1800nm、约1200nm至约1800nm、约1500nm至约1800nm、约100nm至约1500nm、约100nm至约1200nm、约100nm至约1000nm、约100nm至约800nm、约100nm至约700nm、约100nm至约600nm、约100nm至约500nm、约100nm至约400nm、约100nm至约300nm、约200nm至约1500nm、约300nm至约1200nm、约500nm至约1000nm、约600nm至约800nm、约200nm至约400nm、约400nm至约600nm、或约300nm至约500nm)。在一些情况下,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括cx-8998的组合物以达到约200nm的cx-8998的mec。在一些情况下,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括cx-8998的组合物以达到约209nm的cx-8998的mec。在一些情况下,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括cx-8998的组合物以达到约215nm的cx-8998的mec。在一些情况下,在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括一种或多种cav3拮抗剂如cx-8998的组合物时,组合物可有效维持cav3拮抗剂的mec(例如在哺乳动物的血液中和/或在从哺乳动物获得的血样、例如血浆样品中)达任何适当时间量。例如,cx-8998的mec在哺乳动物中可维持至少约12小时(例如约12小时、约13小时、约14小时、约15小时、约16小时、约17小时、约18小时、约19小时、约20小时、约21小时、约22小时、约23小时、约24小时、约25小时或约26小时)。例如,cx-8998的mec可维持约10小时至约15小时(例如约10小时至约14小时、约10小时至约13小时、约10小时至约12小时、约10小时至约11小时、约11小时至约15小时、约12小时至约15小时、约13小时至约15小时、约14小时至约15小时、约11小时至约14小时、约12小时至约13小时、约11小时至约12小时、或约13小时至约14小时)。在一些情况下,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括cx-8998的组合物以维持cx-8998的mec达约12小时。例如,cx-8998的mec可维持约22小时至约26小时(例如约22小时至约25小时、约22小时至约24小时、约22小时至约23小时、约23小时至约26小时、约24小时至约26小时、约25小时至约26小时、约23小时至约25小时、或约23小时至约24小时)。在一些情况下,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括cx-8998的组合物以维持cx-8998的mec达约24小时。在一些情况下,在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括cx-8998的组合物时,组合物可在哺乳动物的血浆中有效地达到cx-8998的最大安全浓度(例如充分耐受(例如引起最少、减少或不引起副作用和/或不良事件)的cx-8998的最大浓度,所述副作用和/或不良事件是例如眩晕、头痛、亢奋、注意力障碍、感觉异常、幻觉、失眠、口腔干燥、味觉障碍、感觉迟钝、嗜眠、嗜睡、睡眠障碍、恶心、呕吐、静坐不能、意识水平下降、晕厥、记忆障碍、焦虑、不安定、疲劳、易怒、便秘、耳鸣、厌食、情绪障碍、性无能、复视、眼球震颤、困倦、麻疹样皮疹、粒细胞减少、粒细胞缺乏、红细胞发育不全和不发育)。例如,cx-8998的最大安全浓度可为约1000nm至约1800nm(例如约1100nm至约1800nm、约1200nm至约1800nm、约1300nm至约1800nm、约1400nm至约1800nm、约1500nm至约1800nm、约1600nm至约1800nm、约1700nm至约1800nm、约1000nm至约1700nm、约1000nm至约1600nm、约1000nm至约1500nm、约1000nm至约1400nm、约1000nm至约1300nm、约1000nm至约1200nm、约1000nm至约1100nm、约1100nm至约1700nm、约1200nm至约1600nm、约1300nm至约1500nm、约1100nm至约1300nm、约1200nm至约1400nm、约1300nm至约1500nm、约1400nm至约1600nm、或约1500nm至约1700nm)。在一些情况下,在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括一种或多种cav3拮抗剂如cx-8998的组合物时,组合物可有效维持cmax(例如在哺乳动物的血浆中达到的最大浓度)低于cav3拮抗剂的耐受性阈值。例如,cx-8998的cmax可小于约1800nm(例如约1700nm、约1600nm、约1500nm、约1400nm、约1300nm、约1200nm、约1100nm、约1000nm、约990nm、约980nm、约970nm、约960nm、约950nm、约940nm、约930nm、约920nm、约910nm或约900nm)。例如,cx-8998的cmax可为约900nm至约1800nm(例如约1000nm至约1800nm、约1200nm至约1800nm、约1400nm至约1800nm、约1500nm至约1800nm、约1600nm至约1800nm、约900nm至约1700nm、约900nm至约1500nm、约900nm至约1400nm、约900nm至约1300nm、约900nm至约1200nm、约900nm至约1100nm、约1000nm至约1500nm、或约1200nm至约1400nm)。在一些情况下,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括cx-8998的组合物以达到约900nm的cx-8998的cmax。在一些情况下,可向哺乳动物(例如患有或处于患上一种或多种运动障碍风险下的哺乳动物)施用包括cx-8998的组合物以达到约1000nm的cx-8998的cmax。在一些情况下,在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括一种或多种cav3拮抗剂如cx-8998的组合物时,组合物可有效维持约3,000nm*小时至约10,000nm*小时的cav3拮抗剂的曲线下面积(auc)。例如,cx-8998的auc可为约3,000nm*小时至约9,000nm*小时、约3,000nm*小时至约8,000nm*小时、约3,000nm*小时至约7,000nm*小时、约3,000nm*小时至约6,000nm*小时、约3,000nm*小时至约5,000nm*小时、约3,000nm*小时至约4,000nm*小时、约4,000nm*小时至约10,000nm*小时、约5,000nm*小时至约10,000nm*小时、约6,000nm*小时至约10,000nm*小时、约7,000nm*小时至约10,000nm*小时、约8,000nm*小时至约10,000nm*小时、约9,000nm*小时至约10,000nm*小时、约4,000nm*小时至约9,000nm*小时、约5,000nm*小时至约8,000nm*小时、约6,000nm*小时至约7,000nm*小时、约4,000nm*小时至约6,000nm*小时、约5,000nm*小时至约7,000nm*小时、约6,000nm*小时至约8,000nm*小时、或约7,000nm*小时至约9,000nm*小时。auc可在任何适当时间(例如在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括cx-8998的组合物之后的任何适当时间)测量。auc可持续任何适当时间间隔(例如在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括cx-8998的组合物之后的任何适当持续时间)进行测量。例如,auc可持续约12小时(auc0-24或auc24)至约36小时(auc0-36或auc36;例如,约12小时至约32小时、约12小时至约28小时、约12小时至约24小时、约12小时至约20小时、约12小时至约18小时、约18小时至约36小时、约22小时至约36小时、约24小时至约36小时、约15小时至约32小时、约18小时至约24小时、约12小时至约15小时、约15小时至约18小时、约18小时至约22小时、约22小时至约26小时、约26小时至约30小时、或约30小时至约32小时)进行测量。在一些情况下,auc可持续约24小时(例如auc0-24或auc24)进行测量。在一些情况下,在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括一种或多种cav3拮抗剂如cx-8998的组合物时,组合物可有效维持哺乳动物中cav3拮抗剂的血浆浓度(例如平均血浆浓度)介于cx-8998的mec(例如约400nmcx-8998)与cx-8998的cmax(例如约1000nm)之间。例如,在向哺乳动物施用包括cx-8998的组合物时,组合物可有效维持哺乳动物中cx-8998的平均血浆浓度为约400nmcx-8998至约1000nmcx-8998。例如,在向哺乳动物施用包括cx-8998的组合物时,组合物可有效维持哺乳动物中cx-8998的平均血浆浓度如图74中所示。在包括cx-8998的组合物包括被配制用于延迟和/或持续释放cx-8998的第一组分和任选地被配制用于立即释放cx-8998的第二组分的情况下,被配制用于延迟和/或持续释放cx-8998的第一组分可有效维持cx-8998的血浆浓度(例如平均血浆浓度)介于cx-8998的mec(例如约400nmcx-8998)与cx-8998的cmax(例如约1000nm)之间,使得组合物可有效维持cx-8998的浓度介于cx-8998的mec与cx-8998的cmax之间,并且被配制用于立即释放cx-8998的第二组分在存在时可有效地快速(例如在小于约60分钟内)达到血浆浓度(例如平均血浆浓度)至少为cx-8998的mec(例如至少约400nmcx-8998)。例如,在向哺乳动物施用包括被配制用于延迟和/或持续释放cx-8998的第一组分和任选地被配制用于立即释放cx-8998的第二组分的包括cx-8998的组合物时,组合物可在小于约60分钟内有效地达到cx-8998的平均血浆浓度,并且可有效维持哺乳动物中cx-8998的平均血浆浓度为约400nmcx-8998至约1000nmcx-8998。通过包括cx-8998的本文所述组合物达到的包括cx-8998的平均血浆浓度可维持任何适当时间量(例如在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括cx-8998的组合物之后的任何适当持续时间)。例如,约400nm至约1000nm的cx-8998的平均血浆浓度可维持至少约12小时(例如约12小时、约13小时、约14小时、约15小时、约16小时、约17小时、约18小时、约19小时、约20小时、约21小时、约22小时、约23小时、约24小时、约25小时或约26小时)。在一些情况下,约400nmcx-8998至约1000nmcx-8998的平均血浆浓度可维持约12小时至约36小时(例如约12小时至约32小时、约12小时至约28小时、约12小时至约24小时、约12小时至约20小时、约12小时至约18小时、约18小时至约36小时、约22小时至约36小时、约24小时至约36小时、约15小时至约32小时、约18小时至约24小时、约12小时至约15小时、约15小时至约18小时、约18小时至约22小时、约22小时至约26小时、约26小时至约30小时、或约30小时至约32小时)。例如,约400nmcx-8998至约1000nmcx-8998的平均血浆浓度可维持约18小时。例如,约400nmcx-8998至约1000nmcx-8998的平均血浆浓度可维持约24小时。例如,在向哺乳动物施用包括cx-8998的组合物时,组合物可有效维持哺乳动物中cx-8998的平均血浆浓度为约400nmcx-8998至约1000nmcx-8998达约24小时(例如可具有约400nm*小时和约1000nm*小时的auc达约24小时)。在一些情况下,在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括一种或多种cav3拮抗剂如cx-8998的组合物时,组合物可有效维持cav3拮抗剂的最大浓度(cmax)除以施用后24小时的cav3拮抗剂的平均血浆浓度(例如)为小于5.0(例如约5、约4.5、约4、约3.5、约3、约2.5、约2、约1.5或约1.0)。例如,在向哺乳动物施用包括cx-8998的组合物时,组合物可有效维持哺乳动物中cx-8998的为约1.0至约5.0(例如约1.0至约4.5、约1.0至约4.0、约1.0至约3.5、约1.0至约3.0、约1.0至约2.5、约1.0至约2.0、约1.5至约5.0、约2.0至约5.0、约2.5至约5.0、约3.0至约5.0、约3.5至约5.0、约4.0至约5.0、约1.5至约4.5、约2.0至约4.0、约1.5至约2.5、约2.5至约3.5、或约3.5至约4.5)。在一些情况下,在向患有或处于患上一种或多种运动障碍风险下的哺乳动物施用包括一种或多种cav3拮抗剂如cx-8998的组合物时,组合物可有效地达到cmax低于由包括一种或多种cav3拮抗剂如cx-8998的立即释放组合物(例如包括相同剂量的一种或多种cav3拮抗剂的立即释放组合物)所达到的cmax。例如,在向哺乳动物施用包括cx-8998的组合物时,组合物可有效维持cx-8998的cmax比由包括cx-8998的立即释放组合物所达到的cmax低约30%至约70%(例如约30%至约60%、约30%至约50%、约30%至约40%、约40%至约70%、约50%至约70%、约60%至约70%、或约40%至约60%)。在一些情况下,治疗患有或处于患上如本文所述一种或多种运动障碍风险下的哺乳动物(例如通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)的方法还可包括将哺乳动物鉴定为患有或处于患上一种或多种运动障碍风险下。可使用任何适当方法将哺乳动物鉴定为患有运动障碍。将哺乳动物鉴定为患有或处于患上运动障碍风险下的方法可包括(但不限于)回顾哺乳动物的病史、回顾哺乳动物的家族史、执行体格检查(例如神经学检查,包括检查腱反射、肌肉强度和肌张力、感知某些感觉的能力、姿势和协调性和/或步态)、实验室检验(例如包括针对甲状腺疾病检查哺乳动物的血液和/或尿液、代谢问题、药物副作用、酒精水平和/或可引起震颤的化学物质的水平的实验室检验)、成像(例如功能成像)、表现测试(例如,用以评价震颤自身的表现测试,包括从玻璃杯饮水、保持手臂伸展、书写和/或绘制螺线)、和/或多巴胺转运蛋白扫描。在一些情况下,将哺乳动物鉴定为患有或处于患上运动障碍风险下可包括基因测试。例如,测试存在或不存在与运动障碍相关的一种或多种基因变体可用于将哺乳动物鉴定为患有或处于患上运动障碍(例如原发性震颤)风险下。例如,与运动障碍(例如原发性震颤)相关的基因变体可如别处所述(参见例如odgerel等人,2018biorxivdoi:http://dx.doi.org/10.1101/248443)。在一些情况下,在治疗患有或处于患上如本文所述一种或多种运动障碍(例如原发性震颤、癫痫和/或帕金森病)风险下的哺乳动物(例如通过施用含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)时,治疗还可包括一种或多种额外疗法。疗法可为任何适当疗法(例如,用于治疗一种或多种运动障碍和/或与运动障碍相关的一种或多种症状的疗法),例如施用一种或多种医药剂、施用一种或多种营养制品、物理疗法、职业疗法、手术、超声波丘脑切开术、神经刺激技术、数字治疗剂和替代药物。可用于治疗患有或处于患上原发性震颤和/或与原发性震颤相关的一种或多种症状风险下的哺乳动物的疗法的实例包括(但不限于)施用一种或多种β阻断剂(例如普萘洛尔),施用一种或多种抗癫痫发作药物(例如扑米酮、加巴喷丁、托吡酯、普瑞巴林、唑尼沙胺和乙琥胺),施用一种或多种安定药(例如阿普唑仑和可那氮平),注射a型奥那肉毒毒素(onabotulinumtoxina)(保妥适(botox)),物理疗法,职业疗法,手术(例如深脑刺激),超声波丘脑切开术(例如聚焦/引导超声波丘脑切开术),神经刺激(例如,跨颅磁刺激)和数字治疗剂(例如,诸如震颤消除佩戴物等装置和诸如震颤勺等用具)。在一些情况下,可用于治疗患有或处于患上原发性震颤和/或与原发性震颤相关的一种或多种症状风险下的哺乳动物的疗法可如别处所述(参见例如美国神经病学会关于原发性震颤的治疗指南,例如可在aan.com/guidelines/home/guidelinedetail/492获得的那些)。可用于治疗患有或处于患上帕金森病和/或与帕金森病相关的一种或多种症状风险下的哺乳动物的疗法的实例包括(但不限于)施用一种或多种多巴胺激动剂(例如卡比多巴-左旋多巴、普拉克索、罗匹尼罗、罗替戈汀和阿朴吗啡),施用一种或多种单胺氧化酶b(maob)抑制剂(例如司来吉兰、雷沙吉兰和沙非酰胺),施用一种或多种儿茶酚o-甲基转移酶(comt)抑制剂(例如恩卡朋和托卡朋),施用一种或多种抗副交感神经药(例如苯托品和三己芬迪),施用金刚烷胺,物理疗法,职业疗法和手术(例如,深脑刺激)。在一些情况下,可用于治疗患有或处于患上帕金森病和/或与帕金森病相关的一种或多种症状风险下的哺乳动物的疗法可如别处所述(参见例如美国神经病学会关于帕金森病的治疗指南,例如可在网站movementdisorders.org/mds-files1/resources/pdfs/treatmentsformotorsymptomsofpd-2018.pdf获得的那些)。在患有或处于患上一种或多种运动障碍风险下的哺乳动物用含有一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物治疗并且用一种或多种额外疗法治疗的情况下,一种或多种额外疗法可同时或单独地施用。例如,首先施用含有一种或多种t型钙通道拮抗剂(例如cx-8998)的组合物,并且其次施用一种或多种额外疗法,或反之亦然。本文件也提供了鉴定本文所述组合物(例如药学上可接受的组合物)(例如含有一种或多种t型钙通道拮抗剂如cx-8998的组合物)中总活性部分(tam)的存在、不存在或量的方法。如本文所用的术语“tam”是指组合物中存在的活性组分的总和。例如,当组合物包括cx-8998时,tam可包括cx8998、m01、m02、m03和/或m04中的一者或多者。例如,当组合物包括cx-8998时,方法可包括鉴定cx8998和代谢物m01、m02、m03和m04的存在、不存在或量。在一些情况下,质谱(ms)方法可用于量化(例如,同时量化)cx-8998、m01、m02、m03和/或m04。可使用任何适当ms方法。在一些情况下,液相色谱-串联ms(lc/ms/ms)生物分析方法可用于同时量化cx-8998和代谢物m01、m02、m03和m04。例如,lc/ms/ms可用于量化与cx-8998相关的tam。在一些情况下,鉴定组合物中tam(例如cx8998、m01、m02、m03和/或m04)的存在、不存在或量可有效地测定系统性能适合性、量化范围、校准线性、测定准确度和精密度和/或测定回收率的验收标准。例如,当组合物包括cx-8998时,鉴定cx8998、m01、m02、m03和/或m04的存在、不存在或量可有效地测定系统性能适合性、量化范围、校准线性、测定准确度和精密度以及测定回收率的验收标准。本文件还提供了含有本文所述的一种或多种材料的试剂盒。例如,本文所述试剂盒中提供的材料可用于治疗患有或处于患上如本文所述一种或多种运动障碍(例如原发性震颤、癫痫和/或帕金森病)风险下的哺乳动物(例如人类)。在一些情况下,包括一种或多种t型钙通道拮抗剂(例如一种或多种cav3拮抗剂如cx-8998)的组合物(例如药学上可接受的组合物)可与包装材料组合并且以试剂盒形式销售。例如,试剂盒可包括包含提供于单位剂量或多剂量容器中的本文所述一种或多种t型钙通道拮抗剂的组合物。容器可具有任何适当大小(例如60cc容器)。容器可为任何适当材料(例如高密度聚乙烯(hdpe),例如白色hdpe)。容器可包括封盖(例如儿童安全封盖)。封盖可为任何适当材料(例如聚丙烯,例如白色、33mm聚丙烯)。在一些情况下,瓶中不包括干燥剂。在一些情况下,瓶中不包括棉花。在一些情况下,试剂盒可包括呈片剂或胶囊(例如,用于口服施用的片剂或胶囊)形式的包括本文所述cx-8998的药学上可接受的组合物,其具有作为持续释放组合物的组分(例如第一组分)和任选地作为立即释放组合物的组分(例如第二组分)。在一些情况下,所述试剂盒中包括的包装材料通常含有阐述可如何使用组合物以例如治疗患有或处于患上如本文所述一种或多种运动障碍(例如原发性震颤、癫痫和/或帕金森病)风险下的哺乳动物(例如人类)的说明书或标记。例如,试剂盒可包括指示哺乳动物在服用组合物之前持续至少约4小时(例如约4小时、约5小时、约6小时、约7小时、约8小时、约9小时、约10小时、约11小时或约12小时)不能进食的说明书或标记。例如,试剂盒可包括指示哺乳动物与食物一起服用组合物的说明书或标记。例如,试剂盒可包括指示哺乳动物在睡醒的约4小时内(例如,睡醒后约4小时、睡醒后约3小时、睡醒后约2小时、睡醒后约1小时、睡醒后约30分钟或睡醒后立即)服用组合物的说明书或标记。在一些情况下,这种试剂盒中包括的包装材料通常含有阐述组合物可如何储存(例如,储存在约15℃至约30℃、或约20℃至约25℃之间)的说明书或标记。在以下实施例中进一步阐述本发明,所述实施例并不限制权利要求书中所述的本发明范围。实施例原发性震颤等级评估量表(tetras)震颤研究组于2008年首次发布trg原发性震颤等级评估量表(tetras)(elble等人,2008movdisord.23(增刊1):s1-6)。tetras由9项目表现子量表和12项目日常生活活动(adl)子量表组成。将tetras开发为对et的快速临床评估,其不需要笔和纸以外的设备。表现子量表的施用花费少于10分钟。所述量表采用客观指标来减少经验评定者的偏差。为了评价tetras的评定者间可靠性,elble等人(2012movdisord.27(12):1567-1569)录制了50份tetras检查,包括对44名患有et的患者和6个对照的评估。et的严重程度从轻度到重度不等。十名专家对视频中的患者进行了2次评定,评定隔开1至2个月的间隔。在10名评定者中,有6名参与tetras的开发,而4名从未使用所述量表。使用具有绝对一致性定义的双向随机效应类内相关性(icc)计算量表的评定者间可靠性。头部和上肢震颤的评定者内和评定者间icc在0.86至0.96的范围内,并且总评分的icc为0.94和0.96。声音、面部、躯干和腿的icc较不稳健(elble等人,2012movdisord.27(12):1567-1569)。etras表现子量表在临床实践中被广泛使用,并且具有高内容效度和强评定者间可靠性。tetrasadl与表现评分高度相关,并且上肢功能的tetras等级与上肢震颤的传感器测量(加速度测定法)密切相关(mostile等人,2010movdisord.25(12):1938-1943)。tetras也显示对随时间的震颤变化敏感(voller等人,2014movdisord.29(4):555-558)。评分越高,震颤越严重,评分减小是震颤改善。实施例1.如由研究者评定的tetras-ps从基线至第28天的显著变化tetras表现子量表表现子量表量化头部、面部、声音、肢体和躯干的震颤。每一项目以0至4等级量表评定,其中上肢震颤的评分允许0.5分增量。具体幅值范围(以厘米测量)定义震颤等级。评定者首先估算震颤的最大幅值,并且然后指定相应等级。个别等级评分的总和提供总体表现评分,在0至64范围内。完整tetras表现子量表如下所示。评分为0-4。对于大部分项目,评分仅由整数定义,但如果认为等级介于两个整数等级之间并且无法与整数核对,则可使用0.5的增量。每增加0.5个等级,专门定义用于评估上肢姿势和运动性震颤以及点近似任务(项目4和8)。除站立震颤外,所有检查项目都在患者舒适就座的情况下实施。对于每一项目,对在检查期间的任何时刻看到的最高幅值进行评分。指示患者不要试图抑制震颤,而是让其出现。1.头部震颤:头部左右完全旋转,并且然后在中间位置观察10s。然后指示患者完全向左凝视并且然后向右凝视,将头部完全置于中间。应将鼻子用作评估的标志,并在检查期间对最大幅值的偏移进行评定。0=无震颤1=轻微震颤(<0.5cm)2=轻度震颤(0.5-<2.5cm)3=中度震颤(2.5-5cm)4=重度或毁损面容的震颤(>5cm)2.面部(包括颌)震颤:微笑、闭上眼睛、张开嘴巴、撅起嘴唇。对涉及最多的面部解剖结构的最高幅值进行评分,无论其是在休息或活动期间发生。反复眨眼或眼睛振抖不应被视为面部震颤的一部分。0=无震颤1=轻微;几乎不可感知的震颤2=轻度:可觉察的震颤3=中度:明显震颤,存在于大部分自主面部收缩中4=重度:肉眼可见的毁损面容的震颤3.声音震颤:首先要求受试者发出延长的“啊”声和“咿”声,各自持续5秒。然后通过询问患者“您如何度过您的平均一天?”评估正常交谈期间的谈话0=无震颤1=轻微:在啊和咿期间震颤,并且在谈话期间无震颤2=轻度:在“啊”和“咿”中震颤并且在谈话中最小震颤3=中度:谈话中明显震撼,完全可以理解4=重度:一些单词难以理解4.上肢震颤:在三个动作期间对震颤进行评估:水平向前伸姿势、横向“翼拍打”姿势和手指-鼻子-手指测试。对每一上肢进行单独评估和评分。水平向前伸姿势保持5秒。横向翼拍打姿势保持20秒。手指-鼻子-手指运动执行三次。应当沿着任何单一面在最大位移点处使用手的最大位移点估算幅值评估。例如,可以拇指或第五个手指来评估绕腕部定桩的纯外旋-内转震颤的幅值。a)向前伸开姿势性震颤:受试者应将其手臂向前,稍微向中线横移,并平行于地面。腕部也应笔直,并展开手指,以使其彼此不接触。b)横向“翼拍打”姿势性震颤:受试者将其手臂平行于地面展开并弯曲肘部,以使两只手彼此不完全接触并处于鼻子的水平面。展开手指以使其彼此不接触。姿势应保持20秒。c)运动性震颤:受试者仅伸出其食指。他们然后触摸位于其触及的最大范围内的设定物体或检查者的手指,其位于相同的高度(与地面平行)并且在中线的稍微外侧。然后,受试者触摸自己的鼻子(如果震颤严重,则触摸下巴),并且来回重复三遍。仅评估沿着最大震颤幅值轨迹的位置。这通常在鼻子处或在四肢完全伸展的点处。对于所有三个手震颤等级,0=无震颤1=震颤几乎不可见1.5=震颤可见,但小于1cm,2=震颤为1-<3cm幅值2.5=震颤为3-<5cm幅值3=震颤为5-<10cm幅值3.5=震颤为10-<20cm幅值4=震颤为>20cm幅值5.下肢震颤:将每一下肢平行于地面水平举起,各5秒。然后对每条腿实施标准足跟至胫动作,实施3次。对任一动作中的最大震颤进行评分,并且仅对具有最大震颤的肢体进行评分。震动可能存在于肢体的任何部位,包括脚。0=无震颤1=轻微:几乎不可感知2=轻度,在任何点小于1cm3=中度震颤,在任何点小于5cm4=重度震颤,大于5cm6.阿基米德螺线:展示如何绘制大约占无网格线标准(信)纸页面1/4的阿基米德螺线。螺旋线应间隔约1.3cm(0.5英寸)。然后要求受试者临摹螺线。对每只手单独测试和评分。使用原子笔。应该握住笔,使得肢体无一部位接触桌子。将纸固定在桌子上适于患者的绘制风格的位置。对螺线中的震颤、而非肢体的运动进行评分。0=正常1=轻微:震颤几乎不可见。2=轻度:明显震颤3=中度:图的部分不可识别。4=重度:图不可识别7.手写:请患者仅使用惯用手来写标准句子“这是我的最佳手写样品”。患者必须草书(即,不打印)。他们无法用另一只手握住或稳定其手。使用原子笔。将纸固定在桌子上适于患者的书写风格的位置。对书写中的震颤、而非肢体的运动进行评分。0=正常1=轻微:由于几乎不可见的震颤而造成的不整洁。2=轻度:清晰,但具有相当大的震颤。3=中度:一些词难以辨认。4=重度:完全难以辨认8.点逼近任务:检查者画一个点或x,并且指示受试者握住笔尖,“尽可能靠近点(或x的中心)而不要接触它(理想地约1mm)达10秒”。每只手单独评分。0=无震颤1=震颤几乎不可见1.5=震颤可见,但小于1cm,2=震颤为1-<3cm幅值2.5=震颤为3-<5cm幅值3=震颤为5-<10cm幅值3.5=震颤为10-<20cm幅值4=震颤为>20cm幅值9.站立震颤:受试者站立,如果可能,无辅助站立。膝分开10-20cm并且以10-20°弯曲。臂朝下放在受试者的侧面。在腿或躯干上的任何点处对震颤进行评估0=无震颤1=几乎不可感知的震颤2=明显但轻度震颤,不引起不稳定,3=中度震颤,损害站姿的稳定性4=重度震颤,在无辅助情况下不能站立主要研究者(或次要研究者)对tetras表现子量表进行评分,在功效的统计分析中利用由研究者提供的tetras表现子量表评分。使用协方差分析(ancova)模型执行tetras表现子量表的功效分析,所述模型具有固定的治疗作用、伴随的抗震颤药物使用、部位类型和tetras表现子量表的基线值。tetras表现量表相比于基线的平均变化指示,cx-8998组与安慰剂不同(图1和表1)。所有测试都是使用ancova模型的最小二乘法(ls)平均值和在α=0.05显著性水平下的双侧检验来实施。如果数据指示偏离正态分布,则实施相应秩检验。表1.如由医师评定的从基线至第15天和第28天的tetras-ps变化实施例2.tetras-adl从基线至第15天和第28天的显著变化tetras日常生活活动子量表adl子量表包括别处阐述的量表中所评估的许多项目(参见例如fahn等人,“clinicalratingscalefortremor,”parkinson'sdiseaseandmovementdisorders中的第225-234页(jankovic等人编辑),baltimore:williams&wilkins,1993;louis,2000archneurol.57(10):1522-1524;以及bain等人,1993jneurolneurosurgpsychiatry56(8):868-873),包括吃喝、穿衣和个人卫生、运送物品和精细动作技能。每一项目以0至4量表评定,其中0代表正常活动,并且4代表重度异常。个别评分的总和提供总体评分,在0至48范围内。完整的tetrasadl子量表如下。日常生活活动子量表对震颤对日常活动的影响进行评定(0-4评分)。1.说话0=正常1=轻微声音震颤,仅在“紧张”时2=轻度声音震颤。所有词都容易理解。3=中度声音震颤。一些词难以理解。4=重度声音震颤。大部分词难以理解。2.用勺子进食0=正常1=轻微异常。存在震颤,但不干扰用勺子进食。2=轻度异常。洒出一点。3=中度异常。洒出许多或改变策略以例如使用两只手或俯身完成任务。4=重度异常。不能用勺子进食。3.从玻璃杯饮水0=正常1=轻微异常。存在震颤,但不干扰从玻璃杯饮水。2=轻度异常。洒出一点。3=中度异常。洒出许多或改变策略以例如使用两只手或俯身完成任务。4=重度异常。不能从玻璃杯饮水或使用吸管或吸管杯。4.卫生0=正常。1=轻微异常。存在震颤,但不干扰卫生。2=轻度异常。有些困难但能完成任务。3=中度异常。除非改变策略,例如使用两只手或使用受影响较小的手,否则不能做大部分精细任务,例如涂口红或剃须。4=重度异常。不能独立地完成卫生活动。5.穿衣0=正常。1=轻微异常。存在震颤,但不干扰穿衣。2=轻度异常。能够做任何事情,但由于震颤有些困难。3=中度异常。除非使用策略(例如使用维可牢搭链(velcro)、在穿上之前扣衬衫或避免穿有鞋带的鞋子),否则无法完成大部分穿衣。4=重度异常。不能独立地穿衣。6.倾倒0=正常。1=轻微异常。存在震颤,但不干扰倾倒。2=轻度异常。必须极为小心避免洒出,但可能偶尔洒出。3=中度异常。必须使用两只手或使用其他策略来避免洒出。4=重度异常。不能倾倒。7.运送食物托盘、盘子或类似物品0=正常1=轻微异常。存在震颤,但不干扰运送食物托盘、盘子或类似物品。2=轻度异常。必须极为小心避免洒出食物托盘上的物品。3=中度异常。使用策略(例如紧紧地抵在身体上)进行运送。4=重度异常。不能运送食物托盘或类似物品。8.使用钥匙0=正常1=轻微异常。存在震颤,但可用一只手毫不困难地插入钥匙。2=轻度异常。通常会错过目标,但用一只手仍会常规地将钥匙放入锁中。3=中度异常。需要使用两只手或其他策略来将钥匙放入锁中。4=重度异常。无法将钥匙放入锁中。9.书写0=正常1=轻微异常。存在震颤,但不干扰书写。2=轻度异常。由于震颤而难以书写3=中度异常。在不使用策略(例如用另一只手握住书写手、以不同的方式握住笔或使用大笔)的情况下,不能书写。4=重度异常。不能书写。10.工作。如果患者退休,则询问其否仍在工作。如果患者是家庭主妇,则询问与家务有关的问题:0=正常。1=轻微异常。存在震颤,但不影响在工作或在家中的表现。2=轻度异常。震颤干扰工作;能够做任何事情,但有错误。3=中度异常。无法在不使用策略(例如更换工作或使用特殊设备)的情况下继续工作。4=重度异常。不能实施任何工作或家庭工作。11.任务受影响最大(姓名任务,例如,使用计算机鼠标、书写等)的总体失能任务0=正常1=轻微异常。存在震颤,但不影响任务。2=轻度异常。震颤干扰任务,但仍能够实施任务。3=中度异常。能进行任务,但必须使用策略。4=重度异常。不能进行任务。12.社交影响0=无1=意识到震颤,但其不影响生活方式或职业生活。2=在一些社交场合或职业会议上因震颤而感到尴尬。3=由于震颤而避免参与一些社交场合或职业会议。4=由于震颤而避免参与大部分社交场合或职业会议。使用与tetras-ps所述相同类型的ancova模型,在α=0.05显著性水平下使用双侧检验对tetras-adl执行分析(图2和表2)。表2.tetras-adl从基线至第15天和第28天的变化。实施例3.tetras总评分从基线至第15天和第28天的显著变化tetras总评分总tetras评分定义为tetras表现子量表总评分与tetrasadl子量表评分的总和。tetras总评分将患者报告的结果和震颤幅值的客观测量值组合成单一评分。使用与tetras-ps所述相同类型的ancova模型,在α=0.05显著性水平下使用双侧检验对tetras-adl执行分析(图3和表3)。表3.总tetras从基线至第15天和第28天的变化实施例4.tetras-ps螺线任务(项目6)从基线至第28天的显著变化tetras-ps子量表的阿基米德螺线项目(项目6)将对功能性震颤影响的常规临床评估纳入对震颤严重程度的tetras评分中。研究者展示如何使用原子笔绘制一条约占无网格线标准(信)纸页面1/4的阿基米德螺线。螺旋线应间隔约1.3cm(0.5英寸)。研究者然后要求受试者临摹螺线。每只手单独测试和评分。握住笔,使得肢体无一部位接触桌子,并且将纸固定在桌子上适于患者的绘制风格的位置。对螺线中的震颤、而非肢体的运动进行评分。0=正常;1=轻微:震颤几乎不可见;2=轻度:明显震颤;3=中度:图的部分不可识别;4=重度:图不可识别汇总两只手的tetras-ps子量表项目6的评分并且计算第15天和第28天的相比于基线的平均变化(图4和表4)。通过曼-惠特尼(mann-whitney)非参数t检验(双尾)计算统计显著性。表4.tetras-ps螺线项目从基线至第15天和第28天的变化。实施例5.通过数字螺线描记法(imotor)测量的功能性震颤频率从基线至第15天和第28天的显著变化由apptomics公司开发的imotor是专门设计用于客观地量化运动障碍患者的运动功能的应用程序(参见例如mitsi等人,2017front.neurol.8:273)。所述应用程序需要患者完成测量运动功能的一个评价周期。对于imotor的阿基米德螺线项目,要求受试者在基于平板电脑的应用程序上使用数字记录针绘制螺线。迅速指示受试者描绘以螺线开始处开始的灰色中心虚线,而无需将记录针抬离屏幕。测试未计时。要求受试者完成任务两次,一次用右手并且一次用左手。通过imotor算法基于中线的记录针交叉点的数目和时间计算功能性震颤频率(以hz计)。计算惯用手的从基线至第15天和第28天的平均变化。通过曼-惠特尼非参数t检验(双尾)计算统计显著性(图5和表5)。表5.通过imotor的功能性震颤频率从基线至第15天和第28天的变化。实施例6.第28天的临床整体改善印象(cgi-i)与安慰剂的显著差异临床整体印象量表开发临床整体印象量表(cgi)用于nimh赞助的临床试验中,以简要、独立地评估临床医师对在开始研究药物之前和之后患者的整体功能的看法(guy(编辑),ecdeuassessmentmanualforpsychopharmacology.rockville,md:usdepartmentofhealth,education,andwelfarepublichealthservicealcohol,drugabuse,andmentalhealthadministration;1976)。cgi是考虑到所有可用的信息的汇总量度,包括对患者的病史、社会心理状况、症状、行为以及症状对患者发挥功能的能力的影响的知识。cgi具有两种形式:cgi严重程度(其对基线的疾病严重程度进行评定)和cgi改善(其对相比于基线的变化进行评定)。cgi改善(cgi-i)cgi-i由从基线cgi-s的总改善或变化的单一7分评定组成,无论所述变化是否完全是由于药物治疗引起。评定者基于以下问题选择一个反应:“与治疗开始时的受试者状况相比,您的受试者发生了多少变化?”评分为:1=极大改善;2=很大改善;3=最小改善;4=无变化;5=最小恶化;6=很大恶化;7=极大恶化。使用描述性统计汇总由cgi-i测量的治疗成功率。使用ancova模型评估治疗组之间的差异,所述模型具有固定的治疗作用、抗震颤药物的使用和部位类型(图6和表6)。表6.治疗组的第28天的平均cgi-i评分。实施例7.在第15天和第28天的患者整体变化印象(pgic)与安慰剂的显著差异总体变化评定(grc)量表被设计以量化患者随时间的改善或恶化,通常用于确定干预的作用或绘制疾患的临床病程图。grc量表要求患者评估其当前的健康状态,在治疗开始时回顾所述状态,并且然后计算两者之间的差异。grc量表的简单性使其易于管理,并且适用于广泛的患者(参见例如kamper等人,2009thejournalofmanual&manipulativetherapy17(3):163-170)。患者整体变化印象(pgic)是由一个问题组成的7分grc:“关于您的原发性震颤,与开始服用研究药物时相比,您现在如何形容自己?”受试者将选择以下答案之一:“极大恶化、很大恶化、最小恶化、无变化、最小改善、很大改善、极大改善。”使用描述性统计汇总由pgic测量的治疗成功率。使用ancova模型评估治疗组之间的差异,所述模型具有固定的治疗作用、抗震颤药物的使用和部位类型(图7和表7)。表7.治疗组的第15天和第28天的平均pgic评分。实施例8.目标达成量表(gas)与安慰剂的显著差异目标达成量表(gas)是涉及在医师与患者之间开发一套书面目标并用于监测患者的进展的工具。gas于1968年开发(参见例如kiresuk和sherman,1968communitymentalhealthjournal4:443-453)并且已用于具有身体病症的患者,所述身体病症包括痉挛(参见例如ashford等人,2006physiotherresint.11(1):24-34)和多发性硬化(khan等人,2008archphysmedrehabil.89(4):652-9)。gas根据个别患者期望的目标而非普遍应用的健康状态来组织讨论。目标定向的护理会促使患者明确哪些健康目标对其最为重要。然后,患者和临床医师可监测达到所述目标的进展。受试者鉴定基线的3个健康目标。目标的实例是从杯子饮水、扣衬衫或书写能力。目标可为主动的或被动的。所有目标都将根据个体进行量身定制,其中每个受试者需要基于3分量表(1=相当重要;2=非常重要;3=极为重要)在重要性水平上对基线的每个目标进行评定。对于研究者而言,在设定目标之前必须考虑达到每个目标的可行性,并根据需要进行相应调整。一旦设定每个目标,即要求研究者根据另一个3分量表(1=大概;2=可能;3=可疑)来对达到每个设定目标的难度进行评定。对于每个目标的进展按5分量表评分:-2=比基线差(不利的结果);-1=与基线相比无变化;0=达到预定的目标(预期结果);+1=优于预期结果;+2=最佳预计结果。gas从最差结果至最佳结果分别映射至值-2至2。使用ancova模型评估治疗组之间的差异,所述模型具有固定的治疗作用、抗震颤药物的使用和部位类型(图8和表8)。总体gas评分=50+(10*重量总和)/(0.7*重量总和**2)的平方根+[0.3*(重量总和)**2]。表8.目标达成评分的相比于安慰剂的平均值差异。实施例9.第15天和第28天对抗震颤药物满意的受试者的百分比相对于安慰剂的显著增加(quest子项目)quest是30项问卷,其促成5个子量表(身体、社会心理、沟通、嗜好/休闲和工作/财务)和总评分,加上3个关于在治疗的最后一个月内性功能以及对震颤控制和药物副作用的满意度的项目。初步报告为其可靠性和有效性提供了初步支持。内部一致性对于4个量表和总评分极佳至优异,并且对于工作/财务量表中等高(参见例如等人,2005parkinsonismrelatdisord.11(6):367-373)。这些可靠性系数也支持quest的构建体有效性。在基线、第15天和第28天,汇总对以下问题做出肯定回答的受试者百分比:“在过去一个月中,您对抗震颤药物对震颤的控制满意吗?(是/否)”。使用费舍尔精确检验(fisher'sexacttest)计算与安慰剂相比的统计学显著性(图9和表9)。表9.在被问到:在过去一个月中,您对抗震颤药物对震颤的控制满意吗?(是/否)时,患者满意。实施例10.第15天和第28天引起临床功效的cx-8998血浆浓度cns疾病模型中的cx-8998功效已展现在300-700nm的人类等效浓度下的cx-8998功效。人类药学-eeg数据的剂量反应分析确认,在血浆cx8998浓度大于200至300nm(bid4mg)时,以剂量和浓度依赖性方式发生α功率约25%或更大的降低(和cns靶标接合的间接标记)。建模表明,8mg的单剂量在投药后约12小时将导致α功率降低25%或更多。不良事件数据的剂量反应分析表明,在大于800nm的浓度下,cns和精神病学不良事件的发生率会增加。根据健康志愿者的人类pk曲线,在进食条件下,以10mgbid剂量在t-calm中靶向介于200nm与800nm之间的cx-8998的最终稳态浓度。在第2次访视(4mgbid剂量下的谷浓度)、第3次访视(8mgbid剂量下的谷浓度)和第4次访视(10mgbid下的谷浓度)在施用cx-8998之前,对所有t-calm受试者抽取血样。另外,在第4次访视时,尽可能接近投药后4小时、但在投药后4-6小时的窗口内收集投药后样品(即,在第4次访视时总共收集两个pk样品)。cavion已开发并验证液相色谱-串联质谱(lc/ms/ms)生物分析方法根据最近的fda指南(fda,guidanceforindustry:bioanalyticalmethodvalidation,2018年5月,可在fda.gov/downloads/drugs/guidances/ucm070107.pdf获得)用于同时量化大鼠、狗和人类血浆中的cx-8998和代谢物(m01、m02、m03和m04)。测定表现令人满意并且符合系统性能适合性、量化范围、校准线性、测定准确度和精密度以及测定回收率的验收标准。使用这些“五合一”生物分析方法对收集于t-calm中的样品中的cx-8998及其4种代谢物进行量化(图10和表10)。表10.cx-8998血浆浓度(nm)。实施例11.bid投用cx-8998后的cx-8998代谢物暴露cx-8998代谢物m01-m04是活性cav3拮抗剂。使用“五合一”生物分析方法对收集于t-calm中的样品中的cx-8998代谢物m01、m02、m03和m04进行量化(图11和表11)。表11.cx-8998血浆浓度(nm)。这些数据表明,包括一种以上cav3拮抗剂的组合(例如cx-8998加上其活性代谢物)的组合物与运动障碍的治疗有关。实施例12.tetras和cgi-i的反应者分析利用反应者分析阐述基于特定量表的截止值展现对治疗的反应的某些阈值的受试者比例。表12汇总了三个tetras量表(表现子量表、日常生活活动和总评分)中达到相比于基线改善至少5分的受试者比例、以及使用cgi-i评分为改善(最小改善、很大改善和极大改善)的受试者比例。通过费舍尔精确检验执行统计分析。表12.cx-8998血浆浓度(nm)。如通过在tetras终点改善至少5分或临床整体改善印象任何水平所定义的实现反应的受试者比例。通过费舍尔精确检验进行统计分析。下表显示每个终点的粒度数据。实施例13.cx-8998血浆浓度与反应之间的关系使用希尔(hill)方程评价如通过tetras-ps相比于基线的变化所测量的cx-8998的cmin暴露水平与反应(e)之间的关系:反应=emax*cmin^b/(cmin^b+ec50^b)估计emax和ec50值为约-4.1和250nm;然而,“b”的值在统计上不显著。pk/pd发现的概述:在稳态条件下使用cx-8998、分析物或总活性部分(tam;于所有测量的cav3拮抗剂中的暴露的总和,例如cx-8998和所有其代谢物的总暴露)的组合的浓度值(cmin)和使用每次研究访视的谷(t=0小时)或投药后(t=4小时;仅第28天)浓度样品来进行评价。评价的药效学变量(反应)包括tetras表现子量表、tetras日常生活活动、tetras总评分相比于基线的变化。分析发现,浓度范围并不低至足以完全限定pk/pd关系,这表明在200至800nm的预计治疗范围内已达到完全药理学作用。tetrasps的变化示于图12中并且代表使用其他pd标记获得的类似结果(例如,总tetras评分的变化)。实施例14.患者群体入选t-calm的患者群体的平均基线tetras-ps评分为28.4分,平均基线tetras-adl评分为26分,并且从et诊断开始的平均时间为23年。另外,大多数患者(64%)也同时服用抗震颤药物,而未服用et药物的患者则对一线和二线et疗法难治或不耐受。因此,在对现有疗法难治的中度至重度et群体中,在护理标准之上观测到功效。表13.患者人口统计。实施例15.不良事件概况这是通过系统器官分类和优先项的与研究药物有关的最常见(>2%)紧急治疗不良事件的汇总,以确认cx-8998安全性和耐受性概况的良性性质。表14.不良事件汇总。实施例16.利用剂量滴定的对不良事件的耐受性t-calm采用以下剂量滴定方案:第1周为4mgbid(8mg/天)、第2周为8mgbid(16mg/天)以及第3周和第4周为10mgbid(20mg/天)。尽管在第2周和第3周开始增加剂量,但不良事件报告在投药的第一周后有所减少。这显示患者快速耐受cx-8998有关的cns和精神病学不良事件(图13)。使用meddrav20.0编码不良事件。仅报告了自研究药物起始后的发作日期和时间至药物中断后30天的紧急治疗不良事件。对于经历超过一次相同编码事件的受试者,提供单一事件。不良事件归因于基于发作日期的研究周,即始于第1次访视或在第1次访视之后并且在第2次访视之前的事件被分类为第1研究周。表15.其他系统器官分类中的不良事件。实施例17.在有效剂量下无心血管安全性发现心电图离群值的综合分析揭示无cvs安全性信号。心电图结果计算为每个时间点一式三份记录的平均值。将基线定义为第1天投药前获得的一式三份值的平均值。如果该值不可用,则使用筛选时获得的一式三份记录的平均值。图14显示电子心电图离群值的汇总。cx-8998与安慰剂之间未观测到差异(图15)。实施例18.亚组分析主要和次要功效终点的探索性亚组分析包括由基线参数形成的以下亚组:1.性别:男性和女性2.在签署知情同意书时的年龄,如通过以下所定义:受试者至多65岁和受试者>65岁3.基线严重程度,如通过中心评定的tetras表现评分基线值所评估:大于和小于中值。4.基线震颤不对称性,定义为任一tetras表现子量表项目4a(姿势性震颤)、4b(翼拍打型震颤)或4c(运动性震颤)的左侧与右侧之间相差>1分:存在不对称性和不存在不对称性5.tetras表现姿势性震颤(子量表项目4a)对运动性震颤(子量表项目4c)的基线比率,如通过总姿势性震颤(左手和右手的总和)除以总运动性震颤(左手和右手的总和)的比率所定义:大于和小于中值比率6.kinesiaone姿势性震颤对运动性震颤的基线比率,类似于上述比率定义和分组7.基线静止性震颤,如定义为在基线时通过kinesiaone测量的总静止评分(左手和右手静止的总和)大于(存在)或小于(不存在)中值总静止性震颤评分:存在静止性震颤和不存在静止性震颤8.在第1天同时服用原发性震颤药物:是和否,也将对关于服用β-阻断剂的受试者的亚组进行检查9.在研究开始时使用扑米酮,如定义为在筛选时段之前两周内或在筛选时段期间中断扑米酮使用的受试者:服用扑米酮和不服用扑米酮显示代表结果的包括tetras-ps(图16)和tetras-adl(图17)的个别终点。利用cav3拮抗剂治疗可显示改善增加的子集包括女性、具有较高严重程度的震颤的患者和未同时服用抗震颤药物的受试者。静止性震颤程度较低和姿势性震颤对运动性震颤的比率较低的受试者也可达到更大程度的改善。实施例19.利用分离m02和m04的单一生物分析型测定测量人类血浆中的cx-8998和代谢物的分析方法针对人类血浆k2edta中cx-8998和m01、m02、m03和m04浓度的同时量化开发、限定和验证液相色谱串联质谱(lc/ms/ms)方法。为了开发一种用于同时量化人类血浆中分析物cx-8998、m01、m02、m03和m04的五合一测定法,能够在合理运行时间内保留和分离所有化合物的hplc方法是必需的。此外,由于m02和m04的等压性质,需要基线分离对这些代谢物进行准确量化。在约400g/mol的mw和介于3与5之间的计算的logp值情况下,所有5种化合物都是有待通过反相色谱分析的最初候选者。基于分析物之间的结构差异,cx-8998、m01、m03和m02或m04之间的分离并不困难。挑战是开发一种能够在m02及其等压位置异构体m04之间达到基线拆分的条件。测试了使用agilentzorbax扩展c18、agilenteclipsexdbc18和phenomenexsynergimaxrp柱的各种早期筛选(数据未示出);然而,没有一个能够令人满意地分离m02和m04。最终,phenomenexlunac18柱在m02和m04之间显示最高拆分能力,并被选择用于进一步的hplc方法优化。最初对phenomenexlunac18色谱法进行了研究。在使用lunac18(30x2.0mm,3μm)柱的初步试验期间,在m02与m04之间仅达到部分分离,并且m01和m03的峰宽度为次最佳的(图18)。用以减小峰宽度并增加峰拆分的优化包括以下方面的调整:样品溶剂组成(降低有机含量以增强保留)流动相调节剂(降低次要相互作用)离子配对(改变保留行为以改善峰形)有机流动相的溶剂强度(降低强度以增强保留和选择性变化)然而,无一产生令人满意的m02与m04之间的分离。此外,峰宽度仍是次最佳的。phenomenexlunaphenyl-hexyl色谱法:推断主要基于疏水相互作用(例如c18)的色谱的强度不足以达到所需分离。引入其他相互作用类型可增强选择性。苯基-己基柱基于其富电子芳族苯基-己基相提供独特静电、氢键结和偶极-偶极相互作用。假设这些独特化学方法可产生足够特异性来分离m02和m04。观测到与基于c18的系统相比,分析物之间的峰拆分略微好一些,然而,这种柱类型(30x2mm,3μm)展现不需要的次要相互作用,从而导致差的峰形(图19)。phenomenexkinetexpfp色谱法:与传统基于c18或苯基-己基反相柱相比,高电负性五氟苯基(pfp)键结相提供替代选择性(疏水、π-π相互作用、偶极-偶极、氢键结和形状选择性),这部分是由于电子缺陷性苯基环和对卤素有关化合物的高保留。使用这种柱化学类型(100x2.1mm,2.6μm,和图20)可将所有五个分析物从血浆样品中接近基线分离。需要非常长的运行时间来充分分离所有分析物,因此,这种方法时间过长,并且将对前瞻性临床血浆测试样品的分析在逻辑上具有挑战性。理想运行时间应为15分钟或更少。研究若干色谱优化方法以进一步减少运行时间:提高柱温度以能够使用更高的流速而不妨碍柱效率较短的柱长度不幸的是,这些变化导致m02与m04之间的拆分劣化。方法运行时间的缩短是不可能的。样品衍生化开发:研究分析物衍生化作为色谱分离m02和m04并同时维持合理方法运行时间的一种替代方法。由于m02与m04之间的结构差异仅取决于一个羟基(m02中为3°-oh;m04中为1°-oh)的位置,因此应该可与1°-oh基团选择性反应,而立体阻碍3°-oh基团保持未反应。这应提供更强的结构差异,以允许在色谱法期间分离。审查来自文献的各种oh-基团衍生化方法,并且选择和测试了4种,包括伯醇部分的选择性酰化和丹磺酰化。表16.色谱优化参数。酸性条件下的乙酰化是选择性的,但似乎以低反应产率形成m04反应产物的复杂混合物。利用丹磺酰化观测到最小衍生化,并且与tfa的反应产生m04反应产物的复杂混合物(数据未示出)。在碱性条件下用乙酸酐进行的乙酰化似乎对m04具有选择性并且显示高反应产率(图21)。由于与试剂的反应,m04的信号强度似乎比其他分析物低100倍。检测到在8.48分钟时形成m04产物峰(与在7分钟时未反应的m04峰相比),并且m/z为439.4,这对应于单独乙酰化m04(m04-ac)实体的质量。这通过在204m/z处观测到子片段得以证实,其与对所有其他分析物观测到的子片段一致。m04乙酰化反应的动力学:随后实施血浆反应和稳定性时程实验,以确定在50℃下至多2小时的反应动力学。独立地且以含有其他分析物(与乙酸酐和吡啶等体积)的混合物形式评价2.5μg/ml的m04的反应动力学,以验证反应效率。在50℃下在存在或不存在其他分析物下,在约30至60分钟之间的反应中,衍生反应达到m04-ac形成的平台期。约30分钟的反应后,约6%的m04仍未反应(图22)。表17.m04乙酰化反应。反应时间(min)剩余的m04峰面积%0100.0255.9384.6702.81302.3为了减少剩余的未反应的m04量,将吡啶和乙酸酐体积分别增加至多2.5倍和5倍(图23)。当血浆:吡啶:乙酸酐体积比率为1:2.5:5时,达到最高产率(未反应的m04%为约2.2%)。选择这种条件进行方法验证。由于分析物不稳定性的潜在问题,未研究提高反应温度作为提高反应产率的手段。在50℃下在等体积的乙酸酐和吡啶存在下,还评价了2.5μg/ml的cx-8998、m01、m02和m03的稳定性。总之,cx-8998、m01、m02和m03似乎在50℃下反应长达130分钟稳定(图24)。在对色谱法进行优化之后,实施以下五合一lc/ms/ms测定用于量化人类血浆k2edta中1至2000ng/ml的cx-8998、m01、m02、m03和m04浓度。样品制备程序:参考先前验证的涉及蛋白质沉淀的cx-8998、m01和m02的方法(merck,westpoint,sbp261),采取以下程序并进行限定:1.在1:1dih2o:acn中制备参考物和内标溶液的系列稀释液2.将各10μl参考标准和内标溶液添加至0.2ml人类血浆k2edta3.利用0.6mlacn沉淀血浆蛋白质4.离心并获得上清液5.于50℃下蒸干上清液6.在超声波处理下将样品重构于0.1ml二氯甲烷中达5分钟7.于50℃下利用各20μl*乙酸酐和吡啶衍生化30分钟8.于50℃下蒸干9.在超声波处理下将样品重构于含0.1%甲酸的1:1dih2o:acn中达10分钟*方法限定后,对体积进行优化,以使用100μl乙酸酐和50μl吡啶。这是微小修饰,除了m04-ac的更高反应产率外,对测定表现无预计的影响。因此,这些体积比率未重新限定。色谱参数表18.hplc梯度时间表。时间%b流速(ml/min)0.00200.50.10200.50.11250.54.50290.54.51360.58.50400.58.51980.510.00980.510.01980.811.50980.811.51200.813.90200.813.91200.814.00200.8质谱参数:使用micromass软件版本4.0控制的micromass串联三重四极杆质谱仪监测分析物。代表性ms参数(esi阳性,mrm模式)如下:表19.质谱参数。色谱法。注射人类血浆样品中的10ng/mlcx-8998、m01、m02、m03和m04后的代表性色谱性能示于图25中。在这项研究中,开发五合一lc/ms/ms测定法用于同时量化人类血浆k2edta中的cx-8998及其代谢物m01、m02、m03和m04。由于m02和m04的等压性质,传统的基于c18的反相系统无法经由疏水相互作用实现完全分离。评价了其他各种替代反相柱,并且利用五氟苯基(pfp)键结柱达到近乎完全分离,但运行时间过长。因此,成功开发一种对m04具有特异性的乙酰化衍生化方法,所述方法能够在合理的测定运行时间内对所有五个分析物进行令人满意的色谱分离。随后对用于同时量化所有五个分析物的lc/ms/ms测定进行限定,对于人类血浆中的所有分析物,校准范围为1至2000ng/ml。测定表现令人满意并且符合系统性能适合性、量化范围、校准线性、测定准确度和精密度以及测定回收率的验收标准。对于cx-8998观测到轻微色谱残留。观测到一些参考标准杂质;然而,水平可忽略并且对校准的影响很小。观测到较低的未反应m04的残留水平,从而导致m02检测受到干扰。总之,由于所有干扰的总和,对m02量化的总偏差仅为7.1%,因此预计将产生最小的影响。在方法验证期间,确定以样品中不同m04比率量化m02的测定法的准确度限值。实施例20.非临床研究中人类治疗暴露与毒理学发现之间的宽的安全界限cx-8998及其代谢物m01和m02的安全界限是基于稳态下10mgbid投药的估计人类血浆浓度(css)和曲线下面积(auc24)值,从对于总(结合和未结合)cx-8998、m01和m02于noael剂量(分别300毫克/千克/天和30毫克/千克/天)下大鼠和狗中90天重复剂量毒理学研究中的暴露来计算。另外,也计算了活性组分(cx-8998、m01和m02)或所谓“总活性部分”(tam)的暴露的总和的安全界限。雄性和雌性中总cx-8998auc24的计算安全界限在大鼠中分别为19倍和41倍,并且在狗中分别为24倍和26倍。雄性和雌性中cx-8998css的计算总界限在大鼠中分别为39倍和91倍,并且在狗中分别为68倍和74倍。在大鼠中,雄性中的界限比雌性中低约2倍,而在狗中,2种性别间的界限类似。对于cx-8998、m01和m02,总auc24(雌性/雄性)的计算安全界限在大鼠中分别为41倍/19倍、13倍/6.8倍和62倍/31倍,并且在狗中分别为26倍24倍、5.6倍/6.5倍和25倍/26倍(表20)。对于cx-8998、m01和m02,总css(雌性/雄性)的计算安全界限在大鼠中分别为91倍/39倍、21倍/13倍和104倍/56倍,并且在狗中分别为74倍/68倍、8倍/10倍和46倍。总auc24tam的计算安全界限在雌性和雄性大鼠中分别为32倍和16倍并且在雌性和雄性犬中分别为17倍和16倍。总csstam的计算安全界限在雌性和雄性大鼠中分别为62倍和31倍并且在雌性和雄性犬中分别为39倍和37倍。表20.基于大鼠和狗第90天tk评估和稳态下人类估计暴露的总界限(以倍数表示)实施例21.cx-8998制剂开发将cx-8998配制成1mg、2mg、3mg、4mg、8mg、12mg和18mg胶囊并用作单一早晨剂量。具有不同剂量的胶囊提供高度灵活的滴定时间表,并且允许理想的投药方案。在人类受试者和模拟的胃肠道中评价cx-8998立即释放胶囊的性能,其中使用900ml酸性介质模拟胃。cx-8998在酸性介质中从立即释放制剂的快速释放示于图26中。总之,血浆曲线遵循2隔室pk模型,其在2-18mg剂量范围内与剂量成比例。在老年患者中观测到终末消除速率和再分布二者轻微变化(图29)。开发了计算机模型以生成理论释放曲线,所述曲线将在稳态下产生介于最小有效浓度与最大耐受浓度之间的期望血浆浓度值。根据独立的数据集对模型进行验证,以预测cx-8998在胃肠道中以及系统地在年轻和老年男性中的行为。在老年男性中以2mg、4mg、8mg、12mg和18mg单一早晨剂量实施优化。老年男性中的pk参数如下所示。参数2隔室cl(l/h)2.6576vc(l/kg)0.18219k12(1/h)1.495k21(1/h)0.28064在2mg、4mg、8mg、12mg或18mg胶囊的单一早晨剂量后老年患者中cx-8998的血浆浓度示于图31中。老年患者的预测和观测的输出相关性示于图32中。在健康男性中利用1mg、3mg、8mg和12mg单一早晨剂量同时实施优化。年轻男性的pk参数如下所示。参数2隔室cl(l/h)3.9515vc(l/kg)0.32511k12(1/h)0.21431k21(1/h)0.13625健康志愿者中与不同剂量相关的最大cx-8998暴露如下:在1mg、3mg、8mg和12mg胶囊的单一早晨剂量后年轻患者中cx-8998的血浆浓度示于图33中。年轻患者的预测和观测的输出相关性示于图34中。在1mg、3mg、8mg和12mg胶囊的多个单一早晨剂量后年轻患者中cx-8998的血浆浓度示于图35中。年轻和老年受试者中12mg/天的单一日剂量和多个日剂量的模拟释放曲线的比较示于图28中。使用计算机模型评价从不同基质的释放曲线,以通过优化基质或通过组合不同释放曲线(例如,立即释放(ir)+持续释放(sr)或ir+延迟释放(dr))的珠粒来确定目标释放曲线。从渗透泵片剂的零级释放示于图38中。从胶囊中具有包衣珠粒的基质片剂的一级释放示于图39中。从具有ir释放包衣的渗透泵片剂的爆发和零级释放示于图40中。从具有ir包衣的基质片剂中ir和缓慢释放珠粒的混合物的爆发和一级释放示于图41中。ir珠粒和ph6肠溶包衣珠粒的混合物从具有肠溶包衣和ir包衣的ir片剂的具有3小时延迟的双重爆发示于图42中。ir珠粒和ph7肠溶包衣珠粒的混合物从具有肠溶包衣和ir包衣的ir片剂的具有6小时延迟的双重爆发示于图10中。基于模型,年轻成年男性的理想释放曲线是约6mg爆发,接着在12小时内以几乎恒定速率约12mg(图36),并且老年男性的理想释放曲线是约7mg爆发,接着在12小时内以轻微减小速率约12mg(图37)。利用多种类型的持续释放和肠溶包衣开发持续释放(sr)珠粒。使用典型聚合物(例如,羟丙基甲基纤维素或卡波普)开发一些sr基质珠粒以达到持续释放。使用ph独立性持续释放渗透泵开发一些sr基质珠粒,以达到从片剂的零级释放。使用ir珠粒的延迟释放(dr)包衣开发一些sr基质珠粒,以在ph6或7下获得释放,并且在较高ph值下利用cx-8998的不良溶解性来提供持续释放。使用ir珠粒的erdragitrs/rl30d包衣开发一些sr基质珠粒,以实现一种依赖于包衣缓慢腐蚀,从而维持药物从ir基质中的释放的方法。实施例22.每日两次(bid)cx-8998对95名患有et的患者实施对抗震颤药物(例如普萘洛尔)反应不充分的随机化、双盲、安慰剂对照、平行组研究(nct03101241)。经过同意和筛选后,受试者随机分配以接受口服剂量的cx-8998(滴定至10mgbid)或安慰剂达28天。在基线(第1次访视)、第15天(第3次访视)和第28天(第4次访视)时收集临床评定量表、患者报告的结果和震颤和数字螺线描记法(spirography)的加速计记录。cx-8998的血浆浓度显示可预测的稳态暴露,所述暴露从每日两次递送的多个不同剂量达到目标水平(图10)。研究者评定的tetras(原发性震颤评定评估量表)表现子量表(p=0.027)、tetrasadl(p=0.049)、tetras总评分(p=0.007)和临床医师整体改善印象(p=0.001)显著改善(图44)。无重要安全性和耐受性问题。基于耐受性概况的剂量上限:至多10mgbid产生与较低cnsae发生率相关的浓度。s形关系表明,cnsae的频率可能会在约700-800nm的cx-8998血浆浓度下开始显著增加(图45)。所述研究在et中达到概念验证,并且鉴定关键研究设计参数,包括主要和次要功效终点、患者群体和滴定方案。cx-8998安全并且在et患者中耐受性良好。预计这些结果使得能够在et中后期开发cx-8998。开发了包括cx-8998和cx-8998代谢物的总活性部分(tam)的cx-8998制剂。例如,tam可为针对血浆蛋白结合(fu)和相对于cx-8998的效能进行调整的cx-8998、m01和m02浓度的总和(cu,ss_tam),如表21中所示。表21.从cx-899810mgbid模拟的cx-8998、m01、m02和总活性部分(tam)的css。cx-899810mgbid施用后血浆和tam中总cx-8998和未结合的cx-8998的目标暴露水平和pk曲线示于图46中。实施例23.目标cx-8998浓度cx-8998的示例性目标浓度如附录a中所确立。实施例24.功效终点和数字生物标记选择的研究设计和方法t-calm研究评估至多10mgbid的剂量的cx-8998用于降低et的严重程度(幅值)的功效。主要t-calm研究的任选/额外组分是t-calm数字子研究,其评价了用于准确量化et患者的运动功能变化的3种不同数字监测平台的可行性。示例性t-calm设计示于图47中。t-calm研究是概念验证、多中心、双盲、随机化、安慰剂对照、平行组试验。筛选时段长达4周。在执行任何研究程序之前,患者必须阅读并签署知情同意书。由于cx-8998可能受cyp3a代谢的影响,排除使用扑米酮(一种强cyp3a4诱导物)。因此,对服用扑米酮的患者进行6周筛选,以允许安全地中断所述药物。在研究期间,允许稳定剂量的除扑米酮以外的单一抗震颤药物作为护理标准。将符合筛选标准的患者随机分配到治疗a或b组。a组接受至多10mgbid的滴定剂量的cx-8998。b组接受匹配安慰剂。随机化研究参与者进入4周双盲剂量滴定时段,接着在最后一个剂量的研究药物后进行1周的安全性随访。在基线(第1天)时,在施用研究治疗之前,患者进行安全性和震颤评价。在第一周期间,患者每日两次接受4mg研究药物或匹配安慰剂。在第8天(第2周),每日两次在诊疗所对患者进行安全性评估并且剂量滴定至8mg(或匹配安慰剂)。在第15天(第3周),患者每日两次向诊疗所报告安全性和功效评价并且最终剂量滴定至10mg(或匹配安慰剂)。最终功效访视是第28天(第4周)。最终安全性访视发生在第35天(第5周)。在第8天、第15天和第28天投药前以及第28天投药后约4小时采集血样,用于cx-8998的血浆浓度测量。如果不可耐受的不良事件(ae)在任何剂量下都明显,则可在第8天或第15天或那些排定的访视之前的任何时间将剂量降低至下一最低剂量。如果最低排定的剂量(4mgbid)不可耐受,则可将其降低至2mgbid。减少剂量后,不允许增加剂量。如果患者不能耐受剂量减少,则将其退出治疗。经筛选并且符合主要t-calm研究的合格标准的患者具有另外参与t-calm数字子研究的选择。子研究中入选的患者以与主要研究相同的比例(1:1)随机分配到cx-8998或安慰剂。在签署知情同意书后,患者可选择使用两个数字工具imotor或kinesia360中的一者或两者,或使用kinesiaone执行额外测试,以客观地测量运动功能。子研究的安全性评估的投药方案和时间表与主要研究相同。在筛选访视时,子研究的imotor组中的患者在研究人员在场的情况下完成评价。子研究的kinesia360组中的患者在筛选后佩戴设备以收集数据达2天并且然后将设备返回至研究地点。这2天收集的数据用作kinesia360的基线运动评估。在基线(第1天)时,基线imotor评价在投药前和完成主要t-calm安全性和震颤评估后进行。在第1周和第2周结束时,收集安全性和kinesia360和imotor评价。在第4周访视结束时,收集两个设备的最终功效测量。最终安全性访视是在第5周结束时。研究招收未经标准护理方法充分治疗的中度至重度et患者群体。主要t-calm研究的关键合格标准如下:1.获得所有研究参与者的签名知情同意书2.招收18-75岁的男性和女性3.包括患有中度至重度et并且在65岁之前最初诊断出的患者4.在tetras-ps中,3个动作的至少一个上肢的震颤严重程度评分为至少25.筛选时tetras-ps评分至少为156.服用稳定剂量时,最多允许同时服用一种抗震颤药物;排除使用强cyp诱导物扑米酮7.排除手术干预主要t-calm研究的纳入/排除标准的完整列表可在clinicaltrials.gov上以注册号nct03101241在线获得。任选的t-calm数字子研究的合格标准如下:1.患者必须符合主要t-calm研究方案的所有合格标准2.患者必须能够遵守现场人员评估的使用者要求3.除非患者同意参与任选的t-calm数字子研究,否则不会向其发放数字设备/下载下文列示主要t-calm研究的主要、次要和探索性功效终点:主要终点是tetras表现子量表从基线至第28天的变化。次要终点是tetras日常生活活动时间表和kinesiaone评分从基线至第28天的变化。存在若干探索性终点。测量总tetras评分(独立视频评定者)和kinesiaone从基线至第15天和第28天的变化。评价tetras表现子量表(独立视频评定者)和kinesiaone评分从基线至第15天的变化。治疗结束时的治疗成功率将通过原发性震颤问卷(quest)中的患者整体变化印象(pgic)、临床整体改善印象(cgi-i)、目标达成量表(gas)和生活质量进行测量。t-calm数字子研究存在2个探索性终点。kinesia360测量震颤幅值从基线至第15天和第28天的变化。imotor测试评价5个简单运动功能,包括从基线至第15天和第28天的数字螺线描记法。在主要t-calm研究中利用若干表现量表和客观生物统计监测。这些工具生成相关且收敛的功效数据,其可更准确地定义患者对et疗法的反应。原发性震颤评定评估量表(tetras;参见例如elble等人,2012movdisord.27:1567-1569)由9项目表现子量表和12项目日常生活活动(adl)子量表构成。这些子量表使用笔和纸方法对et进行快速临床评价(<10分钟)。表现子量表利用5分等级量表测量头部、面部、声音、四肢和躯干的震颤幅值(严重程度)以及功能测试,包括手写、画螺线并在点上方握笔,其中0代表无震颤,并且4指示重度震颤。个别评分的总和产生0至64的总体表现子量表评分。tetras表现子量表由临床现场的研究者和独立视频评定者评分。独立视频评定者的评分统计分析为从基线至第28天的变化,并且用作主要功效终点。选择最佳评定方法(研究者对独立视频评定的tetras表现子量表)进行后期开发。表现子量表数据也用于第15天的功效的探索性分析。adl子量表评价日常生活活动,例如说话、吃、喝、穿衣、个人卫生、书写和运送物品。患者对每一项目评分为0(正常活动)至4(重度异常)。总体评分在0至48的范围内并且分析为从基线至第28天的变化作为次要功效终点。关于从基线至第15天和第28天的变化的总tetras评分(表现子量表和adl的总和)评价为探索性终点。在t-calm中部署kinesiaone平台作为震颤严重程度的数字标记。kinesiaone被fda批准用于监测帕金森病的运动症状严重程度,仅有关于et患者的震颤评估的有限数据。算法得出的评分主要是在帕金森病算法中开发并在et中得到有限验证(参见例如hoffman等人,2011confprocieeeengmedbiolsoc.2011:4378-81)。在tetras表现子量表完成后,将kinesiaone设备放在每名et患者的食指上并在诊疗所中佩戴。患者在左侧和右侧执行四项任务,以评估休息、姿势、动力学和侧翼拍打震颤。从基线至第28天的评分的kinesiaone评分变化评价为次要功效终点。kinesiaone数据也用于探索性分析关于加速度测量法评分从基线至第15天的变化和关于第15天和第28天的幅值测量相比于基线的变化的功效。一致手指传感器放置和一致任务执行是有效kinesiaone评分的关键因素。由于et对日常活动和健康的有害影响,执行若干生活质量评估,以更准确地评估患者对病症的感知以及从基线至治疗结束(第28天)药物疗法的影响。这些问卷和量表中的每一者都需要来自et患者的大量输入,并且所述数据用于解决探索性功效终点。原发性震颤问卷(quest)中的生活质量(参见例如等人,2005parkinsonismrelatdisord.11:367-373)用于评价et对et患者从基线至第28天日常生活的影响。问卷含有30个项目,其涉及5个子量表(身体、心理、沟通、嗜好/休闲和工作/财务)和总评分。还存在额外3个与性能力、对震颤控制的满意度和药物疗法的副作用有关的项目。如果et的治疗程序是有益的,则无论是症状性或治愈性,患者都可能对quest产生积极反应。quest包括预计在28天时间框内不会改变的问题。然而,一些项目(例如对震颤控制的满意度)可产生有见地的数据。临床整体印象量表(cgi)产生临床医师对患者在研究药物之前和之后功能的感知(参见例如guy,assessmentmanualforpsychopharmacology.rockville,md:departmentofhealth,education,andwelfarepublichealthservicealcohol,drugabuse,andmentalhealthadministration,1976)。cgi总体评分考虑患者病史,社会心理状况、症状和关于功能能力的行为。cgi改善(cgi-i)将涉及从基线cgi-严重程度(cgi-s)的总改善或变化的的单一7分评定。临床医师评定者将基于以下问题选择一种反应(从1=极大改善至7=极大恶化):“与您在治疗开始时的患者疾患相比,您的患者改变了多少?”患者整体变化印象(pgic)将量化关于et治疗患者对随时间的改善或衰退的印象(参见例如guy,assessmentmanualforpsychopharmacology.rockville,md:departmentofhealth,education,andwelfarepublichealthservicealcohol,drugabuse,andmentalhealthadministration,1976)。患者将使用pgic量表来评估当前健康状况与治疗开始之间的差异,并计算差异。7分量表将询问问题:“关于您的et,与开始服用研究药物时相比,您现在如何形容自己?”患者回答了7个答案之一,从极大恶化至极大改善。目标达成量表(gas)(参见例如kiresuk等人,1968communitymenthealthj.4:443-453)要求医师与et患者之间进行互动,以开发一套书面的个别患者期望目标以跟踪治疗进展。在基线时,每名患者建立3个个别健康目标并将每个目标评定为相当重要=1、非常重要=2或极其重要=3。临床医师将每个目标的困难程度评定为大概=1、可能=2或可疑=3。在研究期间,利用从比基线=-2差至最佳预期结果=+2的5分量表对进展进行评分。t-calm数字子研究中采用两个额外生物统计监测工具,以探索其测量et患者的运动功能变化的能力。所述数据用于探索性功效终点。kinesia360(greatlakesneurotechnologies,cleveland,oh,usa)(参见例如pullam等人,2014parkinsonismrelatdisord.20:37-40)是利用腕和踝传感器客观地且连续地对运动数据制表的家庭监测系统。kinesia360套件含有具有安装kinesia360应用程序、两个可佩戴传感器和充电设备的智能型手机。传感器每天捕获每名et患者的腕和踝的3维线性加速度和角速度。每天结束时,将运动数据从智能型手机上传至中央服务器。处理数据以详细说明震颤的发生和严重程度,以及患者的日常活动水平。imotor(apptomics,inc.,wellesleyhills,ma,usa)(参见例如mitsi等人,2017front.neurol.13:273)是客观测量异常运动患者的运动功能的基于平板电脑的应用程序。仅在排定的访视期间执行imotor测试。要求每名et患者在平板电脑上执行5个简单任务(手指轻敲、手轻敲、手内转和外旋、对轻度刺激的反应以及利用数字记录针进行螺线绘制)。每个任务的时间限制为30秒并且将进行两次(每只手进行一次)。所有紧急治疗不良事件都以系统器官分类和优先项编码至第20版药事管理的医学辞典(medicaldictionaryforregulatoryactivities,meddra)中并且由治疗组展示于频率表中。不良事件的特征在于严重程度最大、与药物有关的不良事件、严重不良事件和导致研究中断的不良事件。其他安全性评估包括体格检查、神经学检查、生命体征、临床实验室检验(血液学、化学和尿液分析)、尿液药物筛选、妊娠测试、心电图(ecg)、哥伦比亚自杀严重程度等级量表(c-ssrs)、艾普沃斯嗜睡量表(epworthsleepinessscale,ess)和迈阿密大学帕金森病幻觉问卷(um-pdhq)(参见例如papapetropoulos等人,2008bmcneurol.8:21)。所有统计分析都基于预先指定的统计分析计划利用9.4版或更高版本的sas系统执行。基于可比较设计的2期研究,约92名患者的样本大小提供适当支持,以在主要t-calm研究中提供有关cx-8998的概念验证安全性和功效数据。无正式样本大小测定。建议将至少30名患者随机分配到cx-8998或安慰剂。实施例25.cx-8998(一种t型钙通道的选择性调节剂)在患有原发性震颤的患者中的安全性和功效原发性震颤(et)是显著损害生活质量(qol)的使人衰弱的病症。当前的药物治疗并非最佳的。t-calm评价et患者中选择性t型钙通道调节剂cx-8998的安全性和功效。该实施例阐述2期、随机化、双盲、概念验证试验(t-calm),其评价18-75岁的et患者中滴定至每天两次10mg的cx-8998。患者患有中度至重度et,并允许稳定剂量的最多1个同时抗震颤药物(排除扑米酮)。研究参与者随机分组(1:1)接受28天的cx-8998或安慰剂。主要和次要终点分别是tetras-表现量表(ps)和tetras-日常生活活动(adl)评分从基线至第28天的变化。探索性终点包括整体变化印象(cgi)、整体目标达成(gas)和qol评分。监测安全性和耐受性。t-calm以clinicaltrials.gov,nct03101241暂存。t-calm被设计以提供适当支持以使得能够为cx-8998在未充分治疗的et中的进一步发展做出决定。小心地定义合格标准,以确保选择充分定义的et患者同类组。利用验证的表现量表、客观生物统计工具和生活质量问卷的临床医师测量和患者报告的具有临床意义且收敛的结果用作评估药物功效的关键终点。t-calm试验的结果表明,cx-8998基于若干临床有关和融合的功效终点降低了震颤严重程度,并且cx-8998具有有利的安全性和耐受性概况。方法研究设计和参与者t-calm被设计为2期、概念验证、多中心(22个美国站点)、双盲、随机化、安慰剂对照试验以评价在滴定至10mgbid的目标剂量达28天后cx-8998在中度至重度et的患者中的功效、安全性和耐受性。t-calm研究的关键合格标准是签署知情同意书,18-75岁的男性和女性,在65岁之前最初诊断出et,在tetras-ps项目4的3个动作(上肢保持向前且水平,上肢横向且水平伸展并且肘部弯曲,以及双手在下巴附近彼此靠近并且手指-鼻子或下巴-鼻子移动)中的任一者期间在至少一个上肢中震颤严重程度评分为至少2,筛选时tetras-ps总评分至少为15,无抗震颤药物,或者仅一种同时抗震颤药物的稳定剂量。由于扑米酮是可影响cx-8998的代谢的强cyp诱导物,因此排除扑米酮。也排除因et进行外科手术干预的患者。纳入/排除标准的完整列表可在clinicaltrials.gov以注册号nct03101241在线获得。随机化和掩蔽患者以1:1比率随机分配以接受cx-8998或安慰剂。通过同时使用抗震颤药物和研究部位(子研究参与或非参与)对随机化进行分层。随机代码由不参与研究执行的解盲统计学家编写。在研究完成和解盲之前,赞助者、患者、研究者以及参与研究执行或数据分析的任何其他人员都不知道治疗分配。将cx-8998配制于与赋形剂的共混物混合的活性药物的胶囊中。将匹配的安慰剂配制于具有可比较的赋形剂的共混物的相同胶囊中。程序研究示意图示于图47a中。在签署知情同意书后,合格受试者随机分配以在4周双盲投药期间接受至多10mgbid的滴定剂量的cx-8998或匹配的安慰剂,接着进行1周的安全性随访。在第1周期间,受试者接受cx-89984mgbid(或匹配的安慰剂bid),接着在第8天在诊疗所中评估安全性并且剂量增加至8mgbid或匹配的安慰剂bid。在第15天(第3周),在诊疗所对患者进行安全性和功效评价,以及对最终剂量滴定至10mgbid或匹配的安慰剂bid进行评价。最终功效访视发生在第28天(第4周)。如果需要,在第1周和第2周期间允许降低至下一最低剂量,并且仅允许降低1次。不能耐受剂量减少的患者退出研究。不允许重新调整剂量。安全性程序涉及收集在第20版药事管理的医学辞典(meddra)中编码的所有teae(紧急治疗不良事件)。不良事件按最大严重程度、与药物有关的不良事件、严重不良事件和导致研究中断的不良事件分类。其他安全性评估包括体格检查、神经学检查、生命体征、临床实验室检验(血液学、化学和尿液分析)、筛选时尿液药物测试、妊娠测试、心电图(ecg)、哥伦比亚自杀严重程度等级量表(c-ssrs)、艾普沃斯嗜睡量表(ess)和迈阿密大学帕金森病幻觉问卷(um-pdhq)。结果t-calm功效和安全性终点的方法的概述提供于表22中。原发性震颤评定评估量表(tetras)由9项目表现子量表(ps)和12项目日常生活活动(adl)子量表组成。主要终点是tetras-ps评分从基线至第28天的变化。由在临床现场的每名患者的研究者(实时)和五个独立视频评定者对tetras-ps进行评分,所述视频评定者审查每名患者的录像带并进行评分。对两组评分进行统计分析。在不存在先例的情况下,这种双重方法的目标是为后期临床开发选择最佳评定方法(研究者与独立视频评定者tetras-ps)。次要终点是tetras-adl子量表评分从基线至第28天的变化,通过kinesiaone测量的加速度测量法评分从基线至第28天的变化,以及安全性和耐受性:心电图(ecg)参数相比于基线的变化、临床安全性实验室评估、哥伦比亚自杀严重程度等级量表(c-ssrs)、艾普沃斯嗜睡量表(ess)、迈阿密大学帕金森病幻觉问卷(um-pdhq)、生命体征、退出研究数目(%)和由于ae的退出研究数目(%)。还包括以下探索性终点:tetras总评分从基线至第15天和第28天的变化;tetras-ps评分从基线至第15天的变化、tetras-adl子量表评分和kinesiaone;在第15天和第28天的评分和治疗成功率,其是由原发性震颤(quest)问卷中患者整体变化印象(pgic)、临床整体印象(cgi)量表、目标达成量表(gas)和生活质量所测量。表22.t-calm功效和安全性终点的评价方法1elble等人,mov.disord.2008;23(增刊1):s1-6。2elble等人,movdisord.2012;27(12):1567-1569。3mostile等人,movdisord.2010;25(12):1938-1943。4等人,parkinsonismrelatdisord.2005;11(6):367-373。5guy,assessmentmanualforpsychopharmacology.rockville,md:departmentofhealth,education,andwelfarepublichealthservicealcohol,drugabuse,andmentalhealthadministration,1976。6kiresuk等人,communitymenthealthj.1968;4(6):443-453。7posner等人,amjpschiatry2011;168(12):1266-1277。8kenderska等人,sleepmedrev.2014;18(4):321-331。9papapetropoulos等人,bmcneurol.2008;8:21。统计分析所有统计分析都利用9.4版或更高版本的sas系统实施。每个治疗组43名患者的样本大小具有至少90%的检定力,以检测cx-8998与安慰剂之间tetras-ps评分从基线至第28天的变化的主要终点的5.5分差异,假定标准偏差为7.5并且α=0.05。这种计算是基于两种独立方式的wilcoxon-mann-whitney检验并假定每个治疗组具有正常但未确认的标准偏差的正态分布。计划招收约92名患者,以确保有86名患者纳入功效分析中。意图治疗(itt)分析集含有所有随机化患者并且用于患者处置和人口统计。安全性分析集(sas)具有所有随机化患者,其都接受至少1个剂量的研究药物。全分析集(fas)包括所有接受至少1个剂量的研究药物并且具有基线和至少1个基线后功效评估的患者。fas用于所有功效评估。利用fas和协方差分析(ancova)模型分析主要功效终点,所述模型具有固定的治疗作用、抗震颤药物使用、研究部位和基线tetras-ps评分。测试是使用ancova模型的最小二乘法(ls)平均值和在α=0.05显著性水平下的双侧检验来实施。使用多次插补估计丢失第28天的tetras-ps评分的患者的丢失数据。类似地分析次要和探索性功效终点。次要和探索性分析的p值被视为标称。结果t-calm研究时段为2017-2018年,其中首次患者访视的日期为2017年8月29日,最后一次患者访视的日期为2018年7月16日。患者的处置提供于图47b中。itt分析集包括95名随机分配以接受cx-8998(n=48)或安慰剂(n=47)的患者。所述研究由cx-8998组中的37名(77%)患者和安慰剂组中的42名(89%)患者完成。cx-8998治疗的患者中有8名(17%)发生了因不良事件引起的中断,而安慰剂治疗的患者中有3名(6%)发生了中断。主要功效分析集(预先指定的mitt)包括cx-8998组中的37名受试者和安慰剂组中的44名受试者,其接受至少一个剂量的研究药物并且完成基线和至少一个基线后功效评估。基线时人口统计特征示于表23中。男性和女性患者在cx-8998和安慰剂组中几乎相等分布。cx-8998与安慰剂组之间的年龄、种族和族群类似。基线时et特征示于表24中。针对大部分et基线特征匹配cx-8998和安慰剂组。唯一的例外是62%的安慰剂组使用β-阻断剂药物,与之相比,cx-8998组为44%。表23.基线时的人口统计特征(itt分析集)itt=意图治疗。表24.基线时原发性震颤特征(itt分析集)adl=日常生活活动;itt=意图治疗;tetras=原发性震颤评定评估量表;tetras-ps=tetras表现子量表。1通过从签署知情同意书的年龄减去原发性震颤发作的年龄,估计从原发性震颤发作起的时间。2基线定义为在开始研究药物之前或之后≤15分钟获得的最后一个非丢失值。3这个亚组用于探索性分析。4震颤不对称性定义为以下tetras-ps项目中的任一者的右侧与左侧之间的差异>1分:4a(姿势性震颤)、4b(翼拍打震颤)或4c(运动性震颤)。5姿势性震颤(子量表项目4a)对运动性震颤(子量表项目4c)的比率定义为如由tetras-ps提供的总姿势性震颤(左右手总和)评分除以总运动性震颤(左右手总和)评分的比率。除扑米酮外,研究期间允许患者最多使用单一伴随抗震颤药物。cx-8998组中28名患者(58%)使用这些药物,并且安慰剂组中33名患者(70%)使用这些药物。cx-8998(10%)和安慰剂(17%)组中的一些患者正在服用1种以上伴随抗震颤药物(例如苯二氮卓、β-阻断剂)来治疗共病疾患,例如焦虑、失眠和高血压。在cx-8998(29%)和安慰剂(36%)组中,非选择性β-阻断剂(例如普萘洛尔)是最常用的抗震颤药物。20名cx-8998和14名安慰剂患者未使用伴随的抗震颤药物,并且这些患者中的11名(12%)由研究现场报告为出于功效或耐受性原因尝试并且中断护理标准药物(普萘洛尔、扑米酮或二者)。其余23名患者无抗震颤药物的使用史,或无法获得信息。cx-8998(99.3%)和安慰剂(97.7%)的符合研究药物施用相当。作为主要终点的由独立视频评定者评分的tetras-ps中从基线至第28天的ls平均值(±se)变化对于cx-8998为-1.8±0.81并且对于安慰剂为-2.3±0.78,并且差异不显著(p=0.696)(表25)。当研究者(本人)对相同终点进行评分时,与安慰剂(-2.8±0.77)相比,通过cx-8998(-4.8±0.80)的平均评分显著提高(p=0.017),如图48a和表25中所示。敏感性分析确认主要终点(由独立视频评定者和研究者测量)的发现。研究者评分的主要终点的预先指定的亚组分析显示,在患有更重度et的患者中反应更稳健(图48b和表26)。与基线时未服用伴随抗震颤药物的亚组相比,基于伴随的抗震颤药物使用的预先指定的亚组分析显示出明显更大的改善。(图48c和图48d以及表26)。表25.tetras-ps子量表主要终点ancova=协方差分析;ci=置信区间;ls=最小二乘法;tetras=原发性震颤评定评估量表;tetras-ps=tetras表现子量表。1tetras-ps量化头部、面部、声音、四肢和躯干的震颤。总体子量表评分在0至64范围内。评分降低指示改善。2如由独立视频评定者评分的tetras-ps总评分从基线至第28天的变化是方案指定的主要功效终点。tetras-ps也由研究者实时评分,并且进行事后分析(使用用于独立视频评定者评估的相同方法),如同它是主要终点一样。3通过多次插补取代丢失值。使用ancova模型估算ls平均值、标准误差、相比于安慰剂的差异、95%ci和p值,所述模型具有治疗作用、抗震颤药物使用、部位类型和tetras-ps总评分的基线值。4使用procmi输入第28天子量表上的丢失项目,并且产生10个数据集用于分析。使用procmianalyze将10个数据集的ancova模型的结果合并,以产生ls平均值、相比于安慰剂的差异、95%ci和p值。5对排序数据实施分析;使用未排序数据估计ls平均值、标准误差、相比于安慰剂的差异和95%ci,并且使用排序数据计算p值。表26.tetras-ps预先指定的子集分析ancova=协方差分析;ci=置信区间;ls=最小二乘法;tetras=原发性震颤评定评估量表;tetras-ps=tetras表现子量表。1tetras-ps量化头部、面部、声音、四肢和躯干的震颤。总体子量表评分在0至64范围内。评分降低指示改善。2如由独立视频评定者评分的tetras-ps总评分从基线至第28天的变化是方案指定的主要功效终点。tetras-ps也由研究者实时评分,并且进行事后分析(使用用于独立视频评定者评估的相同方法),如同它是主要终点一样。5对排序数据实施分析;使用未排序数据估计ls平均值、标准误差、相比于安慰剂的差异和95%ci,并且使用排序数据计算p值。在第15天(探索性终点)和第28天(次要终点)评价tetras-adl子量表评分,并且与安慰剂相比,cx-8998组的治疗组间相比于基线的变化的差异显示在两个时间点显著降低。如图49a中所提供,cx-8998和安慰剂在第15天的ls平均值(±se)分别为-4.5±0.87和-1.4±0.85(p=0.005),并且在第28天为-4.5±1.12和-1.6±1.08(p=0.049)。第28天的次要终点的预先指定的亚组分析显示随着et严重程度增加,cx-8998患者中的反应更大(图49b)。与tetras-ps相比,与基线时服用伴随抗震颤药物的亚组中的安慰剂相比,基于伴随抗震颤药物使用的tetras-adl评分的预先指定亚组分析显示显著更大改善(图49c)。为了估计tetras-adl改善的临床可检测变化,对获得如通过cgi-i或pgic作为锚定测量的最小改善评分的患者亚组执行tetras-adl评分的事后分析。这种分析揭示,tetras-adl评分相比于基线降低3至4分代表对于临床医师可检测的变化,而评分相比于基线降低2至4分代表对于患者可觉察的变化(表27)。表27.基于cgi-i和pgic的tetras-adl有意义的差异的事后分析adl=日常生活活动;sd=标准偏差;tetras=原发性震颤评定评估量表;cgi-i=临床整体改善印象;pgic=患者整体改善印象。1tetras-adl子量表评估例如吃喝、穿衣和个人卫生、运送物品以及更精细动作技能的项目。每个项目以0至4的量表评定,其中0=正常活动并且4=重度异常。个别评分的总和提供总体评分,其在0至48的范围内。与安慰剂相比,cx-8998的ls平均值(±se)总tetras评分(由研究者评定的tetras-ps评分加上由患者评定的tetras-adl子量表评分)在第15天(-7.5±1.42和-3.7±1.38,p=0.040)和第28天(-9.0±1.66和-4.2±1.60,p=0.029)分别显著提高(图49d)。当tetras-ps评分由独立视频评定者评定时,cx-8998组的总tetras评分有改善的趋势,但在第15天或第28天在统计上不显著。第15天(探索性终点)和第28天(次要终点)的kinesiaone评分(三轴加速度测量法和回旋测量法)在任一时间点都未显示cx-8998与安慰剂之间的显著差异。(表28)表28.kinesiaone评分ancova=协方差分析;ci=置信区间;ls=最小二乘法。1kinesiaone评分表示为左右手的评分的总和。每一测试的值在0(无震颤)至4(重度震颤)范围内。总的整体评分是所有个别项目的总和。2使用ancova模型估算ls平均值、标准误差、相比于安慰剂的差异、95%ci和p值,所述模型具有治疗作用、抗震颤药物使用、部位类型和加速度测量法总评分的基线值。3对排序数据实施分析;使用未排序数据估计ls平均值、标准误差、相比于安慰剂的差异和95%ci,并且使用排序数据计算p值。cx-8998的第28天(探索性终点)的ls平均值(±se)cgi-i评分(1.0±0.13)相比于安慰剂(0.4±0.13)显著更大(改善),p=0.001(图50a和表29)。在第28天,用cx-8998治疗的患者的23%被评定为“很大改善/极大改善”,与之相比,利用安慰剂仅为7%(图50b)。平均pgic评分在第15天(探索性终点)显著更大(改善)(p=0.003)并且往往在第28天(探索性终点)显著改善(p=0.089)。(表30)cx-8998患者的第28天(探索性终点)的平均gas评分(44.8±1.72)相比于安慰剂(40.0±1.67)显著更大(改善),p=0.034。关于gas的详细结果(包括对达到1个或更多或2个或更多目标的患者比例的事后分析)提供于表31中。表29.cgi-i1重新调整cgi-i的大小,以允许更直观地解释结果。2使用协方差分析(ancova)模型估计最小二乘法(ls)平均值、标准误差、相比于安慰剂的差异、95%置信区间(ci)和p值,所述模型具有治疗作用、抗震颤药物使用和部位类型。表30.gic1重新调整pgic的大小,以允许更直观地解释结果。2使用协方差分析(ancova)模型估计最小二乘法(ls)平均值、标准误差、相比于安慰剂的差异、95%置信区间(ci)和p值,所述模型具有治疗作用、抗震颤药物使用和部位类型。表31.gas1总体gas评分=50+(10*重量总和)/(0.7*重量总和**2)的平方根+[0.3*(重量总和)**2]。在基线下指定重量并且分配为1=相当重要,2=非常重要,3=极为重要。2使用协方差分析(ancova)模型估计最小二乘法(ls)平均值、标准误差、相比于安慰剂的差异、95%置信区间(ci)和p值,所述模型具有治疗作用、抗震颤药物使用和部位类型。quest问卷揭示,与安慰剂相比,更高百分比的cx-8998患者在第15天(36%对9%)和第28天(18%对9%)经历来自其治疗的副作用并且更高百分比的cx-8998患者在第15天(38%对11%)和第28天(38%对14%)表示对症状控制满意(表32)。在第15天或第28天的维度评分中,cx-8998组与安慰剂组之间无显著差异(数据未示出)。表32.与抗震颤药物有关的原发性震颤生活质量问卷(quest)一般问题cx-8998组中58%的患者中至少存在1个teae,与之相比,安慰剂为49%。如所预计,cx-8998组中的teae主要是神经病学和精神病学teae。两组中teae主要为轻度或中度。在研究的第一周期间,大多数报告cx-8998组中的teae,而在整个研究中,报告安慰剂组中的teae(表33)。cx-8998组中最常报告的与药物有关的teae是眩晕、头痛、亢奋、注意力障碍、感觉异常、幻觉(幻想,大多是闭着眼睛时可见)、失眠和口腔干燥。眩晕、恶心、嗜眠和呕吐是利用安慰剂的最常见的与药物有关的teae(表33)。表33.任一治疗组中两个或更多个受试者中通过(安全性分析集)报告的紧急治疗不良事件的汇总1不良事件映射是基于meddra(药事管理的医学辞典),版本20.1辞典。经历相同事件超过一次的受试者对于优先项计数一次。经历系统器官分类内1个以上事件的受试者在系统器官分类中仅被计数一次。2系统器官分类中受试者的数量和百分比表示具有系统器官分类中的至少1个不良事件(优先项)的所有受试者。仅显示在神经系统、精神病学系统和胃肠系统中任一治疗组中2个或更多受试者中报告的不良事件。在服用cx-8998的一个患者中报告酒精戒断综合征、重度抑郁症和自杀意念的严重不良事件(sae)。该患者具有长期酒精依赖和抑郁症的病史,其具有自杀意图但其在筛选期间未揭露出来。认为这些sae与研究药物无关。利用安慰剂未发生sae。cte-8998患者中有8名(17%)因teae而中断研究治疗,与之相比,安慰剂组中有2名(4%)。在6%的cx-8998患者和2%的安慰剂患者中执行由于teae引起的剂量减少,并且其通常发生在治疗的前2周内(表34)。表34.中断和剂量减少bid=每日两次;meddra=药事管理的医学辞典。1不良事件映射是基于meddra(药事管理的医学辞典),版本20.0辞典。经历相同事件超过一次的受试者对于优先项计数一次。经历系统器官分类内1个以上事件的受试者在系统器官分类中仅被计数一次。2减少剂量后不允许重新调整剂量。3在第4次访视时受试者药物供应不足。4所有受试者的事件均被认为与研究药物有关。分析检查所观测到的tetras表现子评分的差异是否是由于项目的差异评分所致,以及这些项目可阐明不一致评定背后的因素的程度(图51)。图51的上图显示项目的差异评分(独立视频评定[cr]减去研究者评定[pr]),其中负评分指示独立视频评定者(cr)的评分低于研究者(pr)。如图51所指示,首先,所有项目的独立视频评定者的评分均比研究者低。其次并且更重要的是,一些项目显示出特别大的差异评分。这些项目与震颤、站立、面部和舌头的伸出有关(tp2:面部震颤;tp3:声音震颤;tp5al:左下肢伸展;tp5ar:右下肢伸展;tp9:站立)。与这些观测结果一致,在个别项目上比较独立视频评定和研究者评定的组的anova显示19个项目中有14个在统计学上显著差异(p值<0.05)。与腿震颤、站立和精细震颤有关的所有项目都达到显著性。有趣的是,差异似乎是系统性的,其中特定项目的差异评分幅值在访视间保持一致。例如,项目5al和5ar在所有访视中均显示较大差异评分(图51,下图)。cx-8998和安慰剂组的临床实验室参数中未检测到有临床意义的差异。在生命体征、ecg或神经和体格检查中未发生临床上显著的异常。实施例26.cx-8998盐酸盐和游离碱制剂cx-8998ir制剂尽可能快地释放尽可能多的cx-8998。使用挤出/球形化制备示例性cx-8998ir制剂,以生成含有cx-8998盐酸盐或cx-8998游离碱的珠粒。在一个实验中,含有cx-8998盐酸盐或cx-8998游离碱的制剂如表35中所示。表35.具有盐酸盐和游离碱cx-8998的珠粒制剂(wt%)释放曲线示于图54a中。含有微晶纤维素(其通常用于其他活性成分的ir制剂中)的制剂释放约80%的cx-8998,并且花了一个小时才达到这种释放水平。在一个实验中,含有cx-8998游离碱的制剂如表36中所示。表36.具有盐酸盐和游离碱cx-8998的珠粒制剂(wt%)组分cx-8998制剂cx8998游离碱3.2微晶纤维素8.0乳糖一水合物80.6淀粉羟乙酸钠3.5交联羧甲基纤维素钠3.5硬脂酸镁0.50硬脂酸0.50聚山梨醇酯800.2释放曲线示于图54b中。含有乳糖一水合物(例如,并且不含微晶纤维素)的制剂在20分钟内释放大于80%的cx-8998。实施例27.无微晶纤维素的cx-8998珠粒制剂微晶纤维素通常是通过挤出球形化形成的药物珠粒中的关键成分。表37中提供不含微晶纤维素并快速释放cx-8998的制剂。图55中提供这种制剂的溶解结果。表37.不含微晶纤维素的cx-8998药物珠粒制剂组分组成((核心珠粒wt的wt%))cx-8998(盐酸盐)5.0乳糖一水合物57.7交聚维酮25.0无水柠檬酸8.0月桂基硫酸钠(sls)2.0羟丙基纤维素(hpc)2.0丁基化羟基苯甲醚(bha)0.3丁基化羟基甲苯(bht)0.1将bha和bht在一个烧杯中溶解于乙醇中,同时将柠檬酸、sls和hpc在第二烧杯中溶解于水中。将两种溶液组合并喷涂于在v型混合器中共混的其余成分上,以形成糊状物。然后将糊状物经由挤出机挤出,并且立即将挤出物转移至球化机中。将所得湿药物珠粒在流化床干燥器中干燥。实施例28.cx-8998延迟释放(dr)珠粒制剂cx-8998dr制剂可为在施用后从其释放cx-8998有很大延迟的任何组合物。可通过施加在6.0-6.5范围内的ph值下靶向溶解的ph敏感性包衣来控制从该实施例的组合物释放cx-8998。表38中提供所述组合物。表38.不含微晶纤维素的cx-8998药物珠粒制剂如实施例27中所述制备核心珠粒。将干燥的珠粒装入带有würster插件的流化床干燥器中并用悬浮于95%异丙醇中的上文列举的包衣成分喷涂。继续喷涂直至包衣的质量为无包衣珠粒的质量的约20%为止。将珠粒在流体珠粒中干燥,直到残余溶剂含量可忽略不计为止。实施例29.cx-8998延迟释放珠粒制剂(稍后释放)靶向另一延迟释放珠粒制剂以稍后在肠道中释放。表39中提供了这种制剂的组成。表39.不含微晶纤维素的cx-8998药物珠粒制剂如实施例27中所述制备核心珠粒。将干燥的珠粒装入带有würster插件的流化床干燥器中并用悬浮于纯化水中的上文列举的包衣成分喷涂。继续喷涂直至包衣的质量为无包衣珠粒的质量的约20%为止。将珠粒在流体珠粒中干燥。实施例30.每日两次投用的8mgcx-8998受控释放胶囊通过组合实施例27的立即释放珠粒与实施例28的延迟释放珠粒来产生受控释放药物产品。0号硬明胶胶囊每个胶囊人工填充约128mg立即释放珠粒和约104mg延迟释放珠粒。由此填充的每个胶囊含有8mg当量的cx-8998游离碱。实施例31.每日一次投用的8mgcx-8998受控释放胶囊通过组合实施例27的立即释放珠粒与实施例29的延迟释放珠粒来产生受控释放药物产品。0号硬明胶胶囊每个胶囊人工填充约85mg立即释放珠粒和约158mg延迟释放珠粒。由此填充的每个胶囊含有8mg当量的cx-8998游离碱。实施例32.每日两次投用的20mgcx-8998受控释放胶囊通过组合实施例27的立即释放珠粒与实施例28的延迟释放珠粒来产生受控释放药物产品。00号硬明胶胶囊每个胶囊人工填充约320mg立即释放珠粒和约260mg延迟释放珠粒。由此填充的每个胶囊含有20mg当量的cx-8998游离碱。实施例33.每日一次投用的20mgcx-8998受控释放胶囊通过组合实施例27的立即释放珠粒与实施例29的延迟释放珠粒来产生受控释放药物产品。0号硬明胶胶囊每个胶囊人工填充约212mg立即释放珠粒和约395mg延迟释放珠粒。由此填充的每个胶囊含有20mg当量的cx-8998游离碱。实施例34.mr120mg胶囊的溶解将实施例32中所述的胶囊在usp2型溶解装置中在含有0.1n盐酸中的3%tergitol的介质中测试2小时并且随后在含有ph6.8磷酸盐缓冲剂中的3%tergitol的介质中测试12小时。表40中提供平均值数据。表40.20mg“mr1”胶囊的平均溶解数据小时标签声明的溶解%1502602.56838649761021010214102实施例35.mr220mg胶囊的溶解将实施例33中所述的胶囊在usp2型溶解装置中在含有0.1n盐酸中的3%tergitol的介质中测试2小时并且随后在含有ph6.8磷酸盐缓冲剂中的3%tergitol的介质中测试12小时。表41中提供平均值数据。表41.20mg“mr2”胶囊的平均溶解数据实施例36.cx-8998制剂药代动力学评价实施例27-29的珠粒制剂的溶解。ir、mr1和mr2的溶解速率示于图56中。于40℃/75%rh下储存10天或于5℃下储存10天后的ir、mr1和mr2的溶解速率示于图57中。对珠粒制剂溶解速率进行pk建模。将ir、mr1和mr2珠粒的体外释放曲线插入基于ir数据开发的pk模型中。珠粒比率和总剂量的pk建模是针对老年和非老年(“年轻”)受试者的单独2隔室模型实施。可预测来自不同比率的ir和mr1以及ir和mr2的pk。总剂量可基于cmax、cmin或cave的目标优化。在7天时程内和在稳态下老年人中ir和mr1组合的溶解速率示于图58中。60%ir/40%mr1展现快速摄取(低tmax)和钝化的cmax。在7天时程内和在稳态下老年人中ir和mr2/cr7组合的溶解速率示于图59中。60%ir/40%mr2/cr7展现快速摄取(低tmax)、钝化的cmax和延长持续时间。在7天时程内和在稳态下非老年人中ir和mr1组合的溶解速率示于图60中。60%ir/40%mr1展现钝化的cmax,但与ir相比,作用持续时间无改善。ir和mr2/cr7组合在非老年人中经7天时程和在稳态下的溶解速率示于图61中。60%ir/40%mr2/cr7对于非老年受试者展现良好比率。实施例37.cx-8998组合原型制剂使用相对生物利用度研究来确认bid与qd(持续释放)之间的曲线相似并且达到治疗最小值,并且bid和qd避免理想范围内的目标浓度中的最大耐受浓度仅持续清醒时间。使用具有40%ir、25%mr1(ph6)和35%mr2/cr7(ph7)的制剂以25mg、15mg和8mg的剂量实施模拟。各种剂型的模拟溶解速率示于图62中。表42.25mgqd剂型的cmax表43.15mgqd剂型的cmax实施例38.cx-8998制剂的临床药代动力学实施生物利用度研究以评估人类受试者中各种cx-8998制剂的药代动力学。方法这是与原始ir胶囊制剂相比2种cr测试制剂的安全性、耐受性和pk的开放标签、单剂量、随机化、三向交叉、单中心研究,最多15名受试者。在第一剂量的研究药物施用之前28天内,将对受试者进行研究合格性筛选。符合研究合格标准的受试者将在第一剂量的研究药物施用前一天(时段1/第-1天)的下午被收入临床研究部门。继续符合研究合格标准的受试者将随机接受治疗顺序并且将以随机化次序接受8mg剂量的以下cx-8998治疗中的每一者,每次治疗与上一次治疗之间的间隔为最少7天的洗脱期:·在禁食条件下原始ir胶囊(4个胶囊,每个含有2mgcx-8998(治疗a),·在禁食条件下cr测试制剂#1(含有8mg呈ir和1型调节释放[mr1]珠粒形式的cx-8998的多颗粒胶囊制剂)(治疗b),和·在禁食条件下cr测试制剂#2(含有8mg呈ir和2型调节释放[mr2]珠粒形式的cx-8998的多颗粒胶囊制剂)(治疗c)。·在进食条件下cr测试制剂#2(含有8mg呈ir和2型调节释放[mr2]珠粒形式的cx-8998的多颗粒胶囊制剂)(治疗d)。治疗a、b和c将间隔最少7天的洗脱期。接受治疗a、b和c的受试者也可接受任选的额外治疗,所述额外治疗由在进食条件下cr测试制剂#2(治疗d)组成。治疗d的施用与治疗a、b和c的施用可间隔约4周以允许分析前3个治疗的生物分析样品。1治疗以下是在tfl中使用的研究治疗缩写和排序的列表。缩写:cr=受控释放;ir=立即释放;mr1=1型调节释放;mr2=2型调节释放。2样本大小调整在2个先前的单一剂量pk研究中,12mg单一剂量的cx-8998后的平均受试者间变异系数(cv%)对于最大观测血浆浓度(cmax)在21%至35%范围内并且对于曲线下面积(auc)在22%至26%范围内(研究pn001/第1部分和pn002/第2部分)。假定受试者内可变性为18%(即,35%的最大cv%为约50%),则总共13名受试者将足以估计cx-8998的生物利用度,具有>80%检定力、5%机率,并且μ=1.0。所述研究最多招收15名受试者。可根据赞助者的决定替换由于除治疗有关的不良事件(teae)以外的原因而过早退出研究的受试者,以确保至少13名受试者完成研究。结果在15个年龄在35-75岁范围的受试者中执行单一剂量交叉临床试验。基线下的人口统计特征示于表44中。表44.基线下的研究受试者人口统计每名受试者以随机化次序接受8mg剂量的cx-8998、m01或m02立即释放胶囊,以及实施例30(mr1胶囊)和实施例31(mr2胶囊)的每一种胶囊,剂量之间有7天的洗脱期。与立即释放剂量相比,cr胶囊产生具有类似auc的较低cmax值。从研究获得的药代动力学参数的值提供于表45-48和图63中。表45.cx-8998的pk参数tmax的中值表46.m02的pk参数tmax的中值表47.m01的pk参数tmax的中值表48.来自mr1和mr2胶囊的cx-8998的生物利用度参数评价单一剂量施用cx-8998后的不良事件。与研究药物有关并且在单一8mg剂量的cx-8998后在两名或更多名受试者中观察到的紧急治疗不良事件提供于表49和图64中。表49.完全同类组(n=15)中观测到的不良事件。*调节释放原型制剂1(对于bid为mr1):60%ir珠粒,40%cr(ph6)珠粒,单一胶囊,被设计以达到cmax相对于ir的25%降低和高于阈值的8小时覆盖**调节释放原型制剂2(对于qd为mr2):40%ir珠粒,60%cr(ph7)珠粒、单一胶囊,被设计以达到cmax45%降低,并且高于阈值的12-16小时覆盖鉴于cx-8998的药代动力学的不良事件示于图66-68中。对于ir制剂,cns不良事件和精神病学不良事件通常在投药后2小时内发生,并且许多cns不良事件与处于或低于cx-8998的平均浓度的浓度相关(图65)。对于mr1制剂,cns不良事件和精神病学不良事件通常在投药后2小时内发生,cx-8998的浓度通常低于利用ir制剂观测到的浓度,这表明产生cns不良事件和精神病学不良事件的浓度阈值低于400nm(图66)。对于mr2制剂,cns不良事件和精神病学不良事件通常在以低于400nm的cx-8998峰浓度投药后2小时内发生(图67)。鉴于cx-8998的治疗期的不良事件示于图68中。没有明确的证据表明有周期效应。评价在施用单一剂量(1×8mg)ircx-8998、mr1cx-8998或mr2cx-8998后cx-8998、代谢物m01和代谢物m02的血浆浓度。数据示于表50-52和图72中。未观测到制剂对auc比率有影响。cmax影响与母体减少一致。表50.单一8mg剂量的ircx-8998后的代谢物m01和m02浓度表51.单一8mg剂量的mr1cx-8998后的代谢物m01和m02浓度表52.单一8mg剂量的mr2cx-8998后的代谢物m01和m02浓度实施例39.进食对禁食人类受试者中cx-8998制剂的临床药代动力学实施生物利用度研究,以评价处于进食状态的人类受试者中cx-8998制剂的药代动力学与处于禁食状态的人类受试者中cx-8998制剂的药代动力学的比较。方法如实施例38、治疗d中所述实施所述方法。结果评价在施用单一剂量(1×8mg)的mr2cx-8998后的cx-8998的血浆浓度。数据示于表53以及图74和图75中。表53中列出的p值用于双尾t检验。表53.cx-8998制剂的药代动力学患者是在进食状态或禁食状态下施用cx-8998会影响cx-8998的早期吸收,但似乎不会显著影响稍后吸收。患者在服用cx-8998时的进食状态似乎不会显著影响cx-8998的总体暴露,如通过auc所测量。实施例40.示例性实施方案实施方案1.一种口服剂型,所述口服剂型包含:立即释放组分,所述立即释放组分包含约2mg至约20mg的具有以下结构的化合物:和延迟释放组分,所述延迟释放组分包含约2mg至约20mg的所述化合物或其代谢物。实施方案2.如实施方案1所述的口服剂型,所述口服剂型还包含所述化合物的一种或多种代谢物,其中所述代谢物选自由以下组成的组:及其组合。实施方案3.如实施方案2所述的实施方案1的口服剂型,其中所述口服剂型包含胶囊,并且其中所述立即释放组分和所述延迟释放组分包含于所述胶囊内。实施方案4.如实施方案3所述的口服剂型,其中所述胶囊是明胶胶囊。实施方案5.如实施方案4所述的口服剂型,其中所述明胶胶囊是硬明胶胶囊。实施方案6.如实施方案3至实施方案5中任一项所述的口服剂型,其中所述胶囊是4号胶囊。实施方案7.如实施方案1至实施方案6中任一项所述的口服剂型,其中所述立即释放组分呈多个珠粒形式。实施方案8.如实施方案7所述的口服剂型,其中所述珠粒还包含乳糖一水合物、交聚维酮、柠檬酸和月桂基硫酸钠。实施方案9.如实施方案8所述的口服剂型,其中所述珠粒包含以重量计约5%所述化合物、约60%乳糖一水合物、约25%交聚维酮、约8%柠檬酸和约2%月桂基硫酸钠。实施方案10.如实施方案7至实施方案9中任一项所述的口服剂型,其中所述珠粒是通过挤出和球形化形成。实施方案11.如实施方案1至实施方案10中任一项所述的口服剂型,其中所述延迟释放组分呈多个用具有ph5.5的ph的肠溶聚合物包覆的珠粒形式。实施方案12.如实施方案11所述的口服剂型,其中所述珠粒还包含乳糖一水合物、交聚维酮、柠檬酸和月桂基硫酸钠。实施方案13.如实施方案12所述的口服剂型,其中所述珠粒包含以重量计约5%所述化合物、约60%乳糖一水合物、约25%交聚维酮、约8%柠檬酸和约2%月桂基硫酸钠。实施方案14.如实施方案11至实施方案13中任一项所述的口服剂型,其中所述珠粒是通过挤出和球形化并使用流化床喷涂方法涂布的膜来形成。实施方案15.如实施方案1至实施方案14中任一项所述的口服剂型,其中所述立即释放组分包含约10mg所述化合物,并且其中所述延迟释放组分包含约10mg所述化合物。实施方案16.如实施方案1至实施方案14中任一项所述的口服剂型,其中所述立即释放组分包含约7mg所述化合物,并且其中所述延迟释放组分包含约13mg所述化合物。实施方案17.如实施方案1至实施方案16中任一项所述的口服剂型,其中所述立即释放组分被配制以在递送至人类时在45分钟内释放所述立即释放组分中存在的所述化合物的至少80%。实施方案18.如实施方案1至实施方案17中任一项所述的口服剂型,其中所述延迟释放组分被配制以在递送至人类时提供约100nm至约300nm所述化合物的最小血浆水平。实施方案19.如实施方案18所述的口服剂型,其中所述化合物的所述最小血浆水平维持约12小时。实施方案20.如实施方案18所述的口服剂型,其中所述化合物的所述最小血浆水平维持约24小时。实施方案21.如实施方案18至实施方案20中任一项所述的口服剂型,其中所述化合物的所述最小血浆水平是约200nm。实施方案22.如实施方案1至实施方案21中任一项所述的口服剂型,其中所述延迟释放组分被配制以在递送至人类时提供约900nm至约1800nm所述化合物的最大血浆水平。实施方案23.如实施方案21所述的口服剂型,其中所述化合物的所述最大血浆水平是约900nm。实施方案24.如实施方案21所述的口服剂型,其中所述化合物的所述最大血浆水平是约1000nm。实施方案25.一种治疗患有运动障碍的人类的方法,所述方法包括:向所述人类施用口服剂型,所述口服剂型包含:立即释放组分,所述立即释放组分包含约2mg至约16mg的具有以下结构的化合物:和延迟释放组分,所述延迟释放组分包含约2mg至约16mg所述化合物;其中所述口服剂型是每日一次施用。实施方案26.如实施方案25所述的方法,所述口服剂型还包含所述化合物的一种或多种代谢物,其中所述代谢物选自由以下组成的组:及其组合。实施方案27.如实施方案25或实施方案26所述的方法,其中所述人类是成人。实施方案28.如实施方案27所述的方法,其中所述成人是60岁或以上。实施方案29.如实施方案25至实施方案28中任一项所述的方法,其中所述运动障碍是原发性震颤。实施方案30.如实施方案25至实施方案28中任一项所述的方法,其中所述运动障碍是帕金森病。实施方案31.如实施方案25至实施方案30中任一项所述的方法,其中所述口服剂型是在早晨施用。实施方案32.如实施方案25至实施方案31中任一项所述的方法,其中所述口服剂型是在睡醒后4小时内施用。实施方案33.如实施方案25至实施方案32中任一项所述的方法,其中所述口服剂型的所述延迟释放组分被配制以在肠ph下在所述延迟释放组分内释放所述化合物。实施方案34.如实施方案25至实施方案33中任一项所述的方法,其中所述人类在施用所述口服剂型之前已禁食至少4小时。实施方案35.如实施方案25至实施方案34中任一项所述的方法,其中所述化合物可有效减少或消除所述运动障碍的一种或多种症状。其他实施方案应了解,尽管已结合本发明的详细说明阐述本发明,但上述说明旨在阐释而非限制本发明的范围,本发明的范围是由所附权利要求书的范围限定。其他方面、优点和修改均在所附权利要求书的范围内。4非临床研究4.1非临床测试材料多个不同批次的原料药(cx-8998)是由merck合成并用于所有非临床研究中,只是大鼠和狗中的90天毒理学研究除外,其使用由cavion合成的新的批料。原料药是通常与研究中所用的对照物/媒介物混合的粉末。对于体外研究,产生100%二甲亚砜(dmso)中的储备溶液,并将其连续稀释以用于测试物施用至适当缓冲剂/介质中,使dmso的最终浓度为0.1%至0.5%。一些体外和体内代谢和排泄研究使用来自158.4μci/mg于甲醇中的储备溶液的14c标记的cx-8998,所述储备溶液用未标记物质稀释至在相关媒介物中适于测定的比活性。体内研究使用多种不同媒介物制剂,包括0.5或1%甲基纤维素、90%聚乙二醇(peg)400、80%peg-200、10%tween-80和30%环糊精。关键毒理学研究使用0.5%甲基纤维素/10%聚山梨醇酯-80作为口服施用的媒介物。4.2非临床药理学4.2.1主要药效学4.2.1.1体外研究在荧光成像读板仪(flipr)测定中评估对cav3.3功能抑制的体外活性,所述测定通过人类胚肾(hek)293细胞中异源表达的通道来测量钙流入量的抑制。在去极化条件(富集不活化状态下通道的比例)下,cx-8998在flipr测定中展现3.6nm的ic50(产生半数最大抑制的浓度)(图1,上图)。然而,在超极化条件(富集封闭或休息通道状态)下,这种测定中的ic50位移45倍至161nm,表明cx-8998是cav3.3的状态依赖性拮抗剂。这是利用标准全细胞电压钳测定确认的,所述测定使用-80mv的保持电位(vh),显示对于cav3.3,cx-8998的ic50为84nm,并且当vh=-100mv时位移29倍至ic50为2.4μm(图1,下图)。使用表达cav3.1(vh=-80mv,ic50=69nm)和cav3.2(vh=-80mv,ic50=96nm)的细胞的类似电生理学实验显示,所述化合物以状态依赖性方式强效抑制所有cav3家族成员。这些结果表明,cx-8998是所有3个cav3同种型的强效选择性拮抗剂。图1:体外cav3活性的cx-8998抑制flipr=荧光成像读板仪。来源:merck非临床报告pd001。cx-8998由细胞色素p450(cyp)酶cyp3a4和cyp2c9体内代谢成4种不同产物(命名为m01、m02、m03和m04)(参见章节4.3.5)。代谢主要经由异丙基部分上的羟基化从而产生叔醇和伯醇m02和m04,以及异丙基部分的脂族脱氢以形成m01。叔醇m04的进一步氧化导致形成羧酸衍生物m03。cx-8998的4种代谢物是个别地合成并且在flipr测定中针对cav3.3活性的抑制进行评价。这些研究显示与m01和cx-8998的相似水平的效能和状态依赖性(表1)。m02和m04的效能是cx-8998的约1/5,但保持高水平的状态依赖性,而m03的效能和状态依赖性显著低于cx-8998或其他代谢物。表1:cav3.3flipr测定中cx-8998和代谢物的效能flipr=荧光成像读板仪;nd=未进行。来源:merck非临床报告pd001、pk001附录和pk003;cavion非临床报告427-pdsfeb-2018-perkin-iii-r009。4.2.1.2体内研究cx-8998在多种体内啮齿类动物模型中显示活性,所述模型可预测震颤、癫痫、疼痛和心理运动活动中的治疗作用。cx-8998也在啮齿类动物和非人类灵长类动物体内模型中显示作用。4.2.1.2.1失神型癫痫的wistaralbinoglaxo/rijswijk大鼠模型在失神型癫痫的wistaralbinoglaxo/rijswijk(wag/rij)大鼠模型中评价cx-8998,其中大鼠展示特征在于在脑皮层电图记录中高幅值、低频率棘波放电的自发癫痫发作。cx-8998剂量依赖性抑制癫痫发作的累积时间,而媒介物治疗无作用(图2)。在1mg/kg的剂量下,cx-8998治疗使投药后4小时的癫痫发作耗费的累积时间减少55%。在10mg/kg的剂量下,在投药后4小时和15小时,ck-8998将癫痫发作的累积时间分别抑制77%和54%。在10mg/kg的剂量下,癫痫发作频率也剂量依赖性地被抑制高达50%。来自不同结构系列的其他t型拮抗剂在这种模型中也显示类似功效(broicher,2007;blumenfeld,2008;broicher,2008;shipe,2008;yang,2008;uebele,2009;russo,2010;russo,2011)。图2:雄性wag/rij大鼠中cx-8998对癫痫发作时间的影响植入遥测监视器以同时记录脑皮层电图的雄性wistaralbinoglaxo/rijswijk(wag/rij)大鼠接受口服媒介物或cx-8998(0.1-10mg/kg);收集投药后24小时的记录并与投药前24小时的基线记录进行比较。利用自动化癫痫发作分析软件对数据进行评分、平均化并归一化,以实现治疗比较。来源:merck非临床报告pd002。以至多10mg/kg的剂量经口施用cx-8998的代谢物(m01、m02、m03和m04),以研究代谢物在失神型癫痫的wag/rij模型中的功效(merck非临床报告pd002)。10mg/kg剂量下的抑制百分比对于cx-8998为77%并且对于m01、m02、m03和m04分别为71%、67%、71%和60%。与cx-8998一样,m02展现癫痫发作累积时间的剂量依赖性抑制。以10mg/kg的剂量投药后4小时,代谢物m01、m03和m04抑制癫痫发作的频率和持续时间。4.2.1.2.2来自strasbourg模型的遗传失神型癫痫的大鼠在来自失神型癫痫的strasbourg(gaers)大鼠模型的遗传失神型癫痫大鼠中评价cx-8998,其中大鼠展示特征在于在脑皮层电图记录中双侧、同步棘波放电的原发性复发性全身性非惊厥性癫痫发作。相对于相同动物中的媒介物治疗,10mg/kg剂量的cx-8998完全抑制癫痫发作的数量和频率(图3)。100mg/kg的临床相关剂量的乙琥胺仅部分抑制该同类组中癫痫发作的数量和频率。gaers模型被认为是临床相关的非惊厥性癫痫发作的黄金标准模型,并且被认为可预测在人类中的作用并且在cav3.2中具有导致通道功能升高的突变(proft,2017)。来自不同结构系列的其他t型拮抗剂在该模型中显示功效(tringham,2012)。图3:雄性gaers大鼠中cx-8998对癫痫发作的影响来自植入遥测监视器以同时记录脑皮层电图的strasbourg(gaers)大鼠的雄性遗传失神型癫痫大鼠接受口服媒介物、乙琥胺(100mg/kg)或cx-8998(10mg/kg);收集投药后90分钟的记录并与投药前20分钟的基线记录进行比较。来源:cavion非临床报告uscav18-1g。4.2.1.2.3硼替佐米诱发的外周神经病变的大鼠模型在硼替佐米诱发的外周神经病变的模型中评价cx-8998,包括对神经传导速度(ncv)、触觉敏感性和蛋白酶体抑制的影响。用cx-8998治疗引起触觉敏感性在统计上显著的剂量依赖性减少和尾部ncv改善,而不损害硼替佐米诱发的蛋白酶体抑制(图4)。图4:在硼替佐米诱发的化学治疗神经病变的大鼠模型中cx-8998对神经传导速度的影响每周3次经由尾静脉静脉内施用硼替佐米(bz,0.2mg/kg),持续8周。从第29天开始至第8周以3(cx3)、10(cx10)或30(cx30)mg/kg的剂量每日通过口服灌胃施用cx-8998。对照动物接受媒介物施用。在第58天利用肌电图装置(myto2abnneuro,firenze,italy)测定沿尾部神经的神经传导速度(ncv)。##p<0.0001(相对于对照);****p<0.0001(相对于bz)。来源:cavion非临床报告15.14。4.2.1.2.4安非他命的心理运动作用的减弱评价cx-8998减弱安非他命的心理运动活化作用的能力。所述测定对具有抗精神病药潜力的化合物敏感,这是因为典型和非典型抗精神病药而非镇静剂抑制安非他命的反应。cx-8998在该模型中以剂量依赖性方式显著抑制安非他命诱发的过动(图5)。图5:抗精神病药功效的大鼠预测模型中cx-8998对安非他命诱发的心理运动活动的影响amp=安非他命;mpk=mg/kg;v=媒介物。在cx-8998施用后60分钟通过口服灌胃以1、3或10mg/kg的剂量皮下施用安非他命(amp,1.5mg/kg)。在43.2cm×43.2cmmed-associates装置中从cx-8998施用后30分钟至安非他命施用后90分钟监测自发活动。监视随时间的行进距离(右图)并且对在安非他命时段后90分钟内的总行进距离求和(左图)。*指示与媒介物-媒介物治疗显著不同,而α指示与媒介物-安非他命治疗显著不同。来源:mercktta包装说明。4.2.1.2.5对睡眠架构的影响在啮齿类动物和非人类灵长类动物睡眠架构的研究中研究了cx-8998的体内活性(merck非临床报告pd002)。cav3通道在皮质丘脑环路神经元中大量表达,所述神经元成为导致定义警惕状态的脑波图(eeg)变化的充分表征的振荡活性的基础。静息膜电位和cav3通道的状态决定了2种不同网络活动中的1种:与去极化和清醒状态相关的紧张性活动,或与超极化和慢波睡眠相关的爆发活动。在大鼠睡眠研究中,cx-8998显示慢波睡眠剂量依赖性增加和在睡眠时段早期快速眼动(rem)睡眠减少以及在睡眠时段晚期的rem睡眠增加。在恒河猴睡眠研究中,cx-8998显示活跃清醒显著减少,其中在睡眠时段早期的慢波睡眠(δ睡眠)增加并且在睡眠时段晚期的rem睡眠持续增加。因此,两项研究均显示cx-8998影响清醒、慢波睡眠和rem睡眠。4.2.1.2.6cx-8998与其他cav3拮抗剂的cav3拮抗剂药理性质的比较已报道若干其他cav3拮抗剂在震颤、癫痫、疼痛、增重和睡眠架构的非临床模型中具有活性。大鼠中cx-8998的药理性质与其他cav3拮抗剂的药理性质的比较提供于表2中。表2:啮齿类动物中cav3拮抗剂药理学性质的汇总auc最后=从时间零至最后可测量的浓度的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;nr=未报告;t1/2=终末半衰期;tmax=达到最大血浆药物浓度的时间;tta=t型钙通道拮抗剂。a来自uebele,2009;kraus,2010;reger,2011;francois,2013。b来自shipe,2008。c来自barrow,2010。在大鼠中的哈马灵(harmaline)诱发的震颤模型中以至多10mg/kg的剂量水平评价cav3拮抗剂tta-p2和tta-q6。tta-p2以剂量依赖性方式将哈马灵施用之前的生理震颤和哈马灵诱发的震颤归一化(图6)。图6:大鼠中哈马灵震颤的tta-p2归一化分配给媒介物或3个上升剂量中的1个的tta-p2的大鼠在经口施用后展现对哈马灵诱发的震颤的剂量依赖性反应。在施用哈马灵之前,大鼠也展现对生理震颤减轻的剂量依赖性反应(shipe,2008)。tta-q6也展现在控制哈马灵诱发的震颤中的功效(shipe,2008)。4.2.2次要药效学在超过170个酶和受体结合位点、离子通道和转运蛋白的组中测定cx-8998的体外受体活性概况(merck非临床报告pd001附录)。在10μm的浓度下未观测到cx-8998的脱靶效应,除了大麻素2受体(cb2)之外,其显示10μm下53%的结合抑制和5.34μm的ic50计算值。在由87个酶和受体结合位点组成的类似筛选中评价cx-8998的两种代谢物。在10μmm01的浓度下鉴定出人类过氧化物酶体增殖子活化受体γ(pparγ)结合(54%抑制)和豚鼠腺苷转运蛋白的79%抑制的潜力。m01对腺苷转运蛋白的功能抑制的ic50值被测定为2.92μm(ab75238-1207060)。在10μmm02的浓度下,在87个测试目标中未观测到显著结果(即,≥50%抑制或刺激)(ab75238-1207061)。4.2.3安全性药理学4.2.3.1对心血管系统的影响4.2.3.1.1人类胚肾细胞中的体外herg通道测定使用标准全细胞电压钳技术(研究06-4794)测试cx-8998对在中国仓鼠卵巢(cho)-k1细胞中异源表达的herg(人类ether-à-go-go相关基因)的影响。用cx-8998治疗引起对herg电流的抑制作用相对较弱(ic50=21μm,并且ic20=4μm),其在短暂洗脱后在很大程度上可逆。在patchxpress高通量测定中使用相同细胞高达30μm的剂量的cx-8998对herg的影响的评估提供类似结果(ic50=28μm,并且ic20=8.7μm)(研究06-4720)。4.2.3.1.2人类胚肾细胞中的体外nav1.5测定在使用在hek细胞中稳定表达的人类心脏电压依赖性钙通道(nav1.5)的测定中,高达30μm的浓度的cx-8998对nav1.5电流仅显示轻微抑制作用(22%-26%)(研究06-4736)。4.2.3.1.3狗中的体内研究在latinsquare设计研究中在施用2、20和300mg/kg的单一口服剂量的cx-8998后在有意识的狗中利用遥测术测量结果评价cx-8998对心血管功能的影响,所述心血管功能包括心率、收缩压、舒张压、平均动脉压、心电图参数(pr、qrs和qt间隔)和核心体温,各剂量之间有7天的洗脱期(研究06-5634)。在1、3和10mg/kg的累积静脉内(iv)剂量的cx-8998后,在被麻醉的阴道切除的狗中还评价了cx-8998对心血管功能(血压、心率、血流量和心电图[ecg]间隔)的影响(研究06-5065)。在口服施用2mg/kg的cx-8998之后,对任何参数均未观测到治疗相关影响(投药后2小时的血浆浓度:约3.4μm)。在20和300mg/kg剂量水平(最大血浆药物浓度[cmax]:分别为约30μm和60μm)下,观测到剂量依赖性的心率增加和qt间隔相应减小。在投药后约2小时,在300mg/kg剂量水平下观测到心率峰值增加(+47次/分钟,48%)和qt间隔减小(-14msec,7%),这与最大血浆药物浓度的预计时间(tmax)一致,并且在24至36小时内逐渐恢复至基线。在施用20mg/kg剂量水平后至多2小时,瞬时观测到心率(+28次/分钟,30%)和qt间隔(-27msec,12%)的峰值变化。在任何剂量的cx-8998下,均未观测到对血压;pr、qrs或qtc间隔;或核心体温的治疗相关影响。基于20mg/kg剂量水平下的心血管变化的短暂性质和300mg/kg剂量水平下治疗相关变化,20mg/kg被视为无可见有害作用水平(no-observed-adverse-effectlevel,noael)。无可见作用水平(no-observed-effectlevel,noel)被测定为2mg/kg。在iv施用1或3mg/kg的cx-8998(cmax:分别为9.0μm和22μm)后,未发现cx-8998对心血管参数的治疗相关影响。在iv施用10mg/kgcx-8998(cmax:60μm)后观测到平均动脉压减小15%并且观测到pr间隔增加7%;观测到心率、血流量、或qrs或qtc间隔无变化。4.2.3.2对中枢和周围神经系统作用的影响在施用5、15和30mg/kg的单一口服剂量的cx-8998后使用测试的功能观测组合(functionalobservationalbattery,fob)在有意识的雌性大鼠中评价cx-8998的急性神经行为作用(研究06-5644)。cx-8998在5mg/kg的剂量下对神经行为功能无影响。在投药后2小时在30mg/kg剂量水平下观测到异常姿势和步态、脚张开宽度增加、体温降低和热板潜伏时间延长,但在投药后24小时不明显。在15mg/kg剂量水平下,仅观测到脚张开宽度增加。noel被测定为5mg/kg,这展现投药后2小时的血浆浓度为1.1μm。在施用30、300和2000mg/kg的单一口服剂量的cx-8998后在4周glp(良好实验室实践)毒理学研究(研究06-1088;也参见章节4.4.3.1.2)的第1天在雌性大鼠中评价神经行为功能。在所有剂量下均观测到治疗相关发现,并且包括在≥30毫克/千克/天的剂量下尿池的数量和脚张开度增加、体温和直立次数降低和出现异常步态,以及在300和2000毫克/千克/天剂量水平下热板潜伏期增加、张力减退、异常接近反应和姿势、异常呼吸音并且缺乏耳廓反应。基于低发病率、最小严重程度和这种剂量下的反应类型,fob(包括尿池的数量和脚张开度增加、体温和直立次数降低以及异常步态)的低可见作用水平(loel)为30毫克/千克/天(对于雌性大鼠,从投药时间至投药后24小时的血浆浓度-时间曲线下的血浆面积[auc24]/cmax为43μm·hr/21μm)。总之,这些fob研究中在与人类暴露有关的剂量下治疗相关发现的幅值小,可逆并且与镇静一致。4.2.3.3对呼吸系统的影响使用全身体积描记法在施用30、300和2000mg/kg的单次口服剂量的cx-8998后,在有意识的雌性大鼠中评价cx-8998对呼吸功能的影响(研究06-5634)。在施用≥30mg/kg的剂量后,cx-8998引起潮气量和每分钟通气量略有下降。在300和2000mg/kg的剂量(估计cmax,分别为约28和34μm)下,观测到penh(气道阻力指数)增加(144%-212%),这指示上呼吸道或下呼吸道中气道阻力潜在增加。在有意识的雌性大鼠中,呼吸功能的cx-8998的noel为<30mg/kg;然而,鉴于观测到的变化幅值最小并且在30mg/kg剂量水平下对penh未观测到治疗相关影响,30mg/kg剂量被认为是这项研究的noael。在30分钟内在iv施用6mg/kg后在异氟烷麻醉的雄性大鼠中还评价了cx-8998对气道阻力的影响(研究06-5001)。在30分钟iv输注期间或在整个125分钟观测期内未观测到气道阻力变化。在施用6mg/kgiv剂量的cx-8998后,cx-8998的cmax为24μm。4.3动物中的药代动力学和产物代谢4.3.1分析方法和验证如相关美国食品药品管理局(fda)指导文件(fdaguidanceforindustry,2001)中所述,使用merck开发的经验证的生物分析方法对cx-8998、m01和m02的血浆浓度进行量化。确定测定的有效性和重现性,并且检查校准标准和质量控制样品的精密度和准确度。显示在整个量化范围内回收率是足够且一致的。在测试的不同批次空白血浆中未观测到干扰。在储存和测定条件下,分析物在血浆中稳定。在这些条件下,2种醇代谢物(m02和m04)共洗脱,但正相方法揭示,所有物种中所存在的m02都大大超过m04。这些方法用于由merck执行的非临床和临床研究中。cavion已开发并验证用于根据最近的fda指南(fdaguidanceforindustry,2018)同时量化大鼠、狗和人类血浆中的cx-8998和代谢物(m01、m02、m03和m04)的液相色谱-串联质谱(lc/ms/ms)生物分析方法。测定表现令人满意并且符合系统性能适合性、量化范围、校准线性、测定准确度和精密度以及测定回收率的验收标准。这些“5合1”生物分析方法用于量化样品中的cx-8998及其4种代谢物,所述样品收集于大鼠和狗中的glp90天毒理学研究和进行中的cavion赞助的2期临床研究中。4.3.2吸收和药代动力学在大鼠、狗和恒河猴中的研究中研究了cx-8998及其代谢物的药代动力学概况以及吸收、分布、代谢和排泄(adme)作为探索性和关键毒理学研究的一部分(参见章节4.4)。cx-8998显示在单一剂量施用后在大鼠和狗中口服生物利用度可接受(分别为42%和76%),但在恒河猴中由于显著肝首过代谢(约95%的肝提取率)而口服生物利用度较低(<3%)(表3)。这通过在口服施用后在大鼠和狗中的暴露比在猴中高来反映,如通过3个物种中的cmax和血浆浓度-时间曲线下面积(auc)所测量。cx-8998在大鼠和狗中迅速被吸收,其中平均tmax值分别为0.5小时和1.4小时。在猴中的吸收较慢,其中平均cmax达到8.3小时的平均值。表3:在cx-8998的静脉内和口服剂量后大鼠、狗和恒河猴中cx-8998的药代动力学参数的汇总auc=血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;cl=清除率;f=生物利用度;iv=静脉内;t1/2=终末半衰期;tmax=达到最大血浆药物浓度的时间。a经30分钟在二甲亚砜中施用cx-8998。b结果表示为2只动物(大鼠)的平均值或3只动物(狗和猴)的平均值(sd)。c经由口服灌胃在0.5%或1%甲基纤维素中施用cx-8998。d结果表示为平均值(sd)。来源:研究pk001。在口服施用cx-8998后的所有物种中,代谢物m01和m02的暴露都是显著的(表4)。m01的平均auc24值约为cx-8998的平均auc24值的20%至70%,其中在大鼠中观测到最大暴露。在猴中观测到m02相对于cx-8998的最大暴露,其中对于25mg/kg口服剂量的cx-8998,auc24的代谢物/母体比率为17,并且对于75mg/kg口服剂量的cx-8998为44。大鼠和狗中m02的平均auc24值约为cx-8998的平均auc24值的1倍至2倍。表4:在单一口服剂量的cx-8998后大鼠、狗和恒河猴中对cx-8998和代谢物(m01和m02)的暴露的汇总auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积。a经由口服灌胃在0.5%或1%甲基纤维素中施用cx-8998。b结果表示为2只动物的平均值。来源:研究pk001。4.3.3毒代动力学4.3.3.1大鼠4.3.3.1.1雌性大鼠中的剂量范围发现研究在探索性7天剂量范围发现毒理学研究中经由口服灌胃以10、100或500毫克/千克/天的剂量向雌性大鼠施用(研究06-2515;章节4.4.3.1.1)。在雌性大鼠中,对cx-8998的暴露在10至500毫克/千克/天的范围内不与剂量成比例(表5)。表5:7天口服剂量范围发现毒理学研究中雌性大鼠中的cx-8998的关键毒代动力学参数auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;tmax=达到最大血浆药物浓度的时间。a值表示为平均值(sd)。bcx-8998经由口服灌胃施用7天来源:研究06-2515。4.3.3.1.2雄性和雌性大鼠中的4周口服毒性研究在4周毒理学研究中经由口服灌胃向雄性和雌性大鼠施用30、300或2000毫克/千克/天的cx-8998(研究06-1088;章节4.4.3.1.2)。雌性大鼠中的暴露比雄性大鼠中大(表6)。在雄性或雌性大鼠中,在30至2000毫克/千克/天cx-8998的剂量范围内未观测到剂量比例性。表6:4周口服毒理学研究中雄性和雌性大鼠中cx-8998的关键毒代动力学参数auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;tmax=达到最大血浆药物浓度的时间。a值表示为平均值(sd)或中值(tmax);值代表第28天的数据。bcx-8998经由口服灌胃施用28天。来源:研究06-1088。4.3.3.1.3雄性和雌性大鼠中的90天口服毒性研究在90天毒理学研究中经由口服灌胃向雄性和雌性大鼠施用5、15、100或300毫克/千克/天的cx-8998(研究20131957;章节4.4.3.1.4)。雌性大鼠中的暴露比雄性大鼠中更大(表7)。表7:90天口服毒理学研究中雄性和雌性大鼠中cx-8998的关键毒代动力学参数auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;nc=未计算;tmax=达到最大血浆药物浓度的时间;t1/2=终末半衰期。a值表示为平均值或中值(tmax);值代表第90天的数据。bcx-8998经由口服灌胃施用90天。来源:研究20131957。在所有剂量水平中,雄性大鼠在第1天的cx-8998及其代谢物的平均auc值始终高于第45天或第90天的相应值。相反,在第90天,除300毫克/千克/天的最高剂量外,在所有剂量水平下,雌性大鼠中cx-8998和代谢物的水平最高。m01、m02、m03和m04都是大鼠中的重要cx-8998代谢物,相对于cx-8898的血浆暴露,观测到的血浆暴露为10%或大于10%(表8)。两种性别在第90天的血浆auc值从最高至最低是cx-8998,接着是m02,接着是m01,接着是m03,接着是m04。表8:90天口服毒理学研究中雄性和雌性大鼠中对cx-8998代谢物m01、m02、m03和m04的暴露的汇总auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积。a值表示为平均值并且代表第90天的数据。b从表7中每个性别的cx-8998auc24值计算的比率。来源:研究20131957。4.3.3.2狗4.3.3.2.1比格犬(beagledog)中的剂量范围发现研究在10天的探索性剂量范围发现毒理学研究中,向1只雄性和1只雌性比格犬施用3、30和300mg/kg的递增口服剂量的cx-8998(研究06-1055;章节4.4.3.2.1)。每个剂量施用3天,各剂量之间无洗脱期。雌性犬中的暴露通常大于雄性犬(表9)。在雄性或雌性犬中,暴露在3至300毫克/千克/天的范围内不与剂量成比例。表9:10天口服毒理学研究中雄性和雌性犬中cx-8998的关键毒代动力学参数auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;tmax=达到最大血浆药物浓度的时间。an=每种性别1个。b经由口服灌胃各自施用3、30和300mg/kg的递增口服剂量的cx-8998,持续3天。所报告的值是每个剂量水平施用的最后一天(即,对于3、30和300mg/kg剂量分别为第3天、第6天和第9天)的值。来源:研究06-1055。4.3.3.2.2比格犬中的4周口服毒理学研究在4周毒理学研究中经由口服灌胃向雄性和雌性比格犬施用2、20或800毫克/千克/天的cx-8998(研究06-1087;章节4.4.3.2.2)。在雄性或雌性犬中,在第1天或第28天在2至800毫克/千克/天cx-8998的剂量范围内未观测到剂量比例性。表10:4周口服毒理学研究中雄性和雌性犬中cx-8998的关键毒代动力学参数auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;id=数据不足;tmax=达到最大血浆药物浓度的时间。a值表示为平均值(sem)。bcx-8998经由口服灌胃施用28天。来源:研究06-1087。4.3.3.2.3比格犬中的90天口服毒理学研究在90天毒理学研究中经由口服灌胃向雄性和雌性犬施用10、30或300/100毫克/千克/天的cx-8998(研究20131958;章节4.4.3.2.3)。第90天的cmax和auc的平均值通常随剂量增加而增加,在最高剂量水平下,在雄性犬中比在雌性犬中观测到更大暴露(表11)。表11:90天口服毒理学研究中雄性和雌性犬中cx-8998的关键毒代动力学参数auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;tmax=达到最大血浆药物浓度的时间。a值表示为平均值或中值(tmax);值代表第90天的数据。bcx-8998经由口服灌胃施用90天。来源:研究20131958。cx-8998和代谢物(m01、m02、m03和m04)的auc24值在各研究天中在10和30毫克/千克/天下在雄性与雌性比格犬之间始终相当(表12)。然而,在100毫克/千克/天剂量水平下,cx-8998和代谢物(m01、m02、m03和m04)在第90天的auc24值在雄性比格犬中比在雌性比格犬中高3倍至5倍。表12:90天口服毒理学研究中雄性和雌性犬中cx-8998代谢物m01、m02、m03和m04的关键毒代动力学参数auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积。a值表示为平均值并且代表第90天的数据。b从表11中每种性别的cx-8998auc24值计算的比率。来源:研究20131958。4.3.4分布稳态下的平均分布体积(vdss)在大鼠(1.3l/kg)、狗(0.8l/kg)和猴(0.9l/kg)类似(研究pk001)。cx-8998显示在大鼠、狗、猴和人类血浆中分别约99.2%、99.7%、98.8%和99.6%的体外血浆蛋白质结合和分别0.6:0.7的血液/血浆分配比(研究pk003)。m01代谢物显示在大鼠、狗、猴和人类血浆中分别99.6%、99.7%、99.6%和>99.9%的体外血浆蛋白质结合。m02代谢物的蛋白质结合少得多,在大鼠、狗、猴和人类血浆中分别显示71%、82%、83%和86%的值。值得注意的是,cx-8998(包括代谢物m01和m02)在人类血浆中的血浆蛋白结合要比在大鼠血浆中高。cx-8998显示极少至不显示体外(大鼠和人类)和体内(大鼠)的不可逆蛋白质结合。cx-8998容易分布至大鼠的脑以及狗的脑和脑脊髓液(csf)。在雄性大鼠中执行探索性研究,以确定在口服施用25mg/kg于1%甲基纤维素中以及于90%peg400中的cx-8998后2小时,cx-8998及其代谢物的脑渗透(研究pk001)。cx-8998在脑中的暴露约为在血浆中的暴露的50%(表13)。m01代谢物的峰值血浆浓度与母体的峰值血浆浓度相当,而m02/m04代谢物的峰值血浆浓度较低,并且m03代谢物的峰值血浆浓度高于母体的峰值血浆浓度。m01和m02/04代谢物的脑/血浆比率比cx-8998略低,而m03代谢物的脑/血浆比率比cx-8998低约50倍。表13:在口服施用25mg/kg的cx-8998后两小时雄性大鼠中cx-8998和代谢物的血浆和脑浓度peg=聚乙二醇。an=每组2只大鼠;值表示测量结果的平均值。b用色谱法一起量化m02和m04。来源:研究pk001附录。在探索性剂量范围发现毒性研究中,向1只雄性和1只雌性比格犬施用3、30和300mg/kg的递增口服剂量的cx-8998(研究06-1055)。每个剂量施用3天,各剂量之间无洗脱期。在第10天施用最后的300mg/kg剂量后约24小时,将动物处死,收集脑和csf样品,并测定cx-8998、m01和m02/m04的浓度。对cx-8998、m01和m02/m04的中枢神经系统(cns)暴露在雌性犬中通常比在雄性犬中更大(表14)。cx-8998的脑/血浆比率在雄性犬与雌性犬之间相当;然而,观测到代谢物的比率有所不同,观测到雌性犬中的比率高于雄性犬。cx-8998和m01的csf/血浆比率通常与其在狗血浆中的低游离分数(0.3%)一致。m02的csf/血浆比率在两只狗之间变化更大,但总体上比cx-8998或m01更大,并且与这种代谢物在狗血浆中的更大游离分数(18%)一致。表14:口服施用300mg/kgcx-8998后24小时雄性和雌性比格犬中cx-8998及其代谢物的脑和脑脊髓液浓度(血浆比率)csf=脑脊髓液。a括号中的值是脑或csf与血浆的浓度比率。b用色谱法一起量化m02和m04。来源:研究pk001附录。4.3.5代谢cx-8998的代谢已在来自大鼠、狗、猴和人类组织的合并的肝微粒体制剂以及新鲜分离或低温保存的肝细胞中以及利用重组cyp酶(包括cyp2a6、cyp2c8、cyp2c9、cyp2c19、cyp2d6、cyp3a4和cyp3a5)进行体外评价(研究pk003)。cx-8998的代谢主要由cyp3a和cyp2c酶家族介导,以形成4种主要产物(m01、m02、m03和m04)。代谢主要经由异丙基部分上的羟基化从而产生叔醇(m02)、伯醇(m04),以及异丙基部分的脂族脱氢(m01)。2种醇(m02和m04)的进一步氧化导致形成其相应的二氧化代谢物,以及m04的羧酸衍生物(命名为m03)。在大鼠肝微粒体中,m02和m04的形成广泛(m02>m04);然而,自来自狗、猴或人类组织的制剂中,m04(以及随后的m03)的形成并不显著。在人类、猴和狗肝细胞制剂中检测到氧化代谢物的葡萄糖醛酸苷缀合物。cx-8998在大鼠和狗中广泛地体内代谢,并且使用验证的5合1生物分析测定可以显著水平检测到所有4种代谢物。在1期临床研究中,已在人类中以显著水平测量到m01和m02二者。研究cx-8998-cln2-001(t-calm;章节5.4.2.1)中从患有原发性震颤的患者收集的药代动力学样品的初步未核定数据确认这些发现并且展现,m03和m04的水平在人类中不显著。与其相应预期人类临床自由暴露相比,cx-8998的所有4种代谢物在非临床毒理学物种中由宽暴露界限涵盖。在所测试的重组cyp中,cyp3a4、cyp3a5、cyp2c9、cyp2c19和cyp2d6有效地催化m01和m02的形成。然而,cyp3a5、cyp2c19和cyp2d6在人类肝微粒体中的丰度(2%-5%)较对于主要贡献者cyp3a4(约30%)和cyp2c9(约20%)较低。m01和m02二者对于cyp2c9反应都显示较低km*值(约3μm),表明在低底物浓度下cyp2c9的贡献比cyp3a4更显著;与m02相比,m01的这种观测结果更为明显(研究pk003)。cyp3a家族的贡献通过抗人类cyp3a抗体对m01和m02的反应的显著抑制(在1μmcx-8998下>80%)进一步得以证实。抗人类cyp3a和cyp2c抗体的组合几乎完全抑制代谢(在10μmcx-8998下>90%)。因此,cyp3a4和cyp2c9是负责人类中cx-8998的氧化代谢的主要酶。*km(米氏常数(michaelisconstant))是反应速率为半数最大值的底物浓度。cx-8998和m01在利用人类、猴和大鼠p-糖蛋白的双向转运测定中未显示作为底物或抑制剂的任何活性,而m02在大鼠和猴分析中显示增加的流出量(研究pk003)。cx-8998并非抗乳癌蛋白质(bcrp)测定的底物,但cx-8998和m01二者在高浓度(30μm)下均抑制57%的转运。在浓度高达30μm时,m02不会抑制bcrp介导的转运。尽管这项研究并不排除将m01和m02作为bcrp的底物,但该系统中这些代谢物的转运最可能受限(研究2266n-1703)。4.3.6排泄cx-8998在大鼠和恒河猴中的血浆清除率相对较高(分别为29ml/min/kg和16ml/min/kg;表3),但在狗中较低(1.5ml/min/kg),其中相应地在大鼠和猴中的半衰期(分别为0.8小时和1.3小时)相对于狗(7.3小时)较短。在将用14c标记的cx-8998(20mg/kg)口服施用于大鼠后,在大鼠尿液和胆汁样品中回收到大部分放射性(分别为43%和46%),其中约5%的放射性留在粪便中(研究pk003)。小于1%的总剂量作为未改变的药物排泄于粪便中,表明广泛代谢。72小时后标记的回收率几乎完全(约96%),表明良好口服吸收。所有排泄物中的放射性迅速下降,其中大多数(>85%的总回收放射性)的剂量在第一天即排泄出。口服施用后,大鼠血浆的主要循环组分包括m01、m02、m03和m04,其中这些代谢物的其他代谢产物代表次要组分。m02的二氧化代谢物主要通过尿排泄而清除,而m03及其相应葡萄糖醛酸苷作为胆汁和尿液中的主要组分排泄出。4.3.7药代动力学药物相互作用cx-8998主要通过cyp酶cyp3a4和cyp2c在人类肝微粒体中代谢,以形成4种主要产物m01、m02、m03和m04。cx-8998、m01和m02并非人类肝微粒体中cyp1a2、cyp3a4、cyp2c8、cyp2c9、cyp2c19或cyp2d6的强效抑制剂(ic50>20μm)(merck非临床报告pk003和2266n-1702)。cx-8998、m01和m02对cyp3a4或cyp2b6的时间依赖性抑制并不显著。cx-8998和代谢物m01和m02显示诱导cyp3a4mrna或活性的低潜力,其中在原代人类肝细胞中于10μm下为对照作用的≤10%。一致地,cx-8998、m01和m02对人类孕烷x受体(pxr)的活化分别适中,其中在10μm下为对照作用的≤34%。此外,cx-8998、m01和m02并未显著活化大鼠和猴pxr(于10μm下分别为对照的约4%-6%和6%-22%)。因此,cx-8998显示诱导人类cyp3a4的低潜力。cyp2b6mrna的产生是由人类肝细胞中30μm的cx-8998(在3个供体中为3.8倍至8.6倍)诱导;m01和m02也是轻度至中度诱导物(研究2266n-1701)。4.4毒理学4.4.1利用cx-8998执行的毒理学研究cx-8998的毒理学已在一系列非临床研究中得以表征,所述研究包括在大鼠和狗中持续时间高达90天的关键重复剂量毒理学研究以及体外和体内遗传毒理学研究。在大鼠中28天关键雌性生育力研究以及大鼠和兔中初步胚胎-胎儿发育研究中获得关于生殖和发育毒理学的信息。表15中汇总cx-8998的重复剂量毒理学研究。表15:cx-8998的非临床重复剂量研究fob=功能观测组合;gd=妊娠天数;glp=良好实验室实践;noael=无可见有害作用水平;noel=无可见作用水平;qd=每日一次。4.4.2单一剂量研究4.4.2.1狗1只雄性和1只雌性犬中的单一剂量(800mg/kg,通过口服灌胃)探索性毒代动力学研究(研究06-1092)未显示死亡率或治疗相关征象;然而,雄性犬在投药后1小时内经历呕吐。对cx-8998的暴露(auc24)在雌性犬中比在雄性犬中高(3341μm·hr对1325μm·hr)。cx-8998的tmax在雄性犬中发生的时间比在雌性犬中发生的时间晚(24小时对8小时),但这可能受到在投药后立即发作呕吐的影响。两只狗的m01和m02的tmax为8小时;雌性犬中m03的tmax为8小时并且在雄性犬中为24小时。4.4.2.2兔在未怀孕的dutchbelted兔中实施探索性单一剂量毒代动力学研究(研究08-7027)。将10只雌性兔分配为3组,每组3只动物,并且以剂量递增方案施用3、30、100、300或1000mg/kg的单一剂量的cx-8998。所有兔都存活直至研究终止而无临床征象。对于3、30、100、300和1000mg/kg剂量,平均cx-8998暴露量(auc24)分别为0.75、6.65、13.4、74.0和119μm·h。平均cmax值分别为0.18、1.0、1.45、3.87和6.47μm。4.4.3重复剂量研究4.4.3.1大鼠4.4.3.1.1雌性大鼠中的剂量范围发现研究在7天探索性剂量范围发现研究(研究06-2515)中,经由口服灌胃向雌性大鼠施用10、100或500毫克/千克/天。在死亡率、体征、食物消耗、体重、血清生物化学(第8天)、器官重量、或者肉眼可见或组织形态学检查中未观测到治疗相关发现。在高达500毫克/千克/天的剂量下,未观测到指示剂量限制性毒性的治疗相关发现。4.4.3.1.2雄性和雌性大鼠中的4周口服毒理学研究在4周毒理学研究(研究06-1088)中,经由口服灌胃向雄性和雌性大鼠施用0(媒介物)、30、300或2000毫克/千克/天的cx-8998。还在第1天使用测试的fob在雌性大鼠中评价了神经行为功能。在所有剂量下均观测到治疗相关发现,并且包括在≥30毫克/千克/天的剂量下尿池的数量和脚张开度增加、体温和直立次数降低和出现异常步态,以及在300和2000毫克/千克/天剂量水平下热板潜伏期增加、张力减退、异常接近反应和姿势、异常呼吸音并且缺乏耳廓反应。基于低发病率、最小严重程度和该剂量下的反应类型,fob(包括尿池的数量和脚张开度增加、体温和直立次数降低以及异常步态)的loel为30毫克/千克/天(对于雌性大鼠,血浆auc24/cmax值为43μm·hr/21μm)。研究期间的所有其他临死前发现仅在高剂量(2000毫克/千克/天)下才可见,并且包括死亡率、体征(主要可听见的呼吸音但呼吸加重、腹部胀大、活动减少和摸起来发凉)、体重减轻(间歇)/平均体重增加减少(雄性)、食物消耗减少以及血液学、血清生物化学和尿液分析发现异常。仅在高剂量下才能观测到肉眼可见的死后发现(肝大小增加以及前列腺和精囊大小减小)。雌性中在所有剂量下和雄性中在中剂量和高剂量下器官重量都发生变化,并且构成卵巢(所有剂量)、前列腺(300和2000毫克/千克/天)和垂体(两种性别,300和2000毫克/千克/天)重量减小,以及肝(两种性别都为2000毫克/千克/天)和甲状腺(雄性为2000毫克/千克/天)重量增加。雌性中在所有剂量下和雄性中在中剂量和高剂量下观测到组织学发现;这些发现包括肾小管上皮变性(两种性别都为2000毫克/千克/天)、肝和甲状腺肥大(雄性为300毫克/千克/天并且两种性别为2000毫克/千克/天)、鼻和鼻咽发炎(两种性别都为2000毫克/千克/天)、胃细胞浸润和溃疡(雄性或雌性为2000毫克/千克/天)、前列腺低分泌(300和2000毫克/千克/天)、精囊较低分泌(2000毫克/千克/天)和子宫大小变小、阴道上皮变薄和卵巢黄体减少(雌性中所有剂量)。针对泌乳素激素对对照和2000毫克/千克/天剂量组中雌性大鼠的垂体腺进行染色(免疫组织化学),而在2000毫克/千克/天组中的雌性中可见染色减少。高剂量组中动物的死亡原因是由于鼻咽发炎(由于投药制剂的ph和投药期间的反流)。在2000毫克/千克/天剂量水平(合并性别的雌性大鼠的血浆auc24/cmax值,546μm·hr/30.2μm)下存在剂量限制性毒性的明显证据,如由死亡率、显著体征和鼻咽中明显发炎的组织学变化、以及肾中上皮小管变性所证明。在测试的最低剂量(30毫克/千克/天;雄性的血浆auc24/cmax值,35μm·hr/13μm)下血浆暴露下雄性大鼠中的发现限于fob作用,所述作用可逆并且可在诊疗所中进行监测;在这种暴露下在雄性中未观测到肉眼可见的病理学、器官重量或组织学发现。雄性中的noael为30毫克/千克/天并且雌性中的noael为<30毫克/千克/天(基于卵巢重量和黄体减少,在后续研究中显示为可逆)。两种性别中的noel都为<30毫克/千克/天。4.4.3.1.3雌性大鼠中的4周口服毒理学研究,8周恢复期在雌性大鼠中实施具有8周恢复期的4周口服毒理学研究以确定在10毫克/千克/天下对于雌性生殖器官毒性是否存在noel;在1000毫克/千克/天下雌性生殖器官毒性的潜在可逆性;以及cx-8998及其2种代谢物(m01和m02)的毒代动力学曲线(研究07-0034)。经由口服灌胃向大鼠施用0(媒介物)、10或1000毫克/千克/天的cx-8998,持续4周。所有大鼠存活至排定的终止。仅在1000毫克/千克/天的剂量下可见治疗相关的临死前发现,并且包括体征(主要可听见的呼吸音和流涎但呼吸加重、喘息、尿液染色、活动减少、皮肤碰撞减少、摸起来发凉和外观邋遢)、体重增加减少(相对于并行对照减少约10%)和食物消耗。在恢复期内,所有与治疗相关的临死前发现都消退,指示可逆性。死后发现限于1000毫克/千克/天剂量组。在临时验尸时,在100毫克/千克/天剂量水平下分别在6和9只大鼠中存在以下肉眼可见的发现:较低的卵巢和子宫大小;较低的器官重量(相对于并行对照,卵巢重量相对于脑重量减少38%;相对于并行对照,垂体重量相对于脑重量减少40%);以及组织形态学发现(在8只大鼠中卵巢黄体较少并且有时较小和子宫大小较小;并且在7只大鼠中阴道粘膜厚度减小的特征在于粘膜上皮厚度变薄,有时粘液化)。在最终(恢复)验尸时,在该剂量水平下在生殖器官中无肉眼可见的或组织形态学发现,展现在这些组织中恢复,并且垂体重量存在恢复的趋势(最终验尸时相对于并行对照,垂体重量相对于脑重量仅减少14%)。可归因于cx-8998的死后变化和雌性生殖组织变化(卵巢、子宫和阴道)的noel为10毫克/千克/天。在8周恢复期内展现1000毫克/千克/天剂量水平下雌性生殖组织变化的可逆性。对于cx-8998、m01和m02,noel(10毫克/千克/天)下的平均auc24值分别为13.1、8.11和31.7μm·h,并且对于cx-8998、m01和m02,noel下的平均cmax值分别为5.91、2.66和5.40μm。在noel下对于大鼠中的生殖作用,与在禁食条件下每日两次(bid)8mg剂量下的估计人类稳态暴露相比,cx-8998、m01和m02的安全界限为2倍或更大。在模拟“进食”投药条件(其中cmax降低45%)下,对于10mgbid剂量下的cx-8998(母药)存在2倍或更大的安全界限。4.4.3.1.4雄性和雌性大鼠中的90天口服毒理学研究,具有30天恢复期在雄性和雌性大鼠中实施90天口服毒理学研究,恢复期为30天(对于主要研究,每种性别n=10;对于恢复期,每种性别n=5),以确定cx-8998和4种代谢物(m01、m02、m03和m04)的毒性和毒代动力学(参见章节4.3.3.1.3)(研究20131957)。经由口服灌胃向大鼠施用0(媒介物)、5、15、100或300毫克/千克/天的cx-8998,持续至少90天。评价以下各项:临床征象、体重、体重增加、食物消耗、眼科、临床病理学参数(血液学、凝血、临床化学和尿液分析)、毒代动力学参数、肉眼可见的验尸发现、器官重量和组织病理学检查。在所述研究中未观测到与测试物有关的死亡率。在雄性和雌性动物中从第1天至第9天在≥100毫克/千克/天下注意到与cx-8998有关的瞬时活性降低。在雄性中在300毫克/千克/天下注意到与cx-8998有关的体重增加减少,并导致第90天的绝对平均体重比对照低4%。还注意到相关食物消耗减少高达19%。在投药期结束(第90天)时,相对于对照,在300毫克/千克/天剂量组的雄性中,注意到cx-8998有关的血清甘油三酯水平减少46%;较低的血清甘油三酯水平与观测到的食物消耗减少相关。截至30天无投药期结束,注意到体重增加、食物消耗和血清甘油三酯水平恢复。这些变化不被认为是不利的。在这项研究期间,眼科检查、血液学参数、凝血参数或尿液分析数据没有与cx-8998有关的变化。不存在与cx-8998有关的肉眼可见的观察、绝对或相对器官重量变化、或在主要或恢复终止时观测到的微观发现。总之,雄性和雌性大鼠在高达300毫克/千克/天的剂量下充分耐受通过每日一次口服灌胃施用cx-8998达最少90天。未观测到与cx-8998有关的死亡率或任何全身性毒性的证据,并且未鉴定出靶器官。在研究的第1天至第9天期间而非在第9天之后,在100和300毫克/千克/天下在动物中活动暂时性减少明显。在投药期结束时,300毫克/千克/天剂量组中的雄性中观测到体重增加(4%)和食物消耗(19%)的非不利减少,其伴随着血清甘油三酯水平减少(46%),但在30天恢复期后注意到这些作用逆转。这些作用没有伴随着肉眼可见的病理学或组织学变化,并且未被认为是不利的。基于这些结果,雄性和雌性大鼠中的noael被测定为300毫克/千克/天。在300毫克/千克/天下,在第90天,雄性和雌性大鼠中的平均cx-8998cmax分别为22500nm和52900nm,并且在第90天,雄性和雌性大鼠中的平均cx-8998auc24分别为261000nm·hr和566000nm·hr。4.4.3.2狗4.4.3.2.1比格犬中的剂量范围发现研究在10天的探索性剂量范围发现毒理学研究中,向1只雄性和1只雌性比格犬施用3、30和300mg/kg的递增口服剂量的cx-8998(研究06-1055)。每种剂量施用3天,各剂量之间无洗脱期。在第10天施用最后的300mg/kg剂量后约24小时,将动物处死,并测定血浆、脑(额叶皮层和丘脑)和csf中cx-8998、m01和m02/m04的浓度。在任何剂量下都未观测到治疗相关发现。cx-8998和m02/m04(在生物分析性测定中共洗脱)在血浆中的浓度比在额叶皮层、丘脑或csf中更大(示于表14中)。4.4.3.2.2比格犬中的4周口服毒理学研究在4周毒理学研究中经由口服灌胃向雄性和雌性比格犬施用0(媒介物)、2、20或800毫克/千克/天的cx-8998(研究06-1087)。仅在800毫克/千克/天剂量水平下观测到治疗相关发现,并且包括1只雌性死亡(由于体重减轻/体征而较早被处死)和体征(例如,呕吐、不稳步态、发抖、活动减少、流涎、耳朵摸起来发凉)。观测到平均体重减轻,以及血清生物化学和血液学参数的微小变化,这些变化被认为是继发于体征、压力和/或体重减轻。也观测到器官重量差异(较高的肝重量和较低的睾丸和前列腺重量)和组织学发现(肝中肝细胞的嗜酸性粒细胞细胞质体、精管上皮变性和睾丸中精子生成较少以及前列腺不成熟)。透射电子显微镜揭示,肝细胞中的嗜酸性粒细胞体与光滑内质网一致(表明酶诱导)。睾丸/前列腺的发现与800毫克/千克/天剂量组中雄性的体重减轻引起的延迟发育一致。基于死亡率、与神经系统作用一致的体征以及呕吐接着是体重减轻,在800毫克/千克/天下(合并性别的平均auc24/cmax值,804μm·hr/49.8μm)存在明显剂量限制性毒性。所述研究的noel为20毫克/千克/天(合并性别的平均auc24/cmax值,361μm·hr/38.9μm)。与大鼠中noel下的血浆暴露相比,狗中noel下的血浆暴露(auc24)代表在临床预计的那些内cx-8998、m01和m02的更宽的安全界限(分别为30倍、6倍和23倍)(即,大鼠是更敏感的物种)。毒代动力学评价揭示,在整个研究过程中在800毫克/千克/天剂量下对代谢的诱导作用(cx-8998浓度降低和m01浓度增加),与肝微观发现一致。4.4.3.2.3比格犬中的90天口服毒理学研究,具有30天恢复期在雄性和雌性比格犬中实施90天口服毒理学研究,恢复期为30天(对于主要研究,每种性别n=4;对于恢复期,每种性别n=2),以确定cx-8998和4种代谢物(m01、m02、m03和m04)的毒性和毒代动力学(参见章节4.3.3.2.3)(研究20131958)。经由口服灌胃向狗施用0(媒介物)、10、30或300/100毫克/千克/天的cx-8998,持续至少90天。评价以下参数:临床征象、体重、体重增加、食物消耗、眼科、心电图、临床病理学参数(血液学、凝血、临床化学和尿液分析)、毒代动力学参数、肉眼可见的验尸发现、器官重量和组织病理学检查。在研究中未观测到与测试物有关的死亡率;所有动物都存活至排定的安乐死。与测试物有关的临床征象由以下组成:在第1天在雄性中在≥10毫克/千克/天下、在第1天在雌性中在≥30毫克/千克/天下和在第2天在雌性中在300毫克/千克/天下震颤(即,发抖);在第1天在雄性和雌性中在30毫克/千克/天下和在第1天至第40天在雄性和雌性中在300毫克/千克/天下活动减少;在第1天在雄性和雌性中在30和300毫克/千克/天下喘息;从第8天至第64天在雄性中在10毫克/千克/天下、从第8天至第15天在雌性中在10毫克/千克/天下和从第1天至第91天在两种性别中在30和300/100毫克/千克/天下流涎;以及从大约第21天开始在雄性和雌性中在300毫克/千克/天下外观瘦弱。从第1天至第29天在雄性和雌性中在300毫克/千克/天下发生与测试物有关的体重减轻和/或体重增加减少,导致绝对体重比对照中分别低至多13%和7%。体重变化与研究的第一周内的平均食物消耗显著减少(比对照低至多23%)以及个体动物的食欲降低有关,并且随后与外观瘦弱和身体状况下降有关,其需要在300毫克/千克/天剂量组中的许多动物中补充饲料。在第43天开始,整个组的cx-8998的剂量从300毫克/千克/天减少至100毫克/千克/天。在300/100毫克/千克/天剂量组的雄性和雌性中,在剩余投药阶段和/或恢复期期间剂量减少后,注意到体重恢复。第91天的血液学参数中与测试物有关的变化由以下组成:红细胞质量(红细胞、血红素和血容比)和红细胞分布宽度值在300/100毫克/千克/天剂量组中的雄性中比在对照中分别低28%和11%。一般来说,所述值超出归一化和实际历史对照范围,并且在30天恢复期后仍保持不变。临床化学参数的与测试物有关的变化被认为是非不利的,这是因为它们通常具有较小幅值并且处于或接近归一化历史对照范围。除300/100毫克/千克/天剂量组中的雄性中的血清甘油三酯和白蛋白和300/100毫克/千克/天剂量组中的雌性中的血清钾之外,注意到所有参数都完全恢复。凝血参数不存在与测试物有关的变化。眼科检查、心电图、尿液分析数据或肉眼可见的病理学发现都不存在与测试物有关的变化。在300/100毫克/千克/天下在两种性别中注意到与测试物有关的较高平均绝对和相对肝重量,其在微观上与最小的肝细胞肥大相关。肝变化被认为是适应性和非不利的。在30天无药物期后,注意到完全恢复。总之,狗在至多30毫克/千克/天的剂量下充分耐受通过每日一次口服灌胃施用cx-8998达最少90天。未观测到死亡率。施用300毫克/千克/天的cx-8998导致体重减轻和/或体重增加减少,其与食物消耗减少和外观瘦弱/身体状况下降有关,其在大多数动物中需要补充饲料。随后在第43天开始将剂量水平减少至100毫克/千克/天,并且在其余研究期间体重、食物消耗和身体状况得以改善。在300/100毫克/千克/天下注意到适应性和非不利的最小程度肝细胞肥大,并且其与较高的平均绝对和相对肝重量相关。基于这些结果,雄性和雌性犬中cx-8998的noael为30毫克/千克/天。在30毫克/千克/天下,在第90天,雄性和雌性犬中的平均cx-8998cmax分别为39400nm和43400nm,并且在第90天,雄性和雌性犬中的平均auc24分别为335000nm·hr和363000nm·hr。4.4.4基因毒性(诱变性)在执行用以检测诱变性、dna链断裂(在至多20μm的剂量下阴性碱性洗脱测定)、染色体改变(在至多130μm的剂量下阴性)或骨髓中的微核诱导(在至多2000mg/kg的口服剂量下阴性)(研究06-8055、06-8056、06-8684和06-8923)的测定中,cx-8998既无诱变性(在至多6000μg/板的剂量下阴性ames测定)也无遗传毒性。4.4.5生殖毒性确定的胚胎毒性/畸形学(区段ii)研究和出生前/出生后发育(区段iii)研究尚未完成。4.4.5.1对生育力的影响在同居之前、在同居期间和至第7妊娠日(gd),在每日经口施用0(媒介物)、10、30、100或1000毫克/千克/天的cx-8998(每个剂量组20至28只crl:cd[spague-dawley]大鼠)达28天后评价cx-8998对父本(f0)雌性大鼠的生育力的影响(研究07-7370)。在100毫克/千克/天组中在第28天评价cx-8998、m01和m02的毒代动力学曲线。在gd15至gd17将所有存活动物安乐死;检查子宫内容物的胚胎/胎儿存活率,并且对黄体进行计数。1000毫克/千克/天组中的六只雌性由于与测试物有关的体征和体重减轻而被安乐死。在1000毫克/千克/天剂量组中观测到许多与测试物有关的体征;也观测到在交配前和妊娠期间平均母体体重增加的测试物有关的减少,以及在交配和妊娠期间平均食物消耗减少。不存在与测试物有关的生殖毒性的证据。基于这些发现,大鼠雌性生育力参数的noel≥1000毫克/千克/天。一般毒性参数的noel为100毫克/千克/天4.4.5.2对早期胚胎-胎儿发育的影响4.4.5.2.1大鼠在大鼠中执行探索性口服毒理学研究,以评估cx-8998的潜在发育毒性,并协助确定的发育毒理学研究中的剂量选择(研究07-7175)。在gd6至gd20经由口服灌胃向大鼠施用0(媒介物)、10、30、100、300或1000毫克/千克/天的cx-8998(每个剂量组10只)。1000毫克/千克/天剂量组由于过量母体毒性而在gd13至16终止。在30、100和300毫克/千克/天剂量组中观测到平均母体体重增加和平均食物消耗的治疗相关减少。在100和300毫克/千克/天剂量组中观测到活体胎儿体重的治疗相关下降;在10或30毫克/千克/天剂量组中未观测到发育毒性。基于这些发现,在≥30毫克/千克/天的剂量下观测到母体毒性,并且在1000毫克/千克/天的剂量下认为母体毒性是过量的(导致该剂量组的早期终止)。在≥100毫克/千克/天的剂量下观测到发育毒性(活体胎儿体重下降)。4.4.5.2.2兔在兔中执行探索性口服毒理学研究,以评估cx-8998的母体和发育毒性,并协助确定的发育毒性研究中的剂量选择(研究07-7185)。在gd7至gd20经由口服灌胃向兔施用0(媒介物)、30、100、300或1000毫克/千克/天的cx-8998(每个剂量组10只)。将所有存活动物安乐死,并且检查子宫内容物。发现1000毫克/千克/天剂量组中的一只动物在gd11死亡,并且1000毫克/千克/天剂量组中的其余动物由于过多的体重减轻和母体毒性在gd10和gd11被安乐死。在30和100毫克/千克/天剂量组中从gd7至gd9,观测到平均母体体重的轻微治疗相关下降。在高达300毫克/千克/天的任何剂量水平下都不存在发育毒性的证据。基于这些发现,在≥30毫克/千克/天的剂量下观测到母体毒性,并且在1000毫克/千克/天的剂量下认为母体毒性是过量的(导致该剂量组的早期终止)。在30、100或300毫克/千克/天的剂量下未观测到发育毒性的证据。4.4.6致癌性尚未执行致癌性研究。4.5安全性的非临床评估毒理学研究中对cx-8998的反应可合理地分为(1)明显的毒性,(2)行为影响,和(3)激素影响。对于这3个类别,应单独考虑对人类风险的评估。4.5.1明显的毒性cx-8998在大鼠和狗中的非临床毒理学研究中显示有利的安全性概况。在非临床物种中为预期无临床暴露的>80倍的暴露下出现呈死亡/濒死形式的直接毒性。大鼠中的濒死归因于与投药制剂的ph和投药期间的返流有关的鼻咽发炎,这在人类中将不期望发生。在致死剂量下,大鼠中也出现不良的肾和胃病灶。狗中的死亡率与不良临床征象相关。这些作用的noael剂量是预期无临床暴露的至少39倍(大鼠)和约20倍(狗)。4.5.2神经行为影响动物中与cx-8998治疗相关的神经行为变化(fob作用和临床征象)可能与预期的药理学有关。这种属性是基于以下事实:cx-8998是cns活性化合物,并且这些影响在药理学活性剂量下出现并且在低于药理学活性剂量下不出现。这些影响是可监测的并且对临床发展的风险有限。4.5.3激素影响在28天研究中在大鼠中可见与cx-8998有关的对激素反应器官的影响,但在90天研究中不可见。在28天研究中,在所有剂量(30、300和200毫克/千克/天)下观测到指示延长动情间期ii的状态的雌性生殖器官变化。在8周休药期后,这些变化完全可逆。影响是低发生率和严重程度,并且在暴露量大于预期临床游离auc的100倍时,并没有转化为对大鼠生育力或生殖的影响。相比之下,在随后的90天研究中在高达300毫克/千克/天的剂量下,雌性大鼠中的激素反应组织中无变化,尽管对生殖器官进行了详细阶段性组织学分析。28天研究中的激素破坏的机制尚未知,并且也不清楚为什么在2项研究中观测到反应差异。可能使动物适应治疗的持续时间不断增加的治疗(在28天内6-7个发情周期对90天内18-25个发情周期)。或者,注意到,口服投药制剂的ph在28天研究中非常低,导致在高剂量下严重程度足以导致死亡的鼻咽发炎和胃溃疡。因此,可能会发生继发于疼痛和痛苦的压力,其引起对激素调节的间接影响(whirledge,2010年)。在90天研究中,ph升高至更可耐受的水平,并且这可防止压力和继发性激素反应。大鼠中激素反应组织的变化可能反映变化的激素状态/激素破坏,并且因此不可能代表这些靶器官的直接毒性。另外,在雌性生育力研究中在高达1000毫克/千克/天的cx-8998的剂量下,交配、生育力或产仔参数均无变化,在所述研究中,在同居之前、同居期间以及至gd7施用cx-8998达28天。另外,在临床试验中已施用cx-8998的受试者中尚未报告与激素破坏有关的不良事件。在大鼠和兔中的经口探索性发育毒性研究中,并未发现在临床上可用于人类的剂量将阻止有生育潜力的女性入选临床试验。然而,尚未完成确定的胚胎毒性/畸形学(区段ii)研究和出生前/出生后发育性(区段iii)研究,因此应采取适当的预防措施,以避免在临床试验期间怀孕。4.5.4安全界限基于稳态下10mgbid投药的估计人类游离血浆浓度(css)和auc24值,从游离(未结合)cx-8998、m01和m02的noael剂量(分别300毫克/千克/天和30毫克/千克/天)下大鼠和狗中90天重复剂量毒理学研究中的暴露来计算cx-8998及其代谢物m01和m02的安全界限。另外,还计算了活性组分(cx-8998、m01和m02)、总活性部分(tam)的暴露的效能归一化总和的安全界限。在90天毒理学研究中,cx-8998和代谢物展现舒适的安全界限。对于auc24,跨分析物和物种计算的安全界限通常在大鼠中>25倍并且在狗中≥16倍,而对于cmax,通常在大鼠中>50倍并且在狗中≥23倍。雄性大鼠中的界限一般低于雌性大鼠,而狗中两种性别之间的界限类似。对于cx-8998、m01和m02,游离auc24的计算安全界限(雌性/雄性)在大鼠中分别为81倍/37倍、>49倍/>26倍和129倍/65倍,并且在狗中分别为19倍/18倍、>16倍/>19倍和33倍(表16)。对于cx-8998、m01和m02,游离cmax的计算安全界限在大鼠中分别为184倍/78倍、>81倍/>51倍和216倍/117倍,并且在狗中分别为57倍/51倍、>23倍/>29倍和59倍。游离auc24tam的计算安全界限在雌性和雄性大鼠中分别为114倍和57倍,并且在雌性和雄性犬中为29倍。游离cmaxtam的计算安全界限在雌性和雄性大鼠中分别为193倍和101倍并且在雌性和雄性犬中分别为54倍和53倍。总之,根据研究方案和当地法规指南,关于cx-8998记录的非临床毒性未提供进行临床试验的禁忌症。表16:基于auc24和cmax的人类中的安全界限auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;bid=每日两次;cmax=最大血浆药物浓度;noael=无可见有害作用水平;tam=总活性部分。a大鼠=300毫克/千克/天中90天重复剂量毒理学研究的noael。b狗=30毫克/千克/天中90天重复剂量毒理学研究的noael。c在大鼠中,cx-8998、m01和m02的游离分数分别为0.8%、0.4%和29%;在狗中,cx-8998、m01和m02的游离分数分别为0.3%、0.3%和18%。d在人类中,cx-8998、m01和m02的游离分数分别为0.4%、<0.1%和14%。etam是从cx-8998、m01和m02的效能归一化游离暴露计算来源:计算出的安全界限,90天毒理学备忘录,2018年6月26日。5人类中的影响5.1利用cx-8998执行的临床研究的列表已在健康男性和女性受试者中4个1期研究中以及在患有精神分裂症的急性精神病性患者中1个2a期研究中研究了cx-8998(以前称为mk-8998,merckandcompany,inc)。这些试验是随机化、双盲、安慰剂对照研究,其探索1至24mg的单一剂量的cx-8998(正常健康受试者)、2至18mg的多个剂量每日一次持续7天(正常健康受试者)和8mgbid持续28天(患有精神分裂症的患者)。已在诊疗所中完成患有原发性震颤的成人中cx-8998的4周随机化、双盲、安慰剂对照、剂量滴定研究,并且患有伴有失神型癫痫发作的全身性癫痫综合征的青少年和年轻成人中cx-8998的4周多中心、开放标签、剂量滴定研究正在进行中。计划患有pdt的患者中的开放标签数字子研究(以评估数字评估工具在pdt客观测量中的效用)和患有pdt的患者中cx-8998的4周多中心、随机化、双盲、安慰剂对照、剂量滴定研究。表17提供临床研究的设计特征。表17:cx-8998的临床研究bid=每日两次;eeg=脑波图;mds-uprds=运动障碍协会赞助的统一帕金森病等级量表修订版;panss=阳性和阴性综合征量表;pd=药效学;pdt=帕金森病震颤;pk=药代动力学;serdas=癫痫发作相关的失能评估量表;tetras=原发性震颤评定评估量表。a在晚上饭后4小时服用所述剂量。b计划所述研究以包括8名受试者;第九名受试者随机化以代替时段1后中断研究的受试者。5.2药代动力学和药物代谢5.2.1单一剂量药代动力学5.2.1.1cx-8998的关键单一剂量药代动力学参数在4个1期研究(pn001、pn002/部分i、pn003和pn005)中评价cx-8998的单一剂量药代动力学。通过表18中的研究汇总cx-8998的平均(sd)药代动力学参数。表18:cx-8998的平均(sd)单一剂量药代动力学参数(研究pn001、pn002、pn003和pn005)aucinf=从时间零至无限大的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;t1/2=终末半衰期;tmax=达到最大血浆药物浓度的时间。a除非另外指示,否则剂量是在过夜禁食后的早晨施用。b中值。c调和平均值和假标准偏差。5.2.1.2cx-8998的单一剂量药代动力学曲线cx-8998在1至24mg的剂量范围内的单一剂量药代动力学的特征在于快速吸收和双相消除(图7)。图7:在禁食条件下1至24mg的单一口服剂量的cx-8998后健康男性受试者中cx-8998的平均血浆浓度(研究pn001/部分i)在禁食条件下,cx-8998的cmax出现在投药后0.8至2小时的中值(研究pn001/部分i;表18)。1、3、4、8、12、16、20和24mgcx-8998的单一剂量施用导致cx-8998的暴露水平(cmax和从时间零至无限大的的血浆浓度-时间曲线下面积[aucinf])近似成比例增加。在健康年轻男性受试者(年龄为20至40岁;研究pn001)中,cx-8998的平均终末半衰期(t1/2)范围为9.7至13.5小时,但在中年和老年健康男性受试者(年龄为55至75岁;研究pn002)中则延长,平均t1/2范围为18.8至22.6小时;并且中年和老年健康女性受试者(年龄55至75岁;研究pn005)中延长,平均t1/2范围为30.3小时。5.2.1.3食物影响1mg剂量的cx-8998与高脂膳食一起施用导致口服吸收显著延迟(研究pn001/部分i;图8),其体现为平均cmax明显降低(进食56nm对禁食104nm)和中值tmax延迟0.8小时(禁食)至4.0小时(进食)(表18)。图8:在禁食和进食条件下1mg的单一口服剂量的cx-8998后健康男性受试者中cx-8998的平均血浆浓度(研究pn001/部分i)进食/禁食条件的几何平均值比率和几何平均值比率的值范围对于cmax为0.55(0.40-0.76)并且对于aucinf为0.95(0.77-1.35)。与禁食状态相比,cx-8998与食物一起施用导致cmax降低45%,而未显著降低cx-8998的auc。这些结果指示,cx-8998可与食物一起服用以降低cmax,目的是在不牺牲cx-8998的生物利用度的情况下改善其早期暴露耐受性。5.2.1.4剂量比例性在禁食条件下,1至24mg的单一口服剂量的cx-8998导致cx-8998的平均暴露水平(cmax和aucinf)近似成比例增加,无论cx-8998是在早晨或晚上施用(研究pn001/部分i;图9)。总之,cx-8998的单一剂量药代动力学在1至24mg的剂量范围内呈线性。图9:在禁食条件下早晨或晚上施用cx-8998后针对剂量的cx-8998cmax和aucinf的平均值(±sd)(研究pn001/部分i)aucinf=从时间零至无限大的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度。5.2.1.5使用探索性测定的代谢物概况使用探索性测定对1至24mg范围内的单一口服剂量(研究pn001/部分i)后cx-8998的2种代谢物(m01和m02)的血浆浓度进行量化。与cx-8998所观测到的比例关系一致,12、16和24mgcx-8998的单一剂量施用导致m01和m02的平均aucinf逐步增加。在16mg晚上剂量的cx-8998后m01和m02相对于cx-8998的单一剂量血浆浓度曲线示于图10中。图10:在单一口服16mg晚上剂量cx-8998(研究pn001/部分i)后cx-898和代谢物(m01和m02)的平均血浆浓度曲线m01和m02的这些结果与后续研究中使用更确定的代谢物测定的代谢物发现一致(参见章节5.2.3)。5.2.2多剂量药代动力学cx-8998的单一剂量和多剂量药代动力学的特征在于在每日一次投用2、4、8、12和18mgcx-8998后快速吸收和双相消除(研究pn002/部分ii;图11)。在开始投用所有剂量水平后的2至3天之内似乎达到稳态谷浓度。图11:在健康男性受试者中每日一次投用cx-8998(2、4、8、12和18mg)后cx-8998的平均血浆浓度(研究pn002/部分ii)*由于4名受试者过早中断研究,第8天的18mg剂量,n=2;对于第1天和第7天的所有其他剂量以及第1天的18mg剂量,n=6。随着剂量增加,在第1天和第7天cx-8998的暴露水平(cmax和auc)近似成比例增加(表19)。平均稳态cmax范围为每日一次施用2mg后的215nm至每日一次施用18mg后的1498nm。auc24的累积比率指示截至第7天cx-8998有24%至36%的适度累积。在第7天最后一个剂量的cx-8998后的平均t1/2范围为11.0至13.5小时,这与健康年轻男性受试者中t1/2的单一剂量估计值(9.7-13.5小时)一致。表19:在向健康男性受试者每日一次施用cx-8998后第1天和第7天的平均(sd)cx-8998药代动力学参数(研究pn002/部分ii)auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;t1/2=终末半衰期;tmax=达到最大血浆药物浓度的时间。a中值。b调和平均值和假标准偏差。c第7天/第1天auc24的几何平均值比率。基于暴露水平(即cmax和auc)与剂量之间的比例关系以及各剂量之间t1/2和累积比率的值的一致性,cx-8998的多剂量药代动力学似乎在2至18mg/天的剂量范围内呈线性。5.2.3代谢人类肝微粒体的体外研究已显示,cx-8998经历广泛cyp代谢,主要由异丙基部分的脂族脱氢以形成m01代谢物或通过异丙基部分上的羟基化以形成m02代谢物(章节4.3.5)。cx-8998的代谢是由cyp3a和cyp2c酶介导。然而,显示cx-8998既不是主要cyp同种型的抑制剂(在人类微粒体中的ic50>25nm),也不是cyp3a4代谢的诱导物,如在人类原代肝细胞培养物中所确定。同样,cx-8998不是人类p-糖蛋白的底物。使用确定的(经验证)测定对研究pn002、pn003和pn005中获得的样品的m01和m02的血浆浓度进行量化。研究pn002/部分ii中第一8mg剂量的cx-8998后cx-8998、m01和m02的血浆浓度曲线示于图12中。图12:在第1天第一8mg剂量的cx-8998后cx-8998和代谢物(m01和m02)的平均(sd)浓度(研究pn002/部分ii)研究pn002/部分ii中施用第一8mg剂量的cx-8998后m01和m02的血浆浓度曲线(图12)似乎与每一分析物的单一剂量血浆浓度曲线一致,如从研究pn001/部分i(图10)中的探索性代谢物测定所获得,其中cx-8998的吸收和消除相对于m01和m02的形成和消除的较缓慢速率来说更快速。m01和m02的稳态cmax值低于cx-8998,m01的平均cmax比率范围为0.36至0.57并且m02为0.26至0.33(表20)。然而,m01的总体暴露水平高于m02和cx-8998的总体暴露水平,这是由于m01的平均t1/2(19.0-25.8小时)比m02(12.2-15.4小时)和cx-8998(11.0-13.5小时)更长。表20:在第7天在每日一次施用cx-8998达7天后cx-8998代谢物(m01和m02)的平均(sd)药代动力学参数(研究pn002/部分ii)auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;t1/2=终末半衰期。a代谢物对母体(cx-8998)的几何平均值比率。b调和平均值和假标准偏差。在第7天的24小时投药间隔中,相对于cx-8998,m01和m02的血浆浓度曲线保持相对平坦(图13)。在施用最后一个剂量的cx-8998后12、16和24小时,m01的平均血浆浓度高于cx-8998,这是由于cx-8998更快消除。在施用最后一个剂量的cx-8998后12、16和24小时,m02的平均血浆浓度与cx-8998相当,这表明m02的t1/2比其消除速率更依赖于其形成速率(即翻转动力学)。图13:在第7天在每日一次施用8mg的cx-8998达7天后cx-8998和代谢物(m01和m02)的平均(sd)浓度(研究pn002/部分ii)基于cx-8998、m01和m02的效能和游离分数以及代谢物/母体的最大cmax比率(表20),cx-8998对m01的相对效能将为1.00至0.27,并且cx-8998对m02的相对效能将为1.00至2.98。使用测量非临床物种中观测到的cx-8998和所有4种代谢物(m01、m02、m03和m04)的经验证的5合1方法的来自进行中的研究cx-8998-cln2-001(t-calm;章节5.4.2.1)的初步未核定生物分析数据,确认i期研究的发现,并且展现m03和m04暴露在人类中并不显著。5.2.4特殊群体中的药代动力学5.2.4.1年龄和体重cx-8998的平均cmax和aucinf值在中年和老年男性受试者中比在年轻男性受试者中分别高28%和50%,并且在中年和老年女性受试者中分别高6%和68%(表21)。类似地,m01和m02暴露在中年和老年男性和女性受试者中比在年轻男性受试者中高约1.5倍至2倍(即50%-100%)。然而,应注意,使用探索性测定以确定年轻男性受试者中m01和m02的浓度(研究pn001),而使用确定的(经验证)测定以确定中年和老年受试者中m01和m02的浓度(研究pn002/部分i和pn005)。表21:根据年龄组的单一8mg剂量的cx-8998后cx-8998和代谢物(m01和m02)的平均(sd)药代动力学参数(研究pn001/部分i、pn002/部分i和pn005)aucinf=从时间零至无限大的血浆浓度-时间曲线下面积;cmax=最大血浆药物浓度;t1/2=终末半衰期;tmax=达到最大血浆药物浓度的时间。a平均值±sd年龄:27.3±6.4岁。b平均值±sd年龄:65.8±8.1岁。c平均值±sd年龄:64.9±5.6岁。d基于研究pn001/部分i中12mg剂量的cx-8998的代谢物数据的探索性测定将m01和m02的cmax和aucinf值相对于8mg进行剂量归一化。来源:cx-8998模拟备忘录,2018年6月26日。对完成的4个1期研究(pn001、pn002、pn003和pn005)的数据实施荟萃分析,以检查年龄和体重对cx-8998暴露的影响。鉴于cx-8998剂量与auc之间的比例关系,使用104名受试者(34名在研究pn001中施用1、3、4、8、12、16、20或24mgcx-8998的受试者、35名来自研究pn002的施用2、4、8、12或18mgcx-8998的受试者、28名来自研究pn003的施用12mgcx-8998的受试者和7名来自研究pn005的施用8mgcx-8998的受试者)的共同表示为剂量归一化总体暴露(dn-auc)的aucinf(或在投药间隔内的血浆浓度-时间曲线下的面积[aucτ],在适用的情况下)的剂量归一化值。这种分析的结果(图14)显示,年龄占使用二阶多项式与cx-8998的dn-auc相关的药代动力学可变性的大约22%(r2=0.220),其中中年和老年男性和女性受试者展示比年轻男性受试者高的cx-8998暴露水平。体重与dn-auc之间未观测到明确关系,无论所测试的模型如何,体重占cx-8998的药代动力学可变性的不到2%(r2=0.017)。图14:cx-8998的dn-auc与年龄和体重之间的关系(研究pn001、pn002、pn003和pn005的合并数据)dn-auc=剂量归一化血浆浓度-时间曲线下面积。各组阐释年龄与dn-auc关系(r2=0.220)以及对于体重与dn-auc之间测试的线性关系(r2=0.017)中所示的二阶多项式的回归线和95%置信区间(y(x)=0.177*x2-6.520*x+731,其中x=年龄(岁))这种荟萃分析的结果指示,cx-8998的总体暴露水平在中年和老年受试者中高于年轻受试者,而与cx-8998的剂量无关,但体重并非cx-8998的药代动力学可变性的显著贡献因素。5.2.4.2性别通过比较中年和老年男性受试者(研究pn002/部分i)与中年和老年女性受试者(研究pn005)之间在施用8mg剂量的cx-8998后cx-8998及其代谢物(m01和m02)的血浆浓度来检查性别对cx-8998的药代动力学的影响。cx-8998、m01和m02的血浆浓度曲线在中年和老年男性和女性受试者中类似,如图15中所示。图15:在单一8mg剂量的cx-8998后中年和老年男性(研究pn002/部分i)以及中年和老年女性(研究pn005)受试者中cx-8998和代谢物(m01和m02)的平均(sd)浓度cx-8998、m01和m02的最大暴露(cmax)在中年和老年女性受试者中比在中年和老年男性受试者中分别低18%、低12%和高2%(表21)。cx-8998、m01和m02的总体暴露(aucinf)在中年和老年女性受试者中比在中年和老年男性受试者中分别高12%、低2%和低11%。鉴于健康中年和老年男性和女性受试者之间的比较血浆浓度曲线和药代动力学参数,cx-8998、m01或m02的药代动力学似乎无任何临床上重要的性别差异。5.2.4.3肾或肝损害尚未对具有显著肾或肝损害的受试者执行cx-8998的研究。在具有肾病(肌酸酐清除率<39ml/min)或肝病(血清丙氨酸转氨酶[alt]或天冬氨酸转氨酶[ast]水平>2×正常值上限[uln])的历史的受试者中应小心使用cx-8998。5.2.5药物相互作用尚未利用cx-8998实施临床药物-药物相互作用研究。cx-8998主要由cyp酶cyp3a4和cyp2c9代谢。cx-8998不抑制人类肝微粒体中的cyp3a4、cyp2c8、cyp2c9、cyp2d6、cyp1a2、cyp2c19或cyp2b6(产生半数最大抑制的浓度[ic50],>20-25μm),并且在10μm的浓度下并非cyp3a4或cyp2b6的时间依赖性抑制剂。cx-8998不诱导cyp3a4,但却是cyp2b6的弱诱导物。cx-8998不是p-糖蛋白或bcrp的底物,并且不显著抑制地高辛(digoxin)的转运。5.2.6cx-8998的模拟药代动力学曲线实施使用健康年轻男性受试者(研究pn001/部分i)中在进食条件下1mg的单一剂量的cx-8998的数据的非参数叠加的建模和模拟,以针对在进食条件下服用的cx-8998的bid剂量的临床相关范围预测cx-8998的暴露水平。模拟是基于以下假定:在较高剂量下可维持药代动力学线性(如研究pn001/部分i;图7中所示),并且利用1mg剂量观测到的食物影响(即,钝化cmax而不影响auc;表18)在较高剂量下将一致。对于4、6、8和10mgbid剂量的cx-8998,从第一剂量至稳态的模拟预测,每个剂量的cx-8998的cmax将落在200至800nm的潜在“目标暴露范围”内(图16)。图16:在进食条件下每日两次投用后cx-8998对稳态的模拟药代动力学曲线基于进食条件下1mg剂量的cx-8998的模拟cx-8998药代动力学曲线。4、6、8和10mg每日两次(bid)模拟剂量的auc值对应于研究pn001/部分i中每日一次(qd)剂量的所观测的血浆浓度-时间曲线下面积(auc)值。来源:cx-8998模拟备忘录,2018年6月26日。cx-89988、m01和m02的稳态药代动力学暴露参数(css和auc24)的估计值是源自年轻健康男性受试者(研究pn001/部分i)、老年健康男性受试者(研究pn002/部分i)和老年健康女性受试者(pn005)中的单一剂量数据。如从10mgbid剂量的cx-8998预测的年轻男性受试者、中年和老年男性受试者以及中年和老年女性受试者中的cx-8998、m01和m02的平均、最小和最大css值示于表22中;平均、最小和最大auc24值示于表23中。表22:在禁食条件下每日两次施用10mgcx-8998后cx-8998、m01和m02的估计平均(最小,最大)稳态浓度aucinf=从时间零至无限大的血浆浓度-时间曲线下面积;css=稳态下的浓度。acss是源自剂量归一化aucinf(表21中)。css=1.25×aucinf÷τ,其中投药间隔(τ)等于12小时并且10mg/8mg=1.25。来源:cx-8998模拟备忘录,2018年6月26日。表23:在禁食条件下每日两次施用10mgcx-8998后cx-8998、m01和m02的估计平均(最小,最大)稳态auc24auc24=从时间零至投药后24小时的血浆浓度-时间曲线下面积;aucinf=从时间零至无限大的血浆浓度-时间曲线下面积;css=稳态下的浓度。a稳态auc24是源自剂量归一化aucinf(表21中),剂量为每24小时20mg。auc24=1.25×aucinf*2,其中投药间隔是每24小时两次并且10mg/8mg=1.25。来源:cx-8998模拟备忘录,2018年6月26日。5.31期研究5.3.1研究pn001(健康男性受试者中的两部分研究)5.3.1.1研究pn001/部分i5.3.1.1.1研究设计研究pn001/部分i是24名年龄为20至40岁的健康男性受试者中cx-8998的安全性、耐受性和药代动力学的随机化、双盲、安慰剂对照、5时段、单一、升高剂量研究。各8名受试者的两个交替组(a组和b组)在5个治疗期中在禁食状态下施用单一、升高剂量的1、3、4、8、12和16mg的cx-8998(每个剂量水平6名受试者)或安慰剂(每个剂量水平2名受试者)。a组中的受试者也随高脂早餐一起接受1mgcx-8998或安慰剂。8名受试者的额外组(c组)在3个治疗期内接受单一、升高剂量的16、20和24mg的cx-8998或安慰剂,其中在每个治疗期中相同6名受试者接受cx-8998并且相同2名受试者接受安慰剂。cx-8998或安慰剂是在一些治疗期中的过夜禁食后于早晨服用和在一些时段中的晚餐后禁食最少4小时后于晚上(就寝前)服用。通过不良事件、生命体征和反复临床和实验室评价贯穿始终监测安全性。5.3.1.1.2紧急治疗不良事件高达16mg的单一剂量通常耐受良好。在≥3mg的剂量下,观测到与神经系统有关的不良事件(例如,嗜眠、松弛、情绪变化、集中力差、视觉变化、感觉异常)。由于集中力差、头痛、情绪变化、焦虑、不安定和晚上投药后的生动梦境,20和24mg的剂量耐受性较差。所有不良事件都是轻度或中度和重度并且短暂的。表24中汇总2名或更多名受试者中报告的不良事件。表24:单一剂量的cx-8998后两名或更多名受试者中报告的紧急治疗不良事件(研究pn001/部分i)meddra=药事管理的医学辞典。acx-8998是在高脂早餐后施用。所有其他剂量是在禁食状态下施用。bcx-8998是在晚餐后4小时施用。所有其他剂量是在早晨施用。c“任何剂量”栏是基于接受任何剂量的cx-8998的受试者数目。任何治疗组中经历同一事件超过一次的受试者仅对所述事件计数一次。在接受cx-8998的受试者中未报告严重不良事件。一名安慰剂治疗的受试者经历严重不良事件(扁桃体周脓肿)。所述受试者入院接受口服和静脉内抗生素治疗并且由于该事件而中断研究。5.3.1.1.3其他安全性评估如通过床旁肺活量测量法所评估,在生命体征、安全性实验室评估、ecg参数或1秒内用力呼气量(fev1)中未观测到临床上重要的趋势。5.3.1.1.4结论在这项研究中,健康男性受试者对高达16mg的单一口服剂量耐受良好。由于集中力差、头痛、情绪变化、焦虑、不安定和因晚上投药而梦境生动,20和24mg的剂量耐受性较差。5.3.1.2研究pn001/部分ii5.3.1.2.1研究设计研究pn001/部分ii是12名年龄在22至39岁的清醒健康男性受试者中单一剂量的cx-8998对量化eeg参数的影响的随机化、双盲、安慰剂对照、3时段交叉研究。将受试者随机分配到以下一系列的3种治疗:安慰剂、5mgcx-8998和18mgcx-8998。每个治疗期与前一治疗期相隔大约2周洗脱期,并且所有治疗都是在过夜禁食后的早晨施用。在每个治疗期内,在投药前和投药后的选定时间点持续高达24小时记录脑波图。还实施了认知和主观评估,包括karolinska嗜睡量表和bond-lader视觉模拟量表。5.3.1.2.2药效学数据在安静清醒期间,健康成人的脑后部区域中可见α节律。其起源于丘脑中并且由cav3通道活性驱动(schreckenberger,2004)。激动的清醒个体可能不会展示α节律。清醒eeg的分析揭示,α频带中的eeg功率密度谱具有统计学显著的剂量依赖性变化(图17)。在18mg剂量的整个24小时投药后评价期间,在闭眼姿势期间通常观测到α功率降低,并且跨电极位点通常一致。图17:α带(电极位点oz、闭眼状态)的清醒eeg绝对功率分析(研究pn001/部分ii)ci=置信区间;eeg-脑波图;gm=几何平均值;mk-8998=cx-8998;pb=安慰剂。剂量反应分析(图18)表明,在血浆cx-8998浓度大于200至300nm(4mgbid)时,α功率以剂量和浓度依赖性方式降低约25%或更大。图18:eegα功率的药代动力学/药效学分析(研究pn001/部分ii)bid=每日两次;eeg=脑波图;mk=cx-8998;pbo=安慰剂;pd=药效学。除了eeg药效学作用外,在5和18mg剂量的cx-8998后,在bond和lader视觉模拟量表、karolinska嗜睡量表和认知药物研究镇静组合(其包括反应时间测试和数字警觉测试)中观测到与嗜睡增加一致的剂量依赖性趋势。临床量表中可见的α节律和趋势的剂量依赖性减少与cav3通道(靶标)接合一致,并且用作cx-8998的重要生物活性的证据。5.3.1.2.3安全性数据5.3.1.2.3.1紧急治疗不良事件通常对5和18mg的单一剂量的cx-8998耐受良好,其中神经系统、精神病学和一般性病症是最常报告的事件。表25中汇总2名或更多名受试者中报告的不良事件。表25:单一剂量的cx-8998后两名或更多名受试者中报告的紧急治疗不良事件(研究pn001/部分ii)meddra=药事管理的医学辞典。a经历同一事件超过一次的受试者仅对所述事件计数一次。b研究药物是在禁食条件下于早晨施用。所有不良事件的强度均为轻度或中度。未报告严重不良事件。5.3.1.2.3.2其他安全性评估在生命体征、安全性实验室评估或ecg参数中未观测到临床上重要的趋势5.3.1.2.4结论在这项研究中,健康男性受试者对5和18mg的单一剂量耐受良好。清醒eeg展现,在200至800nm的cx-8998浓度下,α带的绝对功率以剂量依赖性方式降低25%或更大。5.3.2研究pn002(健康男性受试者中的两部分研究)5.3.2.1研究pn002/部分i5.3.2.1.1研究设计研究pn002/部分i是9名健康中年和老年男性(年龄为55-75岁)中cx-8998的随机化、双盲、安慰剂对照、5时段、升高、单一剂量研究。*在时段1至5中过夜禁食后,受试者随机分配以接受2、4、8、12和18mg的单一口服剂量的cx-8998或匹配安慰剂,以最低剂量开始并且进行至最高剂量。在每一时段内,根据计算机生成的随机化时间表分配利用cx-8998(n=6)或安慰剂(n=2)治疗的部署。在时段1中的2mg剂量施用后,治疗期与洗脱期间隔最少1周,并且在随后的治疗期之后间隔最少2周洗脱期。在整个研究中通过不良事件、临床实验室检验、生命体征、ecg和ecg遥测术监测临床安全性。5.3.2.1.2安全性数据5.3.2.1.2.1紧急治疗不良事件通常对高达18mg的单一剂量的cx-8998耐受良好。最常报告的不良事件是松弛、头晕、头痛、头昏目眩、眩晕和情绪改变,所有都是短暂的并且严重程度通常为轻度或中度。表26中汇总2名或更多名受试者中报告的不良事件。所述研究计划包括8名受试者。第九名受试者随机化以代替在时段1后中断的受试者(由于施用2mg剂量后qtc间隔延长,研究者认为其与研究药物无关)。表26:单一剂量的cx-8998后两名或更多名中年或老年男性受试者中报告的紧急治疗不良事件(研究pn002/部分i)meddra=药事管理的医学辞典。a经历同一事件超过一次的受试者仅对所述事件计数一次。未报告严重不良事件。5.3.2.1.2.2其他安全性评估在临床实验室、生命体征、ecg或ecg遥测术中未观测到剂量依赖性作用。5.3.2.1.3结论这项研究中的中年和老年男性受试者(年龄为55-75岁)通常对2至18mg范围内的单一剂量的cx-8998耐受良好。即使在18mg的最高单一剂量下也未观测到剂量限制性毒性。5.3.2.2研究pn002/部分ii5.3.2.2.1研究设计研究pn002/部分ii是40名健康男性受试者(年龄为18-45岁)中多剂量的cx-8998的安全性和药代动力学的随机化、双盲、安慰剂对照、升高剂量、多剂量研究。受试者随机分配到5个多剂量治疗组中的1组,并且在a、b、c、d或e组中在过夜禁食后分别接受2、4、8、12或18mg的剂量的cx-8998或安慰剂,每日一次,持续7天。在每个治疗组内,6名受试者随机分配到cx-8998,并且2名受试者随机分配到安慰剂。在整个研究中通过不良事件、临床实验室检验、生命体征、ecg和ecg遥测术监测临床安全性。5.3.2.2.2安全性数据5.3.2.2.2.1紧急治疗不良事件通常对高达12mg并且每日一次持续7天的多个剂量耐受良好。最常见的与药物有关的不良事件为头痛、头昏目眩、疲倦、感到高兴或愉悦、感到情绪激动、疲倦、嗜眠、松弛、异常或生动梦境或白日梦以及感觉异常/感觉迟钝。在18mg/天的剂量下,情绪或知觉障碍的不良事件更为频繁并且强烈,并且暴露于所述剂量的6名受试者中有4名因不良事件而过早中断治疗。表27中汇总至少2名受试者中报告的不良事件。表27:单一剂量的cx-8998后两名或更多名受试者中报告的紧急治疗不良事件(研究pn002/部分ii)meddra=药事管理的医学辞典。a经历同一事件超过一次的受试者仅对所述事件计数一次。在施用计划的7个每日剂量的18mgcx-8998的第一个剂量后,一名受试者经历严重不良事件(情绪波动)并中断治疗。在接受少于计划的7个每日剂量的18mgcx-8998后,另外3名受试者由于不良事件也过早中断治疗。这4名受试者的叙述显示如下:受试者0001-1020(情绪波动)是44岁的健康白人男性,在接受单一18mg剂量的cx-8998后,其报告多个与神经系统有关的不良事件,包括感觉昏昏沉沉、手脚感觉湿冷、醉酒感、腿不宁、全身发抖、感觉不开心(情绪化)和感觉电脉冲穿过身体。随着日子进行,报告其他不良事件,包括情绪波动、感觉恐惧、间歇地大笑、动手困难、感觉虚弱、味觉增强、指尖失去精细感觉、在皮肤层面(全身)上一些感觉缺失和灵魂出窍感(outofbodyfeeling)。所有不良事件的严重程度均为轻度或中度。大部分事件在投药后数分钟至数小时开始,并且全部都在投药后第3天消退。受试者经历的不良事件被阐述为“情绪波动”,并导致投药中断。情绪波动的事件被归类为严重不良事件,因为其导致临床研究部门中的住院监测延长。受试者0001-1021(攻击性)是25岁健康白人男性,在接受7个计划的18mg剂量的cx-8998的第六个剂量后,其报告感觉有攻击性。另外,其报告对另一名受试者感觉愤怒、感觉晕眩/醉酒、白日梦强烈、发抖/颤抖和出汗。所有不良事件的严重程度均为轻度,并且全部都被认为可能与研究药物有关。未施用最后一个剂量的cx-8998。研究药物中断后所有不良事件都消退。受试者0001-1114(情绪障碍)是28岁的健康白人男性,其在计划的7天治疗中有6天接受18mgcx-8998。受试者经历了与神经系统有关的严重程度逐渐增加的事件,包括头昏目眩、亢奋、疲倦、头痛、难以跟随思维、思想和情感脆弱、感觉乏味和肌肉无力。所有不良事件的严重程度均为轻度或中度,并且全部都被认为可能与研究药物有关。未施用最后一个剂量的cx-8998。研究药物中断后所有不良事件都消退。受试者0001-1115(晕厥)是25岁健康白人男性,在接受7个计划剂量的18mgcx-8998中的第一个剂量后约2小时,其经历晕厥发作,伴有恶心和呕吐。血氧饱和度和血清葡萄糖水平在正常限值内,但事件发生时受试者的血压较低(76/43mmhg,脉搏率为59bpm)。遥测术监测未显示心脏节律异常或临床重要心动过缓的证据。所述事件由研究者判断为严重程度为中度,并且可能与研究药物有关。由于持续的恶心以及虚弱和嗜睡的感觉,所述受试者中断研究。在7个计划剂量的2mgcx-8998中的第一个剂量后约2小时站立时,另一名受试者感到恶心并晕倒。晕倒后1分钟,其血压为131/64mmhg,脉搏率为50bpm,血氧饱和度为97%,并且其血清葡萄糖水平在正常限值内。当时受试者未进行遥测。研究者认为所述事件为血管迷走神经反应(严重程度为中度)并且评估所述事件与研究药物无关。受试者继续研究并完成计划的7天治疗。5.3.2.2.2.2其他安全性评估在生命体征、安全性实验室或ecg参数中未观测到临床上重要的趋势。5.3.2.2.3结论在这项研究中,健康男性受试者对7天每日一次的2、4、8和12mg的多个口服剂量耐受良好。每日一次18mg的多个剂量耐受较差,其中在完成7天治疗期之前,6名受试者中有4名由于不良事件而中断治疗。基于这些结果,每日一次施用7天时,cx-8998的最大耐受剂量为12mg。5.3.3研究pn003(健康男性受试者中的两部分研究)5.3.3.1.1研究设计研究pn-003是28名年龄为40至80岁的健康男性受试者中cx-8998对多睡眠图(psg)终点(包括慢波活动和快速眼动睡眠)的影响的多中心、2部分、适应性设计、随机化、双盲、安慰剂对照、单一剂量、交叉研究。在研究的部分i/时段1中,受试者接受12mgcx-8998的开放标签治疗,而无psg记录。在部分i/时段2和3中,根据随机化、双盲、2时段、交叉设计,受试者在一个时段中接受单一口服12mg剂量的cx-8998,并且在另一时段中接受安慰剂,以比较cx-8998与安慰剂对psg终点的影响。与治疗期隔开至少2周的洗脱期。cx-8998或安慰剂是在每名受试者的正常就寝时间(由睡眠日记确定)之前30分钟和晚饭结束后4小时服用。在每名受试者的习惯就寝时间开始记录psg,并持续8小时。除了psg记录外,第二天也利用睡眠质量的主观测量(leeds睡眠评价问卷)和嗜睡(karolinska嗜睡量表)以及认知和运动任务对受试者进行评价,以评估次日早晨的残余影响。在24名受试者完成研究的部分i后,对主要终点(睡眠的前三分之一中的慢波活动)实施期中分析(章节5.3.3.1.2)。所述研究被适应性设计,以允许在期中分析后重新估计样本大小。根据期中分析的结果,如果达到功效标准(慢波活动增加,p<0.05)或无效标准(p>0.25),则可停止研究。如果任一标准都不符合,则指明重新估计样本大小,并有能力招收至多另外24名受试者。药效学终点符合无效标准(即,睡眠周期的前三分之一中的慢波活动未增加,而是实际上平均减少30%);因此,未招收额外受试者。所有28名入选受试者都完成研究的部分i,并且在至少2周的洗脱期后,在研究的部分ii中重新随机分配以接受单一剂量的cx-8998(4或8mg)或安慰剂。cx-8998或安慰剂是在每名受试者的正常就寝时间之前30分钟和晚饭结束后4小时服用。作为研究的部分i,在每名受试者的习惯就寝时间开始psg记录并持续8小时,并且第二天利用睡眠质量的主观测量(leeds睡眠评价问卷)和嗜睡(karolinska嗜睡量表)以及认知和运动任务对受试者进行评价,以评估次日早晨的残余影响。5.3.3.1.2药效学数据单一12mg剂量的cx-8998在夜晚的前三分之一明显降低慢波活动,其中相对于安慰剂,慢波活动降低约30%(表28)。表28:在睡眠期的前三分之一期间慢波活动(μv2/hz)的观察汇总统计学(研究pn003)ci=置信区间。治疗间p值=0.9999(无治疗差异的无假设检验,相对于cx-8998>安慰剂的单侧替代检验)。所观测到的相比于基线的倍数变化的几何平均值比率(cx-899812mg/安慰剂)为0.70,置信区间为90%(0.62,0.79),并且相应p值为0.9999(无治疗差异的无假设检验,相对于cx-8998>安慰剂的单侧替代检验)。5.3.3.1.3安全性数据5.3.3.1.3.1紧急治疗不良事件在这项研究中,通常对高达12mg的单一剂量的cx-8998耐受良好。所有报告的不良经历都是暂时性的并且通常为轻度强度。表29汇总在施用cx-8998(部分i和ii)之后在至少2名受试者中报告的不良事件。表29:在单一剂量的cx-8998后两名或更多名受试者中报告的紧急治疗不良事件(研究pn003/部分i和部分ii)meddra=药事管理的医学辞典。a表中仅包括2个或更多个暴露于cx-8998的受试者中报告的事件。bn=研究药物的暴露数目,而非受试者数目。每一栏中的受试者被视为个体观测结果。在暴露期间经历同一事件超过一次的受试者仅计数一次;然而,如果受试者在多次暴露中经历同一事件,则针对每次暴露对所述事件进行计数。视觉和/或听觉障碍或幻觉(在4或12mg的单一晚上剂量的cx-8998后报告)通常阐述为看到几何形状、颜色、光和/或移动以及听到如来自电话或电子音乐的音调。视觉障碍往往随着睁眼而停止,并随着闭眼而重现,并且通常不会干扰受试者。研究治疗是在受试者的正常就寝时间之前30分钟施用,并且视觉和/或听觉障碍或幻觉通常在投药后大约30分钟发作(通常接近tmax),并且在几分钟内或在夜间消退。未报告严重不良事件,并且无一受试者因不良事件而中断治疗。四名受试者未完成所有4个治疗期,原因如下:时段1后撤回同意(1名受试者),时段2后由于已满足入选目标而由发起人退出(1名受试者),时段3后由于已满足研究目标退出(1名受试者),和在施用12mg剂量后呕吐导致对研究药物的低暴露而退出(1名受试者)。5.3.3.1.3.2其他安全性评估未观测到安全性实验室参数或生命体征的临床显著趋势。8mg剂量下9次暴露之一(暴露的11.1%)和12mg剂量下54次暴露中的3次(暴露的5.6%)导致qtc间隔从基线增加≥30且≤60msec。未发生相比于基线≥60msec的变化,并且没有受试者的qtc间隔≥500msec。5.3.3.1.4结论对4、8和12mg的单一剂量的cx-8998耐受良好。最常见的不良事件是头痛、生动梦境以及视觉和/或听觉障碍或幻觉(通常阐述为看到几何形状、颜色、光和/或移动以及听到如来自电话或电子音乐的音调)。单一12mg剂量的cx-8998后,观测到慢波活动减少30%、延迟睡眠发作并减少睡眠维持。未满足施用cx-8998后慢波活动会增加的假设。利用单一12mg剂量的cx-8998观测到的模式与对睡眠发作和维持的有利作用不一致。5.3.4研究pn005(健康女性受试者中的单一剂量研究)5.3.4.1.1研究设计研究pn005是9名中年和老年女性受试者(年龄为55-75岁)中cx-8998的安全性、耐受性和药代动力学的随机化、双盲、安慰剂对照、单一剂量研究。受试者在过夜禁食后随机分配以接受单一口服8mg剂量的cx-8998(n=7)或安慰剂(n=2)。在整个研究中通过不良事件、临床实验室检验、生命体征、ecg和ecg遥测术监测临床安全性。5.3.4.1.2安全性数据5.3.4.1.2.1紧急治疗不良事件在这项研究中,健康中年和老年女性通常对单一8mg剂量的cx-8998耐受良好。与精神病学有关的事件(包括轻度大笑(2名受试者)、神经质(1名受试者)、思绪奔涌(1名受试者)和烦恼(1名受试者))是施用cx-8998后最常报告的不良事件。表30中汇总研究期间报告的不良事件。表30:中年和老年女性受试者中单一剂量的cx-8998后报告的紧急治疗不良事件(研究pn005)meddra=药事管理的医学辞典。a经历同一事件超过一次的受试者仅对所述事件计数一次。未报告严重不良事件。5.3.4.1.2.2其他安全性评估在生命体征、安全性实验室或ecg参数中未观测到临床上重要的趋势。一名受试者在与施用cx-8998后1天和4天时血清alt升高(从基线时的28iu/l至投药后1天的41iu/l、至投药后4天的59iu/l;参考范围为6-40iu/l),研究者评估为可能与研究药物有关。每次评价时血清ast、碱性磷酸酶和胆红素的值均在正常限值内,并且截至投药后第2周,受试者的血清alt值已恢复至正常限值内。5.3.4.1.3结论在这项研究中,中年和老年女性(年龄为55-75岁)对单一8mg剂量的cx-8998耐受良好。5.42期研究5.4.1研究pn004(患有精神分裂症的急性精神病性患者)5.4.1.1.1研究设计研究pn004是用以评价患有精神分裂症的急性精神病性患者中cx-8998的安全性和功效的2a期、4周、多中心、随机化、双盲(内部盲化)、安慰剂和活性剂对照、平行组研究。受试者完成安慰剂引入期并且然后以2:2:1比率随机分配以接受:·在第1至7天随食物一起口服cx-8998bid6mg,并且然后在第8至28天口服8mgcx-8998bid(n=86),根据研究者的判断,基于个体受试者的耐受性,在第9天或之后将剂量降低至6mgbid,·在第1至28天安慰剂bid(n=83),或·在第1至7天奥氮平5mgbid并且然后在第8至28天的早晨5mg以及在晚上10mg(n=47)。主要功效终点是第4周时正性与负性综合征量表(panss)总评分相比于基线的平均变化;次要功效终点包括第4周时panss总评分中的反应者率和第4周时临床整体印象-疾病严重程度量表相比于基线的变化。通过不良事件、实验室评价、ecg、holter监测、生命体征、simpson-angus量表、barnes静坐不能等级量表、异常不随意运动量表和哥伦比亚自杀严重程度等级量表(c-ssrs)监测安全性。5.4.1.1.2功效数据未观测到cx-8998或活性比较剂奥氮平与安慰剂之间对第4周时panss总评分相比于基线的平均变化的主要功效终点或对任何次要终点的影响在统计学上的显著差异。5.4.1.1.3安全性数据5.4.1.1.3.1紧急治疗不良事件精神病学、神经系统和与胃肠有关的不良事件是在施用cx-8998后最常报告的事件;然而,在cx-8998治疗组中仅报告失眠的频率≥5。表31中汇总研究期间至少2名受试者中报告的不良事件。表31:患有精神分裂症的两名或更多名受试者中报告的紧急治疗不良事件(研究pn004)meddra=药事管理的医学辞典。a经历同一事件超过一次的受试者仅对所述事件计数一次。86名受试者中有4名(4.7%)报告了眩晕和晕眩。对这些受试者的holter监测的审查未揭示任何数据可证实临床上显著的节律异常或心动过缓,这可能是眩晕和晕眩的原因。眩晕和晕眩在所有4名受试者中都自发消退并且不需要施用并行疗法。受试者并未因这些事件而中断用cx-8998的治疗。在cx-8998治疗的受试者中,未报告严重不良事件。cx-8998治疗的受试者中有9名(10.5%)由于不良事件而中断治疗,与之相比,安慰剂治疗的受试者中有1名(1.2%);然而,cx-8998治疗组中9名中断中的4名是由于精神分裂症恶化。以下提供由于不良事件而中断治疗的cx-8998治疗的受试者的叙述:受试者0006-00007(全身性虚弱)是cx-8998治疗组中55岁的白人男性,其在22岁时被诊断出患有精神分裂症并且先前有4次精神病住院。受试者是以前吸烟者,但无物质滥用史。在2009年12月11日服用第一剂量的cx-8998(6mg)。在2009年12月18日开始,受试者报告全身性虚弱,并且中断了用研究药物的治疗。不良事件于2009年12月21日消退。研究者评估不良事件的强度为中度并且与研究药物有关。这名受试者未报告其他不良事件。受试者0013-00002(全身性强直阵挛性癫痫发作)是cx-8998治疗组中33岁的白人男性,其在13岁时被诊断出患有精神分裂症并且先前有5次精神病住院。受试者无烟草使用或物质滥用史。在2009年6月02日服用第一剂量的研究药物(6mg)。受试者于2009年6月11日具有全身性强直阵挛性癫痫发作的不良事件,其于同日消退。中断研究药物。研究者将不良事件的强度归为重度并且与研究药物有关。受试者于2009年6月07日也具有失眠的不良事件(强度为中度并且与研究药物无关)。受试者0014-00007(右束支传导阻断)是22岁的白人男性,其在15岁时被诊断出患有精神分裂症并且先前有3次精神病住院。受试者是目前吸烟者,但无物质滥用史。在筛选访视(第-9天)时,其ecg显示“v1中的rsr”和qrs持续时间为103msec。在第-3天安慰剂导入投用之前,其ecg显示窦性心动过速(心率95bpm;qrs持续时间104msec)和室内传导缺陷(机器解释)。在2009年10月19日服用第一剂量的cx-8998(6mg)。受试者于2009年10月22日具有右束支传导阻断的不良事件,qrs持续时间为144msec。在2009年10月23日中断研究药物。2009年10月26日的ecg显示不良事件消退,qrs持续时间为88msec。研究者将不良事件的强度归为轻度并且与研究药物有关。这名受试者未报告其他不良事件。受试者0017-00005(心电图qtc间隔延长)是cx-8998治疗组中49岁的白人女性,其在40岁时被诊断出患有精神分裂症并且先前有7次精神病住院。受试者无烟草使用或物质滥用史。在筛选(第-8天)时,利用导程v5测量其qt间隔(由于导程错位),并记录以下值:qtcf间隔420msec;qtcb间隔422msec;并且心率62bpm。在投药前一天(第-1天),其ecg显示qtcf间隔为452msec,qtcb间隔为439msec,并且心率为51bpm。在2009年7月16日施用第一剂量的cx-8998(6mg)后,ecg显示qtcf间隔为458msec,qtcb间隔为441msec,并且心率为48bpm,其与第-1天的测量结果相当。这报导为qtcf延长(轻度,与研究药物有关)和窦性心动过缓(轻度,与研究药物有关)的不良事件。第4天的ecg显示qtcf间隔为454msec,qtcb间隔为448msec,并且心率为55bpm。第6天的另一ecg显示,受试者的qtc间隔延长,qtcf间隔为483msec,qtcb间隔为477msec,并且心率为56bpm。延长的qtc间隔报告为不良事件(中度,与研究药物有关),并且中断研究药物。该不良事件于2009年7月23日消退(qtcf间隔440msec;qtcb间隔432msec;并且心率54bpm)。受试者还经历了以下不良事件:在2009年7月16日qtcf延长(轻度,于2009年7月21日消退,与研究药物有关);在2009年7月16日窦性心动过缓(轻度,于2009年7月20日消退,与研究药物有关);在2009年7月22日静脉穿刺部位血肿(轻度,消退,与研究药物无关);在2009年7月22日红细胞计数减少(轻度,于2009年7月28日消退,与研究药物有关);在2009年7月22日血红素减少(轻度,于2009年7月28日消退,与研究药物有关);以及在2009年7月22日血容比减小(轻度,于2009年7月28日消退,与研究药物有关)。受试者0017-0008(精神分裂症)是cx-8998治疗组中43岁的白人女性,其在24岁时被诊断出患有精神分裂症并且先前有10次精神病住院。受试者无烟草使用或物质滥用史。在2009年9月24日服用第一剂量的研究药物(6mg)。受试者于2009年10月15日具有精神分裂症的不良事件(研究者术语:精神分裂症恶化),其在2009年10月20日消退。在2009年10月06日中断研究药物。研究者将不良事件的强度归为中度并且与研究药物有关。受试者在2009年10月01日也具有(恶化)精神分裂症的不良事件(轻度,于2009年10月04日消退,与研究药物有关)。受试者0019-00008(精神分裂症)是cx-8998治疗组中43岁的白人男性,其在26岁时被诊断出患有精神分裂症并且先前有12次精神病住院。受试者是目前吸烟者,但无物质滥用史。在2009年9月22日服用第一剂量的研究药物(6mg)。受试者于2009年9月30日具有精神分裂症的不良事件(研究者术语:精神分裂症恶化),其于2009年10月12日消退。受试者中断研究。研究者将不良事件的强度归类为中度并且与研究药物无关。受试者于2009年9月22日也具有失眠的不良事件(轻度,于2009年10月14日消退,与研究药物无关)。受试者0019-00009(精神分裂症)是cx-8998治疗组中41岁的白人女性,其在24岁时被诊断出患有精神分裂症并且先前有6次精神病住院。受试者无烟草使用或物质滥用史。在2009年10月01日服用第一剂量的研究药物(6mg)。受试者于2009年10月19日具有精神分裂症的不良事件(研究者术语:精神分裂症恶化),其于2009年10月31日消退。受试者中断研究。研究者将不良事件的强度归类为中度并且与研究药物无关。受试者于2009年9月25日也具有失眠的不良事件(轻度,于2009年10月25日消退,与研究药物无关)。受试者0020-00002(心理运动兴奋性)是cx-8998治疗组中23岁的白人男性,其在19岁时被诊断出患有精神分裂症并且先前有5次精神病住院。受试者是目前吸烟者,但无物质滥用史。于2009年10月01日服用第一剂量的研究药物(6mg)。受试者于2009年10月25日具有心理运动兴奋性的不良事件,其于2009年12月24日消退。受试者中断研究。研究者将不良事件的强度归类为中度并且与研究药物无关。这名受试者未报告其他不良事件。受试者0021-00006(精神分裂症)是cx-8998治疗组中43岁的白人男性,其在22岁时被诊断出患有精神分裂症并且先前有10次精神病住院。受试者无烟草使用或物质滥用史。在2009年10月27日服用第一剂量的研究药物(6mg)。受试者在2009年11月20日具有急性精神分裂症发作的不良事件,其在2009年12月24日消退。受试者中断研究。研究者将不良事件的强度归为轻度并且与研究药物有关。这名受试者未报告其他不良事件。5.4.1.1.3.2其他安全性评估在生命体征、安全性实验室或ecg参数中未观测到临床上重要的趋势。5.4.1.1.4结论在根据滴定投药方案(6mgbid持续7天,增加至8mgbid持续21天)以8mgbid的剂量施用时,参与这项研究的患有精神分裂症的急性精神病受试者对cx-8998耐受良好。最常报告精神病学和神经系统有关的事件,但在≥5%的cx-8998治疗受试者中仅报告失眠。研究结果未提供证据表明cx-8998提供了精神分裂症的改善,如通过第4周时的panss总评分的变化的主要终点或任何次要功效终点来测量。5.4.2进行中的研究5.4.2.1研究cx-8998-cln2-001(t-calm;患有原发性震颤的患者)研究cx-8998-cln2-001是2期、多中心、随机化、双盲、安慰剂对照、剂量滴定研究,以评估剂量至多10mgbid的cx-8998减少与原发性震颤相关的震颤的严重程度(如通过原发性震颤评定评估量表[tetras]和kinesiaone加速度测量法所评估)的功效和安全性。所述研究包括长达4周的筛选期;4周双盲、剂量滴定治疗期;最后一个剂量的研究药物后1周的随访期;以及1周后拨打随访电话。至多106名受试者以1:1比率随机分配以接受cx-8998或安慰剂。随机分配到cx-8998的受试者在第1周内接受4mgcx-8998bid(8mg/天),在第2周内接受8mgcx-8998bid(16mg/天),以及在第3周和第4周内接受10mgcx-8998bid(20mg/天);随机分配到安慰剂的受试者接受相应数量的安慰剂胶囊。安全性评估包括不良事件、实验室评估、生命体征、ecg、c-ssrs和艾普沃斯嗜睡量表。截止到本文件的日期(2018年6月27日),所述研究已在95名受试者随机化的诊疗所中完成。5.4.2.2研究cx-8998-cln2-002(t波;患有伴有失神型癫痫发作的全身性癫痫综合征的患者)研究cx-8998-cln2-002是进行中的2a期、多中心、开放标签、剂量滴定研究,以评估患有伴有失神型癫痫发作的全身性癫痫综合征的青少年和年轻成人(年龄为16-55岁)中剂量至多10mgbid的cx-8998的安全性、耐受性和功效。所述研究包括长达4周的筛选期;4周双盲、剂量滴定治疗期;以及最后一个剂量的研究药物后1周的随访期。将招收至多15名受试者。受试者将在第1天和第2天接受2mgcx-8998bid并且在第3至8天接受4mgcx-8998bid,在第9至14天接受6mgcx-8998bid,在第15至20天接受8mgcx-8998bid,在第21至26天接受10mgcx-8998bid,并且然后在第27天的早晨接受10mgcx-8998。安全性评估包括不良事件、实验室评估、生命体征和ecg。5.4.3计划的研究5.4.3.1研究cx-8998-cln2-001,开放标签数字子研究#2研究cx-8998-cln2-001、开放标签数字子研究#2是主要t-calm原发性震颤研究的组分(cx-8998-cln2-001;章节5.4.2.1)。所述研究被设计以使用如在主要t-calm原发性震颤研究中所使用的评估时间表评估数字评估工具在pdt的客观测量中的效用。将对三种数字工具进行评估:1)运动障碍协会赞助的统一帕金森病等级量表修订版(mds-uprds),2)imotor应用程序,以及3)连续可佩戴震颤监测设备(任选评估是基于设备的可用性)。所述研究的目的是在开始研究cx-8998-cln2-003(章节5.4.3.2)之前,鉴定最适当的pdt特异性数字监测生物标记。具体来说,这项研究将通过探索以下方面来启动数字药物开发工具(dddt)评价的过程:·临床测量与生物统计监测工具参数之间的相关性,·生物统计监测工具变量之间的相关性,以及·不同生物统计监测工具检测治疗作用的能力。将招募约12名患有pdt的受试者进行数字子研究。受试者将在第1周内接受4mgcx-8998bid(8mg/天),在第2周内接受8mgcx-8998bid(16mg/天),并且在第3周和第4周内接受10mgcx-8998bid(20mg/天)。5.4.3.2研究cx-8998-cln2-003(t-calm-pdt;患有帕金森病震颤的患者)研究cx-8998-cln2-003是计划的2期、多中心、随机化、双盲、安慰剂对照、剂量滴定研究,以评估至多10mgbid的剂量的cx-8998用以降低pdt的严重程度(如通过mds-uprds、tetras和kinesiaone加速度测量法所评估)的功效和安全性。所述研究包括长达4周的筛选期;4周双盲、剂量滴定治疗期;最后一个剂量的研究药物后1周的随访期;以及3周后拨打随访电话。至多60名受试者将随机分配以接受cx-8998或安慰剂。随机分配到cx-8998的受试者将在第1周内接受4mgcx-8998bid(8mg/天),在第2周内接受8mgcx-8998bid(16mg/天),以及在第3周和第4周内接受10mgcx-8998bid(20mg/天);随机分配到安慰剂的受试者将接受相应数量的安慰剂胶囊。安全性评估包括不良事件、实验室评估、生命体征、ecg、c-ssrs和艾普沃斯嗜睡量表。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1