一种生物基因序列的概要数据生成方法及系统
1.本公开涉及生物数据处理技术领域,特别涉及一种生物基因序列的概要数据生成方法及系统。
背景技术:
2.本部分的陈述仅仅是提供了与本公开相关的背景技术,并不必然构成现有技术。
3.随着测序技术的发展,生物基因数据库的规模越来越大。从刚开始的公开基因库数据总量不到五千万条核苷酸序列,到现如今一个测序仪器测序一次便可产生超过一万亿条序列,数据规模急剧增加,新测序技术的数据产生能力已经超越了“摩尔定律”。为了高效的处理基因数据,mash、dashing等工具相继被开发出来。在这类工具中,对于基因数据的处理转化为对基因序列中一系列公共子序列片段(k
‑
mer,长度为k的子字符串)的处理。一般会应用均匀、确定性的哈希算法将这些子序列映射成哈希值,然后对哈希值进行处理,采用一定方法形成相应的sketch(即一个能够代表或者总结原始数据的紧凑的、近似的数据摘要),这个sketch相比于原基因数据要小得多,原基因数据可能有几个g的大小,而sketch仅有几kb或者几十kb,这样就达到了数据降维的效果。形成sketch后,后续利用不同序列的sketch进行比对,可以得到jaccard系数、distance等,从而进行相应的研究。而minhash、hyperloglog是其中比较具有代表性的sketch算法。
4.minhash最初是为了检测近乎重复的网页和图像而开发的,后应用于生物信息学领域。它依赖于哈希函数的均一性和确定性,即对于输入的一系列数据经过哈希函数之后,需要均匀随机的分布在输出数据的范围之内,并且对于相同的输入应该保证有相同的输出。minhash算法处理序列数据的方式是对每条序列构建一个sketch集合,通过集合之间的jaccard相似性来估计序列的相似性。其对序列数据的处理方式更详细一些的描述为通过滑动窗口的方式,将序列切分为一个个的k
‑
mer,通过哈希函数将k
‑
mer作为输入,输出为一个哈希值,这就完成了字符串到整数的映射。所有得到的哈希值构建成一个集合,通过对于这些集合之间相似性的计算来代表原始序列的相似性。但是由于全部的k
‑
mer所映射的哈希值所构成的集合大小和序列长度呈线性关系,所以为了达到对数据量的压缩目的,对两条序列所对应的全部哈希值所构成的集合进行计算jaccard相似度的时候,如果两个集合是通过相同的哈希函数构建出来的,根据哈希函数的均一性和确定性,经过哈希映射之后的哈希值在集合中的分布是随机的,于是两个集合拥有相同的最小哈希值的概率和两个集合交集的大小与并集大小的比值是相等的,两个集合之间交集和并集的比值正是jaccard相似性的定义。这也是minhash的基本思路。
5.基于hyperloglog的sketch算法近年来也被应用于生物序列分析之中,hyperloglog是基于数据位模式(bit
‑
pattern)来估计集合的势(集合中元素的个数)。hyperloglog的思想就是通过一个具有均一性和确定性的哈希函数对原始数据求哈希值,然后通过哈希值中最长的前导零加一个1的值去估计集合中有多少个不同的元素。构建sketch时,先对于序列中的每个元素k
‑
mer计算哈希值,然后取出这个哈希值的前缀p位作
为sketch中的索引,索引到寄存器数组相应的位置处,剩余的q位根据前导零的个数去估计每个寄存器集合的大小。当获得一个新的更长的前导零,就更新当前寄存器中的数据。最后通过对每个寄存器存储的值求调和平均数来估计原始集合势的大小。
6.发明人发现,在minhash sketch和hyperloglog sketch相关应用中,对于哈希值的计算是其中的计算密集区域之一,整个程序的性能受限于对于哈希值的求解,目前还缺少性能更优的求解方法;同时,在哈希函数计算之前,对于输入内容k
‑
mer的整理和哈希函数计算之后对于哈希值列表的处理是程序另外的热点部分,现有sketch算法缺乏对哈希求解前和求解后的数据优化处理。
技术实现要素:
7.为了解决现有技术的不足,本公开提供了一种生物基因序列的概要数据生成方法及系统,基于单指令多数据流simd改进的多种哈希函数建基因序列的哈希值列表,采用向量化的实现方式,计算速度更快,能够实现生物基因序列的更高效处理。
8.为了实现上述目的,本公开采用如下技术方案:
9.本公开第一方面提供了一种生物基因序列的概要数据生成方法。
10.一种生物基因序列的概要数据生成方法,包括以下过程:
11.获取待处理的基因序列;
12.将待处理的基因序列利用滑动窗口进行k
‑
mer分解,每次按顺序切分出一个定长k
‑
mer,并得到其基因序列的反向互补链,将m个k
‑
mer以及其反向互补链的k
‑
mer分别封装进向量,采用二进制掩码形式将正向k
‑
mer和反向k
‑
mer进行向量化对比,留下字符值较小的k
‑
mer,将留下的m个k
‑
mer进行向量化转置操作;
13.将转置操作得到的向量输入到基于单指令多数据流simd改进的哈希函数中,得到各个向量对应的哈希值;
14.继续滑动窗口获取新的子序列k
‑
mer,重复上述操作,直到待处理基因序列的所有k
‑
mer都计算出对应的哈希值,根据所有的哈希值构建待处理基因序列的哈希值列表;
15.根据哈希值列表,生成待处理的基因序列的概要数据。
16.进一步的,将输入的转置之后的m个向量,以32位或64位的整数倍为一个单位再次进行划分,每个向量每满n位,就单独划分出来进行一组向量化的位运算操作,其中n为32或64的整数倍;
17.所有满n位的部分处理完毕,最后剩余的不足n位的tail部分进行单独的向量化的位运算操作;
18.将运算结果进行整合得到结果向量,将得到的结果向量通过相应指令解析得到m个序列的哈希值。
19.进一步的,将m个k
‑
mer数据载入m个向量中,利用向量化方式进行转置操作。
20.进一步的,对tail部分的数据,采用对应的掩码将switch语句替换。
21.进一步的,哈希函数计算过程中,利用多路展开并行操作进行填充。
22.进一步的,采用minhash sketch算法生成概要数据。
23.更进一步的,k
‑
mer输入到哈希函数之前,在正向kmer和反向kmer对比过程中,采用位操作的方式代替switch语句。
24.更进一步的,k
‑
mer输入到哈希函数之前,基于simd改进得到k
‑
mer对比算法,利用二进制掩码,采用向量化指令进行比较。
25.更进一步的,生成哈希值列表之后,在优先队列的实现过程中采用robin
‑
hood
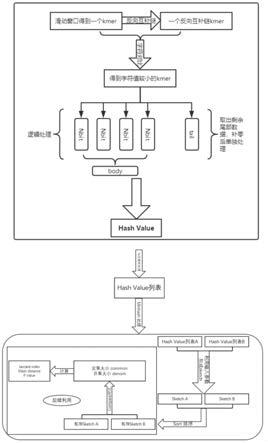
‑
hashing来代替unordered_map方法。
26.进一步的,采用hyperloglog sketch算法生成概要数据。
27.更进一步的,在sketch生成阶段distance阶段采用openmp多线程。
28.更进一步的,对于并集集合,在内部循环中使用单指令多数据流对多个相邻操作数的向量执行算术和位运算。
29.本公开第二方面提供了一种生物基因序列的概要数据生成系统。
30.一种生物基因序列的概要数据生成系统,包括以下过程:
31.数据获取模块,被配置为:获取待处理的基因序列;
32.k
‑
mer分解模块,被配置为:将待处理的基因序列利用滑动窗口进行k
‑
mer分解,每次按顺序切分出一个定长k
‑
mer,并得到其基因序列的反向互补链,将m个k
‑
mer以及其反向互补链的k
‑
mer分别封装进向量,采用二进制掩码形式将正向k
‑
mer和反向k
‑
mer进行向量化对比,留下字符值较小的k
‑
mer,将留下的m个k
‑
mer进行向量化转置操作;
33.哈希计算模块,被配置为:将转置操作得到的向量输入到基于单指令多数据流simd改进的哈希函数中,得到各个向量对应的哈希值;
34.哈希值列表生成模块,被配置为:继续滑动窗口获取新的子序列k
‑
mer,重复上述操作,直到待处理基因序列的所有k
‑
mer都计算出对应的哈希值,根据所有的哈希值构建待处理基因序列的哈希值列表;
35.概要数据生成模块,被配置为:根据哈希值列表,生成待处理的基因序列的概要数据。
36.本公开第三方面提供了一种计算机可读存储介质,其上存储有程序,该程序被处理器执行时实现如本公开第一方面所述的生物基因序列的概要数据生成方法中的步骤。
37.本公开第四方面提供了一种电子设备,包括存储器、处理器及存储在存储器上并可在处理器上运行的程序,所述处理器执行所述程序时实现如本公开第一方面所述的生物基因序列的概要数据生成方法中的步骤。
38.与现有技术相比,本公开的有益效果是:
39.1、本公开所述的方法、系统、介质或电子设备,基于单指令多数据流simd改进的多种哈希函数建基因序列的哈希值列表,采用向量化的实现方式,计算速度更快,能够实现生物基因序列的更高效处理。
40.2、本公开所述的方法、系统、介质或电子设备,采用将m个数据载入m个向量中的形式,然后利用向量化手段对其进行转置操作,造成m个数据无法全部载入且无法取得m个数据相同位置处的32/64位的问题。
41.3、本公开所述的方法、系统、介质或电子设备,利用相应的掩码将switch语句替换掉,避免了分支预测失效,并使其能够充分利用向量化操作,大大提升了程序性能。
42.4、本公开所述的方法、系统、介质或电子设备,利用多路展开并行操作进行填充,使得依赖降低,进一步提升了哈希函数的性能。
附图说明
43.构成本公开的一部分的说明书附图用来提供对本公开的进一步理解,本公开的示意性实施例及其说明用于解释本公开,并不构成对本公开的不当限定。
44.图1为本公开实施例1提供的现有技术中的minhash sketch方法流程图。
45.图2为本公开实施例1提供的改进的minhash sketch方法流程图。
46.图3为本公开实施例1提供的现有技术中的hyperloglog sketch方法流程图。
47.图4为本公开实施例1提供的改进的hyperloglog sketch方法流程图。
具体实施方式
48.下面结合附图与实施例对本公开作进一步说明。
49.应该指出,以下详细说明都是例示性的,旨在对本公开提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本公开所属技术领域的普通技术人员通常理解的相同含义。
50.需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本公开的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
51.在不冲突的情况下,本公开中的实施例及实施例中的特征可以相互组合。
52.实施例1:
53.本公开实施例1提供了一种生物基因序列的概要数据生成方法,对于哈希值的计算,本实施例提供了基于单指令多数据流simd改进的多种哈希函数,包括murmurhash3、cityhash、xxhash、wanghash,利用这些哈希函数构建基因序列的哈希值列表,针对不同情况选择不同哈希函数,使适用性更广;采用向量化的实现方式,使其速度更快。
54.原始哈希函数计算哈希值的方法如下:
55.对于待处理的序列数据,利用滑动窗口生成一个k
‑
mer,然后对该k
‑
mer进行处理,获得其反向互补链(dna一般呈现双链结构,是由两条单链盘绕形成的,两条单链具有互补特性,即每两个碱基之间组成碱基对,这种配对具有特异性,即a只能与t形成配对,c只能与g形成配对,称为碱基的互补配对。其中两条互补的单链在方向上是相反的,每条单链称为另一条单链的反向互补链),然后两k
‑
mer对比,获得字符值较小的k
‑
mer,然后将此k
‑
mer传入哈希函数进行操作,将其分成body部分和tail部分,分别进行处理。在之后,原始算法对body部分每次处理32/64位的整数倍,剩下的不足以构成32/64位整数倍的,作为tail部分单独进行处理,对其进行移位操作然后再进行相应的位运算。
56.例如一个k
‑
mer,k=21,其转化为二进制为168位,假设哈希函数每次操作64位,则body部分为前128位,每次处理64位,tail部分为剩余的40位,对其移位,达到补零补充到64位的效果,再进行相应的位运算,最终获得相应的哈希值。
57.本实施例中,针对已有的hash算法进行了单指令多数据流(simd)的向量化操作,具体实现如下:
58.获取待处理的基因序列;
59.将待处理的基因序列利用滑动窗口进行k
‑
mer分解,每次按顺序切分出一个定长
k
‑
mer,并得到其基因序列的反向互补链,然后将m个k
‑
mer以及其反向互补链的k
‑
mer分别封装进向量,采用二进制掩码形式将正向k
‑
mer和反向k
‑
mer进行向量化对比,即对于每对正向、反向k
‑
mer,选取其中字符值较小的k
‑
mer,最终留下了m个字符值较小的k
‑
mer,将留下的m个k
‑
mer进行向量化转置操作;
60.选取合适的哈希函数,将转置操作得到的向量输入到基于单指令多数据流simd改进的哈希函数中;
61.将输入的转置之后的m个向量根据不同哈希函数的特点,以32/64位的整数倍为一个单位再次进行划分,因为m个k
‑
mer是等长的,所以这m个向量能够找到一个统一的二次划分:每个向量每满n位(n为32/64的整数倍)就单独划分出来进行一组向量化的位运算操作;
62.所有满n位的部分处理完毕,最后剩余的不足n位的部分(即tail部分)进行单独的向量化的位运算操作;
63.将运算结果通过向量化技术进行整合,然后将得到的结果向量通过相应指令解析得到m个序列的哈希值并存储;
64.继续滑动窗口获取新的子序列k
‑
mer,重复上述操作,直到待处理基因序列的所有k
‑
mer都计算出对应的hash值,从而构建出待处理基因序列的哈希值列表。
65.一般m的取值为8,k
‑
mer的默认长度为21.对于序列可以设定不同的k
‑
mer长度,从而切分出的k
‑
mer数量也不一样。
66.本实施例中还采用了如下改进:
67.(1)原始哈希函数每次接收一个数据,而基于simd的哈希函数要求一次性处理m个数据,因此需要对数据的输入进行合并,将其载入成向量的形式。并且原始哈希函数计算过程中是分为body和tail部分,tail部分单独处理,body部分以32/64位为一个基本操作单位,多次循环进行处理。因此基于simd的哈希函数在body部分也必须一次性处理m*(32/64)位,且必须是这m个数据中的每个数据相同位置处的32/64位,也就是说第一个向量载入的应该是这m个数据的前32/64位。但是如果按照原本向量的载入形式,那么其会从头开始载入,载入第一个数据、第二个数据
···
,由于最多载入512bit,由于一个数据一般会超过64bit,这就势必造成m个数据无法全部载入,且无法取得m个数据相同位置处的32/64位。所以载入的数据必须进行重构。
68.本实施例中,采用将m个数据载入m个向量中的形式,然后利用向量化手段对其进行转置操作,解决了上述问题。
69.(2)原始哈希函数单独处理tail部分的数据,一般采取switch
‑
case分支判断方式,这很可能带来很大的开销。
70.本实施例利用相应的掩码将switch语句替换掉,避免了分支预测失效,并使其能够充分利用向量化操作,大大提升了程序性能。
71.(3)哈希函数计算过程中存在一定的依赖关系,这种依赖关系会降低向量化指令的速度。本实施例利用多路展开并行操作进行填充,使得依赖降低,进一步提升了函数的性能。
72.形成sketch的算法过程中,minhash sketch和hyperloglog sketch关于哈希值的计算部分是一致的,在其之后的部分有所不同。
73.如图1所示,其中minhash sketch原始算法后续流程为:
74.获取一个待处理基因序列的哈希值列表;
75.选定合适的sketch大小s,用优先队列对哈希值列表进行处理,获取s个最小的哈希值,形成该基因序列的sketch;如果需要更新当前sketch,需要将两次获得的最小哈希值列表进行合并,重新选取最小的s个值作为新的sketch。
76.重复上述步骤,直到待处理基因序列都形成所对应的sketch;
77.形成sketch后,本实施例可以进行多方面利用,比如比较两个基因序列所对应的各自的sketch,求二者的jaccard系数,即求|s(a∪b)∩s(a)∩s(b)|与|s(a∪b)|的比值,这是jaccard系数的无偏估计。
78.其中,s(a)表示基因序列a的sketch,s(b)表示基因序列b的sketch,s(a∪b)表示两个sketch的并集中s个最小的哈希值所重新形成的一个集合。
79.根据minhashsketch计算jaccard系数等,进而判断其相似度。sketch方法在基因组组装、对宏基因组样本做聚类,亦或者使用不同平台测序数据(拼接好的和未拼接的)实时搜索基因数据库等方面中都得到有效利用。
80.除了哈希函数的优化,对于哈希函数的输入数据的整理和输出哈希值列表的处理还包括如下改进,如图3所示:
81.(1)k
‑
mer传入哈希函数之前,需要求其反向互补链,在生成反向互补链部分,原始算法(如图1)流程中是通过switch
‑
case分支语句预测来构成反向互补链的,而这部分预测错误会有惩罚,带来性能下降。
82.本实施例改进了原始算法的switch
‑
case分支语句预测,本实施例采用位操作的方式代替switch语句,避免了分支预测失效,极大地提高了反向互补链的生成速度。
83.(2)k
‑
mer传入哈希函数之前,得到反向互补链之后,还需要对其进行对比,求得字符值较小的k
‑
mer,然后将此k
‑
mer传入哈希函数求取哈希值。正向k
‑
mer与其反向互补链k
‑
mer比较的部分,原始算法中是调用memcmp函数来进行对比,从而留下字符值小于预设值的k
‑
mer。
84.但是逐个对比速度会比较慢,本实施例基于simd改进得到性能更好的k
‑
mer对比算法,即前面哈希函数算法优化中所述,利用二进制掩码,采用向量化指令进行比较,获得了较好的性能加速比。
85.(3)哈希函数生成哈希值列表之后,在优先队列的实现过程中采用robin
‑
hood
‑
hashing来代替unordered_map方法,提高构建过程中所用的数据结构的性能。
86.(4)对于生成sketch之后的后续应用中,如果求mash距离,原始算法采用基于merge的算法,而本实施例采用基于block的思想,扩展了原始算法,使其速度更快。
87.(5)不同的序列构建sketch的过程没有相互依赖,可以采用多线程实现,在多核平台上,应该将数量设置为可用的最大cpu核数,以获得最佳性能,并且线程亲和性和线程绑定模式上,本实施例采用compact或者scatter模式会比defult模式拥有更佳的性能。
88.如图2所示,hyperloglog sketch原始算法流程为:
89.获取一个待处理基因序列的哈希值列表;
90.构建大小为m的hll数组,初始值为0,对哈希值列表进行处理,将每个哈希值分为前缀p和后缀q,前缀p索引数组位置,后缀q计算该位置处的lzc(前导0计数,即q中从左到右直到遇到最左边的1为止的位数),然后与数组该位置处已存储的lzc进行对比,留存较大的
lzc值,最终得到数组该位置处最大的lzc值即lzc
max
。处理完哈希值列表,得到hyperloglog的sketch;
91.重复上述步骤,直到待处理基因序列都形成所对应的sketch;
92.形成sketch后,本实施例可以利用形成的sketch,估计计算不同集合的基数,其公式为:
[0093][0094]
其中,e为估算的基数,m为数组的大小,α
m
为校正因子,中m
j
为1+lzc
max
,还可以求两个基因序列的jaccardindex,获得其相似度,可用于一系列聚类等操作。
[0095]
对于其中的性能优化部分,除了如上所述的哈希函数优化部分,还包括如下改进,如图4所示:
[0096]
(1)本实施例使用了openmp多线程,形成sketch和求其distance阶段都很容易扩展到100个线程,并且获得了较好的加速比。
[0097]
(2)在应用部分,实现了基于单指令多数据流simd的优化。在算法之中,对于并集和交集基数的估计是相似性度量的重要组成部分,对其进行优化能很好的加快速度。因此,对于集合并集,在内部循环中使用单指令多数据流(simd),能够对多个相邻操作数的向量执行快速算术和位运算,将重要的循环使用simd指令,从而加快了速度,采用基于avx512的指令集会比基于sse2指令集提速20%左右。对集合基数和集合交集基数的统计过程很难用此方法,使用手动循环展开来加速这些内部循环。
[0098]
实施例2:
[0099]
本公开实施例2提供了一种生物基因序列的概要数据生成系统,包括以下过程:
[0100]
数据获取模块,被配置为:获取待处理的基因序列;
[0101]
k
‑
mer分解模块,被配置为:将待处理的基因序列利用滑动窗口进行k
‑
mer分解,每次按顺序切分出一个定长k
‑
mer,并得到其基因序列的反向互补链,将m个k
‑
mer以及其反向互补链的k
‑
mer分别封装进向量,采用二进制掩码形式采用二进制掩码形式将正向k
‑
mer和反向k
‑
mer进行向量化对比,即对于每对正向、反向k
‑
mer,选取其中字符值较小的k
‑
mer,最终留下了m个字符值较小的k
‑
mer,将留下的m个k
‑
mer进行向量化转置操作;
[0102]
哈希计算模块,被配置为:将转置操作得到的向量输入到基于单指令多数据流simd改进的哈希函数中,得到各个向量对应的哈希值;
[0103]
哈希值列表生成模块,被配置为:继续滑动窗口获取新的子序列k
‑
mer,重复上述操作,直到待处理基因序列的所有k
‑
mer都计算出对应的哈希值,根据所有的哈希值构建待处理基因序列的哈希值列表;
[0104]
概要数据生成模块,被配置为:根据哈希值列表,生成待处理的基因序列的概要数据。
[0105]
所述系统的工作方法与实施例1提供的生物基因序列的概要数据生成方法相同,这里不再赘述。
[0106]
实施例3:
[0107]
本公开实施例3提供了一种计算机可读存储介质,其上存储有程序,该程序被处理
器执行时实现如本公开实施例1所述的生物基因序列的概要数据生成方法中的步骤。
[0108]
实施例4:
[0109]
本公开实施例4提供了一种电子设备,包括存储器、处理器及存储在存储器上并可在处理器上运行的程序,所述处理器执行所述程序时实现如本公开实施例1所述的生物基因序列的概要数据生成方法中的步骤。
[0110]
本领域内的技术人员应明白,本公开的实施例可提供为方法、系统、或计算机程序产品。因此,本公开可采用硬件实施例、软件实施例、或结合软件和硬件方面的实施例的形式。而且,本公开可采用在一个或多个其中包含有计算机可用程序代码的计算机可用存储介质(包括但不限于磁盘存储器和光学存储器等)上实施的计算机程序产品的形式。
[0111]
本公开是参照根据本公开实施例的方法、设备(系统)、和计算机程序产品的流程图和/或方框图来描述的。应理解可由计算机程序指令实现流程图和/或方框图中的每一流程和/或方框、以及流程图和/或方框图中的流程和/或方框的结合。可提供这些计算机程序指令到通用计算机、专用计算机、嵌入式处理机或其他可编程数据处理设备的处理器以产生一个机器,使得通过计算机或其他可编程数据处理设备的处理器执行的指令产生用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的装置。
[0112]
这些计算机程序指令也可存储在能引导计算机或其他可编程数据处理设备以特定方式工作的计算机可读存储器中,使得存储在该计算机可读存储器中的指令产生包括指令装置的制造品,该指令装置实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能。
[0113]
这些计算机程序指令也可装载到计算机或其他可编程数据处理设备上,使得在计算机或其他可编程设备上执行一系列操作步骤以产生计算机实现的处理,从而在计算机或其他可编程设备上执行的指令提供用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的步骤。
[0114]
本领域普通技术人员可以理解实现上述实施例方法中的全部或部分流程,是可以通过计算机程序来指令相关的硬件来完成,所述的程序可存储于一计算机可读取存储介质中,该程序在执行时,可包括如上述各方法的实施例的流程。其中,所述的存储介质可为磁碟、光盘、只读存储记忆体(read
‑
only memory,rom)或随机存储记忆体(random accessmemory,ram)等。
[0115]
以上所述仅为本公开的优选实施例而已,并不用于限制本公开,对于本领域的技术人员来说,本公开可以有各种更改和变化。凡在本公开的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本公开的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1