环烷-1,3-二胺衍生物的制作方法
本发明涉及抑制menin与mll蛋白之间的相互作用的低分子化合物或其药学上可接受的盐。
背景技术:
在婴儿白血病和一些预后不良的白血病病例中观察到了mll(混合谱系白血病)基因的染色体易位。由于染色体易位,mll在其氨基末端与70个或更多个各种易位伴侣基因融合,以表达mll融合蛋白。野生型mll构成转录调节复合物,其修饰染色质结构,特别是在组蛋白h3的第4位上使赖氨酸甲基化,并且在参与造血和发育的基因簇(例如hox基因簇等)的转录调控中起着极其重要的作用。同时,通过染色体易位诱导表达的mll融合蛋白丧失了组蛋白甲基化酶活性,但永久激活了参与细胞分化控制的基因簇(例如hox和meis1基因等)。结果,引发异常的细胞生长和造血细胞分化诱导的抑制,这导致白血病的发作。具有mll基因突变的白血病的预后较差,目前用于白血病治疗的标准治疗方法还不够有效。因此,强烈期望开发新的治疗方法。
menin是一种肿瘤抑制蛋白,其被鉴定为多发性内分泌肿瘤1型(men1)的致病因素,men1是常染色体显性遗传性肿瘤综合征之一,其特征在于多个内分泌器官中发生肿瘤。menin是一种广泛表达的核蛋白,可与多种蛋白相互作用,并参与各种细胞过程。人们认为,menin的生物学功能可以是抑制肿瘤的或促进肿瘤的,并取决于细胞环境。menin与mll1的酰胺末端相互作用,并充当增加hox和meis1等基因簇的转录的致癌辅因子。已知对于由mll融合蛋白引起的一系列基因簇的异常激活和白血病的发作,menin与mll融合蛋白之间的相互作用是必不可少的(非专利文献1和2)。因此,期望menin和mll融合蛋白之间的相互作用的抑制有助于治疗和/或预防涉及mll基因的染色体易位的白血病和其他伴随hox和meis1基因的恒定表达的白血病/血液癌。因此,例如,就提供针对癌症治疗的新选择而言,产生抑制menin与mll融合蛋白之间的相互作用的药物是极其重要的。
已知有多种对menin与mll蛋白之间的相互作用具有抑制活性的化合物(专利文献1-4,非专利文献3-5)。
文献列表
专利文献
专利文献1:wo2017/161028小册子
专利文献2:wo2018/053267小册子
专利文献3:wo2018/109088小册子
专利文献4:wo2018/024602小册子
非专利文献
非专利文献1:chen等人,proc.natl.acad.sci.,2006,103,1018-1023;
非专利文献2:yokoyama等人,cell,2005,123,207-218;
非专利文献3:grembecka等人,nat.chem.biol.,2012,8,277-284;
非专利文献4:shi等人,blood,2012,120,4461-4469;
非专利文献5:borkin等人,cancercell,2015,27,589-602。
发明概述
发明要解决的问题
本发明提供了一种新型低分子化合物,其对menin和mll蛋白之间的相互作用具有抑制作用(以下有时称为menin-mll抑制作用),其可用作治疗和/或预防依赖于menin和mll蛋白之间的相互作用的疾病的药物。
解决问题的方法
本发明人为了开发menin-mll抑制剂而对新型低分子化合物进行了研究,发现本发明中公开的具有特定结构的化合物或其药学上可接受的盐具有menin-mll抑制作用,并可用作治疗和/或预防依赖于menin和mll蛋白之间的相互作用的疾病(例如,癌症或糖尿病)的药物,并且基于这些发现完成了本发明。到目前为止,本发明中公开的化合物或其药学上可接受的盐是未知的,并且它们的药理活性也是未知的。
本发明涉及以下[1]至[92]。
[1]由以下通式(1)表示的化合物或其药学上可接受的盐:
[化学式1]
其中
虚线圆圈表示环是芳族的,
r1和r2各自独立地是氢原子或c1-6烷基,
r3和r4之一是氢原子、羟基、卤素原子、c1-6烷氧基、二(c1-6烷基)氨基甲酰基或噁唑基,和
r3和r4中的另一个是氢原子、羟基、卤素原子或c1-6烷氧基,
r5是氢原子、c1-6烷基或羟基c1-6烷基,
r6是氢原子、c1-6烷基、卤素原子、c1-6烷氧基、氨基或c1-6烷基氨基,
r7和r8与r7所键合的碳原子以及r8所键合的碳原子一起形成以下式(2a)至(2c)中的任何一个:
[化学式2]
其中
虚线圆圈表示环是芳族的,
标有a的碳原子是与r8键合的碳原子,
标有b的碳原子是与r7键合的碳原子,
x是ch或氮原子,且
r9是卤代c1-6烷基、c3-8环烷基、c3-8环烷基c1-6烷基、c1-6烷氧基c1-6烷基或氧杂环丁烷基,或者
r7是氢原子,且r8是下式(3):
[化学式3]
其中
*表示键合位点,
r10是二(c1-6烷基)氨基甲酰基、(c1-6烷基)嘧啶基、(c1-6烷基)苯基或(c1-6烷基)吡唑基,
r11是氢原子或卤素原子,且
r12是卤素原子,
m是1或0,
n是1或2,
环q1是在环中任选地含有一个氮原子的6元芳环(该芳环任选地具有一个或两个独立地选自以下组a的取代基)、在环中含有一个或两个独立地选自氮原子和硫原子的杂原子的5元芳族杂环(该芳族杂环任选地具有一个独立地选自以下组a的取代基)、任选地具有一个独立地选自以下组a的取代基的c3-8环烷环、任选地具有一个独立地选自以下组a的取代基的c4-8环烯环、在环中含有一个氮原子的4至8元饱和杂环(该饱和杂环任选地具有一个独立地选自以下组a的取代基)或在环中含有一个氮原子的9元双环芳族杂环(该双环芳族杂环任选地具有一个或两个独立地选自以下组b的取代基),和
w是下式(4a)或(4b):
[化学式4]
其中
*表示键合位点,
环q2是在环中任选地含有一个氮原子的6元芳环(该芳环任选地具有1-3个独立地选自以下组c的取代基)、在环中含有两个氮原子的6元芳族杂环(该芳族杂环任选地具有1-3个独立地选自以下组c的取代基)、在环中含有1-3个独立地选自氮原子、氧原子和硫原子的杂原子的5元芳族杂环(该芳族杂环任选地具有一个独立地选自以下组c的取代基)、在环中含有1-3个独立地选自氮原子和氧原子的杂原子的9或10元双环芳族或部分不饱和杂环(该双环芳族或部分不饱和杂环任选地具有一个或两个独立地选自以下组d的取代基)、在环中含有一个或两个独立地选自氧原子和氮原子的杂原子的5至8元饱和杂环(该饱和杂环任选地具有一个独立地选自以下组e的取代基)或任选地具有一个独立地选自以下组e的取代基的c3-8环烷环,
环q3是在环中含有一个氮原子或一个氧原子的4至8元饱和杂环(该饱和杂环任选地具有一个c1-6烷基磺酰基)或在环中任选地含有一个氮原子的6元芳环(该芳环任选地具有一个独立地选自以下组f的取代基),
y是单键或氧原子,且
z是单键、氧原子、-nh-、-so2-、c1-6亚烷基、*-r13-nhc(=o)-**、*-r14-o-**或*-r15-nh-**,其中*键合至环q2,**键合至环q1,并且r13、r14和r15各自独立地是c1-6亚烷基,
组a:卤素原子、羟基、c1-6烷基、c1-6烷氧基、羟基c1-6烷氧基、乙烯基磺酰基氨基(c1-6烷基)氨基甲酰基和丙-2-烯酰基氨基(c1-6烷基)氨基甲酰基,
组b:氰基、c1-6烷基、卤素原子和c1-6烷氧基,
组c:卤素原子、c1-6烷基、c1-6烷氧基、c1-6烷基(c1-6烷基磺酰基)氨基、氰基、c1-6烷基磺酰基、c1-6烷基氨基、二(c1-6烷基)氨基、卤代c1-6烷基、c1-6烷氧基c1-6烷氧基、卤代c1-6烷氧基、c1-6烷基磺酰基c1-6烷基、二(c1-6烷基)氨磺酰基、c1-6亚烷基二氧基、(c1-6烷基)氨基甲酰基、羟基c1-6烷基、2-c3-6烯酰基氨基、c1-6烷基(2-c3-6烯酰基)氨基、羟基、氧代基团、(2h3)甲氧基和双[(2h3)甲基]氨基,
组d:卤素原子、c1-6烷基和c1-6烷基磺酰基,
组e:氧代基团、羟基和c1-6烷氧基,和
组f:卤素原子和c1-6烷氧基。
[2]根据[1]的化合物或其药学上可接受的盐,其中r1为氢原子或甲基。
[3]根据[1]的化合物或其药学上可接受的盐,其中r1为氢原子。
[4]根据[1]至[3]中任一项的化合物或其药学上可接受的盐,其中r2为氢原子或甲基。
[5]根据[1]至[3]中任一项的化合物或其药学上可接受的盐,其中r2为氢原子。
[6]根据[1]至[5]中任一项的化合物或其药学上可接受的盐,其中式(1)中由下式(5)表示的部分为下式(5a)或(5b):
[化学式5]
[化学式6]
其中
*键合至与r2键合的氮原子,
**键合至与r5键合的氮原子,
r16为氢原子、卤素原子、羟基、二(c1-6烷基)氨基甲酰基、噁唑-2-基或c1-6烷氧基,
r17为氢原子或卤素原子,且
r18为c1-6烷氧基。
[7]根据[1]至[5]中任一项的化合物或其药学上可接受的盐,其中式(1)中由下式(5)表示的部分为下式(6a)或(6b):
[化学式7]
[化学式8]
其中
*键合至与r2键合的氮原子,
**键合至与r5键合的氮原子,且
r19为氢原子、羟基、二甲基氨基甲酰基、噁唑-2-基或甲氧基。
[8][1]至[5]中任一项的化合物或其药学上可接受的盐,其中式(1)中由下式(5)表示的部分为下式(7a):
[化学式9]
[化学式10]
其中
*键合至与r2键合的氮原子,
**键合至与r5键合的氮原子,
r20为氢原子或羟基,且
r21为氢原子、羟基或c1-6烷氧基。
[9]根据[1]至[5]中任一项的化合物或其药学上可接受的盐,其中式(1)中由下式(5)表示的部分是下式(8a)至(8e)中的任何一个:
[化学式11]
[化学式12]
其中
*键合至与r2键合的氮原子,
**键合至与r5键合的氮原子,
r22为氢原子、羟基或甲氧基,
r23为羟基或甲氧基,且
r24为氢原子或羟基。
[10]根据[1]至[5]中任一项的化合物或其药学上可接受的盐,其中式(1)中由下式(5)表示的部分是下式(9a)至(9c)中的任何一个:
[化学式13]
[化学式14]
其中
*键合至与r2键合的氮原子,且
**键合至与r5键合的氮原子。
[11]根据[1]至[10]中任一项的化合物或其药学上可接受的盐,其中r5为氢原子、甲基、乙基或2-羟乙基。
[12]根据[1]至[10]中任一项的化合物或其药学上可接受的盐,其中r5为甲基。
[13]根据[1]至[12]中任一项的化合物或其药学上可接受的盐,其中r6为氢原子、甲基、氯原子、甲氧基、氨基或甲基氨基。
[14]根据[1]至[13]中任一项的化合物或其药学上可接受的盐,其中r7和r8与r7所键合的碳原子以及r8所键合的碳原子一起形成下式(10a):
[化学式15]
其中
虚线圆圈表示环是芳族的,
标有a的碳原子是与r8键合的碳原子,且
标有b的碳原子是与r7键合的碳原子。
[15][1]至[13]中任一项的化合物或其药学上可接受的盐,其中r7为氢原子,且r8为下式(11a)或(11b):
[化学式16]
其中
*表示键合位点,
r25为二异丙基氨基甲酰基、4-异丙基嘧啶-5-基、2-异丙基苯基或1-异丙基吡唑-5-基,且
r26为二异丙基氨基甲酰基。
[16]根据[1]至[15]中任一项的化合物或其药学上可接受的盐,其中环q1为以下(i)至(vii)中的任何一个:
(i)任选地具有一个或两个独立地选自上述组a的取代基的苯环;
(ii)任选地具有一个或两个独立地选自上述组a的取代基的吡啶环;
(iii)1,3-噻唑环或吡唑环(该1,3-噻唑环或吡唑环任选地具有一个独立地选自上述组a的取代基);
(iv)任选地具有一个独立地选自上述组a的取代基的环己烷环;
(v)任选地具有一个独立地选自上述组a的取代基的环己烯环;
(vi)任选地具有一个独立地选自上述组a的取代基的哌啶环;或者
(vii)任选地具有一个或两个独立地选自上述组b的取代基的吲哚环。
[17]根据[1]至[15]中任一项的化合物或其药学上可接受的盐,其中
m为1,且
环q1为下式(12a)至(12h)中的任何一个:
[化学式17]
其中
*键合至z,
**键合至与r1键合的碳原子,
r27为氢原子、卤素原子、c1-6烷氧基或c1-6烷基,
j为氮原子或cr29,
r29为卤素原子,且
r28为氢原子或c1-6烷基。
[18]根据[1]至[15]中任一项的化合物或其药学上可接受的盐,其中
m为1,且
环q1为下式(13a)或(13b):
[化学式18]
其中
*键合至z,
**键合至与r1键合的碳原子,且
r30为氢原子、氟原子、甲基或甲氧基。
[19]根据[1]至[18]中任一项的化合物或其药学上可接受的盐,其中环q2为以下(i)至(vii)中的任何一个:
(i)任选地具有1-3个独立地选自上述组c的取代基的苯环;
(ii)任选地具有1-3个独立地选自上述组c的取代基的吡啶环;
(iii)哒嗪环、吡嗪环或嘧啶环(该哒嗪环、吡嗪环或嘧啶环任选地具有1-3个独立地选自上述组c的取代基);
(iv)吡唑环、咪唑环、1,3-噻唑环、1,3-噁唑环或4h-1,2,4-三唑环(该吡唑环、咪唑环、1,3-噻唑环、1,3-噁唑环或4h-1,2,4-三唑环任选地具有一个独立地选自上述组c的取代基);
(v)异喹啉环、吲唑环、苯并咪唑环、1h-吡咯并[2,3-c]吡啶环、1h-吡咯并[3,2-c]吡啶环、呋喃并[3,2-b]吡啶环、1h-吡唑并[3,4-c]吡啶环或二氢吲哚环(该异喹啉环、吲唑环、苯并咪唑环、1h-吡咯并[2,3-c]吡啶环、1h-吡咯并[3,2-c]吡啶环、呋喃并[3,2-b]吡啶环、1h-吡唑并[3,4-c]吡啶环或二氢吲哚环任选地具有一个或两个独立地选自上述组d的取代基);
(vi)吡咯烷环、哌啶环、吗啉环或氮杂环庚烷环(该吡咯烷环、哌啶环、吗啉环或氮杂环庚烷环任选地具有一个独立地选自上述组e的取代基);或者
(vii)任选地具有一个独立地选自上述组e的取代基的环己烷环。
[20]根据[1]至[18]中任一项的化合物或其药学上可接受的盐,其中
w为上式(4a);和
环q2为下式(14a)至(14f)中的任何一个:
[化学式19]
其中
*表示键合位点,
t为ch或氮原子,
r31为氢原子、c1-6烷氧基、卤代c1-6烷氧基或(2h3)甲氧基,
r32为氢原子、c1-6烷基、卤素原子、c1-6烷氧基、氰基、二(c1-6烷基)氨基、卤代c1-6烷基、c1-6烷基氨基、c1-6烷基磺酰基、c1-6烷氧基c1-6烷氧基、卤代c1-6烷氧基、羟基c1-6烷基、c1-6烷基(2-c3-6烯酰基)氨基、(2h3)甲氧基或双[(2h3)甲基]氨基,或
r31和r32一起形成亚乙基二氧基,
r33和r35各自独立地为氢原子、卤素原子、c1-6烷氧基、c1-6烷基(c1-6烷基磺酰基)氨基、(c1-6烷基)氨基甲酰基、二(c1-6烷基)氨磺酰基、2-c3-6烯酰基氨基或c1-6烷基磺酰基c1-6烷基,
r34为氢原子或卤素原子,
r36为卤素原子,
r37为c1-6烷氧基,
r38为卤素原子,
r39为c1-6烷基或c1-6烷基磺酰基,
r40为c1-6烷基或c1-6烷基磺酰基,
u1为ch或氮原子,
u2为cr41或氮原子,且
r41为氢原子或卤素原子。
[21]根据[1]至[18]中任一项的化合物或其药学上可接受的盐,其中
w为上式(4a);和
环q2为下式(15a)至(15c)中的任何一个:
[化学式20]
其中
*表示键合位点,
r42为甲基、氯原子、甲氧基、氰基、二甲基氨基或双[(2h3)甲基]氨基,
r43为甲氧基或(2h3)甲氧基,且
r44为氯原子、甲氧基、甲氧基乙氧基、二甲基氨基、二氟甲氧基或(2h3)甲氧基。
[22]根据[1]至[18]中任一项的化合物或其药学上可接受的盐,其中
w为上式(4a);和
环q2为下式(16a)至(16g)中的任何一个:
[化学式21]
其中*表示键合位点。
[23]根据[1]至[18]中任一项的化合物或其药学上可接受的盐,其中
w为上式(4b);和
环q2为下式(17a)或(17b):
[化学式22]
其中
*键合至y,且
**键合至z。
[24]根据[1]至[19]和[23]中任一项的化合物或其药学上可接受的盐,其中
w为上式(4b);和
环q3为下式(18a)至(18d)中的任何一个:
[化学式23]
其中
*表示键合位点,
r45为氢原子或卤素原子,
r46为c1-6烷基磺酰基,且
v为氮原子或ch。
[25]根据[1]至[19]和[23]中任一项的化合物或其药学上可接受的盐,其中
w为上式(4b);和
环q3为苯基、氮杂环丁烷-1-基、3-吡啶基、6-氯-3-吡啶基、四氢吡喃-3-基或1-甲基磺酰基-4-哌啶基。
[26][1]至[19]和[23]至[25]中任一项的化合物或其药学上可接受的盐,其中
w为上式(4b);和
y为单键或氧原子。
[27]根据[1]至[26]中任一项的化合物或其药学上可接受的盐,其中z为单键、-nh-、氧原子、-so2-、-ch2-、*-ch2-nhc(=o)-**、*-ch2ch2-o-**或*-ch2-nh-**,其中*键合至环q2,并且**键合至环q1。
[28]根据[1]至[26]中任一项的化合物或其药学上可接受的盐,其中z为单键。
[29]根据[1]的化合物或其药学上可接受的盐,其中
r1为氢原子;
r2为氢原子;
下式(5)表示的部分是下式(9a)至(9c)中的任何一个:
[化学式24]
[化学式25]
其中
*键合至与r2键合的氮原子,且
**键合至与r5键合的氮原子;
r5为甲基;
r6为氢原子、甲基、氯原子、甲氧基、氨基或甲基氨基;
r7和r8与r7所键合的碳原子以及r8所键合的碳原子一起形成下式(10a):
[化学式26]
其中
虚线圆圈表示环是芳族的,
标有a的碳原子是与r8键合的碳原子,且
标有b的碳原子是与r7键合的碳原子,或
r7为氢原子,且r8为下式(11a)或(11b):
[化学式27]
其中
*表示键合位点,
r25为二异丙基氨基甲酰基、4-异丙基嘧啶-5-基、2-异丙基苯基或1-异丙基吡唑-5-基,且
r26为二异丙基氨基甲酰基;
m为1;
环q1为下式(13a)或(13b):
[化学式28]
其中
*键合至z,
**键合至与r1键合的碳原子,且
r30为氢原子、氟原子、甲基或甲氧基;
w为上式(4a),且
环q2为下式(15a)至(15c)中的任何一个:
[化学式29]
其中
*表示键合位点,
r42为甲基、氯原子、甲氧基、氰基、二甲基氨基或双[(2h3)甲基]氨基,
r43为甲氧基或(2h3)甲氧基,且
r44为氯原子、甲氧基、甲氧基乙氧基、二甲基氨基、二氟甲氧基或(2h3)甲氧基,或
w为上式(4b),
环q2为下式(17a)或(17b):
[化学式30]
其中
*键合至y,且
**键合至z,
环q3为苯基、氮杂环丁烷-1-基、3-吡啶基、6-氯-3-吡啶基、四氢吡喃-3-基或1-甲基磺酰基-4-哌啶基,和
y为单键或氧原子;和
z为单键。
[30]根据[1]的化合物或其药学上可接受的盐,其中
r1为氢原子;
r2为氢原子;
下式(5)表示的部分是下式(9a)至(9c)中的任何一个:
[化学式31]
[化学式32]
其中
*键合至与r2键合的氮原子,且
**键合至与r5键合的氮原子;
r5为甲基;
r6为氢原子、甲基、氯原子、甲氧基、氨基或甲基氨基;
r7和r8与r7所键合的碳原子以及r8所键合的碳原子一起形成下式(10a):
[化学式33]
其中
虚线圆圈表示环是芳族的,
标有a的碳原子是与r8键合的碳原子,且
标有b的碳原子是与r7键合的碳原子;
m为1;
环q1为下式(13a)或(13b):
[化学式34]
其中
*键合至z,
**键合至与r1键合的碳原子,且
r30为氢原子、氟原子、甲基或甲氧基;
w为上式(4a);和
环q2为下式(16a)至(16g)中的任何一个:
[化学式35]
其中*表示键合位点;和
z为单键。
[31]选自下组的任何化合物或其药学上可接受的盐:
5-[4-({[(1r,3r,4s)-3-羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]-3-甲氧基吡啶-2-甲腈,
(1r,2s,4r)-4-[({4-[1-(甲磺酰基)-1h-吲唑-4-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-[({4-[6-(二甲基氨基)-5-甲氧基吡啶-3-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5-甲氧基-6-甲基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(1h-咪唑-1-基)苯基]甲基}氨基)-2-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(6-氯-5-甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(6-氟-5-甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(6-氯-5-甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
2-[(4-{[(1s,2r,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-羟基环戊基](甲基)氨基}嘧啶-5-基)氧基]-5-氟-n,n-二(丙-2-基)苯甲酰胺,
(1r,2s,4r)-2-{[2-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)环戊-1-醇,
(1r,3s)-n3-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺,
(1r,2s,4r)-4-[({4-[6-(二甲基氨基)-5-甲氧基哒嗪-3-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
6-[4-({[(1r,3r,4s)-3-羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]-4-甲氧基哒嗪-3-甲腈,
(1s,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-[({4-[5-甲氧基-6-(2-甲氧基乙氧基)哒嗪-3-基]苯基}甲基)氨基]-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(4,5-二甲氧基吡啶-2-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-[({4-[6-(二甲基氨基)-5-甲氧基吡啶-3-基]苯基}甲基)氨基]-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-[({4-[6-(二氟甲氧基)-5-甲氧基哒嗪-3-基]苯基}甲基)氨基]-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-{[(4-{5,6-双[(2h3)甲基氧基]哒嗪-3-基}苯基)甲基]氨基}-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(6-{双[(2h3)甲基]氨基}-5-甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,和
(1r,2s,4r)-4-{[(4-{5,6-双[(2h3)甲基氧基]哒嗪-3-基}苯基)甲基]氨基}-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇。
[32]menin与一种或多种选自mll1、mll2、mll融合蛋白和mll部分串联重复蛋白的蛋白之间的相互作用的抑制剂,其包含作为活性成分的根据[1]至[31]中任一项的化合物或其药学上可接受的盐。
[33]药物组合物,其包含根据[1]至[31]中任一项的化合物或其药学上可接受的盐和药学上可接受的载体。
[34]根据[33]的药物组合物,其用于治疗和/或预防可通过抑制menin与一种或多种选自mll1、mll2、mll融合蛋白和mll部分串联重复蛋白的蛋白之间的相互作用而治疗和/或预防的疾病。
[35]根据[33]的药物组合物,其用于治疗和/或预防糖尿病。
[36]根据[33]的药物组合物,其用于治疗和/或预防癌症。
[37]根据[36]的药物组合物,其中所述癌症是血液癌、前列腺癌、乳腺癌、肝细胞瘤或小儿神经胶质瘤。
[38]根据[36]的药物组合物,其中所述癌症是血液癌。
[39]根据[38]的药物组合物,其中所述血液癌是急性骨髓性白血病(aml)或急性淋巴细胞性白血病(all)。
[40]一种治疗和/或预防糖尿病的方法,其包括施用根据[1]至[31]中任一项的化合物或其药学上可接受的盐。
[41]一种治疗和/或预防癌症的方法,其包括施用根据[1]至[31]中任一项的化合物或其药学上可接受的盐。
[42]根据[1]至[31]中任一项的化合物或其药学上可接受的盐,其用于治疗和/或预防癌症。
[43]根据[1]至[31]中任一项的化合物或其药学上可接受的盐用于制备用于治疗和/或预防癌症的药物的用途。
[44]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇或其药学上可接受的盐。
[45]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇琥珀酸盐。
[46]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇苯磺酸盐。
[47]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇马来酸盐。
[48]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇富马酸盐。
[49]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇或其药学上可接受的盐。
[50]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇富马酸盐。
[51]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇粘酸盐。
[52]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇己二酸盐。
[53]根据[1]的化合物,其为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇琥珀酸盐。
[54]根据[1]的化合物的晶体,其中所述化合物为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇琥珀酸盐,其在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在选自4.66±0.2、7.02±0.2、14.10±0.2、16.68±0.2、17.46±0.2、18.68±0.2、21.34±0.2、24.52±0.2、25.54±0.2和28.22±0.2的衍射角(2θ)处具有至少5个峰。
[55]根据[1]的化合物的晶体,其中所述化合物为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基]环戊-1-醇苯磺酸盐,其在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在选自10.92±0.2、11.70±0.2、12.40±0.2、15.00±0.2、17.38±0.2、18.16±0.2、22.18±0.2、22.62±0.2、23.86±0.2和24.20±0.2的衍射角(2θ)处具有至少5个峰。
[56]根据[1]的化合物的晶体,其中所述化合物为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基]环戊-1-醇马来酸盐,其在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在选自4.64±0.2、7.02±0.2、7.46±0.2、11.14±0.2、14.04±0.2、16.76±0.2、18.54±0.2、19.76±0.2、21.26±0.2和22.62±0.2的衍射角(2θ)处具有至少5个峰。
[57]根据[1]的化合物的晶体,其中所述化合物为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇富马酸盐,其在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在选自4.80±0.2、7.94±0.2、9.66±0.2、11.56±0.2、14.56±0.2、17.62±0.2、18.14±0.2、20.46±0.2、21.36±0.2和24.46±0.2的衍射角(2θ)处具有至少5个峰。
[58]根据[1]的化合物的晶体,其中所述化合物为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,其在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在选自7.14±0.2、8.76±0.2、12.26±0.2、14.30±0.2、17.52±0.2、23.40±0.2、24.40±0.2、24.86±0.2、25.34±0.2和25.90±0.2的衍射角(2θ)处具有至少5个峰。
[59]根据[1]的化合物的晶体,其中所述化合物为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇富马酸盐,其在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在选自8.06±0.2、12.22±0.2、12.52±0.2、15.14±0.2、17.54±0.2、18.56±0.2、20.08±0.2、23.48±0.2、24.28±0.2和25.00±0.2的衍射角(2θ)处具有至少5个峰。
[60]根据[1]的化合物的晶体,其中所述化合物为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇粘酸盐,其在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在选自6.56±0.2、9.44±0.2、9.94±0.2、13.20±0.2、18.22±0.2、18.86±0.2、19.60±0.2、22.68±0.2、25.10±0.2和28.70±0.2的衍射角(2θ)处具有至少5个峰。
[61]根据[1]的化合物的晶体,其中所述化合物为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇己二酸盐,其在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在选自5.88±0.2、6.20±0.2、9.18±0.2、10.34±0.2、12.50±0.2、13.70±0.2、15.66±0.2、17.82±0.2、18.48±0.2和22.16±0.2的衍射角(2θ)处具有至少5个峰。
[62]根据[1]的化合物的晶体,其中所述化合物为(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇琥珀酸盐,其在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在选自4.60±0.2、6.60±0.2、7.74±0.2、8.02±0.2、9.26±0.2、11.16±0.2、12.00±0.2、12.44±0.2、13.22±0.2和19.66±0.2的衍射角(2θ)处具有至少5个峰。
[63]menin与一种或多种选自mll1、mll2、mll融合蛋白和mll部分串联重复蛋白的蛋白之间的相互作用的抑制剂,其包含作为活性成分的根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体。
[64]药物组合物,其包含根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体和药学上可接受的载体。
[65]根据[64]的药物组合物,其用于治疗和/或预防可通过抑制menin与一种或多种选自mll1、mll2、mll融合蛋白和mll部分串联重复蛋白的蛋白之间的相互作用而治疗和/或预防的疾病。
[66]根据[64]的药物组合物,其用于治疗和/或预防糖尿病。
[67]根据[64]的药物组合物,其用于治疗癌症。
[68]根据[67]的药物组合物,其中所述癌症是血液癌、前列腺癌、乳腺癌、肝细胞瘤或小儿神经胶质瘤。
[69]根据[67]的药物组合物,其中所述癌症是血液癌。
[70]根据[69]的药物组合物,其中所述血液癌是急性骨髓性白血病(aml)或急性淋巴细胞性白血病(all)。
[71]治疗和/或预防糖尿病的方法,其包括施用根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体。
[72]治疗癌症的方法,其包括施用根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体。
[73]根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐,或根据[54]至[62]中任一项的晶体,其用于治疗癌症。
[74]根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体用于制备用于治疗癌症的药物的用途。
[75]根据[69]的药物组合物,其中所述血液癌是具有npm1突变的急性骨髓性白血病(aml)。
[76]药物组合物,其包含选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物,以及根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体,所述药物和所述化合物或其药学上可接受的盐或晶体组合施用。
[77]根据[76]的药物组合物,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物和根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体分别作为活性成分包含在不同制剂中并在相同时间或不同时间施用。
[78]根据[76]的药物组合物,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物和根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体包含在单一制剂中。
[79]根据[76]至[78]中任一项的药物组合物,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的药物是维奈托克。
[80]根据[76]至[78]中任一项的药物组合物,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物是阿扎胞苷。
[81]根据[76]至[78]中任一项的药物组合物,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物是阿糖胞苷。
[82]根据[72]的方法,其中所述根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体与选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物组合施用。
[83]根据[82]的方法,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物和根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体分别作为活性成分包含在不同的制剂中并在相同时间或不同时间施用。
[84]根据[82]的方法,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物和根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体包含在单一制剂中。
[85]根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体,其与选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物组合施用。
[86]根据[85]的化合物或其药学上可接受的盐或晶体,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物和根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体分别作为活性成分包含在不同制剂中并在相同时间或不同时间施用。
[87]根据[85]的化合物或其药学上可接受的盐或晶体,其中选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物和根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体包含在单一制剂中。
[88]根据[85]至[87]中任一项的化合物或其药学上可接受的盐或晶体,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物为维奈托克。
[89]根据[85]至[87]中任一项的化合物或其药学上可接受的盐或晶体,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物为阿扎胞苷。
[90]根据[85]至[87]中任一项的化合物或其药学上可接受的盐或晶体,其中所述选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物为阿糖胞苷。
[91]诱导白血病细胞分化的组合物,其包含根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体。
[92]诱导白血病细胞分化的方法,其包括施用根据[1]至[31]和[44]至[53]中任一项的化合物或其药学上可接受的盐或根据[54]至[62]中任一项的晶体。
发明效果
本发明的化合物或其药学上可接受的盐对menin和mll蛋白之间的相互作用表现出抑制作用。具体而言,可以将包含本发明的化合物或其药学上可接受的盐和药学上可接受的载体的药物组合物施用于哺乳动物(人、牛、马、猪等)或鸟类(鸡等)以治疗和/或预防依赖于menin和mll蛋白之间的相互作用的疾病。依赖于menin和mll蛋白之间的相互作用的疾病的实例包括癌症和糖尿病。癌症的实例包括血液癌、骨髓增生异常综合征、前列腺癌、乳腺癌、肝细胞瘤和小儿神经胶质瘤,优选血液癌,更优选急性骨髓性白血病(aml)和急性淋巴细胞性白血病(all)。
附图简述
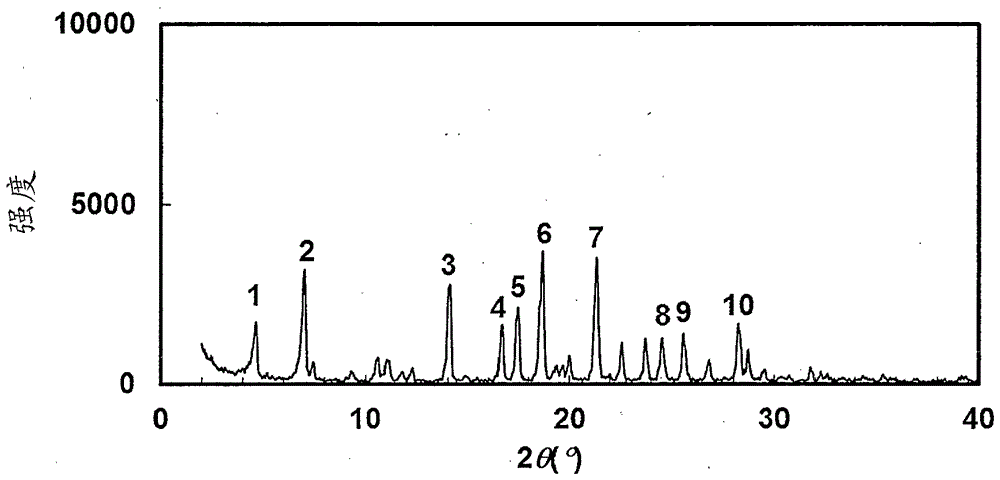
图1是实施例131中获得的晶体的粉末x射线衍射图。纵轴表示以计数/秒(cps)为单位的衍射强度(强度),横轴表示衍射角2θ的值。
图2是实施例132中获得的晶体的粉末x射线衍射图。纵轴表示以计数/秒(cps)为单位的衍射强度(强度),横轴表示衍射角2θ的值。
图3是实施例133中获得的晶体的粉末x射线衍射图。纵轴表示以计数/秒(cps)为单位的衍射强度(强度),横轴表示衍射角2θ的值。
图4是实施例134中获得的晶体的粉末x射线衍射图。纵轴表示以计数/秒(cps)为单位的衍射强度(强度),横轴表示衍射角2θ的值。
图5是实施例135中获得的晶体的粉末x射线衍射图。纵轴表示以计数/秒(cps)为单位的衍射强度(强度),横轴表示衍射角2θ的值。
图6是实施例136中获得的晶体的粉末x射线衍射图。纵轴表示以计数/秒(cps)为单位的衍射强度(强度),横轴表示衍射角2θ的值。
图7是实施例137中获得的晶体的粉末x射线衍射图。纵轴表示以计数/秒(cps)为单位的衍射强度(强度),横轴表示衍射角2θ的值。
图8是实施例138中获得的晶体的粉末x射线衍射图。纵轴表示以计数/秒(cps)为单位的衍射强度(强度),横轴表示衍射角2θ的值。
图9是实施例139中获得的晶体的粉末x射线衍射图。纵轴表示以计数/秒(cps)为单位的衍射强度(强度),横轴表示衍射角2θ的值。
图10是显示用实施例25、27、26或22的化合物处理7天后,活细胞中表达髓样细胞分化抗原gr-1的细胞的比率的图。纵轴表示活细胞中表达髓样细胞分化抗原gr-1的细胞的百分比,横轴表示各化合物及其浓度(nm)。
图11是显示用实施例25、27、26或22的化合物处理7天后活细胞中表达ckit的细胞的比率的图。纵轴表示活细胞中表达ckit的细胞的百分比,横轴表示各化合物及其浓度(nm)。
图12是显示实施例25的化合物和5aza的组合作用对人aml细胞系molm-13细胞的体外生长的作用的图。纵轴表示细胞生长(%),横轴表示实施例25的化合物的浓度(nm)。符号黑色圆圈表示仅实施例25的化合物,符号黑色三角形表示实施例25的化合物+5aza(2.5μm),符号黑色正方形表示实施例25的化合物+5aza(5μm),以及符号x表示实施例25的化合物+5aza(10μm)。误差条指示sd。
图13是显示实施例25的化合物和arac的组合作用对人aml细胞系molm-13细胞的体外生长的作用的图。纵轴表示细胞生长(%),横轴表示实施例25的化合物的浓度(nm)。符号黑色圆圈表示仅实施例25的化合物,符号黑色三角形表示实施例25的化合物+arac(25nm),符号黑色正方形表示实施例25的化合物+arac(50nm),以及符号x表示实施例25的化合物+arac(100nm)。误差条指示sd。
图14是显示实施例25的化合物和维奈托克的组合作用对人aml细胞系molm-13细胞的体外生长的作用的图。纵轴表示细胞生长,横轴表示实施例25的化合物的浓度(nm)。符号黑色圆圈表示仅实施例25的化合物,符号黑色三角形表示实施例25的化合物+维奈托克(39nm),符号黑色正方形表示实施例25的化合物+维奈托克(78nm),以及符号x表示实施例25的化合物+维奈托克(156nm)。误差条指示sd。
具体实施方式
除非另有定义,否则本文使用的所有技术和科学术语都具有与本发明所属领域的普通技术人员通常理解的相同的含义。
在本发明中,“卤素原子”是指氟原子、氯原子、溴原子或碘原子。
在本发明中,“c1-6烷基”是指具有1-6个碳原子的直链或支链烷基。其实例包括甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、异戊基、2-甲基丁基、新戊基、1-乙基丙基、正己基、4-甲基戊基、3-甲基戊基、2-甲基戊基、1-甲基戊基、3,3-二甲基丁基、2,2-二甲基丁基、1,1-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基,2,3-二甲基丁基,2-乙基丁基等。
在本发明中,“c1-6烷氧基”是指其中上述“c1-6烷基”键合至氧原子的基团。其实例包括甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、正戊氧基、异戊氧基、2-甲基丁氧基、正己氧基等。
在本发明中,(c1-6烷基)氨基甲酰基是指其中氨基甲酰基的一个氢原子被上述“c1-6烷基”替代的基团。其实例包括甲基氨基甲酰基、乙基氨基甲酰基、丙基氨基甲酰基、异丙基氨基甲酰基、仲丁基氨基甲酰基、1-乙基丙基氨基甲酰基等。
在本发明中,“二(c1-6烷基)氨基甲酰基”是指其中氨基甲酰基的两个氢原子被相同或不同的两个上述“c1-6烷基”替代的基团。其实例包括二甲基氨基甲酰基、乙基(甲基)氨基甲酰基、甲基(丙基)氨基甲酰基、二乙基氨基甲酰基、二丙基氨基甲酰基、二异丙基氨基甲酰基、仲丁基(戊基)氨基甲酰基等。
在本发明中,“羟基c1-6烷基”是指其中上述“c1-6烷基”中的一个氢原子被羟基替代的基团。其实例包括羟甲基、1-羟基乙基、2-羟基乙基、1-羟基丙基、2-羟基丙基、1-羟基异丙基、1-羟基丁基、2-羟基丁基、1-羟基戊基、2-羟基戊基、1-羟基己基等。
在本发明中,“羟基c1-6烷氧基”是指其中上述“c1-6烷氧基”的一个氢原子被羟基替代的基团。其实例包括羟基甲氧基、2-羟基乙氧基、2-羟基丙氧基、3-羟基丙氧基、2-羟基-1-甲基-乙氧基、3-羟基丁氧基、2-羟基丁氧基、2-羟基戊氧基、5-羟基戊氧基、4-羟基己氧基等。
在本发明中,“c1-6烷基氨基”是指其中氨基的一个氢原子被上述“c1-6烷基”替代的基团。其实例包括甲基氨基、乙基氨基、正丙基氨基、异丙基氨基、正丁基氨基、仲丁基氨基、叔丁基氨基、正戊基氨基等。
在本发明中,“二(c1-6烷基)氨基”是指其中氨基的两个氢原子被相同或不同的两个上述“c1-6烷基”替代的基团。其实例包括二甲基氨基、甲基(乙基)氨基、甲基(丙基)氨基[例如,n-甲基-n-(1-丙基)氨基等]、甲基(丁基)氨基[例如,n-(1-丁基)-n-甲基氨基等]、甲基(戊基)氨基、甲基(己基)氨基、二乙基氨基、乙基(丙基)氨基[例如,[n-乙基-n-(1-丙基)氨基等]、乙基(丁基)氨基、二丙基氨基、丙基(丁基)氨基、二丁基氨基、二戊基氨基、二己基氨基等。
在本发明中,“卤代c1-6烷基”是指其中上述“c1-6烷基”的1-3个氢原子被上述“卤素原子”替代的基团。其实例包括氟甲基、二氟甲基、三氟甲基、氯甲基、二氯甲基、三氯甲基、1-氟乙基、1-氯乙基、2-氟乙基、1,2-二氟丙基、2,2,2-三氟乙基等。
在本发明中,“c3-8环烷基”是指3至8元单环饱和烃基(环)。其实例包括环丙基、环丁基、环戊基、环己基、环庚基和环辛基。
在本发明中,“c3-8环烷基c1-6烷基”是指其中上述“c1-6烷基”的一个氢原子被上述“c3-8环烷基”替代的基团。其实例包括环丙基甲基、2-环丁基乙基、3-环戊基丁基、3-环庚基-2-甲基-丁基等。
在本发明中,“c1-6烷氧基c1-6烷基”是指其中上述“c1-6烷基”的一个氢原子被上述“c1-6烷氧基”替代的基团。其实例包括甲氧基甲基、乙氧基甲基、正丙氧基甲基、异丙氧基甲基、2-甲氧基乙基、2-乙氧基乙基、1-丙氧基乙基、1-异丙氧基乙基等。
在本发明中,“氧杂环丁烷基”是指氧杂环丁烷-3-基或氧杂环丁烷-2-基。
在本发明中,“(c1-6烷基)嘧啶基”是指其中嘧啶基的一个氢原子被上述“c1-6烷基”替代的基团。其实例包括4-异丙基嘧啶-5-基、5-甲基嘧啶-2-基、5-仲丁基嘧啶-4-基、4-戊基嘧啶-5-基等。
在本发明中,“(c1-6烷基)苯基”是指其中苯基的一个氢原子被上述“c1-6烷基”替代的基团。其实例包括3-甲苯基、2-乙基苯基、2-异丙基苯基、4-(2,3-二甲基丁基)苯基等。
在本发明中,“(c1-6烷基)吡唑基”是指其中吡唑基的一个氢原子被上述“c1-6烷基”替代的基团。其实例包括3-甲基-1h-吡唑-4-基、2-异丙基吡唑-3-基、4-异戊基-1h-吡唑-5-基、3-己基吡唑-1-基等。
在本发明中,“c1-6烷基磺酰基”是指其中上述“c1-6烷基”与磺酰基的硫原子键合的基团。其实例包括甲基磺酰基、乙基磺酰基、正丙基磺酰基、异丙基磺酰基、正丁基磺酰基、仲丁基磺酰基、叔丁基磺酰基、正戊磺酰基等。
在本发明中,“c1-6亚烷基”是指具有1至6个碳原子的直链或支链亚烷基。其实例包括亚甲基、亚乙基[-(ch2)2-]、三亚甲基[-(ch2)3-]、四亚甲基、五亚甲基、六亚甲基、甲基亚甲基[-ch(ch3)-]、甲基亚乙基[-ch(ch3)ch2-或-ch2ch(ch3)-]、乙基亚乙基[-ch(ch2ch3)ch2-或-ch2ch(ch2ch3)-]、1,2-二甲基亚乙基[-ch(ch3)ch(ch3)-]、1,1,2,2-四甲基亚乙基[-c(ch3)2c(ch3)2-]等。
在本发明中,“乙烯基磺酰基氨基(c1-6烷基)氨基甲酰基”是指其中氨基甲酰基的一个氢原子被以下“乙烯基磺酰基氨基(c1-6烷基)”替代的基团。其实例包括(乙烯基磺酰基氨基)甲基氨基甲酰基、2-(乙烯基磺酰基氨基)乙基氨基甲酰基、3-(乙烯基磺酰基氨基)丙基氨基甲酰基、2-[(乙烯基磺酰基氨基)甲基]丁基氨基甲酰基等。
在本发明中,“乙烯基磺酰基氨基(c1-6烷基)”是指其中上述“c1-6烷基”的一个氢原子被以下“乙烯基磺酰基氨基”替代的基团。其实例包括(乙烯基磺酰基氨基)甲基、1-(乙烯基磺酰基氨基)乙基、3-(乙烯基磺酰基氨基)丙基、3-甲基-4-(乙烯基磺酰基氨基)丁基等。
在本发明中,“乙烯基磺酰基氨基”是指其中氨基的一个氢原子被以下“乙烯基磺酰基”替代的基团。
在本发明中,“乙烯基磺酰基”是指其中乙烯基与磺酰基的硫原子键合的基团。
在本发明中,“丙-2-烯酰基氨基(c1-6烷基)氨基甲酰基”是指氨基甲酰基的一个氢原子被以下“丙-2-烯酰基氨基(c1-6烷基)”替代的基团。其实例包括(丙-2-烯酰基氨基)甲基氨基甲酰基、2-(丙-2-烯酰基氨基)乙基氨基甲酰基、3-(丙-2-烯酰基氨基)丙基氨基甲酰基、[2-甲基-3-(丙-2-烯酰基氨基)丙基]氨基甲酰基、2-(丙-2-烯酰基氨基)戊基氨基甲酰基等。
在本发明中,“丙-2-烯酰基氨基(c1-6烷基)”是指其中上述“c1-6烷基”的一个氢原子被以下“丙-2-烯酰基氨基)替代的基团”。其实例包括(丙-2-烯酰基氨基)甲基、2-(丙-2-烯酰基氨基)乙基、3-(丙-2-烯酰基氨基)丙基、2-甲基-3-(丙-2-烯酰基氨基)丙基、4-(丙-2-烯酰基氨基)丁基、6-(丙-2-烯酰基氨基)己基等。
在本发明中,“丙-2-烯酰基氨基”是指其中氨基的一个氢原子被以下“丙-2-烯酰基”替代的基团。
在本发明中,“丙-2-烯酰基”是指其中乙烯基与羰基的碳原子键合的基团。
在本发明中,“c1-6烷基(c1-6烷基磺酰基)氨基”是指其中氨基的两个氢原子被上述“c1-6烷基”及上述“c1-6烷基磺酰基”替代的基团。其实例包括甲基(甲基磺酰基)氨基、乙基(异丙基磺酰基)氨基、丁基磺酰基(丙基)氨基、己基磺酰基(异丁基)氨基等。
在本发明中,“c1-6烷氧基c1-6烷氧基”是指其中上述“c1-6烷氧基”的一个氢原子被上述“c1-6烷氧基”替代的基团。其实例包括甲氧基甲氧基、乙氧基甲氧基、正丙氧基甲氧基、异丙氧基甲氧基、甲氧基乙氧基、乙氧基乙氧基、正丙氧基乙氧基、异丙氧基乙氧基等。
在本发明中,“卤代c1-6烷氧基”是指其中上述“c1-6烷氧基”的一个或两个氢原子被上述“卤素原子”替代的基团。其实例包括氟甲氧基、二氟甲氧基、氯甲氧基、二氯甲氧基、1-氟乙氧基、1-氯乙氧基、2-氟乙氧基、1,2-二氟丙氧基等。
在本发明中,“c1-6烷基磺酰基c1-6烷基”是指其中上述“c1-6烷基”的一个氢原子被上述“c1-6烷基磺酰基”替代的基团。其实例包括甲基磺酰基甲基、甲基磺酰基乙基、乙基磺酰基甲基、正丙基磺酰基甲基、异丙基磺酰基甲基、正丁基磺酰基甲基、仲丁基磺酰基甲基、叔丁基磺酰基甲基、叔丁基磺酰基乙基、正戊基磺酰基甲基等。
在本发明中,“二(c1-6烷基)氨磺酰基”是指其中以下“氨磺酰基”的两个氢原子被相同或不同的两个上述“c1-6烷基”替代的基团。其实例包括二甲基氨磺酰基、乙基(甲基)氨磺酰基、乙基(异丙基)氨磺酰基、二丁基氨磺酰基、己基(异戊基)氨磺酰基等。
在本发明中,“氨磺酰基”是指氨基与磺酰基的硫原子键合。
在本发明中,“c1-6亚烷基二氧基”是指其中上述“c1-6烷基”的两个氢原子被氧基(-o-)替代的基团。其实例包括亚甲基二氧基(-och2o-)、亚乙基二氧基[-o(ch2)2o-]、三亚甲基二氧基[-o(ch2)3o-]、甲基亚乙基二氧基[-och(ch3)ch2o-或-och2ch(ch3)o-]等。
在本发明中,“2-c3-6烯酰基氨基”是指其中氨基的一个氢原子被以下“2-c3-6烯酰基”替代的基团。其实例包括丙-2-烯酰基氨基、2-甲基丙-2-烯酰基氨基、3-甲基丁-2-烯酰基氨基、[(e)-戊-2-烯酰基]氨基、[(e)-3-甲基戊-2-烯酰基]氨基等。
在本发明中,“2-c3-6烯酰基”是指其中以下的“1-c2-5烯基”与羰基的碳原子键合的基团。其实例包括丙-2-烯酰基、(e)-丁-2-烯酰基、3-甲基丁-2-烯酰基、(e)-己-2-烯酰基、2-甲基丙-2-烯酰基、3-甲基-2-亚甲基-丁酰基等。
在本发明中,“1-c2-5烯基”是指具有2至5个碳原子的直链或支链烯基(烯基的键合位点存在于不饱和碳原子上)。其实例包括(e)-丙-1-烯基、2-甲基丙-1-烯基、(e)-戊-1-烯基、异丙烯基、1-亚甲基丁基、(z)-1-乙基丙-1-烯基等。
在本发明中,“c1-6烷基(2-c3-6烯酰基)氨基”是指其中氨基的两个氢原子被上述“c1-6烷基”及上述“2-c3-6烯酰基”替代的基团。其实例包括甲基(丙-2-烯酰基)氨基、乙基(2-甲基丙-2-烯酰基)氨基、3-甲基丁-2-烯酰基(丙基)氨基、异丙基-[(e)-戊-2-烯酰基]氨基等。
在本发明中,“(2h3)甲氧基”是指其中甲氧基的三个氢原子全部被氘(2h;d)替代的基团。
在本发明中,“双[(2h3)甲基]氨基”是指其中二甲基氨基的六个氢原子全部被氘(2h;d)替代的基团。
在本发明中,“当环具有氧代基团时”是指其中氧代基团键合至成环原子的情况。例如,当吡啶环具有氧代基团时,其实例包括1h-吡啶-2-酮环、4h-吡啶-3-酮等,并且当哒嗪环具有氧代基团时,其实例包括1h-哒嗪-6-酮环等。
在本发明中,“在环中任选地含有一个氮原子的6-元芳环”是指除碳原子外还任选地含有一个氮原子作为成环原子的6-元单环芳环。其实例包括苯环和吡啶环。对于环q1,“在环中任选地含有一个氮原子的6-元芳环”优选为苯环或吡啶环,更优选为苯环。对于环q2,“在环中任选地含有一个氮原子的6-元芳环”优选为苯环或吡啶环,更优选为吡啶环。对于环q3,“在环中任选地含有一个氮原子的6-元芳环”优选为苯环或吡啶环,更优选为吡啶环。
在本发明中,“在环中含有一个或两个独立地选自氮原子和硫原子的杂原子的5元芳族杂环”是指除碳原子外还含有一个或两个杂原子(氮原子或硫原子)作为成环原子的5元单环芳环。其实例包括噻吩环、1,2-噻唑环、1,3-噻唑环、吡咯环、吡唑环和咪唑环。对于环q1,“在环中含有一个或两个独立地选自氮原子和硫原子的杂原子的5元芳族杂环”优选为1,3-噻唑环或吡唑环。
在本发明中,“c3-8环烷环”是指3至8元单环饱和烃环。其实例包括环丙烷环、环丁烷环、环戊烷环、环己烷环、环庚烷环和环辛烷环。对于环q1,“c3-8环烷环”优选为环己烷环。对于环q2,“c3-8环烷环”优选为环己烷环。
在本发明中,“c4-8环烯环”是指在环中具有一个双键的4至8元单环不饱和烃环。其实例包括环丁烯环、环戊烯环、环己烯环、环庚烯环和环辛烯环。对于环q1,“c4-8环烯环”优选为环己烯环。
在本发明中,“在环中含有1个氮原子的4至8元饱和杂环”是指除碳原子外还含有1个氮原子作为成环原子的4至8元单环饱和环。其实例包括氮杂环丁烷环、吡咯烷环、哌啶环、氮杂环庚烷环和氮杂环辛烷环。对于环q1,“在环中含有1个氮原子的4至8元饱和杂环”优选为哌啶环。
在本发明中,“环中含有1个氮原子的9元双环芳族杂环”是指除碳原子外还含有1个氮原子作为成环原子的9元双环稠合芳环。其实例包括吲哚环、异吲哚环、吲嗪环等。对于环q1,“在环中含有1个氮原子的9元双环芳族杂环”优选为吲哚环。
在本发明中,“在环中含有2个氮原子的6-元芳族杂环”是指除碳原子外还含有2个氮原子作为成环原子的6-元单环芳环。其实例包括哒嗪环、吡嗪环和嘧啶环。对于环q2,“在环中含有2个氮原子的6-元芳族杂环”优选为哒嗪环、吡嗪环或嘧啶环。
在本发明中,“在环中含有1-3个独立地选自氮原子,氧原子和硫原子的杂原子的5元芳族杂环”是指除碳原子外还含有1-3个杂原子(氮原子、氧原子或硫原子)作为成环原子的5元单环芳环。其实例包括吡咯环、呋喃环、噻吩环、咪唑环、吡唑环、1,2-噁唑环、1,3-噁唑环、1,2-噻唑环、1,3-噻唑环、4h-1,2,4-三唑环、1h-1,2,3-三唑环、1,2,4-噁二唑环、1,3,4-噻二唑环等。对于环q2,“在环中含有1-3个独立地选自氮原子、氧原子和硫原子的杂原子的5元芳族杂环”优选为咪唑环、吡唑环、1,3-噻唑环、1,3-噁唑环或4h-1,2,4-三唑环。
在本发明中,“在环中含有1-3个独立地选自氮原子和氧原子的杂原子的9或10元双环芳族或部分不饱和杂环”是指衍生自除碳原子外还含有1-3个杂原子(氮原子或氧原子)作为成环原子的9或10元双环稠合芳环,其在双环的一部分中任选地具有饱和键。其实例包括吲哚环、苯并呋喃环、吲唑环、苯并咪唑环、1h-吡咯并[2,3-c]吡啶环、1h-吡咯并[3,2-c]吡啶环、呋喃并[3,2-b]吡啶环、1h-吡唑并[3,4-c]吡啶环、喹啉环、异喹啉环、1,8-萘啶环、二氢吲哚环等。对于环q2,“在环中含有1-3个独立地选自氮原子和氧原子的杂原子的9或10元双环芳族或部分不饱和杂环”优选为异喹啉环、吲唑环、苯并咪唑环、1h-吡咯并[2,3-c]吡啶环、1h-吡咯并[3,2-c]吡啶环、呋喃并[3,2-b]吡啶环、1h-吡唑并[3,4-c]吡啶环或二氢吲哚环。
在本发明中,“在环中含有一个或两个独立地选自氧原子和氮原子的杂原子的5至8元饱和杂环”是指除碳原子外还含有一个或两个杂原子(氮原子或氧原子)作为成环原子的5至8元单环饱和环。其实例包括吡咯烷环、哌啶环、哌嗪环、吗啉环、氮杂环庚烷环、氧杂氮杂环庚烷环、1,4-氧杂氮杂环庚烷环、1,4-二氮杂环辛烷环等。对于环q2,“在环中含有一个或两个独立地选自氧原子和氮原子的杂原子的5至8元饱和杂环”优选为吡咯烷环、哌啶环、吗啉环或氮杂环庚烷环。
在本发明中,“在环中含有1个氮原子或1个氧原子的4至8元饱和杂环”是指除碳原子外还含有1个杂原子(氮原子或氧原子)作为成环原子的4至8元单环饱和环。其实例包括氮杂环丁烷环、氧杂环丁烷环、吡咯烷环、四氢呋喃环、哌啶环、四氢吡喃环、氮杂环庚烷环等。对于环q3,“在环中含有1个氮原子或1个氧原子的4至8元饱和杂环”优选为氮杂环丁烷环、四氢吡喃环或哌啶环。
在本发明中,“在环中含有氮原子的杂环”是指除碳原子外还含有氮原子作为成环原子的杂环。其实例包括哌啶环、氮杂环庚烷环等。
在本发明中,二羟硼基是指其中硼烷基的两个氢原子均被羟基替代的基团。
在本发明中,二烷氧基硼烷基是指其中硼烷基的两个氢原子均被烷氧基(例如,甲氧基、乙氧基等)替代的基团。其实例包括二甲氧基硼烷基、二乙氧基硼烷基等。
在本发明中,二氧杂硼杂环戊基是指源自由与上述二烷氧基硼烷基的硼原子键合的两个烷氧基与硼原子一起形成的环的基团。其实例包括4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基等。
在本发明中,menin是指被鉴定为多发性内分泌肿瘤1型(men1)的致病因素的肿瘤抑制蛋白,其是遍在表达的核蛋白,其参与dna加工、改性蛋白、蛋白修饰染色质和与许多转录因子的相互作用(agarwal等人;hormmetabres,2005,37(6):369-374)。
在本发明中,mll蛋白是指mll1、mll2、mll融合蛋白或mll部分串联重复蛋白。
在本发明中,mll1是指属于mll(混合谱系白血病)家族的甲基转移酶之一的mll1(也称为kmt2a)蛋白。在本发明中,术语mll1基因表示编码该蛋白的基因。
在本发明中,mll2是指属于mll(混合谱系白血病)家族的甲基转移酶之一的mll2(也称为kmt2d)蛋白。在本发明中,术语mll2基因表示编码该蛋白的基因。
在本发明中,mll融合蛋白是指由mll基因的染色体易位引起的嵌合基因的转录和表达所产生的嵌合蛋白。
在本发明中,mll部分串联重复(ptd)蛋白是指由mll基因的染色体重复引起的异常基因的转录和表达所产生的异常蛋白。
在本发明中,menin与选自mll1、mll2、mll融合蛋白和mll部分串联重复蛋白的一种或多种蛋白之间的相互作用是指由menin形成的蛋白分子与mll1、mll2、mll融合蛋白或mll部分串联重复蛋白之间的相互作用。当在同一系统中存在两种或更多种类型的mll蛋白时,由menin独立形成的蛋白分子与每种mll蛋白之间可能共存两种或更多种相互作用。
在本发明中,术语“肿瘤”和“癌症”可互换使用。此外,在本发明中,可以将肿瘤、恶性肿瘤、癌症、恶性瘤、癌、肉瘤等统称为“肿瘤”或“癌症”。此外,术语“肿瘤”和“癌症”还包括在某些情况下分类为癌前期的病理状况,例如骨髓增生异常综合征。
如本文所用,术语“治疗”及其衍生词是指在发展疾病等的患者中缓解、减轻疾病、病、病症等(以下称为“疾病等”)的临床症状或或延迟其恶化。
如本文所用,术语“预防”及其衍生词是指在可能发展疾病等但尚未发展或担心治疗后疾病等复发的哺乳动物中阻止、抑制、控制、减慢或停止疾病等的临床症状的发作。或。
在本发明中,“bcl-2抑制剂”是指与具有抗凋亡作用的蛋白bcl-2结合以抑制抗凋亡作用并因此诱导凋亡以发挥抗癌作用的药物。
在本发明中,“bcl-2抑制剂”优选为维奈托克。
在本发明中,“嘧啶抗代谢物”是指具有与嘧啶碱基相似的部分结构且抑制核酸生物合成以防止肿瘤细胞的生长和分裂并因此发挥抗癌作用的药物。
在本发明中,“嘧啶抗代谢物”优选为阿糖胞苷。
在本发明中,“dna甲基转移酶抑制剂”是指抑制催化dna的转甲基化的酶并因此发挥抗癌作用的药物。
在本发明中,“dna甲基转移酶抑制剂”优选为阿扎胞苷。
在本发明中,维奈托克是4-(4-{[2-(4-氯苯基)-4,4-二甲基环己-1-烯-1-基]甲基}哌嗪-1-基)-n-[(3-硝基-4-{[(噁烷-4-基)甲基]氨基}苯基}磺酰基]-2-[(1h-吡咯并[2,3-b]吡啶-5-基)氧基]苯甲酰胺(cas登记号:1257044-40-8),也称为venclexta(注册商标)、venclyxto(注册商标)或维奈托克。其很容易作为商业产品获得。
在本发明中,阿扎胞苷为4-氨基-1-β-d-呋喃核糖基-1,3,5-三嗪-2(1h)-酮(cas登记号:320-67-2),并且也被称为5-阿扎胞苷或5aza或vidaza(注册商标)。其很容易作为商业产品获得。
在本发明中,阿糖胞苷为1-β-d-阿拉伯呋喃糖基胞嘧啶(cas登记号:147-94-4),并且也称为ara-c或arac,或称为cylocide(注册商标)。其很容易作为商业产品获得。
在本发明中,维奈托克、阿扎胞苷或阿糖胞苷可以是游离形式、溶剂化物、各种药学上可接受的盐中的任何一种,或者是包含有各种药学上可接受的载体的药物组合物的形式,等等。
在本发明中,术语“组合施用”是指在一定时间段内两种药物均被摄入到待施用对象体内。两种药物可以以单一制剂的形式施用,或者各自可以分别配制并分别施用。当它们分别配制时,它们的施用时机没有特别限制,并且它们可以在相同时间、在不同时间以一定间隔或在不同天施用。
当它们在不同时间或不同天施用时,其施用顺序没有特别限制。通常,根据它们各自的施用方法来施用它们的制剂,从而可以以相同数量的剂量或不同数量的剂量来施用制剂。另外,当它们分别配制时,制剂的各个施用方法(施用途径)可以彼此相同,或者可以通过不同的施用方法(施用途径)来施用制剂。而且,两种药物不必同时存在于体内,并且可以在一定时间段内(例如,一个月,优选一周,更优选几天,甚至更优选一天)摄入体内。施用一种活性成分时,另一种活性成分可能已从体内消失。
下面将描述本发明化合物中合适的取代基。
r1优选为氢原子或甲基。r1更优选为氢原子。
r2优选为氢原子或甲基。r2更优选为氢原子。
r3和r4之一优选为氢原子、羟基、氟原子、甲氧基、二甲基氨基甲酰基或噁唑-2-基,更优选为氢原子或羟基。r3和r4中的另一个优选为氢原子、羟基、氟原子或甲氧基,更优选为氢原子或羟基。
式(1)中由下式(5)表示的部分优选为下式(5a)或(5b)。
[化学式36]
[化学式37]
其中*键合至与r2键合的氮原子,**键合至与r5键合的氮原子,r16为氢原子、卤素原子、羟基、二(c1-6烷基)氨基甲酰基、噁唑-2-基或c1-6烷氧基,r17为氢原子或卤素原子,且r18为c1-6烷氧基。
式(1)中由下式(5)表示的部分更优选为下式(6a)至(6d)中的任一个,还更优选为(6a)或(6b)。
[化学式38]
[化学式39]
其中*键合至与r2键合的氮原子,**键合至与r5键合的氮原子,且r19为氢原子、羟基、二甲基氨基甲酰基、噁唑-2-基或甲氧基。
式(1)中由下式(5)表示的部分优选为下式(7a)。
[化学式40]
[化学式41]
其中*键合至与r2键合的氮原子,**键合至与r5键合的氮原子,r20为氢原子或羟基,且r21为氢原子、羟基或c1-6烷氧基。
式(1)中由下式(5)表示的部分更优选为下式(8a)至(8f)中的任一个,还更优选为(8a)至(8e)中的任一个。
[化学式42]
[化学式43]
其中*键合至与r2键合的氮原子,**键合至与r5键合的氮原子,r22为氢原子、羟基或甲氧基,r23为羟基或甲氧基,且r24为氢原子或羟基。
式(1)中由下式(5)表示的部分最优选为下式(9a)至(9c)中的任何一个。下式(9a)至(9c)中,优选为(9b)或(9c),更优选为(9b)。
[化学式44]
[化学式45]
其中*键合至与r2键合的氮原子,并且**键合至与r5键合的氮原子。
r5优选为氢原子、甲基、乙基或2-羟乙基。r5更优选为甲基。
r6优选为氢原子、甲基、氯原子、甲氧基、氨基或甲基氨基。r6更优选为氢原子、氯原子、甲氧基、氨基或甲基氨基。r6仍更优选为氢原子、氯原子、甲氧基或甲基氨基。
关于r7和r8,优选地,r7和r8与r7所键合的碳原子以及r8所键合的碳原子一起形成下式(2a)或(2b)。
[化学式46]
其中虚线圆圈表示环是芳族的,标有a的碳原子是与r8键合的碳原子,标有b的碳原子是与r7键合的碳原子,x是ch或氮原子,且r9为卤代c1-6烷基、c3-8环烷基、c3-8环烷基c1-6烷基、c1-6烷氧基c1-6烷基或氧杂环丁烷基。
r9优选为2,2,2-三氟乙基、环丙基、环丙基甲基、甲氧基甲基或氧杂环丁烷-3-基。r9更优选为2,2,2-三氟乙基、环丙基或环丙基甲基。r9仍更优选为2,2,2-三氟乙基。
关于r7和r8,更优选地,r7和r8与r7所键合的碳原子以及r8所键合的碳原子一起形成下式(10a)。
[化学式47]
其中虚线圆圈表示环为芳族的,标有a的碳原子是与r8键合的碳原子,标有b的碳原子是与r7键合的碳原子。
关于r7和r8,优选地,r7为氢原子,并且r8为下式(3)。
[化学式48]
其中*表示键合部位,r10为二(c1-6烷基)氨基甲酰基、(c1-6烷基)嘧啶基、(c1-6烷基)苯基或(c1-6烷基)吡唑基,r11为氢原子或卤素原子,且r12为卤素原子。
r10优选为二异丙基氨基甲酰基、4-异丙基嘧啶-5-基、2-异丙基苯基或1-异丙基吡唑-5-基。r11优选为氢原子或氟原子。r12优选为氟原子。
关于r7和r8,更优选r7为氢原子,并且r8为下式(11a)或(11b)。
[化学式49]
其中*表示键合部位,r25为二异丙基氨基甲酰基、4-异丙基嘧啶-5-基、2-异丙基苯基或1-异丙基吡唑-5-基,且r26为二异丙基氨基甲酰基。
m优选为1。
n优选为1。
环q1优选为以下(i)至(vii)中的任何一个。
(i)任选地具有一个或两个独立地选自上述组a的取代基的苯环;
(ii)任选地具有一个或两个独立地选自上述组a的取代基的吡啶环;
(iii)1,3-噻唑环或吡唑环(1,3-噻唑环或吡唑环任选地具有一个独立地选自上述组a的取代基);
(iv)任选地具有一个独立地选自上述组a的取代基的环己烷环;
(v)任选地具有一个独立地选自上述组a的取代基的环己烯环;
(vi)任选具有一个独立地选自上述组a组的取代基的哌啶环;或者
(vii)任选地具有一个或两个独立地选自上述组b的取代基的吲哚环。
当m为0时,则环q1更优选为以下(i)至(iv)中的任何一个。
(i)任选地具有一个或两个独立地选自上述组a的取代基的苯环;
(ii)1,3-噻唑环或吡唑环,各自任选地具有一个独立地选自上述组a的取代基;
(iii)任选地具有一个独立地选自上述组a的取代基的环己烷环;或者
(iv)任选地具有一个独立地选自上述组b的取代基的吲哚环。
当m为0时,则环q1还更优选为苯基、4-羟基苯基、4-[3-(丙-2-烯酰基氨基)丙基氨基甲酰基]苯基、4-[3-(乙烯基磺酰基氨基)丙基氨基甲酰基]苯基、3-氟-4-(2-羟基乙氧基)苯基、噻唑-5-基、环己基或2-氰基-1h-吲哚-5-基。
当m为1时,则环q1更优选为以下(i)至(vii)中的任何一个。
(i)任选地具有一个独立地选自上述组a的取代基的苯环;
(ii)任选地具有一个独立地选自上述组a的取代基的吡啶环;
(iii)任选地具有一个独立地选自上述组a的取代基的吡唑环;
(iv)任选地具有一个独立地选自上述组a的取代基的环己烷环;
(v)任选地具有一个独立地选自上述组a的取代基的环己烯环;
(vi)任选地具有一个独立地选自上述组a的取代基的哌啶环;或者
(vii)任选地具有一个或两个独立地选自上述组b的取代基的吲哚环。
当m为1时,则环q1还更优选为下式(12a)至(12h)中的任何一个。
[化学式50]
其中*与z键合,**键合至与r1键合的碳原子,r27为氢原子、卤素原子、c1-6烷氧基或c1-6烷基,j为氮原子或cr29,r29为卤素原子,且r28为氢原子或c1-6烷基。
当m为1时,则环q1最优选为下式(13a)或(13b)。
[化学式51]
其中*与z键合,**键合至与r1键合的碳原子,并且r30为氢原子、氟原子、甲基或甲氧基。
环q2优选为以下(i)至(vii)中的任何一个。
(i)任选地具有1-3个独立地选自上述组c的取代基的苯环;
(ii)任选地具有1-3个独立地选自上述组c的取代基的吡啶环;
(iii)哒嗪环、吡嗪环或嘧啶环(所述哒嗪环、吡嗪环或嘧啶环任选地具有1-3个独立地选自上述组c的取代基);
(iv)吡唑环、咪唑环、1,3-噻唑环、1,3-噁唑环或4h-1,2,4-三唑环(所述吡唑环、咪唑环、1,3-噻唑环、1,3-噁唑环或4h-1,2,4-三唑环任选地具有一个独立地选自上述组c的取代基);
(v)异喹啉环、吲唑环、苯并咪唑环、1h-吡咯并[2,3-c]吡啶环、1h-吡咯并[3,2-c]吡啶环、呋喃并[3,2-b]吡啶环、1h-吡唑并[3,4-c]吡啶环或二氢吲哚环(所述异喹啉环、吲唑环、苯并咪唑环、1h-吡咯并[2,3-c]吡啶环、1h-吡咯并[3,2-c]吡啶环、呋喃并[3,2-b]吡啶环、1h-吡唑并[3,4-c]吡啶环或二氢吲哚环任选地具有一个或两个独立地选自上述组d的取代基;
(vi)吡咯烷环、哌啶环、吗啉环或氮杂环庚烷环(所述吡咯烷环、哌啶环、吗啉环或氮杂环庚烷环任选地具有一个独立地选自上述组e的取代基);或者
(vii)任选地具有一个独立地选自上述组e的取代基的环己烷环。
当w为上式(4a)时,则环q2更优选为下式(14a)至(14f)中的任何一个。
[化学式52]
其中*表示键合部位,t为ch或氮原子,r31为氢原子、c1-6烷氧基、卤代c1-6烷氧基或(2h3)甲氧基,r32为氢原子、c1-6烷基、卤素原子、c1-6烷氧基、氰基、二(c1-6烷基)氨基、卤代c1-6烷基、c1-6烷基氨基、c1-6烷基磺酰基、c1-6烷氧基c1-6烷氧基、卤代c1-6烷氧基、羟基c1-6烷基、c1-6烷基(2-c3-6烯酰基)氨基、(2h3)甲氧基或双[(2h3)甲基]氨基,或r31和r32一起形成亚乙基二氧基,r33和r35各自独立地为氢原子、卤素原子、c1-6烷氧基、c1-6烷基(c1-6烷基磺酰基)氨基、(c1-6烷基)氨基甲酰基、二(c1-6烷基)氨磺酰基、2-c3-6烯酰基氨基或c1-6烷基磺酰基c1-6烷基,r34为氢原子或卤素原子,r36为卤素原子,r37为c1-6烷氧基,r38为卤素原子,r39为c1-6烷基或c1-6烷基磺酰基,r40为c1-6烷基或c1-6烷基磺酰基,u1为ch或氮原子,u2为cr41或氮原子,且r41为氢原子或卤素原子。
r31优选为氢原子、甲氧基、二氟甲氧基或(2h3)甲氧基。
r32优选为氢原子、甲基、氟原子、氯原子、甲氧基、氰基、二甲基氨基、三氟甲基、甲基氨基、甲基磺酰基、甲氧基乙氧基、二氟甲氧基、羟甲基、甲基(丙-2-烯酰基)氨基、(2h3)甲氧基或双[(2h3)甲基]氨基。
r33和r35各自独立地优选为氢原子、氟原子、甲氧基、丙-2-烯酰基氨基、甲基(甲基磺酰基)氨基、甲基氨基甲酰基、二甲基氨磺酰基或甲基磺酰基甲基。
r34优选为氢原子或氟原子。
r36优选为氟原子。
r37优选为甲氧基。
r38优选为氟原子。
r39优选为甲基或甲基磺酰基。
r40优选为甲基或甲基磺酰基。
r41优选为氢原子或氟原子。
当w为上式(4a)时,则环q2更优选为5,6-二甲氧基吡嗪-2-基、4,5-二甲氧基嘧啶-2-基、4-吡啶基、2,4-二氟-3-甲氧基-苯基、4,5-二甲氧基-2-吡啶基、吗啉代基、噁唑-2-基、4h-1,2,4-三唑-3-基、5-氧代吡咯烷-2-基、2-氧代吡咯烷-1-基、环己基、2-甲氧基噻唑-5-基、呋喃并[3,2-b]吡啶-6-基、吲哚-1-基、3-羟基-1-哌啶基、氮杂环庚烷-1-基、4-氯-1h-吡咯并[3,2-c]吡啶-7-基、1-甲基吡唑并[3,4-c]吡啶-4-基、苯并咪唑-1-基、4-异喹啉基、1-(二氟甲基)-4-甲氧基-6-氧代哒嗪-3-基或6-氧代-1h-吡啶-3-基。
当w为上式(4a)时,则环q2仍更优选为下式(15a)至(15c)中的任何一个。
[化学式53]
其中*表示键合部位,r42为甲基、氯原子、甲氧基、氰基、二甲基氨基或双[(2h3)甲基]氨基,r43为甲氧基或(2h3)甲氧基,且r44为氯原子、甲氧基、甲氧基乙氧基、二甲基氨基、二氟甲氧基或(2h3)甲氧基。
当w为上式(4a)时,则环q2最优选为下式(16a)至(16g)中的任何一个。
[化学式54]
其中*表示键合部位。
当w为上式(4b)时,则环q2更优选为下式(17a)或(17b)。
[化学式55]
其中*与y键合,且**与z键合。
当w为上式(4b)时,则环q3优选为下式(18a)至(18d)中的任何一个。
[化学式56]
其中*表示键合部位,r45为氢原子或卤素原子,r46为c1-6烷基磺酰基,且v为氮原子或ch。
当w为上式(4b)时,则环q3更优选为苯基、氮杂环丁烷-1-基、3-吡啶基、6-氯-3-吡啶基、四氢吡喃-3-基或1-甲基磺酰基-4-哌啶基。
当w为上式(4b)时,则y优选为单键或氧原子。
z优选为单键、-nh-、氧原子、-so2-、-ch2-、*-ch2-nhc(=o)-**、*-ch2ch2-o-**或*-ch2-nh-**,其中*键合至环q2,且**键合至环q1。
z更优选为单键。
w优选为上式(4a)。
本发明的化合物优选为选自以下化合物或其药学上可接受的盐(优选盐酸盐、琥珀酸盐、苯磺酸盐、马来酸盐、富马酸盐、粘酸盐或己二酸盐,更优选琥珀酸盐、苯磺酸盐、马来酸盐、富马酸盐、粘酸盐或己二酸盐)的化合物:
5-[4-({[(1r,3r,4s)-3-羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]-3-甲氧基吡啶-2-甲腈,
(1r,2s,4r)-4-[({4-[1-(甲磺酰基)-1h-吲唑-4-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-[({4-[6-(二甲基氨基)-5-甲氧基吡啶-3-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5-甲氧基-6-甲基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(1h-咪唑-1-基)苯基]甲基}氨基)-2-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(6-氯-5-甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(6-氟-5-甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(6-氯-5-甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
2-[(4-{[(1s,2r,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-羟基环戊基](甲基)氨基}嘧啶-5-基)氧基]-5-氟-n,n-二(丙-2-基)苯甲酰胺,
(1r,2s,4r)-2-{[2-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)环戊-1-醇,
(1r,3s)-n3-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺,
(1r,2s,4r)-4-[({4-[6-(二甲基氨基)-5-甲氧基哒嗪-3-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
6-[4-({[(1r,3r,4s)-3-羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]-4-甲氧基哒嗪-3-甲腈,
(1s,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-[({4-[5-甲氧基-6-(2-甲氧基乙氧基)哒嗪-3-基]苯基}甲基)氨基]-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(4,5-二甲氧基吡啶-2-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-[({4-[6-(二甲基氨基)-5-甲氧基吡啶-3-基]苯基}甲基)氨基]-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-[({4-[6-(二氟甲氧基)-5-甲氧基哒嗪-3-基]苯基}甲基)氨基]-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇,
(1r,2s,4r)-4-{[(4-{5,6-双[(2h3)甲氧基]哒嗪-3-基}苯基)甲基]氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,
(1r,2s,4r)-4-({[4-(6-{双[(2h3)甲基]氨基}-5-甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇,和
(1r,2s,4r)-4-{[(4-{5,6-双[(2h3)甲基氧基]哒嗪-3-基}苯基)甲基]氨基}-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇。
本发明的化合物更优选为选自以下化合物或其药学上可接受的盐(优选盐酸盐、琥珀酸盐、苯磺酸盐、马来酸盐、富马酸盐、粘酸盐或己二酸盐,更优选琥珀酸盐、苯磺酸盐、马来酸盐、富马酸盐、粘酸盐或己二酸盐)的化合物:
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇和
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇。
就menin-mll抑制作用、溶解度、细胞膜通透性、口服吸收、血药浓度、代谢稳定性、组织可转移性、生物利用度、体外活性、体内活性、药物作用的快速发作、药物作用的可持续性、物理稳定性、药物相互作用、毒性等而言,本发明的化合物或其药学上可接受的盐或本发明的晶体具有优异的性质,并且可用作药物。
在一个实施方案中,本发明涉及一种药物组合物,其包含作为活性成分的由通式(1)表示的化合物或其药学上可接受的盐或本发明的晶体,其用于治疗和/或预防可通过抑制mll蛋白和menin之间的相互作用而治疗和/或预防的疾病。
在另一个实施方案中,本发明涉及一种用于治疗和/或预防糖尿病的方法,其包括施用由通式(1)表示的化合物或其药学上可接受的盐或本发明的晶体。
在另一个实施方案中,本发明涉及一种用于治疗和/或预防癌症的方法,其包括施用由通式(1)表示的化合物或其药学上可接受的盐或本发明的晶体。
待治疗的疾病没有特别限制,只要其依赖于menin和mll蛋白之间的相互作用即可,其实例包括癌症和糖尿病(优选癌症)。
待治疗的癌症的类型没有特别限制,只要其被证实对本发明的化合物敏感。其实例包括血液癌、脑瘤(例如,小儿神经胶质瘤等)、头/颈区癌、食道癌、胃癌、阑尾癌、结肠癌、肛门癌、胆囊癌、胆管癌、胰腺癌、胃肠道间质瘤、肺癌、肝癌(例如肝细胞瘤等)、间皮瘤、甲状腺癌、肾癌、前列腺癌、神经内分泌肿瘤、黑色素瘤、乳腺癌、子宫内膜癌、宫颈癌、卵巢癌、骨肉瘤、软组织肉瘤、卡波氏肉瘤、肌肉瘤、膀胱癌和睾丸癌。优选血液癌、前列腺癌、乳腺癌、肝细胞瘤和小儿神经胶质瘤,更优选血液癌。
血液癌的实例包括混合谱系白血病(mll)、与mll相关的白血病、与mll相关联的白血病、mll阳性白血病、mll诱发的白血病、重排混合谱系白血病(mll-r)、与mll重排相关的白血病(mll基因的重排、mll重排白血病)、mll扩增白血病、mll部分串联重复白血病(mll-ptd白血病)、与hox和meis1基因恒定表达相关的其他白血病/血液癌、急性白血病、慢性白血病、惰性白血病、淋巴母细胞性白血病、淋巴细胞性白血病、髓系白血病、骨髓性白血病、儿童期白血病、急性成淋巴细胞性白血病(all)、急性髓系白血病(aml)、急性粒细胞性白血病、急性非淋巴细胞性白血病、慢性淋巴细胞性白血病(cll)、慢性骨髓性白血病(cml)、治疗相关白血病、骨髓增生异常综合征(mds)、骨髓增生性疾病(mpd)、骨髓增生性肿瘤(mpn)、多发性骨髓瘤、骨髓增生异常、浆细胞瘤、皮肤t-细胞淋巴瘤、淋巴样肿瘤、艾滋病相关淋巴瘤、蕈样肉芽肿(granulomafungoides)、alibert-bazin综合征、塞泽里氏综合征、毛细胞白血病(hcl)、t细胞幼淋巴细胞白血病(t-pll)、大颗粒淋巴细胞白血病、脑膜白血病、霍奇金淋巴瘤、非霍奇金淋巴瘤(恶性淋巴瘤)、华氏巨球蛋白血症等。更优选急性髓系白血病(aml)和急性成淋巴细胞性白血病(all)。
p53是抑制癌变的重要因素之一,并且已经在大约一半的人类癌症中观察到p53基因的缺失或突变。已知p53中的突变可能会促进癌症(功能获得性p53突变),并且通过允许具有menin-mll抑制作用的化合物作用于表达功能获得性p53突变的癌细胞系来抑制细胞生长(zhu等人,nature,2015,525,206-211)。由于本发明的化合物或其药学上可接受的盐具有menin-mll抑制作用,因此对于治疗和/或预防表达功能获得性p53突变的癌症是有效的。表达功能获得性p53突变的癌症的实例包括血液癌、脑瘤、头/颈区癌、食道癌、胃癌、阑尾癌、结肠癌、肛门癌、胆囊癌、胆管癌、胰腺癌、胃肠道间质瘤、肺癌、肝癌、间皮瘤、甲状腺癌、肾癌、前列腺癌、神经内分泌肿瘤、黑色素瘤、乳腺癌、子宫内膜癌、宫颈癌、卵巢癌、骨肉瘤、软组织肉瘤、卡波氏肉瘤、肌肉瘤、膀胱癌和睾丸癌。
已知menin与mll融合蛋白之间的相互作用对于表达几种下游癌基因(例如白血病相关的基因,例如hox、meis1、myc等)至关重要(borkin等人,cancercell,2015,27,589-602)。由于本发明的化合物或其药学上可接受的盐具有menin-mll抑制作用,因此对于显示hox基因、meis1基因、myc基因等的表达特性的白血病有效。
由于本发明的化合物或其药学上可接受的盐或本发明的晶体具有menin-mll抑制作用,因此其优选用于依赖于menin与mll蛋白之间的相互作用的疾病。依赖于menin和mll蛋白之间的相互作用的疾病的实例包括血液癌、前列腺癌、乳腺癌、肝细胞瘤、小儿神经胶质瘤和糖尿病(例如,参见以下文献:血液癌(a1,a2,a3,a4),骨髓增生异常综合征(a1,a3),前列腺癌(b),乳腺癌(c1,c2,c3),肝细胞瘤(d),小儿神经胶质瘤(e),糖尿病(f1,f2,f3))。
a1,yokoyama等人,cell,2005,123,207-218。
a2,borkin等人,cancercell,2015,27,589-602。
a3,cierpickiandgrembecka.futuremedchem.2014,447-462。
a4,kuehnmw等人,cancerdiscovery,2016,1166-1181。
b,malik等人,nat.med.,2015,21,344-352。
c1,dreijerink等人,cancerres.,2006,66,4929-4935。
c2,imachi等人,breastcancerres.treat.,2010,122,395-407。
c3,zhu等人,nature,2015,525,206-211。
d,xu等人,proc.natl.acad.sci.usa.,2013,110,17480-17485。
e,fumato等人,science,2014,346,1529-1533。
f1,wu等人,curr.mol.med.,2008,8(8),805-815。
f2,chamberlain等人,j.clin.invest.,2014,124,4093-4101。
f3,yang等人,proc.natl.acad.sci.usa.,2010,107,20358-20363。
在另一个实施方案中,本发明涉及药物组合物,其包含选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物,以及本发明的化合物或其药学上可接受的盐或本发明的晶体,所述药物和所述化合物或其药学上可接受的盐或晶体组合施用。
在另一个实施方案中,本发明涉及一种治疗癌症的方法,其包括与选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物组合施用本发明的化合物或其药学上可接受的盐或本发明的晶体。
在另一个实施方案中,本发明涉及本发明的化合物或其药学上可接受的盐或本发明的晶体,其与选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物组合施用。
选自bcl-2抑制剂、dna甲基转移酶抑制剂和嘧啶抗代谢物的一种药物和本发明的化合物或其药学上可接受的盐或本发明的晶体可以作为活性成分分别包含在不同制剂中。当它们作为活性成分分别包含在不同制剂中时,它们的制剂可以在相同时间或不同时间施用。
在另一个实施方案中,本发明涉及用于诱导白血病细胞分化的组合物,其包含本发明的化合物或其药学上可接受的盐或本发明的晶体。
在另一个实施方案中,本发明涉及诱导白血病细胞分化的方法,其包括施用本发明的化合物或其药学上可接受的盐或本发明的晶体。
在本发明的化合物中,取决于取代基的类型和组合,当本发明的化合物具有不对称碳原子时,可以存在几何异构体例如顺式形式和反式形式、互变异构体或旋光异构体例如l-形式和d-形式(例如对映异构体或非对映异构体)。除非另有说明,否则本发明的化合物包括所有这些异构体及其任何比率的混合物。
在本发明中,药学上可接受的盐包括药学上可接受的酸加成盐和药学上可接受的碱加成盐。
当本发明的化合物具有诸如氨基等的碱性基团时,通常可以形成药理学上可接受的酸加成盐。酸加成盐的实例包括氢卤酸盐,例如氢氟酸盐、盐酸盐、氢溴酸盐、氢碘酸盐等;无机酸盐,例如硝酸盐、高氯酸盐、硫酸盐、磷酸盐等;低级烷磺酸盐,例如甲磺酸盐、三氟甲磺酸盐、乙磺酸盐等;芳基磺酸盐,例如苯磺酸盐、对甲苯磺酸盐等;有机酸盐,例如乙酸盐、苹果酸盐、富马酸盐、琥珀酸盐、柠檬酸盐、酒石酸盐、草酸盐、马来酸、粘酸、己二酸盐等;以及氨基酸盐,例如鸟氨酸盐、谷氨酸盐、天冬氨酸盐等,优选氢卤酸盐、芳基磺酸盐和有机酸盐。本发明化合物的酸加成盐优选为盐酸盐、琥珀酸盐、苯磺酸盐、马来酸盐、富马酸盐、粘酸盐或己二酸盐,更优选琥珀酸盐、苯磺酸盐、马来酸盐、富马酸盐、粘酸盐或己二酸盐。
本发明化合物的酸加成盐包括可以通过将要添加到本发明化合物中的酸与本发明的化合物以任意比率合并而形成的酸加成盐。例如,盐酸盐包括可形成的盐,例如一盐酸盐、二盐酸盐、三盐酸盐等;富马酸盐包括可形成的盐,例如一富马酸盐、1/2富马酸盐等;琥珀酸盐包括可形成的盐,例如一琥珀酸盐、2/3琥珀酸盐、1/3琥珀酸盐等。
当本发明的化合物具有诸如羧基等的酸性基团时,通常可以形成药理学上可接受的碱加成盐。碱加成盐的实例包括碱金属盐,例如钠盐、钾盐、锂盐等;碱土金属盐,例如钙盐、镁盐等;无机盐,例如铵盐等;以及有机胺盐,例如二苄胺盐、吗啉盐、苯基甘氨酸烷基酯盐、乙二胺盐、n-甲基葡糖胺盐、二乙胺盐、三乙胺盐、环己胺盐、二环己胺盐、n,n'-二苄基乙二胺盐、二乙醇胺盐、n-苄基-n-(2-苯基乙氧基)胺盐、哌嗪盐、四甲基铵盐和三(羟甲基)氨基甲烷盐等。
本发明的化合物可以以非溶剂化物或溶剂化物的形式存在。溶剂化物没有特别限制,只要其是药理学上可接受的,并且特别优选为水合物、乙醇化物等。另外,当通式(1)表示的化合物中存在氮原子时,该化合物可以为n-氧化物形式。这样的溶剂化物和n-氧化物形式包括在本发明的范围内。此外,本发明的化合物可以包含一种或多种非天然丰度的同位素作为构成化合物的原子。同位素的实例包括氘(2h;d)、氚(3h;t)、碘-125(125i)、碳-14(14c)等。此外,本发明的化合物可以用放射性同位素例如氚(3h)、碘-125(125i)或碳-14(14c)进行放射性标记。放射性标记的化合物可用作治疗剂或预防剂、研究试剂(例如检测试剂)或诊断剂(例如体内图像诊断剂)。包含任何比率的任何放射性或非放射性同位素的本发明化合物都包括在本发明的范围内。
众所周知,含有一个或多个氘原子(2h;d)作为构成化合物的氢原子的低分子化合物可以表现出可作为药物使用的特性(例如,药物功效,安全性等)(sanderson,nature,2009,doi:10.1038/458269a,maltais等人,j.med.chem.,2009,52,7993-8001)。还预期其中引入一个或多个氘原子代替构成该化合物的氢原子的本发明化合物显示与上述相同的效果。
在本发明中,晶体是指具有通过规则地三维重复组成原子或分子而形成的内部结构的固体,并且与不具有这种规则的内部结构的无定形固体或无定形物质相区分。通过使用粉末x射线晶体分析等可以确认本发明的化合物或其盐为结晶状态。通常,由于测量装置、样品或样品制备的不同,粉末x射线衍射中的峰值可能会固有地发生变化,因此衍射角(2θ)可以在大约±0.2(度)的范围内变化。因此,可以理解,本发明的衍射角的值包括落入约±0.2的范围内的数值。因此,在粉末x射线衍射中,本发明的范围不仅包括具有完全相同的衍射角(2θ)的晶体,而且还包括具有在±0.2的范围内的相同衍射角的晶体。在此,衍射角(2θ)的单位是度(也称为“°”),在衍射角(2θ)的数值的描述中可以省略该单位。
在本发明中,晶体包括通式(1)表示的化合物的晶体,通式(1)表示的化合物的水合物晶体,通式(1)表示的化合物的溶剂化物晶体,通式(1)表示的化合物的药学上可接受的盐的晶体,通式(1)表示的化合物的药学上可接受的盐的水合物晶体和通式(1)表示的化合物的药学上可接受的盐的溶剂化物晶体。本发明的水合物晶体可以是例如0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9、3.0、3.1、3.2、3.3、3.4、3.5、3.6、3.7、3.8、3.9、4.0、4.1、4.2,4.3、4.4、4.5、4.6、4.7、4.8、4.9或5.0水合物的形式,并且根据湿度可增加或减少水合水。
本发明的晶体(以下有时称为“本发明的实施例131的晶体”、“本发明的实施例132的晶体”、“本发明的实施例133的晶体”、“本发明的实施例134的晶体”、“本发明的实施例135的晶体”、“本发明的实施例136的晶体”、“本发明的实施例137的晶体”、“本发明的实施例138的晶体”或“本发明的实施例139的晶体”)可以稳定地作为药物生产中使用的活性药物成分的晶体来供给,并且具有优异的吸湿性或稳定性。这些晶体形式的差异特别通过粉末x射线衍射来区分。
本发明的实施例131的晶体在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在4.66±0.2、7.02±0.2、14.10±0.2、16.68±0.2、17.46±0.2、18.68±0.2、21.34±0.2、24.52±0.2、25.54±0.2和28.22±0.2的衍射角(2θ)处具有峰。
本发明的实施例131的晶体优选是单琥珀酸盐。
本发明的实施例131的晶体优选为非水合物。
本发明的实施例132的晶体在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在10.92±0.2、11.70±0.2、12.40±0.2、15.00±0.2、17.38±0.2、18.16±0.2、22.18±0.2、22.62±0.2、23.86±0.2和24.20±0.2的衍射角(2θ)处具有峰。
本发明的实施例132的晶体优选为单苯磺酸盐。
本发明的实施例132的晶体优选为三水合物。
本发明的实施例133的晶体在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在4.64±0.2、7.02±0.2、7.46±0.2、11.14±0.2、14.04±0.2、16.76±0.2、18.54±0.2、19.76±0.2、21.26±0.2和22.62±0.2的衍射角(2θ)处具有峰。
本发明的实施例133的晶体优选为单马来酸盐。
本发明的实施例133的晶体优选为非水合物。
本发明的实施例134的晶体在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在4.80±0.2、7.94±0.2、9.66±0.2、11.56±0.2、14.56±0.2、17.62±0.2、18.14±0.2、20.46±0.2、21.36±0.2和24.46±0.2的衍射角(2θ)处具有峰。
本发明的实施例134的晶体优选为单富马酸盐。
本发明的实施例134的晶体优选为四水合物。
本发明的实施例135的晶体在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在7.14±0.2、8.76±0.2、12.26±0.2、14.30±0.2、17.52±0.2、23.40±0.2、24.40±0.2、24.86±0.2、25.34±0.2和25.90±0.2的衍射角(2θ)处具有峰。
本发明的实施例135的晶体优选为三水合物。
本发明的实施例136的晶体在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在8.06±0.2、12.22±0.2、12.52±0.2、15.14±0.2、17.54±0.2、18.56±0.2、20.08±0.2、23.48±0.2、24.28±0.2和25.00±0.2的衍射角(2θ)处具有峰。
本发明的实施例136的晶体优选为单富马酸盐。
本发明的实施例136的晶体优选为二水合物。
本发明的实施例137的晶体在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在6.56±0.2、9.44±0.2、9.94±0.2、13.20±0.2、18.22±0.2、18.86±0.2、19.60±0.2、22.68±0.2、25.10±0.2和28.70±0.2的衍射角(2θ)处具有峰。
本发明的实施例137的晶体优选为单粘酸盐。
本发明的实施例137的晶体优选为三水合物。
本发明的实施例138的晶体在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在5.88±0.2、6.20±0.2、9.18±0.2、10.34±0.2、12.50±0.2、13.70±0.2、15.66±0.2、17.82±0.2、18.48±0.2和22.16±0.2的衍射角(2θ)处具有峰。
本发明的实施例138的晶体优选为单己二酸盐。
本发明的实施例138的晶体优选为三水合物。
本发明的实施例139的晶体在通过用铜kα线(λ=1.54埃)辐照获得的粉末x射线衍射图中在4.60±0.2、6.60±0.2、7.74±0.2、8.02±0.2、9.26±0.2、11.16±0.2、12.00±0.2、12.44±0.2、13.22±0.2和19.66±0.2的衍射角(2θ)处具有峰。
本发明的实施例139的晶体优选为单琥珀酸盐。
本发明的实施例139的晶体优选为2.5水合物。
本发明包括可在生物体内的生理状态下由于酶、胃酸等的反应而转化为通式(1)表示的化合物(为作为本发明药物组合物的活性成分)的化合物,即,可通过酶促氧化、还原、水解等转化为通式(1)表示的化合物的化合物;以及可通过由于胃酸等的水解等转化为通式(1)表示的化合物的化合物,作为“药学上可接受的前药化合物”。
当通式(1)表示的化合物包含氨基时,前药的实例包括通过使氨基进行酰化、烷基化或磷酸化而获得的化合物(例如,通过使氨基进行二十碳酰化、丙氨酰化、戊基氨基羰基化、(5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲氧基羰基化、四氢呋喃化、吡咯烷基甲基化、特戊酰氧基甲基化或叔丁基化而获得的化合物)等。当通式(1)表示的化合物含有羟基时,前药的实例包括通过使羟基进行酰化、烷基化、磷酸化或硼化而获得的化合物(例如,通过使羟基进行乙酰化、棕榈酰化、丙酰化、特戊酰化、琥珀酰化、富马酰化、丙氨酰化或二甲基氨基甲基羰基化而获得的化合物)等。当通式(1)表示的化合物含有羧基时,前药的实例包括通过使羧基进行酯化或酰胺化而获得的化合物(例如,通过使羧基进行乙基酯化、苯基酯化、羧甲基酯化、二甲基氨基甲基酯化、特戊酰氧基甲基酯化、乙氧基羰基氧基乙基酯化或甲基酰胺化而获得的化合物)等。
本发明的前药可以根据本身已知的方法由通式(1)表示的化合物制备。本发明的前药还包括可以在生理学条件下转化为通式(1)表示的化合物的化合物,如在hirokawashoten出版(1990)的“iyakuhinnokaihatsu(developmentofpharmaceuticals)”,vol.7,designofmolecules,第163-198页中所述。
[制备方法]
接下来,将描述通式(1)表示的化合物的典型制备方法。本发明的化合物可以根据各种制备方法来制备,以下所示的制备方法仅是一些实例,不应将本发明解释为限于这些。
通式(1)表示的化合物、其药学上可接受的盐及其合成中间体可以通过采用各种已知的制备方法,利用基于基本骨架或取代基类型的特性来制备。已知方法的实例包括在“organicfunctionalgrouppreparations”,第2版,academicpress,inc.,1989,“comprehensiveorganictransformations”,第2版,vchpublishersinc.,1999等中描述的方法。
在制备中,取决于化合物中所含官能团的类型,在制备技术中在原料或中间体阶段用适当的保护基保护该官能团或用可容易地转化为该官能团的基团替代有时可能是有效的。
官能团的实例包括氨基、羟基、甲酰基、羰基和羧基等,并且保护基的实例包括在“protectivegroupsinorganicsynthesis”(第5版,wiley,p.g.wuts撰写,2014年)中描述的保护基。
可以根据用于制备化合物的制备方法的反应条件适当地选择保护基或可以容易地转化为官能团的基团。
根据这种方法,在引入基团并进行反应之后,如果需要,可以通过除去保护基或将该基团转化为期望的基团来获得期望的化合物。
化合物的前药可以通过在原料或中间体的阶段引入特定的基团来制备,或者如上述保护基的情况那样,通过使获得的化合物引入该基团来制备。可以通过采用本领域技术人员已知的常规方法,例如酯化、酰胺化、脱水、氢化等来进行制备前药的反应。
通式(1)表示的化合物可以例如根据以下方法a至e来制备。方法a至方法e中使用的合成中间体可以例如根据以下方法f至y来制备。
当在以下方法a至y的每个步骤中用作反应中的反应底物的化合物具有抑制所需反应的官能团或部分结构(例如氨基、羟基、甲酰基、羰基、羧基、环状化合物上的杂原子等)时,可以根据需要适当地向其中引入保护基或从中除去引入的保护基。这样的保护基没有特别限制,只要其是通常使用的保护基即可,并且可以是例如上述“protectivegroupsinorganicsynthesis(2014年第5版)”中所述的保护基。可以根据上述文献中描述的常规方法进行引入和除去这些保护基的反应。
在方法a至y的每种化合物中,取决于化合物中包含的官能团的类型,可以在原料或中间体阶段用可以容易地转化为该官能团的基团替代该官能团。可以根据已知方法在适当的阶段进行向期望的官能团的转化。已知方法的实例包括在上述文献“organicfunctionalgrouppreparations”、“comprehensiveorganictransformations”等中描述的方法。
在以下方法a至y的每种化合物中,以非溶剂化物、盐或各种溶剂化物(例如水合物)中的任一种的形式分离和纯化。盐可以根据常规方法制备。盐的实例包括盐酸盐、硫酸盐等;有机胺盐;以及钠盐、钾盐等。
在以下方法a至y的每个步骤中用于反应的溶剂没有特别限制,只要其不抑制反应而是部分地溶解起始原料即可,并且例如选自以下溶剂组。溶剂组包括脂族烃,例如正己烷、正戊烷、石油醚和环己烷;芳烃,例如苯、甲苯和二甲苯;卤代烃,例如二氯甲烷(亚甲基氯)、氯仿、四氯化碳、二氯乙烷、氯苯和二氯苯;醚,例如乙醚、二异丙醚、四氢呋喃(thf)、二噁烷、二甲氧基乙烷和二乙二醇二甲醚;酮,例如丙酮、甲乙酮、甲基异丁基酮和环己酮;酯,例如乙酸乙酯、乙酸丙酯、乙酸丁酯;腈,例如乙腈、丙腈、丁腈和异丁腈;羧酸,例如乙酸和丙酸;醇,例如甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇、2-甲基-1-丙醇和2-甲基-2-丙醇;酰胺,例如甲酰胺、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺、n-甲基-2-吡咯烷酮、六甲基磷酰三胺;亚砜,例如二甲基亚砜(dmso)和四氢噻吩1,1-二氧化物;水;及其混合物。
在以下方法a至y的每个步骤中用于反应的酸没有特别限制,只要其不抑制反应即可,并且选自以下酸组。酸组包括无机酸,例如盐酸、氢溴酸、氢碘酸、磷酸、硫酸和硝酸;有机酸,例如乙酸、丙酸、三氟乙酸和五氟丙酸;有机磺酸,例如甲磺酸、三氟甲磺酸、对甲苯磺酸和樟脑磺酸;和路易斯酸,例如三溴化硼、溴化铟(ⅲ)、三氟化硼、氯化铝(ⅲ)和三氟甲磺酸三甲基甲硅烷基酯。
在以下方法a至y的每个步骤中用于反应的碱没有特别限制,只要其不抑制反应即可,并且选自以下碱组。碱组包括碱金属碳酸盐,例如碳酸锂、碳酸钠、碳酸钾和碳酸铯;碱金属碳酸氢盐,例如碳酸氢锂、碳酸氢钠和碳酸氢钾;碱金属氢氧化物,例如氢氧化锂、氢氧化钠和氢氧化钾;碱土金属氢氧化物,例如氢氧化钙和氢氧化钡;碱金属氢化物,例如氢化锂、氢化钠和氢化钾;碱金属氨基化物,例如氨基锂、氨基钠和氨基钾;碱金属醇盐,例如甲醇锂、甲醇钠、乙醇钠、叔丁醇钠和叔丁醇钾;烷基氨基锂,例如二异丙基氨基锂;甲硅烷基氨基化物,例如双三甲基甲硅烷基氨基锂和双三甲基甲硅烷基氨基钠;烷基锂,例如正丁基锂、仲丁基锂和叔丁基锂;烷基卤化镁,例如甲基氯化镁类(甲基氯化镁)、甲基溴化镁类(甲基溴化镁)、甲基碘化镁类(甲基碘化镁)、乙基氯化镁类(乙基氯化镁)、乙基溴化镁类(乙基溴化镁)、异丙基氯化镁类(异丙基氯化镁)、异丙基溴化镁类(异丙基溴化镁)和异丁基氯化镁类(异丁基氯化镁);和有机胺,例如三乙胺(tea)、三丁胺、n,n-二异丙基乙胺(dipea)、1-甲基哌啶、4-甲基吗啉、4-乙基吗啉、吡啶、甲基吡啶、4-二甲基氨基吡啶、4-吡咯烷子基吡啶、2,6-二叔丁基-4-甲基吡啶、喹啉、n,n-二甲基苯胺、n,n-二乙基苯胺、1,5-二氮杂双环[4,3,0]-5-壬烯(dbn)、1,4-二氮杂双环[2,2,2]辛烷(dabco)、1,8-二氮杂双环[5,4,0]-7-十一碳烯(dbu)和咪唑。
在以下方法a至y的每个步骤的反应中,反应温度取决于溶剂、起始原料、试剂等,并且反应时间取决于溶剂、起始原料、试剂,反应温度等。
在以下方法a至y的每个步骤的反应中,完成反应后,通过常规方法从反应混合物中分离出每个步骤的目标化合物。例如,通过以下方法获得目标化合物:(i)如果需要,过滤掉不溶性物质例如催化剂等,(ii)通过向反应混合物中加入水和与水不混溶的溶剂(例如二氯甲烷、乙醚、乙酸乙酯等)来萃取目标化合物,(iii)用水洗涤有机层,并用干燥剂例如无水硫酸钙等干燥所得产物,和(iv)蒸发溶剂。如果需要,可以通过常规方法,例如重结晶、再沉淀、蒸馏或使用硅胶、氧化铝等的柱色谱法(包括正相色谱法和反相色谱法)进一步纯化所获得的目标化合物。通过标准分析技术,例如元素分析、nmr、质谱、ir分析等鉴定所获得的目标化合物,并且因此可以分析其组成或纯度。或者,每个步骤中获得的目标化合物可以不经纯化直接用于下一反应。
在以下方法a至y的每个步骤中,可以通过使用旋光胺例如(r)-(+)-或(s)-(-)-1-苯乙胺等或旋光羧酸例如(+)-或(-)-10-樟脑磺酸等的分步重结晶或通过使用旋光柱的分离,来分离和纯化旋光异构体。
通式(1)表示的化合物的氘(2h;d)取代基可以例如通过在以下方法a至y中的适当阶段采用本领域技术人员通常使用的方法来制备。本领域技术人员通常使用的方法的实例包括nature,2007,446,526-529.,angew.chem.int.ed.,2007,46,7744-7765.,j.med.chem.,2009,52,7993-8001.等中描述的方法。
用于制备本发明化合物的方法a至y中使用的原料和试剂可以是已知化合物,或者可以根据已知方法或与其类似的方法由已知化合物作为起始原料来制备。已知的起始原料化合物也可以从商业供应商处购买。
本文使用的缩写
boc:叔丁氧基羰基
cbz:苄氧羰基
alloc:烯丙氧基羰基
ns:2-硝基苯磺酰基(nosyl)
mom:甲氧基甲基
tms:三甲基甲硅烷基
otf:三氟甲基磺酰氧基
tr:三苯甲基
pmb:对甲氧基苄基
bop:六氟磷酸(苯并三唑-1-基氧基)三(二甲基氨基)鏻
hatu:1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐
comu:六氟磷酸n-[1-(氰基-2-乙氧基-2-氧代乙叉氨基氧基)二甲基氨基(吗啉代)]脲鎓
edc:1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐
hobt:1-羟基苯并三唑
dppa:二苯基磷酰基叠氮化物。
通式(1)表示的化合物可以根据以下所示的方法来制备。
方法a
[化学式57]
在该方案中,pg为氨基的保护基,其实例包括boc基团、cbz基团、ns基团、alloc基团等。也可以使用上述“protectivegroupsinorganicsynthesis”等中描述的保护基。lg为离去基团,其实例包括卤素原子、对甲苯磺酰基等。
步骤a-1是从中间体i和中间体ii获得中间体iii的步骤。该步骤可以通过在对反应呈惰性的溶剂(例如异丙醇等)中在碱(例如dipea等)存在下加热中间体i和中间体ii来进行。
步骤a-2是去除pg的步骤。当pg为cbz基团时,可以通过在对反应呈惰性的溶剂(例如乙腈等)中用酸(例如三甲基碘硅烷等)处理中间体iii来进行该步骤。当pg为boc基团时,可以通过在对反应呈惰性的溶剂(例如二氯甲烷等)中用酸(例如盐酸等)处理中间体iii来进行该步骤。当pg为ns基团时,该步骤可通过使中间体iii与硫醇(例如异丙基苯硫醇等)和碱(例如碳酸铯等)在对反应呈惰性的溶剂(例如thf和甲醇的混合溶剂等)中反应来进行。另外,也可以应用上述“protectivegroupsinorganicsynthesis”中描述的方法。
步骤a-3是从中间体iv和中间体v获得其中r2是氢原子的通式(1)表示的化合物的步骤。该步骤可以通过使中间体iv和中间体v与还原剂(例如三乙酰氧基硼氢化钠、氰基硼氢化钠等)在对反应呈惰性的溶剂(例如二氯甲烷、二氯乙烷等)中反应来进行。也可以使用催化剂例如四异丙氧基钛等来促进反应。
步骤a-4是将其中r2为氢原子的通式(1)表示的化合物的r2转化为c1-6烷基的步骤。该步骤可以通过使化合物与烷基化剂(例如三氟甲磺酸甲酯等)和碱(例如吡啶等)在对反应呈惰性的溶剂(例如二氯甲烷等)中反应来进行。
中间体iii也可以使用中间体i和中间体ii'制备(方法b)。
方法b
[化学式58]
在该方案中,pg如上所定义。
步骤b可以通过使中间体i和中间体ii'与缩合剂(例如bop试剂等)和碱(例如dbu等)在对反应呈惰性的溶剂(例如乙腈等)中反应、然后加热混合物来进行。
通式(1)表示的化合物也可以由中间体iv和中间体v'制备(方法c)。中间体v'可以例如根据cancercell,2015,27,589-602中描述的方法制备。
方法c
[化学式59]
在该方案中,lg如上所定义。
步骤c-1是从中间体iv和中间体v'获得其中r2是氢原子的通式(1)表示的化合物的步骤。该步骤可以通过使中间体iv和中间体v'与碱(例如碳酸钾等)在对反应呈惰性的溶剂(例如dmf等)中反应来进行。当中间体v'的结构中存在保护基时,也可以在步骤c-1之后在合适的反应条件(例如,与四氯化锡等反应的方法,在例如乙腈等溶剂中,在boc基团作为保护基的情况下)下进行脱保护以转化为所需的结构。
步骤c-2是将其中r2为氢原子的通式(1)表示的化合物的r2转化为c1-6烷基的步骤。该步骤可以以与步骤a-4相同的方式进行。当中间体v'的结构中存在保护基时,也可以在步骤c-2之后在合适的反应条件(例如,与四氯化锡等反应的方法,在例如乙腈等溶剂中,在boc基团作为保护基的情况下)下进行脱保护以转化为所需的结构。
上述方法a至c中所示的每个步骤不必以相同的顺序进行,只要它不影响反应底物和反应产物即可,例如,其可以按照以下顺序进行(方法d)。
方法d
[化学式60]
在该方案中,pg和lg如上所定义。
当通式(1)表示的化合物由以下化合物(1')表示时,它也可以通过以下中间体viii(方法e)制备。
方法e
[化学式61]
在该方案中,re1是可以在下述交叉偶联反应(步骤e-2)中反应的取代基,例如卤素原子(例如溴、碘等)、三氟甲基磺酰氧基(otf基团)等。re2是二羟硼基、二烷氧基硼烷基(例如二甲氧基硼烷基等)、二氧杂硼杂环戊基(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基等)等。z1为-nh-或单键。
步骤e-1是由中间体iv和化合物1e制备中间体viii的步骤。该步骤可以以与步骤a-3相同的方式进行。
步骤e-2是从中间体viii和化合物2e和3e中的任何一种获得其中r2是氢原子的通式(1')表示的化合物的步骤。该步骤可通过在氮气气氛下在金属催化剂(例如双(三苯基膦)二氯化钯等)和碱(例如碳酸钾等)的存在下,在对反应呈惰性的溶剂(例如二甲氧基乙烷和水的混合溶剂等)中加热中间体viii和化合物2e和3e中的任何一种来进行。
步骤e-3是将其中r2为氢原子的通式(1')表示的化合物中的r2转化为c1-6烷基的步骤。该步骤可以以与步骤a-4相同的方式进行。
下面将描述各中间体的制备方法。
将描述中间体i和中间体i'的制备方法。以下所示的制备方法仅是实例,不应解释为局限于这些方法。
[化学式62]
在该方案中,pg如上所定义。
如果需要,中间体i或中间体i'也可以通过将(a)在氮原子上引入单独的保护基和(b)除去不必要的保护基的两个步骤适当地组合而相互转化。这些步骤是保护基的一般转化反应,并且可以例如通过采用上述“protectivegroupsinorganicsynthesis”中描述的方法来进行。
当中间体i由以下化合物i-1、i-2、i-3或i-4表示时,其可以例如根据方法f或i制备。起始原料1f是已知的,或根据已知方法或与其类似的方法使用已知化合物作为起始原料来制备。已知化合物可从商业供应商处购买。已知文献的实例包括tetrahedronasymmetry.2013,24,651-656,tetrahedron.2004,60,717-728,bioorg.med.chem.2006,14,2242-2252,tetrahedron.2017,73,1381-1388等。化合物1f也可以根据方法g合成。
方法f
[化学式63]
在该方案中,rf1和rf2各自独立地为c1-6烷基或被保护的羟基c1-6烷基(例如2-[叔丁基(二甲基)甲硅烷基]氧基乙基等)。pg和pg'各自独立地是在氮原子上引入的彼此不同的保护基,其实例包括boc基团、cbz基团、alloc基团等,也可以使用上述“protectivegroupsinorganicsynthesis(2014年第5版)等中描述的保护基。
步骤f-1是从化合物1f合成化合物2f的步骤。该步骤可以通过将化合物1f与叠氮化剂(例如二苯基磷酰叠氮化物(dppa)等)、碱(例如三乙胺等)和醇(例如苄醇、烯丙醇等)在对反应呈惰性的溶剂(例如甲苯等)中一起加热来进行。
步骤f-2是从化合物2f合成中间体i-1或i-2的步骤(如果需要,要除去的保护基可以是pg或pg)。可以在与步骤a-2相同的条件下进行这两个步骤。
当中间体i或i'的r5为rf1或rf2时,可以通过进行步骤f-2、然后进行步骤f-3至f-5来制备中间体i-3或i-4。
步骤f-3是从中间体i-1合成化合物3f-1的步骤,或者是从中间体i-2合成化合物3f-2的步骤。这两个步骤都可以通过,例如,使中间体i-1或中间体i-2与硝基苯磺酰化试剂(例如2-硝基苯磺酰氯等)和碱(例如dipea等)在对反应呈惰性的溶剂(例如二氯甲烷等)中反应来进行。
步骤f-4是从化合物3f-1合成化合物4f-1的步骤,或者是从化合物3f-2合成化合物4f-2的步骤。例如,这两个步骤可通过使化合物3f-1或化合物3f-2与烷基化剂(例如碘甲烷等)和碱(例如碳酸钾等)在对反应呈惰性的溶剂(例如dmf等)中反应来进行。
步骤f-5是从化合物4f-1合成中间体i-3的步骤,或者是从化合物4f-2合成中间体i-4的步骤。这两个步骤可以例如通过使化合物4f-1或化合物4f-2与硫醇(例如异丙基苯硫醇等)和碱(例如碳酸铯等)在对反应呈惰性的溶剂(例如thf和甲醇的混合溶剂等)中反应来进行。
当在r3或r4中存在具有保护基的官能团时,可以在进行步骤f-2的同时去除保护基。
如果需要,中间体i-1至i-4还可通过将(a)在氮原子上引入单独的保护基和(b)除去不必要的保护基的两个步骤适当地组合而相互转化为所需的化合物,所述化合物可用作中间体i或其他步骤的起始原料。这些步骤是保护基的一般转化反应,并且可以例如通过采用上述“protectivegroupsinorganicsynthesis”中描述的方法来进行。
当通式(1f)表示的化合物由以下化合物1f-1表示时,其可以根据方法g制备。起始原料是已知的,或者使用已知化合物作为起始原料根据已知方法或与其类似的方法制备。
方法g
[化学式64]
在该方案中,pg'如上所定义。rg1是羧基的保护基(例如甲基、乙基、mom基等)。rg2为c1-6烷基(例如甲基、乙基等)或羟基的保护基(例如mom等)。
步骤g-1可以通过使化合物1g-1或化合物1g-2与烷基化剂(例如氯甲基甲基醚等)和碱(例如dipea等)在反应促进剂(例如碘化钠等)存在下在对反应呈惰性的溶剂(例如二甲氧基乙烷等)中反应、然后加热混合物来进行。在该步骤中,化合物1g-1或化合物1g-2可以用作原料。虽然该步骤的反应条件取决于底物,但是该步骤也可以通过使化合物1g-1或化合物1g-2与烷基化剂(例如碘甲烷等)在金属催化剂(例如氧化银(i))和添加剂(例如分子筛等)存在下在对反应呈惰性的溶剂(例如二氯甲烷等)中反应、然后加热混合物来进行。将化合物1g-1转化为化合物2g-1的步骤也可以分两个步骤进行:羧基的保护和羟基的保护。
步骤g-2是由化合物2g-1或化合物2g-2合成化合物1f-1的步骤。该步骤例如可以通过在对反应呈惰性的溶剂(例如甲醇与thf的混合溶剂等)中用碱(例如氢氧化钠水溶液等)处理化合物2g-1或化合物2g-2来进行。
中间体i'-1可以如下制备(方法h)。可以例如根据方法f合成原料化合物1h。
方法h
[化学式65]
在该方案中,pg和pg'如上所定义。rh为c1-6烷基。
可以以与步骤g-1相同的方式进行步骤h-1。
可以以与步骤a-2相同的方式进行步骤h-2。
当中间体i由以下化合物i-5表示时,其也可以如下制备(方法i)。原料化合物1i可以例如根据方法g合成。
方法i
[化学式66]
在该方案中,ri1和ri2各自独立地为c1-6烷基。
步骤i-1是从化合物1i获得化合物2i的步骤。该步骤可以通过使化合物1i与化合物7i在缩合剂(例如edc等)、催化剂(例如hobt等)和碱(例如三乙胺等)的存在下在对反应呈惰性的溶剂(例如二氯甲烷等)中反应来进行。
步骤i-2是从化合物2i获得化合物3i的步骤。该步骤可以通过在对反应呈惰性的溶剂(例如1,4-二噁烷等)中用酸(例如盐酸等)处理化合物2i来进行。
步骤i-3是从化合物3i获得化合物4i的步骤。该反应可以通过使化合物3i与羧酸(例如4-硝基苯甲酸等)在膦化合物(例如三苯基膦等)和偶氮二甲酸酯化合物(例如偶氮二甲酸二异丙酯等)的存在下在对反应呈惰性的溶剂(例如thf等)中反应来进行。
步骤i-4是从化合物4i获得化合物5i的步骤。该步骤可以通过在对反应呈惰性的溶剂(例如乙醇等)中用碱(例如碳酸钾等)处理化合物4i来进行。
步骤i-5是从化合物5i获得化合物6i的步骤。该步骤可以通过使化合物5i与二苯基磷酰基叠氮化物(dppa)在偶氮二甲酸酯化合物(例如偶氮二甲酸二异丙酯(diad)等)存在下在对反应呈惰性的溶剂(例如thf等)中反应来进行。
步骤i-6是从化合物6i获得中间体i-5的步骤。该步骤可以通过使化合物6i与还原剂(例如三苯基膦等)在对反应呈惰性的溶剂(例如thf等)中反应、然后用水处理所得物并将其加热来进行。
接下来,将描述中间体ii和ii'的制备方法。以下所示的制备方法仅是实例,不应解释为局限于这些方法。
[化学式67]
中间体ii和ii'是已知的,或根据已知方法或与其类似的方法使用已知化合物作为起始原料来制备。已知文献的实例包括nat.chem.biol.2012,8,277-284.,cancercell.2015,27,589-602.,j.med.chem.2016,59(3),892-913.,wo2017//214367,wo2016/195776,wo2012/097013,j.heterocyclicchem.2005,42(4),509-513.,j.med.chem.2001,44(17),2695-2700等。
当中间体ii由以下化合物ii-1或ii'-1表示时,其也可以根据以下方法制备(方法j)。原料化合物1j是已知的,或根据已知方法或与其类似的方法使用已知化合物作为起始原料来制备。已知文献的实例包括wo2004/007491。
方法j
[化学式68]
在该方案中,rj1为氯原子或甲氧基。rj2和rj3都是碳原子,或者rj2是硫原子并且rj3是键。
步骤j-1是从化合物1j获得化合物2j的步骤。该步骤可以通过使化合物1j与(三氟甲基)三甲基甲硅烷(ruppert试剂)和作为氟离子源的试剂(例如四丁基氟化铵等)在对该反应呈惰性的溶剂(例如thf等)中反应来进行。
步骤j-2是从化合物2j获得化合物3j的步骤。该步骤可以通过在对反应呈惰性的溶剂(例如四氢呋喃等)中用酸(例如盐酸等)处理化合物2j来进行。
步骤j-3是从化合物3j获得化合物4j的步骤。该步骤可以通过使化合物3j与氯代硫羰碳酸苯酯和碱(例如tea等)在对反应呈惰性的溶剂(例如二氯甲烷等)中反应来进行。
步骤j-4是从化合物4j获得中间体ii-1的步骤。该步骤可以通过使化合物4j与自由基还原剂(例如氢化三丁基锡等)和自由基引发剂(例如偶氮二(异丁腈)等)在对反应呈惰性的溶剂(例如甲苯等)中反应来进行。
步骤j-5是从其中rj1为甲氧基的中间体ii-1获得其中rj1为羟基的中间体ii'-1的步骤。该步骤可通过在对反应呈惰性的溶剂(例如thf等)中用酸(例如盐酸等)处理其中rj1为甲氧基的中间体ii-1来进行。
当中间体iii由以下化合物iii-1或iii-2表示时,其也可以根据以下方法(方法k)制备。原料化合物1k是已知的,或根据已知方法或与其类似的方法使用已知化合物作为起始原料来制备。已知文献的实例包括eur.j.org.chem.2013,17,3477-3493。
方法k
[化学式69]
可以以与步骤f-1至f-5相同的方式进行步骤k-1至k-5,并且可以以与步骤a-1相同的方式进行步骤k-6。在步骤k-6的纯化过程中,还可以通过使用旋光柱例如chiralpak(注册商标,daicelco.,ltd.)-ia、ib、ic、id等除去在一系列步骤中作为副产物产生的各种异构体。作为展开剂,可以使用正己烷、乙醇、异丙醇等。
步骤k-7是从化合物7k获得中间体iii-1和iii-2的步骤。该步骤可以通过使化合物7k与催化剂(例如四氧化锇等)和氧化剂(例如4-甲基吗啉n-氧化物等)在对反应呈惰性的溶剂(例如丙酮和水的混合溶剂等)中反应来进行。
当中间体iii由以下化合物iii-3表示时,其也可以根据以下方法(方法l)制备。原料化合物11是已知的,或根据已知方法或与其类似的方法使用已知化合物作为起始原料来制备。已知文献的实例包括bioorg.med.chem.2006,14,2242-2252。
方法l
[化学式70]
可以以与步骤i-3至i-6相同的方式进行步骤l-1至步骤l-4,可以以与步骤a-1相同的方式进行步骤l-5,和可以以与步骤g-2相同的方式进行步骤l-6。
步骤l-7是从化合物4l获得化合物5l的步骤。该步骤可以通过使化合物4l与化合物7l在缩合剂(例如comu等)和碱(例如dipea等)的存在下在对反应呈惰性的溶剂(例如dmf等)中反应来进行。
步骤l-8是从化合物5l获得化合物6l的步骤。该步骤可以通过在对反应呈惰性的溶剂(例如二氯甲烷等)中用酸(例如三氟乙酸等)处理化合物5l来进行。
步骤l-9是从化合物6l获得中间体iii-3的步骤。该步骤可以通过使化合物61与膦化合物(例如三苯基膦等)、六氯乙烷和碱(例如三乙胺等)在对该反应呈惰性的溶剂(例如二氯甲烷等)中反应来进行。
当中间体iii由以下化合物iii-4表示时,其可以根据以下方法(方法m)制备。化合物1m可以根据已知方法(wo2017//214367)合成。硼酸(3m)是已知化合物,或者可以根据已知方法或与其类似的方法使用已知化合物作为起始原料来制备。
方法m
[化学式71]
在该方案中,rm1为芳环基。
步骤m-1是从中间体i和化合物1m获得化合物2m的步骤。该步骤可以以与步骤a-1相同的方式进行。
步骤m-2是从化合物2m获得中间体iii-4的步骤。该步骤可以通过在惰性气体气氛下在对反应呈惰性的溶剂(例如二噁烷和水的混合溶剂等)中使用金属催化剂(例如四(三苯基膦)钯等)和碱(例如碳酸钠等)加热化合物2m来进行。在此步骤中,可以使用硼酸酯代替硼酸(3m)。
当中间体iii由以下化合物iii-5表示时,其可以例如根据以下方法(方法n)制备。化合物1n可以以与方法a的步骤a-1相同的方式制备。
方法n
[化学式72]
在该方案中,rn1为c1-6烷基、对甲氧基苄基等。
步骤n是从化合物1n获得中间体iii-5的步骤。该步骤可以通过使化合物1n与胺(例如甲胺、对甲氧基苄基胺等)在加热下(合意地,使用微波反应器等加热到溶剂的沸点以上)在对反应呈惰性的溶剂(例如丁腈等)中反应来进行。
接下来,将描述中间体v的制备方法。中间体v是已知的,或根据已知方法或与其类似的方法使用已知化合物作为起始原料制备。已知文献的实例包括cancercell.2015,27,589-602,j.med.chem.2016,59(3),892-913,wo2007/118041,wo2014/164749等。
由于中间体v在环q1上具有由r1c(=o)-(甲酰基或c1-6烷基羰基)表示的官能团,因此其也可以衍生自具有可容易地转化为该官能团的基团(在甲酰基的情况下,其实例包括羟甲基、c1-6烷氧基羰基、羧基等,并且在c1-6烷基羰基的情况下,其实例包括乙酰基(acetyl)、乙酰基(ethanoyl)等)的前体。
中间体v也可以根据方法o至方法y制备。
[化学式73]
中间体v由以下化合物v-1表示,其也可以例如根据方法o制备。
方法o
[化学式74]
在该方案中,ro1为卤素原子(例如氯、溴或碘)或三氟甲基磺酰氧基。ro2是可容易地转化为甲酰基的官能团,其实例包括羟甲基、c1-6烷氧基羰基、羧基、缩醛基等。环q4是在环中含有氮原子的杂环(该杂环任选地具有一个或多个取代基),其实例包括哌啶环等。
步骤o-1是从化合物1o和2o获得化合物3o的步骤。该步骤可通过在惰性气体气氛下在金属催化剂(例如三(二亚苄基丙酮)二钯(0)和(±)-2,2'-双(二苯基膦基)-1,1'-联萘等)和碱(例如叔丁醇钠、碳酸铯、三乙胺等)的存在下在对反应呈惰性的溶剂(例如甲苯等)中加热化合物1o和2o来进行。
步骤o-2是将化合物3o转化为中间体v-1的步骤。例如,当ro2为羟甲基时,其可以通过使化合物3o与氧化剂(氯铬酸吡啶鎓、dess-martin氧化剂、氧化锰(iv)等)在对反应呈惰性的溶剂(例如二氯甲烷、氯仿、dmso等)中反应来进行。当ro2为另一官能团(例如c1-6烷氧基羰基、羧基、缩醛基等)时,可以根据上述“organicfunctionalgrouppreparations”、“comprehensiveorganictransformations”等中描述的方法进行该官能团向甲酰基的转化。
当中间体v由以下化合物v-2表示时,其也可以例如根据方法p制备。
方法p
[化学式75]
在该方案中,rp1是卤素原子(例如氯、溴或碘)或三氟甲基磺酰氧基。rp2是可容易地转化为甲酰基的官能团,其实例包括羟甲基、c1-6烷氧基羰基、羧基、缩醛基等。环q5是在环中含有氮原子的杂环(该杂环任选地具有一个或多个取代基),其实例包括二氢吲哚环等。
步骤p-1是从化合物1p和化合物2p获得化合物3p的步骤。该步骤可以以与步骤o-1相同的方式进行。
步骤p-2是从化合物3p获得中间体v-2的步骤。例如,当rp2为缩醛基时,其可以通过在对反应呈惰性的溶剂(例如thf等)中用酸(例如盐酸等)处理化合物3p来进行。
中间体v由以下化合物v-3表示,其也可以例如根据方法q制备。
方法q
[化学式76]
在该方案中,rq1和rq2中的一个是氨基,并且rq1和rq2中的另一个是卤素原子(例如氯、溴或碘)或三氟甲基磺酰氧基。rq3为甲酰基或可容易地转化为甲酰基的官能团,其实例包括羟甲基、c1-6烷氧基羰基、羧基、缩醛基等。
步骤q-1是从化合物1q和化合物2q获得化合物3q的步骤。该步骤可以通过在惰性气体气氛下在金属催化剂(例如三(二亚苄基丙酮)二钯(0)和叔丁基膦的组合,或四(三苯基膦)钯(0)等)和碱(例如叔丁醇钠、碳酸钠等)的存在下在对反应呈惰性的溶剂(例如甲苯、二噁烷和水的混合溶剂,二甲氧基乙烷和水的混合溶剂等)中加热化合物1q和化合物2q来进行。
步骤q-2是从化合物3q获得中间体v-3的步骤。例如,当rq3为缩醛基时,其可以通过在对反应呈惰性的溶剂(例如thf等)中用酸(例如盐酸等)处理化合物3q来进行。当rq3是甲酰基时,不需要该步骤。
中间体v由以下中间体v-4表示,其也可以例如根据方法r制备。
方法r
[化学式77]
在该方案中,rr1和rr2中的一个是卤素原子(例如氯、溴或碘)或三氟甲基磺酰氧基,并且rr1和rr2的另一个是二羟硼基、二烷氧基硼烷基(例如二甲氧基硼烷基等)、二氧杂硼杂环戊基(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基等)等。rr3为甲酰基或可容易地转化为甲酰基的官能团,其实例包括羟甲基、c1-6烷氧基羰基、羧基、缩醛基等。
步骤r-1是从化合物1r和化合物2r获得化合物3r的步骤。该步骤可以通过在惰性气体气氛下在金属催化剂(例如三(二亚苄基丙酮)二钯(0)和叔丁基膦的组合,或四(三苯基膦)钯(0)等)和碱(例如叔丁醇钠、碳酸钠、磷酸三钾等)的存在下在对反应呈惰性的溶剂(例如甲苯、二噁烷和水的混合溶剂,二甲氧基乙烷和水的混合溶剂等)中加热化合物1r和化合物2r来进行。
步骤r-2是从化合物3r获得中间体v-4的步骤。例如,当rr3为c1-6烷氧基羰基时,可以根据以下方法v进行该步骤。当rr3为甲酰基时,不需要该步骤。
方法o至方法r中使用的原料化合物是已知的,或根据已知方法或与其类似的方法使用已知化合物作为起始原料制备。已知文献的实例包括wo2014/078813,synlett.2015,26(7),953-959,j.med.chem.2014,57(19),8086-8098,eur.j.inorg.chem.2015,28,4666-4677,wo2004/108690,wo2015/0291572,wo2010/141796,wo2013/093849,j.med.chem.2014,57(19),8086-8098,j.org.chem.2014,79,10311-10322,wo2011/109267,wo2007/013673,tetrahedron.2015,71(49),9240-9244,“organicfunctionalgrouppreparations”,第2版,academicpress,inc.,1989,“comprehensiveorganictransformations”,vchpublishersinc.,1989等。
化合物1r也可以根据以下方法s或方法t制备。
当化合物1r由以下化合物2s或3s表示时,其可以例如根据方法s制备。
方法s
[化学式78]
在该方案中,rs1、rs2和rs3各自独立地为c1-6烷基(例如甲基、乙基等)、芳环基团(例如吡啶基等)。rs2和rs3任选地与rs2和rs3所键合的氮原子一起形成环。
步骤s-1是从化合物1s获得化合物2s的步骤。该步骤可以通过使化合物1s与化合物4s在碱(例如氢化钠、碳酸钾等)存在下在对反应呈惰性的溶剂(例如甲苯、dmf等)中反应来进行。
步骤s-2是从化合物1s获得化合物3s的步骤。该步骤可以通过使化合物1s与化合物5s在碱(例如二异丙胺等)存在下在对反应呈惰性的溶剂(例如thf等)中反应来进行。
当化合物1r由以下化合物3t至5t中的任一个表示时,其也可以例如根据方法t制备。
方法t
[化学式79]
步骤t-1是从化合物1s获得化合物1t和化合物2t的步骤。该步骤可以通过使化合物1s与碱(例如乙酸钾等)在对该反应呈惰性的溶剂(例如乙酸和水的混合溶剂等)中反应、然后加热混合物来进行。例如,通过利用溶解度的差异,可以将所得的区域异构体1t和2t彼此分离。
步骤t-2是从化合物1t获得化合物3t的步骤。该步骤可以通过使化合物1t与碱(例如氢氧化钾水溶液等)和二氟甲基化剂(例如二氟甲基三氟甲磺酸等)在对反应呈惰性的溶剂(例如乙腈等)中反应来进行。
步骤t-3是从化合物2t获得化合物4t和5t的步骤。该步骤可以以与步骤t-2相同的方式进行。所得异构体4t和5t可以例如通过使用硅胶柱色谱法等的方法彼此分离。
当中间体v由以下化合物v-5表示时,其可以例如根据以下方法(方法u)制备。
方法u
[化学式80]
在该方案中,环q6是在环中含有氮原子的杂环(该杂环任选地具有一个或多个取代基),并且其实例包括哌啶环、氮杂环庚烷环等。
步骤u是从化合物1u和化合物2u获得中间体v-5的步骤。该步骤可以通过使化合物1u和化合物2u与胺(例如dipea等)在对反应呈惰性的溶剂(例如二氯甲烷等)中反应来进行。
当中间体v由以下化合物v-6表示时,其也可以例如根据以下方法(方法v)制备。起始原料是已知的,或根据已知方法或与其类似的方法使用已知化合物作为起始原料来制备。已知文献的实例包括wo2014/114186,wo2005/108399,j.med.chem.2016,59(18),8233-8262,wo2005/032488,science.2016,352(6291),1304-1308,bioorg.med.chem.let.2006,16(19),4987-4993,helveticachimicaacta.2007,90(6),1043-1068,tetrahedron.2015,71(49),9240-9244,“organicfunctionalgrouppreparations”,第2版,academicpress,inc.,1989,“comprehensiveorganictransformations”,vchpublishersinc.,1989等。
方法v
[化学式81]
在该方案中,rv1为氢原子或烷基(例如甲基、乙基等)。
步骤v-1是从化合物1v获得化合物2v的步骤。该步骤可以例如通过使化合物1v与还原剂(例如氢化铝锂、硼氢化锂等)在对反应呈惰性的溶剂(例如thf等)中反应来进行。
步骤v-2是从化合物2v获得中间体v-6的步骤。该步骤可以例如通过使化合物2v与氧化剂(例如氯铬酸吡啶鎓、dess-martin氧化剂、氧化锰(iv)等)在对反应呈惰性的溶剂(例如二氯甲烷、氯仿、dmso等)中反应来进行。
步骤v-3是从化合物1v获得中间体v-6的步骤。该步骤可以通过使化合物1v与还原剂(例如二异丁基氢化铝等)在对反应呈惰性的溶剂(例如二氯甲烷等)中反应来进行。
当中间体v由以下化合物v-7表示时,其可以例如根据方法w制备。
方法w
[化学式82]
在该方案中,rw1为任选地具有一个或多个取代基的c1-6烷基(例如3-(叔丁氧基羰基氨基)丙基、1-甲基吡唑-4-基甲基等)。
步骤w是从化合物1w和2w获得中间体v-7的步骤。该步骤可以以与步骤i-1相同的方式进行。
当中间体v具有官能团时,在通过已知方法将该官能团转化为所需官能团之后,也可以使用中间体v。已知方法的实例包括在“organicfunctionalgrouppreparations”、“comprehensiveorganictransformations”和“protectivegroupsinorganicsynthesis(第5版,2014)”中描述的方法,在j.med.chem.2014,57(18),7590-7599,synlett.2015,26(7),953-959,wo2013/013503,angew.chem.int.ed.2015,54(33),9668-9672,org.lett.2012,14(14),3700-3703,wo2017//100668等中描述的方法,并且可以进行各种反应,例如硝基还原、acroylation、磺酰化、甲基化、脱保护等。
当通式(1)表示的化合物具有官能团时,该官能团也可以通过已知方法转化为所需的官能团。例如,可以进行保护或脱保护、官能团的转化或修饰等。已知方法的实例包括在“organicfunctionalgrouppreparations”,“comprehensiveorganictransformations”和“protectivegroupsinorganicsynthesis(第5版,2014)”中描述的方法,以及在j.med.chem.2014,57(18),7590-7599,angew.chem.int.ed.2017,56(21),5886-5889等中描述的方法。
通过上述方法制备的化合物可以通过已知方法分离和纯化,例如,萃取、沉淀、蒸馏、色谱法、分步重结晶、重结晶等。
可以通过使用以下实验例1中所述的化学发光alphalisa(注册商标,perkinelmer)方法来测定menin和mll之间的无细胞结合的抑制活性。或者,作为评估蛋白-蛋白相互作用的一般方法,可以使用时间分辨荧光共振能量转移(tr-fret)方法(其中通过fret(荧光共振能量转移)检测从具有长寿命荧光的供体结合蛋白到受体结合蛋白的荧光能量转移)、表面等离子体共振(spr)方法等。
本发明化合物或其药学上可接受的盐的细胞生长抑制活性可以通过使用本领域技术人员常规使用的生长抑制试验方法来检查。例如,如以下实验例2所述,可以通过将在有受试化合物存在下获得的细胞生长程度与在没有受试化合物存在下获得的细胞生长程度进行比较,来检查细胞生长抑制活性。可以例如通过使用用于测量活细胞的测试系统来检查生长程度。测量活细胞的方法的实例包括[3h]-胸苷摄取检测、brdu方法和mtt检测。
体内的抗肿瘤活性可以通过本领域技术人员常规使用的抗肿瘤试验方法来检查。例如,如以下实验例3-1至3-4中所述,将各种肿瘤细胞移植到小鼠、大鼠等中,并且在确认移植的细胞的植入之后,将本发明的化合物口服或静脉内施用。在几天或几周后,比较非施用组和化合物施用组中的肿瘤生长,从而可以确认根据本发明的体内抗肿瘤活性。
本发明的化合物或其药理学上可接受的盐可以与其他抗肿瘤剂一起使用。其实例包括烷化剂、抗代谢物、抗肿瘤抗生素、抗肿瘤植物成分、brm(生物反应调节剂)、激素、维生素、抗肿瘤抗体、分子靶标药物、其他抗肿瘤剂等。
更具体地,烷化剂的实例包括烷化剂例如氮芥、氮芥n-氧化物、苯丁酸氮芥等;基于氮丙啶的烷化剂,例如卡波醌、噻替派等;基于环氧化物的烷化剂,例如二溴甘露糖醇、二溴卫矛醇等;基于亚硝基脲的烷化剂,例如卡莫司汀、洛莫司汀、司莫司汀、盐酸尼莫司汀、链脲菌素、氯脲霉素、雷莫司汀等;以及白消安、甲苯磺酸英丙舒凡、达卡巴嗪等。
抗代谢物的实例包括嘌呤抗代谢物,例如6-巯基嘌呤、6-硫鸟嘌呤、硫肌苷等;嘧啶抗代谢物,例如氟尿嘧啶、替加氟、替加氟-尿嘧啶、卡莫氟、去氧氟尿苷、溴尿苷、阿糖胞苷、依诺他滨等;叶酸抗代谢物,例如甲氨蝶呤、三甲曲沙等;等等。
抗肿瘤抗生素的实例包括丝裂霉素c、博来霉素、培洛霉素、柔红霉素、aclarbicin、阿霉素、伊达比星、吡柔比星、thp-阿霉素、4'-表阿霉素或表柔比星、色霉素a3和放线菌素d等。
抗肿瘤植物成分的实例包括长春花生物碱,例如长春地辛、长春新碱、长春花碱等;紫杉烷类,例如紫杉醇、多西紫杉醇等;和表鬼臼毒素,例如依托泊苷、替尼泊苷等。
brm的实例包括肿瘤坏死因子、消炎痛等。
激素的实例包括氢化可的松、地塞米松、甲基泼尼松龙、泼尼松龙、普拉睾酮、倍他米松、曲安西龙、羟甲烯龙、诺龙、美替诺龙、磷雌酚、乙炔雌二醇、氯地孕酮、美雄烷、甲羟孕酮等。
维生素的实例包括维生素c、维生素a等。
抗肿瘤抗体和分子靶标药物的实例包括维奈托克、曲妥珠单抗、利妥昔单抗、西妥昔单抗、尼妥珠单抗、地诺单抗、贝伐单抗、英夫利昔单抗、伊匹单抗、纳武单抗、派姆单抗、阿维单抗、匹地利珠单抗、阿特珠单抗、雷莫芦单抗、甲磺酸伊马替尼、达沙替尼、吉非替尼、埃罗替尼、奥西替尼、舒尼替尼、拉帕替尼、达拉非尼、曲美替尼、考比替尼、帕唑帕尼、帕博西尼、帕比司他、索拉非尼、克唑替尼、维罗非尼、奎扎替尼、硼替佐米、卡非佐米、伊沙佐米、米哚妥林、吉列替尼等。
其他抗肿瘤剂的实例包括顺铂、卡铂、奥沙利铂、他莫昔芬、来曲唑、阿那曲唑、依西美坦、托瑞米芬柠檬酸盐、氟维司群、比卡鲁胺、氟他胺、米托坦、亮丙瑞林、醋酸戈舍瑞林、喜树碱、异环磷酰胺、环磷酰胺、美法仑、l-门冬酰胺酶、醋葡醛内酯、裂褶菌多糖、溶链菌制剂、丙卡巴肼、哌泊溴烷、新制癌菌素、羟基脲、乌苯美司、沙利度胺、来那度胺、泊马度胺、艾瑞布林、维甲酸、云芝多糖等。
本发明的药物组合物包含本发明的化合物或药学上可接受的盐和药学上可接受的载体,并且可以作为各种注射剂例如静脉内注射剂、肌内注射剂、皮下注射剂等施用,或通过各种方法例如口服施用、透皮施用等施用。药学上可接受的载体是指参与将本发明化合物或含有本发明化合物的组合物从一个器官运输到另一器官的药学上可接受的物质(例如赋形剂、稀释剂、添加剂、溶剂等)。。
通过使用常规制剂中使用的添加剂,例如载体、赋形剂等,可以制备含有本发明化合物或其药理学上可接受的盐作为有效成分的制剂。本发明化合物的施用可以是片剂、丸剂,、胶囊、颗粒剂、粉剂、液体等形式的口服施用,或注射剂(例如静脉内注射剂、肌内注射剂等)、栓剂、透皮剂、鼻用剂、吸入剂等形式的肠胃外施用。考虑到症状和施用对象的年龄、性别等,根据个体情况适当确定本发明化合物的剂量和剂数。对于向成人口服施用,剂量通常为每剂0.001mg/kg至100mg/kg,对于向成人静脉内施用,剂量通常为每剂0.0001mg/kg至10mg/kg。剂数通常为每天一次至六次,或每天一次至每七天一次。
用于本发明的口服施用的固体制剂可以是片剂、粉剂、颗粒剂等。可以根据常规方法通过将一种或多种活性物质与惰性赋形剂、润滑剂、崩解剂、溶解助剂等混合来制备这种制剂。赋形剂可以是例如乳糖、甘露糖醇或葡萄糖。润滑剂可以是例如硬脂酸镁。崩解剂可以是例如羧甲基淀粉钠。如果需要,片剂或丸剂可以用糖衣或胃溶性或肠溶性包衣剂包衣。
用于口服施用的液体制剂可以是药学上可接受的乳液、液体、混悬液、糖浆、酏剂等。这样的制剂包含通常使用的惰性溶剂(例如纯净水、乙醇),并且可以进一步包含增溶剂、润湿剂、悬浮剂、甜味剂、调味剂、芳香剂或防腐剂。
用于肠胃外施用的注射剂可以是无菌的水性或非水性液体、混悬液或乳液。用于注射剂的水性溶剂可以是例如蒸馏水或生理盐水溶液。用于注射剂的非水性溶剂可以是例如丙二醇、聚乙二醇、植物油例如橄榄油、醇例如乙醇或聚山梨酯80(药典名称)。这样的制剂可以进一步包含张度剂、防腐剂、润湿剂、乳化剂、分散剂、稳定剂或溶解辅助剂。这种制剂可以例如通过细菌保留过滤器过滤、与杀菌剂混合或暴露于辐射而被灭菌。或者,也可以使用通过在使用前将无菌固体组合物溶解或悬浮在无菌水或注射溶剂中而获得的组合物。
实施例
在下文中,将参考参考例和实施例更详细地描述本发明,但是本发明的范围不限于这些实施例,并且这些实施例在任何意义上均不受限制地解释。另外,除非另有说明,否则本文中使用的试剂、溶剂和起始原料可容易地从市售来源获得。
质子核磁共振谱(1h-nmr)使用jeol制造的400mhz核磁共振光谱仪或varian制造的400mhz核磁共振光谱仪进行测定。光谱数据示出显著峰,显示化学位移(其显示为相对于四甲基硅烷峰的相对ppm(δ))、质子数和峰分裂的多重性(其显示为s:单峰;d:双峰;t:三重峰;q:四重峰;quint:五重峰;m:多重峰;br:宽;brs:宽单峰等),以及进一步地(如果指定),耦合常数j值(单位:hz)。
使用电喷雾电离法(esi)或大气压化学电离法(apci)测量质谱(msm/z)。显示了有关通过反相高效液相色谱柱(agilentsystem;柱:develosilcombi-rp-5,2.0x50mm,cadenzacd-c18、3.0x75mm,或zorbaxsb-c18,1.8μm,2.1x50mm;溶剂:含0.1%甲酸的乙腈/水系统,或含0.01%三氟乙酸的乙腈/水系统)后获得的最大电离峰(几乎在所有情况下对应于最大uv吸收峰)的质谱数据。
通过使用市售的填充柱和自动制备纯化系统(例如,biotage制造的sp1,yamazen制造的epclc-w-prep2xy,shokoscience制造的purif-α2等)进行硅胶柱色谱法,并且仅描述了用于流动相的多种类型的溶剂。洗脱在通过薄层色谱法(tlc)的观察下进行。作为tlc板,使用merck制造的硅胶60f254或60nh2f254s、wakopurechemicalindustries,ltd.制造的nh2硅胶60f254板或fujisilysiachemicalltd.制造的chromatorexnhtlc。作为展开剂,使用在柱色谱法中使用的流动相。作为检测方法,采用uv检测器或显色剂。在参考例和实施例中,“氨基硅胶”是指其表面被具有氨基的官能团化学修饰的硅胶(例如,purif-pack(注册商标,shokoscientific)-ex,nh系列等)。
制备薄层色谱法(ptlc)通过使用merck制造的硅胶60f254板或wakopurechemicalindustries,ltd.制造的硅胶70pf254板或nh2硅胶60f254板进行,并仅描述了用于流动相的多种类型的溶剂。
通过使用nomurachemical制造的反相柱(develosilcombi-rp-5)进行制备型高效液相色谱法(制备型hplc),并且将含0.1%甲酸的乙腈/水用作流动相。
这些色谱法中使用的溶剂量和溶剂比、转化时间和梯度方法在此未描述。但是,本文中使用的纯化和/或分离方法可以通过化学合成的常识和/或技术来重现。
在实施例中,粉末x射线衍射测量的设备和测量条件如下:
型号:rigakurintttr-iii
样品架:非反射样品架
样品量:适量
x射线产生条件:50kv,300ma
波长:1.54å(cu-kα射线)
测量温度:室温
扫描速度:20°/min
扫描范围:2至40°
采样宽度:0.02°
分析程序:用刮刀取几毫克的受试化合物,放在非反射样品架上,并用药品包装纸弄平。之后,在上述条件下分析峰型(peakpattern)。
在以下参考例和实施例中,为方便起见,根据以下标准描述外消旋物、旋光物质、几何异构体和空间符号。(1)使用楔形线(从纸平面向前的键)、虚线(从纸平面向后的键)、实线(纸平面中的键)或波浪线(未指定构型的键)表示结构式中的空间符号。(2)当与化合物的结构式一起描述“外消旋物”时,表明由该结构式表示的化合物是外消旋物(与对映异构体的等量混合物)。当与化合物的结构式一起描述“非对映异构体混合物”时,表明由该结构式表示的化合物是非对映异构体的混合物(源自波浪线表示的键的构型)。当与化合物的结构式一起描述“顺式-反式混合物”时,表明由该结构式表示的化合物是几何异构体的混合物。另外,除非在结构式和化合物名称中另有说明,否则表示由结构式表示的化合物是单一的立体异构体(包括旋光物质)。(3)当在化合物名称后附加“(外消旋)”时,表示由该化合物名称所表示的化合物是外消旋的(与对映异构体的等量混合物)。当在化合物名称后附加“(非对映异构体混合物)”时,表示由该化合物名称所表示的化合物是非对映异构体的混合物(源自波浪线表示的键的构型)。当在化合物名称后附加“(顺式-反式混合物)”时,表示由该化合物名称所表示的化合物是几何异构体的混合物。
在以下参考例和实施例中,与化合物的结构式一起描述的“hcl”的符号以及在化合物名称后附加的“盐酸”表示由该结构式表示的化合物或由该化合物名称表示的化合物和氯化氢可以以各种比率形成盐酸盐,而不一定以1:1比率形成(例如,包括一盐酸盐、二盐酸盐、三盐酸盐等)。
参考例a-1
[(1r,3r,4s)-3-羟基-4-(甲基氨基)环戊基]氨基甲酸苄酯
步骤1(1r,4s)-4-氨基环戊-2-烯-1-甲酸甲酯盐酸盐
[化学式83]
在0℃下经50分钟向(1s)-(+)-2-氮杂双环[2.2.1]庚-5-烯-3-酮(cas:130931-83-8)(98.6g)和甲醇(300ml)的混合物中加入亚硫酰氯(cas:7719-09-7)(40ml),并将混合物在0℃搅拌2小时。减压浓缩反应混合物,将获得的固体悬浮在乙酸乙酯中,并通过过滤收集,得到标题化合物(158g),为固体。
步骤2(1r,4s)-4-[(叔丁氧基羰基)氨基]环戊-2-烯-1-甲酸甲酯
[化学式84]
在0℃下向上述步骤1中获得的化合物(158g)、thf(700ml)和水(280ml)的混合物中加入二碳酸二叔丁酯(cas:24424-99-5)(194g)和碳酸钠(cas:497-19-8)(104g),并将混合物在室温搅拌过夜。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并减压浓缩,得到标题化合物(214g),为油状物。
步骤3(3as,5s,6s,6as)-6-溴-2-氧代六氢-2h-环戊[d][1,3]噁唑-5-甲酸甲酯
[化学式85]
在0℃下向上述步骤2中获得的化合物(214g)、thf(700ml)和水(70ml)的混合物中加入n-溴代琥珀酰亚胺(cas:128-08-5)(174g),并将混合物在室温搅拌过夜。减压浓缩反应溶液,并向其中加入乙酸乙酯。有机层用1n盐酸、饱和碳酸氢钠水溶液、饱和硫代硫酸钠水溶液和饱和盐水洗涤,经无水硫酸钠干燥,并减压浓缩。将残余物悬浮在乙酸乙酯/正己烷中,并通过过滤收集固体。再次将获得的固体悬浮在乙酸乙酯/正己烷中,并通过过滤收集,得到标题化合物(113g),为固体。减压浓缩滤液,将残余物悬浮在乙酸乙酯/正己烷中,并通过过滤收集固体。再次将获得的固体悬浮在乙酸乙酯/正己烷中,并通过过滤收集。将乙酸乙酯/正己烷和水加入到获得的固体中,并将混合物进行液体分离。有机层经无水硫酸钠干燥,并减压浓缩,得到标题化合物(11.0g),为固体。总共获得固体形式的标题化合物(124g)。
步骤4(3r,4s)-4-[(叔丁氧基羰基)氨基]-3-羟基环戊-1-烯-1-甲酸
[化学式86]
在0℃下向上述步骤3中获得的化合物(124g)、甲醇(580ml)和水(580ml)的混合物中加入氢氧化钾(cas:1310-58-3)(106g),并将混合物在90℃搅拌18小时。减压浓缩反应溶液,于0℃向其中加入thf(180ml)和二碳酸二叔丁酯(cas:24424-99-5)(102g),并将混合物在室温搅拌4小时。混合物在0℃用5n盐酸和2n盐酸中和,并用乙酸乙酯萃取五次。合并的有机层经无水硫酸钠干燥,并在减压下蒸发溶剂。将获得的固体悬浮在乙酸乙酯/正己烷中,通过过滤收集,并且在减压下干燥以得到标题化合物。浓缩滤液,并将获得的固体悬浮在乙酸乙酯/正己烷中,通过过滤收集,并在减压下干燥,得到标题化合物。总共获得固体形式的标题化合物(96.9g)。
步骤5(3s,4r)-3-[(叔丁氧基羰基)氨基]-4-羟基环戊烷-1-甲酸(非对映异构体混合物)
[化学式87]
将上述步骤4中获得的化合物(96.9g)、10%湿钯/碳(10.4g)和甲醇(780ml)的混合物在氢气气氛下搅拌7.5小时。将反应溶液通过硅藻土过滤。减压浓缩滤液,得到标题化合物(105g),为固体。
步骤6(1r,3s,4r)-3-[(叔丁氧基羰基)氨基]-4-羟基环戊烷-1-甲酸2-甲基丙-2-胺盐
[化学式88]
将上述步骤5中获得的化合物(105g)、叔丁胺(42.5ml)、甲醇(90ml)和叔丁基甲基醚(675ml)的混合物在冰箱中放置3天。通过过滤收集沉淀的固体,并在减压下干燥,得到标题化合物(108g),为固体。
步骤7(1r,3s,4r)-3-[(叔丁氧基羰基)氨基]-4-羟基环戊烷-1-甲酸
[化学式89]
在0℃将上述步骤6中获得的化合物(108g)和水(200ml)的混合物用2n盐酸(150ml)调节至ph=3。混合物用二氯甲烷/甲醇=10/1萃取3次,合并的有机层经无水硫酸钠干燥,并在减压下浓缩,得到标题化合物(63.9g),为固体。
步骤8(1r,3s,4r)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环戊烷-1-甲酸甲氧基甲酯
[化学式90]
将上述步骤7中获得的化合物(61.6g)、氯甲基甲基醚(cas:107-30-2)(56.7ml)、dipea(cas:7087-68-5)(262ml)、碘化钠(75.2g)和1,2-二甲氧基乙烷(1000ml)的混合物加热回流1小时。使反应溶液冷却至室温,向其中加入饱和盐水和水,并将混合物用乙酸乙酯萃取两次。合并的有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(66.5g),为油状物。
步骤9(1r,3s,4r)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环戊烷-1-甲酸
[化学式91]
将上述步骤8中获得的化合物(66.5g)、1n氢氧化钠水溶液(400ml)、甲醇(20ml)和thf(580ml)的混合物在室温下搅拌2小时。在0℃下向反应溶液中加入1n盐酸,并将混合物用乙酸乙酯萃取两次。合并的有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(53.6g),为油状物。
步骤10[(1r,3s,4r)-4-(甲氧基甲氧基)环戊烷-1,3-二基]双氨基甲酸苄酯叔丁酯
[化学式92]
将上述步骤9中获得的化合物(53.6g)、二苯基磷酰基叠氮化物(dppa)(cas:26386-88-9)(51.9ml)、tea(cas:121-44-8)(33.4ml)和甲苯(730ml)的混合物在90℃下搅拌30分钟。向反应溶液中加入苯甲醇(38.3ml),并将混合物在90℃搅拌2.5小时。使反应溶液冷却至室温,向其中加入水,并将混合物用乙酸乙酯萃取两次。合并的有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(63.2g),为油状物。
步骤11[(1r,3s,4r)-3-氨基-4-羟基环戊基]氨基甲酸苄酯盐酸盐
[化学式93]
051(2)。
将上述步骤10中获得的化合物(120g)、氯化氢(4mol/l,1,4-二噁烷溶液,420ml)和甲醇(420ml)的混合物在室温下搅拌30分钟。减压浓缩反应溶液,得到标题化合物(87.2g),为固体。
步骤12{(1r,3r,4s)-3-羟基-4-[(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸苄酯
[化学式94]
将上述步骤11中获得的化合物(87.2g)、2-硝基苯磺酰氯(74.5g)、dipea(159ml)和二氯甲烷(1000ml)的混合物在0℃下搅拌1小时。向反应溶液中加入水,并将混合物用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将乙酸乙酯加入到残余物中,并通过过滤收集固体,得到固体形式的标题化合物(33.0g)。浓缩滤液,将乙酸乙酯加入到残余物中,并通过过滤收集固体,得到标题化合物(25.2g),为固体。浓缩滤液,并将残余物通过氨基硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(40.9g),为固体。总共获得固体形式的标题化合物(99.1g)。
步骤13{(1r,3r,4s)-3-羟基-4-[甲基(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸苄酯
[化学式95]
在0℃下向上述步骤12中获得的化合物(87.2g)和dmf(630ml)的混合物中添加碳酸铯(74.2g)和碘甲烷(21.3ml),并将混合物在室温下搅拌2小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取两次。将合并的有机层用水和饱和盐水洗涤3次,经无水硫酸钠干燥,并在减压下浓缩。向残余物中加入乙酸乙酯/正己烷,并通过过滤收集固体,得到标题化合物(80.9g),为固体。
步骤14[(1r,3r,4s)-3-羟基-4-(甲基氨基)环戊基]氨基甲酸苄酯
[化学式96]
将上述步骤13中获得的化合物(40.4g)、碳酸铯(52.7g)、4-异丙基苯硫醇(16.8ml)、thf(140ml)和甲醇(140ml)的混合物在室温下搅拌3小时。将氨基硅胶加入到反应溶液中,并将混合物在减压下浓缩。残余物通过氨基硅胶柱色谱法(正己烷/乙酸乙酯,然后乙酸乙酯/甲醇)纯化,得到油状物。向其中加入乙酸乙酯/正己烷,并通过过滤收集固体,得到标题化合物(22.0g),为固体。
参考例a-2
[(1s,3r)-3-氨基环己基]氨基甲酸叔丁酯
步骤1(1r,3s)-环己烷-1,3-二基双氨基甲酸苄酯叔丁酯
[化学式97]
使用(1r,3s)-3-(叔丁氧基羰基氨基)环己烷甲酸(cas:222530-34-9),以与参考例a-1的步骤10中相同的方式获得标题化合物。
步骤2[(1s,3r)-3-氨基环己基]氨基甲酸叔丁酯
[化学式98]
将上述步骤1中获得的化合物(12.37g)悬浮在乙醇(120ml)中,向其中加入10%湿钯/碳(4.0g),并将混合物在氢气气氛下在室温下搅拌4小时。过滤除去钯催化剂,将滤液减压浓缩,得到标题化合物(8.26g),为固体。其直接用于下一步。
参考例a-3
n-[(1r,3s)-3-氨基环戊基]-n-甲基-2-硝基苯-1-磺酰胺
步骤1(1r,3s)-环戊烷-1,3-二基双氨基甲酸苄酯叔丁酯
[化学式99]
使用(-)-(1r,3s)-n-boc-3-氨基环戊烷甲酸(cas:161660-94-2),以与参考例a-1的步骤10中相同的方式得到标题化合物。
步骤2{(1s,3r)-3-[(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸叔丁酯
[化学式100]
将上述步骤1中获得的化合物(19.0g)悬浮在乙醇(280ml)中,向其中加入10%湿钯/碳(2.5g),并将混合物在氢气气氛下于室温搅拌1.5小时。通过过滤除去钯,并在减压下浓缩滤液。向其中加入二氯甲烷(230ml)和dipea(14.8ml),并在冰冷却下向其中加入2-硝基苯磺酰氯(12.6g)。在室温搅拌2小时后,向反应溶液中加入二氯甲烷和水,并将混合物进行液体分离。有机层经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(12.77g),为固体。
步骤3{(1s,3r)-3-[甲基(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸叔丁酯
[化学式101]
使用在上述步骤2中获得的化合物,以与参考例a-1的步骤13中相同的方式获得标题化合物。
步骤4n-[(1r,3s)-3-氨基环戊基]-n-甲基-2-硝基苯-1-磺酰胺
[化学式102]
将上述步骤3中获得的化合物(2.04g)溶解于二氯甲烷(20ml)中,向其中加入氯化氢(4mol/l,1,4-二噁烷溶液,20ml),并将混合物在室温下搅拌1小时。减压蒸发溶剂,将二氯甲烷和饱和碳酸氢钠水溶液加入到残余物中,并将混合物进行液体分离。用二氯甲烷萃取水层。合并有机层,经无水硫酸钠干燥,并在减压下浓缩。所得残余物通过氨基硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(1.21g),为油状物。
参考例a-4
[(1s,3r)-3-(甲基氨基)环戊基]氨基甲酸叔丁酯
[化学式103]
使用参考例a-3的步骤3中获得的化合物,以与参考例a-1的步骤14中相同的方式获得标题化合物。
参考例a-5
[(1s,3r)-3-(乙基氨基)环戊基]氨基甲酸叔丁酯
步骤1{(1s,3r)-3-[乙基(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸叔丁酯
[化学式104]
使用参考例a-3的步骤2中获得的化合物,以与参考例a-1的步骤13中相同的方式获得标题化合物,除了使用溴乙烷(cas:74-96-4)代替碘甲烷。
步骤2[(1s,3r)-3-(乙基氨基)环戊基]氨基甲酸叔丁酯
[化学式105]
使用在上述步骤1中获得的化合物,以与参考例a-1的步骤14中相同的方式获得标题化合物。
参考例a-6
(1r,3s,5r)-3-氨基-5-(甲氧基甲氧基)环己烷-1-甲酸苄酯(外消旋物)
步骤1(1s,3r,5s)-3-{[(苄氧基)羰基]氨基}-5-羟基环己烷-1-甲酸甲氧基甲酯(外消旋物)
[化学式106]
将根据文献(bioorg.med.chem.2006,14,2242-2252)中描述的方法合成的(1s,3r,5s)-3-{[(苄氧基)羰基]氨基}-5-羟基环己烷-1-甲酸(外消旋物)(11.4g)、dipea(20.4ml)和二氯甲烷(390ml)的混合物用冰冷却,向其中加入氯甲基甲基醚(3.08ml),并将混合物在室温下搅拌3小时。将饱和氯化铵水溶液加入到反应溶液中,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并减压浓缩,得到标题化合物(11.8g),为油状物。所得化合物无需纯化即可直接用于下一步。
步骤2(1s,3r,5s)-3-{[(苄氧基)羰基]氨基}-5-(甲氧基甲氧基)环己烷-1-甲酸甲氧基甲酯(外消旋物)
[化学式107]
将上述步骤1中获得的化合物(11.8g)、dipea(18.2ml)、氯甲基甲基醚(3.4ml)和二氯甲烷(350ml)的混合物在室温搅拌2小时。将反应溶液用冰冷却,向其中加入氯甲基甲基醚(10.5ml),并将混合物在室温下再搅拌18小时。将饱和氯化铵水溶液加入到反应溶液中,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(7.88g),为油状物。
步骤3(1s,3r,5s)-3-{[(苄氧基)羰基]氨基}-5-(甲氧基甲氧基)环己烷-1-甲酸(外消旋物)
[化学式108]
使用在上述步骤2中获得的化合物(7.88g),以与参考例a-1的步骤9中相同的方式获得标题化合物。
步骤4[(1r,3s,5s)-5-(甲氧基甲氧基)环己烷-1,3-二基]双氨基甲酸苄酯丙-2-烯-1-基酯(外消旋物)
[化学式109]
将上述步骤3中获得的化合物(6.65g)、二苯基磷酰基叠氮化物(5.52ml)、tea(3.57ml)和甲苯(98.6ml)的混合物在90℃搅拌30分钟。向其中添加烯丙醇(1.40g),将混合物在相同温度下搅拌5小时,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(6.2g),为固体。
步骤5(1r,3s,5r)-3-氨基-5-(甲氧基甲氧基)环己烷-1-甲酸苄酯(外消旋物)
[化学式110]
将上述步骤4中获得的化合物(335mg)、二氯甲烷(8ml)、吡咯烷(0.177ml)和四(三苯基膦)钯(0)(20mg)的混合物在室温下搅拌30分钟,并减压浓缩。残余物通过硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(93mg),为固体。
参考例a-7
[(1s,3r,5s)-3-氨基-5-(二甲基氨基甲酰基)环己基]氨基甲酸苄酯(外消旋物)
步骤1[(1s,3r,5r)-3-(二甲基氨基甲酰基)-5-(甲氧基甲氧基)环己基]氨基甲酸苄酯(外消旋物)
[化学式111]
在室温下向参考例a-6的步骤3中获得的化合物(1.59g)和二氯甲烷(15ml)的混合物中加入二甲胺盐酸盐(cas:506-59-2)(0.48g)、无水hobt(cas:123333-53-9)(0.77g)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(cas:1892-57-5)(1.10g)和tea(1.31ml)。将该反应溶液温热至室温,并搅拌18小时。向该反应溶液中加入乙酸乙酯(30ml),并依次用饱和氯化铵水溶液(10ml)、饱和碳酸氢钠水溶液(10ml)和饱和盐水(10ml)洗涤混合物,有机层经无水硫酸钠干燥,并减压浓缩。残余物通过硅胶柱色谱法(甲醇/二氯甲烷)纯化,得到标题化合物(1.65g),为油状物。
步骤2[(1s,3r,5r)-3-(二甲基氨基甲酰基)-5-羟基环己基]氨基甲酸苄酯(外消旋物)
[化学式112]
在室温下,向上述步骤1中获得的化合物(1.65g)和1,4-二噁烷(5ml)的混合物中加入氯化氢(4mol/l,1,4-二噁烷溶液,5.66ml)。将该反应溶液在室温搅拌2小时,并向其中加入水(20ml)和乙酸乙酯(30ml)。将该混合物用水(10ml)洗涤两次,并依次用饱和碳酸氢钠水溶液(10ml)和饱和盐水(20ml)洗涤,有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(甲醇/二氯甲烷)纯化,得到标题化合物(1.12g),为油状物。
步骤34-硝基苯甲酸(1s,3s,5r)-3-{[(苄氧基)羰基]氨基}-5-(二甲基氨基甲酰基)环己基酯(外消旋物)
[化学式113]
在冰冷却下向上述步骤2中获得的化合物(1.06g)、4-硝基苯甲酸(cas:62-23-7)(0.66g)、三苯基膦(cas:14264-16-5)(纯度97%,1.04g)和thf(15ml)的混合物中加入偶氮二甲酸二异丙酯(40%甲苯溶液)(cas:2446-83-5)(2.1ml)。将该混合物温热至室温,并在室温搅拌5.5小时。向其中加入饱和碳酸氢钠水溶液(20ml)、乙酸乙酯(20ml)和饱和盐水(20ml),并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(甲醇/二氯甲烷)纯化,得到标题化合物(1.55g),为油状物。
步骤4[(1s,3r,5s)-3-(二甲基氨基甲酰基)-5-羟基环己基]氨基甲酸苄酯(外消旋物)
[化学式114]
在室温下向上述步骤3中获得的化合物(1.55g)和乙醇(22ml)的混合物中加入碳酸钾(cas:584-08-7)(1.37g),并将混合物搅拌2小时。向其中加入饱和碳酸氢钠水溶液(20ml)、乙酸乙酯(20ml)和饱和盐水(20ml),并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(甲醇/二氯甲烷)纯化,得到标题化合物(0.710g),为固体。
步骤5[(1s,3r,5r)-3-叠氮基-5-(二甲基氨基甲酰基)环己基]氨基甲酸苄酯(外消旋物)
[化学式115]
在冰冷却下向上述步骤4中获得的化合物(0.513g)、三苯基膦(cas:14264-16-5)(0.752g)、二苯基磷酰基叠氮化物(0.412ml)和thf(15ml)的混合物中加入偶氮二甲酸二异丙酯(40%甲苯溶液)(1.50ml)。将该反应溶液温热至室温,并搅拌3小时。向该反应溶液中加入乙酸乙酯(30ml),并将混合物用饱和盐水(10ml)洗涤两次。有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(甲醇/二氯甲烷)纯化,得到标题化合物(0.553g),为油状物。
步骤6[(1s,3r,5s)-3-氨基-5-(二甲基氨基甲酰基)环己基]氨基甲酸苄酯(外消旋物)
[化学式116]
在冰冷却下向上述步骤5中获得的化合物(0.553g)和thf(15ml)的混合物中加入三苯基膦(cas:14264-16-5)(0.840g)。将该反应溶液温热至室温,并搅拌30分钟。向该反应溶液中加入水(10ml),将混合物静置19小时,加热至85℃。并搅拌2小时。将反应溶液在减压下浓缩。将乙醇(20ml)加入残余物中,并将混合物在减压下浓缩。进行三次这些浓缩操作,得到粗制标题化合物(0.511g),为油状物。该产物无需纯化即用于下一步。
参考例a-8
[(1s,3r)-3-氨基环己基]氨基甲酸苄酯盐酸盐
步骤1(1r,3s)-环己烷-1,3-二基双氨基甲酸苄酯叔丁酯
[化学式117]
使用(1s,3r)-3-[(叔丁氧基羰基)氨基]环己烷-1-甲酸(cas:222530-34-9),以与参考例a-1的步骤10中相同的方式获得标题化合物。
步骤2[(1s,3r)-3-氨基环己基]氨基甲酸苄酯盐酸盐
[化学式118]
将上述步骤1中获得的化合物(1.90g)溶于氯化氢(4mol/l,1,4-二噁烷溶液,54.5ml),并将溶液在室温下搅拌30分钟。将反应溶液在减压下浓缩,残余物用乙酸乙酯进行浆化洗涤,得到标题化合物(1.54g),为固体。
参考例a-9
(1s,3r,5s)-3-氨基-5-{[(苄氧基)羰基]氨基}环己烷-1-甲酸甲酯(外消旋物)
步骤14-硝基苯甲酸(1s,3s,5r)-3-{[(苄氧基)羰基]氨基}-5-(甲氧基羰基)环己基酯(外消旋物)
[化学式119]
使用根据文献(bioorg.med.chem.,2006,14,2242-2252)中描述的方法合成的(1r,3s,5r)-3-(苄氧基羰基氨基)-5-羟基-环己烷甲酸甲酯(外消旋物),以与参考例a-7的步骤3中相同的方式获得标题化合物。
步骤2(1r,3s,5s)-3-{[(苄氧基)羰基]氨基}-5-羟基环己烷-1-甲酸甲酯(外消旋物)
[化学式120]
使用上述步骤1中获得的化合物,以与参考例a-7的步骤4中相同的方式获得标题化合物。
步骤3(1r,3r,5s)-3-叠氮基-5-{[(苄氧基)羰基]氨基}环己烷-1-甲酸甲酯(外消旋物)
[化学式121]
使用上述步骤2中获得的化合物,以与参考例a-7的步骤5中相同的方式获得标题化合物。
步骤4(1s,3r,5s)-3-氨基-5-{[(苄氧基)羰基]氨基}环己烷-1-甲酸甲酯(外消旋物)
[化学式122]
使用上述步骤3中获得的化合物,以与参考例a-7的步骤6中相同的方式获得标题化合物。
参考例a-10
[(1s,2s,5s)-5-氨基-2-甲氧基环己基]氨基甲酸叔丁酯
步骤1(1r,3s,4s)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环己烷-1-甲酸乙酯
[化学式123]
将根据文献(tetrahedron2017,73,1381-1388)中描述的方法合成的(1r,3s,4s)-3-[(叔丁氧基羰基)氨基]-4-羟基环己烷-1-甲酸乙酯(13.84g)、二甲氧基乙烷(155ml)、碘化钠(cas:7681-82-5)(6.97g)、氯甲基甲基醚(10.5ml)和dipea(49ml)的混合物在150℃下回流加热2小时。向反应溶液中加入饱和盐水和水,混合物用乙酸乙酯萃取,有机层经无水硫酸钠干燥,并减压浓缩。残余物通过硅胶柱色谱法(二氯甲烷/乙酸乙酯)纯化,得到标题化合物(14.43g),为固体。
步骤2(1s,3s,4s)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环己烷-1-甲酸
[化学式124]
将上述步骤1中合成的化合物(14.43g)溶解于乙醇(150ml),向其中加入乙醇钠(cas:141-52-6)(浓度20%,乙醇溶液,24ml),并将混合物在50℃下搅拌40小时。向反应溶液中加入1n盐酸乙醇溶液(cas:7647-01-0)(61ml),并将混合物减压浓缩。将水添加到获得的残余物中,并将混合物用二氯甲烷萃取。有机层经无水硫酸钠干燥,并在减压下浓缩。将残余物进行硅胶柱色谱法(二氯甲烷/乙酸乙酯)。将获得的残余物悬浮在正己烷/乙酸乙酯中,并通过过滤收集固体,得到含有标题化合物的混合物(4.11g),为固体。
步骤3[(1s,3s,4s)-4-(甲氧基甲氧基)环己烷-1,3-二基]双氨基甲酸苄酯叔丁酯
[化学式125]
将上述步骤2中获得的化合物(4.11g)、1,4-二噁烷(66ml)、二苯基磷酰基叠氮化物(cas:26386-88-9)(3.5ml)和tea(2.5ml)的混合物在90℃搅拌2小时。然后,向其中加入苯甲醇(cas:100-51-6)(2.2ml),并将混合物在90℃搅拌5小时。向反应溶液中加入饱和盐水和水,并将混合物用乙酸乙酯萃取。有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(二氯甲烷/乙酸乙酯)纯化,得到标题化合物(2.64g),为固体。
步骤4[(1s,3s,4s)-4-羟基环己烷-1,3-二基]双氨基甲酸苄酯叔丁酯
[化学式126]
将上述步骤3中获得的化合物(0.595g)溶解在1,4-二噁烷(18ml)中,向其中加入氯化氢(4mol/l,1,4-二噁烷溶液,36ml),并将混合物在室温下搅拌3小时。然后,在减压下蒸发溶剂,并将残余物干燥。将thf(10ml)、tea(0.57ml)和二碳酸二叔丁酯(cas:24424-99-5)(0.418g)在thf(6.0ml)中的溶液添加至所得残余物中,并将混合物在室温下搅拌2.5小时。将饱和碳酸氢钠水溶液加入到反应溶液中,并将混合物用二氯甲烷萃取。有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(0.403g),为固体。
步骤5[(1s,3s,4s)-4-甲氧基环己烷-1,3-二基]双氨基甲酸苄酯叔丁酯
[化学式127]
将上述步骤4中获得的化合物(0.350g)、二氯甲烷(14ml)、氧化银(i)(cas:20667-12-3)(0.677g)、分子筛3a(cas:308080-99)-1)(0.460g,在180℃减压干燥2小时后使用)和碘甲烷(cas:74-88-4)(1.2ml)的混合物在45℃下剧烈搅拌9小时。通过硅藻土过滤反应溶液,并在减压下浓缩滤液。将获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(0.269g),为固体。
步骤6[(1s,2s,5s)-5-氨基-2-甲氧基环己基]氨基甲酸叔丁酯
[化学式128]
将上述步骤5中获得的化合物(0.267g)、5%湿钯/碳(ph)(cas:7440-05-3)(0.304g)和乙醇(12ml)的混合物在氢气气氛下在室温下搅拌3.5小时。将反应溶液过滤,并将滤液在减压下浓缩。所得残余物通过氨基硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(0.161g),为固体。
参考例a-11
[(1r,3s,5s)-3-氨基-5-甲氧基环己基]氨基甲酸苄酯(外消旋物)
步骤1(1s,3r,5s)-3-{[(苄氧基)羰基]氨基}-5-甲氧基环己烷-1-甲酸甲酯(外消旋物)
[化学式129]
向根据文献(bioorg.med.chem.2006,14,2242-2252)中描述的方法合成的(1s,3r,5s)-3-{[(苄氧基)羰基]氨基}-5-羟基环己烷-1-甲酸(外消旋物)(0.498g)和二氯甲烷(17.0ml)的溶液中加入氧化银(i)(1.57g)、分子筛3a(0.249g)和碘甲烷(2.11ml),并将混合物在40℃搅拌3小时。向其中加入另外的碘甲烷(2.11ml),并将混合物在40℃搅拌7.5小时。通过硅藻土过滤除去不溶物,用二氯甲烷和甲醇洗涤,并将滤液减压浓缩。所得粗产物通过柱色谱法(甲醇/二氯甲烷)纯化,得到标题化合物(0.480g),为油状物。
步骤2(1s,3r,5s)-3-{[(苄氧基)羰基]氨基}-5-甲氧基环己烷-1-甲酸(外消旋物)
[化学式130]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤9中相同的方式获得标题化合物。所获得的粗产物直接用于下一步。
步骤3[(1r,3s,5s)-5-甲氧基环己烷-1,3-二基]双氨基甲酸苄酯丙-2-烯-1-基酯(外消旋物)
[化学式131]
使用上述步骤2中获得的化合物,以与参考例a-6的步骤4中相同的方式获得标题化合物。
步骤4[(1r,3s,5s)-3-氨基-5-甲氧基环己基]氨基甲酸苄酯(外消旋物)
[化学式132]
使用上述步骤3中获得的化合物,以与参考例a-6的步骤5中相同的方式获得标题化合物。
参考例a-12
[(1r,2r,4s)-2-(甲氧基甲氧基)-4-(甲基氨基)环戊基]氨基甲酸叔丁酯
步骤1(1s,3r,4r)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环戊烷-1-甲酸甲酯
[化学式133]
使用(1s,3r,4r)-3-[(叔丁氧基羰基)氨基]-4-羟基环戊烷-1-甲酸甲酯(cas:321744-16-5),以与参考例a-6的步骤2中相同的方式获得标题化合物。
步骤2(1s,3r,4r)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环戊烷-1-甲酸
[化学式134]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤9中相同的方式获得标题化合物。
步骤3[(1s,3r,4r)-4-(甲氧基甲氧基)环戊烷-1,3-二基]双氨基甲酸苄酯叔丁酯
[化学式135]
使用上述步骤2中获得的化合物,以与参考例a-1的步骤10中相同的方式获得标题化合物。
步骤4[(1r,2r,4s)-4-氨基-2-(甲氧基甲氧基)环戊基]氨基甲酸叔丁酯
[化学式136]
使用上述步骤3中获得的化合物,以与参考例a-2的步骤2中相同的方式获得标题化合物。
步骤5{(1r,2r,4s)-2-(甲氧基甲氧基)-4-[(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸叔丁酯
[化学式137]
使用上述步骤4中获得的化合物,以与参考例a-1的步骤12中相同的方式获得标题化合物。
步骤6{(1r,2r,4s)-2-(甲氧基甲氧基)-4-[甲基(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸叔丁酯
[化学式138]
使用上述步骤5中获得的化合物,以与参考例a-1的步骤13中相同的方式获得标题化合物。
步骤7[(1r,2r,4s)-2-(甲氧基甲氧基)-4-(甲基氨基)环戊基]氨基甲酸叔丁酯
[化学式139]
使用上述步骤6中获得的化合物,以与参考例a-1的步骤14中相同的方式获得标题化合物。
参考例a-13
[(1r,2s,4s)-2-(甲氧基甲氧基)-4-(甲基氨基)环戊基]氨基甲酸叔丁酯
步骤1(1s,3r,4s)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环戊烷-1-甲酸甲酯
[化学式140]
使用(1r,2s,4s)-n-boc-1-氨基-2-羟基环戊烷-4-甲酸甲酯(cas:321744-14-3),以与参考例a-6的步骤2中相同的方式获得标题化合物。
步骤2(1s,3r,4s)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环戊烷-1-甲酸
[化学式141]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤9中相同的方式获得标题化合物。
步骤3[(1s,3r,4s)-4-(甲氧基甲氧基)环戊烷-1,3-二基]双氨基甲酸苄酯叔丁酯
[化学式142]
使用上述步骤2中获得的化合物,以与参考例a-1的步骤10中相同的方式获得标题化合物。
步骤4[(1r,2s,4s)-4-氨基-2-(甲氧基甲氧基)环戊基]氨基甲酸叔丁酯
[化学式143]
使用上述步骤3中获得的化合物,以与参考例a-2的步骤2中相同的方式获得标题化合物。
步骤5{(1r,2s,4s)-2-(甲氧基甲氧基)-4-[(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸叔丁酯
[化学式144]
使用上述步骤4中获得的化合物,以与参考例a-1的步骤12中相同的方式获得标题化合物。
步骤6{(1r,2s,4s)-2-(甲氧基甲氧基)-4-[甲基(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸叔丁酯
[化学式145]
使用上述步骤5中获得的化合物,以与参考例a-1的步骤13中相同的方式获得标题化合物。
步骤7[(1r,2s,4s)-2-(甲氧基甲氧基)-4-(甲基氨基)环戊基]氨基甲酸叔丁酯
[化学式146]
使用上述步骤6中获得的化合物,以与参考例a-1的步骤14中相同的方式获得标题化合物。
参考例a-14
[(1r,3s,4s)-3-羟基-4-(甲基氨基)环戊基]氨基甲酸苄酯
步骤1(1r,3s,4s)-3-(乙酰氧基)-4-[(叔丁氧基羰基)氨基]环戊烷-1-甲酸甲酯
[化学式147]
将(1r,3s,4r)-3-[(叔丁氧基羰基)氨基]-4-羟基环戊烷-1-甲酸甲酯(cas:321744-23-4)(0.300g)溶解于thf(2.3ml),向其中加入二苯基-2-吡啶基膦(0.487g)、1,1'-(偶氮二羰基)二哌嗪(0.394g)和乙酸(0.132ml),并将混合物在室温下搅拌2小时。将2n盐酸和乙酸乙酯加入到反应溶液中,并将混合物进行萃取操作。有机层用饱和碳酸氢钠水溶液和饱和盐水洗涤,并经无水硫酸钠干燥。减压蒸发溶剂,残余物用正己烷/乙酸乙酯=3/1的混合溶剂洗涤,由此通过过滤除去不溶物。减压浓缩滤液,并将残余物通过硅胶色谱法(乙酸乙酯/二氯甲烷)纯化,得到标题化合物(0.230g),为固体。
步骤2(1r,3s,4s)-3-[(叔丁氧基羰基)氨基]-4-羟基环戊烷-1-甲酸
[化学式148]
将上述步骤1中获得的化合物(46.6g)溶解在thf(406ml)中,向其中加入氢氧化锂水合物(51.9g)、水(309ml)和甲醇(77ml),并将混合物在室温下搅拌3小时。向反应溶液中加入冰块,然后向其中加入5n盐酸(263ml)。向其中加入氯化钠和10%甲醇/二氯甲烷溶液,并对混合物进行萃取。将10%甲醇/二氯甲烷溶液加入到水层中,并将混合物进行萃取。合并的有机层经无水硫酸钠干燥,并减压蒸发溶剂,得到标题化合物(36.8g),为固体。
步骤3(1r,3s,4s)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环戊烷-1-甲酸甲氧基甲酯
[化学式149]
使用上述步骤2中获得的化合物,以与参考例a-1的步骤8中相同的方式获得标题化合物。
步骤4(1r,3s,4s)-3-[(叔丁氧基羰基)氨基]-4-(甲氧基甲氧基)环戊烷-1-甲酸
[化学式150]
使用上述步骤3中获得的化合物,以与参考例a-1的步骤9中相同的方式获得标题化合物。
步骤5[(1r,3s,4s)-4-(甲氧基甲氧基)环戊烷-1,3-二基]双氨基甲酸苄酯叔丁酯
[化学式151]
使用上述步骤4中获得的化合物,以与参考例a-1的步骤10中相同的方式获得标题化合物。
步骤6[(1r,3s,4s)-3-氨基-4-羟基环戊基]氨基甲酸苄酯
[化学式152]
使用上述步骤5中获得的化合物,以与参考例a-3的步骤4中相同的方式获得标题化合物。
步骤7{(1r,3s,4s)-3-羟基-4-[(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸苄酯
[化学式153]
使用上述步骤6中获得的化合物,以与参考例a-1的步骤12中相同的方式获得标题化合物。
步骤8{(1r,3s,4s)-3-羟基-4-[甲基(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸苄酯
[化学式154]
使用上述步骤7中获得的化合物,以与参考例a-1的步骤13中相同的方式获得标题化合物。
步骤9[(1r,3s,4s)-3-羟基-4-(甲基氨基)环戊基]氨基甲酸苄酯
[化学式155]
使用上述步骤8中获得的化合物,以与参考例a-1的步骤14中相同的方式获得标题化合物。
参考例a-15
[(1r,3r,4s)-3-羟基-4-(甲基氨基)环戊基]氨基甲酸叔丁酯
步骤1{(1r,3r,4s)-3-羟基-4-[甲基(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸叔丁酯
[化学式156]
将参考例a-1的步骤13中获得的化合物(20.6g)溶解于乙腈(305ml)中,并在冰冷却下向其中滴加碘代三甲基甲硅烷(18.8ml),并将混合物在相同温度下搅拌1小时。向反应溶液中加入1n盐酸乙醇溶液(45.8ml),并将混合物在相同温度下搅拌30分钟,并在减压下浓缩。将残余物溶于乙醇(15ml)中,并向其中加入thf(153ml)、碳酸钠(24.3g)和二碳酸二叔丁酯(15.0g),并将混合物在室温下搅拌3小时。将乙酸乙酯和饱和盐水加入到反应溶液中,并将混合物进行萃取操作。有机层经无水硫酸钠干燥,并在减压下蒸发溶剂。残余物通过硅胶色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(17.1g),为固体。
步骤2[(1r,3r,4s)-3-羟基-4-(甲基氨基)环戊基]氨基甲酸叔丁酯
[化学式157]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤14中相同的方式获得标题化合物。
参考例a-16
[(1s,2r,4r)-4-氨基-2-甲氧基环戊基]甲基氨基甲酸叔丁酯
步骤1(1r,3s,4r)-3-[(叔丁氧基羰基)氨基]-4-甲氧基环戊烷-1-甲酸甲酯
[化学式158]
将(1r,3s,4r)-3-(叔丁氧基羰基氨基)-4-羟基-环戊烷甲酸甲酯(cas:321744-23-4)(3.55g)、二氯甲烷(60ml)、氧化银(i)(cas:20667-12-3)(9.18g)、分子筛3a(cas:308080-99-1)(4.73g,在180℃减压干燥2小时后使用)和碘甲烷(cas:74-88-4)(16.2ml)的混合物在55℃剧烈搅拌42小时。通过硅藻土过滤反应溶液,并在减压下浓缩滤液。所获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(3.25g),为油状物。
步骤2(1r,3s,4r)-3-[(叔丁氧基羰基)氨基]-4-甲氧基环戊烷-1-甲酸
[化学式159]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤9中相同的方式获得标题化合物。
步骤3[(1r,3s,4r)-4-甲氧基环戊烷-1,3-二基]双氨基甲酸苄酯叔丁酯
[化学式160]
使用上述步骤2中获得的化合物,以与参考例a-1的步骤10中相同的方式获得标题化合物。
步骤4{(1r,3r,4s)-3-甲氧基-4-[(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸苄酯
[化学式161]
使用上述步骤3中获得的化合物,以与参考例a-1的步骤11和步骤12中相同的方式获得标题化合物。
步骤5{(1r,3r,4s)-3-甲氧基-4-[甲基(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸苄酯
[化学式162]
使用上述步骤4中获得的化合物,以与参考例a-1的步骤13中相同的方式获得标题化合物。
步骤6[(1r,3r,4s)-3-甲氧基-4-(甲基氨基)环戊基]氨基甲酸苄酯
[化学式163]
使用上述步骤5中获得的化合物,以与参考例a-1的步骤14中相同的方式获得为粗产物的标题化合物。
步骤7{(1r,3s,4r)-3-[(叔丁氧基羰基)(甲基)氨基]-4-甲氧基环戊基}氨基甲酸苄酯
[化学式164]
将上述步骤6中获得的化合物(4.82g)溶解在thf(40ml)中,向其中加入二碳酸二叔丁酯(cas:24424-99-5)(3.02g)在thf(20ml)中的溶液和tea(cas:121-44-8)(2.3ml),并将混合物在室温下搅拌16小时。减压蒸发溶剂,并将所得残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(3.15g),为油状物。
步骤8[(1s,2r,4r)-4-氨基-2-甲氧基环戊基]甲基氨基甲酸叔丁酯
[化学式165]
将上述步骤7中获得的化合物(3.15g)、5%湿钯/碳(ph)(cas:7440-05-3)(3.41g)和乙醇(100ml)的混合物在氢气气氛下在室温下搅拌3.5小时。将反应溶液过滤,并将滤液在减压下浓缩。所得残余物通过氨基硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(1.78g),为油状物。
参考例a-17
[(1s,2s,4r)-4-氨基-2-甲氧基环戊基]甲基氨基甲酸叔丁酯
步骤1{(1r,3s,4s)-3-[(叔丁氧基羰基)(甲基)氨基]-4-羟基环戊基}氨基甲酸苄酯
[化学式166]
使用参考例a-14的步骤9中获得的化合物,以与参考例a-16的步骤7中相同的方式获得标题化合物。
步骤2{(1r,3s,4s)-3-[(叔丁氧基羰基)(甲基)氨基]-4-甲氧基环戊基}氨基甲酸苄酯
[化学式167]
将上述步骤1中获得的化合物(1.10g)溶解在thf(37ml)中,向其中加入碘甲烷(0.31ml)和氢化钠(cas:7646-69-7)(纯度55%,0.205g),并将混合物在0℃搅拌1小时。然后,向其中加入另外的碘甲烷(0.10ml)和氢化钠(纯度55%,0.067g),并将混合物在0℃搅拌1小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/丙酮)纯化,得到标题化合物(0.827g),为油状物。
步骤3[(1s,2s,4r)-4-氨基-2-甲氧基环戊基]甲基氨基甲酸叔丁酯
[化学式168]
使用上述步骤2中获得的化合物,以与参考例a-16的步骤8中相同的方式获得标题化合物。
参考例a-18
[(1r,5s)-5-氨基-3,3-二氟环己基]氨基甲酸叔丁酯
步骤1[(1r,3s)-5,5-二氟环己烷-1,3-二基]双氨基甲酸苄酯叔丁酯
[化学式169]
使用(1s,5r)-5-[(叔丁氧基羰基)氨基]-3,3-二氟环己烷-1-甲酸(cas:2227198-19-6),以与参考例a-1的步骤10中同样的方式获得标题化合物。
步骤2[(1r,5s)-5-氨基-3,3-二氟环己基]氨基甲酸叔丁酯
[化学式170]
向上述步骤1中获得的化合物(596mg)在乙醇(20ml)中的溶液中加入10%湿钯/碳催化剂(400mg),并将混合物在氢气气氛下在室温搅拌4小时。氮置换后,将混合物通过硅藻土过滤,并将滤液减压浓缩,得到标题化合物(445mg),为油状物。所得化合物直接用于下一步。
参考例a-19
{(1s,3r)-3-[(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)氨基]环戊基}氨基甲酸叔丁酯
步骤1{(1s,3r)-3-[(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)(2-硝基苯-1-磺酰基)氨基]环戊基}氨基甲酸叔丁酯
[化学式171]
使用参考例a-3的步骤2中获得的化合物,以与参考例a-1的步骤13中相同的方式获得标题化合物,除了使用(2-溴乙氧基)-叔丁基二甲基甲硅烷(cas:86864-60-0)代替碘甲烷。
步骤2{(1s,3r)-3-[(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)氨基]环戊基}氨基甲酸叔丁酯
[化学式172]
将上述步骤1中获得的化合物(585mg)、4-叔丁基苯硫醇(0.362ml)、碳酸钾(595mg)和dmf(5.4ml)的混合物在40℃下搅拌6小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(428mg),为油状物。
参考例b-1
4-氯-2-甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶
步骤12-氨基-5-(2,2,2-三氟乙基)噻吩-3-甲酰胺
[化学式173]
向4,4,4-三氟丁醛水合物(2.00g)、2-氰基乙酰胺(cas:107-91-5)(1.75g)和硫(668mg)中加入dmf(14ml),并在冰冷却下向其中滴加tea(3.46ml)。滴加完成后,将混合物温热至室温,搅拌10小时,并静置过夜。将乙酸乙酯和饱和盐水加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(3.00g),为固体。
步骤22-甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4(3h)-酮
[化学式174]
向上述步骤1中获得的化合物(2.95g)中加入乙酸(13ml)和原乙酸三乙酯(cas:78-39-7)(10ml),并将混合物加热回流3小时。然后,将混合物在微波反应器中加热(在150℃持续5.5小时)。将饱和碳酸氢钠水溶液和乙酸乙酯加入到反应溶液中,并将混合物进行液体分离。将水层用乙酸乙酯萃取3次,有机层经无水硫酸钠干燥,并在减压下浓缩。将获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯,二氯甲烷/甲醇)纯化,得到标题化合物(0.86g),为固体。
步骤34-氯-2-甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶
[化学式175]
向2-甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4(3h)-酮(0.86g)中加入氧氯化磷(10.33g)和dmf(0.03ml),并将混合物在110℃下搅拌3.5小时。将反应溶液一点一点地添加到二氯甲烷和冰的混合物中,并将混合物剧烈搅拌1小时,并且进行液体分离。用水洗涤有机层,经无水硫酸钠干燥,并在减压下浓缩。所得残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(0.89g),为油状物。
参考例b-2
4-氯-6-(环丙基甲基)噻吩并[2,3-d]嘧啶
步骤16-(环丙基甲基)噻吩并[2,3-d]嘧啶-4(3h)-酮
[化学式176]
向3-环丙基丙醛(cas:5618-02-0)(968mg)、2-氰基乙酰胺(829mg)和硫(316mg)中加入dmf(10ml),并将混合物在冰盐浴中充分冷却。向其中滴加tea(1.64ml)。滴加完成后,将混合物温热至室温,并搅拌10小时。减压蒸发溶剂。将乙酸(10ml)和原甲酸三乙酯(6.8ml)加入到残余物中,并将混合物加热回流3.5小时。减压蒸发溶剂,并将正己烷/乙酸乙酯=1/1的混合溶剂加入到残余物中。过滤收集固体,并用正己烷/乙酸乙酯=1/1的混合溶剂洗涤,得到标题化合物(1.32g),为固体。
步骤24-氯-6-(环丙基甲基)噻吩并[2,3-d]嘧啶
[化学式177]
使用上述步骤1中获得的化合物,以与参考例b-1的步骤3中相同的方式获得标题化合物。
参考例b-3
4-氯-6-环丙基噻吩并[2,3-d]嘧啶
[化学式178]
将6-溴-4-氯噻吩并[2,3-d]嘧啶(100mg)、环丙基硼酸(79.0mg)、碳酸钠(144mg)、甲苯(1.5ml)、水(0.5ml)和四(三苯基膦)钯(0)(56.0mg)的混合物在氮气气氛下在110℃搅拌。向反应溶液中加入水,并将混合物用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(36.8mg),为固体。
参考例b-4
2,4-二氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶
步骤16-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-2,4(1h,3h)-二酮
[化学式179]
将参考例b-1的步骤1中获得的化合物(6.20g)溶解在1,4-二噁烷(100ml)中,并向其中加入三光气(3.55g),并将混合物加热回流6小时。使反应溶液冷却至室温,并在减压下浓缩。残余物用二氯甲烷进行浆化洗涤,得到标题化合物(2.41g),为固体。
步骤22,4-二氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶
[化学式180]
将上述步骤1中获得的化合物(2.41g)悬浮在氧氯化磷(11.5ml)中,向其中加入dmf(0.030ml),并将混合物在110℃加热下搅拌4小时。使反应溶液冷却至室温,用二氯甲烷洗涤,倒入冰水中,并将混合物在室温下剧烈搅拌1小时。有机层经无水硫酸钠干燥,并在减压下蒸发溶剂。残余物通过硅胶色谱法(二氯甲烷/正己烷)纯化,得到标题化合物(1.42g),为固体。
参考例b-5
4-氯-2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶
步骤12-(甲基硫基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4(3h)-酮
[化学式181]
将参考例b-1的步骤1中获得的化合物(40.0g)、乙醇(500ml)和乙基黄原酸钾(85.8g)的混合物在氮气气氛下加热回流19小时。减压浓缩反应溶液,并将残余物用二氯甲烷(200ml)进行浆化洗涤,并通过过滤收集固体(101g)。在冰冷却下,向获得的固体(101g)和dmf(2.09l)的混合物中加入碘甲烷(13.0ml),并将混合物在室温下搅拌1小时。向其中加入碘甲烷(8.5ml),并将混合物再搅拌30分钟。向反应溶液中加入水,并通过过滤收集沉淀的固体,得到标题化合物(14.5g),为固体。将乙酸乙酯加入到滤液中,并将混合物进行萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。向其中加入水,并通过过滤收集沉淀的固体,得到标题化合物(3.85g),为固体。滤液用盐酸酸化,并用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(17.1g),为固体。
步骤22-(甲磺酰基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4(3h)-酮
[化学式182]
在冰冷却下向上述步骤1中获得的化合物(42.2g)和thf(600ml)的混合物中加入oxone(cas:10058-23-8)(278g)和水(600ml)的混合物,并将混合物在室温搅拌4小时。向反应溶液中加入水,并通过过滤收集沉淀的固体,得到标题化合物(43.7g),为固体。
步骤32-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4(1h)-酮
[化学式183]
向上述步骤2中获得的化合物(14.3g)中加入甲醇(450ml)和碳酸钾(12.7g),并将混合物加热回流2小时。向其中添加另外的碳酸钾(6.33g),并将混合物加热回流1小时。将反应溶液浓缩至约一半至三分之一的体积,并用2n盐酸酸化。向其中加入乙酸乙酯,将混合物进行液体分离,并将水层用乙酸乙酯萃取。合并有机层,用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将乙醚添加到获得的残余物中,并通过过滤收集固体,得到标题化合物(7.85g),为固体。
步骤44-氯-2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶
[化学式184]
向上述步骤3中获得的化合物(8.39g)在氧氯化磷(36.9ml)中的悬浮液中加入dmf(8滴),并将混合物在室温下搅拌2.5小时,然后在60℃下搅拌15分钟。使反应溶液冷却,并一点一点地添加到碳酸氢钠和冰水的混合物中,用过的容器用二氯甲烷洗涤。将混合物搅拌10分钟,向其中加入二氯甲烷,并将混合物进行液体分离。用饱和碳酸氢钠水溶液将水层调节至ph=约9,并用二氯甲烷萃取。合并的有机层用饱和碳酸氢钠水溶液洗涤,经硫酸钠干燥,并减压浓缩。将获得的残余物通过硅胶柱色谱法(乙酸乙酯/正己烷)纯化,得到标题化合物(7.85g),为固体。
参考例b-6
4-氯-6-(2,2,2-三氟乙基)噻吩并[3,2-d]嘧啶
步骤14-氯-6-{2,2,2-三氟-1-[(三甲基甲硅烷基)氧基]乙基}噻吩并[3,2-d]嘧啶
[化学式185]
将4-氯噻吩并[3,2-d]嘧啶-6-甲醛(cas:875340-14-0)(500mg)溶于thf(6ml),向其中加入(三氟甲基)三甲基甲硅烷(cas:81290-20-2)(0.558ml),然后在冰冷却下向其中加入四丁基氟化铵(1.0mol/l,thf溶液)(0.126ml)。使混合物冷却至室温,并搅拌1小时。将乙酸乙酯和饱和盐水加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(294mg),为油状物。
步骤21-(4-氯噻吩并[3,2-d]嘧啶-6-基)-2,2,2-三氟乙-1-醇
[化学式186]
将上述步骤1中获得的化合物(260mg)溶解在thf(15ml)中,向其中加入1n盐酸(1.1ml),并将混合物在室温搅拌15分钟。将乙酸乙酯和饱和碳酸氢钠水溶液加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并减压浓缩,得到标题化合物(206mg),为固体。
步骤3硫代碳酸o-[1-(4-氯噻吩并[3,2-d]嘧啶-6-基)-2,2,2-三氟乙基]酯o-苯酯
[化学式187]
向氯硫羰碳酸苯酯(cas:1005-56-7)(77mg)的二氯甲烷溶液(1ml)中添加上述步骤2中获得的化合物(100mg)和tea(0.067ml),将混合物在室温下搅拌2小时。将水和二氯甲烷加入到反应溶液中,并将混合物进行液体分离。有机层经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(125mg),为固体。
步骤44-氯-6-(2,2,2-三氟乙基)噻吩并[3,2-d]嘧啶
[化学式188]
将上述步骤3中获得的化合物(2.42g)溶于甲苯(120ml)中,向其中加入氢化三丁基锡(cas:688-73-3)(3.45ml)和2,2'-偶氮二(异丁腈)(cas:78-67-1)(393mg),并将混合物在80℃搅拌2小时。减压蒸发溶剂,并将残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(1.22g),为固体。
参考例b-7
6-(2,2,2-三氟乙基)喹唑啉-4(3h)-酮
步骤1(4-甲氧基喹唑啉-6-基)甲醇
[化学式189]
将4-氯喹唑啉-6-甲酸甲酯(cas:152536-17-9)(3.00g)和thf(30ml)的混合物冷却至-50℃,向其中缓慢加入氢化二异丁基铝(cas:1191-15-7)(1.0mol/l,甲苯溶液,29.0ml),并经1.5小时将混合物温热至0℃。然后,使混合物温热至室温,并搅拌20小时。然后,将反应溶液冷却至0℃,缓慢地向其中加入氢化二异丁基铝(1.0mol/l,甲苯溶液,15.0ml),并将混合物在相同温度下搅拌15分钟,然后在室温下搅拌2小时。将反应溶液再次冷却至0℃,向其中加入酒石酸钾钠水溶液(2mol/l,75ml)。使混合物温热至室温,并搅拌过夜。反应混合物用乙酸乙酯和二氯甲烷萃取,有机层经无水硫酸钠干燥,并减压浓缩。将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯)。将获得的残余物溶于甲醇,并将溶液减压浓缩,得到标题化合物(1.46g,含有杂质(纯度约60%)),为固体。该化合物无需进一步纯化即用于下一步反应。
步骤24-甲氧基喹唑啉-6-甲醛
[化学式190]
将上述步骤1中获得的化合物(1.45g,含有杂质(纯度约60%))、二氯甲烷(60ml)和戴斯-马丁氧化剂(cas:87413-09-0)(3.51g)的混合物在室温下搅拌3小时。将饱和碳酸氢钠水溶液加入到反应溶液中,并将混合物用二氯甲烷萃取。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯)。将获得的固体溶解在乙酸乙酯中,并将溶液用饱和碳酸氢钠水溶液、水和饱和盐水洗涤,经无水硫酸钠干燥,并减压浓缩,得到标题化合物(0.893g,含有杂质(纯度约60%)),为固体。该化合物无需进一步纯化即用于下一步反应。
步骤34-甲氧基-6-{2,2,2-三氟-1-[(三甲基甲硅烷基)氧基]乙基}喹唑啉
[化学式191]
在冰冷却下向上述步骤2中获得的化合物(0.890g,含有杂质(纯度约60%))和thf(23ml)的混合物中加入(三氟甲基)三甲基甲硅烷(1.1ml)和氟化铯(cas:13400-13-0)(0.0336g)。将混合物在相同温度下搅拌10分钟,然后在室温下搅拌5小时。然后,向其中添加另外的氟化铯(0.240g),并将混合物在室温下搅拌3.5小时。然后,向其中添加另外的氟化铯(0.240g),并将混合物在室温下搅拌14小时。然后,向其中添加另外的(三氟甲基)三甲基甲硅烷(0.37ml)和氟化铯(0.250g),并将混合物在室温搅拌5小时。向反应溶液中加入饱和盐水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(0.633g),为固体。
步骤42,2,2-三氟-1-(4-甲氧基喹唑啉-6-基)乙-1-醇
[化学式192]
使用上述步骤3中获得的化合物,以与参考例b-6的步骤2中相同的方式获得标题化合物。
步骤5硫代碳酸o-苯酯o-[2,2,2-三氟-1-(4-甲氧基喹唑啉-6-基)乙基]酯
[化学式193]
使用上述步骤4中获得的化合物,以与参考例b-6的步骤3中相同的方式获得标题化合物。
步骤64-甲氧基-6-(2,2,2-三氟乙基)喹唑啉
[化学式194]
使用上述步骤5中获得的化合物,以与参考例b-6的步骤4中相同的方式获得标题化合物。
步骤76-(2,2,2-三氟乙基)喹唑啉-4(3h)-酮
[化学式195]
将上述步骤6中获得的化合物(0.111g)溶解在thf(2.8ml)中,并在冰冷却下向溶液中加入1n盐酸(1.4ml)。经5.5小时将反应溶液温热至室温,并向其中加入饱和碳酸氢钠水溶液。反应混合物用二氯甲烷萃取。将获得的水层用1n盐酸酸化,并用二氯甲烷萃取。合并所有有机层,经无水硫酸钠干燥,并减压浓缩,得到标题化合物(0.101g),为固体。
参考例b-8
6-(甲氧基甲基)噻吩并[2,3-d]嘧啶-4(3h)-酮
[化学式196]
以与参考例b-2的步骤1中同样的方式获得标题化合物,除了使用3-甲氧基丙醛(1.00g)(cas2806-84-0)代替3-环丙基丙醛。
参考例b-9
6-(氧杂环丁烷-3-基)噻吩并[2,3-d]嘧啶-4(3h)-酮
[化学式197]
以与参考例b-2的步骤1中相同的方式获得标题化合物,除了使用根据文献(wo2014/049133a1)中描述的方法合成的(氧杂环丁烷-3-基)乙醛代替3-环丙基丙醛。
参考例b-10
6-[(4-氯嘧啶-5-基)氧基]-2,3-二氟-n,n-二(丙-2-基)苯甲酰胺
步骤12,3-二氟-6-甲氧基-n,n-二(丙-2-基)苯甲酰胺
[化学式198]
在冰冷却下,向2,3-二氟-6-甲氧基苯甲酸(cas:773873-26-0)(2.0g)、二异丙胺(3.00ml)和二氯甲烷(28ml)的混合物中添加hatu(4.85g),将混合物在室温搅拌5小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(2.41g),为固体。
步骤22,3-二氟-6-羟基-n,n-二(丙-2-基)苯甲酰胺
[化学式199]
将上述步骤1中获得的化合物(2.41g)和二氯甲烷(17.8ml)的混合物冷却至-78℃,并经30分钟向其中滴加三溴化硼(约1mol/l,二氯甲烷溶液)(17.8ml)。滴加完成后,将混合物在0℃搅拌30分钟。冷却至-78℃后,向其中滴加甲醇(5ml)。温热后,向反应溶液中加水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(2.41g),为固体。
步骤32,3-二氟-n,n-二(丙-2-基)-6-[(嘧啶-5-基)氧基]苯甲酰胺
[化学式200]
将上述步骤2中获得的化合物(2.41g)、5-溴嘧啶(cas:4595-59-9)(4.24g)、dmf(44.5ml)和碳酸铯(8.70g)的混合物在120℃搅拌12小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(812mg),为固体。
步骤42,3-二氟-6-[(1-氧代-1λ5-嘧啶-5-基)氧基]-n,n-二(丙-2-基)苯甲酰胺
[化学式201]
将上述步骤3中获得的化合物(810mg)溶于二氯甲烷(24.2ml),在冰冷却下向其中加入3-氯过氧苯甲酸(含有30%水)(1.79g),并将混合物在0℃搅拌1小时,然后在室温下搅拌20小时。将反应溶液用冰冷却,并向其中加入饱和硫代硫酸钠水溶液。有机层用饱和碳酸氢钠水溶液洗涤两次,经无水硫酸钠干燥,并减压浓缩,得到标题化合物,为油状物(849mg)。所得化合物直接用于下一步反应。
步骤56-[(4-氯嘧啶-5-基)氧基]-2,3-二氟-n,n-二(丙-2-基)苯甲酰胺
[化学式202]
在冰冷却下,向tea(0.502ml)和氯仿(1.3ml)的混合物中加入氧氯化磷(0.92g)。向其中滴加上述步骤4中获得的化合物(849mg)在氯仿(12ml)中的溶液。将混合物加热至65℃,并搅拌4.5小时。冷却后,将反应溶液一点一点地加入到冰冷却的饱和碳酸氢钠水溶液中,并将混合物剧烈搅拌30分钟。用饱和碳酸氢钠将混合物的ph调节至7至8,并进行液体分离。用二氯甲烷萃取水层,并将有机层合并,经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(236mg),为固体。
参考例c-1
(1r,3s)-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺
步骤1[(1s,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸苄酯
[化学式203]
将参考例a-8的步骤2中获得的化合物(11.6g)溶解于2-丙醇(407ml)中,并将向其中加入根据文献(cancercell2015,27,589-602)中描述的方法合成的4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶(cas:1628317-85-0)(10.3g)和dipea(21.6ml),并将反应溶液加热回流3天。使反应溶液冷却至室温,并在减压下浓缩。残余物通过硅胶色谱法(二氯甲烷/乙酸乙酯)纯化,得到标题化合物(13.2g),为固体。
步骤2(1r,3s)-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺
[化学式204]
将上述步骤1中获得的化合物(12.9g)溶解于二氯甲烷(92.6ml)中。在冰冷却下向其中滴加三甲基碘硅烷(5.33ml)。将反应溶液温热至室温,并搅拌2小时。向反应溶液中加入甲醇(0.500ml),并将混合物减压浓缩。残余物通过氨基硅胶色谱法(二氯甲烷/甲醇)纯化,得到标题化合物(4.00g),为油状物。
参考例c-2
(1r,3s)-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
步骤1[(1s,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸叔丁酯
[化学式205]
将参考例a-2的步骤2中获得的化合物(6.20g)和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶(6.72g)溶解于2-丙醇(90ml)中,向其中加入dipea(9.27ml),将混合物加热回流7小时。将乙酸乙酯和饱和盐水加入到反应溶液中,将混合物进行液体分离,并将水层用乙酸乙酯萃取。合并有机层,用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(7.50g),为固体。
步骤2(1r,3s)-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
[化学式206]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-3
(1r,3r)-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
步骤1[(1r,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式207]
使用[(1r,3r)-3-氨基环戊基]氨基甲酸叔丁酯(cas:1009075-44-8)和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1r,3r)-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式208]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-4
(1r,3s)-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
步骤1[(1s,3r)-3-{甲基-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式209]
使用参考例a-4中获得的化合物和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中同样的方式获得标题化合物。
步骤2(1r,3s)-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式210]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-5
(1r,3s)-n1-乙基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
步骤1[(1s,3r)-3-{乙基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式211]
使用参考例a-5的步骤2中获得的化合物和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1r,3s)-n1-乙基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式212]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-6
(1r,3s)-n1-甲基-n1-[6-(2,2,2-三氟乙基)喹唑啉-4-基]环戊烷-1,3-二胺盐酸盐
步骤1[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)喹唑啉-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式213]
将参考例b-7的步骤7中得到的化合物(0.0974g)、bop(cas:56602-33-6)(0.250g)、乙腈(8.5ml)和dbu(cas:6674-22-2)(0.145ml)的混合物在室温下搅拌7分钟。然后,向其中加入参考例a-4中获得的化合物(0.218g)在乙腈(8.5ml)中的溶液,并将混合物在60℃搅拌1小时。减压浓缩反应溶液,并将所得残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(0.178g),为油状物。
步骤2(1r,3s)-n1-甲基-n1-[6-(2,2,2-三氟乙基)喹唑啉-4-基]环戊烷-1,3-二胺盐酸盐
[化学式214]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-7
n4-[(1r,3s)-3-氨基环戊基]-n2,n4-二甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-2,4-二胺盐酸盐
步骤1[(1s,3r)-3-{[2-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊基]氨基甲酸叔丁酯
[化学式215]
使用参考例a-4中获得的化合物和参考例b-4的步骤2中获得的化合物,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2[(1s,3r)-3-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式216]
将上述步骤1中得到的化合物(800mg)悬浮于丁腈(6ml)中,向其中加入40%甲胺水溶液(0.741ml),并将混合物在微波反应器中在150℃加热下搅拌1小时45分钟。将乙酸乙酯和饱和盐水加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将二氯甲烷/正己烷=1/3的混合溶剂添加至获得的残余物中,并通过过滤收集固体,得到标题化合物(650mg),为固体。
步骤3n4-[(1r,3s)-3-氨基环戊基]-n2,n4-二甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-2,4-二胺盐酸盐
[化学式217]
使用上述步骤2中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-8
(1r,3s)-n1-[6-(甲氧基甲基)噻吩并[2,3-d]嘧啶-4-基]-n1-甲基环戊烷-1,3-二胺盐酸盐
步骤1[(1s,3r)-3-{[6-(甲氧基甲基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊基]氨基甲酸叔丁酯
[化学式218]
使用参考例a-4中获得的化合物和参考例b-8中获得的化合物,以与参考例c-6的步骤1中相同的方式获得标题化合物。
步骤2(1r,3s)-n1-[6-(甲氧基甲基)噻吩并[2,3-d]嘧啶-4-基]-n1-甲基环戊烷-1,3-二胺盐酸盐
[化学式219]
使用上述步骤1中获得的化合物(140mg),以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-9
2-{[(1r,3s)-3-氨基环戊基][6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}乙-1-醇盐酸盐
步骤1[(1s,3r)-3-{(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式220]
使用参考例a-19的步骤2中获得的化合物和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤22-{[(1r,3s)-3-氨基环戊基][6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}乙-1-醇盐酸盐
[化学式221]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。所得化合物无需纯化即直接用于下一步。
参考例c-10
(1r,3s)-n1-甲基-n1-[2-甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
步骤1[(1s,3r)-3-{甲基[2-甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式222]
使用参考例a-4中获得的化合物和参考例b-1的步骤3中获得的化合物,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1r,3s)-n1-甲基-n1-[2-甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式223]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-11
(1r,3s)-n1-甲基-n1-[6-(氧杂环丁烷-3-基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
步骤1[(1s,3r)-3-{甲基[6-(氧杂环丁烷-3-基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式224]
使用参考例a-4的步骤1中获得的化合物和参考例b-9中获得的化合物,以与参考例c-6的步骤1中相同的方式获得标题化合物。
步骤2(1r,3s)-n1-甲基-n1-[6-(氧杂环丁烷-3-基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式225]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。所得化合物无需纯化即直接用于下一步。
参考例c-12
(1r,3s)-n1-(6-环丙基噻吩并[2,3-d]嘧啶-4-基)-n1-甲基环戊烷-1,3-二胺盐酸盐
步骤1{(1s,3r)-3-[(6-环丙基噻吩并[2,3-d]嘧啶-4-基)(甲基)氨基]环戊基}氨基甲酸叔丁酯
[化学式226]
使用参考例a-4中获得的化合物和参考例b-3中获得的化合物,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1r,3s)-n1-(6-环丙基噻吩并[2,3-d]嘧啶-4-基)-n1-甲基环戊烷-1,3-二胺盐酸盐
[化学式227]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。所得化合物无需纯化即直接用于下一步。
参考例c-13
(1s,3r)-5,5-二氟-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
步骤1[(1r,5s)-3,3-二氟-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸叔丁酯
[化学式228]
使用参考例a-18的步骤2中获得的化合物和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1s,3r)-5,5-二氟-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
[化学式229]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-14
(1r,2s,4r)-4-氨基-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
步骤1[(1r,3r,4s)-3-羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸苄酯
[化学式230]
将参考例a-1的步骤14中获得的化合物(43.3g)、4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶(cas:1628317-85-0)(43.5g)、dipea(57.1ml)和2-丙醇(820ml)的混合物在90℃下搅拌5小时。将反应溶液在减压下浓缩,向其中添加水,并将混合物用乙酸乙酯/二氯甲烷/甲醇萃取。有机层经无水硫酸钠干燥,并在减压下浓缩。向残余物中加入乙酸乙酯/正己烷,并通过过滤收集固体,得到标题化合物(64.2g),为固体。浓缩滤液,将乙酸乙酯/正己烷加入到残余物中,并通过过滤收集固体,得到标题化合物(26.5g),为固体。将滤液浓缩,并将残余物通过硅胶柱色谱法(二氯甲烷/乙酸乙酯)纯化。将乙酸乙酯加入到获得的固体中,并通过过滤收集固体,得到标题化合物(3.67g),为固体。
步骤2(1r,2s,4r)-4-氨基-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
[化学式231]
在0℃下向上述步骤1中获得的化合物(8.98g)和乙腈(90ml)的混合物中加入三甲基碘硅烷(8.1ml),并将混合物在0℃搅拌30分钟。向反应溶液中加入1n盐酸和水,并将混合物用乙酸乙酯洗涤。将有机层用1n盐酸萃取,将2n氢氧化钠水溶液加入到合并的水层中,并将混合物用二氯甲烷/甲醇萃取九次。合并的有机层经无水硫酸钠干燥,并减压浓缩,得到标题化合物(6.53g),为固体。
步骤3(1r,2s,4r)-4-氨基-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
[化学式232]
将上述步骤2中获得的化合物(6.53g)、1n盐酸(21ml)和乙腈(60ml)的混合物在室温下搅拌10分钟。减压浓缩反应溶液,向其中加入乙腈,并通过过滤收集固体,得到标题化合物(5.83g),为固体。浓缩滤液,向其中加入乙腈,并通过过滤收集固体,得到标题化合物(1.13g),为固体。
参考例c-15
(1r,2s,4r)-4-氨基-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇盐酸盐
步骤1[(1r,3r,4s)-3-羟基-4-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊基]氨基甲酸叔丁酯
[化学式233]
向参考例a-15的步骤2中获得的化合物(3.33g)和参考例b-5的步骤4中获得的化合物(4.09g)中加入2-丙醇(49ml)和dipea(7.56ml),将混合物加热回流3小时,并在室温下放置过夜。将乙酸乙酯和饱和盐水加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(5.05g),为固体。
步骤2(1r,2s,4r)-4-氨基-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇盐酸盐
[化学式234]
在室温下搅拌上述步骤1中获得的化合物(810mg)、二氯甲烷(7ml)和氯化氢(4mol/l,1,4-二噁烷溶液,7ml)的混合物。向其中加入少量甲醇以制成溶液,将溶液进一步搅拌,并在减压下浓缩。将残余物悬浮在正己烷/乙醇中,并通过过滤收集固体,得到标题化合物(724mg),为固体。
参考例c-16
(1s,2s,4r)-4-氨基-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
步骤1[(1r,3s,4s)-3-羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸苄酯
[化学式235]
使用参考例a-14的步骤9中获得的化合物和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1s,2s,4r)-4-氨基-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
[化学式236]
使用上述步骤1中获得的化合物,以与参考例c-1的步骤2中相同的方式获得标题化合物。
参考例c-17
(1r,3s,5r)-3-氨基-n,n-二甲基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酰胺(外消旋物)
步骤1[(1s,3s,5r)-3-(二甲基氨基甲酰基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸苄酯(外消旋物)
[化学式237]
使用参考例a-7的步骤6中获得的化合物和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1r,3s,5r)-3-氨基-n,n-二甲基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酰胺(外消旋物)
[化学式238]
使用上述步骤1中获得的化合物,以与参考例c-1的步骤2中相同的方式获得标题化合物。
参考例c-18
(1s,3r,5r)-5-甲氧基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺(外消旋物)
步骤1[(1r,3s,5s)-3-甲氧基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸苄酯(外消旋物)
[化学式239]
使用参考例a-11的步骤4中获得的化合物和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1s,3r,5r)-5-甲氧基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺(外消旋物)
[化学式240]
使用上述步骤1中获得的化合物,以与参考例c-14的步骤2中相同的方式获得标题化合物。
参考例c-19
(1r,3r,5s)-3-氨基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇(外消旋物)
步骤1[(1r,3s,5s)-3-(甲氧基甲氧基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸苄酯(外消旋物)
[化学式241]
使用参考例a-6的步骤5中获得的化合物和4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1r,3r,5s)-3-氨基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇(外消旋物)
[化学式242]
使用上述步骤1中获得的化合物,以与参考例c-1的步骤2中相同的方式获得标题化合物。
参考例c-20
(1r,2s,4r)-4-氨基-2-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
步骤1[(1r,3s,4r)-3-{[2-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}-4-羟基环戊基]氨基甲酸苄酯
[化学式243]
使用参考例a-1的步骤14中获得的化合物和参考例b-4的步骤2中获得的化合物,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2[(1r,3r,4s)-3-羟基-4-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸苄酯
[化学式244]
使用上述步骤1中获得的化合物,以与参考例c-7的步骤2中相同的方式获得标题化合物。
步骤3(1r,2s,4r)-4-氨基-2-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
[化学式245]
使用上述步骤2中获得的化合物,以与参考例c-14的步骤2中相同的方式获得标题化合物。
参考例c-21
(1r,2s,4r)-4-氨基-2-{[2-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇盐酸盐
步骤1[(1r,3s,4r)-3-{[2-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}-4-羟基环戊基]氨基甲酸叔丁酯
[化学式246]
将参考例c-20的步骤1中获得的化合物(8.41g)、乙腈(100ml)和三甲基碘硅烷(6.71ml)的混合物在冰冷却下搅拌1小时。向其中加入甲醇(10ml),将混合物搅拌20分钟,并在减压下蒸发溶剂。将thf(100ml)、水(100ml)、碳酸钠(6.93g)和二碳酸二叔丁酯(5.70g)添加到获得的油中,并将混合物在室温搅拌1小时。将水和乙酸乙酯加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将二氯甲烷/正己烷添加到获得的残余物中,并通过过滤收集固体,得到标题化合物(6.51g),为固体。浓缩滤液,并将残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(0.903g),为固体。
步骤2(1r,2s,4r)-4-氨基-2-{[2-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇盐酸盐
[化学式247]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例c-22
(1s,2s,4r)-4-氨基-2-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
步骤1[(1r,3s,4s)-3-{[2-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}-4-羟基环戊基]氨基甲酸苄酯
[化学式248]
使用参考例a-14的步骤9中获得的化合物和参考例b-4的步骤2中获得的化合物,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2[(1r,3s,4s)-3-羟基-4-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸苄酯
[化学式249]
使用上述步骤1中获得的化合物,以与参考例c-7的步骤2中相同的方式获得标题化合物。
步骤3(1s,2s,4r)-4-氨基-2-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
[化学式250]
使用上述步骤2中获得的化合物,以与参考例c-14的步骤2中相同的方式获得标题化合物。
参考例c-23
(1r,2s,4r)-4-氨基-2-{[2-{[(4-甲氧基苯基)甲基]氨基}-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇
步骤1[(1r,3r,4s)-3-羟基-4-{[2-{[(4-甲氧基苯基)甲基]氨基}-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊基]氨基甲酸苄酯
[化学式251]
将参考例c-20的步骤1中获得的化合物(1.57g)、4-甲氧基苄胺(1.97ml)、dipea(2.66ml)和丁腈(9.0ml)的混合物在微波反应器中于150℃加热搅拌1.5小时。滤出反应溶液中的不溶物,将水加入滤液中,并将混合物用乙酸乙酯萃取。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(氯仿/甲醇),得到标题化合物(1.62g),为固体。
步骤2(1r,2s,4r)-4-氨基-2-{[2-{[(4-甲氧基苯基)甲基]氨基}-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇
[化学式252]
使用上述步骤1中获得的化合物,以与参考例c-14的步骤2中相同的方式获得标题化合物。
参考例c-24
2-[(4-{[(1s,2r,4r)-4-氨基-2-羟基环戊基](甲基)氨基}嘧啶-5-基)氧基]-5-氟-n,n-二(丙-2-基)苯甲酰胺
步骤1{(1r,3s,4r)-3-[(5-{2-[二(丙-2-基)氨基甲酰基]-4-氟苯氧基}嘧啶-4-基)(甲基)氨基]-4-羟基环戊基}氨基甲酸苄酯
[化学式253]
将参考例a-1的步骤14中获得的化合物(0.233g)、根据文献(wo2017/214367)中所述的方法制备的2-[(4-氯嘧啶-5-基)氧基]-5-氟-n,n-二(丙-2-基)苯甲酰胺(0.308g)、2-丙醇(3.50ml)和dipea(0.305ml)的混合物在微波反应器中于110℃搅拌30分钟,然后在相同条件下搅拌2小时。向其中加入参考例a-1的步骤14中获得的化合物(0.0463g),将混合物在微波反应器中于110℃搅拌2小时,并使其在室温下静置16小时。向其中加入参考例a-1的步骤14中获得的化合物(0.0463g),并将混合物在微波反应器中于110℃搅拌2小时。将反应溶液在减压下浓缩,用水稀释,并用乙酸乙酯萃取。有机层用饱和盐水洗涤,并经无水硫酸钠干燥。在减压下蒸发溶剂,并将获得的残余物进行硅胶柱色谱法(正己烷/乙酸乙酯,然后是乙酸乙酯/甲醇),得到标题化合物(0.475g),为固体。
步骤22-[(4-{[(1s,2r,4r)-4-氨基-2-羟基环戊基](甲基)氨基}嘧啶-5-基)氧基]-5-氟-n,n-二(丙-2-基)苯甲酰胺
[化学式254]
使用上述步骤1中获得的化合物,以与参考例a-2的步骤2中相同的方式获得标题化合物。
参考例c-25
(1r,2s,4r)-4-氨基-2-[(5-{4-氟-2-[4-(丙-2-基)嘧啶-5-基]苯氧基}嘧啶-4-基)(甲基)氨基]环戊-1-醇
步骤1[(1r,3s,4r)-3-{[5-(2-溴-4-氟苯氧基)嘧啶-4-基](甲基)氨基}-4-羟基环戊基]氨基甲酸苄酯
[化学式255]
使用参考例a-1的步骤14中获得的化合物和根据文献(wo2017/214367)中描述的方法制备的5-(2-溴-4-氟苯氧基)-4-氯嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2{(1r,3s,4r)-3-[(5-{4-氟-2-[4-(丙-2-基)嘧啶-5-基]苯氧基}嘧啶-4-基)(甲基)氨基]-4-羟基环戊基}氨基甲酸苄酯
[化学式256]
将上述步骤1中获得的化合物(140mg)、4-异丙基嘧啶-5-硼酸(cas:913835-27-5)(52.5mg)、四(三苯基膦)钯(0)(30.4mg)、碳酸钠(112mg)、水(1.1ml)和1,4-二噁烷(3.3ml)的混合物在90℃下搅拌1.5小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯)。将正己烷/乙酸乙酯添加到所得残余物中,并通过过滤收集所得固体,得到标题化合物(92.5mg),为固体。
步骤3(1r,2s,4r)-4-氨基-2-[(5-{4-氟-2-[4-(丙-2-基)嘧啶-5-基]苯氧基}嘧啶-4-基)(甲基)氨基]环戊-1-醇
[化学式257]
使用上述步骤2中获得的化合物,以与参考例c-14的步骤2中相同的方式获得标题化合物。
参考例c-26
(1r,2s,4r)-4-氨基-2-[(5-{[5-氟-2'-(丙-2-基)[1,1'-联苯]-2-基]氧基}嘧啶-4-基)(甲基)氨基]环戊-1-醇
步骤1{(1r,3s,4r)-3-[(5-{[5-氟-2'-(丙-2-基)[1,1'-联苯]-2-基]氧基}嘧啶-4-基)(甲基)氨基]-4-羟基环戊基}氨基甲酸苄酯
[化学式258]
使用参考例c-25的步骤1中获得的化合物和2-异丙基苯基硼酸(cas:89787-12-2),以与参考例c-25的步骤2中相同的方式获得标题化合物。
步骤2(1r,2s,4r)-4-氨基-2-[(5-{[5-氟-2'-(丙-2-基)[1,1'-联苯]-2-基]氧基}嘧啶-4-基)(甲基)氨基]环戊-1-醇
[化学式259]
使用上述步骤1中获得的化合物,以与参考例c-14的步骤2中相同的方式获得标题化合物。
参考例c-27
(1r,2s,4r)-4-氨基-2-[(5-{4-氟-2-[1-(丙-2-基)-1h-吡唑-5-基]苯氧基}嘧啶-4-基)(甲基)氨基]环戊-1-醇
步骤1{(1r,3s,4r)-3-[(5-{4-氟-2-[1-(丙-2-基)-1h-吡唑-5-基]苯氧基}嘧啶-4-基)(甲基)氨基]-4-羟基环戊基}氨基甲酸苄酯
[化学式260]
使用参考例c-25的步骤1中获得的化合物和1-异丙基吡唑-5-硼酸(cas:839714-33-9),以与参考例c-25的步骤2中相同的方式获得标题化合物。
步骤2(1r,2s,4r)-4-氨基-2-[(5-{4-氟-2-[1-(丙-2-基)-1h-吡唑-5-基]苯氧基}嘧啶-4-基)(甲基)氨基]环戊-1-醇
[化学式261]
使用上述步骤1中获得的化合物,以与参考例c-14的步骤2中相同的方式获得标题化合物。
参考例c-28
6-[(4-{[(1s,2r,4r)-4-氨基-2-羟基环戊基](甲基)氨基}嘧啶-5-基)氧基]-2,3-二氟-n,n-二(丙-2-基)苯甲酰胺
步骤1{(1r,3s,4r)-3-[(5-{2-[二(丙-2-基)氨基甲酰基]-3,4-二氟苯氧基}嘧啶-4-基)(甲基)氨基]-4-羟基环戊基}氨基甲酸苄酯
[化学式262]
使用参考例a-1的步骤14中获得的化合物和参考例b-10的步骤5中获得的化合物,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤26-[(4-{[(1s,2r,4r)-4-氨基-2-羟基环戊基](甲基)氨基}嘧啶-5-基)氧基]-2,3-二氟-n,n-二(丙-2-基)苯甲酰胺
[化学式263]
使用上述步骤1中获得的化合物,以与参考例c-14的步骤2中相同的方式获得标题化合物。
参考例c-29
[(1s,3r)-3-{[6-(环丙基甲基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊基]氨基甲酸叔丁酯
[化学式264]
向[(1s,3r)-3-(甲基氨基)环戊基]氨基甲酸叔丁酯(200mg)和参考例b-2的步骤2中获得的化合物(210mg)中加入2-丙醇(5ml)和dipea(0.325ml),并将混合物加热回流6小时。将乙酸乙酯和饱和盐水加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(205mg),为油状物。
参考例c-30
(1r,2r,4s)-2-氨基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
步骤1[(1r,2r,4s)-2-(甲氧基甲氧基)-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式265]
将参考例a-12的步骤7中得到的化合物(0.370g)、4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶(0.358g)、dipea(0.469ml)和2-丙醇(15.0ml)的混合物在100℃下搅拌10小时。将混合物在减压下浓缩,并将获得的残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(0.592g),为固体。
步骤2(1r,2r,4s)-2-氨基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
[化学式266]
将上述步骤1中得到的化合物(0.162g)、氯化氢(4mol/l,1,4-二噁烷溶液,5ml)和甲醇(5ml)的混合物在室温下搅拌1个小时。浓缩混合物,并将残余物干燥,得到标题化合物(0.138g),为固体。其直接用于下一步。
参考例c-31
(1s,2r,4s)-2-氨基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
步骤1[(1r,2s,4s)-2-(甲氧基甲氧基)-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯
[化学式267]
将[(1r,2s,4s)-2-(甲氧基甲氧基)-4-(甲基氨基)环戊基]氨基甲酸叔丁酯(0.414g)、4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶(0.400g)、2-丙醇(15.0ml)和dipea(0.525ml)的混合物在100℃下搅拌4小时。将混合物在减压下浓缩,并将获得的残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(0.662g),为固体。
步骤2(1s,2r,4s)-2-氨基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
[化学式268]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。该化合物直接用于下一步反应。
参考例c-32
(1r,3s,5r)-5-(1,3-噁唑-2-基)-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺(外消旋物)
步骤1(1s,3s,5r)-3-{[(苄氧基)羰基]氨基}-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酸甲酯(外消旋物)
[化学式269]
使用参考例a-9的步骤4中获得的化合物和根据文献(cancercell2015,27,589-602)中描述的方法制备的氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1s,3s,5r)-3-{[(苄氧基)羰基]氨基}-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酸(外消旋物)
[化学式270]
将上述步骤1中获得的化合物(1.00g)、thf(15.0ml)和氢氧化锂一水合物(210mg)的混合物在室温搅拌2小时。将反应混合物在减压下浓缩,将残余物在0℃用2n盐酸(2.00ml)酸化(ph3-4),并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,并经无水硫酸钠干燥。减压蒸发溶剂,得到标题化合物(0.949g),为固体。
步骤3[(1s,3s,5r)-3-[(2,2-二甲氧基乙基)氨基甲酰基]-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸苄酯(外消旋物)
[化学式271]
在0℃下向上述步骤2中获得的化合物(0.455g)在dmf(4.50ml)中的溶液中依次加入comu(cas:1075198-30-9)(0.471g)、dipea(0.234ml)和氨基乙醛缩二甲醇(cas:22483-09-6)(0.118ml),并将混合物在室温搅拌16小时。将反应混合物倒入水中,并用乙酸乙酯/乙醚的混合溶剂萃取。依次用水(三遍)和饱和盐水洗涤有机层,并经无水硫酸钠干燥。在减压下蒸发溶剂,并将获得的残余物进行硅胶柱色谱法(正己烷/乙酸乙酯,随后乙酸乙酯/甲醇),得到标题化合物(0.491g),为固体。
步骤4[(1s,3s,5r)-3-[(2-氧代乙基)氨基甲酰基]-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸苄酯(外消旋物)
[化学式272]
在室温下向上述步骤3中获得的化合物(0.491g)在二氯甲烷(80.0ml)中的溶液中加入三氟乙酸(24.0ml),并将混合物在相同温度下搅拌16小时。将混合物在加热回流下搅拌另外2小时。减压蒸发溶剂,将乙酸乙酯和饱和碳酸氢钠水溶液加入到获得的残余物中,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,并经无水硫酸钠干燥。减压蒸发溶剂,并将残余物干燥,得到标题化合物,为固体。其直接用于下一步。
步骤5[(1s,3s,5r)-3-(1,3-噁唑-2-基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸苄酯(外消旋物)
[化学式273]
在室温下向上述步骤4中获得的化合物(0.382g)在二氯甲烷(7.00ml)中的溶液中加入三苯基膦(0.273g)、六氯乙烷(cas:67-72-1)(0.247g)和tea(0.193ml),将混合物搅拌3天。将反应混合物在减压下浓缩,并将残余物进行硅胶柱色谱法(乙酸乙酯/甲醇),得到包含目标产物的混合物,为油状物。该化合物无需进一步纯化即用于下一步。
步骤6(1r,3s,5r)-5-(1,3-噁唑-2-基)-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺(外消旋物)
[化学式274]
在0℃下向上述步骤5中获得的化合物在乙腈中的溶液(15.0ml)中加入三甲基碘硅烷(0.239ml),并将混合物在室温搅拌2小时。向其中加入另外的三甲基碘硅烷(0.239ml),并将混合物搅拌0.5小时。在减压下蒸发溶剂,并将获得的残余物进行氨基硅胶柱色谱法(正己烷/乙酸乙酯,然后是乙酸乙酯/甲醇),得到标题化合物(0.0472g),为油状物。
参考例c-33
[(1r,2r,3s,4s)-2,3-二羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯(异构体a)
[(1r,2s,3r,4s)-2,3-二羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯(异构体b)
步骤1(1r,3s)-环戊-4-烯-1,3-二基双氨基甲酸叔丁酯丙-2-烯-1-基酯
[化学式275]
使用根据文献(eur.j.org.chem.2013,17,3477-3493)中描述的方法合成的(1s,4r)-4-[(叔丁氧基羰基)氨基]环戊-2-烯-1-甲酸(1.00g),以与参考例a-6的步骤4中相同的方式获得标题化合物。
步骤2[(1r,4s)-4-氨基环戊-2-烯-1-基]氨基甲酸叔丁酯
[化学式276]
将上述步骤2中获得的化合物(2.87g)、二氯甲烷(15ml)、吡咯烷(0.366ml)和四(三苯基膦)钯(0)(41mg)的混合物在室温搅拌1小时。将混合物在减压下浓缩,并将残余物通过硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(1.35g,含有少量的立体异构体),为油状物。
步骤3{(1r,4s)-4-[(2-硝基苯-1-磺酰基)氨基]环戊-2-烯-1-基}氨基甲酸叔丁酯
[化学式277]
使用上述步骤2中获得的化合物,以与参考例a-1的步骤12中相同的方式获得标题化合物(含有少量的立体异构体)。
步骤4{(1r,4s)-4-[甲基(2-硝基苯-1-磺酰基)氨基]环戊-2-烯-1-基}氨基甲酸叔丁酯
[化学式278]
使用上述步骤3中获得的化合物,以与参考例a-1的步骤13中相同的方式获得标题化合物(含有少量的立体异构体)。
步骤5[(1r,4s)-4-(甲基氨基)环戊-2-烯-1-基]氨基甲酸叔丁酯
[化学式279]
将上述步骤4中获得的化合物(887mg)、4-叔丁基苯硫醇(0.751ml)、碳酸钾(1.23g)和dmf(11.2ml)的混合物在40℃下搅拌2.5小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将残余物通过硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(498mg,含有少量的立体异构体),为油状物。
步骤6[(1r,4s)-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-2-烯-1-基]氨基甲酸叔丁酯
[化学式280]
向上述步骤5中获得的化合物(622mg)和2-丙醇(23.4ml)的混合物中加入dipea(0.816ml),并将混合物在90℃搅拌过夜。将混合物在减压下浓缩,并将残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到固体(800mg)。通过手性柱(daicel,chiralpak(注册商标,daicelcorporation)ia,正己烷/2-丙醇)纯化得到标题化合物(541mg),为固体。
步骤7[(1r,2r,3s,4s)-2,3-二羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯(异构体a)
[(1r,2s,3r,4s)-2,3-二羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基甲酸叔丁酯(异构体b)
[化学式281]
将上述步骤6中获得的化合物(172mg)、四氧化锇(4%水溶液)(0.051ml)、4-甲基吗啉n-氧化物(70.5mg)、丙酮(3.6ml)和水(0.40ml)的混合物在室温下搅拌4小时。将另外的4-甲基吗啉n-氧化物(70.5mg)和四氧化锇(4%水溶液)(0.13ml)加入到反应溶液中,并将混合物搅拌另外3小时。将硫代硫酸钠水溶液加入到反应溶液中,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(异构体a,较早洗脱的组分)(44.9mg),和标题化合物(异构体b,较晚洗脱的组分)(56.4mg),分别为固体。
异构体a(较早洗脱的组分)
异构体b(较晚洗脱的成分)
参考例d-1
5-甲酰基-1h-吲哚-2-甲腈
[化学式282]
将氢化钠(纯度55%,628mg)置于反应容器中,对该容器进行氮置换,并在冰冷却下向其中加入thf(30ml)。向其中逐滴添加根据文献(wo2014/164749)中描述的方法合成的5-溴-1h-吲哚-2-甲腈(1.59g)的thf溶液(42ml),并将混合物在室温下搅拌15分钟。将反应溶液冷却至-78℃,经20分钟向其中滴加叔丁基锂(1.65mol/l,正戊烷溶液,10.9ml),并将混合物在相同温度下搅拌45分钟。经5分钟向其中滴加dmf(2.8ml),并将混合物在-78℃搅拌45分钟。向其中滴加乙酸(4.1ml),并将混合物温热至室温。将乙酸乙酯和0.5n盐酸水溶液加入到反应溶液中。通过过滤除去不溶物,并对滤液进行液体分离。用乙酸乙酯萃取水层,并将有机层合并,用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(946mg),为固体。
参考例d-2
5-甲酰基-4-甲基-1-{[1-(三苯基甲基)-1h-吡唑-4-基]甲基}-1h-吲哚-2-甲腈
步骤15-甲酰基-4-甲基-1h-吲哚-2-甲腈
[化学式283]
使用根据文献(cancercell2015,27,589-602)中描述的方法合成的5-溴-4-甲基-1h-吲哚-2-甲腈(1.22g),以与参考例d-1中相同的方式获得标题化合物。
步骤25-甲酰基-4-甲基-1-{[1-(三苯基甲基)-1h-吡唑-4-基]甲基}-1h-吲哚-2-甲腈
[化学式284]
在氩气气氛下,在0℃下,向上述步骤1中获得的化合物(0.250g)、碳酸铯(0.619g)和dmf(50ml)的混合物中加入根据文献(j.med.chem.2016,59(3),892-913)中描述的方法合成的4-(溴甲基)-1-(三苯基甲基)-1h-吡唑(0.821g)和dmf(90ml)的混合物,并将混合物在室温搅拌1小时。将水添加到反应溶液中,并将混合物用乙酸乙酯/乙醚萃取。通过过滤收集有机层的固体,得到标题化合物(0.369g),为固体。浓缩滤液,将乙醚加入到残余物中,并通过过滤收集固体,得到标题化合物(0.193g),为固体。
参考例d-3
4-溴-1-(甲磺酰基)-1h-吡咯并[2,3-c]吡啶
[化学式285]
在冰冷却下向4-溴-1h-吡咯并[2,3-c]吡啶(cas:69872-17-9)(401mg)、dipea(1.05ml)和二氯甲烷(15ml)的混合物中滴加甲磺酰氯(0.281ml)。将混合物在室温搅拌10分钟,在冰冷却下将饱和碳酸氢钠水溶液加入到反应溶液中,并将混合物用乙酸乙酯萃取。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(394mg),为固体。
参考例d-4
4-溴-1-甲基-1h-吡唑并[3,4-c]吡啶
[化学式286]
在冰冷却下向4-溴-1h-吡唑并[3,4-c]吡啶(cas:1032943-43-3)(262mg)、碳酸铯(863mg)和dmf(6.5ml)的混合物中滴加碘甲烷(cas:74-88-4)(0.107ml)。将混合物在室温搅拌20小时,在冰冷却下将水加入到反应溶液中,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(氯仿/甲醇),得到标题化合物(160mg),为油状物。
参考例d-5
4-溴-1-(甲磺酰基)-1h-吲唑
[化学式287]
在冰冷却下,向氢化钠(cas:7646-69-7)(纯度55%,734mg)和dmf(100ml)的悬浮液中一点一点地加入4-溴吲唑(3.00g),混合物在室温下搅拌15分钟,在冰冷却下向其中滴加甲磺酰氯(2.27g)。将混合物在室温下搅拌3小时,并在冰冷却下用1n盐酸将反应溶液弱酸化,并用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(2.93g),为固体。
参考例d-6
4-溴-6-氟-1-(甲磺酰基)-1h-吲唑
[化学式288]
使用4-溴-6-氟-1h-吲唑(cas:885520-35-4),以与参考例d-5中相同的方式获得标题化合物。
参考例d-7
n-(3-溴苯基)-n-甲基甲磺酰胺
[化学式289]
使用3-溴-n-甲基苯胺,以与参考例d-3中相同的方式获得标题化合物。
参考例d-8
1-溴-2,4-二氟-3-甲氧基苯
[化学式290]
使用3-溴-2,6-二氟苯酚,以与参考例d-4中相同的方式获得标题化合物。
参考例d-9
(5-溴-3-甲氧基吡啶-2-基)甲醇
[化学式291]
在冰冷却下将5-溴-3-甲氧基吡啶-2-甲醛(1.22g)、硼氢化钠(0.214g)和乙醇(20.0ml)的混合物搅拌1小时。向其中加入1n盐酸,并用乙酸乙酯萃取混合物。合并有机层,用饱和盐水洗涤,经无水硫酸镁干燥,并在减压下浓缩。将获得的残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(0.819g),为固体。
参考例d-10
5-溴-3-甲氧基-n-甲基吡啶-2-胺
[化学式292]
在0℃下向5-溴-3-甲氧基-吡啶-2-胺(1.00g)和thf(25.0ml)的混合物中加入氢化钠(纯度55%,0.431g),并将混合物搅拌10分钟。向其中加入碘甲烷(0.370ml),并将混合物在相同温度下搅拌1小时。将冰加入到反应溶液中,并将反应混合物用乙醚萃取。依次用水和饱和盐水洗涤有机层,经无水硫酸钠干燥,过滤,并在减压下浓缩。将获得的残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(0.960g),为固体。
参考例d-11
4-[6-(二甲基氨基)-5-甲氧基吡啶-3-基]苯甲醛
步骤15-溴-3-甲氧基-n,n-二甲基吡啶-2-胺
[化学式293]
在冰冷却下向5-溴-3-甲氧基吡啶-2-胺(3.00g)和thf(75ml)的混合物中加入氢化钠(纯度55%,2.58g)。10分钟后,向其中加入碘甲烷(2.77ml),并将混合物在相同温度下搅拌1小时,然后在室温下搅拌16小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(3.41g),为油状物。
步骤24-[6-(二甲基氨基)-5-甲氧基吡啶-3-基]苯甲醛
[化学式294]
将5-溴-3-甲氧基-n,n-二甲基吡啶-2-胺(3.41g)、4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)苯甲醛(3.49g)、四(三苯基膦)钯(0)(1.71g)、碳酸钠(6.26g)、水(62ml)和1,4-二噁烷(184ml)的混合物在氮气气氛下在100℃搅拌3小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化。将获得的残余物悬浮在正己烷/乙酸乙酯中,并通过过滤收集固体,得到标题化合物(2.37g),为固体。
参考例d-12
5-溴-3-甲氧基-n,n-双[(2h3)甲基]吡啶-2-胺
[化学式295]
以与参考例d-11的步骤1中相同的方式获得标题化合物,除了使用碘甲烷-d3(cas:865-50-9)代替碘甲烷。
参考例d-13
6-氯-4-甲氧基-n,n-二甲基哒嗪-3-胺
[化学式296]
将根据文献(j.org.chem.2014,79,10311-10322)中描述的方法合成的3,6-二氯-4-甲氧基哒嗪(cas:70952-62-4)(0.506g)、thf(2.1ml)和二甲胺(cas:124-40-3)(浓度2.0mol/l,thf溶液,2.1ml)的混合物在室温下搅拌21小时。然后,向其中添加另外的二甲胺(cas:124-40-3)(浓度2.0mol/l,thf溶液,6.4ml),并将混合物在室温下搅拌21小时。然后,向其中添加另外的二甲胺(cas:124-40-3)(浓度2.0mol/l,thf溶液,8.4ml),并将混合物在室温下搅拌24小时。将反应混合物减压浓缩,并将残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(0.110g),为固体。
参考例d-14
6-氯-4-甲氧基-3-(2-甲氧基乙氧基)哒嗪
[化学式297]
在冰冷却下,向氢化钠(纯度55%,203mg)和甲苯(40ml)的悬浮液中一点一点地加入2-甲氧基乙醇(0.330ml),将混合物在室温下搅拌15分钟,并且在冰冷却下一点一点地加入3,6-二氯-4-甲氧基-哒嗪(750mg)。将混合物在室温搅拌4小时,在冰冷却下用1n盐酸将反应溶液弱酸化,并用乙酸乙酯萃取。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(295mg),为固体。
参考例d-15
6-氯-3-{[1-(甲磺酰基)哌啶-4-基]氧基}-4-甲氧基哒嗪
[化学式298]
在冰冷却下,向氢化钠(纯度55%,156mg)在甲苯(10ml)中的溶液中加入1-(甲基磺酰基)哌啶-4-醇(561mg),并将混合物搅拌15分钟。在相同温度下向其中加入3,6-二氯-4-甲氧基-哒嗪(400mg),并将混合物在80℃搅拌2.5小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(288mg),为固体。
参考例d-16
4-{5-甲氧基-6-[(吡啶-3-基)氧基]哒嗪-3-基}苯甲醛
步骤16-氯-4-甲氧基-3-[(吡啶-3-基)氧基]哒嗪,3-氯-4-甲氧基-6-[(吡啶-3-基)氧基]哒嗪
[化学式299]
将3,6-二氯-4-甲氧基哒嗪(300mg)、3-羟基吡啶(159mg)、dmf(11.2ml)和碳酸钾(463mg)的混合物在120℃搅拌2小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题两种异构体的混合物(88.6mg),为固体。其直接用于下一步。
步骤24-{5-甲氧基-6-[(吡啶-3-基)氧基]哒嗪-3-基}苯甲醛
[化学式300]
使用上述步骤1中获得的化合物,以与参考例d-11的步骤2中相同的方式获得标题化合物。
参考例d-17
6-氯-4-甲氧基-3-[(氧杂环己烷-3-基)氧基]哒嗪(外消旋物)
[化学式301]
以与参考例d-15中相同的方式获得标题化合物,除了使用3-羟基四氢吡喃代替1-(甲基磺酰基)哌啶-4-醇。
参考例d-18
3-(氮杂环丁烷-1-基)-6-氯-4-甲氧基哒嗪
[化学式302]
将3,6-二氯-4-甲氧基-哒嗪(4.00g)、氮杂环丁烷(4.98g)、thf(40ml)和dipea(2.43ml)的混合物在0℃下搅拌1小时,然后在室温下搅拌16小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。合并有机层,用饱和盐水洗涤,经无水硫酸镁干燥,并在减压下浓缩。将获得的残余物进行硅胶柱色谱法,得到标题化合物(0.498g),为固体。
参考例d-19
6-氯-4-甲氧基哒嗪-3-醇
[化学式303]
将3,6-二氯-4-甲氧基哒嗪(2.00g)和乙酸(40ml)的混合物在110℃下搅拌3.5小时。减压浓缩反应溶液,将残余物悬浮在二氯甲烷中,并通过过滤除去不溶物。浓缩滤液,并将残余物进行硅胶柱色谱法(二氯甲烷/甲醇),得到标题化合物(0.572g),为固体。
参考例d-20
6-氯-5-甲氧基哒嗪-3-醇
[化学式304]
向3,6-二氯-4-甲氧基-哒嗪(2.00g)和乙酸钾(1.21g)的混合物中加入乙酸(29ml)/水(5.8ml),并将混合物在微波反应器中在140℃加热搅拌1小时。反应完成后,将水加入反应溶液中,并通过过滤收集不溶的固体,得到标题化合物(739mg),为固体。
参考例d-21
6-氯-3-(二氟甲氧基)-4-甲氧基哒嗪
[化学式305]
在0℃下向参考例d-19中获得的化合物(0.570g)、50%氢氧化钾水溶液(3.5ml)、水(3.5ml)和乙腈(7.0ml)的混合物中加入三氟甲磺酸二氟甲酯(cas:1885-46-7)(1.35ml),并将混合物在室温搅拌4小时。在0℃下向其中加入三氟甲磺酸二氟甲酯(1.35ml),并将混合物在室温下搅拌1小时,然后在60℃下搅拌1小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(0.130g),为固体。
参考例d-22
3-氯-6-(二氟甲氧基)-4-甲氧基哒嗪(异构体a)
6-氯-2-(二氟甲基)-5-甲氧基哒嗪-3(2h)-酮(异构体b)
[化学式306]
向参考例d-20中获得的化合物(200mg)和乙腈(2.5ml)的混合物中加入50%氢氧化钾水溶液(1.25ml)和水(1.25ml)。在冰冷却下向其中加入三氟甲磺酸二氟甲酯(0.475ml),并将混合物在室温搅拌1小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(异构体a,较早洗脱的组分,139mg),和标题化合物(异构体b,较晚洗脱的组分,46.5mg),分别为固体。
异构体a和异构体b的结构式为估计结构,并且异构体a的结构式和异构体b的结构式可以互换。后续步骤(参考例d-59和参考例d-60,以及例110和111)同样如此。
参考例d-23
6-氯-3,4-二甲氧基哒嗪
[化学式307]
将3,4,6-三氯哒嗪(cas:6082-66-2)(2.57g)溶解在甲醇(50ml)中,并向其中加入甲醇钠(cas:124-41-4)。将混合物在0℃搅拌10分钟,使其温热至室温,并搅拌20小时。将反应溶液在减压下浓缩,向其中加入乙酸乙酯和饱和氯化铵水溶液,并将混合物进行分液操作。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,并在减压下浓缩。将残余物进行硅胶柱色谱法(二氯甲烷/乙酸乙酯),并将获得的固体悬浮在正己烷/乙酸乙酯中。过滤收集不溶物,干燥得到标题化合物(1.10g),为固体。
参考例d-24
6-氯-3,4-双[(2h3)甲基氧基]哒嗪
[化学式308]
以与参考例d-23中相同的方式获得标题化合物,除了使用甲醇-d3代替甲醇。
参考例d-25
5-(4-甲酰基苯基)-3-甲氧基吡啶-2-甲腈
步骤13-氟-5-(4-甲酰基苯基)吡啶-2-甲腈
[化学式309]
将5-溴-3-氟吡啶-2-甲腈(cas:886373-28-0)(2.00g)、(4-甲酰基苯基)硼酸(1.49g)、四(三苯基膦)钯(0)(1.15g)、碳酸钠(3.16g)、水(8.0ml)和1,4-二噁烷(24ml)的混合物在100℃搅拌4小时。反应溶液用乙酸乙酯稀释,依次用水和饱和盐水洗涤,经无水硫酸镁干燥,并减压浓缩。将获得的残余物进行硅胶柱色谱法(正己烷/乙酸乙酯)。浓缩级分,并将获得的固体用正己烷进行浆化洗涤,得到标题化合物(1.28g),为固体。
步骤25-(4-甲酰基苯基)-3-甲氧基吡啶-2-甲腈
[化学式310]
将上述步骤1中获得的化合物(0.250g)、碳酸钾(0.458g)和甲醇(10.0ml)的混合物在70℃下搅拌10分钟。反应溶液用乙酸乙酯稀释,用水和饱和盐水洗涤,经无水硫酸镁干燥,并减压浓缩。将获得的残余物进行硅胶柱色谱法(二氯甲烷/乙酸乙酯),得到标题化合物(0.163g),为固体。
参考例d-26
4-(5,6-二甲氧基哒嗪-3-基)苯甲醛
[化学式311]
将参考例d-23中获得的化合物(1.10g)、4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)苯甲醛(cas:128376-64-7)(2.17g)、四(三苯基膦)钯(0)(cas:14221-01-3)(0.720g)、碳酸钠(1.70g)、1,2-二甲氧基乙烷(30ml)和水(10ml)的混合物在100℃搅拌5小时。使反应溶液冷却至室温,向其中加入饱和盐水,并将混合物用乙酸乙酯萃取。有机层经无水硫酸钠干燥,过滤,并减压浓缩。将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯,然后是乙酸乙酯/甲醇),并将获得的固体悬浮在正己烷/乙酸乙酯中。过滤收集所得固体,干燥,得到标题化合物(1.21g),为固体。
参考例d-27
4-(4-甲酰基苯基)-1h-吡唑-1-甲酸叔丁酯
[化学式312]
将1-boc-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)吡唑(1.43g)、4-溴苯甲醛(150mg)、碳酸铯(3.17g)、[1,1'-双(二苯基膦基)二茂铁]二氯化钯(ii)二氯甲烷加合物(397mg)、1,4-二噁烷(25.6ml)和水(2.56ml)的混合物在70℃下搅拌4小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(842mg),为固体。
下表1-1至表1-12中所述的产物以与参考例d-26或d-27中相同的方式由表中所述的原料1和原料2制备。
参考例d-75
4-[1-(甲磺酰基)-1h-吡唑-4-基]苯甲醛
步骤14-(1h-吡唑-4-基)苯甲醛盐酸盐
[化学式313]
使用参考例d-27中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
步骤24-[1-(甲磺酰基)-1h-吡唑-4-基]苯甲醛
[化学式314]
使用上述步骤1中获得的化合物,以与参考例d-3中相同的方式获得标题化合物。
参考例d-76
4-(5-甲氧基-6-苯氧基哒嗪-3-基)苯甲醛
[化学式315]
使用参考例d-28中获得的化合物和苯酚,以与参考例d-16的步骤1中相同的方式获得标题化合物。
参考例d-77
4'-甲酰基n,n-二甲基[1,1'-联苯]-3-磺酰胺
[化学式316]
使用参考例d-67中获得的化合物,以与参考例a-1的步骤13中相同的方式获得标题化合物。
参考例d-78
4-(5,6-二甲氧基哒嗪-3-基)环己烷-1-甲醛(顺式-反式混合物)
步骤14-(5,6-二甲氧基哒嗪-3-基)环己-3-烯-1-甲醛(外消旋物)
[化学式317]
将参考例d-35中获得的化合物(0.400g)溶解于二氯甲烷(11ml)中,并在-78℃下向其中滴加氢化二异丁基铝(1.22mol/l,二氯甲烷溶液,2.24ml)。将反应溶液在相同温度下搅拌2小时。向反应溶液中加入甲醇(0.400ml),然后向其中加入饱和酒石酸钾钠水溶液,并将混合物温热至室温。用乙酸乙酯萃取混合物,有机层经无水硫酸钠干燥,并在减压下蒸发溶剂。残余物通过硅胶色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(0.178g),为固体。
步骤24-(5,6-二甲氧基哒嗪-3-基)环己烷-1-甲醛(顺式-反式混合物)
[化学式318]
将上述步骤1中得到的化合物(0.175g)溶解于乙酸乙酯(7ml)中,向其中加入10%湿钯/碳(0.300g),并将混合物在室温和氢气气氛下搅拌5小时。将反应溶液过滤,并将滤液减压浓缩,得到标题化合物(0.179g),为油状物。
参考例d-79
n-[6-(4-甲酰基苯基)-4-甲氧基哒嗪-3-基]-n-甲基丙-2-烯酰胺
步骤14-[5-甲氧基-6-(甲基氨基)哒嗪-3-基]苯甲醛
[化学式319]
将参考例d-63中得到的化合物(100mg)、碘甲烷(0.032ml)、碳酸钾(121mg)和dmf(2.2ml)的混合物在70℃搅拌1.5小时。向反应溶液中加入水,并通过过滤收集沉淀的固体,得到标题化合物(62.3mg),为固体。
步骤2n-[6-(4-甲酰基苯基)-4-甲氧基哒嗪-3-基]-n-甲基丙-2-烯酰胺
[化学式320]
在冰冷却下向上述步骤1中获得的化合物(62.3mg)、dipea(0.134ml)和二氯甲烷(2.56ml)的混合物中加入丙烯酰氯(0.0228ml),并将混合物在室温下搅拌1小时。向反应溶液中加入水,并将混合物用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(43.4mg),为固体。
参考例d-80
4-[(吡啶-3-基)氨基]苯甲醛
[化学式321]
将4-溴苯甲醛(150mg)、吡啶-3-胺(91.6mg)、三(二亚苄基丙酮)二钯(0)(37.1mg)、4,5-双(二苯基膦基)-9,9-二甲基呫吨(51.6mg)、碳酸铯(370mg)和1,4-二噁烷(1.62ml)的混合物在氮气气氛下在100℃搅拌过夜。冷却反应溶液,通过硅藻土过滤,并将滤液在减压下浓缩。残余物通过硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(146mg),为固体。
参考例d-81
3-[(吡啶-3-基)氨基]苯甲醛
[化学式322]
以与参考例d-80中相同的方式获得标题化合物,除了使用3-溴苯甲醛代替4-溴苯甲醛。
参考例d-82
4-(2,3-二氢-1h-吲哚-1-基)苯甲醛
步骤11-[4-(1,3-二氧杂环戊烯-2-基)苯基]-2,3-二氢-1h-吲哚
[化学式323]
将二氢吲哚(0.280ml)、2-(4-溴苯基)-1,3-二氧杂环戊烷(cas:10602-01-4)(1.15g)、tea(1.15ml)、(±)-2,2'-双(二苯基膦基)-1,1'-联萘(54.9mg)、三(二亚苄基丙酮)二钯(0)(69.2mg)、碳酸铯(2.46g)和甲苯(20ml)的混合物在氮气气氛下在110℃搅拌5小时。向反应溶液中加入饱和碳酸氢钠水溶液,并用乙酸乙酯萃取混合物。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(342mg),为固体。
步骤24-(2,3-二氢-1h-吲哚-1-基)苯甲醛
[化学式324]
在室温下向上述步骤1中获得的化合物(155mg)在thf(2.3ml)中的溶液中加入1n盐酸(2.32ml),并将混合物在相同温度下搅拌16小时。向反应溶液中加入饱和碳酸氢钠水溶液,并用氯仿萃取混合物。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(115mg),为固体。
参考例d-83
n-[3-(4-甲酰基苯胺基)苯基]丙-2-烯酰胺
步骤1n-[4-(1,3-二氧杂环戊烷-2-基)苯基]-3-硝基苯胺
[化学式325]
将3-硝基苯胺(cas:99-09-2)(4.13g)、2-(4-溴苯基)-1,3-二氧杂环戊烷(5.71g)、叔丁醇钠(cas:865-48-5)(4.31g)、三(二亚苄基丙酮)二钯(0)(1.14g)、三叔丁基膦(10%正己烷溶液)(cas:13716-12-6)(7.42ml)和甲苯(50ml)的混合物在室温下搅拌1小时,然后在80℃下搅拌1小时。将乙酸乙酯和水加入到反应溶液中,并将混合物通过硅藻土过滤。将滤液进行液体分离,并将水层用乙酸乙酯萃取。合并有机层,用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。所得残余物通过氨基硅胶柱色谱法(正己烷/乙酸乙酯)纯化。减压蒸发溶剂,将正己烷/乙醚=1/1的混合溶剂加入到残余物中,并通过过滤收集固体,得到标题化合物(4.08g),为固体。
步骤2n1-[4-(1,3-二氧杂环戊烷-2-基)苯基]苯-1,3-二胺
[化学式326]
向上述步骤1中获得的化合物(1.50g)和10%湿钯/碳(0.300g)中加入乙醇(20ml),并将混合物在氢气气氛下于室温搅拌2小时。过滤除去钯,将滤液减压浓缩,得到标题化合物(1.29g),为固体。其无需进一步纯化即用于下一步反应。
步骤3n-{3-[4-(1,3-二氧杂环戊烷-2-基)苯胺基]苯基}丙-2-烯酰胺
[化学式327]
将上述步骤2中获得的化合物(590mg)溶解于二氯甲烷(15ml)中,向其中添加dipea(1.2ml)和丙烯酰氯(cas:814-68-6)(0.205ml),并将混合物在室温下搅拌1小时。向其中加入二氯甲烷和水,并将混合物进行液体分离。有机层经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(709mg),为固体。
步骤4n-[3-(4-甲酰基苯胺基)苯基]丙-2-烯酰胺
[化学式328]
使用上述步骤3中获得的化合物,以与参考例d-82的步骤2中相同的方式获得标题化合物。
参考例d-84
4-甲酰基-n-[(1-甲基-1h-吡唑-4-基)甲基]苯甲酰胺
[化学式329]
将4-甲酰基苯甲酸(cas:619-66-9)(150mg)、(1-甲基-1h-吡唑-4-基)甲胺(122mg)、hobt(162mg)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(wscd)(230mg)和dmf(8.0ml)的混合物在室温下搅拌15小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(氯仿/甲醇),得到标题化合物(200mg),为固体。
参考例d-85
n-{3-[(乙磺酰基)氨基]丙基}-4-甲酰基苯甲酰胺
步骤1[3-(4-甲酰基苯甲酰胺基)丙基]氨基甲酸叔丁酯
[化学式330]
以与参考例d-84中相同的方式获得标题化合物,除了使用n-(3-氨基丙基)氨基甲酸叔丁酯(cas:75178-96-0)代替(1-甲基-1h-吡唑-4-基)甲胺。
步骤2n-{3-[(乙磺酰基)氨基]丙基}-4-甲酰基苯甲酰胺
[化学式331]
将上述步骤1中获得的化合物(449mg)溶解在二氯甲烷(3ml)中,并向其中加入氯化氢(4mol/l,1,4-二噁烷溶液,3ml)。将混合物在室温搅拌2小时,并在减压下蒸发溶剂。向残余物中加入二氯甲烷(11ml)、dipea(1.92ml)和2-氯乙磺酰氯(cas:1622-32-8)(0.174ml),并将混合物在室温搅拌3小时。减压蒸发溶剂。残余物通过硅胶柱色谱法(乙酸乙酯)纯化,得到标题化合物(110mg),为固体。
参考例d-86
4-[(2-氧代吡咯烷-1-基)甲基]苯甲醛
步骤14-[(2-氧代吡咯烷-1-基)甲基]苯甲酸甲酯
[化学式332]
在冰冷却下,向氢化钠(纯度55%,238mg)和dmf(15ml)的悬浮液中加入2-吡咯烷酮(cas:616-45-5)(557mg),并在30分钟后,在相同温度下向其中滴加4-(溴甲基)苯甲酸甲酯(cas:2417-72-3)(1.00g)在dmf(15ml)中的溶液。将混合物在室温搅拌16小时,将水加入到反应溶液中,并将混合物用乙酸乙酯萃取。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(576mg),为固体。
步骤21-{[4-(羟基甲基)苯基]甲基}吡咯烷-2-酮
[化学式333]
在冰冷却下,向上述步骤1中获得的化合物(576mg)在thf(20ml)中的溶液中一点一点地添加硼氢化锂(cas:16949-15-8)(108mg),并在相同温度下向其中加入甲醇(0.2ml)。将混合物在室温搅拌18小时,然后在50℃搅拌4小时。在冰冷却下,再次向反应溶液中一点一点地添加硼氢化锂(108mg),并将混合物在60℃下搅拌9小时。通过在冰冷却下一点一点地添加,将反应溶液用1n盐酸弱酸化,并用氯仿萃取。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(氯仿/甲醇),得到标题化合物(338mg),为固体。
步骤34-[(2-氧代吡咯烷-1-基)甲基]苯甲醛
[化学式334]
在冰冷却下,向上述步骤2中获得的化合物(336mg)在二氯甲烷(15ml)中的溶液中添加dess-martin氧化剂(cas:87413-09-0)(764mg)。将混合物在室温搅拌1.5小时,将水添加至反应溶液,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(氯仿/甲醇),得到标题化合物(343mg),为油状物。
参考例d-87
4-(5-氧代吡咯烷-2-基)苯甲醛(外消旋物)
步骤14-(5-氧代吡咯烷-2-基)苯甲酸甲酯(外消旋物)
[化学式335]
将5-(4-溴苯基)吡咯烷-2-酮(cas:207989-90-0)(1.00g)、tea(1.15ml)、甲醇(15ml)、[1,1'-双(二苯基膦基)二茂铁]二氯化钯(ii)二氯甲烷加合物(170mg)和dmf(30ml)的混合物在一氧化碳气氛下于90℃搅拌2小时。将饱和氯化铵水溶液加入到反应溶液中,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(氯仿/甲醇),得到标题化合物(342mg),为固体。
步骤25-[4-(羟基甲基)苯基]吡咯烷-2-酮(外消旋物)
[化学式336]
使用上述步骤1中获得的化合物,以与参考例d-86的步骤2中相同的方式获得标题化合物。
步骤34-(5-氧代吡咯烷-2-基)苯甲醛(外消旋物)
[化学式337]
将上述步骤2中获得的化合物(110mg)溶解在氯仿(5ml)中,向其中加入二氧化锰(498mg),并将混合物在室温搅拌1.5小时。向其中加入乙酸乙酯和水,通过过滤除去不溶物,并对滤液进行分液。用乙酸乙酯萃取水层,并将有机层合并,用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(氯仿/甲醇)纯化,得到标题化合物(56mg),为油状物。
参考例d-88
4-(环己基氨基)苯甲醛
步骤14-(环己基氨基)苯甲酸甲酯
[化学式338]
将4-氨基苯甲酸甲酯(cas:619-45-4)(1.13g)、硫酸镁(1.81g)、环己酮(3.01g)、乙酸(7.58ml)、三乙酰氧基硼氢化钠(14.3g)、甲醇(30ml)和二氯甲烷(10ml)的混合物在室温下搅拌36小时,然后在60℃下搅拌9小时。向反应混合物中加入饱和碳酸氢钠水溶液,并用氯仿萃取混合物。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,并在减压下浓缩。将获得的残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(1.16g),为固体。
步骤2[4-(环己基氨基)苯基]甲醇
[化学式339]
使用上述步骤1中获得的化合物,以与参考例d-86的步骤2中相同的方式获得标题化合物。
步骤34-(环己基氨基)苯甲醛
[化学式340]
使用上述步骤2中获得的化合物,以与参考例d-87的步骤3中相同的方式获得标题化合物。
参考例d-89
(4-甲酰基苯基)氨基甲酸叔丁酯苄酯
步骤14-[苄基(叔丁氧基羰基)氨基]苯甲酸甲酯
[化学式341]
在冰冷却下向氢化钠(纯度55%,131mg)和thf(10ml)的悬浮液中加入根据文献(j.med.chem.2016,59(18),8233-8262)中所述方法合成的4-[(叔丁氧基羰基)氨基]苯甲酸甲酯(503mg)在thf(5ml)中的溶液。将混合物在室温搅拌30分钟,并在冰冷却下向其中滴加苄基溴(cas:100-39-0)(0.310ml)。将混合物在室温搅拌15小时,将水添加至反应溶液中,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(正己烷/乙酸乙酯),得到标题化合物(408mg),为固体。
步骤2苄基[4-(羟基甲基)苯基]氨基甲酸叔丁酯
[化学式342]
在冰冷却下,向上述步骤1中获得的化合物(405mg)在thf(10ml)中的溶液中一点一点地添加硼氢化锂(cas:16949-15-8)(78.9mg)。将混合物在室温搅拌1小时,并将混合物在50℃搅拌4小时。通过在冰冷却下一点一点地添加,将反应溶液用1n盐酸弱酸化,并用氯仿萃取。有机层用饱和盐水洗涤,并经无水硫酸钠干燥,得到标题化合物的粗产物(369mg),为油状物。
步骤3苄基(4-甲酰基苯基)氨基甲酸叔丁酯
[化学式343]
使用上述步骤2中获得的化合物,以与参考例d-87的步骤3中相同的方式获得标题化合物。
参考例d-90
1-(5,6-二甲氧基哒嗪-3-基)哌啶-4-甲醛
步骤1[1-(5,6-二甲氧基哒嗪-3-基)哌啶-4-基]甲醇
[化学式344]
将6-氯-3,4-二甲氧基哒嗪(364mg)、4-哌啶甲醇(cas:586-95-8,200mg)、(+/-)-2,2'-双(二苯基膦基)-1,1'-联萘(216mg)、叔丁醇钠(200mg)、甲苯(4ml)和三(二亚苄基丙酮)二钯(0)(159mg)的混合物在氮气气氛下于100℃搅拌4小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(208mg),为固体。
步骤21-(5,6-二甲氧基哒嗪-3-基)哌啶-4-甲醛
[化学式345]
使用上述步骤1中获得的化合物,以与参考例d-86的步骤3中相同的方式获得标题化合物。
参考例d-91
4-(1h-1,2,4-三唑-5-基)苯甲醛
步骤1[4-(1h-1,2,4-三唑-5-基)苯甲醛
[化学式346]
使用4-(1h-1,2,4-三唑-5-基)苯甲酸(cas:876715-40-1),以与参考例d-86的步骤2中相同的方式获得标题化合物。所获得的化合物无需纯化即直接用于下一步骤。
步骤24-(1h-1,2,4-三唑-5-基)苯甲醛
[化学式347]
将上述步骤1中获得的化合物(155mg)、氯铬酸吡啶鎓(381mg)、dmso(3ml)和二氯甲烷(8ml)的混合物在室温下搅拌过夜。向反应溶液中加入饱和碳酸氢钠水溶液,并用乙酸乙酯萃取混合物。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(乙酸乙酯)纯化,得到标题化合物(118mg),为固体。
参考例d-92
4-(3-羟基哌啶-1-磺酰基)苯甲醛(外消旋物)
[化学式348]
向3-羟基哌啶(51.4mg)在二氯甲烷(1ml)中的溶液中加入4-甲酰基苯-1-磺酰氯(80mg)在二氯甲烷(1ml)中的溶液,并将混合物在室温搅拌1小时。向反应溶液中加入水,并将混合物用二氯甲烷萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(102mg),为固体。
参考例d-93
4-(氮杂环庚烷-1-磺酰基)苯甲醛
[化学式349]
以与参考例d-92中相同的方式获得标题化合物,除了使用氮杂环庚烷代替3-羟基哌啶。
参考例d-94
6-(4-甲酰基苯基)-4-甲氧基哒嗪-3-甲腈
[化学式350]
将参考例d-28中获得的化合物(0.200g)、氰化锌(0.0965g)、四(三苯基膦)钯(0)(0.0930g)和n,n-二甲基乙酰胺(3.00ml)的混合物在氮气气氛下在100℃搅拌1小时。反应溶液用乙酸乙酯稀释,依次用水和饱和盐水洗涤,经无水硫酸镁干燥,并减压浓缩。将获得的残余物进行硅胶柱色谱法(二氯甲烷/乙酸乙酯)。将级分减压浓缩,并将获得的固体用二氯甲烷进行浆化洗涤,并干燥得到标题化合物(0.0826g),为固体。
参考例d-95
{4-[(5-甲氧基吡啶-3-基)氨基]苯基}甲醇
步骤1n-[4-({[叔丁基(二甲基)甲硅烷基]氧基}甲基)苯基]-5-甲氧基吡啶-3-胺
[化学式351]
向根据文献(tetrahedron2015,71(49),9240-9244)中描述的方法合成的4-({[叔丁基(二甲基)甲硅烷基]氧基}甲基)苯胺(0.404g)、3-溴-5-甲氧基吡啶(cas:50720-12-2)(0.320g)、三(二亚苄基丙酮)二钯(0)(0.156g)、4,5-双(二苯基膦基)-9,9-二甲基呫吨(cas:161265-03-8)(0.197g)和碳酸铯(1.67g)中加入1,4-二噁烷(2.43ml),并将混合物在氮气气氛下于70℃搅拌2小时。将反应混合物在减压下浓缩,并将残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(0.252g),为固体。
步骤2{4-[(5-甲氧基吡啶-3-基)氨基]苯基}甲醇
[化学式352]
向上述步骤1中获得的化合物(0.252g)在thf(7.31ml)中的溶液中加入1n四丁基氟化铵thf溶液(1.46ml),并将混合物在室温下搅拌过夜。将反应混合物在减压下浓缩,将盐水加入到残余物中,并将混合物用二氯甲烷萃取三次。有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(二氯甲烷/乙酸乙酯)纯化,得到标题化合物(0.117g),为固体。
参考例d-96
4-(5-甲氧基-6-苯氧基哒嗪-3-基)苯甲醛
[化学式353]
将参考例d-28中获得的化合物(100mg)、苯酚(37.8mg)、dmf(2.68ml)和碳酸钾(111mg)的混合物在100℃下搅拌15小时。使反应溶液冷却至室温。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(正己烷/乙酸乙酯)纯化,得到标题化合物(63.9mg),为固体。
参考例e-1
5-({[(1s,3r)-3-(甲基氨基)环戊基]氨基}甲基)-1h-吲哚-2-甲腈
步骤12-氰基-5-[({(1s,3r)-3-[甲基(2-硝基苯-1-磺酰基)氨基]环戊基}氨基)甲基]-1h-吲哚-1-甲酸叔丁酯
[化学式354]
向参考例a-3的步骤4中获得的化合物(1.11g)、根据文献(cancercell2015,27,589-602)中描述的方法制备的5-(溴甲基)-2-氰基-吲哚-1-甲酸叔丁酯(1.24g)和碳酸钾(1.02g)中加入dmf(12ml),并将混合物在室温下搅拌2小时,并放置过夜。将乙酸乙酯和饱和盐水加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将获得的残余物通过硅胶柱色谱法(正己烷/乙酸乙酯,二氯甲烷/甲醇)纯化,得到标题化合物(916mg),为固体。
步骤25-({[(1s,3r)-3-(甲基氨基)环戊基]氨基}甲基)-1h-吲哚-2-甲腈
[化学式355]
将上述步骤1中获得的化合物(916mg)溶于thf(10ml)和甲醇(10ml)的混合溶剂中,向其中加入4-异丙基苯硫醇(0.504ml)和碳酸铯(1.08g),将混合物在室温下搅拌1小时,并放置过夜。将乙酸乙酯和饱和盐水加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,水层用乙酸乙酯萃取五次。合并有机层,经无水硫酸钠干燥,并在减压下浓缩。所得残余物通过氨基硅胶柱色谱法(正己烷/乙酸乙酯,然后二氯甲烷/甲醇)纯化,得到标题化合物(0.281g)。
参考例e-2
(1r,3s,4s)-n1-{[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}-4-甲氧基-n3-甲基环戊烷-1,3-二胺盐酸盐
步骤1[(1s,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-甲氧基环戊基]甲基氨基甲酸叔丁酯
[化学式356]
向参考例a-17的步骤3中获得的化合物(0.460g)和参考例d-56中获得的化合物(0.525g)在二氯甲烷(16ml)中的悬浮液中依次加入三乙酰氧基硼氢化钠(1.35g)和乙酸(0.320ml),并将混合物在室温搅拌17小时。将水(25ml)加入到反应混合物中,并将混合物用二氯甲烷/甲醇(9/1)的混合溶剂萃取两次(80ml,50ml)。合并有机层,并经无水硫酸钠干燥。减压蒸发溶剂,并将所得残余物通过硅胶柱色谱法(二氯甲烷/甲醇)纯化,得到标题化合物(0.733g),为固体。
步骤2(1r,3s,4s)-n1-{[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}-4-甲氧基-n3-甲基环戊烷-1,3-二胺盐酸盐
[化学式357]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物。
参考例e-3
(1s,3s,4s)-n1-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-4-甲氧基环己烷-1,3-二胺
步骤1[(1s,2s,5s)-5-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-甲氧基环己基]氨基甲酸叔丁酯
[化学式358]
在室温下向参考例a-10的步骤6中获得的化合物(0.143g)、参考例d-26中获得的化合物(0.175g)和氯仿(15ml)的混合物中加入原钛酸四异丙酯(0.520ml),并将混合物搅拌1.5小时。向其中加入三乙酰氧基硼氢化钠(0.692g),并将混合物搅拌16小时。向其中加入饱和碳酸氢钠水溶液(5.00ml)和罗谢尔盐水溶液(5.00ml),将混合物搅拌2小时,并将反应混合物用二氯甲烷萃取。有机层经无水硫酸钠干燥,在减压下蒸发溶剂,并将获得的残余物用氨基硅胶柱作为装料柱进行硅胶柱色谱法(乙酸乙酯/甲醇),得到标题化合物(0.179g),为固体。
步骤2(1s,3s,4s)-n1-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-4-甲氧基环己烷-1,3-二胺
[化学式359]
使用上述步骤1,以与a-3的步骤4中相同的方式获得标题化合物。
参考例e-4
[(1s,2r,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-甲氧基环戊基]甲基氨基甲酸叔丁酯
[化学式360]
使用参考例a-16的步骤8中获得的化合物和参考例d-26中获得的化合物,以与参考例e-2的步骤1中相同的方式获得标题化合物。
实施例1:
5-({[(1r,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-1h-吲哚-2-甲腈盐酸盐
步骤12-氰基-5-({[(1r,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-1h-吲哚-1-甲酸叔丁酯
[化学式361]
将参考例c-3的步骤2中获得的化合物(160mg)和根据文献(cancercell2015,27,589-602)中所述的方法制备的5-(溴甲基)-2-氰基-吲哚-1-甲酸叔丁酯(117mg)溶解于dmf(2ml)中,向其中加入碳酸钾(121mg),并将混合物在室温下搅拌过夜。将乙酸乙酯和饱和盐水加入到反应溶液中,并将混合物进行液体分离。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(二氯甲烷/甲醇)纯化,得到标题化合物(133mg),为固体。
步骤25-({[(1r,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-1h-吲哚-2-甲腈
[化学式362]
将上述步骤1中获得的化合物(133mg)溶解于乙腈(5ml)中,并向其中加入氯化锡(iv)(约1.0mol/l,二氯甲烷溶液)(2.3ml),混合物在室温下搅拌1小时。向反应溶液中加入乙酸乙酯和饱和碳酸氢钠水溶液,通过硅藻土过滤除去不溶物,并对滤液进行分液。用乙酸乙酯萃取水层,并将有机层合并,用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。将获得的残余物通过硅胶柱色谱法(二氯甲烷/甲醇)纯化,得到标题化合物(76mg),为固体。
步骤35-({[(1r,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-1h-吲哚-2-甲腈盐酸盐
[化学式363]
将上述步骤2中获得的化合物(76mg)溶解于乙醇(3ml)中,向其中加入1n盐酸(0.178ml),并在减压下蒸发溶剂。将乙醚加入到残余物中,并通过过滤收集固体,得到标题化合物(62mg),为固体。
实施例2:
4-甲基-5-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-1-[(1h-吡唑-4-基)甲基]-1h-吲哚-2-甲腈盐酸盐
步骤14-甲基-5-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-1-{[1-(三苯基甲基)-1h-吡唑-4-基]甲基}-1h-吲哚-2-甲腈
[化学式364]
将参考例d-2的步骤2中得到的化合物(0.362g)、参考例c-4的步骤2中得到的化合物(0.320g)、dipea(0.373ml)、乙酸(0.245ml)和二氯甲烷(15ml)的混合物在氩气气氛下于室温搅拌30分钟。将三乙酰氧基硼氢化钠(0.468g)加入到反应溶液中,并将混合物在室温搅拌过夜。向反应溶液中加入饱和碳酸氢钠水溶液,并用二氯甲烷萃取混合物。有机层经无水硫酸钠干燥,并在减压下浓缩。将残余物进行氨基硅胶柱色谱法(二氯甲烷/甲醇,正己烷/乙酸乙酯),得到标题化合物(0.238g),为油状物。
步骤24-甲基-5-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-1-[(1h-吡唑-4-基)甲基]-1h-吲哚-2-甲腈
[化学式365]
将上述步骤1中获得的化合物(0.228g)、氯化氢(4mol/l,1,4-二噁烷溶液,0.75ml)、甲醇(2.5ml)和二氯甲烷(15ml)的混合物在氩气气氛下于室温下搅拌30分钟。将反应溶液在减压下浓缩,并将残余物进行氨基硅胶柱色谱法(二氯甲烷/甲醇),得到标题化合物(0.139g),为固体。
步骤34-甲基-5-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-1-[(1h-吡唑-4-基)甲基]-1h-吲哚-2-甲腈盐酸盐
[化学式366]
将上述步骤2中得到的化合物(0.129g)、1n盐酸(0.210ml)和乙醇(4.0ml)的混合物在室温下搅拌30分钟。减压浓缩反应溶液,将残余物悬浮在乙醚中,并通过过滤收集,得到标题化合物(0.133g),为固体。
实施例3:
5-({[(1s,3r)-3-{甲基[2-(2,2,2-三氟乙基)[1,3]噻唑并[5,4-d]嘧啶-7-基]氨基}环戊基]氨基}甲基)-1h-吲哚-2-甲腈盐酸盐
步骤15-({[(1s,3r)-3-{甲基[2-(2,2,2-三氟乙基)[1,3]噻唑并[5,4-d]嘧啶-7-基]氨基}环戊基]氨基}甲基)-1h-吲哚-2-甲腈
[化学式367]
将参考例e-1的步骤2中获得的化合物(0.0790g)、根据文献(wo2016/195776)中所述的方法制备的7-氯-2-(2,2,2-三氟乙基)[1,3]噻唑并[5,4-d]嘧啶(75.0mg)、碳酸钾(0.0610g)和dmf(2ml)的混合物在氩气气氛下在室温搅拌1小时。将水添加到反应溶液中,并将混合物用乙酸乙酯/乙醚萃取。有机层经无水硫酸钠干燥,并在减压下浓缩。将残余物进行二醇改性的硅胶柱色谱法(二氯甲烷/甲醇),得到标题化合物(0.0184g),为固体。
步骤25-({[(1s,3r)-3-{甲基[2-(2,2,2-三氟乙基)[1,3]噻唑并[5,4-d]嘧啶-7-基]氨基}环戊基]氨基}甲基)-1h-吲哚-2-甲腈盐酸盐
[化学式368]
使用上述步骤1中获得的化合物,以与实施例2的步骤3中相同的方式获得标题化合物。
实施例4:
5-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[3,2-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-1h-吲哚-2-甲腈
[化学式369]
将参考例e-1的步骤2中获得的化合物(83mg)和参考例b-6的步骤4中获得的化合物(78mg)溶解于2-丙醇(5ml)中,向其中加入dipea(0.108ml),将混合物在70℃下搅拌5小时,并使其在室温下静置过夜。将反应混合物进行硅胶柱色谱法(二醇硅胶,二氯甲烷/甲醇),然后通过chiralflash(注册商标,daicelcorporation)ia(正己烷/ipa)纯化,得到标题化合物。
实施例5:
4-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)喹唑啉-4-基]氨基}环戊基]氨基}甲基)苯酚盐酸盐
步骤1(1r,3s)-n3-[(4-甲氧基苯基)甲基]-n1-甲基-n1-[6-(2,2,2-三氟乙基)喹唑啉-4-基]环戊烷-1,3-二胺
[化学式370]
使用参考例c-6的步骤2中得到的化合物和4-茴香醛(cas:1122-91-4),以与实施例2的步骤1中相同的方式获得标题化合物(含有杂质,纯度约80%)。
步骤24-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)喹唑啉-4-基]氨基}环戊基]氨基}甲基)苯酚
[化学式371]
将上述步骤1中获得的化合物(0.0920g,含有杂质,纯度约80%)溶于二氯甲烷(4.2ml)中,向其中加入三溴化硼(cas:10294-33-4)(1n二氯甲烷溶液,2.6ml),并将混合物在室温搅拌50分钟。将反应溶液冷却至0℃,向其中缓慢加入饱和碳酸氢钠水溶液。将反应混合物在0℃剧烈搅拌5分钟,用二氯甲烷/甲醇(9/1)萃取,有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(二氯甲烷/甲醇)纯化,得到标题化合物(0.0653g),为固体。
步骤34-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)喹唑啉-4-基]氨基}环戊基]氨基}甲基)苯酚盐酸盐
[化学式372]
使用上述步骤2中获得的化合物,以与实施例2的步骤3中相同的方式获得标题化合物。
实施例6:
4-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-n-{3-[(丙-2-烯酰基)氨基]丙基}苯甲酰胺盐酸盐
步骤1{3-[4-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯甲酰胺]丙基}氨基甲酸叔丁酯
[化学式373]
使用参考例c-4的步骤2中获得的化合物和参考例d-85的步骤1中获得的化合物,以与实施例2的步骤1中相同的方式获得标题化合物。
步骤2n-(3-氨基丙基)-4-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯甲酰胺盐酸盐
[化学式374]
使用上述步骤1中获得的化合物,以与参考例a-1的步骤11中相同的方式获得标题化合物(608mg)。
步骤34-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)-n-{3-[(丙-2-烯酰基)氨基]丙基}苯甲酰胺盐酸盐
[化学式375]
向上述步骤2中获得的化合物(250mg)中加入二氯甲烷(5ml)和dipea(0.435ml)。在冰冷却下向其中加入丙烯酰氯(0.022ml),并将混合物在0℃搅拌1小时。将水和二氯甲烷加入到反应溶液中,并将混合物进行液体分离。有机层经无水硫酸钠干燥,并在减压下浓缩。所获得的残余物通过硅胶柱色谱法(二氯甲烷/甲醇)纯化,得到游离形式的标题化合物(37mg),为固体。将其溶于乙醇(3ml)中,向其中加入1n盐酸(0.064ml),并在减压下蒸发溶剂。将乙醚加入到残余物中,并通过过滤收集固体,得到标题化合物(39mg),为固体。
实施例7:
(1r,3s)-n3-{[4-(苄基氨基)苯基]甲基}-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
步骤1苄基[4-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]氨基甲酸叔丁酯
[化学式376]
使用参考例c-4的步骤2中获得的化合物和参考例d-89的步骤3中获得的化合物,以与实施例2的步骤1中相同的方式获得标题化合物。
步骤2(1r,3s)-n3-{[4-(苄基氨基)苯基]甲基}-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺
[化学式377]
在室温下向上述步骤1中获得的化合物(397mg)在二氯甲烷(1.5ml)中的溶液中添加三氟乙酸(cas:76-05-1)(1.5ml)。将混合物在室温搅拌15小时,将反应溶液用饱和碳酸氢钠水溶液碱化,并用氯仿萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行反相高效液相色谱法(水/乙腈/0.1%甲酸),得到标题化合物(301mg),为固体。
步骤3(1r,3s)-n3-{[4-(苄基氨基)苯基]甲基}-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式378]
使用上述步骤2中获得的化合物,以与实施例2的步骤3中相同的方式获得标题化合物。
实施例8:
(1r,3s)-n3-({4-[(5-甲氧基吡啶-3-基)氨基]苯基}甲基)-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
步骤14-[(5-甲氧基吡啶-3-基)氨基]苯甲醛
[化学式379]
向参考例d-94的步骤2中获得的化合物(0.117g)在thf(5.07ml)中的溶液中加入二氧化锰(0.441g),将混合物在室温下搅拌5小时,并使其静置过夜。向其中添加另外的二氧化锰(0.441g),并将混合物在45℃搅拌2小时。使反应混合物冷却至室温,通过硅藻土过滤,并将滤液在减压下浓缩。所获得的标题化合物的粗产物直接用于下一步。
步骤2(1r,3s)-n3-({4-[(5-甲氧基吡啶-3-基)氨基]苯基}甲基)-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式380]
使用上述步骤1中获得的化合物和参考例c-4的步骤2中获得的化合物,以与实施例2的步骤1和步骤3中相同的方式获得标题化合物。
实施例9:
(1r,3s)-n1-[6-(环丙基甲基)噻吩并[2,3-d]嘧啶-4-基]-n3-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-n1-甲基环戊烷-1,3-二胺盐酸盐
[化学式381]
将参考例c-29中获得的化合物(205mg)溶解于二氯甲烷(5ml)中,向其中加入氯化氢(4mol/l,1,4-二噁烷溶液,5ml),并将混合物在室温下搅拌1小时。减压蒸发溶剂。将二氯甲烷(5ml)和dipea(0.266ml)加入到所得残余物中。向其中加入参考例d-65中获得的化合物(119mg)、三乙酰氧基硼氢化钠(324mg)和乙酸(0.132ml),并将混合物在室温搅拌1小时。向其中加入二氯甲烷和饱和碳酸氢钠水溶液,将混合物进行液体分离,并将水层用二氯甲烷萃取。合并有机层,用饱和碳酸氢钠水溶液洗涤,经无水硫酸钠干燥,并在减压下浓缩。通过硅胶柱色谱法(二氯甲烷/甲醇,然后用乙酸乙酯/甲醇)纯化获得的残余物,得到游离形式的标题化合物(40mg)。将其溶于乙醇(3ml)中,向其中加入1n盐酸(0.240ml),并在减压下蒸发溶剂。将乙醚加入到残余物中,并通过过滤收集固体,得到标题化合物(23mg),为固体。
实施例10:
n-(氧杂环丁烷-3-基)-4-({[(1s,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基}甲基)苯甲酰胺盐酸盐
步骤14-({[(1s,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基}甲基)苯甲酸甲酯
[化学式382]
使用参考例c-2的步骤2中获得的化合物和4-甲酰基苯甲酸甲酯(cas:1571-08-0),以与实施例2的步骤1中相同的方式获得标题化合物。
步骤24-({(叔丁氧基羰基)[(1s,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基}甲基)苯甲酸甲酯
[化学式383]
使用上述步骤1中获得的化合物,以与参考例a-16的步骤7中相同的方式获得标题化合物。
步骤34-({(叔丁氧基羰基)[(1s,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基}甲基)苯甲酸
[化学式384]
使用上述步骤2中获得的化合物,以与参考例a-1的步骤9中相同的方式获得标题化合物。
步骤4({4-[(氧杂环丁烷-3-基)氨基甲酰基]苯基}甲基)[(1s,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基甲酸叔丁酯
[化学式385]
使用上述步骤3中获得的化合物和3-氧杂环丁胺,以与参考例c-32的步骤3中相同的方式获得标题化合物。
步骤5n-(氧杂环丁烷-3-基)-4-({[(1s,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基}甲基)苯甲酰胺
[化学式386]
在室温下向上述步骤4中获得的化合物(47.9mg)在二氯甲烷(0.4ml)中的溶液中添加三氟乙酸(cas:76-05-1)(0.2ml)。将混合物在室温搅拌15小时,将反应溶液用饱和碳酸氢钠水溶液碱化,并用氯仿萃取。将有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行反相高效液相色谱法(水/乙腈/0.1%甲酸),得到标题化合物(34.1mg),为固体。
步骤6n-(氧杂环丁烷-3-基)-4-({[(1s,3r)-3-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己基]氨基}甲基)苯甲酰胺盐酸盐
[化学式387]
使用上述步骤5中获得的化合物,以与实施例2的步骤3中相同的方式获得标题化合物。
实施例11:
5-[4-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]吡啶-2(1h)-酮盐酸盐
步骤1(1r,3s)-n3-[(4-碘苯基)甲基]-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺
[化学式388]
使用参考例c-4的步骤2中获得的化合物和4-碘苯甲醛(cas:15164-44-0),以与实施例2的步骤1中相同的方式获得标题化合物。
步骤25-[4-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]吡啶-2(1h)-酮
[化学式389]
将上述步骤1中获得的化合物(0.110g)溶于1,4-二噁烷(2.00ml)中,向其中加入2-羟基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)吡啶(cas:1054483-78-1)(0.0600g)、[1,1'-双(二苯基膦基)二茂铁]钯(ii)二氯甲烷加合物(0.0295g)和饱和碳酸氢钠水溶液(1.00ml),并将混合在微波反应器中于110℃加热搅拌10分钟。使反应混合物冷却至室温,向其中添加水,并将混合物用二氯甲烷萃取两次。减压浓缩有机层,并将残余物通过氨基硅胶色谱法纯化(二氯甲烷/乙酸乙酯/甲醇),得到标题化合物(0.0828g),为固体。
步骤35-[4-({[(1s,3r)-3-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]吡啶-2(1h)-酮盐酸盐
[化学式390]
将上述步骤2中获得的化合物(0.0828g)溶于乙醇(3.00ml),向其中加入1n盐酸(0.161ml),并在减压下蒸发溶剂。将乙醚加入到残余物中,并通过过滤收集固体,得到标题化合物(0.0505g),为固体。
实施例12:
(1r,3s)-n1-甲基-n3-({4-[(1-甲基-1h-吡唑-4-基)氨基]苯基}甲基}-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺
[化学式391]
将实施例11的步骤1中获得的化合物(50.0mg)、1-甲基-1h-吡唑-4-胺(cas:69843-13-6)(16.2mg)、碳酸铯(89.5mg)、4,5-双(二苯基膦基)-9,9-二甲基呫吨(15.9mg)、乙酸钯(ii)(4.8mg)和1,4-二噁烷(1.0ml)的混合物在微波反应器中在氮气气氛下在110℃加热搅拌1小时。向反应溶液中加入水,并将混合物用氯仿萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行反相高效液相色谱法(水/乙腈/0.1%甲酸),得到标题化合物(9.86mg),为固体。
实施例13:
(1r,3s)-n3-{[4-(6'-氯[2,3'-联吡啶]-5-基)苯基]甲基}-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺
[化学式392]
将实施例11的步骤1中获得的化合物(0.269g)溶解在乙二醇二甲醚(2.28ml)中,向其中加入6-氯-3-吡啶基硼酸(cas:444120-91-6)(0.143g)、双(三苯基膦)二氯化钯(ii)二氯甲烷加合物(0.0320g)、碳酸钾(0.189g)和水(0.228ml),并将混合物在氮气气氛下于60℃搅拌4小时。向反应溶液中加入水,并将混合物用二氯甲烷萃取两次。减压浓缩有机层,并将残余物通过硅胶色谱法(二氯甲烷/甲醇)纯化,得到标题化合物(0.0101g),为固体。
实施例14:
(1r,3s)-n3-{[4-(呋喃并[3,2-b]吡啶-6-基)苯基]甲基}-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式393]
使用实施例11的步骤1中获得的化合物和6-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)呋喃并[3,2-b]吡啶(cas:1188539-34-5),以与实施例11的步骤2和步骤3中相同的方法获得标题化合物。
实施例15:
(1r,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇盐酸盐(15a)
(1s,3s,5r)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇盐酸盐(15b)
步骤1(1r,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇(外消旋物)
[化学式394]
使用参考例c-18的步骤2中获得的化合物和参考例d-65中获得的化合物,以与实施例2的步骤1中相同的方式获得标题化合物(390mg)。
步骤2(1r,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇(15a的游离形式)
(1s,3s,5r)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇(15b的游离形式)
[化学式395]
用手性高效液相色谱法(chiralpak(注册商标,daicelcorporation)ia,流动相:正己烷/2-丙醇),将上述步骤1中获得的化合物(390mg)进行光学拆分,分别得到为固体的15a的游离形式(较早洗脱的组分,150mg)和15b的游离形式(较晚洗脱的组分,185mg)。
15a的游离形式(较早洗脱的组分)
15b的游离形式(较晚洗脱的组分)
分离条件(分析)chiralpak(注册商标,daicelcorporation)ia,尺寸0.46cm×15cm,流速1.0ml/min,流动相:正己烷/2-丙醇=20/80至0/100,温度26℃
游离形式的15a保留时间3.0分钟,游离形式的15b保留时间3.6分钟
分离条件(制备)chiralpak(注册商标,daicelcorporation)ia,尺寸2.5cm×25cm,流速20ml/min,流动相正己烷/2-丙醇=50/50,温度40℃
游离形式的15a保留时间5.7分钟,游离形式的15b保留时间7.2分钟。
步骤3(1r,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇盐酸盐(15a)
[化学式396]
将上述步骤2中获得的游离形式的15a(较低极性异构体,149mg)、盐酸(1mol/l,乙醇溶液,0.274ml)和乙醇(2ml)的混合物搅拌,并在减压下浓缩。将乙醇/乙醚溶液加入到残余物中,并通过过滤收集所得固体,得到标题化合物(142mg),为固体。
步骤4(1s,3s,5r)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇盐酸盐(15b)
[化学式397]
将上述步骤9中获得的游离形式的15b(125mg)、盐酸(1mol/l,乙醇溶液,0.23ml)和乙醇(2ml)的混合物搅拌,并在减压下浓缩。将乙醇/乙醚溶液加入到残余物中,并通过过滤收集所得固体,得到标题化合物(116mg),为固体。
实施例16:
(1r,3s,5r)-3-[{[4-(5-甲氧基吡啶-3-基)苯基]甲基}(甲基)氨基]-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己-1-醇
[化学式398]
将实施例15的步骤2中获得的化合物(15b的游离形式)(0.0353g)、二氯甲烷(0.649ml)和吡啶(0.0157ml)的混合物用冰冷却,并向其中加入三氟甲磺酸甲酯(0.0147ml)。将混合物在相同温度下搅拌2小时,逐渐温热至室温,并搅拌1小时。将反应混合物用冰冷却,向其中添加另外的吡啶(0.0261ml)和三氟甲磺酸甲酯(0.0294ml),并将混合物在相同温度下搅拌1小时,然后在室温下搅拌过夜。向反应混合物中加入饱和碳酸氢钠水溶液,并将混合物用乙酸乙酯萃取3次。有机层经无水硫酸钠干燥,并在减压下浓缩。将残余物通过制备型tlc(二氯甲烷/甲醇)和氨基硅胶制备型tlc(二氯甲烷/乙酸乙酯)纯化,得到标题化合物(0.0028g)。
实施例17:
(1s,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-n,n-二甲基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酰胺盐酸盐(17a)
(1r,3s,5r)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-n,n-二甲基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酰胺盐酸盐(17b)
步骤1(1r,3s,5r)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-n,n-二甲基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酰胺盐酸盐(外消旋物)
[化学式399]
在室温下向参考例c-17的步骤2中获得的化合物(0.0375g)、参考例d-65中获得的化合物(0.0240g)和氯仿(2.0ml)的混合物中加入原钛酸四异丙酯(cas:546-68-9)(0.083ml)。将该反应溶液在室温搅拌1.5小时,向其中加入三乙酰氧基硼氢化钠(纯度97%,0.109g),将混合物在室温搅拌6小时,并使其静置16小时。向该反应溶液中加入饱和碳酸氢钠水溶液(10ml)和饱和酒石酸钾钠水溶液(10ml),并将混合物用二氯甲烷(10ml)萃取3次。有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过氨基硅胶柱色谱法(甲醇/乙酸乙酯)纯化,得到油状物(0.0457g)。在室温下向获得的油状物(0.0457g)和乙醇(3.0ml)的混合物中加入1n氯化氢乙醇溶液(0.076ml)。将该反应溶液在室温搅拌2.5小时,并在减压下浓缩。过滤残留物在乙醇(0.05ml)和乙醚(6.0ml)中的悬浮液,并将得到的固体在40℃减压干燥2小时,得到标题化合物(0.0419g),为固体。
步骤2(1s,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-n,n-二甲基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酰胺(17a的游离形式)
(1r,3s,5r)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-n,n-二甲基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酰胺(17b的游离形式)
[化学式400]
在室温下向上述步骤1中获得的化合物(0.0339g)和二氯甲烷(5.0ml)的混合物中加入饱和碳酸氢钠水溶液(5.0ml)。将该混合物在室温下剧烈搅拌1小时,并用二氯甲烷(10ml)萃取3次,将有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过手性高效液相色谱法(chiralpak(注册商标,daicelcorporation)ia,流动相:2-丙醇/正己烷)纯化,分别得到为油状物的游离形式的17a(较早洗脱的组分,0.0137g)和游离形式的17b(较晚洗脱的组分,0.0145g)。
游离形式的17a(较早洗脱的组分)
游离形式的17b(较晚洗脱的组分)
分离条件(分析)chiralpak(注册商标,daicelcorporation)ia,尺寸0.46cm×15cm,流速1.0ml/min,流动相正己烷/2-丙醇=50/50至0/100,温度40℃
游离形式的17a保留时间5.2分钟,游离形式的17b保留时间7.3分钟
分离条件(制备型)chiralpak(注册商标,daicelcorporation)ia,尺寸2.0cm×25cm,流速10ml/min,流动相正己烷/2-丙醇=25/75,温度20℃
游离形式的17a保留时间14.0分钟,游离形式的17b保留时间19.8分钟
步骤3(1s,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-n,n-二甲基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酰胺盐酸盐(17a)
[化学式401]
在室温下向上述步骤2中获得的游离形式的17a(0.0137g)和乙醇(5.0ml)的混合物中加入1n氯化氢乙醇溶液(0.030ml)。将该反应溶液在室温搅拌2.5小时,并在减压下浓缩。过滤残留物在乙醇(0.05ml)和乙醚(6.0ml)中的悬浮液,并将得到的固体在40℃减压干燥2小时,得到标题化合物(0.0120g),为固体。
步骤4(1r,3s,5r)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-n,n-二甲基-5-{[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环己烷-1-甲酰胺盐酸盐(17b)
[化学式402]
在室温下向上述步骤2中获得的游离形式的17b(0.0145g)和乙醇(5.0ml)的混合物中加入1n氯化氢乙醇溶液(0.030ml)。将该反应溶液在室温搅拌2.5小时,并在减压下浓缩。过滤残留物在乙醇(0.05ml)和乙醚(6.0ml)中的悬浮液,并将得到的固体在40℃减压干燥2小时,得到标题化合物(0.0145g),为固体。
实施例18:
(1r,3s,5r)-5-甲氧基-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
(1s,3r,5s)-5-甲氧基-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
步骤1(1r,3s,5r)-5-甲氧基-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基]-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺(外消旋物)
[化学式403]
使用参考例c-18的步骤2中获得的化合物和参考例d-65中获得的化合物,以与参考例e-3的步骤1中相同的方式获得标题化合物。
步骤2(1r,3s,5r)-5-甲氧基-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
(1s,3r,5s)-5-甲氧基-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
[化学式404]
使用手性高效液相色谱法(chralpak(注册商标,daicelcorporation)ia,流动相:正己烷/2-丙醇/乙酸乙酯/二乙胺),将上述步骤1中获得的化合物(0.24g)进行光学拆分,以得到游离形式的18a(较早洗脱的组分,69mg)和游离形式的18b(较晚洗脱的组分,51mg)。
将游离形式的18a(0.069g)溶于甲醇(3.00ml),向其中加入1n盐酸(0.124ml),并在减压下蒸发溶剂。将乙醚添加至残余物,并通过过滤收集固体,得到标题化合物的一种化合物(18a)(0.069g),为固体。
将游离形式的18b(0.051g)溶于甲醇(3.00ml),向其中加入1n盐酸(0.092ml),并在减压下蒸发溶剂。将乙醚加入到残余物中,并通过过滤收集固体,得到标题化合物的另一化合物(18b)(0.054g),为固体。
分离条件(分析)chralpak(注册商标,daicelcorporation)ia,尺寸0.46cm×25cm,流速1.0ml/min,流动相正己烷/2-丙醇/乙酸乙酯/二乙胺=50/30/20/0.1,温度40℃
游离形式的18a保留时间7.2分钟,游离形式的18b保留时间9.8分钟
分离条件(制备型)chralpak(注册商标,daicelcorporation)ia,尺寸2.0cm×25cm,流速5.7ml/min,流动相正己烷/2-丙醇/乙酸乙酯/二乙胺=50/30/20/0.1,温度40℃
游离形式的18a保留时间21分钟,游离形式的18b保留时间27分钟
实施例19:
(1s,3r,5r)-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-5-(1,3-噁唑-2-基)-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
(1r,3s,5s)-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-5-(1,3-噁唑-2-基)-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
步骤1(1s,3r,5r)-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-5-(1,3-噁唑-2-基)-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺(外消旋物)
[化学式405]
使用参考例c-32的步骤6中获得的化合物和参考例d-65中获得的化合物,以与参考例e-3的步骤1中相同的方式获得标题化合物。
步骤2(1s,3r,5r)-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-5-(1,3-噁唑-2-基)-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
(1r,3s,5s)-n1-{[4-(5-甲氧基吡啶-3-基)苯基]甲基}-5-(1,3-噁唑-2-基)-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
[化学式406]
使用手性高效液相色谱法(chralpak(注册商标,daicelcorporation)ia,流动相:正己烷/2-丙醇),将上述步骤1(68mg)中得到的化合物进行光学拆分,得到游离形式的19a(较早洗脱的组分,22mg)和游离形式的19b(较晚洗脱的组分,23mg)。
在室温下向游离形式的19a(22mg)在乙醇(0.500ml)中的溶液中加入2n盐酸(0.0185ml),并将混合物减压浓缩。向其中加入乙醇(1.00ml),共沸浓缩混合物,并将残余物用乙醚固化,得到标题化合物的一种化合物(19a)(23mg),为固体。
在室温下向游离形式的19b(23mg)在乙醇(0.500ml)中的溶液中加入2n盐酸(0.0196ml),并将混合物减压浓缩。向其中加入乙醇(1.00ml),共沸浓缩混合物,并将残余物用乙醚固化,得到标题化合物的另一种化合物(19b)(23mg),为固体。
分离条件(分析)chiralpak(注册商标,daicelcorporation)ia,尺寸0.46cm×15cm,流速1.0ml/min,流动相2-丙醇100%,温度40℃
游离形式的19a保留时间5.4分钟,游离形式的19b保留时间7.7分钟
分离条件(制备型)chiralpak(注册商标,daicelcorporation)ia,尺寸2cm×25cm,流速10ml/min,流动相正己烷/2-丙醇=20/80,温度约20℃
游离形式的19a保留时间19.9分钟,游离形式的19b保留时间32.9分钟
实施例20:
(1s,2r,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊烷-1,2-二醇盐酸盐(20a)
(1r,2s,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊烷-1,2-二醇盐酸盐(20b)
步骤1(1s,2r,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊烷-1,2-二醇
[化学式407]
将参考例c-33的步骤7中得到的异构体a(193mg)、二氯甲烷(5ml)和tfa(5ml)的混合物在室温下搅拌2小时。减压浓缩反应溶液,得到油状物(283mg)。其直接用于下一步。上述油状物(283mg)、参考例d-65中获得的化合物(75.6mg)、dipea(0.218ml)、二氯甲烷(4.2ml)、三乙酰氧基硼氢化钠(265mg)和乙酸(0.143ml)的混合物在室温下搅拌2.5小时。再次向其中加入参考例d-65中获得的化合物(25.0mg),并将混合物再搅拌1.5小时。向反应溶液中加入饱和碳酸氢钠水溶液,并用乙酸乙酯萃取混合物。有机层用饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(乙酸乙酯/甲醇)纯化,然后通过制备型tlc(乙酸乙酯/甲醇)纯化,得到标题化合物(30.3mg),为固体。
步骤2(1s,2r,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基}-5-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊烷-1,2-二醇盐酸盐(20a)
[化学式408]
将上述步骤1中获得的化合物(28.3mg)、氯化氢(1mol/l,乙醇溶液,0.0506ml)和乙醇(1ml)的混合物在室温下搅拌。将混合物在减压下浓缩。将乙醇/乙醚加入到残余物中,通过过滤收集所得固体,并干燥得到标题化合物(25.0mg),为固体。
步骤3(1r,2s,3r,5s)-3-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-5-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊烷-1,2-二醇盐酸盐(20b)
[化学式409]
使用参考例c-33的步骤7中获得的异构体b,以与上述步骤1和步骤2中相同的方式获得标题化合物。
实施例21:
(1r,2s,4r)-2-{[2-氨基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}-4-({[4-(1h-咪唑-1-基)苯基]甲基}氨基)环戊-1-醇盐酸盐
步骤1(1r,2s,4r)-4-({[4-(1h-咪唑-1-基)苯基]甲基}氨基)-2-{[2-{[(4-甲氧基苯基)甲基]氨基}-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇
[化学式410]
使用参考实施例c-23的步骤2中获得的化合物和4-(1h-咪唑-1-基)苯甲醛(cas:10040-98-9),以与实施例2的步骤1中相同的方式获得标题化合物。
步骤2(1r,2s,4r)-2-{[2-氨基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}-4-({[4-(1h-咪唑-1-基)苯基]甲基}氨基)环戊-1-醇
[化学式411]
在室温下向上述步骤1中获得的化合物(137mg)在二氯甲烷(0.55ml)中的溶液中加入三氟乙酸(cas:76-05-1)(1.64ml)。将混合物在60℃搅拌1.5小时,并将反应溶液在减压下浓缩。将获得的残余物进行硅胶柱色谱法(乙酸乙酯/甲醇),得到标题化合物(60.1mg),为固体。
步骤3(1r,2s,4r)-2-{[2-氨基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}-4-({[4-(1h-咪唑-1-基)苯基]甲基}氨基)环戊-1-醇盐酸盐
[化学式412]
将上述步骤2中得到的化合物(0.601g)溶解于乙醇(1.5ml)中,并向其中加入1n盐酸(0.116ml)。减压蒸发溶剂,并将残余物干燥。将获得的残余物悬浮在乙醚(3ml)中,通过过滤收集所得固体,并干燥得到标题化合物(0.0605g),为固体。
实施例22:
5-[4-({[(1r,3r,4s)-3-羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]-3-甲氧基吡啶-2-甲腈盐酸盐
步骤15-[4-({[(1r,3r,4s)-3-羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]-3-甲氧基吡啶-2-甲腈
[化学式413]
将参考例d-25的步骤2中得到的化合物(0.0756g)、参考例c-14的步骤2中得到的化合物(0.110g)、三乙酰氧基硼氢化钠(0.208g)、乙酸(0.092ml)和二氯甲烷(5ml)的混合物在室温搅拌15小时。反应溶液用二氯甲烷稀释,依次用饱和碳酸氢钠水溶液和饱和盐水洗涤,经无水硫酸镁干燥,并减压浓缩。将获得的残余物进行硅胶柱色谱法(乙酸乙酯/甲醇),得到标题化合物(0.0707g),为固体。
步骤25-[4-({[(1r,3r,4s)-3-羟基-4-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊基]氨基}甲基)苯基]-3-甲氧基吡啶-2-甲腈盐酸盐
[化学式414]
将上述步骤1中获得的化合物(0.0707g)、1n盐酸(0.120ml)和乙醇(5.00ml)的混合物在室温下搅拌10分钟,并在减压下浓缩。将获得的残余物悬浮在乙腈(10ml)中,通过过滤收集所得固体,并干燥得到标题化合物(0.0683g),为固体。
实施例23:
(1r,2s,4r)-4-[({4-[1-(甲磺酰基)-1h-吲唑-4-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
步骤1(1r,2s,4r)-4-[({4-[1-(甲磺酰基)-1h-吲唑-4-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
[化学式415]
将参考例c-14的步骤2中获得的化合物(0.0817g)、参考例d-40中获得的化合物(0.0708g)、二氯甲烷(2.5ml)、dipea(0.0411ml)、乙酸(0.0810ml)和三乙酰氧基硼氢化钠(0.150g)的混合物在室温下搅拌2小时。向反应混合物中加入饱和碳酸氢钠水溶液,并用氯仿萃取混合物。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,并在减压下浓缩。使用氨基硅胶柱作为装料柱,将获得的残余物进行硅胶柱色谱法(乙酸乙酯/甲醇),得到标题化合物(0.105g),为固体。
步骤2(1r,2s,4r)-4-[({4-[1-(甲磺酰基)-1h-吲唑-4-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
[化学式416]
将上述步骤1中得到的化合物(0.105g)溶解在乙醇(1.5ml)中,并向其中加入1n盐酸(0.166ml)。减压蒸发溶剂,并将残余物干燥。将获得的残余物悬浮在乙醚(3ml)中,并通过过滤收集所得固体,并干燥得到标题化合物(0.0966g),为固体。
实施例24:
n4-[(1s,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-甲氧基环戊基]-n2,n4-二甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-2,4-二胺盐酸盐
步骤1(1r,3s,4s)-n3-[2-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]-n1-{[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}-4-甲氧基-n3-甲基环戊烷-1,3-二胺
[化学式417]
将参考例e-2的步骤2中得到的化合物(0.440g)、参考例b-4的步骤2中得到的化合物(0.318g)、2-丙醇(22ml)和dipea(cas:7087-68-5)(3.2ml)的混合物在100℃搅拌3小时。将饱和盐水/水(3∶1)加入到反应溶液中,并将混合物用乙酸乙酯萃取。有机层经无水硫酸钠干燥,并在减压下浓缩。残余物通过硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(0.508g),为固体。
步骤2n4-[(1s,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-甲氧基环戊基]-n2,n4-二甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-2,4-二胺
[化学式418]
将上述步骤1中获得的化合物(0.253g)溶于丁腈(2.5ml)中,向其中加入甲胺盐酸盐(cas:593-51-1)(0.109g)和dipea(cas:7087-68-5)(0.42ml),并将混合物在150℃下微波辐照1小时。然后,向其中添加另外的甲胺盐酸盐(cas:593-51-1)(0.106g)和dipea(cas:7087-68-5)(0.40ml),并将混合物在160℃下微波辐照3小时。向反应溶液中加入水,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,在减压下浓缩,并将残余物进行硅胶柱色谱法(二氯甲烷/甲醇)、反相hplc(gilson,水(0.10%甲酸)/乙腈(0.10%甲酸))和氨基硅胶柱色谱法(乙酸乙酯/甲醇)。将获得的油状物溶解在乙醚中,并将溶液用水和饱和盐水洗涤,经无水硫酸钠干燥,并在减压下浓缩,得到标题化合物(0.0773g),为固体。
步骤3n4-[(1s,2s,4r)-4-({[4-(5,6-二甲氧基吡啶-3-基)苯基]甲基}氨基)-2-甲氧基环戊基]-n2,n4-二甲基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-2,4-二胺盐酸盐
[化学式419]
使用上述步骤2中获得的化合物,以与实施例2的步骤3中相同的方式获得标题化合物。
实施例25:
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
步骤1(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
[化学式420]
将参考例c-14的步骤2中得到的化合物(0.102g)、参考例d-26中得到的化合物(0.0844g)、二氯甲烷(3.2ml)、三乙酰氧基硼氢化钠(cas:56553-60-7)(0.215g)和乙酸(0.0500ml)的混合物在室温下搅拌17小时。将水/饱和碳酸氢钠水溶液(2/1)加入到反应溶液中,并将混合物用二氯甲烷/甲醇(9/1)萃取。有机层经无水硫酸钠干燥,过滤,在减压下浓缩,并将残余物通过硅胶柱色谱法(二氯甲烷/甲醇)纯化,得到标题化合物(0.115g),为油状物。
步骤2(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
[化学式421]
将上述步骤1中获得的化合物(0.112g)溶解在乙醇(3.6ml)中,向其中加入1n盐酸(0.173ml),并在减压下蒸发溶剂。将获得的固体悬浮在乙醚中,通过过滤收集得到的固体,并干燥以得到标题化合物(0.104g),为固体。
实施例26:
(1r,2s,4r)-4-[({4-[6-(二甲基氨基)-5-甲氧基吡啶-3-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
步骤1(1r,2s,4r)-4-[({4-[6-(二甲基氨基)-5-甲氧基吡啶-3-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇
[化学式422]
将参考例c-14的步骤3中获得的化合物(0.112g)、二氯甲烷(3.0ml)、参考例d-11的步骤2中获得的化合物(0.080g)、dipea(0.153ml)、乙酸(0.084ml)和三乙酰氧基硼氢化钠(0.190g)的混合物在室温下搅拌16小时。将饱和碳酸氢钠水溶液加入到反应混合物中,并将混合物用乙酸乙酯萃取。有机层用饱和盐水洗涤,经无水硫酸钠干燥,过滤,并在减压下浓缩。使用氨基硅胶柱作为装料柱,将获得的残余物进行硅胶柱色谱法(乙酸乙酯-甲醇),得到标题化合物(0.125g),为固体。
步骤2(1r,2s,4r)-4-[({4-[6-(二甲基氨基)-5-甲氧基吡啶-3-基]苯基}甲基)氨基]-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
[化学式423]
将上述步骤1中获得的化合物(0.122g)溶解在乙醇(2ml)中,并向其中加入5n盐酸水溶液(0.042ml)。减压蒸发溶剂,并将残余物干燥。将获得的残余物悬浮在乙腈(2ml)中,通过过滤收集所得固体,并干燥得到标题化合物(0.117g),为固体。
实施例27:
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇盐酸盐
步骤1(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇
[化学式424]
将参考例c-15的步骤2中得到的化合物(90.0mg)、参考例d-26中得到的化合物(55.9mg)、dipea(0.114ml)、二氯甲烷(2.18ml)、三乙酰氧基硼氢化钠(139mg)和乙酸(0.0624ml)的混合物在室温下搅拌1.5小时。将饱和碳酸氢钠水溶液和二氯甲烷加入到反应溶液中,并将混合物进行液体分离。有机层经无水硫酸钠干燥,并在减压下浓缩。将获得的残余物通过硅胶柱色谱法(乙酸乙酯/甲醇)纯化,得到标题化合物(94.0mg),为固体。
步骤2(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇盐酸盐
[化学式425]
将上述步骤1中获得的化合物(90.0mg)、乙醇(1ml)和1n盐酸乙醇溶液(0.149ml)的混合物在室温下搅拌,并在减压下浓缩。将乙醇/乙醚添加到残余物中,通过过滤收集所得固体,并干燥得到标题化合物(84.1mg),为固体。
使用表中描述的原料1和原料2,以与实施例27的步骤1中同样的方式制备以下表2-1至表2-54中的各产物。当最终形式为盐酸盐时,使用游离产物作为原料,以与实施例27的步骤2中相同的方式制备盐酸盐。在表2-1至表2-54中,当最终形式由两项表示时,实施例的化合物为下项所示的盐化合物(例如,表2-14中的实施例54的化合物为(1s,2s,4r)-2-{甲基[2-(甲基氨基)-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}-4-[({4-[(吡啶-3-基)氨基]苯基}甲基)氨基]环戊-1-醇盐酸盐,其显示在下项中。在实验例中,将上述盐化合物用作实施例的化合物。
实施例126:
(1s,3s,4s)-n1-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-4-甲氧基-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
步骤1(1s,3s,4s)-n1-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-4-甲氧基-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺
[化学式426]
使用参考例e-3的步骤2中得到的化合物和根据文献(cancercell,2015,27,589-602)中所述的方法制备的4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶,以与参考例c-2的步骤1中相同的方式获得标题化合物。
步骤2(1s,3s,4s)-n1-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-4-甲氧基-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环己烷-1,3-二胺盐酸盐
[化学式427]
在室温下向上述步骤1中获得的化合物(0.143g)在乙醇(2.00ml)中的溶液中加入5n盐酸(0.0486ml),在减压下蒸发溶剂,并将残余物干燥。将获得的残余物悬浮在乙醚中,通过过滤收集所得固体,并干燥得到标题化合物(0.142g)。
实施例127:
(1r,3s,4r)-n1-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-4-甲氧基-n3-甲基-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
步骤1(1r,3s,4r)-n1-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-4-甲氧基-n3-甲基-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺
[化学式428]
将参考例e-4中获得的化合物(0.638g)、氯化氢(4mol/l,1,4-二噁烷溶液,13.5ml)和二氯甲烷(15.0ml)的混合物在室温下搅拌35分钟,并浓缩。将残余物干燥,得到固体(0.601g)。将所得固体、根据文献(cancercell,2015,27,589-602)中所述的方法制备的4-氯-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶(0.358g)、dipea(1.41ml)和2-丙醇(20ml)的混合物在80℃搅拌5小时,减压浓缩,并将获得的残余物进行硅胶柱色谱纯化(乙酸乙酯/甲醇),得到标题化合物(0.454g),为固体。
步骤2(1r,3s,4r)-n1-{[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}-4-甲氧基-n3-甲基-n3-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式429]
将上述步骤1中得到的化合物(0.454g)、1n盐酸(0.739ml)和乙醇(3ml)的混合物在室温下搅拌5分钟,并浓缩。将乙腈(10ml)加入到获得的残余物中,通过过滤收集沉淀的固体,并干燥得到标题化合物(0.413g),为固体。
实施例128:
(1r,2s,4r)-4-({[(1s,4s)-4-(5,6-二甲氧基哒嗪-3-基)环己基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇盐酸盐
(1r,2s,4r)-4-({[(1r,4r)-4-(5,6-二甲氧基哒嗪-3-基)环己基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇盐酸盐
[化学式430]
将参考例c-15的步骤2中获得的化合物(0.220g)悬浮在二氯甲烷(5.3ml)中,并向其中添加dipea(0.371ml)。将混合物在室温搅拌1小时。向其中加入参考例d-78的步骤2中获得的化合物(0.140g)和乙酸(0.213ml),并将混合物搅拌15分钟。将三乙酰氧基硼氢化钠(0.384g)加入到混合物中,并将混合物在室温搅拌过夜。将饱和碳酸氢钠水溶液加入到反应溶液中,并将混合物进行萃取操作。有机层经无水硫酸钠干燥,并在减压下蒸发溶剂。残余物通过硅胶色谱法(二氯甲烷/甲醇)纯化,得到固体。通过高效液相色谱法(chiralpak(注册商标,daicelcorporation)ig,流动相:正己烷/乙醇)纯化固体,得到分别为固体的游离形式的128a(较早洗脱的组分,0.026g)和游离形式的128b(较晚洗脱的组分,0.100g)。
游离形式的128a
游离形式的128b
将上述步骤中获得的游离形式的128a(25mg)溶解在乙醇(0.5ml)中,向其中加入1n盐酸乙醇溶液(41μl),并在减压下浓缩混合物。向残余物中加入乙酸乙酯(1ml),并通过过滤收集沉淀的固体,得到标题化合物的一种化合物(128a)(28mg),为固体。
类似地,将游离形式的128b(95.0mg)溶解在乙醇(2ml)中,向其中加入1n盐酸乙醇溶液(0.160ml),并将混合物减压浓缩。向残余物中加入乙酸乙酯(2ml),并通过过滤收集沉淀的固体,得到标题化合物的另一化合物(128b)(0.103g),为固体。
分离条件(分析)chiralpak(注册商标,daicelcorporation)ig,尺寸0.46cm×25cm,流速1.0ml/min,流动相正己烷/乙醇=20/80,温度40℃
游离形式的128a保留时间8.5分钟,游离形式的128b保留时间11.7分钟
分离条件(制备型)chiralpak(注册商标,daicelcorporation)ig,尺寸2cm×25cm,流速15.0ml/min,流动相正己烷/乙醇=20/80,温度40℃
游离形式的128a保留时间9.9分钟,游离形式的128b保留时间12.2分钟。
实施例129
(1r,2s,4r)-4-({[4-(5-甲氧基吡啶-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇盐酸盐
[化学式431]
使用参考例c-14的步骤2中获得的化合物和参考例d-65中获得的化合物,以与实施例27的步骤1和步骤2中相同的方式获得标题化合物。
实施例130
(1r,3s)-n3-{[4-(2-甲氧基-1,3-噻唑-5-基)苯基]甲基}-n1-甲基-n1-[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]环戊烷-1,3-二胺盐酸盐
[化学式432]
使用实施例11的步骤1中获得的化合物和2-甲氧基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)噻唑,以与实施例11的步骤2和步骤3中相同的方式获得标题化合物。
实施例131
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基}-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇琥珀酸盐
将以与实施例25的步骤1中相同的方式获得的(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇(25.8g)悬浮在2-丙醇(103ml)中,向其中加入水(12.9ml)。将琥珀酸(5.56g)加入到混合物中,向其中加入水(12.9ml),并将混合物在室温下搅拌4小时。不溶物通过过滤收集,同时用2-丙醇(150ml)洗涤,并干燥得到为晶体的标题化合物(30.1g)。
所得晶体的粉末x射线衍射示于图1。
表3示出了在粉末x射线衍射(cukα,λ=1.54埃,扫描速度=20°/min)的衍射图图1中当最大峰强度为100时相对强度为35或更高的峰。
实施例132
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇苯磺酸盐
向以与实施例25的步骤1中相同的方式获得的(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇(200.31mg)和苯磺酸(57.89mg)中加入80%含水丙酮(1639μl),并将混合物在室温下搅拌约24小时。收集沉淀的固体,并在室温下干燥过夜,得到为晶体的标题化合物(220.45mg)。
所得晶体的粉末x射线衍射示于图2。
表4示出了在粉末x射线衍射(cukα,λ=1.54埃,扫描速度=20°/min)的衍射图图2中当最大峰强度为100时相对强度为24或更高的峰。
实施例133
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇马来酸盐
向以与实施例25的步骤1中相同的方式获得的(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇(300.14mg)和马来酸(63.66mg)中加入80%含水2-丙醇(5455μl),并将混合物在40℃下搅拌约24小时。收集沉淀的固体,并在室温下干燥过夜,得到为晶体的标题化合物(304.38mg)。
所得晶体的粉末x射线衍射示于图3。
表5示出了在粉末x射线衍射(cukα,λ=1.54埃,扫描速度=20°/min)的衍射图图3中当最大峰强度为100时相对强度为42或更高的峰。
实施例134
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{甲基[6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基]氨基}环戊-1-醇富马酸盐
向以与实施例25的步骤1中相同的方式获得的化合物(200.38mg)和富马酸(42.50mg)中加入80%含水2-丙醇(1638μl),并将混合物在40℃下搅拌约24小时。收集沉淀的固体,并在室温下干燥过夜,得到为晶体的标题化合物(236.21mg)。
所得晶体的粉末x射线衍射示于图4。
表6示出了在粉末x射线衍射(cukα,λ=1.54埃,扫描速度=20°/min)的衍射图图4中当最大峰强度为100时相对强度为51或更高的峰。
实施例135
(1r,2s,4r)-4-({[4-(5,6-二甲氧基吡哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇的晶体
向以与实施例25的步骤1中相同的方式获得的(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇(20.17mg)中加入2-丙醇(81μl)和水(323μl),并将混合物在室温下搅拌约24小时。收集获得的晶体,并在室温下干燥过夜,得到标题化合物(13.89mg)。
所得晶体的粉末x射线衍射示于图5。
表7示出了在粉末x射线衍射(cukα,λ=1.54埃,扫描速度=20°/min)的衍射图图5中当最大峰强度为100时相对强度为12或更高的峰。
实施例136
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇富马酸盐
向以与实施例25的步骤1中相同的方式获得的(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇(20.76mg)和富马酸(4.150mg)中加入80%含水2-丙醇(415μl),并将混合物在室温下搅拌约24小时。收集沉淀的固体,并在室温下干燥过夜,得到为晶体的标题化合物(22.13mg)。
所得晶体的粉末x射线衍射示于图6。
表8示出了在粉末x射线衍射(cukα,λ=1.54埃,扫描速度=20°/min)的衍射图图6中当最大峰强度为100时相对强度为20或更高的峰。
实施例137
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇粘酸盐
向以与实施例25的步骤1中相同的方式获得的(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇(20.10mg)和粘酸(7.421mg)中加入80%含水2-丙醇(402μl),并将混合物在室温下搅拌约24小时。收集沉淀的固体,并在室温下干燥过夜,得到为晶体的标题化合物(16.85mg)。
所得晶体的粉末x射线衍射示于图7。
表9示出了在粉末x射线衍射(cukα,λ=1.54埃,扫描速度=20°/min)的衍射图图7中当最大峰强度为100时相对强度为22或更高的峰。
实施例138
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇己二酸盐
向以与实施例25的步骤1中相同的方式获得的(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇(19.68mg)和己二酸(5.019mg)中加入80%含水2-丙醇(394μl),并将混合物在室温下搅拌约24小时。收集沉淀的固体,并在室温下干燥过夜,得到为晶体的标题化合物(19.75mg)。
所得晶体的粉末x射线衍射示于图8。
表10示出了在粉末x射线衍射(cukα,λ=1.54埃,扫描速度=20°/min)的衍射图图8中当最大峰强度为100时相对强度为10或更高的峰。
实施例139
(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇琥珀酸盐
向以与实施例25的步骤1中相同的方式获得的(1r,2s,4r)-4-({[4-(5,6-二甲氧基哒嗪-3-基)苯基]甲基}氨基)-2-{[2-甲氧基-6-(2,2,2-三氟乙基)噻吩并[2,3-d]嘧啶-4-基](甲基)氨基}环戊-1-醇(20.27mg)中加入80%含水2-丙醇(368μl),向其中加入1mol/l的琥珀酸水溶液(81μl),并将混合物在室温下搅拌约24小时。收集沉淀的固体,并在室温下干燥过夜,得到为晶体的标题化合物(14.41mg)。
所得晶体的粉末x射线衍射示于图9。
表11示出了在粉末x射线衍射(cukα,λ=1.54埃,扫描速度=20°/min)的衍射图图9中当最大峰强度为100时相对强度为26或更高的峰。
(制剂实施例)
制剂实施例1(注射剂)
将1.5重量%的实施例化合物在10体积%的丙二醇中搅拌,然后用注射用水将体积调节至预定值,并将混合物灭菌以得到注射剂。
制剂实施例2(硬胶囊)
将100mg的粉末状实施例化合物、128.7mg乳糖、70mg纤维素和1.3mg硬脂酸镁混合并通过60目筛,然后将所得粉末放入250mg3号明胶胶囊中以得到胶囊。
制剂实施例3(片剂)
将100mg的粉末状实施例化合物、124mg的乳糖、25mg的纤维素和1mg的硬脂酸镁混合并用压片机压片,得到每片250mg的片剂。该片剂可以根据需要包糖衣。
(实验例)
通过以下试验证实了本发明化合物的药理活性。
[实验例1]menin与mll之间的结合抑制活性的评价
将包含实施例1至130中任何化合物的10μl反应溶液(50mmtris-hcl(ph7.5)、50mmnacl、0.01%tritonx-100、0.01%牛血清白蛋白、3mmtcep)、1nmmenin(带flag标签,daiichisankyordnovareco.,ltd.)和10nm生物素化的mll1肽(1-46aa:scrumco.,ltd.)加入到384孔板中,并在室温下反应30分钟。然后,向其中加入10μl的anti-flagalphalisa受体珠(perkinelmerco.,ltd.,al112c)和链霉亲和素包被的alphascreen供体珠(perkinelmerco.,ltd.,6760002)的混合溶液(各10μg/ml),并使混合物在室温下反应1小时。然后,使用读板器(perkinelmerco.,ltd.,envisionxcite)测量alphalisa(注册商标,perkinelmerco.,ltd.)的荧光信号。基于测量的信号,计算实施例1至130的化合物在每种浓度下的结合抑制率,并且使用医学统计分析软件graphpadprism(graphpadsoftware,inc.)分析获得的数据以获得ic50值。
[实验例2]细胞生长抑制活性的评价
补充有10%fbs的rpmi1640培养基用作各细胞(mv-4-11、molm-13细胞、人aml细胞、k562细胞、人cml细胞)的培养基。各细胞均购自美国典型培养物保藏中心(atcc)。用freedomevo150(tecantradingag)稀释并制备各药物(实施例化合物)(公比为2,从10mm或5mm至20μm或10μm的10个浓度)后,使用echo555(labcyteinc.)将药物以40nl/孔接种在384孔组织培养板(#3712,corninginc.)上(终浓度为10μm或5μm至20nm或10nm)。制备的含药板在-30℃下储存℃直到使用,并在使用前解冻。
用10%fbsrpmi1640培养基制备每种细胞的悬浮液以使其为1000个细胞/ml(k562)或10000个细胞/ml(mv-4-11、molm-13),并接种在含药板上(40μl/孔)(第0天)。将细胞再培养3或7天。在添加药物的当天(第0天)和添加药物后3天(第3天)或7天(第7天),将atp测量试剂(celltiter-glo(注册商标,promegacorporation)2.0检测,型号g9242,promegacorporation)以10μl/孔添加到每个孔中。在底面上粘贴黑色贴纸后,使用微板混合脱气机(型号为weltornadofk-62,sakakidengyoco.,ltd.)搅拌混合物(搅拌条件,转数9,旋转7,时间12)。使用酶标仪(型号名称:envision2102-0010,perkinelmerco.,ltd.)的发光检测器测量发光(cps)(n=4)。
作为细胞生长抑制活性的指标,使用excel2010(microsoftcorporation)计算将细胞生长抑制50%的浓度(gi50)。当将无药物组的细胞生长视为100%时,将添加药物的组从第0天至第3天或第7天的细胞生长计算为t/c%。使用中间夹有将细胞生长抑制50%的浓度(t/c%=50%)的两个浓度和t/c%,通过growth函数(指数回归)计算gi50。
实验例1和2的结果示于表12-1至表12-4。
[实验例3-1]mv-4-11细胞皮下移植模型中的抗肿瘤活性的评价
mv-4-11细胞以1×107个细胞/头的比率皮下移植到雌性foxchasescidc.b.17/icr-scid/scidjcl小鼠的右腹部,并在17天后,基于估计的肿瘤体积(长轴×短轴×短轴/2),将小鼠分组为每组6只动物。mv-4-11细胞购自atcc。雌性foxchasescidc.b.17/icr-scid/scidjcl小鼠购自clairejapan。从分组后的第二天起,以25、50或100mg/kg/天的剂量设定以每天一次、持续17天的每一天(qd×17)的方案(schedule)口服施用实施例26的化合物。化合物以在0.5%甲基纤维素(mc)中的悬浮液形式施用。对于无化合物的组,施用0.5%mc作为溶剂。从分组当天至移植后34天(试验结束日期),测量估计的个体肿瘤体积。
[实验例3-2]在mv-4-11细胞皮下移植模型中的抗肿瘤活性的评价
mv-4-11细胞以1×107个细胞/头的比率皮下移植到雌性foxchasescidc.b.17/icr-scid/scidjcl小鼠的右腹部,并在17天后,基于估计的肿瘤体积(长轴×短轴×短轴/2),将小鼠分组为每组6只动物。mv-4-11细胞购自atcc。雌性foxchasescidc.b.17/icr-scid/scidjcl小鼠购自clairejapan。从分组当天起,以12.5、25、50或100mg/kg/天的剂量设定以每天一次、持续18天的每一天(qd×18)的方案口服施用实施例25或27的化合物。化合物以在0.5%mc中的悬浮液形式施用。对于无化合物的组,施用0.5%mc作为溶剂。从分组当天至移植后34天(试验结束日期),测量估计的个体肿瘤体积。
[实验例3-3]在mv-4-11细胞皮下移植模型中的抗肿瘤活性的mv-4-11细胞评价
将mv-4-11细胞以1×107个细胞/头的比率皮下移植到雌性foxchasescidc.b.17/icr-scid/scidjcl小鼠的右腹部,并在14天后,基于估计的肿瘤体积(长轴×短轴×短轴/2),将小鼠分组为每组6只动物。mv-4-11细胞购自atcc。雌性foxchasescidc.b.17/icr-scid/scidjcl小鼠购自clairejapan。从分组后的第二天起,以25、50或100mg/kg/天的剂量设定以每天一次持续17天的每一天(qd×17)的方案口服施用实施例68、25或27的化合物。化合物以在0.5%mc中的悬浮液形式施用。对于无化合物的组,施用0.5%mc作为溶剂。从分组当天至移植后31天(试验结束日期),测量估计的个体肿瘤体积。
[实验例3-4]mv-4-11细胞皮下移植模型中的抗肿瘤活性的评价
将mv-4-11细胞以1×107个细胞/头的比率皮下移植到雌性foxchasescidc.b.17/icr-scid/scidjcl小鼠的右腹部,并在15天后,基于估计的肿瘤体积(长轴×短轴×短轴/2),将小鼠分组为每组6只动物。mv-4-11细胞购自atcc。雌性foxchasescidc.b.17/icr-scid/scidjcl小鼠购自clairejapan。从分组后的第二天起,以25、50或100mg/kg/天的剂量设定以每天一次持续16天的每一天(qd×16)的方案口服施用实施例60或67的化合物。化合物以在0.5%mc中的悬浮液形式施用。对于无化合物的组,施用0.5%mc作为溶剂。从分组当天至移植后32天(试验结束日期),测量估计的个体肿瘤体积。
在每个试验的结束日期,通过以下公式计算实验例3-1至3-4中的mv4:11细胞皮下移植模型中的抗肿瘤活性。
肿瘤生长抑制率%=(1-tvct/tvcc)x100
tvc=(试验结束日期的个体肿瘤体积)-(分组日期的个体肿瘤体积)
tvct:化合物施用组的tvc平均值
tvcc:无化合物组的tvc平均值
实验例3-1至3-4的结果示于表13。
[实验例4]细胞生长抑制活性的评价(npm1突变体)
补充有20%fbs的mem-α培养基用作人aml细胞、oci-aml3细胞的培养基。这些细胞购自deutschesammlungvonmikroorganismenundzellkulturengmbh(dsmz)。用freedomevo150(tecantradingag)稀释并制备各药物(表14中显示的每种实施例化合物)(公比为4,从10mm至38nm的10个浓度)后,使用echo555(labcyteinc.)将药物以40nl/孔接种在384孔组织培养板(#3712,corninginc.)上(终浓度10μm至0.038nm)。制备的含药板在-30℃下储存直到使用,并在使用前解冻。
用10%fbsrpmi1640培养基制备细胞的悬浮液以使其为50000个细胞/ml,并接种在含药板上(40μl/孔)(第0天)。将细胞再培养7天。在添加药物的当天(第0天)和添加药物后7天(第7天),将atp测量试剂(celltiter-glo(注册商标,promegacorporation)2.0检测,型号g9242,promegacorporation)以10μl/孔添加到每个孔中。在底面上粘贴黑色贴纸后,使用微板混合脱气机(型号为weltornadofk-62,sakakidengyoco.,ltd.)搅拌混合物(搅拌条件,转数9,旋转7,时间12)。使用酶标仪(型号名称:envision2104-0010,perkinelmerco.,ltd.)的发光检测器测量发光(cps)(n=4)。
作为细胞生长抑制活性的指标,使用excel2010(microsoftcorporation)计算将细胞生长抑制50%的浓度(gi50)。当将无药物组的细胞生长视为100%时,将添加药物的组从第0天至第7天的细胞生长计算为t/c%。使用中间夹有将细胞生长抑制50%的浓度(t/c%=50%)的两个浓度和t/c%,通过growth函数(指数回归)计算gi50。
实验例4的结果示于表14。
[实验例5]分化诱导活性的评价
通过逆转录病毒感染,将人mll-af9融合基因导入从c57bl6小鼠的骨髓中分离的ckit阳性单核细胞中。通过病毒感染后长期液体培养细胞,建立了获得异常生长能力的mll-af9过表达aml样细胞(ma9细胞)。使用无血清培养基(glutamax,p/s,含有10ng/mlmil-3、50ng/mlmscf和10ng/ml人抑瘤素m的stempro-34培养基)作为培养基。
用培养基制备细胞的悬浮液,使其达到12500个细胞/ml,以2ml/孔接种在6孔组织培养板上,然后以各种浓度(实施例25和27的化合物为5nm或20nm,实施例26的化合物为150nm或300nm,实施例22的化合物为50nm或100nm)向其中加入药物(实施例25、27、26或22的化合物)(2μl/孔)(第0天)。对于对照组,以0.1%的最终浓度向其中添加dmso(2μl/孔)(第0天)。培养7天(第7天)后,收集细胞,并在室温下用含有10%小鼠bdfc阻断剂(bd)的5%fbs/pbs阻断10分钟,然后以终浓度0.4μg/样品向其中添加各种抗体(gr-1-fitc或cd117(ckit)-apc:biolegend)的任一种,并使混合物在冰上反应30分钟。然后,向其中添加死细胞染色染料dapi(0.2mg/ml)以使其达到1μl/样品,并且在遮蔽下在冰上再反应2分钟。然后,通过novocyte流式细胞仪(lms)测量细胞的每种表面抗原的表达水平。用flowjo软件(bectondickinson)分析获得的数据,并绘制除死细胞外的活细胞中表达每种表面抗原的细胞的比率。
实验例5的结果示于图10和图11。
图10是显示用实施例25、27、26或22的化合物处理7天后,活细胞中表达髓样细胞分化抗原gr-1的细胞的比率的图。纵轴表示活细胞中表达髓样细胞分化抗原gr-1的细胞的百分比,横轴表示各化合物及其浓度(nm)。由于与对照组相比,每种化合物均增加了gr-1阳性细胞的比率,因此证明了这些化合物具有诱导ma9细胞分化的作用。
图11是显示用实施例25、27、26或22的化合物处理7天后活细胞中表达ckit的细胞的比率的图。纵轴表示活细胞中表达ckit的细胞的百分比,横轴表示各化合物及其浓度(nm)。与对照组相比,每种化合物均降低了ckit阳性细胞的比率。ckit参与控制造血祖细胞的存活/分化/生长,并且在髓样细胞中的未成熟造血干祖细胞中特别高表达。从图10和图11的结果清楚地表明,这些化合物诱导ma9细胞的分化并降低了造血干祖细胞的比率。
[实验例6]2种药物的组合使用对细胞生长抑制活性的评价
补充有10%fbs的rpmi1640培养基用作molm-13细胞的培养基。这些细胞购自美国典型培养物保藏中心(atcc)。用10%fbsrpmi1640培养基制备molm-13细胞的悬浮液以使其达到25000个细胞/ml,并接种(50μl/孔)在96孔板上(第0天)。实施例25的化合物与各种其他药物(arac(阿糖胞苷)、5aza(阿扎胞苷)或维奈托克)中的任何一种的溶液使用培养液制备(实施例25的化合物:公比为4,由2500nm的最终浓度的5个浓度,arac:公比为2,由200nm的最终浓度的3个浓度,5aza:公比为2,由10000nm的最终浓度的3个浓度,维奈托克:公比为2,由156nm的最终浓度的3个浓度),将每种溶液以25μl/孔添加到每个孔中,并将混合物再培养7天。在添加药物的当天(第0天)和添加药物后的7天(第7天),将atp测量试剂(celltiter-glo(注册商标,promegacorporation)2.0检测,型号g9242,promegacorporation)以50μl/孔添加到每个孔中。用平板混合器搅拌2分钟后,使混合物在室温下静置10分钟或更长时间。然后,用酶标仪(n=4,型号名称envision2104multilabelreader,perkinelmerco.,ltd.)测量每个孔的发光水平。
当将无药物组的细胞生长视为100%时,使用excel2010(microsoftcorporation)对每种药物绘制从第0天至第7天的仅实施例25的化合物的处理组和实施例25的化合物与另一种药物的处理组的细胞生长(%)。
图12是显示实施例25的化合物和5aza的组合作用对人aml细胞系molm-13细胞的体外生长的作用的图。纵轴表示细胞生长(%),横轴表示实施例25的化合物的浓度(nm)。符号黑色圆圈表示仅实施例25的化合物,符号黑色三角形表示实施例25的化合物+5aza(2.5μm),符号黑色正方形表示实施例25的化合物+5aza(5μm),符号x表示实施例25的化合物+5aza(10μm)。误差条指示sd。
图13是显示实施例25的化合物和arac的组合作用对人aml细胞系molm-13细胞的体外生长的作用的图。纵轴表示细胞生长(%),横轴表示实施例25的化合物的浓度(nm)。符号黑色圆圈表示仅实施例25的化合物,符号黑色三角形表示实施例25的化合物+arac(25nm),符号黑色正方形表示实施例25的化合物+arac(50nm),符号x表示实施例25的化合物+arac(100nm)。误差条指示sd。
图14是显示实施例25的化合物和维奈托克的组合作用对人aml细胞系molm-13细胞的体外生长的作用的图。纵轴表示细胞生长,横轴表示实施例25的化合物的浓度(nm)。符号黑色圆圈表示仅实施例25的化合物,符号黑色三角形表示实施例25的化合物+维奈托克(39nm),符号黑色正方形表示实施例25的化合物+维奈托克(78nm),符号x表示实施例25的化合物+维奈托克(156nm)。误差条指示sd。
从这些结果清楚地表明,实施例25的化合物与各种药物中的任何一种的组合使用增强了aml细胞的生长抑制作用。
[实验例7]细胞生长抑制活性的评价
作为每种细胞的培养基,使用补充有10%fbs的rpmi1640培养基(molm-13细胞(人aml细胞))或补充有10%fbs的imdm培养基(k562细胞(人cml细胞),mv-4-11细胞(人aml细胞))。
用各培养基制备每种细胞的悬浮液以使其为25000个细胞/ml(k562,molm-13)或50000个细胞/ml(mv-4-11),并接种在96孔板(50μl/孔)中(第0天)。将药物(实施例131的化合物)的各浓度的溶液(mv-4-11和molm-13:公比为3,由1μm的最终浓度的9个浓度,k562:公比为3,由10μm的最终浓度的9个浓度)或含有0.2%dmso的生长培养基以50μl/孔加入到每个孔中,并将细胞再培养7天。在添加药物的当天(第0天)和添加药物后的7天(第7天),将atp测量试剂(celltiter-glo(注册商标,promegacorporation)2.0检测,型号g9242,promegacorporation)以100μl/孔添加到每个孔中。用平板混合器搅拌2分钟后,使混合物在室温下静置10分钟或更长时间。然后,用酶标仪(n=6,型号名称envision2104multilabelreader,perkinelmerco.,ltd.)测量每个孔的发光水平。
作为细胞生长抑制活性的指标,使用excel2010(microsoftcorporation)计算将细胞生长抑制50%的浓度(gi50)。当将无药物组(dmso组)的细胞生长视为100%时,将添加药物组从第0天至第7天的细胞生长计算为t/c%。通过sigmoidemax模型,将细胞生长抑制50%的浓度(t/c%=50%)计算为gi50。当通过sigmoidemax模型进行的分析未汇集(converge)时(k562细胞),使用夹有50%的两个浓度下的细胞活力,通过线性回归计算gi50。在该测试中,实施例131的化合物的gi50在mv-4-11细胞中为1.97nm,在molm-13细胞中为10.1nm,和在k562细胞中为9110nm。
工业适用性
由于本发明的由通式(1)表示的化合物或其药学上可接受的盐表现出对menin与mll蛋白之间的相互作用的抑制作用,因此其可用于治疗和/或预防依赖于menin与mll蛋白之间的相互作用的疾病。具体而言,本发明的由通式(1)表示的化合物或其药学上可接受的盐可用于治疗和/或预防癌症或糖尿病,优选骨髓增生异常综合征、血液癌、前列腺癌、乳腺癌、肝细胞瘤或小儿神经胶质瘤,更优选白血病。
- 还没有人留言评论。精彩留言会获得点赞!