治疗去势抵抗性和去势敏感性前列腺癌的方法与流程
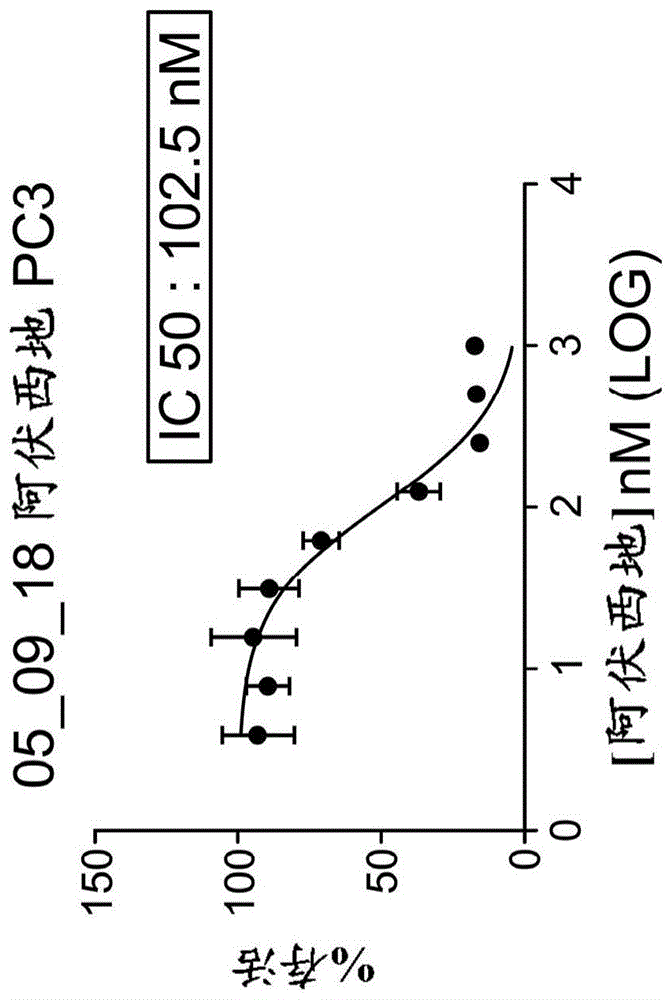
相关申请本申请要求于2019年10月25日提交的u.s.临时申请no.62/926,390、于2019年10月1日提交的u.s.临时申请no.62/909,147和于2018年12月7日提交的u.s.临时申请no.62/776,985的益处。上述申请的全部教导通过援引并入本文。发明概要简言之,本发明的实施方式提供治疗去势抵抗性前列腺癌的方法。本发明的其它实施方式提供治疗去势敏感性前列腺癌的方法。相应地,第一实施方式提供在有需要的受试者中治疗去势抵抗性前列腺癌的方法,包括向受试者给予有效量的具有下述结构(i)的化合物:或其药学上可接受的盐或两性离子形式。第二实施方式提供在有需要的受试者中抑制去势抵抗性前列腺癌进展的方法,包括向受试者给予有效量的具有结构(i)的化合物或其药学上可接受的盐或两性离子形式。第三实施方式提供在有需要的受试者中抑制去势抵抗性前列腺癌组织增殖的方法,包括向受试者给予有效量的具有结构(i)的化合物或其药学上可接受的盐或两性离子形式。第四实施方式提供在患前列腺癌的受试者中预防或抑制去势抵抗性前列腺癌发展的方法,所述方法包括向受试者给予有效量的具有结构(i)的化合物或其药学上可接受的盐或两性离子形式。第五实施方式提供在有需要的受试者中治疗去势敏感性前列腺癌的方法,包括向受试者给予有效量的具有结构(i)的化合物或其药学上可接受的盐或两性离子形式。第六实施方式提供在有需要的受试者中抑制去势敏感性前列腺癌进展的方法,包括向受试者给予有效量的具有结构(i)的化合物或其药学上可接受的盐或两性离子形式。第七实施方式提供在有需要的受试者中抑制去势敏感性前列腺癌组织增殖的方法,包括向受试者给予有效量的具有结构(i)的化合物或其药学上可接受的盐或两性离子形式。本发明实施方式的这些和其它方面将在参考下文详述后变得明显。为此,本文描述了各种参考文献其,其更为详细地描述某些背景信息、程序、化合物和/或组合物,并且通过援引将各自这些文献全部并入本文。附图说明专利或申请文件含有至少一个着色的附图。该专利或专利申请公开带有着色附图的拷贝将应要求且在支付所需费用后由专利局提供。在附图中,相同的标注数字指出相似的要素。图中要素的尺寸和相对位置不一定按比例绘制,并且这些要素中的一些经放大且调整位置以改善图的可辨认性。此外,所绘制的要素的特定形状并不期望提供关于该具体要素的真实形状的任何信息,并且仅是由于在图中易于识别而选用。图1a-1d显示在用阿伏西地治疗之后前列腺癌细胞系的存活性测试(pc3/图1a、vcap/图1b、lncap/图1c和22rv1/图1d),阿伏西地是结构(i)化合物及其药学上可接受的盐和两性离子形式的活性代谢物。图2a显示在血清刺激(在样品收集之前刺激1小时)之后,阿伏西地治疗(3和24小时)对22rv1细胞和lncap细胞中par(ser515/ser81)和arv7表达和总ar(tar)表达的效果。图2b显示在血清刺激(在样品收集之前刺激1时)之后,阿伏西地治疗(24小时)对22rv1细胞中parser81arv7和arv7蛋白质水平的效果。图3显示在血清刺激(在样品收集之前刺激1时)之后,阿伏西地治疗(3和24小时)对22rv1细胞中tmprss2表达的效果。图4显示在血清刺激(在样品收集之前刺激23小时&3小时)之后,阿伏西地治疗(3和24小时)对22rv1细胞中psa表达的效果。图5显示在整个22rv1异种移植研究中各组的平均肿瘤体积。图6显示在整个22rv1异种移植研究中各组的肿瘤体积,表示为肿瘤体积相对对照组的百分比。图7显示在整个22rv1异种移植研究中各组的肿瘤体积的平均百分比变化,表示为肿瘤体积平均百分比变化相对对照组的比率。图8显示在22rv1异种移植研究的第35天各组个体的肿瘤体积。图9显示在整个22rv1异种移植研究中各组的肿瘤生长的平均百分比抑制,与对照组比较。图10显示对于22rv1异种移植研究各组的肿瘤生长抑制的平均百分比变化,与对照组比较。图11显示在整个22rv1异种移植研究中各组的平均体重。图12显示在整个22rv1异种移植研究中各组的平均百分比体重变化。图13显示在22rv1异种移植研究的第35天组1-11(如表1中所定义)的个体体重。图14显示在22rv1异种移植研究的第35天组1-11(如表1中所定义)个体的百分比体重变化。图15显示在整个c4-2异种移植研究中各组的平均肿瘤体积。图16显示在整个lncap异种移植研究中各组的平均肿瘤体积。图17显示在整个c4-2异种移植研究中各组的平均体重。图18显示在整个lncap异种移植研究中各组的平均体重。图19显示在血清刺激之后阿伏西地治疗(3小时)对22rv1细胞中rnapolii磷酸化的效果。图20显示阿伏西地治疗(48小时)对vcap和lncap细胞中psa蛋白质水平的效果。图21显示阿伏西地治疗(48小时)对lncap细胞中细胞死亡的效果,如半胱天冬酶3裂解所指示。图22a显示在pc-3异种移植模型中在给予结构(i)化合物之后多至24小时结构(i)化合物的血浆浓度(上部小图)和肿瘤浓度(下部小图)。图22b显示在口服给予之后4小时结构(i)化合物在pc-3肿瘤中抑制mcl1,如免疫印迹所显示。图22c显示在pc-3小鼠异种移植模型中于1.25mg/kgbid×21,7.5mg/kgq7d×3或15mg/kgq7d×3口服给予的结构(i)化合物对肿瘤生长的效果。图23是图并且显示在实施例12中经同期组群5描述的研究的完整周期。图24a是血浆阿伏西地浓度(ng/ml)vs时间的图,并且显示在每日口服qd剂量给药含有配制剂no.401-01的1-mg强度胶囊之后的第1天阿伏西地在同期组群1患者血浆中的浓度。图24b是血浆阿伏西地浓度(ng/ml)vs时间的图,并且显示在每日口服qd剂量给药含有配制剂no.401-01的1-mg强度胶囊之后第14天阿伏西地在同期组群1患者血浆中的浓度。图24c是血浆阿伏西地浓度(ng/ml)vs时间的图,并且显示在每日口服bid剂量给药含有配制剂no.401-01的1-mg强度胶囊之后第1天阿伏西地在同期组群2患者血浆中的浓度。图24d是血浆阿伏西地浓度(ng/ml)vs时间的图,并且显示在每日口服bid剂量给药含有配制剂no.401-01的1-mg强度胶囊之后第14天阿伏西地在同期组群2患者血浆中的浓度。图24e是血浆阿伏西地浓度(ng/ml)vs时间的图,并且显示在每日口服bid剂量给药6mg配制剂no.401-01之后第1天阿伏西地在同期组群5患者血浆中的浓度。图24f是血浆阿伏西地浓度(ng/ml)vs时间的图,并且显示在每日口服bid剂量给药6mg配制剂no.401-01之后第14天阿伏西地在同期组群5患者血浆中的浓度。图24g是阿伏西地(ng/ml)vs同期组群的图,并且显示在每日口服qd剂量给药含有配制剂no.401-01的1-mg强度胶囊之后第1天和第14天阿伏西地的平均cmax。图24h是阿伏西地(ng*hr/ml)vs同期组群的图,并且显示在每日口服bid剂量给药含有配制剂no.401-01的1-mg强度胶囊之后第1天(auc0-8)和第14天(auc0-8和auc0-24)阿伏西地的曲线下面积(auc)。图24i是阿伏西地平均浓度(nm)vs时间的图,并且显示在24-小时时间段内阿伏西地在同期组群5患者血浆中的平均浓度。图25说明x射线粉末衍射(xrpd)图谱,其得自多晶型形式b的xrpd分析。图26显示多晶型形式b的热流对温度作图的差示扫描量热法输出。图27a是免疫印迹图,并且显示在描述于实施例3的雄激素独立性22rv1模型中裂解的半胱天冬酶3的量随治疗组的变化。图27b是免疫印迹图,并且显示在描述于实施例3的雄激素独立性22rv1模型中mcl-1的量随治疗组的变化。图27c是免疫印迹图,并且显示在描述于实施例3的雄激素独立性22rv1模型中裂解的半胱天冬酶3的量随治疗组的变化。图27d是免疫印迹图,并且显示在描述于实施例3的雄激素独立性22rv1模型中mcl-1的量随治疗组的变化。图27e是免疫印迹图,并且显示在描述于实施例3的22rv1模型中c-myc的量随治疗组的变化。图27f是柱状图,并且显示在图27e免疫印迹中描述的各治疗组中c-myc/肌动蛋白相对媒介物的比率。图27g是免疫印迹图,并且显示在描述于实施例3的22rv1模型中c-myc的量随治疗组的变化。图27h是柱状图,并且显示在图27g免疫印迹中描述的各治疗组中c-myc/肌动蛋白相对媒介物的比率。发明详述在下文的说明书中,描述某些特定细节以便提供对本发明各种实施方式的透彻理解。然而,本领域技术人员将理解本发明可以不用这些细节来实施。除非上下文另有要求,在本说明书和权利要求通篇中,措辞"包含"及其变型比如"含有"和"包涵"解释为开放式的包括含义,也即"包括,但不限于。"在说明书通篇中提及"一种实施方式"或"实施方式"意指该实施方式中描述的特定特征、结构或属性包括在本发明的至少一种实施方式中。从而,在说明书通篇各处中出现的短语"在一种实施方式中"或"在实施方式中"不一定全都指相同的实施方式。另外,特定特征或属性可以以任何适宜的方式在一种或多种实施方式中组合。如本文所用,术语"约"意指所指范围、值或结构的±20%(例如±10%、±5%或±1%),除非另有指定。"去势抵抗性前列腺癌"是指在给予一种或多种雄激素剥夺疗法(adts)之后在受试者中进展的前列腺癌。前列腺癌的进展能够通过下述证实:例如,小于或等于10个月的前列腺特异性抗原倍增时间(psadt)、先有的疾病(例如放射照像进展、临床进展、骨骼相关性事件、前列腺特异性抗原(psa)进展)的进展、和/或受试者中出现新转移,并且一般受雄激素(包括睾酮和二氢睾酮(dht)的一种激素类)驱动。这些雄激素结合至雄激素受体(ar),其是促进前列腺细胞包括前列腺癌细胞的生长和生存的转录活化剂。adt是指抑制雄激素水平(例如手术去势或化学去势)或雄激素信号转导(例如通过降低与雄激素受体的雄激素结合)的疗法,其可以用来减缓前列腺癌的进展。雄激素剥夺疗法一般导致肿瘤负担的暂时降低,伴随血清psa的降低。去势抵抗的机理包括出现不存在雄激素的活性ar变型,包括剪接变型、形成ar的点突变和ar基因放大。去势抵抗能够在症状发作之前在生物化学上通过血清psa的上升滴度表征(miller,etal.,1992j.urol.147,956961)。放射照像进展能够用连续成像来评价,并且通过例如骨骼扫描鉴定确认两个或更多个新骨骼损伤(根据prostatecancerclinicaltrialsworkinggroup2标准)证实。responseevaluationcriteriainsolidtumors(recistv1.1)标准还能够用来评价软组织损伤的放射照像进展。监测前列腺癌包括前列腺癌进展的指南描述于nccnclinicalpracticeguidelinesinoncology:prostatecancer,version4.2019,2019年8月19日,通过援引将其全部有关内容并入本文。"去势抵抗性前列腺癌"在本文中与"雄激素抵抗性前列腺癌"、"雄激素独立性前列腺癌"和"激素抵抗性前列腺癌"可互换地使用。"去势敏感性前列腺癌"是指在给予一种或多种adt之后不进展(例如响应)的前列腺癌。前列腺癌的进展能够根据本文描述的例如关于"去势抵抗性前列腺癌"的标准评价,并且监测前列腺癌包括前列腺癌进展的指南描述于nccnclinicalpracticeguidelinesinoncology:prostatecancer,version4.2019,2019年8月19日,通过援引将其全部有关内容并入本文。"去势敏感性前列腺癌"在本文中与"雄激素敏感性前列腺癌","雄激素依赖性前列腺癌"和"激素敏感性前列腺癌"可互换地使用。"癌"包括"肿瘤"是指细胞的不受控生长和/或异常增加的细胞存活和/或细胞凋亡的抑制,其干扰身体器官和系统的正常功能。"癌"(例如肿瘤)包括实体和非实体癌。患癌或肿瘤的受试者具有受试者身体中存在的可客观测量数量的癌细胞。"癌"包括良性和恶性癌(例如分别良性和恶性的肿瘤),以及休眠的肿瘤或微转移。"药物组合物"是指活性化合物比如结构(i)化合物或其药学上可接受的盐或两性离子形式和本领域一般接受用于将生物学活性化合物递送至哺乳动物例如人类的介质的配制剂。上述介质包括全部药学上可接受的载体,稀释剂或赋形剂。根据本发明的药物组合物的"有效量"是治疗有效量或预防有效量。"治疗有效量"是指以剂量和所需的时间段有效实现所希望的治疗结果的量,所述效果是比如缩小的肿瘤尺寸(例如肿瘤尺寸缩小5%、10%、15%或20%),增加的寿命,前列腺癌生物标记减少(例如psa水平减少0.1ng/ml、0.5ng/ml、1ng/ml或5ng/ml;或psa水平减少至少10%、至少15%、至少20%、至少25%、至少30%、至少40%或至少50%),受试者gleason得分降低(本领域普通技术人员已知的前列腺癌分级系统),或增加的预期寿命。化合物的治疗有效量可以根据因素比如受试者的疾病状态、年龄、性别和体重,和化合物在受试者中引起希望应答的能力而变化。可以调节给药方案以提供最佳的治疗反应。一般地,治疗有效量还是治疗有益效果超过化合物的任何毒性或有害效果的那些。在某些实施方式中,有效量是治疗有效量。"预防有效量"是指以剂量和所需的时间段有效实现所希望的预防结果的量,所述结果是比如肿瘤的延缓发作,增加的寿命,增加的预期寿命,预防或抑制去势抵抗性前列腺癌的发展,抑制去势抵抗性前列腺癌的进展,和/或抑制前列腺癌的转移。预防或抑制去势抵抗性前列腺癌的发展可以通过非去势抵抗性前列腺癌(即响应雄激素剥夺疗法的癌)并不进展成为去势抵抗性(即不响应雄激素剥夺疗法)或者具有进展为去势抵抗的延缓来证实。抑制前列腺癌进展可以通过例如下述来证实:肿瘤尺寸不增加,gleason得分不增加,受试者psa水平不增加,和/或不进展为转移的去势抵抗性前列腺癌(对于患未转移的去势抵抗性前列腺癌的受试者)。一般地,在患病之前或在疾病的更早阶段(例如在癌变得去势抵抗性之前)在受试者中使用预防剂量,从而预防有效量可以小于治疗有效量。在某些实施方式中,有效量是预防有效量。"治疗"或"治疗的"如本文所用涵盖治疗患有关疾病或病况的受试者例如哺乳动物优选人类中的有关疾病或病况,和包括:(i)抑制疾病或病况,例如减缓其进展,停止其发展;(ii)缓解疾病或病况,例如导致疾病或病况消退;和/或(iii)缓解疾病或病况导致的症状,例如缓解疼痛而不应对基础疾病或病况。如本文所用,术语"疾病"和"病况"可以互换地使用或可以是不同的,因为特定弊病(malady)或病况可以不具有已知的致病因素(从而并未建立病原学)和因此其并不被认为是疾病而仅是不希望的病况或综合征,其中临床医师已鉴定出或多或少特定的症状集合。"治疗效果"如本文所用涵盖如上文描述的治疗益处和/或预防益处。预防性效果包括延缓或消除疾病或病况的出现,延缓或消除疾病或病况症状的发作,减缓、停止或逆转疾病或病况的进展,或其任何组合。术语"共同给予"、"与...组合给予"及其语法等价物如本文所用涵盖向受试者比如动物包括人类给予两种或更多种试剂以治疗本文描述的疾病、障碍或病况。在某些实施方式中,两种或更多种试剂的给予使得试剂和/或其代谢物均同时存在于受试者中。共同给予包括同时在分开的组合物中给予,在分开的组合物中于不同的时间给予,或在存在两种或更多种试剂的组合物中给予。"抗癌剂"、"抗肿瘤剂"或"化疗剂"是指用于治疗瘤形成病况的任何试剂。一类抗癌剂包含化疗剂。"化疗"意指通过各种方法向癌症患者给予一种或多种化疗药物和/或其它试剂,所述方法包括静脉内、口服、肌内、腹腔内、膀胱内、皮下、透皮、颊或吸入或以栓剂形式。如本文所用,"受试者"是指动物。"受试者"可以是哺乳动物比如人类,非人灵长类,大鼠,小鼠,牛,马,猪,绵羊,山羊,犬,猫等。受试者可以被怀疑患有去势抵抗性前列腺癌或有患去势抵抗性前列腺癌的风险。去势抵抗性前列腺癌的临床描述是本领域普通技术人员已知的。"哺乳动物"包括人类和家养动物比如实验室动物和家居宠物(例如猫、犬、猪、牛、绵羊、山羊、马、兔),和非家养动物比如野生动物等。如本文所用,"疗法"是指任何癌症治疗(例如化疗、免疫治疗、靶向治疗、激素治疗、放疗)。在某些实施方式中,疗法是化疗。如本文所用,"一线治疗"是指对疾病或病况提供的第一种治疗。如本文所用,"后续治疗"是指在一线治疗之后对疾病或病况提供的任何治疗。在一线治疗包括药物的情况下,后续治疗包含不同于一线治疗药物的一种或多种药物。在某些实施方式中,后续治疗是二线治疗(也即对疾病或病况提供的第二种治疗)。在某些实施方式中,后续治疗是三线治疗(也即对疾病或病况提供的第三种治疗)。在某些实施方式中,后续治疗是四线治疗(也即对疾病或病况提供的第四种治疗)。用于本文描述的治疗(例如,包含一种或多种本文描述的化合物比如结构(i)化合物或其药学上可接受的盐或两性离子形式的治疗的在先治疗比如一线治疗;与一种或多种本文描述的化合物比如结构(i)化合物或其药学上可接受的盐或两性离子形式组合)试剂的实例包括darolutamide,阿帕鲁胺(apalutamide),恩杂鲁胺(enzalutamide),比卡鲁胺,多西他赛,泼尼松,阿比特龙(例如乙酸阿比特龙),甲基泼尼松,二氯化镭223lhrh激动剂(例如亮丙立德、戈舍瑞林、曲普瑞林、组氨瑞林),sipuleucel-tnivolumab,ipilumab,cetrelimab,canerpaturev,prostvac-v,prostvac-f,新抗原dna疫苗,紫杉醇(例如),卡铂,雷莫芦单抗,米托蒽醌和卡巴他赛,或前述任何的药学上可接受的盐,或前述的两种或更多种的组合。用于本文描述的治疗的其它试剂在本公开通篇描述。"放疗"意指用从业者已知的常规方法和组合物将受试者暴露至辐射发射体比如发射α-粒子的放射性核素(例如锕和钍放射性核素),低线性能量转移(let)辐射发射体(也即β发射体),转换电子发射体(例如锶-89和钐-153-edtmp),或高能辐射,包括但不限于x射线、γ射线和中子。本文描述受试者的治疗"失败"的条件是在给予所述治疗之后受试者经诊断患去势抵抗性前列腺癌。例如,用雄激素剥夺疗法的在先治疗可以导致去势抵抗性前列腺癌的发展。在这种情况下,描述受试者的雄激素剥夺疗法失败,原因是在给予雄激素剥夺疗法之后受试者经诊断患去势抵抗性前列腺癌。在又一实例中,预先经诊断患去势抵抗性前列腺癌的受试者可以用去势抵抗性前列腺癌疗法来治疗但不响应该治疗。同样描述该受试者的治疗失败,原因是在给予该治疗后受试者经诊断患去势抵抗性前列腺癌。术语"体内"是指受试者身体内部发生的事件。本文公开的本发明实施方式也意在涵盖全部药学上可接受的结构(i)化合物及其药学上可接受的盐和两性离子形式,其是通过用具有不同的原子质量或质量数的原子替换一个或多个原子从而同位素标记的。能够掺入本公开化合物的同位素的实例包括氢、碳、氮、氧、磷、氟、氯和碘的同位素,各自比如2h、3h、11c、13c、14c、13n、15n、15o、17o、18o、31p、32p、35s、18f、36cl、123i和125i。这些同位素标记的化合物能通过表征例如作用位点或作用模式或与药理学重要的作用位点的结合亲和力用来帮助确定或测量化合物的有效性。某些同位素标记的结构(i)化合物及其药学上可接受的盐和两性离子形式,例如掺入放射性同位素的那些,用于药物和/或底物组织分布研究。放射性同位素氚即3h和碳-14即14c对于该目的特别有用,原因是它们掺入容易且检测手段方便。用更重的同位素比如氘即2h取代可以提供某些治疗优势,原因是更高的代谢稳定性例如增加的体内半衰期或降低的剂量要求,和于是可以在某些环境中优选。用正电子发射同位素比如11c、18f、15o和13n取代能够用于检查底物受体占有率的正电子发射断层摄影术(pet)研究。同位素标记的结构(i)化合物及其药学上可接受的盐和两性离子形式能够一般通过本领域技术人员已知的常规技术用适当的同位素标记的试剂代替原先使用的非标记的试剂来制备。"结晶"如本文所用是指通过组成部分之间具有固定距离的重复三维模式的原子、离子或分子形成的均质固体。晶胞是该模式中的最简重复单元。虽然理想晶体性质均匀,但完美晶体很少存在(如果存在)。"结晶"如本文所用涵盖包括结晶缺陷的晶型,例如一般通过操作(例如制备、纯化)本文描述的晶型而形成的结晶缺陷。虽然存在所述缺陷,但本领域技术人员能够确定化合物样品是否是结晶。在本文描述的化合物(例如晶型)的某些实施方式中,化合物是实质上纯的。如本文所用"实质上纯"在不加进一步品质限定的情况下意指所指的化合物具有大于90重量百分比例如大于90、91、92、93、94、95、96、97、98、或99重量百分比的纯度,以及包括等于约100重量百分比的纯度,按化合物的重量计。剩余物质包含化合物的其它形式,和/或其制备引起的反应杂质和/或处理杂质(例如阿伏西地)。纯度能够用本领域已知的技术评价,例如用hplc测试。"实质上纯"还能够满足"关于具有结构(i)的化合物或其药学上可接受的盐或两性离子形式的其他物理形式实质上纯"或"关于阿伏西地实质上纯"的条件。从而在满足条件的情况下,"实质上纯"意指所指化合物含有小于10%,优选小于5%,更优选小于3%,最优选小于1%重量的所指杂质(例如所指化合物晶型的任何其它物理形式;阿伏西地)。如本文所用,术语"阿伏西地"意指2-(2-氯苯基)-5,7-二羟基-8-[(3s,4r)-3-羟基-1-甲基哌啶-4-基]色烯-4-酮或其盐(例如药学上可接受的盐)(例如,2-(2-氯苯基)-5,7-二羟基-8-[(3s,4r)-3-羟基-1-甲基哌啶-4-基]色烯-4-酮盐酸盐)。"多晶型"如本文所用是指化合物晶型,特征为其分子在晶格中的不同排列。多晶型能够通过分析方法比如x射线粉末衍射(xrpd)、差示扫描量热法(dsc)和热重分析表征。"实质上按照"本文一个或多个分别显示xrpd图样或衍射图或者dsc热分析图的附图的xrpd图谱或dsc热分析图会被本领域技术人员视为代表具有结构(i)的化合物或其药学上可接受的盐或两性离子形式的相同单一晶型,作为具有结构(i)的化合物或其药学上可接受的盐或两性离子形式的样品,用来提供本文提供的一个或多个附图的图样或衍射图或者热分析图。从而,实质上按照的附图的xrpd图谱或dsc热分析图可以与各图之一相同或者更可能可以在某种程度不同于附图中的一个或多个。例如,某种程度不同于附图中一个或多个的xrpd图谱可以不一定显示本文所示的衍射图谱的每一条线和/或可以显示线的外观或强度的轻微变化或线位置的位移。这些差异一般来自与数据获得有关的条件差异或用来获得数据的样品的纯度差异。通过比较样品的xrpd图谱或dsc热分析图和本文公开的相应xrpd图谱或dsc热分析图,本领域技术人员能够确定结晶化合物的样品是否是本文公开形式的相同形式或不同形式。本文提供的晶型还能够基于差示扫描量热法(dsc)和/或热重分析(tga)鉴定。dsc是热分析技术,其中测量提升样品温度所需的热量差异随温度的变化。dsc能够用来检测样品的物理转化比如相变。例如,dsc能够用来检测样品发生结晶、熔化或玻璃化转变的温度。应理解与本文所指定的dsc有关的任何温度,除图或实例中的dsc温度以外,意味着所指定值±5℃或更少。例如,在实施方式或权利要求指定吸热峰在264℃,这应理解为意指264℃±5℃或更少,即259℃至269℃的温度。在优选的实施方式中,dsc温度是所指定值±3℃或更少,在更优选的实施方式中,dsc温度是所指定值±2℃或更少。"药学上可接受的载体、稀释剂或赋形剂"包括但不限于任何助剂,载体,赋形剂,助流剂,甜味剂,稀释剂,防腐剂,染料/着色剂,调味剂增强剂,表面活性剂,润湿剂,分散剂,助悬剂,稳定剂,等渗剂,溶剂或乳化剂,其已被美国食品和药物局批准为在人类或家养动物使用可接受的。"药学上可接受的盐"包括酸和碱加成盐。"药学上可接受的酸加成盐"是指保留游离碱的生物学有效性和在生物学上或其它方面并非不希望的特性并且与无机酸和有机酸形成的那些盐,所述无机酸是比如但不限于盐酸,氢溴酸,硫酸,硝酸,磷酸等,而所述有机酸是比如但不限于乙酸,2,2-二氯乙酸,己二酸,藻酸,抗坏血酸,天冬氨酸,苯磺酸,苯甲酸,4-乙酰胺基苯甲酸,樟脑酸,樟脑-10-磺酸,癸酸,己酸,辛酸,碳酸,肉桂酸,柠檬酸,环己氨磺酸,十二烷基硫酸,乙烷-1,2-二磺酸,乙磺酸,2-羟基乙磺酸,甲酸,富马酸,粘酸,龙胆酸,葡庚糖酸,葡萄糖酸,葡糖醛酸,谷氨酸,戊二酸,2-氧代-戊二酸,甘油磷酸,羟基乙酸,马尿酸,异丁酸,乳酸,乳糖酸,月桂酸,马来酸,苹果酸,丙二酸,苦杏仁酸,甲磺酸,粘酸,萘-1,5-二磺酸,萘-2-磺酸,1-羟基-2-萘甲酸,烟酸,油酸,乳清酸,草酸,棕榈酸,双羟萘酸,丙酸,焦谷氨酸,丙酮酸,水杨酸,4-氨基水杨酸,癸二酸,硬脂酸,琥珀酸,酒石酸,硫氰酸,对-甲苯磺酸,三氟乙酸,十一烯酸,等。"药学上可接受的碱加成盐"是指保留游离酸的生物学有效性和在生物学上或其它方面并非不希望的特性的那些盐。这些盐制备自向游离酸加入无机碱或有机碱。衍生自无机碱的盐包括但不限于钠盐,钾盐,锂盐,铵盐,钙盐,镁盐,铁盐,锌盐,铜盐,锰盐,铝盐等。优选的无机盐铵盐,钠盐,钾盐,钙盐和镁盐。衍生自有机碱的盐包括但不限于伯、仲和叔胺,取代的胺包括天然取代的胺、环状胺和碱性离子交换树脂,比如氨,异丙胺,三甲胺,二乙胺,三乙胺,三丙胺,二乙醇胺,乙醇胺,地阿诺(deanol),2-二甲基氨基乙醇,2-二乙基氨基乙醇,二环己基胺,赖氨酸,精氨酸,组氨酸,咖啡因,普鲁卡因,海巴明(hydrabamine),胆碱,甜菜碱,苯乙苄胺,n,n'-二苄基乙二胺,乙二胺,氨基葡萄糖,甲基葡萄糖胺,可可碱,三乙醇胺,氨丁三醇,嘌呤类,哌嗪,哌啶,n-乙基哌啶,多元胺树脂等的盐。特别优选的有机碱是异丙胺,二乙胺,乙醇胺,三甲胺,二环己基胺,胆碱和咖啡因。"两性离子形式"是指结构(i)化合物的形式,其中至少一个官能团具有正电荷,至少一个官能团具有负电荷,且整个分子的净电荷是零。例如,结构(i)化合物的磷酸基团(-po3h2)可以以阴离子形式存在(例如-po3h-),和结构(i)化合物的氮原子可以以质子化(阳离子)形式存在。具有结构(ii)的化合物:例如是具有结构(i)的化合物的两性离子形式。实施方式包括结构(i)化合物的两性离子及其晶型和多晶型。"互变异构体"是指质子从分子的一个原子移动至相同分子的又一个原子。本发明的实施方式包括结构(i)化合物的互变异构体,即使未特别说明或指定。i.方法在各种实施方式中,本发明提供通过向受试者给予结构(i)化合物或其药学上可接受的盐或两性离子形式或包含其的药物组合物在有需要的受试者中治疗去势抵抗性前列腺癌的方法。在其它实施方式中,本发明提供通过向受试者给予结构(i)化合物或其药学上可接受的盐或两性离子形式或包含其的药物组合物在有需要的受试者中治疗去势敏感性前列腺癌的方法。在第一实施方式中,本发明提供在有需要的受试者中治疗去势抵抗性前列腺癌的方法,所述方法包括向受试者给予有效量的具有下述结构(i)的化合物:或其药学上可接受的盐或两性离子形式。在第二实施方式中,本发明提供在有需要的受试者中抑制去势抵抗性前列腺癌进展的方法,所述方法包括向受试者给予有效量的具有下述结构(i)的化合物或其药学上可接受的盐或两性离子形式。在第三实施方式中,本发明提供在有需要的受试者中抑制去势抵抗性前列腺癌组织增殖的方法,所述方法包括向受试者给予有效量的具有下述结构(i)的化合物或其药学上可接受的盐或两性离子形式。在第四实施方式中,本发明提供在有需要的受试者中治疗去势敏感性前列腺癌的方法,所述方法包括向受试者给予有效量的具有下述结构(i)的化合物或其药学上可接受的盐或两性离子形式。在第五实施方式中,本发明提供在有需要的受试者中抑制去势敏感性前列腺癌进展的方法,所述方法包括向受试者给予有效量的具有下述结构(i)的化合物或其药学上可接受的盐或两性离子形式。在第六实施方式中,本发明提供在有需要的受试者中抑制去势敏感性前列腺癌组织增殖的方法,所述方法包括向受试者给予有效量的具有下述结构(i)的化合物或其药学上可接受的盐或两性离子形式。在第七实施方式中,本发明提供在患前列腺癌的受试者中预防或抑制去势抵抗性前列腺癌发展的方法,所述方法包括向受试者给予具有下述结构(i)的化合物或其药学上可接受的盐或两性离子形式。例如在受试者用雄激素剥夺疗法治疗的情况下可以发生去势抵抗的发展,其基于机理例如:雄激素受体(例如雄激素受体变型7,其是缺少雄激素结合区域的活性变型)的选择性剪接,雄激素受体中的点突变,和/或雄激素受体基因的扩增。在实施方式一至七的某些方面中,受试者已被预先给予雄激素剥夺疗法(也即在给予结构(i)化合物或其药学上可接受的盐或两性离子形式之前)。雄激素剥夺疗法的实例包括外科去势,化学去势(例如用促性腺激素释放激素(gnrh)激动剂比如亮丙瑞林、戈舍瑞林、曲普瑞林、组氨瑞林、布舍瑞林治疗;用gnrh拮抗剂比如地加瑞克治疗),用雄激素受体(ar)拮抗剂治疗和用雄激素受体信号转导抑制剂治疗。在实施方式一至七的某些方面中,所述受试者已被预先给予雄激素受体信号转导抑制剂。如本文所用,"雄激素受体信号转导抑制剂"是指抑制雄激素受体信号转导途径的试剂。雄激素受体信号转导抑制剂的实例包括雄激素受体拮抗剂,比如本文描述的那些。在某些实施方式中,雄激素受体信号转导抑制剂是阿比特龙,阿帕鲁胺或恩杂鲁胺。在实施方式一至七的某些方面中,所述受试者已被预先给予雄激素受体(ar)拮抗剂。ar拮抗剂的实例包括阿比特龙,阿帕鲁胺,恩杂鲁胺,氟他胺,乙酸环丙孕酮,比卡鲁胺,尼鲁米特,arn-509,azd-3514,ezn-4176,odm-201,和tok-001(例如阿比特龙、阿帕鲁胺、恩杂鲁胺)。在实施方式一至七的某些方面中,所述受试者已被预先给予包含阿比特龙,阿帕鲁胺,恩杂鲁胺,氟他胺,乙酸环丙孕酮,比卡鲁胺,尼鲁米特,arn-509,azd-3514,ezn-4176,odm-201或tok-001,或其任何组合(例如阿比特龙,阿帕鲁胺,恩杂鲁胺)的治疗。在实施方式一至七的某些方面中,结构(i)化合物或其药学上可接受的盐或两性离子形式作为一线治疗给予(例如作为单一疗法,在联合疗法中)。在实施方式一至七的某些方面中,结构(i)化合物或其药学上可接受的盐或两性离子形式作为在先治疗(例如一线治疗)之后的后续治疗(例如二线治疗)给予,例如作为单一疗法、在联合疗法中给予,所述在先治疗是比如雄激素剥夺疗法和/或包括雄激素受体信号转导抑制剂(例如雄激素受体拮抗剂比如恩杂鲁胺、阿帕鲁胺或阿比特龙)的治疗。在某些方面,受试者的在先治疗(例如一线治疗)已失败。在实施方式一至三的特定方面中,所述方法是在有需要的受试者中治疗转移的去势抵抗性前列腺癌的方法,并且包括向受试者给予有效量的结构(i)化合物或其药学上可接受的盐或两性离子形式,其中受试者的包含雄激素受体信号转导抑制剂或紫杉烷的在先治疗(例如一线治疗)已失败。在又一方面,受试者没有内脏损伤。在还又一方面,结构(i)化合物或其药学上可接受的盐或两性离子形式作为后续治疗(例如二线治疗)给予。在本文描述的化合物或试剂描述为作为治疗(例如后续治疗、在先治疗)给予的情况下,应理解所指出的治疗包括所描述的化合物或试剂。例如,在结构(i)化合物或其药学上可接受的盐或两性离子形式作为后续治疗给予的情况下,所述后续治疗能够是牵涉结构(i)化合物或其药学上可接受的盐或两性离子形式的单一疗法,或牵涉结构(i)化合物或其药学上可接受的盐或两性离子形式的联合疗法。换言之,本文描述的方法能够包括向有需要的受试者给予包含有效量的结构(i)化合物或其药学上可接受的盐或两性离子形式的治疗(比如说,例如在先治疗之后的后续治疗比如二线治疗)。在第七实施方式的某些方面中,受试者已预先经诊断患前列腺癌,但并未预先经诊断患去势抵抗性前列腺癌。例如,受试者可以最近经诊断患前列腺癌,并且仍未接受雄激素剥夺疗法。受试者可以或可以不同时用雄激素剥夺疗法处理。在实施方式一至七中的某些中,结构(i)化合物作为药学上可接受的盐提供。在其它实施方式中,结构(i)化合物不是盐,例如具有结构(i)或其两性离子形式并且不包括酸或碱平衡离子。在某些实施方式中,结构(i)化合物具有下述结构(ii):在实施方式一至七的某些方面中,受试者具有与去势抵抗有关的雄激素受体变型(例如预先确定的雄激素受体变型),比如点突变或剪接变型。与前列腺癌成为去势抵抗性有关的雄激素受体点突变的实例包括f977l和t878a。与前列腺癌成为去势抵抗性有关的雄激素受体剪接变型的实例包括雄激素受体v7剪接变型,雄激素受体v3剪接变型,雄激素受体v9剪接变型,和雄激素受体v12剪接变型。各种剪接变型比如v7变型和v12变型的外显子应用能够参见例如dehm,s.&tindalld.,endocrrelatcancer.2011oct;18(5):r183-r196。检测剪接变型的方法一般是本领域普通技术人员已知的并且能够参见例如londono,j.,&philipp,s.,bmcmolbiol.2016;17:8doi:10.1186/s12867-016-0060-1。在特别的实施方式中,受试者具有雄激素受体v7剪接变型。在实施方式一至七的方面中,前列腺癌(例如去势抵抗性前列腺癌)是转移的。在实施方式一至七的其它方面中,前列腺癌(例如去势抵抗性前列腺癌)是未转移的。在实施方式一至七的某些方面中,方法还包括监测受试者的前列腺特异性抗原(psa)水平。在年龄依赖性阈值之下的稳定或降低的psa水平(正常psa水平随年龄增加)可以指示有效的治疗。在psa水平保持稳定在低水平(例如低于4.0ng/ml)时,这能够指出治疗有效和/或前列腺癌并未进展。如果psa水平在治疗期间稳定化(例如保持低于4.0ng/ml)、然后开始上升,这可以指出前列腺癌已变得去势抵抗性。在实施方式一至七的某些方面中,方法还包括在给予具有结构(i)的化合物或其药学上可接受的盐或两性离子形式之前检测受试者的psa水平。在实施方式一至七的某些方面中,方法还包括在给予具有结构(i)的化合物或其药学上可接受的盐或两性离子形式之后检测受试者的psa水平。在实施方式一至七的某些方面中,方法还包括在给予具有结构(i)的化合物或其药学上可接受的盐或两性离子形式之前,和在给予具有结构(i)的化合物或其药学上可接受的盐或两性离子形式之后检测受试者的psa水平。在实施方式一至七的某些方面中,与在给予具有结构(i)的化合物或其药学上可接受的盐或两性离子形式之前相比,受试者的psa水平在给予具有结构(i)的化合物或其药学上可接受的盐或两性离子形式之后低至少10%(例如至少15%,至少20%,至少25%,至少30%,至少40%,至少50%)。在某些实施方式中,前列腺癌是mcl-1依赖性的。如本文所用,"mcl-1-依赖性"是指癌亚型,其中骨髓细胞白血病1(mcl-1)是抑制细胞凋亡的主要驱动因素。一般来说,mcl-1依赖性促进癌的存活,并且与治疗抵抗和复发有关。mcl-1依赖性能够例如通过将受试者癌细胞与测试肽接触来评价,如国际公开nos.wo2016/172214和wo2018/119000的描述,通过援引将其全部有关内容并入本文。在某些实施方式中,癌是c-myc-改变的。如本文所用,"c-myc-改变的"是指癌亚型,其中c-myc相对其天然序列被改变,其中其表达相对适当对照(例如相应正常细胞)被放大,并且其中蛋白质水平提示c-myc的过表达。例如,已发现c-myc驱动前列腺癌中的雄激素独立性,并且过表达减弱雄激素受体抑制的抗肿瘤活性。此外,c-myc在雄激素受体敏感性前列腺癌中显著上调。能够是c-myc-改变的癌的实例包括但不限于淋巴瘤(例如伯基特淋巴瘤,b-细胞淋巴瘤,t-细胞淋巴瘤),宫颈癌,结肠癌,卵巢癌,乳腺癌,肺癌,前列腺癌,结直肠癌,胰癌,胃癌和子宫癌。在全部前述方法的某些特定实施方式中,方法包括向受试者口服给予结构(i)化合物或其药学上可接受的盐或两性离子形式,或包含其的药物组合物。本发明的某些方面使用包含结构(i)化合物或其药学上可接受的盐或两性离子形式和药学上可接受的赋形剂和/或载体的组合物。本文描述的方法包括给予如本文描述的结构(i)化合物或其药学上可接受的盐或两性离子形式,或如本文描述的结构(i)化合物或其药学上可接受的盐或两性离子形式的组合物(例如有效量的组合物),或如本文描述的有效量的结构(i)化合物或其药学上可接受的盐或两性离子形式。结构(i)化合物及其药学上可接受的盐和两性离子形式能够通过将磷酸基团加至阿伏西地的游离羟基制备,如u.s.专利公开no.us2016/0340376的描述,通过援引将其全部公开并入本文。应注意结构(i)化合物或其药学上可接受的盐或两性离子形式的剂量可以在不同的实施方式中变化。对于任何特定的受试者,可以根据个体需要和给予或监督给予组合物的人士的专业判断来随时间调节特定的给药方案。本文描述的特定的剂量和剂量范围仅是示范性并且不限制医学从业者可以选择的剂量和剂量范围。结构(i)化合物或其药学上可接受的盐或两性离子形式在组合物中的量可以根据因素比如受试者的疾病状态、年龄、性别和体重变化。可以调节给药方案以提供最佳治疗反应。例如,可以随时间给予数个分开的剂量,或者剂量可以治疗情况的急迫性成比例地减少或增加。在某些实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式可以例如但不限于与一种或多种额外的前列腺癌治疗组合使用。例如,如本文描述的额外的治疗可以作为新辅助(在先)治疗、附加(在其间)治疗、和/或辅助(在后)治疗与手术、辐射(近程放射治疗或外部光束)、高强度聚焦超声(hifu)、雄激素剥夺(即雄激素消融)或任何其它治疗途径使用。在实施方式一至七的某些方面中,受试者被给予一种或多种额外的治疗。在特别的实施方式中,一种或多种额外的治疗是:睾丸切除术,辐射,高光束聚焦超声(hifu),和/或一种或多种额外的有抗癌活性的治疗剂。关于组合治疗,本公开的一种实施方式提供任何一种或多种的结构(i)化合物或其药学上可接受的盐或两性离子形式与用来或可以用来治疗前列腺癌(例如去势抵抗性前列腺癌)的一种或多种目前使用的或实验的额外治疗的组合。还提供包含上述组合的方法、用途和药物组合物。相应地,一种实施方式包含结构(i)化合物或其药学上可接受的盐或两性离子形式与一种或多种具有抗癌活性的药理学治疗在联合疗法中的用途,不受所述药理学治疗的生物学作用机理所限制,包括但不限于直接或间接抑制雄激素受体的药理学治疗(例如雄激素剥夺疗法),实质为细胞毒性的药理学治疗,和干扰雄激素生物学产生或功能的药理学治疗(此后称为"额外治疗剂")。"联合疗法"意指将任何一种或多种结构(i)化合物或其药学上可接受的盐或两性离子形式,和一种或多种额外治疗剂给予相同受试者。在实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式和一种或多种额外治疗剂的药理学效果彼此同时,或如果并不同时则彼此增效(尽管依次而不是同时给药)。所述给药包括但不限于一种或多种结构(i)化合物或其药学上可接受的盐或两性离子形式和一种或多种额外治疗剂作为分开的试剂剂量给药而在剂量给药之前不进行任何混合,以及作为配制剂剂量给药,其包括作为预混配制剂的一种或多种额外治疗剂和与之混合的一种或多种结构(i)化合物或其药学上可接受的盐或两性离子形式。与额外治疗剂组合给予结构(i)化合物或其药学上可接受的盐或两性离子形式以治疗上述疾病状态也包括通过任何剂量给药方法剂量给药,包括但不限于静脉内递送、口服递送、腹膜内递送、肌内递送或肿瘤内递送。在本公开的又一方面,一种或多种额外治疗剂可以在给予结构(i)化合物或其药学上可接受的盐或两性离子形式之前向受试者给予。在又一实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式可以与一种或多种额外治疗剂共同给予。在又一方面,一种或多种额外治疗剂可以在给予结构(i)化合物或其药学上可接受的盐或两性离子形式之后向受试者给予。完全属于本公开范围的是结构(i)化合物或其药学上可接受的盐或两性离子形式的剂量与一种或多种额外治疗剂的剂量比率可以或可以不等于1并且可以相应地变化以实现最佳的治疗益处。额外的治疗包括但不限于具有抗癌效果的任何药理学试剂。例如,额外治疗剂可以包含烷基化剂,比如苯丁酸氮芥,环磷酰胺,顺铂;有丝分裂抑制剂比如多西他赛(紫杉特尔;1,7β,10β-三羟基-9-氧代-5β,20-桥氧基紫衫-11-烯-2α,4,13α-三基4-乙酸酯2-苯甲酸酯13-{(2r,3s)-3-[(叔丁氧羰基)氨基]-2-羟基-3-苯基丙酸酯})或紫杉醇;抗代谢药比如5-氟尿嘧啶,阿糖胞苷,甲氨蝶呤,或培美曲塞;抗肿瘤抗生素比如柔红霉素或多柔比星;皮质类固醇比如泼尼松或甲基泼尼松;或bcl-2抑制剂比如维奈托克(venetoclax)。在全部实施方式(例如实施方式四至七)的某些方面中,额外治疗剂是多西他赛。多西他赛(商品名)是一类称为抗微管试剂的化疗剂。多西他赛用于治疗各种癌,比如转移的前列腺癌。多西他赛治疗常常静脉内给予,和常常包括皮质类固醇比如泼尼松的术前用药。在全部实施方式(例如实施方式一至三和七)的某些方面中,额外治疗剂是维奈托克(gdc-0199,abt199,rg7601,商品名或),其是能够诱导癌细胞中的细胞凋亡的bcl-2抑制剂。维奈托克一般口服给予。额外治疗剂可以是目前由食品和药物局(fda)在美国(或在其他地区由任何其它规则机构)批准用作前列腺癌的药理学治疗,或目前正在实验中用作涉及前列腺癌的临床试验计划的一部分的药理学试剂。例如,额外治疗剂可以包含但不限于:称为恩杂鲁胺或mdv3100(4-(3-(4-氰基-3-(三氟甲基)苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-甲基苯甲酰胺)的化学物质和有关的化合物;称为tok001的化学物质和有关的化合物;称为arn-509的化学物质;称为阿比特龙(或cb-7630;(3s,8r,9s,10r,13s,14s)-10,13-二甲基-17-(吡啶-3-基)2,3,4,7,8,9,10,11,12,13,14,15-十二氢-1h-环戊二烯并[a]菲-3-醇)的化学物质和有关的分子;称为比卡鲁胺(n-[4-氰基-3-(三氟甲基)苯基]-3-[(4-氟苯基)磺酰基]-2-羟基-2-甲基丙酰胺)的化学物质和有关的化合物;称为尼鲁米特(5,5-二甲基-3-[4-硝基-3-(三氟甲基)苯基]咪唑烷-2,4-二酮)的化学物质和有关的化合物;称为氟他胺(2-甲基-n-[4-硝基-3-(三氟甲基)苯基]-丙酰胺)的化学物质和有关的化合物;称为乙酸环丙孕酮(6-氯-1β,2β-二氢-17-羟基-3′h-环丙烯并[1,2]孕-4,6-二烯-3,20-二酮)的化学物质和有关的化合物,其目前用来治疗前列腺癌,称为多西他赛的化学物质和有关的化合物,其目前单独或与泼尼松组合用来治疗前列腺癌,称为贝伐珠单抗的化学物质(avastin),可以用来治疗前列腺癌的单克隆抗体,称为osu-hdac42((s)-(+)-n-羟基-4-(3-甲基-2-苯基丁酰氨基)-苯甲酰胺)的化学物质和有关的化合物;称为vitaxin的化学物质,其可以用来治疗前列腺癌,称为舒尼替尼(n-(2-二乙基氨基乙基)-5-[(z)-(5-氟-2-氧代-1h-吲哚-3-亚基)甲基]-2,4-二甲基-1h-吡咯-3-甲酰胺)的化学物质和有关的化合物,其可以用来治疗前列腺癌,称为zd-4054(n-(3-甲氧基-5-甲基吡嗪-2-基)-2-[4-(1,3,4-噁二唑-2-基)苯基]吡啶-3-磺酰胺)的化学物质和有关的化合物;称为vn/124-1(3β-羟基-17-(1h-苯并咪唑-1-基)雄-5,16-二烯)的化学物质和有关的化合物;称为卡巴他赛(xrp-6258)的化学物质和有关的化合物;称为mdx-010(伊匹木单抗)的化学物质;称为ogx427的化学物质;称为ogx011的化学物质;称为非那雄胺(proscar,propecia;n-(1,1-二甲基乙基)-3-氧代-(5α,17β)-4-氮杂雄-1-烯-17-甲酰胺)的化学物质和有关的化合物;称为度他雄胺(avodart;5α,17β)-n-{2,5二(三氟甲基)苯基}-3-氧代-4-氮杂雄-1-烯-17-甲酰胺)的化学物质和有关的化合物;称为妥罗雄脲((4ar,4bs,6as,7s,9as,9bs,11ar)-1,4a,6a-三甲基-2-氧代-n-(丙-2-基)-n-(丙-2基氨基甲酰基)十六氢-1h-茚并[5,4-f]喹啉-7-甲酰胺)的化学物质和有关的化合物;称为贝氯特来(ly-191,704;(4as,10br)-8-氯-4-甲基-1,2,4a,5,6,10b-六氢苯并[f]喹啉-3-酮)的化学物质和有关的化合物;称为艾宗特来(ly-320,236;(4ar,10br)-8-[(4-乙基-1,3-苯并噻唑-2-基)硫烷基]-4,10b-二甲基-1,4,4a,5,6,10b-六氢苯并[f]喹啉-3(2h)-酮)的化学物质和有关的化合物;称为fce28260的化学物质和有关的化合物;称为skf105,111的化学物质和有关的化合物;称为azd3514的化学物质;称为ezn-4176的化学物质;称为odm-201,sipuleucel-t,卡巴他赛的化学物质;贝伐珠单抗,多西他赛,沙利度胺和泼尼松的组合;和/或阿比特龙。在全部实施方式的某些方面中,额外治疗剂是阻断雄激素结合至雄激素受体的雄激素受体拮抗剂。阻断雄激素结合至雄激素受体的治疗的实例包括恩杂鲁胺和阿帕鲁胺。在特别的实施方式中,额外治疗剂是恩杂鲁胺。恩杂鲁胺(商品名)是用于治疗未转移的去势抵抗性前列腺癌和转移的去势抵抗性前列腺癌的雄激素受体(ar)拮抗剂。恩杂鲁胺治疗可以与去势(手术或化学)组合。在全部实施方式的某些方面中,额外治疗剂是阿比特龙。阿比特龙(商品名)是cyp17a1抑制剂,其显著降低睾酮产生。阿比特龙治疗可以与其它额外的治疗比如皮质类固醇(例如泼尼松)和/或去势(手术或化学)组合。在全部实施方式的某些方面中,额外治疗剂选自下述的至少一种:布罗莫结构域抑制剂,组蛋白甲基转移酶抑制剂,组蛋白脱乙酰基酶抑制剂,或组蛋白脱甲基酶抑制剂。在全部实施方式的某些方面中,额外治疗剂是布罗莫结构域抑制剂,例如布罗莫结构域蛋白质比如brd2,brd3,brd4和/或brdt的抑制剂。在特别的实施方式中,额外治疗剂包含brd4抑制剂。在这些实施方式的某些中,额外治疗剂是jq-1(nature2010dec23;468(7327):1067-73),bi2536(acschem.biol.2014may16;9(5):1160-71;boehringeringelheim),tg101209(acschem.biol.2014may16;9(5):1160-71),otx015(mol.cancerther.november201312;c244;oncoethix),ibet762(jmedchem.2013oct10;56(19):7498-500;glaxosmithkline),ibet151(bioorg.med.chem.lett.2012apr15;22(8):2968-72;glaxosmithkline),pfi-1(j.med.chem.2012nov26;55(22):9831-7;cancerres.2013jun1;73(11):3336-46;structuralgenomicsconsortium)或cpi-0610(constellationpharmaceuticals)。在其它实施方式中,brd抑制剂是ibet762(gsk525762),十-010(tenshatherapeutics),cpi-203(leukemia.28(10):2049-59,2014),rvx-208(proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica.110(49):19754-9,2013),ly294002(acschemicalbiology.9(2):495-502,2014),azd5153(journalofmedicinalchemistry.59(17):7801-17,2016),mt-1(naturechemicalbiology.12(12):1089-10962016)或ms645(proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica.115(31):7949-7954,2018)。在全部实施方式的某些方面中,额外治疗剂是组蛋白甲基转移酶抑制剂。在这些实施方式的某些中,额外治疗剂包含dot1类组蛋白甲基转移酶(dot1l)抑制剂。dot1l是靶向在组蛋白h3的球状区域中的赖氨酸79从而一、二或三甲基化的组蛋白甲基转移酶。在这些实施方式的某些中,额外治疗剂是epz004777,epz-5676(blood.2013aug8;122(6):1017-25)或sgc0946(nat.commun.2012;3:1288),例如,epz-5676。在全部实施方式的某些方面中,额外治疗剂是组蛋白脱乙酰基酶(hdac)抑制剂。hdac蛋白质可以基于对酵母hdac蛋白质的同源性分类,类型i由hdac1,hdac2,hdac3和hdac8构成;类型iia由hdac4,hdac5,hdac7和hdac9构成;类型iib由hdac6和hdac10构成;而类型iv由hdac11构成。在这些实施方式的某些中,额外治疗剂是曲古抑菌素a,伏林司他(proc.natl.acad.sci.u.s.a.1998mar17;95(6):3003-7),吉维司他,阿贝司他(mol.cancerther.2006may;5(5):1309-17),贝林司他(mol.cancerther.2003aug;2(8):721-8),帕比司他(clin.cancerres.2006aug1;12(15):4628-35),瑞诺司他(clin.cancerres.2013oct1;19(19):5494-504),quisinostat(clin.cancerres.2013aug1;19(15):4262-72),缩肽(blood.2001nov1;98(9):2865-8),恩替司他(proc.natl.acad.sci.u.s.a.1999apr13;96(8):4592-7),莫替司他(bioorg.med.chem.lett.2008feb1;18(3):1067-71)或丙戊酸(emboj.2001dec17;20(24):6969-78)。例如,在某些实施方式中,额外治疗剂是帕比司他。在其它实施方式中,额外治疗剂是帕比司他或saha。在全部实施方式的某些方面中,额外治疗剂是组蛋白脱甲基酶抑制剂。在特别的实施方式中,组蛋白脱甲基酶抑制剂是赖氨酸特异性脱甲基酶1a(lsd1)抑制剂。在这些实施方式的某些中,额外治疗剂是hci-2509(bmccancer.2014oct9;14:752),反苯环丙胺或ory-1001(j.clin.oncol31,2013(suppl;abstre13543)。在其它实施方式中,额外治疗剂是hci-2509。在全部实施方式的某些方面中,额外治疗剂是mll-menin抑制剂。menin是致瘤mll融合蛋白的辅因子,并且mll-menin抑制剂阻断两种蛋白的相互作用。mll-menin抑制剂的实例包括mi-453,m-525,和mi-503。在全部实施方式的某些方面中,额外治疗剂是b-细胞受体信号转导拮抗剂(例如,bruton酪氨酸激酶(btk)抑制剂,比如依罗替尼)。在全部实施方式的某些方面中,额外治疗剂是免疫调节剂。特别用于与本公开化合物组合的免疫调节剂包括:afutuzumab(可获自);培非司亭来那度胺(cc-5013,);沙利度胺actimid(cc4047);和irx-2(包括白细胞介素1、白细胞介素2和干扰素γ的人类细胞因子的混合物,cas951209-71-5,可获自irxtherapeutics)。在全部实施方式的某些方面中,额外治疗剂包含嵌合抗原受体t-细胞(car-t)治疗。特别用于与本公开化合物组合的car-t治疗包括:tisagenlecleucel(novartis),axicabtageneciloleucel(kite),和托珠单抗和atlizumab(roche)。在全部实施方式的某些方面中,额外治疗剂是免疫检查点抑制剂(例如,pd-1抑制剂,比如pembrolizumab或nivolumab;pd-l1抑制剂,比如阿特珠单抗,avelumab或durvalumab;ctla-4抑制剂;lag-3抑制剂;或tim-3抑制剂)。用于与本公开化合物组合的其它免疫检查点抑制剂包括:pd-1抑制剂,比如pembrolizumabnivolumabcemiplimabspartalizumab(pdr001),pidilizumab(curetech),medi0680(medimmune),cemiplimab(regn2810),dostarlimab(tsr-042),pf-06801591(pfizer),tislelizumab(bgb-a317),camrelizumab(incshr1210,shr-1210),和amp-224(amplimmune);pd-l1抑制剂,比如阿特珠单抗avelumabdurvalumabfaz053(novartis),和bms-936559(bristol-myerssquibb);和靶向ctla-4的药物,比如伊匹木单抗在各种实施方式中,免疫检查点抑制剂是pd-1抑制剂。在特定的实施方式中,pd-1抑制剂是pembrolizumab,nivolumab或其组合。在特别的实施方式中,pd-1抑制剂是pembrolizumab(也称为lambrolizumab,mk-3475,mk03475,sch-900475,或)。pembrolizumab和其它抗-pd-1抗体公开于hamid,o.,etal.(2013)newenglandjournalofmedicine369(2):134-44,us8,354,509,和wo2009/114335,通过援引将其全部并入。在特别的实施方式中,pd-1抑制剂是nivolumab(也称为mdx-1106,mdx-1106-04,ono-4538,bms-936558,或)。nivolumab(克隆5c4)和其它抗-pd-1抗体公开于us8,008,449和wo2006/121168,通过援引将其全部并入。在一些其它实施方式中,pd-1抑制剂是amp-224(amplimmune),cbt-501(cbtpharmaceuticals),cbt-502(cbtpharmaceuticals),js001(junshibiosciences),ibi308(innoventbiologics),incshr1210(incyte),也称为shr-1210(hengruimedicine),bgba317(beigene),bgb-108(beigene),bat-i306(bio-therasolutions),gls-010(gloriapharmaceuticals;wuxibiologics),ak103,ak104,ak105(akesiobiopharma;hangzhouhansibiologics;hanzhongbiologics),lzm009(livzon),hlx-10(henliusbiotech),medi0680(medimmune),pdf001(novartis),pf-06801591(pfizer),pidilizumab(curetech),regn2810(regeneron),tsr-042(tesaro),也称为anb011,或cs1003(cstonepharmaceuticals)。medi0680(medimmune)也称为amp-514。medi0680和其它抗-pd-1抗体公开于us9,205,148和wo2012/145493,通过援引将其全部并入。pidilizumab也称为ct-011。pidilizumab和其它抗-pd-1抗体公开于rosenblatt,j.,etal.(2011)jimmunotherapy34(5):409-18,us7,695,715,us7,332,582,和us8,686,119,通过援引将其全部并入。在一种实施方式中,抗-pd-1抗体分子是cemiplimab。在一种实施方式中,抗-pd-1抗体分子是sintilimab。在一种实施方式中,抗-pd-1抗体分子是toripalimab。在一种实施方式中,抗-pd-1抗体分子是camrelizumab。其它已知的抗-pd-1抗体分子包括例如wo2015/112800,wo2016/092419,wo2015/085847,wo2014/179664,wo2014/194302,wo2014/209804,wo2015/200119,us8,735,553,us7,488,802,us8,927,697,us8,993,731,和us9,102,727中描述的那些,通过援引将其全部并入。在一种实施方式中,pd-1抑制剂是抗-pd-1抗体分子,如描述于us2015/0210769。在一种实施方式中,抗-pd-1抗体分子包含bap049-克隆-e或bap049-克隆-b的cdr、可变区、重链和/或轻链,公开于us2015/0210769。本文描述的抗体分子能够通过描述于us2015/0210769的媒介、宿主细胞和方法制备,通过援引将其全部并入。在一种实施方式中,pd-1抑制剂是抑制pd-1信号转导途径的肽,例如描述于us8,907,053,通过援引将其全部并入。在一种实施方式中,pd-1抑制剂是免疫粘合素(例如包含稠合至恒定区(例如免疫球蛋白序列的fc区域)的pd-l1或pd-l2的细胞外或pd-1结合部分的免疫粘合素。在一种实施方式中,pd-1抑制剂是amp-224(b7-dcig(amplimmune),例如公开于wo2010/027827和wo2011/066342,通过援引将其全部并入)。在某些实施方式中,免疫检查点抑制剂是pd-l1抑制剂。在一些这种实施方式中,pd-l1抑制剂是阿特珠单抗,avelumab,durvalumab或其组合。在特别的实施方式中,pd-l1抑制剂是阿特珠单抗,也称为mpdl3280a,rg7446,ro5541267,yw243.55.s70,或tecentriqtm。阿特珠单抗和其它抗-pd-l1抗体公开于us8,217,149,通过援引将其全部并入。在特别的实施方式中,pd-l1抑制剂是avelumab,也称为msb0010718c。avelumab和其它抗-pd-l1抗体公开于wo2013/079174,通过援引将其全部并入。在特别的实施方式中,pd-l1抑制剂是durvalumab,也称为medi4736。durvalumab和其它抗-pd-l1抗体公开于us8,779,108,通过援引将其全部并入。在某些实施方式中,pd-l1抑制剂是kn035(alphamab;3dmed),bms936559(bristol-myerssquibb),cs1001(cstonepharmaceuticals),faz053(novartis),shr-1316(hengruimedicine),tqb2450(chiataitianqing),sti-a1014(zhaokepharm;lee'spharm),bgb-a333(beigene),msb2311(mabspacebiosciences),或hlx-20(henliusbiotech)。在一种实施方式中,抗-pd-l1抗体分子是bms-936559(bristol-myerssquibb),也称为mdx-1105或12a4。bms-936559和其它抗-pd-l1抗体公开于us7,943,743和wo2015/081158,通过援引将其全部并入。在某些实施方式中,pd-l1抑制剂是单克隆抗体(例如由hisunpharm制备和用于临床试验)。在一种实施方式中,pd-l1抑制剂是抗-pd-l1抗体分子。在一种实施方式中,pd-l1抑制剂是公开于us2016/0108123的抗-pd-l1抗体分子,通过援引将其全部并入。在一种实施方式中,抗-pd-l1抗体分子包含bap058-克隆o或bap058-克隆n的cdr、可变区、重链和/或轻链,公开于us2016/0108123。其它已知的抗-pd-l1抗体包括例如wo2015/181342,wo2014/100079,wo2016/000619,wo2014/022758,wo2014/055897,wo2015/061668,wo2013/079174,wo2012/145493,wo2015/112805,wo2015/109124,wo2015/195163,us8,168,179,us8,552,154,us8,460,927和us9,175,082中描述的那些,通过援引将其全部并入。在某些实施方式中,免疫检查点抑制剂是ctla-4抑制剂。在某些实施方式中,ctla-4抑制剂是伊匹木单抗。在其它实施方式中,ctla4抑制剂是曲美木单抗。在某些实施方式中,免疫检查点抑制剂是lag-3抑制剂。在某些实施方式中,lag-3抑制剂选自lag525(novartis),bms-986016(bristol-myerssquibb),或tsr-033(tesaro)。在一种实施方式中,lag-3抑制剂是抗-lag-3抗体分子。在一种实施方式中,lag-3抑制剂是公开于us2015/0259420的抗-lag-3抗体分子,通过援引将其全部并入。在一种实施方式中,抗-lag-3抗体分子包含bap050-克隆i或bap050-克隆j的cdr、可变区、重链和/或轻链,公开于us2015/0259420。在一种实施方式中,抗-lag-3抗体分子是bms-986016(bristol-myerssquibb),也称为bms986016。bms-986016和其它抗-lag-3抗体公开于wo2015/116539和us9,505,839,通过援引将其全部并入。在一种实施方式中,抗-lag-3抗体分子是tsr-033(tesaro)。在一种实施方式中,抗-lag-3抗体分子是imp731或gsk2831781(gsk和primabiomed)。imp731和其它抗-lag-3抗体公开于wo2008/132601和us9,244,059,通过援引将其全部并入。在一种实施方式中,抗-lag-3抗体分子是imp761(primabiomed)。其它已知的抗-lag-3抗体包括例如wo2008/132601,wo2010/019570,wo2014/140180,wo2015/116539,wo2015/200119,wo2016/028672,us9,244,059,us9,505,839中描述的那些,通过援引将其全部并入。在一种实施方式中,抗-lag-3抑制剂是可溶的lag-3蛋白质,例如imp321(primabiomed),例如公开于wo2009/044273,通过援引将其全部并入。在某些实施方式中,免疫检查点抑制剂是tim-3抑制剂。在某些实施方式中,tim-3抑制剂是mgb453(novartis)或tsr-022(tesaro)。在一种实施方式中,tim-3抑制剂是抗-tim-3抗体分子。在一种实施方式中,tim-3抑制剂是公开于us2015/0218274的抗-tim-3抗体分子,通过援引将其全部并入。在一种实施方式中,抗-tim-3抗体分子包含abtim3-hum11或abtim3-hum03的cdr、可变区、重链和/或轻链,公开于us2015/0218274。在一种实施方式中,抗-tim-3抗体分子是tsr-022(anaptysbio/tesaro)。在一种实施方式中,抗-tim-3抗体分子包含ape5137或ape5121的一个或多个cdr序列(或全部总cdr序列),重链或轻链可变区序列,或重链或轻链序列。ape5137,ape5121和其它抗-tim-3抗体公开于wo2016/161270,通过援引将其全部并入。在一种实施方式中,抗-tim-3抗体分子是抗体克隆f38-2e2。其它已知的抗-tim-3抗体包括例如wo2016/111947,wo2016/071448,wo2016/144803,us8,552,156,us8,841,418和us9,163,087中描述的那些,通过援引将其全部并入。为了保护普通细胞免于治疗毒性和限制器官毒性,细胞保护性试剂(比如神经保护剂,自由基清除剂,心脏保护剂,蒽环类抗生素外渗中和剂,营养素等)可以作为辅助治疗与本公开化合物组合使用。适宜的细胞保护性试剂包括氨磷汀谷氨酰胺,地美司钠美司钠右雷佐生(或),扎利罗登和亚叶酸(也称为钙亚叶酸,柠胶因子和亚叶酸)。在给药期间或之后,某些患者可以经历对本公开化合物和/或其它治疗剂(例如抗癌剂)的变应性反应。因此,抗过敏剂能够与本公开化合物和/或其它治疗剂(例如抗癌剂)组合给予以最小化变应性反应的风险。适宜的抗过敏剂包括皮质类固醇(knutson,s.,etal.,plosone,doi:10.1371/journal.pone.0111840(2014)),比如地塞米松(例如,),倍氯米松(例如,),氢化可的松(也称为可的松,氢化可的松琥珀酸钠,氢化可的松磷酸钠,以商品名销售,氢化可的松磷酸盐,hydrocort和),泼尼松龙(以商品名和销售),泼尼松(以商品名liquid和销售),甲泼尼龙(也称为6-甲泼尼龙,甲泼尼龙乙酸盐,甲泼尼龙琥珀酸钠,以商品名和销售);抗组胺药,比如苯海拉明(例如,),羟嗪,和赛庚啶;和支气管扩张药,比如β-肾上腺素能药物受体激动剂,沙丁胺醇(例如,),和特布他林在给予本文描述的化合物和/或其它治疗剂(例如抗癌剂)期间和之后某些患者可以经验恶心。因此,镇吐剂能够与本公开化合物和/或其它治疗剂(例如抗癌剂)组合使用以预防恶心(上胃)和呕吐。适宜的镇吐剂包括阿瑞匹坦昂丹司琼格拉司琼hcl劳拉西泮(地塞米松丙氯拉嗪卡索匹坦(和),及其组合。常常开具减轻在治疗期间经历的疼痛的药物来使患者更舒适。普通非处方镇痛药比如还能够与本公开化合物和/或其它治疗剂(例如抗癌剂)组合使用。阿片类物质镇痛药比如氢可酮/对乙酰氨基酚或氢可酮/对乙酰氨基酚(例如,),吗啡(例如,或),羟考酮(例如,或),羟吗啡酮盐酸盐和芬太尼(例如,)能够用于中度或重度疼痛,并且能够与本公开化合物和/或其它治疗剂(例如抗癌剂)组合使用。在第七实施方式的某些方面中,如果受试者也在进行雄激素剥夺疗法或接受阻断雄激素结合至雄激素受体的雄激素受体拮抗剂,那么用结构(i)化合物或其药学上可接受的盐或两性离子形式治疗患前列腺癌的受试者可以预防或抑制去势抵抗的发展。通常,结构(i)化合物或其药学上可接受的盐或两性离子形式应不导致实质毒性地使用。结构(i)化合物或其药学上可接受的盐或两性离子形式的毒性能够用标准技术确定,例如在细胞培养物或实验动物中测试并确定治疗指数,也即ld50(50%群体致死剂量)与ld100(100%群体致死剂量)的比率。然而在某些环境中,比如在严重疾病条件下,可能需要给予实质过量的组合物。滴定研究可以用来确定毒性和非毒性浓度。毒性可以通过检查特定化合物的或组合物的细胞系间特异性来评价。动物研究可以用来提供化合物对其它组织是否具有任何效果的指示。结构(i)化合物或其药学上可接受的盐或两性离子形式在宽剂量范围有效。例如,在治疗成人时,约0.01mg至约1000mg、约0.1mg至约100mg、约0.5mg至约50mg每天和约1mg至约10mg每天的剂量是在某些实施方式中使用的剂量实例。示范性剂量是约0.5mg至约50mg每天。在特别的实施方式中,剂量的范围是约1mg至约60mg(例如,约5mg至约60mg,约10mg至约60mg,约5mg至约50mg,约10mg至约30mg,约10mg至约50mg,约20至约50mg,约25mg至约45mg)每天。在其它实施方式中,剂量是约1mg至约30mg每天,例如约1mg,约2mg,约4mg,约8mg,约12mg,约16mg,约20mg,约22mg,约24mg,约26mg,约28mg或约30mg每天(例如qd给予,bid给予)。在其它实施方式中,剂量是约1mg至约30mg,例如约1mg,约2mg,约4mg,约6mg,约8mg,约11mg,约12mg,约16mg,约20mg,约22mg,约24mg,约26mg,约28mg或约30mg,bid给予。精确剂量将取决于给药途径,结构(i)化合物或其药学上可接受的盐或两性离子形式的给予形式,待治疗的受试者,待治疗的受试者的体重,和主治医师的选择和经验。在实施方式一至七的某些方面中,治疗有效量是约0.5mg至约50mg每天。在实施方式一至七的某些方面中,治疗有效量是约1mg至约60mg(例如约5mg至约60mg,约10mg至约60mg,约5mg至约50mg,约10mg至约30mg,约10mg至约50mg,约20至约50mg,约25mg至约45mg)每天。在实施方式一至七的某些方面中,治疗有效量是约1mg至约30mg每天,例如约1mg,约2mg,约4mg,约8mg,约12mg,约16mg,约20mg,约22mg,约24mg,约26mg,约28mg或约30mg每天(例如qd给予,bid给予)。在实施方式一至七的某些方面中,治疗有效量是约1mg至约30mg,例如约1mg,约2mg,约4mg,约6mg,约8mg,约11mg,约12mg,约16mg,约20mg,约22mg,约24mg,约26mg,约28mg或约30mg,bid给予。在某些实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式以单剂量给予。结构(i)化合物或其药学上可接受的盐或两性离子形式的单剂量还可以用于治疗急性病况。在某些实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式以多剂量给予。在某些实施方式中,剂量给药是每天约一次,两次,三次,四次,五次,六次或多于六次。在一些特定实施方式中,剂量给药是每天两次(bid)。在某些特定实施方式中,剂量给药每天一次(qd)。在其它实施方式中,剂量给药是约每月一次,每两周一次,每周一次,或每隔一天一次。在又一实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式和又一试剂约每天1次至约每天6次一起给予。在又一实施方式中,给予结构(i)化合物或其药学上可接受的盐或两性离子形式和试剂继续少于约7天。在还又一实施方式中给药继续大于约6、10、14、21、28天,2个月,6个月,或1年。在某些情况下,实现连续剂量给药并按需要时长保持(例如直至进展或无法接受的毒性)。结构(i)化合物或其药学上可接受的盐或两性离子形式的给药可以按需要时长继续。在某些实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式给予多于1、2、3、4、5、6、7、14或28天。在某些实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式给予小于28、14、7、6、5、4、3、2或1天。在某些实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式给予连续的21天。在某些实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式连续长期性地给予,例如用于慢性效果的治疗。结构(i)化合物或其药学上可接受的盐或两性离子形式的给药可以以治疗周期进行。如本文所用,"治疗周期"是指期望以有规律计划重复的后接无治疗时间段的治疗时间段。在某些实施方式中,治疗周期是21天治疗周期。在某些实施方式中,治疗周期是28天治疗周期。结构(i)化合物或其药学上可接受的盐或两性离子形式的给药可以包括连续的剂量给药和/或可以包括治疗中断。对于包括治疗中断的剂量给药计划,结构(i)化合物或其药学上可接受的盐或两性离子形式可以以治疗周期给予,所述治疗周期包括连续剂量给药的时间间隔、随后是不给予化合物的治疗中断。治疗中断可以例如是大于1、2、3、4、5、6、7、14或28天。在某些特定实施方式中,剂量给药计划是21天治疗周期包括14天剂量给药,随后是7天治疗中断。在其它特别的实施方式中,剂量给药计划是28天治疗周期包括21天剂量给药(例如bid剂量给药、qd剂量给药),随后是7天治疗中断。换言之,在某些实施方式中,具有结构(i)的化合物或其药学上可接受的盐或两性离子形式在28天治疗周期的前21天给予,并且在28天治疗周期的第22至28天不给予。治疗周期可以重复至少一次,至少两次,至少三次,或至少四次。在某些实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式以剂量给予。由于化合物药代动力学的受试者间可变性,在某些实施方式中提供剂量给药方案的个体化。结构(i)化合物或其药学上可接受的盐或两性离子形式的剂量给药可以参照本公开通过惯例实验发现和/或能够通过本领域普通技术人员衍生。可根据本文描述的方法治疗的癌症的其它实例包括血液癌。能够用具有结构(i)的化合物或其互变异构体或两性离子形式治疗的血液恶性包括白血病和淋巴瘤。在某些实施方式中,血液癌选自急性髓性白血病(aml),小囊淋巴瘤,急性成淋巴细胞白血病(all),慢性淋巴细胞白血病(cll),多发性骨髓瘤(mm)和非霍奇金淋巴瘤(例如aml、小囊淋巴瘤、all、cll和非霍奇金淋巴瘤)。在更特定的实施方式中,血液癌是aml。在其它更特定的实施方式中,血液癌是cll。在更特定的实施方式中,血液癌是mm。在其它特定的实施方式中,血液癌是脊髓发育不良综合征(mds)。实体肿瘤还能够根据本文描述的方法治疗。相应地,在某些实施方式中,癌是实体肿瘤癌。在各种实施方式中,实体肿瘤癌是乳腺癌,膀胱癌,肝癌,胰癌,肺癌,结直肠癌,卵巢癌,前列腺癌,或黑色素瘤。在某些实施方式中,癌是膀胱癌。在某些实施方式中,癌是肺癌。在其它实施方式中,癌是肝癌。在各种实施方式中,实体肿瘤癌是乳腺癌,膀胱癌,肝癌,胰癌,肺癌,结直肠癌,卵巢癌,前列腺癌,或黑色素瘤。在某些实施方式中,癌是膀胱癌。在某些实施方式中,癌是肺癌。在其它实施方式中,癌是肝癌。在某些实施方式中,癌是肉瘤,膀胱癌或肾癌。在某些实施方式中,癌是前列腺癌。在其它实施方式中,癌是膀胱癌,胰癌,结直肠癌,肾癌,非小细胞肺癌,前列腺癌,肉瘤,皮肤癌,甲状腺癌,睾丸癌或外阴癌。在某些实施方式中,癌是子宫内膜癌,胰癌,睾丸癌,肾癌,黑色素瘤,结直肠癌,甲状腺癌,膀胱癌,胰癌,外阴癌,肉瘤,前列腺癌,肺癌或肛门癌。可根据本文描述的方法治疗的癌的其它实例包括但不限于,急性成淋巴细胞白血病(all);急性髓性白血病(aml);肾上腺皮质癌;肾上腺皮质癌,儿童期;aids-相关性癌(例如卡波西肉瘤,aids-相关性淋巴瘤,原发cns淋巴瘤);肛门癌;阑尾癌;星形细胞瘤,儿童期;非典型畸胎样/杆状肿瘤,儿童期,中枢神经系统;皮肤基底细胞癌;胆管癌;膀胱癌;膀胱癌,儿童期;骨癌(包括尤因肉瘤,骨肉瘤和恶性纤维组织细胞瘤);脑肿瘤/癌;乳腺癌;伯基特淋巴瘤;类癌瘤(胃肠道);类癌瘤,儿童期;心脏(心)肿瘤,儿童期;胚胎肿瘤,儿童期;胚细胞瘤,儿童期;原发cns淋巴瘤;宫颈癌;儿童期宫颈癌;胆管上皮癌;脊索瘤,儿童期;慢性淋巴细胞白血病(cll);慢性髓性白血病(cml);慢性骨髓增殖瘤;结直肠癌;儿童期结直肠癌;颅咽管瘤,儿童期;皮肤t-细胞淋巴瘤(例如蕈样真菌病和赛杂瑞综合征);原位导管癌(dcis);胚胎肿瘤,中枢神经系统,儿童期;子宫内膜癌(子宫癌);室管膜瘤,儿童期;食管癌;儿童期食管癌;成感觉神经细胞瘤;尤因肉瘤;颅外胚细胞瘤,儿童期;性腺外胚细胞瘤;眼癌;儿童期眼内黑色素瘤;眼内黑色素瘤;成视网膜细胞瘤;输卵管癌;恶性骨纤维状组织细胞瘤,和骨肉瘤;胆囊癌;胃(胃)癌;儿童期胃(胃)癌;胃肠道类癌瘤;胃肠道间质瘤(gist);儿童期胃肠道间质瘤;胚细胞瘤;儿童期中枢神经系统胚细胞瘤(例如儿童期颅外胚细胞瘤,性腺外胚细胞瘤,卵巢胚细胞瘤,睾丸癌);妊娠滋养细胞疾病;多毛细胞白血病;头颈癌;心脏肿瘤,儿童期;肝细胞(肝)癌;组织细胞增多症,langerhans细胞;霍奇金淋巴瘤;咽下癌;眼内黑色素瘤;儿童期眼内黑色素瘤;胰岛细胞肿瘤,胰神经内分泌肿瘤;卡波西肉瘤;肾(肾细胞)癌;朗格汉斯细胞组织细胞增生症;喉癌;白血病;唇和口腔癌;肝癌;肺癌(非小细胞和小细胞);儿童期肺癌;淋巴瘤;男性乳腺癌;骨恶性纤维组织细胞瘤和骨肉瘤;黑色素瘤;儿童期黑色素瘤;黑色素瘤,眼内(眼);儿童期眼内黑色素瘤;梅克尔细胞癌;恶性间皮瘤;儿童期间皮瘤;转移癌;转移的鳞状颈癌伴原发潜隐性;中线道癌伴nut基因变化;口癌;多发性内分泌腺瘤综合征;多发性骨髓瘤/血浆细胞瘤;蕈样真菌病;骨髓增生异常综合征,脊髓发育不良/骨髓增殖瘤;髓性白血病,慢性(cml);髓性白血病,急性(aml);骨髓增殖瘤,慢性;鼻腔和鼻旁窦癌;鼻咽癌;成神经细胞瘤;非霍奇金淋巴瘤;非小细胞肺癌;口腔癌,唇和口腔癌和口咽癌;骨肉瘤和骨恶性纤维组织细胞瘤;卵巢癌;儿童期卵巢癌;胰癌;儿童期胰癌;胰神经内分泌肿瘤;乳头状瘤病(儿童期喉性);副神经节瘤;儿童期副神经节瘤;鼻旁窦和鼻腔癌;副甲状腺癌;阴茎癌;咽癌;嗜铬细胞瘤;儿童期嗜铬细胞瘤;垂体肿瘤;血浆细胞瘤/多发性骨髓瘤;胸膜肺母细胞瘤;孕期和乳腺癌;原发中枢神经系统(cns)淋巴瘤;原发腹膜癌;前列腺癌;直肠癌;复发癌;肾细胞(肾)癌;成视网膜细胞瘤;横纹肌肉瘤,儿童期;唾腺癌;肉瘤(例如儿童期横纹肌肉瘤,儿童期血管肿瘤,尤因肉瘤,卡波西肉瘤,骨肉瘤(骨癌),软组织肉瘤,子宫肉瘤);赛杂瑞综合征;皮肤癌;儿童期皮肤癌;小细胞肺癌;小肠癌;软组织肉瘤;皮肤鳞状细胞癌;转移的鳞状颈癌伴原发潜隐性;胃(胃)癌;儿童期胃(胃)癌;皮肤t-细胞淋巴瘤(例如蕈样真菌病和赛杂瑞综合征);睾丸癌;儿童期睾丸癌;咽喉癌(例如鼻咽癌,口咽癌,咽下癌);胸腺瘤和胸腺癌;甲状腺癌;肾骨盆和输尿管移行细胞癌;输尿管和肾骨盆,移行细胞癌;尿道癌;子宫癌,子宫内膜;子宫肉瘤;阴道癌;儿童期阴道癌;血管肿瘤;外阴癌;和wilms肿瘤和其它儿童期肾肿瘤。前述癌的转移也能够按照本文描述的方法治疗。从而,在某些实施方式中,癌是转移的癌。在其它实施方式中,癌是未转移的癌。ii.结构(i)化合物的结晶和多晶型形式已发现具有结构(i)的化合物或其互变异构体或两性离子形式能够以各种结晶和/或多晶型形式存在。相应地,一种实施方式提供具有下述结构(i)的化合物的晶型:或其互变异构体或两性离子形式。在某些实施方式中,晶型包含形式b。在某些实施方式中,晶型实质上由形式b组成。在某些实施方式中,晶型由形式b组成。在某些实施方式中,晶型是具有结构(ii)的化合物的晶型。形式b具有结构(ii):并且在某些实施方式中,通过x射线粉末衍射(xrpd)图谱表征,其包含至少三个峰(例如三个峰、至少四个峰、四个峰、至少五个峰、五个峰、六个峰),所述峰位于选自4.8±0.2°,10.8±0.2°,13.7±0.2°,14.9±0.2°,20.0±0.2°和24.6±0.2°的2-θ角。在某些实施方式中,形式b通过xrpd图谱表征,其包含位于下述2-θ角的峰:10.8±0.2°,14.9±0.2°和20.0±0.2°。在某些实施方式中,形式b通过xrpd图谱表征,其包含位于下述2-θ角的峰:4.8±0.2°,10.8±0.2°,14.9±0.2°和20.0±0.2°。在某些实施方式中,形式b通过xrpd图谱表征,其包含位于下述2-θ角的峰:4.8±0.2°,10.8±0.2°,13.7±0.2°,14.9±0.2°和20.0±0.2°。在某些实施方式中,形式b具有实质上按照图25所描述的xrpd图谱。在某些实施方式中,形式b通过dsc热分析图表征,其包含位于约264℃的吸热峰。在某些实施方式中,形式b通过dsc热分析图表征,其实质上按照图26的描述。iii.药物组合物出于给药意图,结构(i)化合物或其药学上可接受的盐或两性离子形式可以作为粗制化学品给予或可以配制为药物组合物。本文描述的方法提供的药物组合物包含结构(i)化合物或其药学上可接受的盐或两性离子形式,和药学上可接受的载体、稀释剂或赋形剂。在实施方式中,结构(i)化合物或其药学上可接受的盐或两性离子形式以有效治疗去势抵抗性前列腺癌的量存在于组合物中,和优选具有患者可接受的毒性。结构(i)化合物或其药学上可接受的盐或两性离子形式的生物利用度能够由本领域技术人员确定,例如如下文实施例描述。适当的浓度和剂量能够由本领域技术人员容易地确定。纯形式或在适当药物组合物中的结构(i)化合物或其药学上可接受的盐或两性离子形式的给药能够经由给药发挥相似作用的试剂的任何接受模式进行。本发明实施方式的药物组合物能够通过将结构(i)化合物或其药学上可接受的盐或两性离子形式与适当的药学上可接受的载体、稀释剂或赋形剂组合制备,并且可以配制成固体、半固体、液体或气态形式的制剂,比如片剂、胶囊、粉末、颗粒剂、软膏剂、溶液、栓剂、注射剂、吸入剂、凝胶、微球和气雾剂。所述药物组合物的典型给予途径包括但不限于口服、局部、透皮、吸入,肠胃外,舌下、颊、直肠、阴道和鼻内。术语肠胃外如本文所用包括皮下注射剂,静脉内,肌内,胸骨内注射或输注技术。配制本发明的药物组合物以便允许其中含有的活性成分在向患者给药组合物后是生物可获得的。向受试者或患者给予的组合物呈一种或多种剂量单元形式,其中例如片剂可以是单剂量单元。制备所述剂型的实际方法对本领域技术人员是已知的或将是明显的;例如参见remington:thescienceandpracticeofpharmacy,20thedition(philadelphiacollegeofpharmacyandscience,2000)。待给予的组合物在任何情况下含有治疗有效量的结构(i)化合物或其药学上可接受的盐或两性离子形式,用于按照本发明教导治疗去势抵抗性前列腺癌。在本文描述的全部实施方式的某些方面中,结构(i)化合物或其药学上可接受的盐或两性离子形式口服给予。某些本发明实施方式的药物组合物可以呈固体或液体形式。在一个方面,载体是粒状的,从而组合物例如呈片剂或粉末形式。载体可以是液体,而组合物是例如口服糖浆、可注射液体或气雾剂,其用于例如吸入给药。在期望用于口服给药的情况下,药物组合物优选呈固体或液体形式,其中半固体、半液体、悬浮液和凝胶形式包括在本文视为固体或液体的形式中。作为口服给药的固体组合物,药物组合物可以配制成粉末,颗粒剂,压制片剂,丸剂,胶囊,口香糖,糯米纸囊剂等形式。上述固体组合物一般地含有一种或多种惰性稀释剂或可食用载体。此外,可以存在下述的一种或多种:粘结剂比如羧甲纤维素,乙基纤维素,微晶纤维素,黄蓍胶或明胶;赋形剂比如淀粉,乳糖或糊精,崩解剂比如藻酸,藻酸钠,primogel,玉米淀粉等;润滑剂比如硬脂酸镁或sterotex;助流剂比如胶体二氧化硅;甜味剂比如蔗糖或糖精;调味试剂比如胡椒薄荷,水杨酸甲酯或橙调味剂;和着色剂。在药物组合物呈胶囊形式例如植物基胶囊比如羟基丙基甲基纤维素(hpmc)胶囊或明胶胶囊的情况下,其除了上述类型的物质之外可以还含有液体载体比如聚乙二醇或油。药物组合物可以呈液体形式,例如酏剂,糖浆,溶液,乳液或悬浮液。液体可以用于口服给药或用于注射递送,作为两种实例。在期望口服给药的情况下,优选的组合物除了本申请化合物之外还含有一种或多种甜味剂,防腐剂,染料/着色剂和调味剂增强剂。在期望注射给予的组合物中,可以包括一种或多种表面活性剂,防腐剂,润湿剂,分散剂,助悬剂,缓冲剂,稳定剂和等渗剂。某些本发明实施方式的液体药物组合物,无论是溶液、悬浮液或其它类似形式,可以包括下述助剂的一种或多种:无菌稀释剂比如注射用水,盐水溶液,优选生理盐水,林格溶液,等渗氯化钠,非挥发油比如合成甘油一或二酯,其可以充当溶剂或悬浮介质,聚乙二醇,甘油,丙二醇或其它溶剂;抗菌剂比如苄醇或羟苯甲酯;抗氧化剂比如抗坏血酸或亚硫酸氢钠;螯合剂比如乙二胺四乙酸;缓冲剂比如乙酸、柠檬酸或磷酸和张力调节剂比如氯化钠或右旋糖。肠胃外制剂能够封闭在玻璃或塑料的安瓿,一次性注射器或多剂量小瓶中。生理盐水是优选的助剂。可注射的药物组合物优选无菌。期望用于肠胃外或口服给药的本发明某些实施方式的液体药物组合物应含有一定量的结构(i)化合物或其药学上可接受的盐,从而将获得适宜的剂量。在某些实施方式中,本发明实施方式的药物组合物可以期望用于局部给药,在该情况下载体可以适宜地包含溶液,乳液,软膏剂或凝胶基底。所述基底例如可以包含下述的一种或多种:凡士林,羊毛脂,聚乙二醇,蜂蜡,矿物油,稀释剂比如水和醇,和乳化剂和稳定剂。增稠剂可以存在于药物组合物用于局部给药。如果期望经皮给药,组合物可以包括透皮贴剂或电离子透入装置。本发明各种实施方式的药物组合物可以期望用于直肠给药,呈例如在直肠中熔化和释放药物的栓剂形式。直肠给药组合物可以含有含油基底作为适宜的无刺激性赋形剂。所述基底包括但不限于羊毛脂,可可油和聚乙二醇。本发明药物组合物的实施方式可以包括各种物质,其修饰固体或液体剂量单元的物理形式。例如,组合物可以包括围绕活性成分形成包衣壳的物质。形成包衣壳的物质一般是惰性的,并且可以选自例如糖、虫胶和其它肠衣试剂。另选地,活性成分可以包入胶囊比如hpmc胶囊中。呈固体或液体形式的某些本发明实施方式的药物组合物可以包括结合至结构(i)化合物或其药学上可接受的盐或两性离子形式和由此帮助化合物递送的试剂。可以起该作用的适宜试剂包括单克隆或多克隆抗体,蛋白质或脂质体。本发明其它实施方式的药物组合物可以由能够作为气雾剂给予的剂量单元组成。术语气雾剂用来表示各种系统,从胶体性质的那些直至由加压包装组成的系统。递送可以通过液化或压缩气体或者通过分配活性成分的适宜泵系统来进行。本发明化合物的气雾剂可以以单相、双相或三相系统递送以便递送活性成分。气雾剂的递送包括必需的容器、活化剂、阀、子容器等,其一起可以构成试剂盒。本领域技术人员无需过分实验就可以确定优选的气雾剂。在某些实施方式中,本发明实施方式的药物组合物可以通过药物领域熟知的方法制备。例如,期望通过注射给予的药物组合物能够通过将结构(i)化合物或其药学上可接受的盐或两性离子形式与无菌蒸馏水组合形成溶液而制备。可以加入表面活性剂促进均质溶液或悬浮液的形成。表面活性剂是化合物,其与结构(i)化合物或其药学上可接受的盐或两性离子形式非共价地相互作用以便促进结构(i)化合物或其药学上可接受的盐或两性离子形式在含水递送系统中的溶解或均匀悬浮。本发明方法包括给予治疗有效量的结构(i)化合物或其药学上可接受的盐或两性离子形式,所述量取决于各种因素变化,包括所用特定化合物的活性;化合物的代谢稳定性和作用长度;患者的年龄、体重、一般健康、性别和膳食;给药模式和时间;排泄速率;药物组合;特定障碍或病况的严重性;和经历治疗的受试者。结构(i)化合物或其药学上可接受的盐或两性离子形式还可以与给药一种或多种额外治疗剂同时、在其之前或之后给予。所述联合疗法包括给药单一药物剂量配制剂,其含有结构(i)化合物或其药学上可接受的盐或两性离子形式和一种或多种额外的活性剂;以及在其本身的分开药物剂量配制剂中给药结构(i)化合物或其药学上可接受的盐或两性离子形式和各活性剂。例如,结构(i)化合物或其药学上可接受的盐或两性离子形式和其它活性剂能够在单一口服剂量组合物比如片剂或胶囊中一起给予患者,或者各试剂在分开的口服剂量配制剂中给予。在使用分开的剂量配制剂的情况下,本发明化合物和一种或多种额外的活性剂能够在本质上相同时间即同时给予,或在分别错开的时间即依次给予;联合疗法理解包括全部这些方案。在某些实施方式中,本发明药物组合物中提供的结构(i)化合物或其药学上可接受的盐或两性离子形式的浓度小于100%,90%,80%,70%,60%,50%,40%,30%,20%,19%,18%,17%,16%,15%,14%,13%,12%,11%,10%,9%,8%,7%,6%,5%,4%,3%,2%,1%,0.5%,0.4%,0.3%,0.2%,0.1%,0.09%,0.08%,0.07%,0.06%,0.05%,0.04%,0.03%,0.02%,0.01%,0.009%,0.008%,0.007%,0.006%,0.005%,0.004%,0.003%,0.002%,0.001%,0.0009%,0.0008%,0.0007%,0.0006%,0.0005%,0.0004%,0.0003%,0.0002%,或0.0001%w/w,w/v或v/v。在某些实施方式中,本发明药物组合物中提供的结构(i)化合物或其药学上可接受的盐或两性离子形式的浓度大于90%,80%,70%,60%,50%,40%,30%,20%,19.75%,19.50%,19.25%19%,18.75%,18.50%,18.25%18%,17.75%,17.50%,17.25%17%,16.75%,16.50%,16.25%16%,15.75%,15.50%,15.25%15%,14.75%,14.50%,14.25%14%,13.75%,13.50%,13.25%13%,12.75%,12.50%,12.25%12%,11.75%,11.50%,11.25%11%,10.75%,10.50%,10.25%10%,9.75%,9.50%,9.25%9%,8.75%,8.50%,8.25%8%,7.75%,7.50%,7.25%7%,6.75%,6.50%,6.25%6%,5.75%,5.50%,5.25%5%,4.75%,4.50%,4.25%,4%,3.75%,3.50%,3.25%,3%,2.75%,2.50%,2.25%,2%,1.75%,1.50%,125%,1%,0.5%,0.4%,0.3%,0.2%,0.1%,0.09%,0.08%,0.07%,0.06%,0.05%,0.04%,0.03%,0.02%,0.01%,0.009%,0.008%,0.007%,0.006%,0.005%,0.004%,0.003%,0.002%,0.001%,0.0009%,0.0008%,0.0007%,0.0006%,0.0005%,0.0004%,0.0003%,0.0002%,或0.0001%w/w,w/v或v/v。在某些实施方式中,本发明药物组合物中提供的结构(i)化合物或其药学上可接受的盐或两性离子形式的浓度范围是大约0.0001%至大约50%,大约0.001%至大约40%,大约0.01%至大约30%,大约0.02%至大约29%,大约0.03%至大约28%,大约0.04%至大约27%,大约0.05%至大约26%,大约0.06%至大约25%,大约0.07%至大约24%,大约0.08%至大约23%,大约0.09%至大约22%,大约0.1%至大约21%,大约0.2%至大约20%,大约0.3%至大约19%,大约0.4%至大约18%,大约0.5%至大约17%,大约0.6%至大约16%,大约0.7%至大约15%,大约0.8%至大约14%,大约0.9%至大约12%,大约1%至大约10%w/w,w/v或v/v。在某些实施方式中,本发明药物组合物中提供的结构(i)化合物或其药学上可接受的盐或两性离子形式的浓度范围是大约0.001%至大约10%,大约0.01%至大约5%,大约0.02%至大约4.5%,大约0.03%至大约4%,大约0.04%至大约3.5%,大约0.05%至大约3%,大约0.06%至大约2.5%,大约0.07%至大约2%,大约0.08%至大约1.5%,大约0.09%至大约1%,大约0.1%至大约0.9%w/w,w/v或v/v。在某些实施方式中,本发明药物组合物中提供的结构(i)化合物或其药学上可接受的盐或两性离子形式的量等于或小于10g,9.5g,9.0g,8.5g,8.0g,7.5g,7.0g,6.5g,6.0g,5.5g,5.0g,4.5g,4.0g,3.5g,3.0g,2.5g,2.0g,1.5g,1.0g,0.95g,0.9g,0.85g,0.8g,0.75g,0.7g,0.65g,0.6g,0.55g,0.5g,0.45g,0.4g,0.35g,0.3g,0.25g,0.2g,0.15g,0.1g,0.09g,0.08g,0.07g,0.06g,0.05g,0.04g,0.03g,0.02g,0.01g,0.009g,0.008g,0.007g,0.006g,0.005g,0.004g,0.003g,0.002g,0.001g,0.0009g,0.0008g,0.0007g,0.0006g,0.0005g,0.0004g,0.0003g,0.0002g,或0.0001g。在某些实施方式中,本发明药物组合物中提供的结构(i)化合物或其药学上可接受的盐或两性离子形式的量大于0.0001g,0.0002g,0.0003g,0.0004g,0.0005g,0.0006g,0.0007g,0.0008g,0.0009g,0.001g,0.0015g,0.002g,0.0025g,0.003g,0.0035g,0.004g,0.0045g,0.005g,0.0055g,0.006g,0.0065g,0.007g,0.0075g,0.008g,0.0085g,0.009g,0.0095g,0.01g,0.015g,0.02g,0.025g,0.03g,0.035g,0.04g,0.045g,0.05g,0.055g,0.06g,0.065g,0.07g,0.075g,0.08g,0.085g,0.09g,0.095g,0.1g,0.15g,0.2g,0.25g,0.3g,0.35g,0.4g,0.45g,0.5g,0.55g,0.6g,0.65g,0.7g,0.75g,0.8g,0.85g,0.9g,0.95g,1g,1.5g,2g,2.5,3g,3.5,4g,4.5g,5g,5.5g,6g,6.5g,7g,7.5g,8g,8.5g,9g,9.5g,或10g。在某些实施方式中,本发明药物组合物中提供的结构(i)化合物或其药学上可接受的盐或两性离子形式的量范围是0.0001-10g,0.0005-9g,0.001-8g,0.005-7g,0.01-6g,0.05-5g,0.1-4g,0.5-4g,或1-3g。实施例实施例1阿伏西地在对雄激素有抗性的前列腺癌细胞中的体外活性结构(i)化合物及其药学上可接受的盐和两性离子形式在体内转化为阿伏西地(参见u.s.专利公开no.us2016/0340376,通过援引将其全部公开并入本文)。因此,在具有对雄激素的变化敏感性的多个前列腺癌细胞系中评价阿伏西地的体外效果。pc3是ar阴性前列腺癌细胞系,几乎不直至不表达psa且对雄激素低敏感性。vcap是对arv7阳性的前列腺癌细胞系,并且能够以雄激素独立性方式生长。lncap是雄激素依赖性前列腺癌细胞系。22rv1是对arv7阳性的前列腺癌细胞系,具有雄激素低敏感性,并且衍生自异种移植物,其在亲代雄激素依赖性cwr22异种移植物的去势诱导消退和复发之后的小鼠中系列传播。前列腺癌细胞系pc3、vcap、lncap和22rv1用对pc3、vcap和lncap3.9nm-1000nm而对22rv1的0.0026nm-1000nm的一系列阿伏西地剂量治疗,并且测量细胞存活。图1a显示阿伏西地治疗后的pc3细胞存活和显示ic50为102.5nm。图1b显示阿伏西地治疗后的vcap细胞存活,ic50为34.55nm。图1c显示阿伏西地治疗后的lncap细胞存活和显示ic50为31.82nm。图1d显示阿伏西地治疗后的22rv1细胞存活和显示ic50为169.4nm。细胞存活可以例如用celltiter-glo根据生产商方案评价。实施例2阿伏西地对血清刺激的前列腺癌细胞的体外效果在评价阿伏西地对血清刺激的前列腺癌细胞的第一实验中,雄激素受体表达通过免疫印迹评价。图2a上部图显示实验方案图解。前列腺癌细胞22rv1或lncap用dmso或阿伏西地(80nm或160nm)治疗3小时或24小时,和在样品收集之前血清刺激一小时(或血清饥饿细胞作为对照)。图2a底部左小图显示阿伏西地治疗(3-小时或24-小时治疗)对血清刺激的22rv1细胞中的下述蛋白质水平的效果:par515;parser81;arv7;总ar(tar);半胱天冬酶-3;和微管蛋白(作为负债对照)。图2a右小图显示阿伏西地治疗(3-小时或24-小时治疗)对在血清刺激的lncap细胞中的下述蛋白质水平的效果:par515;parser81;arv7;总和ar(tar);半胱天冬酶-3;和微管蛋白(作为负载对照)。图2b显示阿伏西地治疗(24-小时治疗)对parser81arv7和arv7蛋白质水平的效果。如图2a和2b中可见,在24小时治疗之后阿伏西地降低磷酸-和总-ar水平。在又一实验中,雄激素受体表达和功能用前列腺癌细胞系22rv1中的跨膜蛋白酶mrna水平、丝氨酸2(tmprss2)mrna水平的定量实时pcr测量来评价,如chen等人.,2012.jbc.287:8571的描述。tmprss1是雄激素应答基因,其受雄激素受体转录地调节的。雄激素应答可以通过在前列腺癌细胞系中加入外源睾酮或通过血清刺激(含有雄激素)驱动。图3显示阿伏西地治疗(3小时或24小时治疗)对血清刺激的22rv1细胞中的tmprss2表达的效果。图3上部图显示实验方案的流程图。22rv1细胞用dmso或阿伏西地(80nm或160nm)治疗3小时或24小时,和在样品收集之前血清刺激一小时(或血清饥饿细胞作为对照)。图3底部小图显示各条件下tmprss2表达的倍数变化。如圆圈所示,阿伏西地抑制tmprss2的血清刺激诱导。图4显示阿伏西地治疗(24-小时治疗)对阿伏西地治疗后3小时或23小时用血清刺激的血清刺激22rv1细胞中的psa表达的效果。图4上部图显示实验方案的流程图。22rv1细胞用dmso或阿伏西地(80nm或160nm)治疗24小时,和在治疗后血清刺激3小时或23小时(或血清饥饿细胞作为对照)。图4底部小图,显示各条件下psa表达的倍数变化。如圆圈所示,阿伏西地抑制psa的血清刺激诱导。实施例3在22rv1异种移植前列腺癌模型中的效力研究该研究目的是评价结构(i)化合物或其药学上可接受的盐或两性离子形式在治疗皮下的22rv1人类前列腺癌异种移植模型中的体内疗效。对于22rv1模型,在雄性balb/c裸鼠中评价结构(i)化合物的效力。在肿瘤接种之前2天以2gy(1gy=100拉德)进行全部小鼠的钴-60辐射。各小鼠右胁腹接种0.1mlpbsmatrigel(1:1)混合物中的1x107肿瘤细胞。肿瘤细胞接种的日期表示为第0天。在平均肿瘤体积达到大约200mm3的情况下进行去势。用氯胺酮/赛拉嗪麻醉小鼠;经由中线阴囊切开进行手术去势,允许双侧访问半阴囊内容物;在暴露各睾丸之后,用6-0vicryl缝合线来结扎精索然后除去睾丸;然后用6-0vicryl缝合线分别封闭精囊和皮肤。在平均肿瘤体积再生长至大约100mm3的情况下开始治疗。在肿瘤达到大约100-200mm3的适当尺寸之后,小鼠随机化分为示于表1的治疗组1-11。各治疗在第15天以剂量给药体积5μl/g开始给予,并继续21天(或对q7d治疗组为22天)。对于q7dx3周治疗组,在随机化后第1、8和15天进行剂量给药,和在随机化后第22天终止研究。组合的治疗间隔各自是0h,而bid间隔是8h。随机化基于"匹配分布"方法(studydirectortm软件,版本3.1.399.19)随机化区组设计来进行。表1.22rv1异种移植研究的治疗组。肿瘤体积在二维随机化之后用测径器每周测量两次,和体积用下式表达为mm3:"v=(lxwxw)/2",其中v是肿瘤体积,l是肿瘤长度(最长肿瘤尺度)和w肿瘤宽度(与l垂直的最长肿瘤尺度)。各组的平均肿瘤体积示于图5。各组的平均肿瘤体积,作为相对对照组平均肿瘤体积的百分比(组1),示于图6。各组肿瘤体积的平均百分比变化,作为相对对照组的肿瘤体积平均百分比变化比率示于图7。示于图7的百分比(%t/c)按平均(t)/平均(c)*100%计算,"t"代表肿瘤体积和"c"代表组1的肿瘤体积.在研究第35天的各组个体的肿瘤体积示于图8。肿瘤生长抑制(tgi)或"抑制":tgi%是抗癌活性的指示和表示为:平均%抑制=(平均(c)-平均(t))/平均(c)*100%。t和c是治疗组和对照组(组1)在给定天分别的平均肿瘤体积(或重量)。各组肿瘤生长与对照组比较的平均百分比抑制示于图9。各组肿瘤生长抑制与对照组比较的平均百分比变化示于图10。示于图10的百分比按%δt/c=(平均(t)-平均(t0))/(平均(c)-平均(c0))*100%计算,"0"指出初始的时间点。如图5-10所示,去势引起肿瘤生长的小减少,其类似下述治疗:维奈托克、恩杂鲁胺和多西他赛,这指出这些药物作为单药治疗在22rv1异种移植模型中没有活性。额外地,两个不同剂量和计划的结构(i)化合物作为单一疗法显示适度的活性。然而,结构(i)化合物与abt199(维奈托克)组合显示在22rv1模型中稳健的抗癌活性,在22rv1模型中展示64%肿瘤生长抑制。在整个研究中,还监测体重。各组平均体重示于图11。各组平均百分比体重变化示于图12。在第35天组1-11(如表1中所定义)的个体体重示于图13。在第35天组1-11个体的百分比体重变化示于图14。在21天治疗时间段之后,在末次剂量给药之后观察小鼠7天,或直至组1的单独肿瘤体积达到3000mm3,或组1的平均肿瘤体积达到2000mm3。在研究末尾测量肿瘤重量,收获肿瘤(qd治疗组为在末次剂量给药之后24小时;bid治疗组为在末次剂量给药之后12小时而q7d治疗组为在末次剂量给药之后6天):对于各肿瘤,1/2用于急冻,和1/2用于ffpe。获自研究(组1、5、6、9、10、11)的组织样品通过珠匀化器匀化(20s,4.5m/s)(fisherbrandbeadmill24匀化器,fisherscientific)和在1%triton缓冲剂(cellsignallingtechnology,cat#9803)随蛋白酶和磷酸酶抑制剂(热fisherscientific,cat#1861281,lot#ta261245)裂解。单独的样品通过于15,000rpm离心清洁,定量和对各治疗组汇集,和蛋白质表达通过免疫印迹分析。将30ng蛋白质载入4-12%凝胶和转移至pvdf膜。阻断抗裂解的半胱天冬酶3(cellsignallingtechnology,cat#9664,lot#21;1:1,000),抗-β-肌动蛋白(proteintech,cat#hrp-60008;1:10,000),和二次染色在5%乳中完成。抗-cmyc(cellsignallingtechnology,cat#4572;1:1,000),和抗-mcl-1(cellsignallingtechnology,cat#4572;1:1,000)抗体在bsa中稀释。图27a和c是免疫印迹图,并且显示裂解的半胱天冬酶3的量随治疗组的变化。图27b和d是免疫印迹图,并且显示mcl-1的量随治疗组的变化。图27e和g是免疫印迹图,并且显示c-myc的量随治疗组的变化。图27f和h是柱状图,并且显示各治疗组中c-myc/肌动蛋白相对媒介物的比率,分别描述在图27e和g的免疫印迹中。与媒介物以及单一治疗组相比,用cmpdstr(i)和维奈托克的组合治疗诱导裂解的半胱天冬酶3。从而,cmpdstr(i)和维奈托克的组合治疗导致的肿瘤生长抑制与肿瘤溶解产物免疫印迹确定的半胱天冬酶3蛋白的增加裂解相应。实施例4在c4-2异种移植前列腺癌模型中的效力研究该研究的目的是评价结构(i)化合物在治疗皮下c4-2人类前列腺癌异种移植模型中的体内疗效。c4-2细胞是建立自雄激素依赖性前列腺癌细胞系lncap的雄激素独立性前列腺癌细胞系。对于c4-2模型,在雄性ncg(nod-prkdcem26cd52il2rgem26cd22nju)小鼠中评价效力。在肿瘤接种之前于2gy(1gy=100拉德)进行全部小鼠的钴-60辐射2天。各小鼠在右胁腹接种0.1mlpbs和matrigel混合物(1:1)中的5x106肿瘤细胞(c4-2细胞)。肿瘤细胞接种日期表示为第0天。在平均肿瘤体积达到大约200mm3时进行去势。小鼠用氯胺酮/赛拉嗪麻醉;经由正中线阴囊切开进行手术去势,允许双侧访问半阴囊内容物;在暴露各睾丸之后,用6-0vicryl缝合线来结扎精索然后除去睾丸;然后用6-0vicryl缝合线分别封闭精囊和皮肤。在去势之后,肿瘤可以缩小(肿瘤消退),从而在平均肿瘤体积再生长至大约100mm3时开始随机化和治疗。如果由于肿瘤消退平均肿瘤体积很小,则基于体重进行随机化。在肿瘤如上文所述达到适当尺寸之后,将小鼠随机化为示于表2的治疗组1-9。各治疗以剂量给药体积5μl/g给予和继续21天(或对q7d治疗组22天)。对q7dx3周治疗组,在随机化后第1、8和15天进行剂量给药,在随机化后第22天终止研究。组合的治疗间隔各自是0h,和bid间隔是8h。随机化基于"匹配分布"方法(studydirectortm软件,版本3.1.399.19)的随机化区组设计进行。表2.c4-2异种移植研究的治疗组。在21天治疗时间段之后,在末次剂量给药之后观察小鼠7天,或直至组1的单独肿瘤体积达到3000mm3,或组1的平均肿瘤体积达到2000mm3。肿瘤体积在二维随机化之后用测径器每周测量两次,和体积用下式表达为mm3:"v=(lxwxw)/2",其中v是肿瘤体积,l是肿瘤长度(最长肿瘤尺度)和w是肿瘤宽度(与l垂直的最长肿瘤尺度)。肿瘤重量在研究末尾测量。肿瘤生长抑制(tgi):tgi%是抗癌活性的指示,和表示为:tgi(%)=100x(1-t/c)。t和c是治疗组和对照组在给定天分别的平均肿瘤体积(或重量)。在研究末尾收获肿瘤(对qd治疗组在末次剂量给药之后24小时;对bid治疗组在末次剂量给药之后12小时和对q7d治疗组在末次剂量给药之后6天):对于各肿瘤,1/2用于急冻,和1/2用于ffpe。实施例5lncapfgc异种移植前列腺癌模型中的效力研究该研究的目的是评价结构(i)化合物在治疗皮下lncapfgc人类前列腺癌异种移植模型中的体内疗效。lncap克隆fgc是建立自前列腺癌细胞系lncap的雄激素依赖性前列腺癌细胞系。对于lncap克隆fgc模型,在雄性ncg(nod-prkdcem26cd52il2rgem26cd22nju)小鼠中评价效力。在肿瘤接种之前1天,各小鼠在左胁腹经皮下植入雄激素丸剂(15mg/丸剂,含丙酸睾酮粉末)。在肿瘤接种之前于2gy(1gy=100拉德)进行全部小鼠的钴-60辐射2天。各小鼠在右胁腹接种在0.1mlpbs与matrigel(1:1)混合物中的5x106肿瘤细胞(lncap,克隆fgc细胞)。肿瘤细胞接种日期表示为第0天。平均肿瘤体积达到大约200mm3时进行去势。小鼠用氯胺酮/赛拉嗪麻醉;经由正中线阴囊切开进行手术去势,允许双侧访问半阴囊内容物;在暴露各睾丸之后,用6-0vicryl缝合线来结扎精索然后除去睾丸;然后用6-0vicryl缝合线分别封闭精囊和皮肤。在去势之后,肿瘤可以缩小(肿瘤消退),从而在平均肿瘤体积再生长至大约200mm3的情况下开始随机化和治疗。如果平均肿瘤体积由于肿瘤消退很小,则基于体重进行随机化。在如上文所述肿瘤达到适当尺寸之后,将小鼠随机化为示于表3的治疗组1-9。各治疗以剂量给药体积5μl/g给予并继续21天(或对q7d治疗组22天)。对q7dx3周治疗组,在随机化后第1、8和15天进行剂量给药,和在随机化后第22天终止研究。组合的治疗间隔各自是0h,和bid间隔是8h。随机化基于"匹配分布"方法(studydirectortm软件,版本3.1.399.19)的随机化区组设计进行。表3.lncapfgc异种移植研究的治疗组。在21天治疗时间段之后,在末次剂量给药之后观察小鼠7天,或直至组1的单独肿瘤体积达到3000mm3,或组1的平均肿瘤体积达到2000mm3。肿瘤体积在二维随机化之后用测径器每周测量两次,和体积用下式表达为mm3:"v=(lxwxw)/2",其中v是肿瘤体积,l是肿瘤长度(最长肿瘤尺度)和w是肿瘤宽度(与l垂直的最长肿瘤尺度)。肿瘤重量在研究末尾测量。肿瘤生长抑制(tgi):tgi%是抗癌活性的指示,和表示为:tgi(%)=100x(1-t/c)。t和c是治疗组和对照组在给定天分别的平均肿瘤体积(或重量)。在研究末尾收获肿瘤(对qd治疗组在末次剂量给药之后24小时;对bid治疗组在末次剂量给药之后12小时和对q7d治疗组在末次剂量给药之后6天):对于各肿瘤,1/2用于急冻,和1/2用于ffpe。实施例6c4-2异种移植前列腺癌模型中的效力研究结果该研究的目的是评价结构(i)化合物在治疗皮下c4-2人类前列腺癌异种移植模型中的体内疗效。c4-2细胞是建立自雄激素依赖性前列腺癌细胞系lncap的雄激素独立性前列腺癌细胞系。对于c4-2模型,在雄性ncg(nod-prkdcem26cd52il2rgem26cd22nju)小鼠中评价效力。各小鼠在右胁腹区域经皮下接种0.1mlpbs与matrigel(1:1)混合物中的5x106肿瘤细胞(c4-2细胞)。肿瘤细胞接种日期表示为第0天。在平均肿瘤体积达到大约200mm3时进行去势。小鼠用氯胺酮/赛拉嗪麻醉;经由正中线阴囊切开进行手术去势,允许双侧访问半阴囊内容物;在暴露各睾丸之后,用6-0vicryl缝合线来结扎精索然后除去睾丸;然后用6-0vicryl缝合线分别封闭精囊和皮肤。在平均肿瘤体积达到大约100-200mm3时随机化未去势的小鼠。在去势之后,在去势小鼠的平均肿瘤体积达到大约100-200mm3时开始随机化。随机化基于"匹配分布"方法(studydirectortm软件,版本3.1.399.19)进行。在研究中使用小鼠并随机分配为表4所示的研究组。表4.c4-2异种移植研究的治疗组。各治疗以剂量给药体积5μl/g给予,在分组后第1天开始(第1天),和继续21天(或对q7d治疗组22天)。对q7dx3周治疗组,在随机化第1、8、和15天进行剂量给药,和在随机化后第22天终止研究。组合的治疗间隔各自是0h,和bid间隔是8h。为了效率,在接种后第38天终止研究。肿瘤体积在二维随机化之后用测径器每周测量两次,和体积用下式表达为mm3:"v=(lxwxw)/2",其中v是肿瘤体积,l是肿瘤长度(最长肿瘤尺度)和w是肿瘤宽度(与l垂直的最长肿瘤尺度)。图15显示在整个c4-2异种移植研究期间的各组的平均肿瘤体积。在研究末尾收获肿瘤(对qd治疗组在末次剂量给药之后24小时;对bid治疗组在末次剂量给药之后12小时和对q7d治疗组在末次剂量给药之后6天):对于各肿瘤,1/2用于急冻,和1/2用于ffpe。在整个研究中监测全部动物的体重。图17显示在整个c4-2异种移植研究期间的各组平均体重。实施例7lncapfgc异种移植前列腺癌模型中的效力研究的结果该研究的目的是评价结构(i)化合物在治疗皮下lncapfgc人类前列腺癌异种移植模型中的体内疗效。lncap克隆fgc是建立自前列腺癌细胞系lncap的雄激素依赖性前列腺癌细胞系。对于lncap克隆fgc模型,在雄性ncg(nod-prkdcem26cd52il2rgem26cd22nju)小鼠中评价效力。在肿瘤接种之前1天,各小鼠在左胁腹经皮下植入雄激素丸剂(15mg/丸,含丙酸睾酮粉末,来自aladdin工业公司,usa)。各小鼠在右胁腹接种0.1mlpbs与matrigel(1:1)混合物中的1x107肿瘤细胞(lncap,克隆fgc细胞)。肿瘤细胞接种日期表示为第0天。在平均肿瘤体积达到大约200mm3时进行去势。小鼠用氯胺酮/赛拉嗪麻醉;经由正中线阴囊切开进行手术去势,允许双侧访问半阴囊内容物;在暴露各睾丸之后,用6-0vicryl缝合线来结扎精索然后除去睾丸;然后用6-0vicryl缝合线分别封闭精囊和皮肤。在平均肿瘤体积达到大约100-200mm3时随机化未去势的小鼠。在去势之后,在去势小鼠的平均肿瘤体积达到大约100-200mm3时开始随机化。随机化基于"匹配分布"方法(studydirectortm软件,版本3.1.399.19)进行。在研究中使用小鼠和随机分配为表5所示的研究组。表5.lncapfgc异种移植研究的治疗组。各治疗以剂量给药体积5μl/g给予,在分组后1天开始(第1天),和继续21天(或对q7d治疗组22天)。对q7dx3周治疗组,在随机化后第1、8和15天进行剂量给药,和在随机化后第22天终止研究。组合的治疗间隔各自是0h,和bid间隔是8h。为了效率,在接种后49天终止研究。肿瘤体积在二维随机化之后用测径器每周测量两次,和体积用下式表达为mm3:"v=(lxwxw)/2",其中v是肿瘤体积,l是肿瘤长度(最长肿瘤尺度)和w是肿瘤宽度(与l垂直的最长肿瘤尺度)。图16显示整个lncap异种移植研究期间的各组的平均肿瘤体积。在研究末尾收获肿瘤(对qd治疗组在末次剂量给药之后24小时;对bid治疗组在末次剂量给药之后12小时和对q7d治疗组在末次剂量给药之后6天):对于各肿瘤,1/2用于急冻,和1/2用于ffpe。在整个研究中监测全部动物的体重。图18显示整个lncap异种移植研究期间的各组平均体重。实施例8阿伏西地抑制22rv1前列腺癌细胞系中的rnapolii磷酸化为了评价阿伏西地对血清刺激的前列腺癌细胞的效果,通过流式细胞术评价rna聚合酶(pol)ii磷酸化。前列腺癌细胞(22rv1细胞)用dmso或阿伏西地(80nm或160nm)治疗三小时,和在样品收集之前血清刺激(10%)一小时。山羊抗-兔iggh&l用作同种型对照。图19通过流式细胞术显示在阿伏西地治疗之后与dmso对照相比的染色细胞的百分比。在用阿伏西地治疗的情况下,在流式细胞术测试中22rv1细胞显示rnapolii磷酸化染色的剂量依赖性降低。实施例9阿伏西地抑制雄激素独立性前列腺癌细胞系lncap和vcap中的psa表达在用25nm或100nm阿伏西地治疗之后48-小时,在前列腺癌细胞系vcap和lncap中测量psa蛋白质水平。在治疗当天,培养基用10ml新鲜培养基替换。vcap和lncap细胞用25nm或100nm阿伏西地治疗48小时。通过刮擦收集细胞,在pbs中洗涤,和在补充蛋白酶和磷酸酶抑制剂的ripa缓冲剂中用超声裂解。将30ng蛋白质载入4-12%凝胶,和转移至硝基纤维素膜。阻断抗-肌动蛋白和二次染色在5%乳中完成。抗-psa抗体在bsa中稀释。图20显示在48小时之后阿伏西地治疗与媒介物组相比抑制psa在vcap和lncap前列腺细胞系中在蛋白质水平的表达。实施例10阿伏西地诱导前列腺癌细胞系中的细胞死亡lncap细胞用100nm阿伏西地在规律的10%血清条件下治疗48小时。刮擦收集细胞,在pbs中洗涤,和在补充蛋白酶和磷酸酶抑制剂的ripa缓冲剂中用超声裂解。将30ng蛋白质载入4-12%凝胶,和转移至pvdf膜。阻断和染色在5%乳中进行。图21显示阿伏西地治疗诱导细胞死亡,如半胱天冬酶3裂解指示,其使用抗裂解的半胱天冬酶3抗体。抗-肌动蛋白用作负载对照。实施例11在pc-3小鼠异种移植模型中,口服阿伏西地前药保留在血浆和肿瘤中,和抑制mcl1和肿瘤生长口服阿伏西地前药(结构(i)化合物)的pk/pd分析在pc-3小鼠异种移植模型中进行。在补充10%胎牛血清的ham’sf12k培养基中,在37℃,在5%co2/空气气氛中,pc-3前列腺肿瘤细胞在体外保持为单层培养基。收获指数生长期的细胞并计数用于肿瘤接种。各小鼠在右胁腹区域经皮下接种0.1mlpbs中的pc-3前列腺肿瘤细胞(5×106细胞),用于肿瘤发展。在平均肿瘤尺寸达到大约300mm3时,将小鼠随机化和划分为研究组。结构(i)化合物于7.5或15mg/kg通过口服灌胃法给予,在剂量给药后对各剂量水平于0、0.5、1、2、4、8、12、16和24小时收集肿瘤和血浆。结构(i)化合物和阿伏西地在肿瘤和血浆中的浓度通过药代动力学分析的lc-ms/ms确定。图22a显示在给药结构(i)化合物之后24小时阿伏西地保留在血浆和肿瘤组织中。来自研究的额外肿瘤样品通过珠匀化器匀化和用含蛋白酶和磷酸酶抑制剂的ripa缓冲剂裂解。汇集各治疗组的样品并且通过免疫印迹用抗-mcl1抗体和抗-β-微管蛋白抗体作为负载对照分析蛋白质表达。图22b显示在口服给药之后4小时结构(i)化合物抑制pc-3肿瘤中的mcl1,如免疫印迹显示。还评价结构(i)化合物对pc-3小鼠异种移植模型中肿瘤生长的效果。在补充10%胎牛血清的ham’sf12k培养基中、在37℃、在5%co2/空气气氛中,pc-3前列腺肿瘤细胞在体外保持为单层培养物。收获指数生长期的细胞并计数用于肿瘤接种。各小鼠在右胁腹区域经皮下接种0.1mlpbs中的pc-3前列腺肿瘤细胞(5×106细胞),用于肿瘤发展。在平均肿瘤尺寸达到大约100mm3时,将小鼠随机化和划分为研究组。结构(i)化合物于1.25mg/kg每日两次(bid)给予21天,或7.5mg/kg或15mg/kg每周一次(q7dx3)口服灌胃。二维肿瘤体积用测径器每周测量两次,和体积用下式表达为mm3:v=(lxwxw)/2,其中v是肿瘤体积,l是肿瘤长度(最长肿瘤尺度)和w是肿瘤宽度(与l垂直的最长肿瘤尺度)。图22c显示口服给予的结构(i)化合物抑制pc-3小鼠异种移植模型中的肿瘤生长。实施例12i期,向患晚期实体肿瘤的患者给予的口服结构(i)化合物的药代动力学(pk)和药效学(pd)剂量逐步增加研究录用晚期转移性或进行性实体肿瘤患者,其抗拒或不耐已知为其病况提供临床益处的已确立的治疗。3-6名患者的同期组群各自接受逐步升高剂量的结构(i)化合物,使用修正的斐波纳契剂量逐步增加途径。一旦确立最佳剂量,可以录用额外患者来确认安全性并探索效力。在最大耐受剂量(mtd)的扩展同期组群中将录用二十名额外的患者。这是进行中的1期、开放标记、剂量逐步增加、安全、pk和pd研究。为口服结构(i)化合物建议的初始剂量和计划是1-mg平剂量每日一次(qd)持续14天,随后是7天不用药物的恢复时间段(各周期=21天)。在至少3名患者的第一同期组群中不存在剂量限制性毒性(dlts),用修正的斐波纳契剂量逐步增加方案增加剂量,并且根据描述于表8的剂量逐步增加计划开始bid剂量给药。同期组群6的第一名患者已于8mg结构(i)化合物bid录用。研究中录用的前14名患者的基线人口统计描述于表6。表6.基线人口统计(n=14itt)图23是描述同期组群5研究完整周期的图。迄今,不存在无法解释的毒性,并且不存在剂量限制性腹泻或中性粒细胞减少症的证据。迄今观察到的级别≥3的随治疗出现的不良事件报告在表7中。表7.级别≥3的随治疗出现的不良事件。meddra优选术语3级4级5级贫血1(7%)--胸痛1(7%)--疼痛1(7%)--肿胀1(7%)--恶性胸膜积液1(7%)--血尿1(7%)--缺氧1(7%)--低血压1(7%)--3名患者的顺序同期组群将继续用根据表8的逐步升高剂量治疗直至建立mtd。表8.a在研究过程期间可能加入额外和/或中间剂量水平。b剂量水平-1代表需要从开始剂量水平减少剂量的患者的治疗剂量。如果开始剂量水平最初就与出乎意料的或无法接受的毒性有关,这还充当更低剂量水平。应注意该情况的剂量给药是每隔一天的单次早晨给药(qod)(不需要傍晚给药)。c应注意同期组群1的剂量给药是每日一次(qd)早晨给药(不需要傍晚给药)。d如果临床上指示,可以研究高于11mgbid的剂量水平。如果在给定剂量水平3名患者中有1名观察到dlt,则将录用多至3名的额外患者和在该剂量水平治疗。在给定剂量水平加入多至3额外患者的情况下,如果那6名患者中仅1名经历dlt,则将剂量增加至下一个剂量水平。如果3-6患者中≥2名在剂量水平经历dlt,则将剂量降低至先前(更低)的剂量水平和将在该剂量水平录用3名额外患者。如果6名患者中任何0或1名患者经历dlt,但是已研究了下一个更高剂量水平,那么目前的剂量将被宣布为mtd并且研究将推进至扩展同期组群。mtd定义为剂量,在此6名患者中≤1名在周期1期间经历dlt,而在下一个更高剂量3至6名患者中至少2名在周期1期间经历dlt。一旦mtd已建立,将在该mtd录用20名额外患者。从在mtd录用的患者收集的数据将用来确认安全性,探索潜在的生物标记,和评价结构(i)化合物活性的潜在信号。全部患者可以于周期1期间提供的相同剂量继续以21天周期(14天活性治疗)接受结构(i)化合物直至他们经历无法接受的毒性或明确的疾病进展。20名患者扩展同期组群中的患者可以以28天周期在mtd接受结构(i)化合物,包括21天活性治疗和随后7天不用药物的恢复时间段,条件是耐受。在逐步增加相期间不允许患者内的结构(i)化合物剂量逐步增加,直至建立mtd。患者符合全部下述选择标准:1.具有晚期转移性或进行性实体肿瘤的组织学上确认的诊断,排除快速细胞更新的肿瘤类型,也即小细胞癌(肺和肺外),炎性乳腺癌(ibc),成神经管细胞瘤,成神经细胞瘤和黑色素瘤伴广泛肝转移(≥50%的肝牵涉;患黑色素瘤和转移至<50%的肝的患者是适宜的)2.抗拒或不耐已知为其病况提供临床益处的已确立的治疗3.患有一种或多种肿瘤,其是如实体肿瘤应答评价标准(recist)v1.1的描述可测量或可评价的4.具有≤1的东方合作肿瘤学集团(ecog)体力状态5.具有≥3个月的预期寿命6.年龄≥18岁7.具有阴性怀孕试验(如果是能生育的女性)8.具有可接受的肝功能:a)胆红素≤1.5x正常上限(uln)(除非与吉伯特氏综合征有关)b)天冬氨酸氨基转移酶(ast/sgot),丙氨酸氨基转移酶(alt/sgpt)和碱性磷酸酶≤2.5xuln**如果存在肝转移,则允许3xuln9.具有可接受的肾功能:计算肌酸酐清除率≥30ml/min10.具有可接受的血液学状态:c)粒细胞≥1500细胞/mm3d)血小板计数≥100,000(plt/mm3)e)血红蛋白≥8g/dl11.具有可接受的凝血状态:f)凝血酶原时间(pt)在1.5x正常限以内g)活化的部分促凝血酶原激酶时间(aptt)在1.5x正常限以内12.未育或同意使用适当的避孕方法。性活性患者及其伴侣在进入研究之前使用有效的避孕方法(激素或屏障节育方法或禁欲)并且在研究参与期间持续并且在末次给予研究药物之后持续至少3个月(男性)和6月(女性)。13.在任何研究相关性程序之前已阅读和签署institutionalreviewboard(irb)-批准的知情同意表(icf)。符合下述排除标准中任一种的患者禁止参与研究:1.病史:充血性心力衰竭(chf);心脏病,在周期1/第1天之前的过去6个月内心肌梗死;超声心动图(echo)测得左心室喷射级分(lvef)<45%,不稳定的心律不齐,或在周期1/第1天之前的14天内的心电图(ecg)缺血证据2.具有校正的qt间隔(用fridericia校正式)(qtcf),男性>450msec和女性>470msec3.具有需要抗惊厥治疗的癫痫发作4.存在症状性中枢神经系统转移病或需要局部治疗比如放疗、手术或在先前2周内增加剂量的类固醇的疾病5.具有严重慢性阻塞性肺疾病伴低氧血症(定义为呼吸空气的静息o2饱和≤90%)6.在周期1/第1天之前2周内已进行大手术7.具有需要系统治疗的活性的不受控的细菌、病毒或真菌感染8.怀孕或哺乳9.在进入研究之前28天或5个半衰期之内(两者中先发生者)(对亚硝基脲类或丝裂霉素c为6周)接受使用放疗、手术、化疗或研究疗法的治疗10.不愿或无法顺从该方案需要的程序11.具有已知的人类免疫缺陷病毒、乙肝或丙肝感染。有目前无活性的慢性肝炎病史的患者是适宜的12.就研究者和/或资助人看来具有能危害方案目标的严重的非恶性疾病(例如肾盂积水,肝衰竭或其它病况)13.目前正接受任何其它研究试剂14.具有对相似结构的化合物、生物学试剂或配制剂已展示的变应性反应15.具有吸收障碍(例如克罗恩病)或已进行胃肠道大手术,其能损伤吸收或能引起短肠综合征伴吸收障碍致腹泻。dlt定义为周期1中观察到的下述事件中任一种,无论研究者属性,除非存在清楚的其它解释:1.3级或更高的发热性中性粒细胞减少症2.4级中性粒细胞减少症≥7个连续天3.4级血小板减少症或3级血小板减少症伴临床上显著出血或需要血小板输注4.3或4级非血液学aes将视为剂量限制,无论本文描述的特定参数之外的持续时间5.4级恶心、呕吐或腹泻,无论持续时间6.由于随治疗出现的不良事件(teaes)或有关的严重实验室试验值使得剂量给药延缓>1周7.任何ast和alt上升>3xuln(如果基线值正常)或≥3x基线值(如果基线值异常)伴血清胆红素水平>2xuln8.任何≥3级的<72小时内未缓解的电解质紊乱(例如高钾血症、低磷酸盐血症、高尿酸血症)9.任何≥3级的肌酸酐升高10.任何5级毒性在研究期间在同期组群1-5中在特定时间点评价结构(i)化合物和阿伏西地的血浆pk参数。根据描述于表9的药代动力学采样计划从同期组群1-5的患者收集血液。表9.药代动力学采样计划a在每日一次(qd)早晨给药接受结构(i)化合物的第一剂量同期组群中录用的患者中不收集傍晚(pm)样品。b在前一天早晨给药之后大约24小时且在当天给药之前(即,周期1/第2天的采样会在第1天早晨给药之后24小时且在第2天早晨给药之前进行)c8-小时样品在临当天傍晚给药之前收集(对接受结构(i)化合物的患者为bid),或在当天给药之后8小时(对接受结构(i)化合物的患者为qd)。pk参数用标准非房室模型方法估计。使用真实样品收集次数而不是计划的收集次数。在定量限之下的血浆浓度视为0。嵌入的缺失血浆浓度(例如两个观察值之间的缺失值)用线性外推估计。这与用梯形法则计算auc相符。其它缺失的血浆浓度从估计pk参数的计算中排除。图24a和24b是血浆阿伏西地浓度(ng/ml)vs时间的图,和显示在每日口服qd剂量给药含有配制剂no.401-01的1-mg强度胶囊之后分别在第1和14天阿伏西地在同期组群1患者血浆中的浓度。在第14天在24小时之后受试者104显示一定的阿伏西地蓄积。受试者102在第14天剂量给药之前中断计划。图24c和24d是血浆阿伏西地浓度(ng/ml)vs时间的图,和显示在每日口服bid剂量给药含有配制剂no.401-01的1-mg强度胶囊之后分别在第1和14天阿伏西地在同期组群2患者血浆中的浓度。对bid仅可检测到1mg的阿伏西地,和对任何样品在任何时间点均未检测到结构(i)化合物。在第1天于8小时和再次于24小时在任何受试者中均不能检测到药物(小于1.0ng/ml阿伏西地)。然而,受试者201和202在第14天存在可检测的阿伏西地蓄积(平均=2.39ng/ml),意味着bid剂量给药在第14天促进保持药物水平。图24e和24f是血浆阿伏西地浓度(ng/ml)vs时间的图,和显示在每日口服bid剂量给药6mg配制剂no.401-01之后分别在第1和14天阿伏西地在同期组群5患者血浆中的浓度。表10报告同期组群5患者在周期1第1和14天的阿伏西地的tmax、cmax和auc(0-24)。表10.图24g是阿伏西地(ng/ml)vs同期组群的图,和显示在每日口服qd剂量给药含有配制剂no.401-01的1-mg强度胶囊之后在第1天和第14天的阿伏西地平均cmax。图24h是阿伏西地(ng*hr/ml)vs同期组群的图,和显示在每日口服bid剂量给药含有配制剂no.401-01的1-mg强度胶囊之后在第1天(auc0-8)和第14天(auc0-8和auc0-24)的阿伏西地的曲线下面积(auc)。在任何时间点均不存在可检测的结构(i)化合物。同期组群2显示从第1天至第14天平均cmax和auc显著增加,展示bidvsqd剂量给药的影响。在第1天同期组群5与同期组群4相比cmax增加46%,在第14天增加69%。auc的相应增加是在第1天52%和在第14天30%。图24i是阿伏西地平均浓度(nm)vs时间的图,和显示在24-小时时间段内同期组群5患者血浆中的阿伏西地平均浓度。通过用结构(i)化合物给予阿伏西地,阿伏西地能够以更低剂量在更长时间内给予,具有更低的毒性和相似的暴露。实施例13多晶型形式b结构(ii)化合物形式b的绝对立体化学、磷酸部分位置以及两性离子性质通过单晶体x射线衍射用下述参数确定:stoestadip.铜kαi辐射,40kv/40ma;mythen1k检测器透射模式,弯曲单色器,0.02°2θ步长,12s步时,1.5-50.5°2θ扫描范围,1°2θ检测器步进,步进扫描模式。各样品(25-40mg粉末)置于用金属垫圈(0.4mm厚,12-mm内径;"夹心元件")隔开的两片乙酸纤维素箔材之间。将夹心元件转移至用乙酸箔材密封的样品架(scell)。在环境空气气氛中获得样品和在测量期间旋转。图25显示得自形式b的xrpd分析的xrpd衍射图。形式b作为无水分子结晶,不存在溶剂包合。键距和键角全部在期望值以内。形式b产生的制表数据提供于表11。表11.形式b的xrpd衍射图的制表数据dsc用tainstruments的taq200/q2000dsc进行,使用缓变方法,卷曲铝样品盘,25℃。加热速率是10℃/分,冲洗气体是氮。图26显示多晶型形式b热流对温度作图的差示扫描量热法输出。形式b能够根据方案1描述的程序合成,描述如下。方案1。步骤1.1:在室温下向清洁和无水的三颈圆底烧瓶(rbf)(3l)加入a-1(90g,0.192mol)和氯苯(774ml)。在室温下向反应烧瓶缓慢加入bbr3(391.5g)。在完成bbr3加入之后,将反应混合物温度缓慢地提高至80-83℃,在相同温度搅拌反应混合物10小时。将反应混合物温度进一步提高至100-103℃,将反应混合物在100-103℃保持5小时。通过tlc和hplc监测反应进程。在完成反应之后,在室温下通过向反应混合物氮鼓泡并保持激烈搅拌除去hbr和甲基溴。反应混合物用甲醇(180ml)/水(90ml)混合物(270ml),随后甲醇(180ml)缓慢地猝灭。在大气压下在25-50℃蒸馏除去溶剂,达到12体积(vol)的目标反应质量体积。然后,在50-55℃用氢氧化钠溶液(48.8g溶于135mldm水)将反应混合物ph调节至3.0±1。再次,在大气压下在50-100℃蒸馏除去溶剂,达到12vol的目标反应体积。然后,在50℃用氢氧化钠溶液(8.5g溶于87mldm水)将反应混合物ph调节至ph8.1±0.2,随后在恒定搅拌下在50℃缓慢加水1小时。让反应混合物缓慢达到室温和在室温下保持3小时。过滤所得固体和用甲醇(315ml)/水(135ml)混合物(3x450ml)随后水(5x450ml)洗涤。在真空炉中在50-55℃干燥固体48小时,获得a-2,是黄色固体(70g,90%)。hplc纯度:99.72%。步骤2.1a:在室温下,在氮气氛下向清洁和无水的三颈rbf(3l)加入a-2(35.0g,0.087mol)和dmf(245ml)。然后,在室温下向反应混合物加入dmap(1.06g,0.0086mol)随后ccl4(66.5g0.434mol)。在室温下向反应混合物加入亚磷酸二叔丁酯(25.5g,0.131mol)。反应混合物在室温下在氮气氛下搅拌24小时。通过hplc监测反应进程。反应混合物冷却至0-5℃,在0-5℃缓慢加dm水(1950ml)30分钟猝灭。然后,将氯仿(1627.5ml)加入反应混合物,将反应混合物在0-5℃搅拌10分钟。分开有机层,在硫酸钠上干燥。减压除去溶剂,同时保持浴温低于45℃。所得残余物与甲苯(4×175ml)共蒸。残余物在高真空下保持45分钟,获得a-10,是淡黄色残余物。(51.0g,98.5%)。hplc纯度:91.48%。步骤2.1b:在室温下向清洁和无水的rbf(1l)加入a-10(51.0g,0.0858mol)和乙酸(102ml)。然后,在25-30℃滴加4nhcl溶液/1,4-二噁烷(102ml)。反应混合物在25-30℃搅拌40分钟。通过tlc监测反应进程。在反应完成之后,在搅拌下将甲苯(2×510ml)加入反应混合物,将反应混合物保持5分钟。停止搅拌,在25-30℃让反应混合物中的固体沉淀5分钟。倾析溶剂,获得半固体。半固体与甲苯(3x123ml)共蒸,获得淡黄色固体。所得淡黄色固体置入清洁rbf,在25-30℃加入甲醇(123ml)随后滴加水(41ml)。反应混合物在25-30℃搅拌2小时,获得淡黄色固体。过滤所得固体和真空干燥10分钟,获得a-11,是淡黄色固体(36.5g,82%)。hplc纯度:97.03%。该物质直接用于步骤3.1不加进一步干燥。步骤3.1:向清洁和无水的三颈500mlrbf加入a-11(34.0g,0.066mol)和acn(51ml)。在25-30℃在搅拌下向反应混合物滴加碳酸氢铵溶液(16.2g溶于170mldm水)30分钟。再次,在25-30℃缓慢加入acn(51ml)30分钟。反应混合物冷却至10-15℃和在10-15℃搅拌60分钟。过滤所得固体,用acn(102ml)洗涤。固体在真空炉中在25-30℃干燥16小时,获得a-4,是淡黄色固体(28.5g,90.10%)。hplc纯度:99.68%。步骤4.1:在室温下向清洁和无水的500-ml三颈rbf加入a-4(7.5g,0.015mol)和甲醇(187.5ml)。在50℃,在氮气氛下向反应混合物缓慢加入乙酸(7.5ml,1.0vol)。反应混合物在50℃在氮气氛下搅拌1小时。反应混合物冷却至室温和搅拌2小时。过滤固体,在真空下干燥,获得5.0ga-5(66.5%),是淡黄色固体。hplc纯度:99.77%。作为进行步骤4.1的备择方法,下述条件能够用来进行多晶型转化。在室温下向清洁和无水的100-ml三颈rbf加入a-4(2.0g,0.004mol),thf(29ml)和dm水(1.7ml)。然后,在室温下向反应混合物加入马来酸(0.44g)。反应混合物在室温下搅拌12小时。过滤所得固体和真空干燥。在室温下将湿固体溶于乙醇(12ml)和搅拌24小时。过滤所得固体,用乙醇(2.5ml)洗涤,真空干燥,获得a-5(1.5g,60%),是淡黄色固体。hplc纯度:99.91%。多晶型形式b用下文所指的组分配制为1-mg强度胶囊,其中百分比按重量/重量计算:为了制备,将结构(i)化合物和所指赋形剂的粉末共混物包囊在#4羟丙基甲基纤维素(hpmc)胶囊中。所得胶囊是即释胶囊。在包囊为胶囊之前,直接共混制备药物产品:将结构(i)化合物研磨入所指赋形剂,随后在手动胶囊填充设备上将胶囊填充在100片的胶囊板中。胶囊包装在铝泡罩包装中,1个胶囊每泡罩和7个胶囊每泡罩片。各片上有3个泡罩留空。实施例14口服结构(i)化合物的ii期研究,向转移的去势抵抗性前列腺癌患者每日给予两次持续21天这是建立结构(i)化合物(例如结构(i)化合物形式b)效力和安全性的2期、开放标记、非随机化、simon2-阶段设计研究,所述化合物在用雄激素信号转导抑制剂前线治疗后又进展的转移的去势抵抗性前列腺癌患者中每日服用一次持续28天周期的21天。在20名患者中的活组织检查子研究使得可以评价患者子集的组织生物标记。将录用六十名(60)患者。数据用来评价效力、确认安全性和探索相关的潜在生物标记。全部患者可以以周期1期间提供的相同剂量以28天周期(21天活性治疗)继续接受结构(i)化合物直至他们经历无法接受的毒性或明确的疾病进展。适宜的患者必须符合全部下述选择标准:1.也具有组织学上或细胞学上确认的前列腺腺癌的男性患者;和:h)对用雄激素剥夺疗法(adt)治疗(或双侧睾丸切除术后状态)有去势抵抗性,睾酮水平小于(<)50纳克每分升(50ng/dl,相当于1.7nmol/l);和:i)根据前列腺癌临床试验工作组3(pcwg3)标准具有放射照像确定的进展,并且正用乙酸阿比特龙或恩杂鲁胺与adt组合治疗2.抗拒或不耐已知为其病况提供临床益处的已确立的治疗3.具有一种或多种肿瘤,其是通过实体肿瘤应答评价标准(recist)v1.1的描述可测量的4.愿意进行两(2)次研究活体检查(仅活组织检查子研究同期组群)5.具有≤1的东方合作肿瘤学集团(ecog)体力状态6.具有≥3个月的预期寿命7.年龄≥18岁8.具有可接受的肝功能:a)胆红素≤1.5x正常上限(uln)(除非与吉伯特氏综合征有关)b)天冬氨酸氨基转移酶(ast/sgot),丙氨酸氨基转移酶(alt/sgpt)和碱性磷酸酶≤2.5xuln**如果存在肝转移,则3xuln是允许的。9.具有可接受的肾功能:计算肌酸酐清除率≥30ml/min10.具有可接受的血液学状态:a)粒细胞≥1500细胞/mm3b)血小板计数≥100,000(plt/mm3)c)血红蛋白≥8g/dl11.具有可接受的凝血状态:a)凝血酶原时间(pt)在1.5x正常限以内b)活化的部分促凝血酶原激酶时间(aptt)在1.5x正常限以内12.未育或同意使用适当的避孕方法。性活性患者及其伴侣必须在进入研究之前使用有效的避孕方法(节育激素或屏障方法或禁欲)并且持续研究参与时间并且在末次给予研究药物之后持续至少3个月(男性)和6个月(女性)。如果在其伴侣参与该研究期间女性变得怀孕或怀疑怀孕,她应立即告知其治疗医师。13.在任何研究-相关性程序之前已阅读和签署institutionalreviewboard(irb)-批准的知情同意表格(icf)。(在为参与研究再筛选患者或者方案修改使进行中的患者护理改变的情况下,必须签署新icf)。符合这些排除标准中任一种的患者将禁止参与该研究:1.病史:充血性心力衰竭(chf);心脏病,在周期1/第1天之前的过去6个月心肌梗死2.具有校正的qt间隔(用fridericia校正式)(qtcf),男性>450msec和女性>470msec3.具有需要抗惊厥治疗的癫痫发作4.存在症状性中枢神经系统转移的疾病或在先前2周以内需要局部治疗比如放疗、手术或增加剂量的类固醇的疾病5.具有严重慢性阻塞性肺疾病伴低氧血症(定义为呼吸空气的静息o2饱和≤90%)6.在周期1/第1天之前的2周内已进行大手术7.具有需要系统治疗的活性、不受控的细菌、病毒或真菌感染8.怀孕或哺乳9.在进行研究之前的28天或5个半衰期内(两者中先发生者)(对亚硝基脲类或丝裂霉素c为6周)接受放疗、手术、化疗或研究治疗10.不愿或无法顺从该方案需要的程序11.具有人类免疫缺陷病毒、乙肝或丙肝的已知感染。有慢性肝炎病史但目前无活性的患者是适宜的12.具有就研究者和/或资助人看来能危害方案目标的严重非恶性疾病(例如肾盂积水、肝衰竭或其它病况)13.目前接受任何其它研究试剂14.具有对相似结构的化合物、生物学试剂或配制剂已展示的变应性反应15.具有吸收障碍(例如克罗恩病等)或已进行胃肠道大手术,其能损伤吸收或能引起短肠综合征伴吸收障碍致腹泻。录用的患者将接受结构(i)化合物(例如作为含有配制剂no.401-01的1-mg胶囊提供,其中结构(i)化合物是结构(i)化合物形式b),每日给予两次(bid),持续28天周期的前21天。成功完成4-周治疗周期而无显著治疗-相关性毒性或进行性疾病证据的患者将继续接受使用相同剂量和剂量给药计划的治疗。效力评价基于pcwg3-调整的recistv1.1指南进行,从而包括客观应答速率(orr),dor,应答类型(例如完成缓解、部分缓解、稳定疾病)和进展时间的评价。orr定义为根据pcwg3-调整的recistv1.1标准的cr或pr患者,相对应答可评价群体的百分比。orr将通过符合orr定义的患者数量和百分比总结,具有相应的精确95%置信区间。口服结构(i)化合物的耐受和毒性通过评价物理检查、生命体征、实验室参数、aes包括dlts和全部死因来评价。随治疗出现的不良事件(teaes)的发生率将在各剂量水平内用medicaldictionaryforregulatoryactivities(meddra)优选术语和主要系统器官分类水平来总结。将对aes子集比如(1)研究者认为涉及研究治疗的那些和(2)严重不良事件(saes)进行相似总结。其它惯例安全评价(例如临床实验室参数和生命体征)将通过结构(i)化合物剂量水平用平均值、标准偏差、中位数、相对基线值的最小和最大变化来总结。潜在肿瘤和外周血液生物标记的pd参数和评价包括但不限于活组织检查和ctc样品中的cdk9-相关性基因(包括c-myc);磷酸-ar;活组织检查和pbmc样品上的磷酸rnapol2;血清psa。从全部患者收集血液以评价结构(i)化合物药效学和潜在生物标记。在基线(在周期1/第1天剂量给药之前)和周期二(2)末尾在参与活组织检查子研究的患者子集中取得活组织检查样品。从全部患者要求最近存档的肿瘤组织(原发和转移位点,如果可获得)以评价潜在生物标记。本说明书或所附申请数据页中提及的全部美国专利、美国专利申请公开、美国专利申请、外国专利、外国专利申请和非专利公开通过援引全部并入本文,条件是与本说明书并不冲突。从前文将认识到,尽管本文出于展示意图已描述本公开的特定实施方式,可以进行各种变型而不偏离本公开的主旨和范围。相应地,本公开并不受所附权利要求之外的限制。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1