光辉性色调剂、静电荷图像显影剂、色调剂盒、处理盒、图像形成设备和图像形成方法与流程
本发明涉及光辉性色调剂、静电荷图像显影剂、色调剂盒、处理盒、图像形成设备和图像形成方法。
背景技术:
近年来,检验了出于形成具有光泽(例如金属光泽)的图像的目的而使用含有光辉性颜料的光辉性色调剂。
为了因此提供减轻含板状金属颜料的色调剂的清洁故障的图像形成设备,公开了一种图像形成设备,其包含:图像保持部件;充电单元,其对图像保持部件的表面充电;静电荷图像形成单元,其在图像保持部件的经充电表面上形成静电荷图像;显影单元,其容纳静电荷图像显影剂并利用静电荷图像显影剂将形成在图像保持部件表面上的静电荷图像显影为色调剂图像,所述静电荷图像显影剂含有色调剂,所述色调剂含有平均纵向长度为5μm~12μm且平均厚度为0.01μm~0.5μm的板状金属颜料,并且具有平均纵向长度为7μm~20μm、且平均厚度为1μm~3μm且平均圆形度为0.5~0.9的扁平形状;转印单元,其将形成在图像保持部件表面上的色调剂图像转印至记录介质的表面上;校准单元,其使转印后残留在图像保持部件表面上的色调剂从图像保持部件表面上升起;清洁单元,其具有用于清洁转印后残留在图像保持部件表面上的色调剂的清洁刮板;和定影单元,其将转印至记录介质表面上的色调剂图像定影(例如,参见专利文献1)。
为了形成具有特殊效果(例如金属效果)的电子照相印刷色调剂图像,公开了无孔干燥型色调剂颗粒,其基本上包含:聚合物粘合剂相;和分散在聚合物粘合剂相中的非导电性金属氧化物颗粒,其中(a)无孔干燥型色调剂颗粒在定影前具有至少15μm至最大40μm的体积加权平均粒径(dvol);(b)无孔干燥型色调剂颗粒中所有非导电性金属氧化物颗粒的至少50重量%具有至少5的长径比和至少2μm至最大50μm的ecd;(c)基于无孔干燥型色调剂颗粒的总重量,非导电性金属氧化物颗粒以至少15重量%至最大50重量%存在;(d)定影前的无孔干燥型色调剂颗粒的dvol和无孔干燥型色调剂颗粒中非导电性金属氧化物颗粒的平均圆当量直径(ecd)之间的比大于0.1至最大10;(e)非导电性金属氧化物基本上包含(i)具有外表面的二氧化硅、氧化铝或云母基材和(ii)一个或多个铁、铬、硅、钛或铝的氧化物层,其各自具有至少30nm至最大700nm的平均干燥层厚度,使得设置在至少一部分基材外表面上的所有氧化物层的总平均干燥厚度为至少30nm至最大1400nm;和(f)至少一个铁、铬、硅、钛或铝的氧化物层形成非导电性金属氧化物颗粒的最外层(例如,参见专利文献2)。
为了提供即使在添加少量着色剂的情况下也能实现光辉性金色印刷且以低成本展现优异带电性和低温定影性的静电潜像显影金色色调剂,公开了一种至少包含粘合剂树脂和着色剂的静电潜像显影金色色调剂,其中所述着色剂是通过将纵向上的平均厚度为2μm~5μm且平均长度为15μm~500μm的板状玻璃片用银涂布而获得的光辉性颜料(例如,参见专利文献3)。
[专利文献1]jp-a-2015-118358
[专利文献2]jp-t-2015-523590
[专利文献3]jp-a-2003-207941
技术实现要素:
本发明的一个目的是提供一种光辉性色调剂,其包含含有粘合剂树脂和扁平状光辉性颜料的色调剂颗粒,并且与光辉性颜料的平均厚度等于或小于0.5μm或大于1.5μm的情形相比,在利用所述光辉性色调剂连续形成光辉性色调剂图像之后利用所述光辉性色调剂之外的色调剂形成色调剂图像时,所述光辉性色调剂防止由于所述光辉性色调剂造成的初始色调剂图像中的图像污染。
根据本发明的第一方面,提供了一种光辉性色调剂,其包含:
色调剂颗粒,所述色调剂颗粒含有粘合剂树脂和具有扁平形状的光辉性颜料,
其中所述光辉性颜料的平均厚度大于0.5μm~1.5μm。
根据本发明的第二方面,在第一方面所述的光辉性色调剂中,
其中所述光辉性颜料的体积平均粒径为1.0μm~25μm。
根据本发明的第三方面,在第一方面所述的光辉性色调剂中,所述光辉性颜料是金属颜料。
根据本发明的第四方面,在第三方面所述的光辉性色调剂中,所述金属颜料是铝颜料。
根据本发明的第五方面,在第一方面所述的光辉性色调剂中,相对于全部所述光辉性色调剂,除无机物之外的甲苯不溶性部分为0.1重量%~50重量%。
根据本发明的第六方面,在第一方面所述的光辉性色调剂中,所述粘合剂树脂是玻璃化转变温度为50℃~80℃的聚酯树脂。
根据本发明的第七方面,在第一方面所述的光辉性色调剂中,所述粘合剂树脂是重均分子量(mw)为5,000~1,000,000的聚酯树脂。
根据本发明的第八方面,在第一方面所述的光辉性色调剂中,所述光辉性颜料的平均厚度的上gsd等于或小于1.4。
根据本发明的第九方面,在第一方面所述的光辉性色调剂中,所述光辉性颜料的平均厚度的下gsd等于或小于1.4。
根据本发明的第十方面,在第一方面所述的光辉性色调剂中,所述光辉性颜料的长径比为3~40。
根据本发明的第十一方面,在第一方面所述的光辉性色调剂中,所述色调剂颗粒的体积平均粒径为3μm~30μm。
根据本发明的第十二方面,在第一方面所述的光辉性色调剂中,所述色调剂颗粒的长径比为3~15。
根据本发明的第十三方面,在第一方面所述的光辉性色调剂中,所述光辉性颜料的体积平均粒径(d50(pig))与所述色调剂颗粒的体积平均粒径(d50(tn))之比(d50(pig)/d50(tn))为0.46~0.93。
根据本发明的第十四方面,在第一方面所述的光辉性色调剂中,所述光辉性颜料的长径比(a(pig))与所述色调剂颗粒的长径比(a(tn))之比(a(pig)/a(tn))为0.71~6.08。
根据本发明的第十五方面,在第一方面所述的光辉性色调剂中,所述粘合剂树脂含有脲改性聚酯树脂。
根据本发明的第十六方面,提供了一种静电荷图像显影剂,其包含:
第一~第十五方面中任一项所述的光辉性色调剂。
根据本发明的第十七方面,提供了一种色调剂盒,其包含:
含有第一~第十五方面中任一项所述的光辉性色调剂的容器,其中所述色调剂盒能够从图像形成设备上拆卸。
根据本发明的第十八方面,提供了一种处理盒,其包含:
显影单元,所述显影单元容纳第十六方面所述的静电荷图像显影剂,并通过利用所述静电荷图像显影剂使在图像保持部件的表面上形成的静电荷图像显影为色调剂图像,
其中所述处理盒能够从图像形成设备上拆卸。
根据本发明的第十九方面,提供了一种图像形成设备,其包含:
图像保持部件;
充电单元,其对所述图像保持部件的表面充电;
静电荷图像形成单元,其在所述图像保持部件的经充电表面上形成静电荷图像;
显影单元,其容纳第十六方面所述的静电荷图像显影剂并通过利用所述静电荷图像显影剂使在所述图像保持部件的表面上形成的所述静电荷图像显影为色调剂图像;
转印单元,其将在所述图像保持部件的表面上形成的所述色调剂图像转印至记录介质的表面;和
定影单元,其将转印到所述记录介质的表面的所述色调剂图像定影。
根据本发明的第二十方面,提供了一种图像形成方法,其包括:
对图像保持部件的表面充电;
在所述图像保持部件的经充电表面上形成静电荷图像;
通过利用第十六方面所述的静电荷图像显影剂将在所述图像保持部件的表面上形成的所述静电荷图像显影为色调剂图像;
将所述图像保持部件的表面上形成的所述色调剂图像转印至记录介质的表面;和
将转印到所述记录介质的表面的所述色调剂图像定影。
根据本发明的第一方面、第五~第七方面、第十三方面和第十四方面,光辉性色调剂包含含有粘合剂树脂和扁平状光辉性颜料的色调剂颗粒,并且与光辉性颜料的平均厚度等于或小于0.5μm或大于1.5μm的情形相比,在利用所述光辉性色调剂连续形成光辉性色调剂图像之后利用所述光辉性色调剂之外的色调剂形成色调剂图像时,所述光辉性色调剂防止由于所述光辉性色调剂造成的初始色调剂图像中的图像污染。
根据本发明的第二方面,与光辉性颜料的体积平均粒径小于1.0μm或大于25μm的情形相比,所述光辉性色调剂进一步防止由于所述光辉性色调剂造成的图像污染。
根据本发明的第三方面,与使用金属颜料之外的颜料作为所述光辉性颜料的情形相比,所述光辉性色调剂进一步防止由于所述光辉性色调剂造成的图像污染。
根据本发明的第四方面,与使用铝颜料之外的颜料作为所述金属颜料的情形相比,所述光辉性色调剂进一步防止由于所述光辉性色调剂造成的图像污染。
根据本发明的第八方面,与光辉性颜料的平均厚度的上gsd大于1.4的情形相比,所述光辉性色调剂进一步防止由于所述光辉性色调剂造成的图像污染。
根据本发明的第九方面,与光辉性颜料的平均厚度的下gsd大于1.4的情形相比,所述光辉性色调剂进一步防止由于所述光辉性色调剂造成的图像污染。
根据本发明的第十方面,与照片光辉性颜料的长径比小于3或大于40的情形相比,所述光辉性色调剂进一步防止由于所述光辉性色调剂造成的图像污染。
根据本发明的第十一方面,与色调剂颗粒的体积平均粒径小于3μm或大于30μm的情形相比,所述光辉性色调剂进一步防止由于所述光辉性色调剂造成的图像污染。
根据本发明的第十二方面,与色调剂颗粒的长径比小于3或大于15的情形相比,所述光辉性色调剂进一步防止由于所述光辉性色调剂造成的图像污染。
根据本发明的第十五方面,与粘合剂树脂不含脲改性聚酯树脂的情形相比,所述光辉性色调剂进一步防止由于所述光辉性色调剂造成的图像污染。
根据本发明的第十六方面,所述静电荷图像显影剂包含光辉性色调剂,所述光辉性色调剂包含含有粘合剂树脂和扁平状光辉性颜料的色调剂颗粒,并且与光辉性颜料的平均厚度等于或小于0.5μm或大于1.5μm的情形相比,在利用所述光辉性色调剂连续形成光辉性色调剂图像之后利用所述光辉性色调剂之外的色调剂形成色调剂图像时,所述光辉性色调剂防止由于所述光辉性色调剂造成的初始色调剂图像中的图像污染。
根据本发明的第十七方面,所述色调剂盒容纳光辉性色调剂,所述光辉性色调剂包含含有粘合剂树脂和扁平状光辉性颜料的色调剂颗粒,并且与光辉性颜料的平均厚度等于或小于0.5μm或大于1.5μm的情形相比,在利用所述光辉性色调剂连续形成光辉性色调剂图像之后利用所述光辉性色调剂之外的色调剂形成色调剂图像时,所述光辉性色调剂防止由于所述光辉性色调剂造成的初始色调剂图像中的图像污染。
根据本发明的第十八方面,所述处理盒容纳包含光辉性色调剂的静电荷图像显影剂,所述光辉性色调剂包含含有粘合剂树脂和扁平状光辉性颜料的色调剂颗粒,并且与光辉性颜料的平均厚度等于或小于0.5μm或大于1.5μm的情形相比,在利用所述光辉性色调剂连续形成光辉性色调剂图像之后利用所述光辉性色调剂之外的色调剂形成色调剂图像时,所述光辉性色调剂防止由于所述光辉性色调剂造成的初始色调剂图像中的图像污染。
根据本发明的第十九方面,所述图像形成设备使用包含光辉性色调剂的静电荷图像显影剂,所述光辉性色调剂包含含有粘合剂树脂和扁平状光辉性颜料的色调剂颗粒,并且与光辉性颜料的平均厚度等于或小于0.5μm或大于1.5μm的情形相比,在利用所述光辉性色调剂连续形成光辉性色调剂图像之后利用所述光辉性色调剂之外的色调剂形成色调剂图像时,所述光辉性色调剂防止由于所述光辉性色调剂造成的初始色调剂图像中的图像污染。
根据本发明的第二十方面,所述图像形成方法使用包含光辉性色调剂的静电荷图像显影剂,所述光辉性色调剂包含含有粘合剂树脂和扁平状光辉性颜料的色调剂颗粒,并且与光辉性颜料的平均厚度等于或小于0.5μm或大于1.5μm的情形相比,在利用所述光辉性色调剂连续形成光辉性色调剂图像之后利用所述光辉性色调剂之外的色调剂形成色调剂图像时,所述光辉性色调剂防止由于所述光辉性色调剂造成的初始色调剂图像中的图像污染。
附图说明
将基于以下附图详细描述本发明的示例性实施方式,其中:
图1是示意性图解本示例性实施方式的示例性色调剂颗粒的截面图;
图2是示意性图解本示例性实施方式的示例性图像形成设备的构造图;和
图3是示意性图解本示例性实施方式的示例性处理盒的构造图。
具体实施方式
下文中,将提供本发明示例性实施方式的光辉性色调剂、静电荷图像显影剂、色调剂盒、处理盒、图像形成设备和图像形成方法的示例性实施方式的详述。
光辉性色调剂
本示例性实施方式的光辉性色调剂(下文中在一些情形中也称作“色调剂”)是包含色调剂颗粒的色调剂,所述色调剂颗粒含有粘合剂树脂和具有扁平形状的光辉性颜料(下文中称作扁平状光辉性颜料),其中光辉性颜料的平均厚度大于0.5μm~1.5μm。
根据本示例性实施方式的色调剂,在利用光辉性色调剂连续形成光辉性色调剂图像之后利用所述光辉性色调剂之外的色调剂形成色调剂图像时,防止初始色调剂图像中由于所述光辉性色调剂造成的图像污染。其原因尚不清楚,但推测如下。
在相关领域内,存在以下情形,当在连续形成光辉性色调剂图像之后用光辉性色调剂之外的色调剂(例如黑色色调剂和彩色色调剂)形成色调剂图像时,光辉性色调剂被出乎意料地置于利用光辉性色调剂之外的色调剂形成色调剂图像上,并且利用光辉性色调剂之外的色调剂形成的色调剂图像被光辉性色调剂污染。
具体而言,具有高热容量的板材(例如所谓的硬纸板)适合作为用于形成光辉性色调剂图像的记录介质。如果在具有高热容量的板材上连续形成光辉性色调剂图像,则设置在图像形成设备中作为定影单元的定影辊和定影带的表面温度趋于降低。如果定影辊和定影带的表面温度降低,则趋于发生光辉性色调剂的定影故障,并且光辉性色调剂趋于残留在定影辊和定影带的表面上(例如,以光辉性色调剂粘附在表面上的状态)。因此,推测如果在连续形成光辉性色调剂图像之后用光辉性色调剂之外的色调剂形成色调剂图像,则残留在定影辊和定影带表面上的光辉性色调剂粘附至利用光辉性色调剂之外的色调剂形成的色调剂图像,并且该色调剂图像被光辉性色调剂污染。该现象趋于在连续形成光辉性色调剂图像之后从第一色调剂图像开始在一些色调剂图像中发生。
作为深入研究的结果,本发明人将光辉性色调剂所含的光辉性颜料的平均厚度设定在预定范围内,使得光辉性色调剂几乎不残留在定影辊和定影带的表面上。由此,推测在利用光辉性色调剂连续形成光辉性色调剂图像后利用光辉性色调剂之外的色调剂形成色调剂图像时,使用本示例性实施方式的色调剂防止了由于光辉性色调剂造成的初始色调剂图像中的图像污染。
在本示例性实施方式的色调剂中,相对于全部色调剂,无机物之外的甲苯不溶性部分优选为0.1重量%~50重量%,更优选为2重量%~30重量%,进一步优选为3重量%~10重量%。
例如,通过以下方法调节甲苯不溶性部分:1)通过向在末端具有反应性官能团的聚合物成分添加交联剂而形成交联结构或支化结构的方法;2)通过向在末端具有离子型官能团的聚合物成分添加多价金属离子而形成交联结构或支化结构的方法;或3)通过用异氰酸酯等进行处理而延长或形成树脂链长度的分支的方法。
在此,甲苯不溶性部分是色调剂组分中除无机物之外的组分,其不溶于甲苯。然而,在色调剂颗粒中含有防粘剂以及光辉性颜料和粘合剂树脂的情形中,所述甲苯不溶性部分表示无机物和防粘剂之外的甲苯不溶部分。换言之,所述甲苯不溶部分表示含有不溶于甲苯的粘合剂树脂成分(具体而言,粘合剂树脂的高分子量成分)作为主要成分(例如,相对整体而言等于或大于90重量%)的不溶性部分。非光辉性颜料和外添剂等以及光辉性颜料对应于无机物。
甲苯不溶性部分是通过以下方法测量的值。
首先,用双酚a型液体环氧树脂和固化剂包埋作为测量目标的色调剂,然后形成切割用样品。随后,使用利用金刚石刀具的切割机(例如ultracutuct(由leica制造))在100℃将样品切割成片。
通过具有能量分散型x射线分析仪(sem-edx)的扫描电子显微镜观察切割成片的样品截面,并且通过能量分散型x射线分析仪(edx)识别存在于色调剂中的无机物(扁平状光辉性颜料(在截面中以针状观察到)和在对色调剂颗粒外部添加外添剂的情形中的外添剂)的组成元素。随后,通过荧光x射线分析仪确定无机物的量(重量%)。
在此,使用具有能量分散型x射线分析仪“emax模型6923h”(由horiba,ltd.制造)的电子显微镜“s-4100”(由horiba,ltd.制造)作为所述具有能量分散型x射线分析仪的扫描电子显微镜,并且作为测量条件将加速电压设定为20kv。相比之下,使用由shimadzucorporation制造的“荧光x射线分析仪xrf-1500”作为所述荧光x射线分析仪,并且作为测量条件将管电压设定为40kv,将管电流设定为90ma,并将测量时间设定为5分钟。
相比之下,将1g称量的色调剂放入由称量的玻璃纤维制成的圆柱形滤纸中,并附接至加热型索氏提取器的提取管。随后,将甲苯倒入烧瓶中并利用覆套式加热器以110℃加热。另外,利用附接至提取管的加热器以125℃加热提取管的外围。以一定的回流速度进行提取,以便一次提取循环在4分钟~5分钟内进行。在提取10小时之后,提取圆柱形滤纸和色调剂残留物,干燥并称重。
然后,基于下式计算色调剂残留量(重量%):色调剂残留量(重量%)=[(圆柱形滤纸量+色调剂残留量)(g)–柱形滤纸量(g)]/色调剂重量(g)×100。色调剂残留物包含无机物(如光辉性颜料和外添剂)以及甲苯不溶性部分。在色调剂颗粒含有防粘剂的情形中,由于通过加热进行了提取,防粘剂可溶于甲苯。
随后,由通过荧光x射线分析仪量化的“无机物(光辉性颜料和在外部添加外添剂的情形中的外添剂)的量(重量%)”和通过加热索氏提取器而提取的“色调剂残留量(重量%)”计算甲苯不溶性部分(重量%)。换言之,由等式“甲苯不溶性部分(重量%)”=“色调剂残留量(重量%)”–“无机物的量(重量%)”计算甲苯不溶性部分(重量%)。
在本示例性实施方式的色调剂中,“光辉性”是指在通过光辉性色调剂形成图像时所观察到的光泽,如金属光泽。
具体而言,本示例性实施方式的色调剂优选具有2~100的比例(x/y),其中,在形成实心图像并用入射光以–45°的入射角照射该图像的情形中,通过变角光度计测量受光角+30°处的反射率x和受光角-30°处的反射率y。
等于或大于2的比例(x/y)表示与入射侧相对侧(正角侧)上的反射大于入射光入射的入射侧(负角侧)上的反射,即,防止了入射光的漫反射。在入射光的漫反射在各方向上发生的情形中,在视觉识别中观察到反射光具有暗色调的颜色。因此,如果比例(x/y)小于2,则存在无法在反射光的视觉识别中观察到光泽并且光辉性劣化的情形。
相比之下,如果比例(x/y)超过100,则存在以下情形,可视觉识别反射光的视角变得极其狭窄,并且由于正反射光成分大,根据视角会观察到黯黑颜色。
就光辉性和色调剂制造性的角度而言,比例(x/y)优选为4~50,更优选为6~20,特别优选为8~15。
通过变角光度计对比例(x/y)的测定
在此,首先将给出入射角和受光角的描述。在本示例性实施方式中,在通过变角光度计进行测量时,将入射角设定为-45°,这是因为对于具有较宽范围的光泽度的图像而言实现了高测量灵敏度。
将受光角设定为-30°和+30,这是由于在有光泽感图像和无光泽感图像的评估中实现了最高的测量灵敏度。
接下来,将给出测量比例(x/y)的方法的描述。
使用由nippondenshokuindustriesco.,ltd.制造的分光式变角色差仪gc5000l作为可变角光度计,从而以相对于作为测量目标的图像(光辉性图像)为-45°的入射角使入射光入射在所述图像上,并且测量受光角+30°处的反射率x和受光角-30°处的反射率y。以20nm的间隔由400nm~700nm的波长范围内的光测量反射率x和反射率y,并且获得各波长处的反射率的平均值。由这些测量结果计算比例(x/y)。
从满足上述比例(a/b)的角度而言,本示例性实施方式的色调剂优选满足以下要求(1)和(2)。
(1)平均圆当量直径d大于色调剂颗粒的平均最大厚度c。
(2)在色调剂颗粒厚度方向上的截面的纵轴方向与光辉性颜料在其观察的纵轴方向之间角度为-30°~+30°的光辉性颜料的比例等于或小于观察到的全部光辉性颜料的60%。
据认为如果色调剂颗粒具有厚度长于圆当量直径的扁平形状(参见图1),则在形成图像的定影工序中,在定影期间由于压力,扁平状色调剂颗粒的扁平表面侧排列为面向记录介质表面。在图1中,2表示色调剂颗粒,4表示光辉性颜料,而l表示色调剂颗粒的厚度。
因此,据认为在色调剂中所含的扁平状(薄片状)光辉性颜料颗粒中满足上述要求(2)“色调剂截面的纵轴方向与光辉性颜料的纵轴方向之间的角度为-30°~+30°”的光辉性颜料颗粒的最大面积表面侧排列为面向记录介质的表面。据认为由于在使光入射在由此形成的图像上的情形中,针对入射光,以漫射形式反射的光辉性颜料的比例被阻止,故实现了上述范围的比例(x/y)。
下文中,将给出本示例性实施方式的色调剂的详细描述。
本示例性实施方式的色调剂包含色调剂颗粒,其至少含有粘合剂树脂和扁平状光辉性颜料。根据需要,本示例性实施方式的色调剂颗粒可含有其他成分。
本示例性实施方式的色调剂可包含含有光辉性颜料和粘合剂树脂的色调剂颗粒,以及外部添加至该色调剂颗粒的外添剂。
色调剂颗粒
所述色调剂颗粒含有粘合剂树脂和扁平状光辉性颜料。根据需要,所述色调剂颗粒可含有其他添加剂,例如防粘剂。
粘合剂树脂
粘合剂树脂的实例包括包含以下单体的均聚物或这些单体中的两种以上类型的共聚物的乙烯基树脂,该单体例如:苯乙烯类(如苯乙烯、对氯苯乙烯或α-甲基苯乙烯);(甲基)丙烯酸酯类(如丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丙酯、丙烯酸正丁酯、丙烯酸十二烷基酯、丙烯酸2-乙基己酯、甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸正丙基、甲基丙烯酸十二烷基酯或甲基丙烯酸2-乙基己酯);烯键式不饱和腈类(如丙烯腈或甲基丙烯腈);乙烯基醚类(如乙烯基甲基醚或乙烯基异丁基醚);乙烯基酮类(如乙烯基甲基酮、乙烯基乙基酮或乙烯基异丙烯基酮);或烯烃类(如乙烯、丙烯和丁二烯)。
粘合剂树脂的实例还包括非乙烯基树脂,例如环氧树脂、聚酯树脂、聚氨酯树脂、聚酰胺树脂、纤维素树脂、聚醚树脂、改性松香,此类非乙烯基树脂与上述乙烯基树脂的混合物,以及通过在这些树脂中任何一种的存在下使乙烯基单体聚合而获得的接枝聚合物。
此类粘合剂树脂可以单独使用一种,或组合使用两种以上。
优选使用聚酯树脂作为粘合剂树脂。
聚酯树脂的实例包括已知聚酯树脂。
聚酯树脂的实例包括多元羧酸和多元醇的缩聚物。可以使用商购聚酯树脂或合成聚酯树脂。
多元羧酸的实例包括脂肪族二羧酸(例如,草酸、丙二酸、马来酸、富马酸、柠康酸、衣康酸、戊烯二酸、琥珀酸、烯基琥珀酸、己二酸或癸二酸)、脂环族羧酸(例如,环己烷二羧酸)、芳香族二羧酸(例如,对苯二甲酸、间苯二甲酸、邻苯二甲酸或萘二甲酸)、它们的酸酐或其低级烷基酯(例如含有1~5个碳原子)。在所述实例中,例如,优选使用芳香族二羧酸作为多元羧酸。
作为多元羧酸,可以与二元羧酸一起使用具有交联结构或支化结构的三元以上的羧酸。三元以上的羧酸的实例包括偏苯三酸、均苯四酸、它们的酸酐或其低级烷基酯(例如含有1~5个碳原子)。
多元羧酸的一种或两种以上可单独或组合使用。
多元醇的实例包括脂肪族二醇(例如,乙二醇、二乙二醇、三乙二醇、丙二醇、丁二醇、己二醇或新戊二醇)、脂环族二醇(例如,环己二醇、环己烷二甲醇或氢化双酚a)、芳香族二醇(如双酚a的氧化乙烯加合物或双酚a的氧化丙烯加合物)。在所述实例中,例如,优选芳香族二醇和脂环族二醇,更优选芳香族二醇作为所述二醇。
作为多元醇,可以与二醇组合使用具有交联结构或支化结构的三元以上的多元醇。三元以上的多元醇的实例包括甘油、三羟甲基丙烷和季戊四醇。
多元醇的一种或两种以上可单独或组合使用。
聚酯树脂的玻璃化转变温度(tg)优选为50℃~80℃,更优选为50℃~65℃。
由通过差示扫描量热法(dsc)获得的dsc曲线来确定玻璃化转变温度。更具体而言,基于jisk7121-1987“塑料转变温度的测试方法”中如何获得玻璃化转变温度的方法中描述的“外推法玻璃化转变起始温度”来测定玻璃化转变温度。
聚酯树脂的重均分子量(mw)优选为5,000~1,000,000,更优选为7,000~500,000。
聚酯树脂的数均分子量(mn)优选为2,000~100,000。
聚酯树脂的分子量分布mw/mn优选为1.5~100,更优选为2~60。
通过凝胶渗透色谱法(gpc)测量重均分子量和数均分子量。使用作为测量设备的由tosohcorporation制造的gpc·hlc-8120gpc、由tosohcorporation制造的柱tskgelsuperhm-m(15cm)和thf溶剂进行gpc分子量测量。使用由单分散苯乙烯标准样品形成的分子量校准曲线从所述测量结果计算重均分子量和数均分子量。
通过已知制备方法获得聚酯树脂。具体而言,通过将聚合温度设定为180℃~230℃获得聚酯树脂,例如,根据需要降低反应体系中的压力,并且使在除去缩合期间生成的水和醇的同时引起反应。
在原料的单体在反应温度下不溶解或不共混的情形中,可添加具有高沸点的溶剂作为增溶剂来促进溶解。在此情形中,缩聚反应在蒸发增溶剂的同时进行。在共聚反应中存在低相容性单体的情形中,优选的是预先使低相容性的单体和酸或醇缩合以便与该单体缩聚,并且随后与主要组分进行缩聚。
在此,上述未改性聚酯树脂之外的聚酯树脂的实例包括改性聚酯树脂。改性聚酯树脂是其中存在酯连接之外的连接基团的聚酯树脂,或者是其中与聚酯树脂成分不同的树脂成分通过共价键或离子键键合的聚酯树脂。改性聚酯的实例包括通过以下方式制备的具有改性末端的树脂:使包含具有与酸基或羟基反应的官能团(例如异氰酸酯基团)的末端的聚酯树脂与活性氢化合物反应。
特别优选脲改性聚酯树脂作为改性树脂。包含脲改性聚酯树脂作为粘合剂树脂利于防止定影部件中出现由于光辉性颜料造成的损坏。这被认为是由于脲改性聚酯具有适当的亲水性并设置在色调剂颗粒中,从而作为甲苯不溶性部分围绕光辉性颜料的边缘部分。就这点而言,相对于全部粘合剂树脂,脲改性聚酯树脂的含量优选为10重量%~30重量%,更优选为15重量%~25重量%。
作为脲改性聚酯树脂,使用通过具有异氰酸酯基团的聚酯树脂(聚酯预聚物)和胺化合物之间的反应(交联反应和伸长反应中的至少一个)获得的脲改性聚酯树脂。脲改性聚酯可含有氨基甲酸酯键和脲键。
具有异氰酸酯基团的聚酯预聚物是多元羧酸与多元醇的缩聚物,其实例包括通过引起具有活性氢的聚酯和多价异氰酸酯化合物之间的反应而获得的预聚物。聚酯中包含的具有活性氢的基团的实例包括羟基(醇式羟基和酚式羟基)、氨基、羧基和巯基,优选使用醇式羟基。
具有异氰酸酯基团的聚酯预聚物中的多元羧酸和多元醇的实例包括与针对聚酯树脂描述的那些多元羧酸和多元醇相似的化合物。
多价异氰酸酯化合物的实例包括:脂肪族聚异氰酸酯(例如四亚甲基二异氰酸酯、六亚甲基二异氰酸酯或2,6-二异氰酸酯甲基己酸酯);脂环族聚异氰酸酯(例如异佛尔酮二异氰酸酯、环己基甲烷二异氰酸酯);芳香族二异氰酸酯(甲代亚苯基(tolylene)二异氰酸酯或二苯甲烷二异氰酸酯);芳香族-脂肪族二异氰酸酯(α,α,α’,α’-四甲基亚二甲苯基二异氰酸酯);异氰尿酸酯;以及通过诸如酚衍生物、肟或己内酰胺等封阻剂将上述聚异氰酸酯封阻而制备的那些。
对于多价异氰酸酯化合物,可单独或组合使用一种或两种以上的封阻剂。
针对多价异氰酸酯化合物的比例,具有羟基的聚酯预聚物中的异氰酸酯基团[nco]和羟基[oh]之间的当量比[nco]/[oh]优选为1/1~5/1,更优选为1.2/1~4/1,进一步优选为1.5/1~2.5.1。如果[nco]/[oh]设定在1/1~5/1,则甲苯不溶性部分在上述范围内的趋势增加,并且趋于防止定影部件中出现的由于光辉性颜料造成的损坏。如果[nco]/[oh]设定为等于或小于5,则趋于防止低温定影性的降低。
在具有异氰酸酯基团的聚酯预聚物中,相对于全部具有异氰酸酯基团的聚酯预聚物,源自于多价异氰酸酯化合物的成分的含量优选为0.5重量%~40重量%,更优选为1重量%~30重量%,进一步优选为2重量%~20重量%。如果源自于多价异氰酸酯化合物的成分的含量被设定为0.5重量%~40重量%,则甲苯不溶性部分在上述范围内的趋势增加,并且趋于防止定影部件中出现的由于光辉性颜料造成的损坏。如果源自于多价异氰酸酯化合物的成分的含量被设定为等于或小于40重量%,则趋于防止低温定影性的降低。
具有异氰酸酯基团的聚酯预聚物的一个分子中所含的异氰酸酯基团数优选平均等于或大于1,更优选为平均1.5~3,进一步优选为平均1.8~2.5。如果每个分子的异氰酸酯基团数被设定为等于或大于1,则反应后的脲改性聚酯树脂的分子量增加,甲苯不溶性部分在上述范围内的趋势增加,并且趋于防止定影部件中出现的由于光辉性颜料造成的损坏。
待与具有异氰酸酯基团的聚酯预聚物反应的胺化合物的实例包括二胺、三元以上的多胺、氨基醇、氨基硫醇、氨基酸以及通过将其氨基封阻而获得的化合物。
二胺的实例包括芳香族二胺(亚苯基二胺、二乙基甲苯二胺或4,4’-二氨基苯甲烷);脂环族二胺(例如4,4-二氨基-3,3’-二甲基二环己基甲烷、二胺环己烷或异佛尔酮二胺);和脂肪族二胺(例如乙二胺、四亚甲基二胺或六亚甲基二胺)。
三元以上的多胺的实例包括二亚乙基三胺和三亚乙基四胺。
氨基醇的实例包括乙醇胺和羟乙基苯胺。
氨基硫醇的实例包括氨乙基硫醇和氨丙基硫醇。
氨基酸的实例包括丙氨酸和氨基己酸。
通过将其氨基封阻而获得的化合物的实例包括由胺化合物(例如二胺、三元以上的多胺、氨基醇、氨基硫醇或氨基酸)和酮化合物(例如丙酮、甲基乙基酮或甲基异丁基酮)获得的酮亚胺化合物以及噁唑啉化合物。
在这些胺化合物中,优选使用酮亚胺化合物。
可单独或组合使用一种或两种以上的胺化合物。
脲改性聚酯树脂可以是通过如下方式在反应后具有调节过的分子量的树脂:为了停止交联反应和伸长反应中的至少一个,用终止剂(下文中也称作“交联/伸长反应终止剂”)调节具有异氰酸酯基团的聚酯树脂(聚酯预聚物)和胺化合物之间的反应(交联反应和伸长反应中的至少一个)。
交联/伸长反应终止剂的实例包括一元胺(例如二乙胺、二丁胺、丁胺或月桂胺)和通过将一元胺封阻而获得的物质(酮亚胺化合物)。
对于胺化合物的比例,具有异氰酸酯基团的聚酯预聚物中的异氰酸酯基团[nco]和胺中氨基[nhx]的当量比[nco]/[nhx]优选为1/2~2/1,更优选为1/1.5~1.5/1,进一步优选为1/1.2~1.2/1。如果[nco]/[nhx]设定在上述范围内,则反应后的脲改性聚酯树脂的分子量增加,甲苯不溶性部分在上述范围内的趋势增加,并且趋于防止定影部件中出现的由于光辉性颜料造成的损坏。
脲改性聚酯树脂的玻璃化转变温度优选为40℃~65℃,进一步优选为45℃~60℃。数均分子量优选为2,500~50,000,进一步优选为2,500~30,000。重均分子量为10,000~500,000,进一步优选为30,000~100,000。
粘合剂树脂的含量优选为40重量%~95重量%,更优选为50重量%~90重量%,进一步优选为60重量%~85重量%。
光辉性颜料
光辉性颜料的实例包括可施加光泽感(如金属光泽)的颜料。光辉性颜料的具体实例包括:包含诸如铝(单独al的金属)、黄铜、青铜、镍、不锈钢或锌等金属粉末的金属颜料;覆盖二氧化钛或氧化铁黄等的云母;覆盖有硫酸钡、层状硅酸盐、层状铝的硅酸盐的具有薄扁平形状的无机结晶基质;单晶板状二氧化钛;碱性碳酸盐;铋酸氯氧化物;天然鸟嘌呤;扁平状玻璃粉;和金属沉积扁平状玻璃粉,并且没有特别限制,只要颜料展现光辉性。
在光辉性颜料的实例中,从镜面反射强度出发,优选使用金属颜料,具体而言,最优选使用铝颜料。
光辉性颜料的形状是扁平形状(薄片形状)。
光辉性颜料的平均厚度大于0.5μm~1.5μm,优选为0.7μm~1.5μm,更优选为1.0μm~1.5μm。光辉性颜料的平均厚度还大于0.5μm~1.5μm,优选大于0.5μm~1.2μm,更优选大于0.5μm~1.0μm。
如果光辉性颜料的平均厚度等于或小于0.5μm,光辉性颜料趋于保持在作为定影单元的定影辊和定影带上。相比之下,如果光辉性颜料的平均厚度大于1.5μm,其变得难以对光辉性色调剂充电,并且初始显影性劣化。
光辉性颜料的体积平均粒径优选为1.0μm~25μm,更优选为2.0μm~20μm,进一步优选为3.0μm~15μm。
光辉性颜料在纵轴方向上的平均长度优选为1.0μm~25μm,更优选为2.0μm~20μm,进一步优选为3.0μm~15μm。
假定光辉性颜料在厚度方向上的平均长度为1时,其在纵轴方向上的平均长度的比例(长径比)优选为3~40,更优选为5~35,进一步优选为8~30。
光辉性颜料的平均厚度的上gsd优选等于或小于1.4,更优选等于或小于1.35,进一步优选等于或小于1.3。
光辉性颜料的平均厚度的下gsd优选等于或小于1.4,更优选等于或小于1.35,进一步优选等于或小于1.3。
光辉性颜料的平均厚度、纵轴方向上的平均长度、长径比、平均厚度的上gsd和平均厚度的下gsd是通过以下方法测得的值。
通过以3g/m2的色调剂施加量将光辉性色调剂定影在记录介质(例如一张纸)上并形成光辉性色调剂图像,光辉性色调剂中所含光辉性颜料的面方向沿着记录介质的面方向定向。利用双酚a型液体环氧树脂和固化剂包埋光辉性色调剂图像,随后形成切割用样品。接下来,使用利用金刚石刀具的切割机(例如超微切片机设备(由leica制造的ultracutuct))来切割所述切割用样品,并形成观察用样品。用透射电子显微镜(tem)观察该观察用样品,并且获得100件光辉性颜料的纵轴方向上的厚度和长度。至于放大率,采用在单个视野中观察到约1~10件光辉性颜料的放大率来进行观察。由获得的值计算光辉性颜料的平均厚度、纵轴方向上的平均长度、长径比、平均厚度的上gsd和平均厚度的下gsd。
光辉性颜料在纵轴方向上的长度是光辉性颜料的截面的外接圆直径。光辉性颜料的厚度是光辉性颜料在与光辉性颜料的的截面的外接圆直径直交的方向上的最大厚度。
当基于数目从较薄厚度开始描绘测量的100件光辉性颜料的厚度的累积分布时,假定(t84/t50)1/2是上gsd,(t50/t16)1/2是下gsd,从而计算平均厚度的上gsd和下gsd,其中对应于16%累积的厚度是t16,对应于50%累积的厚度是t50,且对应于84%累积的厚度是t84。
光辉性颜料的体积平均粒径被定义为,在基于利用诸如multisizerii(由beckmancoulter,inc.制造)等测量设备测得的粒径分布划分的粒径范围(区段)内,通过按体积从较小粒径侧起绘制累积分布而获得的对应于50%累积的粒径。
从光辉性色调剂中提取光辉性颜料的方法没有特别限制,并且例如,可通过以下方式提取光辉性颜料:将光辉性色调剂放入其中溶解了粘合剂树脂的溶剂(例如四氢呋喃)中,使粘合剂树脂溶解,通过离心工序使光辉性颜料沉淀,并收集光辉性颜料。在光辉性色调剂含有添加至其的外添剂的情形中,可在将光辉性色调剂放入溶剂之前通过作为预处理的超声工序将外添剂从色调剂中去除。
例如,相对于100重量份的色调剂颗粒,光辉性颜料的含量优选为1重量份~50重量份,更优选为15重量份~25重量份。
防粘剂
防粘剂的实例包括:烃蜡;天然蜡,如巴西棕榈蜡、米蜡或小烛树蜡;合成、矿物或石油蜡,如褐煤蜡;酯蜡,如脂肪酸酯或褐煤酸酯。防粘剂不限于此。
防粘剂的熔融温度优选为50℃~110℃,更优选为60℃~100℃。
基于jisk7121-1987“塑料转变温度的测试方法”中如何获得熔融温度的方法中描述的“熔融峰温度”由差示扫描量热法(dsc)得到的dsc曲线来获得熔融温度。
例如,相对于全部色调剂颗粒,防粘剂的含量优选为1重量%~20重量%,更优选5重量%~15重量%。
其他添加剂
其他添加剂的实例包括除磁性材料、电荷控制剂、无机粉末和光辉性颜料之外的已知添加剂,例如着色剂。色调剂颗粒含有这些添加剂作为内添剂。
电荷控制剂的实例包括含有季铵盐化合物、苯胺黑化合物、铝、铁或铬的络合物的染料,以及三苯甲烷颜料。
作为无机颗粒,可单独或组合使用一种或两种以上已知的无机颗粒,如二氧化硅颗粒、二氧化钛颗粒、氧化铝颗粒、二氧化铈颗粒或者通过将其表面用疏水化剂进行处理而获得的颗粒。在这些实例中,优选使用具有低于粘合剂树脂的折射率的二氧化硅颗粒。另外,可对二氧化硅颗粒进行各类表面处理,例如,优选使用表面用例如硅烷偶联剂、钛偶联剂或硅油进行了处理的二氧化硅颗粒。
光辉性颜料之外的着色剂的实例包括已知着色剂,并且根据目标色调进行选择。作为其他着色剂,可根据需要使用表面经过处理的着色剂,或者着色剂可与分散剂一起使用。
色调剂颗粒的特性
色调剂颗粒可以是具有单层结构的色调剂颗粒,或者可以是具有由芯(芯颗粒)和覆盖所述芯的覆盖层(壳层)形成的所谓芯壳结构的色调剂颗粒。
具有芯壳结构的色调剂颗粒优选由以下形成:含有光辉性颜料和粘合剂树脂以及必要时的其他添加剂(防粘剂)的芯;和含有粘合剂树脂的覆盖层。
色调剂颗粒的平均最大厚度c和平均圆当量直径d
色调剂颗粒具有扁平形状,并且平均最大厚度c优选地长于平均圆当量直径d。平均最大厚度c与平均圆当量直径d的比例(c/d)更优选为0.001~0.700,该比例进一步优选为0.100~0.600,并且该比例特别优选为0.300~0.450。
如果该比例(c/d)等于或大于0.001,则保证了色调剂的强度,防止了因图像形成期间的应力而引起的破裂,防止由颜料暴露导致的带电减少和由此所致的雾化。另一方面,如果该比例等于或小于0.700,则可实现优异的光辉性。
平均最大厚度c和平均圆当量直径d通过以下方法测量。
将色调剂颗粒置于扁平表面上,随后通过对其施加振动而使其无规则地分散。通过以下方式计算平均最大厚度c和平均圆当量直径d:使用彩色激光显微镜“vk-9700”(由keyencecorporation制造)以便将1000个色调剂颗粒放大1000倍,测量从上侧观察时的光辉性色调剂颗粒的最大厚度c和平面的圆当量直径d,并获得其算术平均值。
色调剂颗粒截面内的纵轴方向和光辉性颜料的纵轴方向之间的角度
在观察色调剂颗粒在厚度方向上的截面的情形中,相对于观察到的全部光辉性颜料,在色调剂颗粒截面的纵轴方向与光辉性颜料的纵轴方向之间角度为-30°~+30°的光辉性颜料的比例(基于数目)等于或大于60%。此外,上述比例进一步优选为70%~95%,特别优选为80%~90%。
如果上述比例等于或大于60%,则可实现更优异的光辉性。
在此,将给出色调剂颗粒截面的观察方法的描述。
利用双酚a型液体环氧树脂和固化剂包埋光辉性色调剂图像,随后形成切割用样品。接下来,使用利用金刚石刀具的切割机(例如超微切片机设备(由制造的ultracutuct))以-100°切割所述切割用样品,并形成观察用样品。利用超高分辨率场致发射型扫描电子显微镜(由hitachihigh-technologiescorporation制造的s-4800)以可在单个视野中观察到约1~10个色调剂颗粒的放大率观察所述观察用样品。
具体而言,观察色调剂颗粒的截面(色调剂颗粒在厚度方向上的截面),利用诸如由mitanicorporation制造的图像分析软件(winroof)等图像分析软件或者观察的图像的输出样品和分度器,计算观察的100个色调剂颗粒中在色调剂颗粒截面的纵轴方向与光辉性颜料的纵轴方向之间角度为-30°~+30°的光辉性颜料的件数,并计算其比例。
“色调剂颗粒截面内的纵轴方向”表示与平均圆当量直径d长于平均最大厚度c的上述色调剂颗粒的厚度方向直交的方向,而“光辉性颜料的纵轴方向”表示光辉性颜料的长度方向。
色调剂的体积平均粒径优选为3μm~30μm,更优选为5μm~20μm。
光辉性颜料的体积平均粒径(d50(pig))与色调剂颗粒的体积平均粒径(d50(tn))之比(d50(pig)/d50(tn))优选为0.46~0.93。
色调剂颗粒的体积平均粒径d50v通过以下方式获得:通过如multisizerii(由beckmancoulter,inc.制造)等测量设备测得粒径分布,在基于该粒径分布划分的粒径范围(区段)内,从较小粒径侧起分别绘制针对体积和数目的累积分布。将对应于16%累积的粒径定义为具有体积d16v和数目d16p,将对应于50%累积的粒径定义为具有体积d50v和数目d50p,并将对应于84%累积的粒径定义为具有体积d84v和数目d84p。利用所述值将体积粒径分布指数(gsdv)计算为(d84v/d16v)1/2。
假定色调剂颗粒在厚度方向上的平均长度为1时,在纵轴方向上的平均长度的比例(长径比)优选为3~15,更优选为5~12。
作为色调剂颗粒的厚度方向上的平均长度和纵轴方向上的平均长度,采用通过上述观察色调剂颗粒截面的方法测得的100个色调剂颗粒的厚度方向上的平均长度和纵轴方向上的平均长度的平均值。
光辉性颜料的长径比(a(pig))与色调剂颗粒的长径比(a(tn))之比为0.71~6.08。
外添剂
外添剂的实例包括无机颗粒。无机颗粒的实例包括sio2、tio2、al2o3、cuo、zno、sno2、ceo2、fe2o3、mgo、bao、cao、k2o、na2o、zro2、cao·sio2、k2o·(tio2)n、al2o3·2sio2、caco3、mgco3、baso4和mgso4。
优选用疏水化剂处理作为外添剂的无机颗粒表面。例如,通过将无机颗粒浸在疏水化剂中进行利用疏水化剂的处理。虽然疏水化剂没有具体限制,其实例包括硅烷偶联剂、硅油、钛酸酯偶联剂和铝偶联剂。疏水化剂的一种或两种以上可单独或组合使用。
例如,相对于100重量份的无机颗粒,疏水化剂的量一般为1重量份~10重量份。
外添剂的实例还包括树脂颗粒和(例如聚苯乙烯、聚甲基丙烯酸甲酯(pmma)或三聚氰胺甲醛树脂等树脂颗粒)和清洁助剂(例如,高级脂肪酸的金属盐,其典型实例包括硬脂酸锌,以及氟高分子量材料的颗粒)。
例如,相对于色调剂颗粒的量,外添剂的量优选为0.01重量%~5重量%,更优选为0.01重量%~2.0重量%。
色调剂颗粒的制备方法
接下来,将给出本示例性实施方式的色调剂的制备方法的描述。
本示例性实施方式的色调剂可以通过以下方式获得:制备含有光辉性颜料的色调剂颗粒,然后对色调剂颗粒外部添加外添剂。
色调剂颗粒可通过干式制备方法(例如,捏合粉碎法)和湿式制备方法(例如,凝集聚并法、悬浮聚合法或溶解悬浮法)中的任意一种来制备。色调剂颗粒的制备方法并不限于这些制备方法,并且可采用已知的制备方法。
例如,溶解悬浮法是通过以下方式制备和获得色调剂颗粒的方法:通过将形成色调剂颗粒的原料(如树脂颗粒和光辉性颜料)溶解或分散在有机溶剂(粘合剂树脂可溶解在其中)中而获得溶液,将该溶液分散在含有颗粒分散剂的水系溶剂中,之后去除有机溶剂。
另外,凝集聚并法是通过进行以下工序获得色调剂颗粒的方法:由形成色调剂颗粒的原料(树脂颗粒和光辉性颜料等)形成凝集体的凝集工序,和将凝集体聚并的聚并工序。
在这些制备方法中,优选的是通过下述溶解悬浮法获得含有脲改性聚酯树脂作为粘合剂树脂的色调剂颗粒。虽然在溶解悬浮法的如下说明中将描述获得含有防粘剂的色调剂颗粒的方法,色调剂颗粒根据需要含有防粘剂。虽然将描述获得含有未改性聚酯树脂和脲改性聚酯树脂作为粘合剂树脂的色调剂颗粒的方法,色调剂颗粒可仅含有脲改性聚酯树脂作为粘合剂树脂。
制备油相溶液的工序
制备油相溶液,其中将包含未改性聚酯树脂、具有异氰酸酯基团的聚酯预聚物、胺化合物、光辉性颜料和防粘剂的色调剂颗粒材料溶解或分散在有机溶剂中(制备油相溶液的工序)。制备油相溶液的工序是通过将色调剂颗粒材料溶解或分散在有机溶剂中而获得色调剂材料混合溶液的工序。
制备油相溶液的方法的实例包括:1)通过将色调剂材料共同溶解或分散在有机溶剂中而制备油相溶液的方法;2)通过预先捏合色调剂材料,然后将捏合的材料溶解或分散在有机溶剂中而制备油相溶液的方法;3)通过将未改性聚酯树脂、具有异氰酸酯基团的聚酯预聚物和胺化合物溶解在有机溶剂中,然后将光辉性颜料和防粘剂分散在有机溶剂中而制备油相溶液的方法;4)通过将光辉性颜料和防粘剂分散在有机溶剂中,然后将未改性聚酯树脂、具有异氰酸酯基团的聚酯预聚物和胺化合物溶解在有机溶剂中而制备油相溶液的方法;5)通过将除了具有异氰酸酯基团的聚酯预聚物和胺化合物之外的色调剂颗粒材料(未改性聚酯树脂、光辉性颜料和防粘剂)溶解或分散在有机溶剂中,然后将具有异氰酸酯基团的聚酯预聚物和胺化合物溶解在有机溶剂中而制备油相溶液的方法;以及6)通过将除了具有异氰酸酯基团的聚酯预聚物或胺化合物之外的色调剂颗粒材料(未改性聚酯树脂、光辉性颜料和防粘剂)溶解或分散,然后将具有异氰酸酯基团的聚酯预聚物或胺化合物溶解在有机溶剂中而制备油相溶液的方法。制备油相溶液的方法不限于此。
用于油相溶液的有机溶剂的实例包括:酯溶剂,如乙酸甲酯或乙酸乙酯;酮溶剂,如甲基乙基酮或甲基异丙基酮;脂肪族烃溶剂,如己烷或环己烷;以及卤化烃,如二氯甲烷、氯仿或三氯乙烷。这种有机溶剂优选地溶解粘合剂树脂,优选地以0重量%~30重量%的比例溶于水,并且优选地具有等于或小于100℃的沸点。在这些有机溶剂中,优选使用乙酸乙酯。
制备悬浮液的工序
接下来,通过将所得油相溶液分散在水相溶液中而制备悬浮液(制备悬浮液的工序)。
随后,与悬浮液的制备一起,在具有异氰酸酯基团的聚酯预聚物和胺化合物之间引起反应。然后,通过该反应形成脲改性聚酯树脂。该反应伴随有分子链的交联反应和伸长反应中的至少一个。具有异氰酸酯基团的聚酯预聚物和胺化合物之间的反应可与去除溶剂的工序一起进行,其将在后面进行描述。
在此,根据聚酯预聚物的异氰酸酯基团和胺化合物之间的反应性选择反应条件,例如,反应时间优选为10分钟~40小时,并优选为2小时~24小时。反应温度优选为0℃~150℃,并优选为40℃~98℃。为了形成脲改性聚酯树脂,可根据需要使用已知催化剂(月桂酸二丁基锡或月桂酸二辛基锡等)。换言之,可对油相溶液或悬浮液添加催化剂。
水相溶液的实例包括通过将诸如树脂颗粒分散剂或无机颗粒分散剂等颗粒分散剂分散在水性溶剂中而获得的水相溶液。水相溶液的实例还包括通过将颗粒分散剂分散在水性溶液并将高分子分散剂溶解在水性溶液中而获得的水相溶液。另外,可对水相溶液添加已知添加剂,如表面活性剂。
水性溶液的实例包括水(例如,一般而言,离子交换水、蒸馏水或纯水)。水性溶液可以是连同水一起含有有机溶剂的溶剂,所述有机溶剂例如为醇(如甲醇、异丙醇或乙二醇)、二甲基甲酰胺、四氢呋喃、溶纤剂(如甲基溶纤剂)或低级酮(如丙酮或甲基乙基酮)。
有机颗粒分散剂的实例包括亲水性有机颗粒分散剂。有机颗粒分散剂的实例包括聚(甲基)丙烯酸烷基酯树脂(如聚甲基丙烯酸甲酯)、聚苯乙烯树脂和聚(苯乙烯丙烯腈)树脂的颗粒。
无机分散剂的实例包括亲水性无机颗粒分散剂。无机颗粒分散剂的具体实例包括二氧化硅、氧化铝、氧化钛、碳酸钙、碳酸镁、磷酸三钙、粘土、硅藻土或膨润土的颗粒,优选使用碳酸钙的颗粒。无机颗粒分散剂的一种或两种以上可单独或组合使用。
可用具有羧基的聚合物处理颗粒分散剂的表面。
具有羧基的聚合物的实例包括选自盐(例如碱金属盐、碱土金属盐、铵盐和胺盐)中的至少一种和α,β-单烯键式不饱和羧酸酯的共聚物,所述盐通过如下方式获得:用碱金属、碱土金属、氨或胺等中和α,β-单烯键式不饱和羧酸或α,β-单烯键式不饱和羧酸中的羧基。具有羧基的聚合物的实例还包括通过以下方式获得的盐(例如碱金属盐、碱土金属盐、铵盐和胺盐):中和α,β-单烯键式不饱和羧酸和α,β-单烯键式不饱和羧酸酯的共聚物中的羧基。具有羧基的聚合物可以单独使用一种,或组合使用两种以上。
α,β-单烯键式不饱和羧酸的代表性实例包括α,β-不饱和单羧酸(如丙烯酸、甲基丙烯酸或巴豆酸)和α,β-不饱和二羧酸(如马来酸、富马酸或衣康酸)。α,β-单烯键式不饱和羧酸酯的代表性实例包括(甲基)丙烯酸的烷基酯、具有烷氧基的(甲基)丙烯酸酯、具有环己基的(甲基)丙烯酸酯、具有羟基的(甲基)丙烯酸酯和聚亚烷基二醇单(甲基)丙烯酸酯。
高分子分散剂的实例包括亲水性高分子分散剂。高分子分散剂的具体实例包括具有羧基且不具有疏水性基团(例如羟基丙氧基或甲氧基)的高分子分散剂(水溶性纤维素醚,例如羧甲基纤维素或羧乙基纤维素)。
去除溶剂的工序
接下来,通过从获得的悬浮液中去除有机溶剂而获得色调剂颗粒分散液(去除溶剂的工序)。去除溶剂的工序是通过去除分散在悬浮液中的水相溶液的液滴中所含的有机溶剂而制备色调剂颗粒的工序。从悬浮液去除有机溶剂可在制备悬浮液的工序之后马上进行,或者可在制备悬浮液的工序完成1分钟以上之后进行。
在去除溶剂的过程中,例如,优选通过将所得悬浮液冷却或加热至0℃~100℃来从悬浮液中去除有机溶剂。
作为去除有机溶剂的具体方法,示例了如下方法。
(1)对悬浮液吹送气流并且将悬浮液表面上的气相强制更新的方法。在此情形中,可将气体吹送到悬浮液中。
(2)降低压力的方法。在此情形中,可通过气体的充填将悬浮液表面上的气相强制更新,或者可将气体进一步吹送到悬浮液中。
通过上述工序获得色调剂颗粒。
在此,在去除溶剂的工序结束之后,获得在对色调剂颗粒分散液中形成的色调剂颗粒进行了已知清洁工序、固液分离工序和干燥工序之后的干燥状态的色调剂颗粒。
从带电性出发,在清洁工序中,优选的是通过离子交换水充分进行置换清洁。
虽然没有特别限制,但从生产率出发,在固液分离工序中,优选的是进行抽滤或压滤等。虽然没有特别限制,但从生产率出发,在干燥工序中,优选的是进行冷冻干燥、闪喷干燥、流化干燥或振动型流化干燥等。
本示例性实施方式的色调剂通过例如对所得干燥状态的色调剂颗粒添加外添剂并将色调剂颗粒与外添剂混合来制造。
混合优选用v型共混器、亨舍尔混合机或lodige混合机等来进行。
此外,根据需要,可利用振动分级器或风力分级器等除去色调剂的粗大颗粒。
在本示例性实施方式中,可以使用凝集聚并法,其中,容易控制色调剂颗粒的形状和粒径,并且色调剂颗粒结构(如芯壳结构)可在较宽范围内控制。下文中,将给出通过凝集聚并法制备色调剂颗粒的方法的详细描述。
本示例性实施方式的凝集聚并法包括通过将形成色调剂颗粒的原料分散而形成树脂颗粒(乳化颗粒)的分散工序、形成树脂颗粒的凝集体的凝集工序和使凝集体聚并的聚并步骤。
分散工序
树脂颗粒分散液可通过常见聚合方法((如乳化聚合法、悬浮聚合法或分散聚合法)制造,或者可通过对利用分散机将水性介质和粘合剂树脂混合而获得的溶液施加剪切力来制造。此时,可通过加热使得树脂成分具有低粘度,从而形成颗粒。另外,可以使用分散剂来使分散的树脂颗粒稳定化。只要树脂是油性并且溶于对水的溶解度相对较低的溶剂,则通过以下方式制备树脂颗粒分散液:将树脂溶解在溶剂中,将颗粒连同分散剂和聚合物电解质一起分散于水中,然后通过加热或减压来蒸发溶剂。
水性介质的实例包括水(如蒸馏水或离子交换水)和醇。优选使用水。
乳化工序中使用的分散剂的实例包括:水溶性聚合物,如聚乙烯醇、甲基纤维素、乙基纤维素、羟乙基纤维素、羧甲基纤维素、聚丙烯酸钠或聚甲基丙烯酸钠;表面活性剂,如阴离子型表面活性剂(例如,十二烷基苯磺酸钠、十八烷基硫酸钠、油酸钠、月桂酸钠或硬脂酸钾)、阳离子型表面活性剂(例如,月桂基胺乙酸盐、硬脂基胺乙酸盐或月桂基三甲基氯化铵)、两性离子型表面活性剂(例如,月桂基二甲基氧化胺)和非离子型表面活性剂(例如,聚氧乙烯烷基醚、聚氧乙烯烷基苯基醚或聚氧乙烯烷基胺);或无机盐(如磷酸三钙、氢氧化铝、硫酸钙、碳酸钙或碳酸钡)。
用于制备乳化液的分散机的实例包括均质器、均相混合机、压力捏合机、挤出机和介质分散机。至于树脂颗粒的尺寸,平均粒径(体积平均粒径)优选等于或小于1.0μm,优选为60nm~300nm,进一步优选为150nm~250nm。如果平均粒径等于或大于60nm,则存在树脂颗粒溶剂凝集的情形,因为树脂颗粒在分散液中趋于不稳定。如果平均粒径等于或小于1.0μm,则存在色调剂的粒径分布变窄的情形。
为了制备防粘剂分散液,将防粘剂与离子型表面活性剂或聚合物电解质(如聚合物酸或聚合物碱)一起分散在水中,以等于或大于防粘剂熔融温度的温度加热所得物,并且利用施加高剪切力的均质器或压力排出型分散机进行分散处理。通过这样的处理,获得防粘剂分散液。在分散处理中,可以向分散液中添加如聚氯化铝等无机化合物。优选的无机化合物的实例包括聚氯化铝、硫酸铝、高碱性聚氯化铝(bac)、聚氢氧化铝和氯化铝。在这些实例中,优选使用聚氯化铝和硫酸铝等。
通过分散处理,获得含有体积平均粒径等于或小于1μm的防粘剂颗粒的防粘剂分散液。防粘剂颗粒的体积平均粒径更优选为100nm~500nm。
如果体积平均粒径等于或大于100nm,防粘剂成分一般容易进入色调剂中,其受到使用的粘合剂树脂的特性影响。如果体积平均粒径等于或小于500nm,则实现了防粘剂在色调剂中的令人满意的分散状态。
为了制备光辉性颜料分散液,可以使用已知的分散方法,并且可以使用如旋转剪切型均质器、具有介质的球磨机、砂磨机、戴诺磨机或超细磨机(ultimizer)等典型分散单元,而且没有限制。光辉性颜料连同离子型表面活性剂和高分子电解质(如高分子酸或高分子碱)一起分散在水中。只要尺寸等于或小于20μm,可容许则分散的光辉性颜料的体积平均粒径。然而,该体积平均粒径优选为3μm~16μm,这是因为该体积平均粒径不破坏凝集性并且可实现光辉性颜料在色调剂中的令人满意的分散。
可通过以下方式制备被粘合剂树脂覆盖的光辉性颜料的分散液:将光辉性颜料和粘合剂树脂分散或溶解和混合在溶剂中,并通过相转化乳化或剪切乳化将所得物分散在水中。
凝集工序
在凝集工序中,通过将树脂颗粒分散液、光辉性颜料分散液和防粘剂分散液等混合而获得混合物,并以等于或小于树脂颗粒的玻璃化转变温度的温度加热混合溶液以便凝集,形成凝集颗粒。在许多情形中,在搅拌的同时将混合溶液的ph控制在酸性,从而形成凝集颗粒。在所述搅拌条件下,可以将比例(c/d)设定在优选范围内。更具体而言,在凝集颗粒形成阶段,通过高速搅拌混合溶液并加热混合溶液可以使比例(c/d)减小,并且通过在更低温度下以降低的速度搅拌混合溶液可以使比例(c/d)增大。另外,ph优选为2~7,此时也可以有效地使用凝集剂。
在凝集工序中,防粘剂可被添加并与各种分散液(如树脂颗粒分散液)一次性混合在一起,或者可被分开和多次添加。
作为凝集剂,优选使用极性与用于分散剂的表面活性剂的极性相反的表面活性剂、无机金属盐以及二价以上的金属络合物。由于可以减少表面活性剂的使用量并增强带电特性,特别优选使用金属络合物。
作为无机金属盐,特别优选使用铝盐及其聚合物。至于无机金属盐的价态,二价无机金属盐比一价无机金属盐更适合,三价无机金属盐比二价无机金属盐更适合,四价无机金属盐比三价无机金属盐更适合,并且在相同价态的情形中,更适合的是聚合型无机金属盐聚合物,以便获得较窄的粒径分布。
在本示例性实施方式中,优选使用含铝的四价无机金属盐的聚合物以获得较窄的粒径分布。
当获得凝集颗粒的所需粒径时,可通过额外添加树脂颗粒而制备具有芯材凝集颗粒的表面被树脂覆盖的构造的色调剂(覆盖工序)。从带电性和显影性出发此情形中的构造是优选的,这是因为防粘剂和光辉性颜料不易暴露于色调剂的表面。在额外添加树脂颗粒分散液的情形中,可添加凝集剂或在额外添加之前进行ph调节。
聚并工序
在聚并工序中,通过以下方式使凝集颗粒聚并:通过在上述凝集工序中的搅拌条件下将凝集颗粒悬浮液的ph增加至3~9来停止凝集的进行,并以等于或高于树脂的玻璃化转变温度的温度进行加热。
在用树脂覆盖芯材、凝集颗粒的情形中,树脂也聚并以覆盖芯材、凝集颗粒。可容许任何加热时间,只要可实现凝结,并且凝结可在一段约0.5小时~约10小时的时间内进行。
在聚并后将所得物冷却,并且获得聚并颗粒。在冷却的过程中,可通过在树脂的玻璃化转变温度附近(玻璃化转变温度±10℃的范围内)降低冷却速率,即进行缓慢冷却,来促进结晶。
对通过聚并获得的聚并颗粒进行如过滤等固液分离工序和必要时的清洁工序和干燥工序,由此获得色调剂颗粒。
出于电荷调节、施加流动性和施加电荷交换性等的目的,无机氧化物等(其代表性实例包括二氧化硅、二氧化钛和氧化铝)可作为外添剂添加并粘附至所得色调剂颗粒。优选的外部添加方法和优选的外添剂量如上所述。
除了上述无机氧化物等以外,还可以添加如电荷控制剂、有机颗粒、润滑剂和研磨剂等其他成分(颗粒)作为外添剂。
虽然电荷控制剂没有特别限制,但优选使用无色或浅色的电荷控制剂。其实例包括季铵盐化合物、苯胺黑化合物、铝或铬的络合物以及三苯甲烷类颜料。
有机颗粒的实例包括乙烯基树脂、聚酯树脂或硅酮树脂等通常用作添加至色调剂表面的外添剂的颗粒。另外,无机颗粒和有机颗粒用作流动助剂或清洁助剂等。
润滑剂的实例包括脂肪酸酰胺,如亚乙基二硬脂酸酰胺或油酸酰胺;和脂肪酸金属盐,如硬脂酸锌和硬脂酸钙。
研磨剂的实例包括二氧化硅、氧化铝和氧化铈。
静电荷图像显影剂
本示例性实施方式的静电荷图像显影剂至少含有本示例性实施方式的色调剂。
本示例性实施方式的静电荷图像显影剂可以是仅包含本示例性实施方式的色调剂的单组分显影剂,也可以是色调剂与载体混合的双组分显影剂。
载体没有特别限制,例举已知载体。载体的实例包括由磁性颗粒制成的芯材的表面被树脂覆盖的覆盖型载体;磁性颗粒分散并共混在基质树脂中的磁性颗粒分散型载体;以及多孔磁性颗粒中浸渍有树脂的树脂浸渍型载体。
磁性颗粒分散型载体和树脂浸渍型载体可以是所述载体的构成颗粒形成芯材并且其表面被树脂覆盖的载体。
磁性粉末的实例包括:磁性金属,如氧化铁、镍或钴;以及磁性氧化物,如铁氧体和磁铁矿。
覆盖用树脂和基质树脂的实例包括聚乙烯、聚丙烯、聚苯乙烯、聚乙酸乙烯酯、聚乙烯醇、聚乙烯缩丁醛、聚氯乙烯、聚乙烯基醚、聚乙烯基酮、氯乙烯-乙酸乙烯酯共聚物、苯乙烯-丙烯酸共聚物、或含有有机硅氧烷键的直链硅酮树脂或其改性物质;氟树脂;聚酯;聚碳酸酯;酚醛树脂;和环氧树脂。覆盖用树脂和基质树脂可含有诸如导电性颗粒等添加剂。
导电性颗粒的实例包括:金属,例如金、银和铜;以及炭黑、二氧化钛、氧化锌、氧化锡、硫酸钡、硼酸铝或钛酸钾等颗粒。
为了覆盖芯材的表面,例举利用覆盖层形成用溶液的方法,所述溶液通过将覆盖用树脂和各种添加剂(按需使用)溶解在适当溶剂中而获得。所述溶剂没有特别限制,并且考虑所用树脂的类型和施加适用性等来进行选择。树脂覆盖方法的具体实例包括:将芯材浸渍在覆盖层形成用溶液中的浸渍法;将覆盖层形成用溶液喷至芯材的表面上的喷雾法;以使芯材通过流动空气而漂浮的状态喷洒覆盖层形成用溶液的流化床法;以及使载体的芯材与覆盖层形成用溶液在捏合涂布机中混合然后除去溶剂的捏合涂布机法。
双组分显影剂中色调剂与载体的混合比(重量比)优选为色调剂:载体=1:100~30:100,更优选为3:100~20:100。
图像形成设备/图像形成方法
将给出本示例性实施方式的图像形成设备和图像形成方法的描述。
本示例性实施方式的图像形成设备包含图像保持部件;对图像保持部件的表面充电的充电单元;在图像保持部件的经充电表面上形成静电荷图像的静电荷图像形成单元;容纳静电荷图像显影剂并利用静电荷图像显影剂使在所述图像保持部件的表面上形成的所述静电荷图像显影为色调剂图像的显影单元;将在所述图像保持部件的表面上形成的所述色调剂图像转印至记录介质的表面上的转印单元;和将转印到所述记录介质表面上的所述色调剂图像定影的定影单元。
本示例性实施方式的静电荷图像显影剂作为所述静电荷图像显影剂施加。
本示例性实施方式的图像形成设备执行图像形成方法(本示例性实施方式的图像形成方法),所述图像形成方法包括:对图像保持部件的表面充电的充电工序;在所述图像保持部件的经充电表面上形成静电荷图像的静电荷形成工序;通过本示例性实施方式的静电荷图像显影剂将在所述图像保持部件表面上形成的静电荷图像显影为色调剂图像的显影工序;将在图像保持部件的表面上形成的所述色调剂图像转印至记录介质的表面上的转印工序;和将转印到记录介质的表面上的色调剂图像定影的定影工序。
作为本示例性实施方式的图像形成设备,适用已知图像形成设备,例如:将在图像保持部件表面上形成的色调剂图像直接转印至记录介质的直接转印型设备;将在图像保持部件表面上形成的色调剂图像一次转印至中间转印部件的表面上,然后将转印至中间转印部件表面上的色调剂图像二次转印至记录介质的中间转印型设备;设置有清洁单元的设备,该清洁单元用于在充电前且转印色调剂图像后清洁图像保持部件的表面;或者设置有除电单元的设备,该设备在充电之前和转印色调剂图像之后通过用除电光照射图像保持部件的表面而消除电荷。
在中间转印型设备的情形中,例如,使用一种结构,所述结构包含其表面上转印有色调剂图像的中间转印部件、将在图像保持部件表面上形成的色调剂图像一次转印至中间转印部件表面上的一次转印单元、和将转印至中间转印部件表面的色调剂图像二次转印至记录介质表面的二次转印部件。
在本示例性实施方式的图像形成设备中,例如,包含显影单元的部分可具有能够从图像形成设备上拆卸的盒式结构(处理盒)。作为所述处理盒,例如,优选使用容纳本示例性实施方式的静电荷图像显影剂且设置有显影单元的处理盒。
下文中将给出本示例性实施方式的图像形成设备的实例的描述。然而,所述图像形成设备并不限于此。将描述附图中示出的主要部件,而省略其他部件的描述。
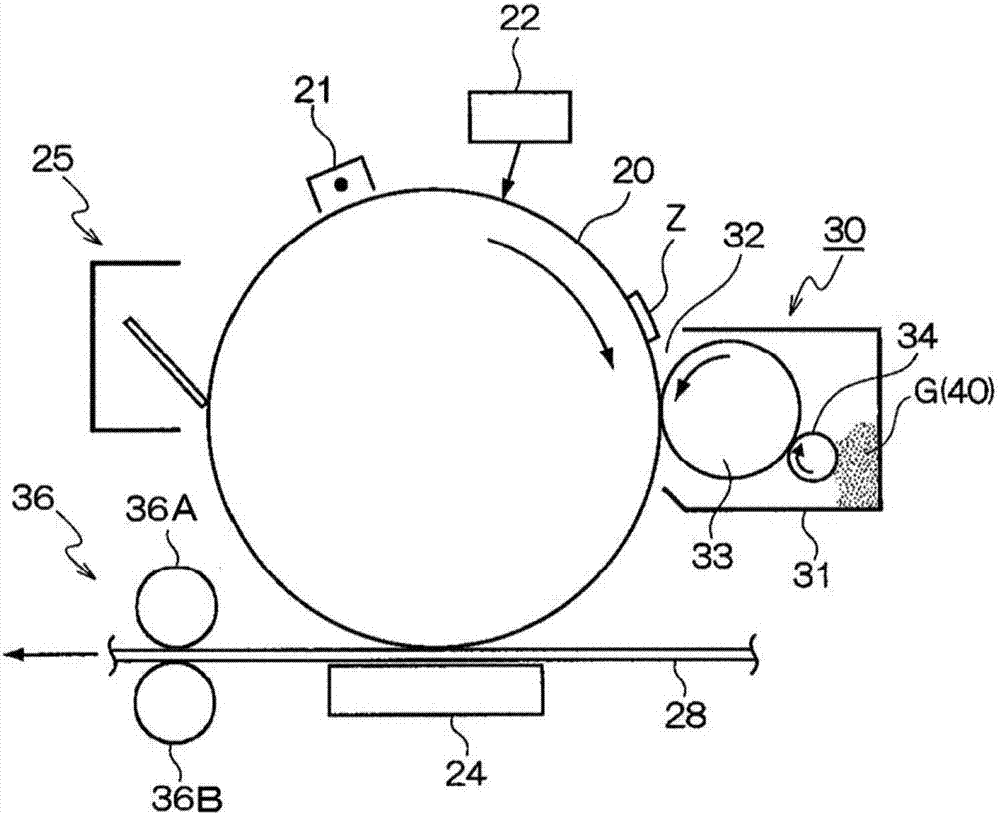
图2是示意性图解本示例性实施方式的示例性图像形成设备的构造图,所述图像形成设备包含施加有本示例性实施方式的静电荷图像显影剂的显影装置。
在图中,本示例性实施方式的图像形成装置包括沿预定方向旋转的作为图像保持部件的感光鼓20。在感光鼓20周围依次设置有:充电装置21,其对感光鼓20充电;曝光装置22,其例如作为在感光鼓20上形成静电荷图像z的静电荷图像形成装置;显影装置30,其使在感光鼓20上形成的静电荷图像z显影为可视图像;转印装置24,其将感光鼓20上可视化的色调剂图像转印至作为记录介质的记录纸28;和清洁装置25,其清除感光鼓20上残留的色调剂。
如图2所示,在本示例性实施方式中,显影装置30具有容纳包含色调剂40的显影剂g的显影外壳31。显影开口32在显影外壳31中打开以面向感光鼓20。作为色调剂保持部件的显影辊(显影电极)33被设置为面向显影开口32。通过对显影辊33施加预定显影偏压,在介于感光鼓20与显影辊33之间的区域(显影区)中形成显影电场。此外,在显影外壳31中提供作为电荷注入部件的电荷注入辊(注入电极)34以便面向显影辊33。具体而言,在本示例性实施方式中,电荷注入辊34还充当对显影辊33供给色调剂40的色调剂供给辊。
此处,可选择电荷注入辊34的旋转方向。不过,考虑到色调剂供给性质和电荷注入特性,电荷注入辊34优选在与显影辊33相对的部分沿与显影辊33相同的方向并在有圆周速度差(例如,1.5倍以上)的条件下旋转,将色调剂40介入电荷注入辊34和显影辊33之间的区域,并在刮擦的同时注入电荷。
接下来将给出本示例性实施方式的图像形成装置的运行的描述。
如果图像形成过程开始,则充电装置21首先对感光鼓20的表面充电,曝光装置22将静电荷图像z写在经充电的感光鼓20上,而显影装置30使静电荷图像z显影为色调剂图像(其为可视图像)。其后,将感光鼓20上的色调剂图像输送至转印部分,并且转印装置24将在感光鼓20上的色调剂图像静电转印至作为记录介质的记录纸28。清洁装置25清洁在感光鼓20上残余的色调剂。其后,设置有定影部件36a(定影带和定影辊等)和加压部件36b的定影装置36将记录纸28上的色调剂图像定影,获得图像。
处理盒/色调剂盒
将给出本示例性实施方式的处理盒的描述。
本示例性实施方式的处理盒是如下处理盒,其包括容纳本示例性实施方式的静电荷图像显影剂并通过该静电荷图像显影剂使在图像保持部件表面上形成的静电荷图像显影形为色调剂图像的显影单元,并且能够从图像形成设备上拆卸。
本示例性实施方式的处理盒并不限于上述构造,并且可具有如下构造,其包含显影装置,且必要时包含选自诸如图像保持部件、充电单元、静电荷图像形成单元和转印单元等其他单元中的至少一个。
虽然将在下面描述本示例性实施方式的处理盒的实例,但处理盒并不限于此。另外,将描述附图中示出的主要部件,而省略其他部件的描述。
图3是示意性图解本示例性实施方式的处理盒的构造图。
图3所示的处理盒200在设置有接轨116和曝光用开口118的外壳117内一体化地组成和保持的例如感光体107(图像保持部件的实例)、在感光体107的周围设置的充电辊108(充电单元的实例)、显影装置111(显影单元的实例)、以及清洁装置113(清洁单元的实例),并且作为处理盒提供。
在图3中,109表示曝光装置(静电荷图像形成单元的实例),112表示转印装置(转印单元的实例),115表示定影装置(定影单元的实例),而300表示记录纸(记录介质的实例)。
接下来,将给出本示例性实施方式的色调剂盒的描述。
本示例性实施方式的色调剂盒可容纳本示例性实施方式的色调剂,并能够从图像形成装置上拆卸。本示例性实施方式的色调剂盒仅必需至少容纳色调剂,并且,例如,显影剂可根据图像形成装置的机制而被容纳在其中。
图2所示的图像形成设备是具有如下构造的图像形成设备,其中色调剂盒(未示出)可自由拆卸,并且显影装置通过未在图中示出的色调剂供给管与色调剂盒连接。在色调剂盒中所容纳的色调剂的量变少时,可更换色调剂盒。
实施例
虽然下面将提供参照实施例对本示例性实施方式的详细描述,但本示例性实施方式不限于这些实施例。在以下描述中,除非另作说明,所有“份”和“%”的描述都是基于重量。
未改性聚酯树脂(1)的制备
·对苯二甲酸:1243份
·双酚a的氧化乙烯加合物:1830份
·双酚a的氧化丙烯加合物:840份
在于180℃加热的同时将上述成分混合,随后向其中加入3份二丁基氧化锡,在于220℃加热混合物的同时将水蒸发,并获得不饱和聚酯树脂。所得不饱和聚酯树脂的玻璃化转变温度tg为60℃,酸值为3mgkoh/g,且羟基值为1mgkoh/g。
聚酯预聚物(1)的制备
·对苯二甲酸:1243份
·双酚a的氧化乙烯加合物:1830份
·双酚a的氧化丙烯加合物:840份
在于180℃加热的同时将上述成分混合,随后向其中加入3份二丁基氧化锡,在于220℃加热混合物的同时将水蒸发,并获得聚酯。将350份所得聚酯、50份甲代亚苯基二异氰酸酯和450份乙酸乙酯倒入容器中,将其混合物在130℃加热3小时,并且获得具有异氰酸酯基的聚酯预聚物(1)(下文中称作异氰酸酯改性聚酯预聚物(1))。
酮亚胺化合物(1)的制备
将50份甲基乙基酮和150份六亚甲基二胺倒入容器中,并将混合物在60℃搅拌,并获得酮亚胺化合物(1)。
光辉性颜料分散液(1)的制备
·平均粒径为4μm的铝雾化粉末(atomizedpowder):100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨15小时,获得光辉性颜料(1)。
·光辉性颜料(1):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(1)的光辉性颜料分散液(1)(固体成分浓度:10%)。光辉性颜料分散液(1)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(2)的制备
·平均粒径为3.5μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨15小时,获得光辉性颜料(2)。
·光辉性颜料(2):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(2)的光辉性颜料分散液(2)(固体成分浓度:10%)。光辉性颜料分散液(2)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(3)的制备
·平均粒径为5μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨15小时,获得光辉性颜料(3)。
·光辉性颜料(3):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(3)的光辉性颜料分散液(3)(固体成分浓度:10%)。光辉性颜料分散液(3)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(4)的制备
·平均粒径为3.5μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨20小时,获得光辉性颜料(4)。
·光辉性颜料(4):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(4)的光辉性颜料分散液(4)(固体成分浓度:10%)。光辉性颜料分散液(4)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(5)的制备
·平均粒径为5.5μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨15小时,获得光辉性颜料(5)。
·光辉性颜料(5):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(5)的光辉性颜料分散液(5)(固体成分浓度:10%)。光辉性颜料分散液(5)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(6)的制备
·珍珠颜料(由merck&co.,inc.制造的扁平状光辉性颜料iriodin121):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(6)的光辉性颜料分散液(6)(固体成分浓度:10%)。光辉性颜料分散液(6)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(7)的制备
·平均粒径为2μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨15小时,获得光辉性颜料(7)。
·光辉性颜料(7):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(7)的光辉性颜料分散液(7)(固体成分浓度:10%)。光辉性颜料分散液(7)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(8)的制备
·平均粒径为9μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨20小时,获得光辉性颜料(8)。
·光辉性颜料(8):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(8)的光辉性颜料分散液(8)(固体成分浓度:10%)。光辉性颜料分散液(8)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(9)的制备
·平均粒径为4μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨10小时(a)。提取170份混合物并通过球磨机进一步研磨5小时(b)。将(a)和(b)混合,获得光辉性颜料(9)。
·光辉性颜料(9):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(9)的光辉性颜料分散液(9)(固体成分浓度:10%)。光辉性颜料分散液(9)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(10)的制备
·平均粒径为4μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨15小时(c)。提取55份混合物并通过球磨机进一步研磨5小时(d)。将(c)和(d)混合,获得光辉性颜料(10)。
·光辉性颜料(10):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(10)的光辉性颜料分散液(10)(固体成分浓度:10%)。光辉性颜料分散液(10)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(11)的制备
·平均粒径为2.5μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨10小时,获得光辉性颜料(11)。
·光辉性颜料(11):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(11)的光辉性颜料分散液(11)(固体成分浓度:10%)。光辉性颜料分散液(11)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(12)的制备
·平均粒径为8.5μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨25小时,获得光辉性颜料(12)。
·光辉性颜料(12):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(12)的光辉性颜料分散液(12)(固体成分浓度:10%)。光辉性颜料分散液(12)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(13)的制备
·平均粒径为10μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨25小时,获得光辉性颜料(13)。
·光辉性颜料(13):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(13)的光辉性颜料分散液(13)(固体成分浓度:10%)。光辉性颜料分散液(13)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(14)的制备
·平均粒径为2.5μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨8小时,获得光辉性颜料(14)。
·光辉性颜料(14):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(14)的光辉性颜料分散液(14)(固体成分浓度:10%)。光辉性颜料分散液(14)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
光辉性颜料分散液(15)的制备
·平均粒径为8.5μm的铝雾化粉末:100份
·矿油精:120份
·硬脂酸:3份
将上述成分混合并通过球磨机研磨28小时,获得光辉性颜料(15)。
·光辉性颜料(15):100份
·乙酸乙酯:500份
将上述成分混合,重复5次将过滤混合物并使该混合物与500份乙酸乙酯进一步混合的操作,随后利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)将混合物分散1小时,并获得其中分散有光辉性颜料(15)的光辉性颜料分散液(15)(固体成分浓度:10%)。光辉性颜料分散液(15)中所含光辉性颜料的平均厚度、体积平均粒径、平均厚度的上gsd、平均厚度的下gsd和长径比将在表1中示出。
防粘剂分散液(1)的制备
·石蜡(熔融温度:89℃):30份
·乙酸乙酯:270份
以在10℃冷却的状态通过微型珠分散器(dcp磨机)将上述成分湿式粉碎,获得防粘剂分散液(1)。
油相溶液(1)的制备
·未改性聚酯树脂(1):136份
·光辉性颜料分散液(1):500份
·乙酸乙酯:56份
将上述成分搅拌和混合,然后向所得混合物中添加75份防粘剂分散液(1),搅拌混合物,获得油相溶液(1)。
苯乙烯丙烯酸树脂颗粒分散液(1)的制备
·苯乙烯:370份
·丙烯酸正丁酯:30份
·丙烯酸:4份
·十二烷硫醇:24份
·四溴化碳:4份
在烧瓶中将通过混合和溶解以上成分而获得的混合物在水溶液中乳化,所述水溶液通过将6份非离子型表面活性剂(由sanyochemicalindustries,ltd.制造的nonipol400)和10份阴离子型表面活性剂(由dskco.,ltd.制造的neogensc)溶解在560份离子交换水中获得,将通过使4份过硫酸铵溶解在50份离子交换水中获得的水溶液倒入混合物中同时使混合物混合10分钟,对混合物进行氮吹扫,随后搅拌烧瓶中内含物的同时在油浴中加热直至内含物的温度达到70℃,并且是乳化聚合原样继续5小时。因此,获得苯乙烯丙烯酸树脂颗粒分散液(1)(树脂颗粒浓度:40%),其中分散有平均粒径为180nm且重均分子量(mw)为15,500的树脂颗粒。苯乙烯丙烯酸树脂颗粒的玻璃化转变温度为59℃。
水相溶液(1)的制备
·苯乙烯丙烯酸树脂颗粒分散液(1):60份
·celogenbs-h(dskco.,ltd.)的2%水溶液:200份
·离子交换水:200份
将上述成分搅拌并混合,获得水相溶液(1)。
实施例1
色调剂颗粒(1)的制备
·油相溶液(1):300份
·异氰酸酯改性聚酯预聚物(1):25份
·酮亚胺化合物(1):0.5份
将以上成分放入容器中,通过用均质器(ika制造的ultra-turrax)搅拌所述成分2分钟而获得油相溶液(1p),随后将1000份水相溶液(1)加入容器中,并通过均质器将混合物搅拌20分钟。随后,通过推进型搅拌器以常压(1atm)将混合物溶液在室温(25℃)下搅拌48小时,通过引起异氰酸酯改性聚酯预聚物(1)和酮亚胺化合物(1)之间的反应而形成脲改性聚酯树脂,去除有机溶剂,获得粒状物。然后,将粒状物用水洗涤,干燥并分级,获得色调剂颗粒(1)。色调剂颗粒的体积平均粒径为12.0μm,且长径比为6.0。
光辉性色调剂(1)的制备
通过样品磨机以10000rpm将100份色调剂颗粒(1)、1.5份疏水性二氧化硅(由nipponaerosilco.,ltd.制造的ry50)和1.0份疏水性二氧化钛(由nipponaerosilco.,ltd.制造的t805)混合30秒。其后,通过具有45μm的筛网的振动分级器将混合物分级,获得光辉性色调剂(1)。
实施例2
以与油相溶液(1)相同的方式获得油相溶液(2),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(2)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(2),不同之处在于使用油相溶液(2)。
实施例3
以与油相溶液(1)相同的方式获得油相溶液(3),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(3)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(3),不同之处在于使用油相溶液(3)。
比较例1
以与油相溶液(1)相同的方式获得油相溶液(4),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(4)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(4),不同之处在于使用油相溶液(4)。
比较例2
以与油相溶液(1)相同的方式获得油相溶液(5),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(5)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(5),不同之处在于使用油相溶液(5)。
实施例4
以与油相溶液(1)相同的方式获得油相溶液(6),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(6)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(6),不同之处在于使用油相溶液(6)。
实施例5
以与油相溶液(1)相同的方式获得油相溶液(7),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(7)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(7),不同之处在于使用油相溶液(7)。
实施例6
以与油相溶液(1)相同的方式获得油相溶液(8),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(8)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(8),不同之处在于使用油相溶液(8)。
实施例7
以与油相溶液(1)相同的方式获得油相溶液(9),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(9)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(9),不同之处在于使用油相溶液(9)。
实施例8
以与油相溶液(1)相同的方式获得油相溶液(10),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(10)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(10),不同之处在于使用油相溶液(10)。
实施例9
以与油相溶液(1)相同的方式获得油相溶液(11),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(11)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(11),不同之处在于使用油相溶液(11)。
实施例10
以与油相溶液(1)相同的方式获得油相溶液(12),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(12)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(12),不同之处在于使用油相溶液(12)。
实施例11
以与油相溶液(1)相同的方式获得油相溶液(13),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(13)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(13),不同之处在于使用油相溶液(13)。
实施例12
以与油相溶液(1)相同的方式获得油相溶液(14),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(14)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(14),不同之处在于使用油相溶液(14)。
实施例13
以与油相溶液(1)相同的方式获得油相溶液(15),不同之处在于在油相溶液(1)的制备中使用光辉性颜料分散液(15)代替光辉性颜料分散液(1)。
另外,以与光辉性色调剂(1)相同的方式获得光辉性色调剂(15),不同之处在于使用油相溶液(15)。
实施例14
粘合剂树脂的合成
·己二酸二甲酯:74份
·对苯二甲酸二甲酯:192份
·双酚a的氧化乙烯加合物:216份
·乙二醇:38份
·四丁氧基钛酸酯(催化剂):0.037份
将以上成分放入经加热和干燥的两颈烧瓶中,在通过将氮气引入容器内而维持惰性气氛的同时提高所述成分的温度,并搅拌所述成分,在160℃下引起7小时共缩聚反应,随后在压力缓慢降低至10托的同时使温度提高至220℃并维持4小时。一旦压力返回常压,则向其中加入9份偏苯三酸酐,再次使压力缓慢降低至10托,随后在220℃下维持1小时,并由此合成粘合剂树脂。
利用差示扫描量热计(由shimadzucorporation制造的dsc-50)在升温速度为10℃/分钟的条件下从室温(25℃)升高至150℃通过测量获得粘合剂树脂的玻璃化转变温度(tg)。将吸热部分处的基线和上升线的延长线之间交点的温度视为玻璃化转变温度。粘合剂树脂的玻璃化转变温度为63.5℃。
树脂颗粒分散液的制备
·粘合剂树脂:160份
·乙酸乙酯:233份
·氢氧化钠水溶液(0.3n):0.1份
将以上成分放入1000ml分离式烧瓶中,在70℃加热,并用three-one马达(由shintoscientificco.,ltd.制造)搅拌,制备树脂混合物溶液。在将该树脂混合物在90rpm进一步搅拌的同时,向其缓慢添加373份离子交换水,对该混合物进行转相乳化,从其中除去溶剂,并由此获得树脂颗粒分散液(固体成分浓度:30%)。树脂颗粒分散液的体积平均粒径为162nm。
防粘剂分散液的制备
·巴西棕榈蜡(由toakaseico.,ltd.制造的rc-160):50份
·阴离子型表面活性剂(由dskco.,ltd.制造的neogenrk):1.0份
·离子交换水:200份
将以上成分混合并在95℃加热,使用均质器(由ika制造的ultra-turraxt50)分散,并利用mantongaulin高压均质器(由mantongaulinmanufacturingco.,inc.制造)进行分散处理360分钟,制备分散有体积平均粒径为0.23μm的防粘剂颗粒的防粘剂分散液(固体成分浓度:20%)。
色调剂(16)的制备
·树脂颗粒分散液:480份
·防粘剂分散液:72份
·光辉性颜料分散液(1):140份
·非离子型表面活性剂(igepalca897):1.40份
将以上原料置于2l圆筒形不锈钢容器中,在施加剪切力的同时通过均质器(由ika制造的ultra-turraxt50)在4000rpm分散并混合10分钟。然后,向其缓慢滴加1.75份的聚氯化铝的10%硝酸水溶液作为凝集剂,通过将均质器的旋转速度设定在5000rpm而将该混合物分散并混合15分钟,由此获得原料分散液。
其后,将原料分散液转移到设置有具有双桨片搅拌桨叶的搅拌器和温度计的聚合槽中,在将搅拌旋转速度设定为810rpm的同时通过覆套式加热器开始加热,并在54℃促进凝集颗粒的生长。此时,通过0.3n的硝酸和1n的氢氧化钠溶液将原料分散液的ph控制在2.2~3.5的范围内。原料分散液在该ph范围内保持约2小时,形成凝集颗粒。
接下来,额外添加100份粘合剂树脂分散液,使粘合剂树脂的树脂颗粒附着于凝集颗粒的表面。将温度进一步升高至56℃,并在通过光学显微镜和multisizerii观察粒径的同时使凝集颗粒组织化。其后,将ph增加至8.0以使凝集颗粒聚并,然后将温度升高至67.5℃。在通过光学显微镜检查出凝集颗粒已聚并之后,在将温度维持在67.5℃的同时将ph降至6.0,在1小时后停止加热,并以1.0℃/分钟的降温速度进行冷却。其后,将颗粒用20μm的筛网分级,用水反复洗涤,并通过真空干燥器干燥,获得色调剂颗粒。所得色调剂颗粒的体积平均粒径为11.5μm。
利用样品磨机以10,000rpm将1.5份疏水性二氧化硅(由nipponaerosilco.,ltd.制造的ry50)对100份所得色调剂颗粒混合和共混30秒。其后,通过具有45μm的筛网的振动分级器将混合物分级,由此制备色调剂。
表1
载体的制备
·铁氧体颗粒(体积平均粒径:35μm):100份
·甲苯:14份
·甲基丙烯酸甲酯-全氟辛基乙基丙烯酸酯共聚物(临界表面张力:24dyn/cm):1.6份
·炭黑(产品名称:vxc-72,由cabotcorporation制造,体积电阻率:等于或小于100ωcm):0.12份
·交联的三聚氰胺甲醛树脂颗粒(平均粒径:0.3μm,不溶于甲苯):0.3份
首先,将炭黑在甲苯中稀释,将混合物添加至甲基丙烯酸甲酯-全氟辛基乙基丙烯酸酯共聚物并在砂磨机中分散。随后,通过搅拌器将除铁氧体颗粒以外的以上各成分分散10分钟,获得覆盖层形成用溶液。随后,将覆盖层形成用溶液和铁氧体颗粒放入脱气型捏合机中,并将混合物在60℃搅拌30分钟。然后,降低压力,将甲苯蒸发,形成树脂覆盖层,由此获得载体。
显影剂的制备
将36份各实施例中获得的光辉性色调剂、414份载体放入2l的v型搅拌机中,将混合物搅拌20分钟,随后用212μm的筛网分级,由此获得显影剂。
色调剂图像的污染
用所得显影剂填充由fujixeroxco.,ltd.制造的“改造机color800press”的显影机。另外,用商购的黑色显影剂填充不同于含有光辉性色调剂的显影剂用显影机的显影机。
该改造机用于通过使用光辉性色调剂在10,000张oktopcoat纸(基重:127,由ojipaperco.,ltd.制造)上输出实心图像,其中光辉性色调剂施加量为4.0g/m2。
在利用光辉性色调剂输出第10,000个实心图像之后,在10秒内输出具有4.5g/m2的黑色色调剂施加量的单个实心图像。
视觉观察通过黑色色调剂施加在实心图像上的光辉性色调剂,并基于以下标准进行评估。获得的结果将在表2中示出。
标准
g1:没有观察到光辉性颗粒。
g2:虽然存在若干个光辉性颗粒,但图像可接受。
g3:存在多个光辉性颗粒,且图像不可接受。
比例(x/y)
使用由nippondenshokuindustriesco.,ltd.制造的分光式变角色差仪gc5000l作为可变角光度计,从而以相对利用光辉性色调剂输出的第10,000个实心图像为-45°的入射角使入射光入射在该实心图像上,并且测量受光角+30°处的反射率x和受光角-30°处的反射率y。
以20nm的间隔由400nm~700nm的波长范围内的光测量反射率x和反射率y,并且获得各波长处的反射率的平均值。由这些测量结果计算比例(x/y)。结果将在表2中示出。
随着比例(x/y)增加,光泽感增加。随着比例(x/y)降低,暗色调增加,且光泽感降低。
表2也示出了色调剂颗粒的体积平均粒径和长径比。
表2
提供对本发明示例性实施方式的前述描述是为了说明和描述的目的。并非试图穷尽本发明或将本发明限制于所披露的精确形式。显然,许多改进和变化对于本领域技术人员将是显而易见的。选择并描述所述实施方式是为了能够最好地解释本发明的原理及其实际用途,由此使得本领域的其他技术人员能够理解适用于预计的特定用途的本发明的各种实施方式和各种改进方案。试图使本发明的范围由下述权利要求及其等同物所限定。
- 还没有人留言评论。精彩留言会获得点赞!
- 基于景深的图像饱和度的处理方法、处理装置和电子装置与流程
- 一种对抗色调映射的高动态范围图像水印方法与流程
- 色调剂、静电荷图像显影剂、色调剂盒、处理盒、图像形成设备和图像形成方法与流程
- 光辉性色调剂、静电荷图像显影剂、色调剂盒、处理盒、图像形成设备和图像形成方法与流程
- 色调剂、静电荷图像显影剂、色调剂盒、处理盒、图像形成设备和图像形成方法与流程
- 色调剂、显影剂、色调剂盒、处理盒、图像形成设备和图像形成方法与流程
- 一种基于平均饱和度先验的图像去雾方法与流程
- 色调剂、显影剂、色调剂盒、显影剂盒、处理盒、图像形成装置和方法与流程
- 对图像进行逆色调映射的方法和设备与流程
- 色调剂、显影剂、色调剂盒、显影剂盒、处理盒、图像形成装置和图像形成方法与流程