一种低成本制备超细钴粉的方法与流程
本发明属于金属微粒制备
技术领域:
,尤其涉及一种低成本制备超细钴粉的方法。
背景技术:
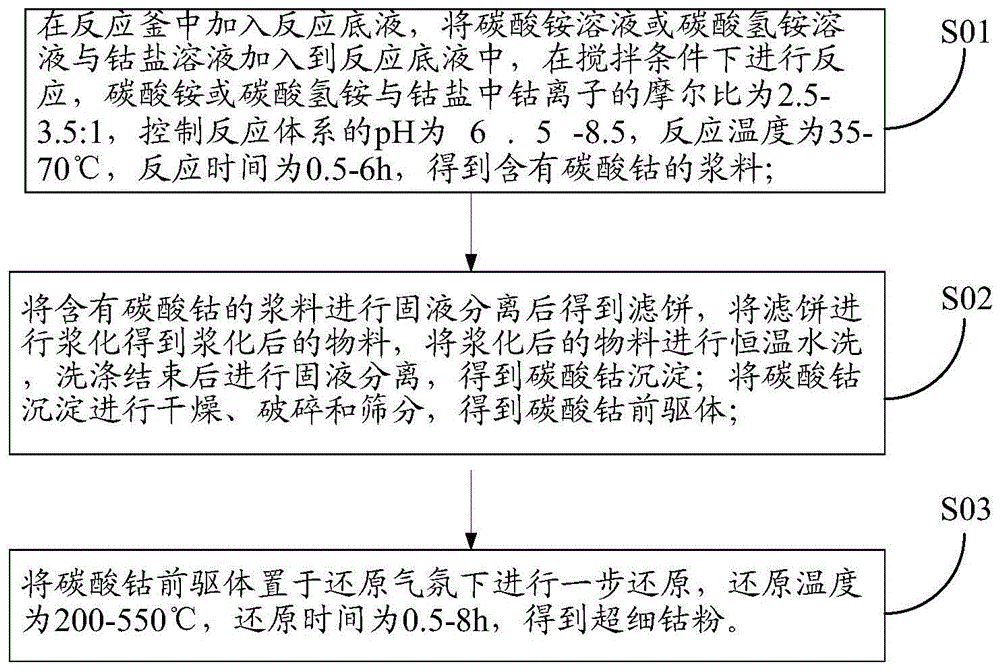
:作为一种优良的粘结剂,钴粉在硬质合金、金刚石工具和粉末冶金等领域有重要作用。随着钴粉粒径减小,可以显著降低硬质合金粉末混料的研磨时间、烧结温度,增强硬质合金抗弯强度、耐磨性和抗裂性等性能,减少钴池和孔隙的出现,因此,目前国内外都把超细钴粉作为硬质合金和金刚石工具等领域的发展方向。由于对超细钴粉的前驱体制备、钴粉还原及后处理等过程要求程度很高,导致超细钴粉的生产成本相对较高,主要体现在以下几方面:(1)、部分工艺采用氢氧化钠、氢氧化钾等碱液作为沉淀剂,会引入较多的金属阳离子杂质,增加了除杂成本。如公开号为CN103624251A、CN103128304A的发明通过氢氧化钠沉淀钴离子,沉淀剂用量为钴离子8倍以上,导致后续除Na较难;(2)、前驱体合成pH低,造成产品收率低。如公开号为CN101829786A的发明在pH5.5-6.8条件下沉钴,反应结束后母液仍呈红色,钴离子含量大于1g/L,在工业大规模生产情况下,钴的损失量大;(3)、前驱体制备中引入添加剂,增加辅料成本的同时,增加了除杂成本。如公开号为CN102689020A的发明中往钴盐中加表面活性剂和铝粉,导致后续除杂成本较高;(4)、前驱体先经过高温煅烧,再高温还原制备钴粉,生产能耗大。如公开号为CN102560100A的发明先经过350-580℃高温煅烧后再还原。公开号为CN101829786A的发明同样经过400-800℃ 煅烧2-8h后得到氧化钴粉末,再经400-550℃还原4-6h;(5)、制备工序繁杂、流程长,增加处理成本。如公开号为CN103028735A的发明先合成碳酸钴,然后利用草酸将碳酸钴转化为小粒径草酸钴,再进行干燥还原,工艺复杂。综上所述,目前生产超细钴粉的工艺普遍存在成本高的问题,若通过合理的工艺,降低除杂、能耗和工艺流程过长等带来的成本,将更有利于超细钴粉的规模化生产,推动超细晶粒硬质合金及金刚石工具等行业更好的发展。技术实现要素:为解决上述问题,本发明提供了一种低成本制备超细钴粉的方法,该制备方法工艺简单,制备成本较低,解决了现有技术制备超细钴粉成本较高的问题。本发明所述超细钴粉指的是Fsss粒度为0.4-1.0μm的钴粉。本发明提供了一种低成本制备超细钴粉的方法,包括以下步骤:(1)在反应釜中加入反应底液,将碳酸铵溶液或碳酸氢铵溶液与钴盐溶液加入到所述反应底液中,在搅拌条件下进行反应,所述碳酸铵或所述碳酸氢铵与所述钴盐中钴离子的摩尔比为2.5-3.5:1,反应体系的pH为6.5-8.5,反应温度为35-70℃,反应时间为0.5-6h,得到含有碳酸钴的浆料;(2)将所述含有碳酸钴的浆料进行固液分离后得到滤饼,将所述滤饼进行浆化得到浆化后的物料,将所述浆化后的物料进行恒温水洗,洗涤结束后进行固液分离,得到碳酸钴沉淀;将所述碳酸钴沉淀进行干燥、破碎和筛分,得到碳酸钴前驱体;(3)将所述碳酸钴前驱体置于还原气氛下进行一步还原,所述还原温度为200-550℃,所述还原时间为0.5-8h,得到超细钴粉。优选地,步骤(1)中所述反应底液为纯水、碳酸盐溶液或碳酸氢盐溶液。更优选地,步骤(1)中当所述反应底液为碳酸盐溶液或碳酸氢盐溶液时,所述反应底液中碳酸盐或碳酸氢盐的浓度大于0g/L小于250g/L。更优选地,所述碳酸盐为碳酸铵,所述碳酸氢盐为碳酸氢铵。优选地,步骤(1)中所述钴盐为氯化钴、醋酸钴、硫酸钴和硝酸钴中的至 少一种。本发明的沉淀剂选用碳酸铵或碳酸氢铵,没有引入金属阳离子杂质,降低了后续的除杂成本。优选地,步骤(1)中,将所述碳酸铵溶液或所述碳酸氢铵溶液与所述钴盐溶液并流加入到所述反应底液中。根据反应实际情况通过改变碳酸铵或碳酸氢铵的流量来调节反应pH。本发明采用并流加料的方式合成碳酸钴时,通过调节所述碳酸铵溶液或所述碳酸氢铵溶液的流量来调节加料过程中pH值,可以避免正流法和逆流法加料过程中pH值的大幅度变化和局部浓度过高现象,有助于产品质量的稳定和连续化生产。优选地,步骤(1)中,所述钴盐溶液中钴离子浓度为50-160g/L。优选地,步骤(1)中,所述碳酸铵溶液或碳酸氢铵溶液的浓度为120-260g/L。步骤(1)中的反应体系的pH为6.5-8.5,在该pH范围内可以制备出粒径更小的碳酸钴,同时可以降低生产成本。优选地,步骤(1)中,所述反应体系的pH为7.0-8.5。优选地,步骤(1)中所述搅拌速度为100-400rpm。本发明在制备碳酸钴前驱体过程中,采用不含金属元素的碳酸铵或碳酸氢铵作为沉淀剂,同时制备过程中没有引入添加剂,降低了原料成本和后续的除杂成本。另外,合成的pH相对于现有技术较高,可以提高钴的产量,降低生产成本。本发明在一定的反应温度和反应时间范围内,可以得到粒径小、比表面积大和分散性好的碳酸钴前驱体,反应充分,后续不需进行陈化工艺,在缩短工艺流程、降低加工成本的同时,产品的性能也得到了更好地控制。优选地,步骤(2)中,采用板框压滤机和微孔过滤器中的至少一种固液分离设备进行所述固液分离。更优选地,利用所述板框压滤机进行固液分离时,压强为18-22MPa。更优选地,利用所述微孔过滤器进行固液分离时,压强为0.1-0.25MPa。固液分离后得到的母液可进入回收系统进行重复利用,得到的滤饼进行浆 化洗涤。本发明所使用固液分离设备是板框压滤机或微孔过滤器、或两种洗涤设备的组合,在固液分离后得到滤饼,无需对滤饼进行洗涤,直接卸料进行后续的浆化洗涤操作。更优选地,步骤(2)中,将所述含有碳酸钴的浆料采用板框压滤机进行固液分离得到滤饼。采用板框压滤机时,压力更大,可以提高固液分离速度,即提升了生产效率。优选地,步骤(2)中,将所述含有碳酸钴的浆料进行固液分离后得到滤饼,将所述滤饼卸料至漏斗式下料槽,然后用纯水冲洗至浆化槽中进行浆化,浆化后,得到浆化后的物料,将所述浆化后的物料通过泵输送至浆洗槽中进行恒温水洗,洗涤结束后进行固液分离。更优选地,所述浆化时间为20min-40min,通过浆化工艺将滤饼分散均匀,有助于将物料通过泵直接输送至浆洗槽进行洗涤,同时,有利于后续除杂洗涤操作。更优选地,步骤(2)中,所述水洗的温度为0-80℃。进一步优选地,步骤(2)中,所述水洗的温度为25-80℃。更优选地,步骤(2)中,所述水洗的时间为0.5-10h。更优选地,所述恒温水洗时进行搅拌,所述搅拌速度为30-200rpm。优选地,所述恒温水洗的次数为1-4次。每次恒温水洗后,都要进行固液分离,然后进行第二次水洗或进入后续工艺。所述水洗温度、洗涤水量、水洗时间以及洗涤次数根据洗涤过程中物料中的Cl-、NO3-、SO42-或CH3COO-等杂质的含量水平决定。优选地,第一次洗涤结束时,以Cl-为例,物料中Cl含量应不高于0.8%,若Cl-含量较高则加大水量、延长洗涤时间;第二次洗涤结束时,物料中Cl-含量应不高于0.08%,若含量高于0.08%,则加大洗水量、延长洗涤时间;若第二次洗涤结束后Cl-含量高于0.1%,则进行第三次洗涤,直至物料中Cl-含量小于0.08%,进入下一个工序。优选地,本发明还可以根据浆料中Cl-含量调整洗涤过程温度,如果Cl-含量高于控制标准,则升高洗涤温度,Cl-含量较低,则适当调低洗涤温度。通过控制洗涤过程温度,在一定程度上可以维持相同洗涤用水量的情况下,增强洗涤效果,进而节约洗涤用水量。现有技术的洗涤方式主要是固液分离后,直接将过滤得到的滤饼或滤渣进行简单地淋洗,或者将滤饼或滤渣浆化后,进行固液分离得到滤饼后再次洗涤滤饼。这些洗涤的方式由于洗涤方式单一、洗涤条件固定,都难以将产物中的杂质完全去除。同时会造成产品颗粒分布不均匀,一致性差,不利于产品质量的稳定。与现有技术中通过检测滤液中杂质(如Cl-)含量以及洗涤时间、洗涤水量固定的方式不同。本发明优选地根据洗涤后物料中的Cl-含量情况控制水洗温度、洗涤水量和水洗时间等因素,在优选的洗涤温度下、洗涤水量下、洗涤时间下以及洗涤次数下可以得到更好的洗涤效果,因此,本发明提供的洗涤方法更有利于节约生产用水及生产时间,同时,可以达到更好的除杂效果,最终显著降低物料中的杂质含量。本发明通过固液分离和洗涤操作可以显著地降低碳酸钴中的杂质含量,同时固液分离和浆化洗涤操作连续化程度高,可以根据含有碳酸钴的浆料中的杂质含量调节洗涤参数,降低产能消耗,增强洗涤效果,有利于规模化生产。优选地,步骤(2)中,所述干燥的方法为离心喷雾干燥、闪蒸干燥或鼓风干燥。更优选地,步骤(2)中,所述离心喷雾干燥的条件为:进料口温度为160-250℃,出料口温度为70-105℃。进一步优选地,所述离心喷雾干燥的条件为:进料口温度为180-220℃,出料口温度为80-100℃。更优选地,所述闪蒸干燥条件为:进料口温度为170-230℃,出料口温度为90-120℃。进一步优选地,步骤(2)中,所述闪蒸干燥条件为:进料口温度为180-220℃, 出料口温度为100-110℃。采用所述离心喷雾干燥或所述闪蒸干燥方法时,物料干燥时间小于5min,相比于现有技术的干燥方式,大大缩短了干燥时间,减少干燥过程对产品性能的影响。优选地,所述鼓风干燥的条件为:干燥温度75-140℃,干燥时间为6-12h。更优选干燥温度为80-120℃。优选地,步骤(2)中,在将所述碳酸钴沉淀干燥后,碳酸钴中的含水量为小于3.0%。步骤(2)中,在将所述碳酸钴沉淀干燥后,当所述碳酸钴的粒径D50>4.5μm时,需要进行破碎操作。优选地,所述破碎的方法为气流破碎和机械破碎中的至少一种。更优选地,步骤(2)中,破碎后,对所述碳酸钴采用200目筛网进行筛分。本发明制得的碳酸钴前驱体的粒径较小,纯度较高。优选地,所述碳酸钴前驱体的D50粒径为0.5-3.0μm,松装密度为0.2-0.5g/cm3,比表面积为40-150m2/g。优选地,所述碳酸钴前驱体中Cl含量小于等于100ppm,Ca、Na、Mg、Fe元素含量小于等于20ppm,Cu、Zn、Al元素含量小于等于10ppm。超细颗粒的合成需要在溶液过饱和条件下进行,在过饱和溶液中,沉淀剂的加入会导致碳酸钴爆炸性成核,形成的碳酸钴颗粒小、比表面积大,难以进行固液分离和除杂。本发明在沉淀反应结束后,将含有碳酸钴的浆料通过固液分离后,使得母液去除完全,减少了杂质残留。之后在一定温度下进行高强度的浆化洗涤,促使吸附在碳酸钴颗粒表面的杂质离子脱附,再经过固液分离,再次去除部分杂质。固液分离和浆化洗涤结束后,物料经过合适条件下的干燥、破碎得到粒径小、纯度高的碳酸钴颗粒。本发明采用钴盐为原料,碳酸铵或碳酸氢铵为沉淀剂,在一定原料和沉淀剂摩尔比、pH、温度范围内,合成含有碳酸钴的浆料。然后将所述含碳酸钴的浆料经过固液分离及浆化洗涤后,进行干燥、破碎、筛分,最终得到杂质含量低、粒径小、比表面积大的碳酸钴粉末。相比其他制备方法,本发明制备碳酸 钴过程中并未引入任何其他添加剂,生产成本低,流程简单,且所得产品中金属元素杂质及阴离子杂质含量均很低,有利于得到粒径小、纯度高的钴粉。优选地,步骤(3)中,将所述碳酸钴前驱体置于多温区管式炉或钢带式还原炉中进行一步还原。更优选地,采用所述多温区管式炉进行一步还原时,还原温度为200-450℃,还原时间为5-8h。进一步优选地,采用所述多温区管式炉进行一步还原时,还原温度控制模式采用非线性控制模式,所述非线性控制模式为梯形温控模式或平台温控模式。进一步优选地,所述非线性控制模式为梯形温控模式时,所述还原过程中在所述多温区管式炉中分为三个温区分别为进炉区、中间区和出炉区,所述进炉区和所述出炉区温度低于所述中间区温度,所述进炉区温度为200-320℃,所述中间温区温度为320-450℃,所述出炉区温度250-350℃,在所述进炉区停留时间为2-4h,在所述中间温区的停留时间为4-6h,在所述出炉区中的停留时间为2-4h。进一步优选地,所述非线性控制模式为平台温控模式时,所述还原过程中在所述多温区管式炉中分为2或3个温区,所述2个温区分别为低温区和高温区,所述3个温区分别为第一低温区、第二低温区和高温区,所述低温区、第一低温区或第二低温区的温度各为200-350℃,停留时间各为3-5h,所述高温区的温度350-450℃,停留时间为2-4h。更优选地,采用所述钢带式还原炉进行一步还原时,还原温度为250-550℃,还原时间为0.5-4h。进一步优选地,采用所述钢带式还原炉进行一步还原时,还原温度控制模式采用非线性控制模式,所述非线性控制模式为梯形温控模式或平台温控模式。进一步优选地,所述非线性控制模式为梯形温控模式时,所述还原过程中在所述多温区管式炉中分为三个温区分别为进炉区、中间区和出炉区,所述进炉区和所述出炉区温度低于所述中间区温度,所述进炉区温度为250-450℃,所述中间温区温度为350-550℃,所述出炉区温度250-400℃,在所述进炉区停留 时间为0.1-2h,在所述中间温区的停留时间为0.3-2h,在所述出炉区中的停留时间为0.1-2h。进一步优选地,所述非线性控制模式为平台温控模式时,所述还原过程中在所述多温区管式炉中分为2或3个温区,所述2个温区分别为低温区和高温区,所述3个温区分别为第一低温区、第二低温区和高温区,所述低温区、第一低温区或第二低温区的温度各为250-400℃,停留时间分别为0.3-3h,所述高温区的温度350-550℃,停留时间为0.2-1h。优选地,步骤(3)中,所述还原气氛为氢气气氛。优选地,步骤(3)中,在所述还原过程中加入氢气,所述氢气的流量是4-50m3/h。本发明在采用非线性温控模式进行一步还原,相对于现有技术的先高温煅烧再还原的技术,降低了氢气的消耗量,生产能耗大大降低。相对于线性温控模式一步还原法,本发明设置了低温段,为物料进入还原阶段提供了一定的过渡期,在较低温度下还原时,有利于保持物料的形貌及还原过程样的均一性,有利于得到性能更均一的钴粉。后期的高温段还原,则保证了钴粉的还原完全,保证了产品质量。优选地,步骤(3)中,将所述碳酸钴还原后,进行破碎、筛分和混料包装,得到超细钴粉。优选地,所述超细钴粉的Fsss粒度为0.4-1.0μm。更优选地,所述超细钴粉的Fsss粒度为0.5-0.8μm。本发明超细钴粉的制备包括前驱体的合成与前驱体的还原。所述前驱体合成过程为液相沉淀法,前驱体的合成过程中未引入添加剂,减少了原料的杂质元素引入量,同时工艺流程简单,大大降低了生产成本,制得了粒径较小、纯度较高的碳酸钴前驱体。所述前驱体的还原采用一步还原法,通过采用合理的温控模式,一步还原制得钴粉,能耗成本及工艺成本都得到有效控制,最终实现了低成本条件下规模化生产超细钴粉的目的。本发明提供的低成本制备超细钴粉的方法,具有如下有益效果:本发明采用液相沉淀—一步还原法制备超细钴粉,通过优化工艺、简化过程,多途径缩减生产成本,最终实现了低成本条件下生产超细钴粉的目的。附图说明图1是本发明一实施例中低成本制备超细钴粉方法的流程示意图;图2为本发明实施例1制备的超细钴粉的扫描电镜(SEM)图;图3为本发明实施例2制备的超细钴粉的扫描电镜(SEM)图;图4为本发明实施例3制备的超细钴粉的扫描电镜(SEM)图。具体实施方式以下所述是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也视为本发明的保护范围。请参考图1,图1是本发明一实施例中低成本制备超细钴粉方法的流程示意图,包括以下步骤:S01、在反应釜中加入反应底液,将碳酸铵溶液或碳酸氢铵溶液与钴盐溶液加入到反应底液中,在搅拌条件下进行反应,碳酸铵或碳酸氢铵与钴盐中钴离子的摩尔比为2.5-3.5:1,控制反应体系的pH为6.5-8.5,反应温度为35-70℃,反应时间为0.5-6h,得到含有碳酸钴的浆料;S02、将含有碳酸钴的浆料进行固液分离后得到滤饼,将滤饼进行浆化得到浆化后的物料,将浆化后的物料进行恒温水洗,洗涤结束后进行固液分离,得到碳酸钴沉淀;将碳酸钴沉淀进行干燥、破碎和筛分,得到碳酸钴前驱体;S03、将碳酸钴前驱体置于还原气氛下进行一步还原,还原温度为200-550℃,还原时间为0.5-8h,得到超细钴粉。本发明超细钴粉的制备包括前驱体的合成与前驱体的还原。所述前驱体合成过程为液相沉淀法,前驱体的合成过程中未引入添加剂,减少了原料的杂质 元素引入量,同时工艺流程简单,大大降低了生产成本,制得了粒径较小、纯度较高的碳酸钴前驱体。所述前驱体的还原采用一步还原法,通过采用合理的温控模式,一步还原制得钴粉,能耗成本及工艺成本都得到有效控制,最终实现了低成本条件下生产超细钴粉的目的。实施例1一种低成本制备超细钴粉的方法,包括以下步骤:(1)、向合成反应釜中加入纯水(反应底液)至搅拌桨处,升高反应釜温度至55℃后,启动搅拌桨,控制转速为220rpm,同时并流加入氯化钴溶液和碳酸氢铵溶液,其中氯化钴溶液中钴离子浓度为80g/L,碳酸氢铵溶液浓度为180g/L,通过碳酸氢铵流量调节pH,控制反应pH在7.5-8.2之间,反应温度为55℃±2℃。反应2.5h后停止加料,得到含有碳酸钴的浆料;碳酸氢铵与氯化钴中钴离子的摩尔比为2.6:1;(2)将含有碳酸钴的浆料输送至板框压滤机进行固液分离,压强为19Mpa,得到母液和滤饼,母液进入回收系统,滤饼卸料至漏斗式下料槽,然后用纯水冲洗至带搅拌浆的浆化槽中,浆化20min后得到浆化后的物料,将浆化后的物料通过泵输送至带搅拌浆的浆洗槽中进行恒温水洗,启动搅拌,搅拌速度为30rpm保持洗涤温度为60℃,洗涤2.5h。重复进行上述固液分离与浆化洗涤1次,洗涤结束后将物料经板框压滤机压干,得到碳酸钴沉淀;将碳酸钴沉淀转移至闪蒸干燥机,进料口温度为210℃,出料口温度为85℃,使得干燥后粉末水分含量<3%,干燥后物料经过气流破碎、筛分后得到碳酸钴前驱体;(3)将碳酸钴前驱体送至还原工段的多温区钢带式还原炉,还原炉中依次设置三个温区,分别为进炉区、中间区和出炉区,进炉区温度为450℃,中间温区温度为550℃,出炉区温度400℃,在进炉区停留时间为0.1h,在中间温区的停留时间为0.3h,在所述出炉区中的停留时间为0.1h。钢带速度为200mm/min,还原后所得钴粉依次经气流破碎机、筛分机、混料包装等设备后得到超细钴粉,超细钴粉的Fsss粒度为0.56μm。实施例1制得的碳酸钴前驱体的D50粒径为2.8μm,松装密度为0.45g/cm3,比表面积为87m2/g;碳酸钴前驱体中Ca、Na、Fe、Mg含量小于15ppm,Cl含量小于60ppm,杂质含量具体参见表1。从表1中可以看出,本实施例1制得的碳酸钴前驱体杂质含量小,纯度高。表1实施例1制得的碳酸钴前驱体中的杂质含量NiCuFeCaMgMnZnPb0.0007%0.0001%0.0008%0.0011%0.0008%0.0002%0.0001%0.0002%CdNaAlSiCrSAsCl0.0001%0.0015%0.0001%0.0005%0.0005%0.0020%0.0001%0.0052%图2为本发明实施例1制备的超细钴粉的扫描电镜(SEM)图;从图2中可以看出,实施例1制得的超细钴粉粒径较小,粒径均匀分布。实施例2一种低成本制备超细钴粉的方法,包括以下步骤:(1)向合成反应釜中加入纯水(反应底液)至搅拌桨处,升高反应釜温度至65℃后,启动搅拌桨,控制转速为220rpm,同时并流加入醋酸钴溶液和碳酸铵溶液,其中醋酸钴溶液中钴离子浓度为140g/L,碳酸铵溶液浓度为240g/L。通过碳酸铵流量调节pH,控制反应pH在7.0-8.0之间,反应温度65℃±2℃。反应1h后停止加料,得到含有碳酸钴的浆料;碳酸铵和醋酸钴中钴离子的摩尔比为3.0:1;(2)将含有碳酸钴的浆料输送至微孔过滤器进行固液分离,压强为0.2Mpa,得到母液和滤饼,母液进入回收系统,滤饼卸料至漏斗式下料槽,然后用纯水冲洗至带搅拌浆的浆化槽中,浆化30min后得到浆化后的物料,将浆化后的物料通过泵输送至带搅拌浆的浆洗槽中进行恒温水洗,启动搅拌,搅拌速度为200rpm,保持洗涤温度为45℃,洗涤5h。重复进行上述固液分离与浆化洗涤3次,洗涤结束后物料经微孔过滤器压干,得到碳酸钴沉淀;将碳酸钴沉淀利用闪蒸干燥机进行干燥。进料口温度为195℃,出料口温度为95℃,使得干燥后粉末水分含量<3%,干燥后物料经过气流破碎、筛分后得到碳酸钴前驱体;(3)将碳酸钴前驱体送至还原工段的多温区管式还原炉,还原炉中依次设置三个温区,分别为进炉区、中间区和出炉区,进炉区温度为320℃,中间温区温度为450℃,出炉区温度350℃,在进炉区停留时间为2h,在中间温区的停留时间为4h,在出炉区中的停留时间为2h。还原后所得钴粉依次经气流破碎机、筛分机、混料包装等设备后得到超细钴粉,超细钴粉的Fsss粒度为0.65μm。实施例2制得的碳酸钴前驱体的D50粒径为3.9μm,松装密度为0.49g/cm3,比表面积为56m2/g;碳酸钴前驱体中Ca、Na、Fe、Mg含量小于17ppm,Cl含量小于75ppm,杂质含量具体参见表2。从表2中可以看出,本实施例2制得的碳酸钴前驱体杂质含量小,纯度高。表2实施例2制得的碳酸钴前驱体中的杂质含量NiCuFeCaMgMnZnPb0.0010%0.0001%0.0006%0.0013%0.0006%0.0001%0.0001%0.0001%CdNaAlSiCrSAsCl0.0001%0.0014%0.0002%0.0003%0.0004%0.0018%0.0001%0.072%图3为本发明实施例2制备的超细钴粉的扫描电镜(SEM)图;从图3中可以看出,实施例2制得的超细钴粉粒径较小,粒径均匀分布。实施例3一种低成本制备超细钴粉的方法,包括以下步骤:(1)、向合成反应釜中加入纯水(反应底液)至搅拌桨处,升高反应釜温度至35℃后,启动搅拌桨,控制转速为400rpm,同时并流加入硫酸钴溶液和碳酸铵溶液,其中硫酸钴溶液中钴离子浓度为100g/L,碳酸铵溶液浓度为200g/L。通过碳铵流量调节pH,控制反应pH在7.2-8.5之间,反应温度35℃±2℃。反应6h后停止加料,得到含有碳酸钴的浆料;碳酸铵和硫酸钴中钴离子的摩尔比为2.5:1;(2)将含有碳酸钴的浆料输送至板框压滤机进行固液分离,压强为20Mpa,得到母液和滤饼,母液进入回收系统,滤饼卸料至漏斗式下料槽,然后用纯水冲洗至带搅拌浆的浆化槽中,浆化30min后得到浆化后的物料,将浆化后的物 料通过泵输送至带搅拌浆的浆洗槽中进行恒温水洗,启动搅拌,搅拌速度为100rpm,保持洗涤温度为75℃,洗涤0.5h。重复进行上述固液分离与浆化洗涤2次,洗涤结束后将物料经微孔过滤器压干,得到碳酸钴沉淀;将碳酸钴沉淀利用离心喷雾机进行干燥,进料口温度为220℃,出料口温度为80℃,使得干燥后粉末水分含量<3%,干燥后物料经过涡轮破碎、筛分后得到碳酸钴前驱体;(3)将碳酸钴前驱体送至还原工段的多温区钢带式还原炉,还原炉中依次设置三个温区,分别为第一低温区、第二低温区和高温区,第一低温区和第二低温区的温度为400℃,停留时间各为0.3h,高温区的温度550℃,停留时间为1h,钢带速度为250mm/min,还原后所得钴粉依次经气流破碎机、筛分机、混料包装等设备后得到超细钴粉,超细钴粉的Fsss粒度为0.45μm。实施例3制得的碳酸钴前驱体的D50粒径为1.6μm,松装密度为0.40g/cm3,比表面积为98m2/g;碳酸钴前驱体中Ca、Na、Fe、Mg含量小于20ppm,Cl含量小于105ppm,杂质含量具体参见表3。从表3中可以看出,本实施例3制得的碳酸钴前驱体杂质含量小,纯度高。表3实施例3制得的碳酸钴前驱体中的杂质含量NiCuFeCaMgMnZnPb0.0013%0.0001%0.0008%0.0015%0.0007%0.0001%0.0002%0.0001%CdNaAlSiCrSAsCl0.0001%0.0015%0.0003%0.0002%0.0003%0.0016%0.0001%0.0103%图4为本发明实施例3制备的超细钴粉的扫描电镜(SEM)图;从图4中可以看出,实施例3制得的超细钴粉粒径较小,粒径均匀分布。实施例4一种低成本制备超细钴粉的方法,包括以下步骤:(1)向合成反应釜中加入纯水(反应底液)至搅拌桨处,升高反应釜温度至45℃后,启动搅拌桨,控制转速为250rpm,同时并流加入硝酸钴溶液和碳酸氢铵溶液,其中硝酸钴溶液中钴离子浓度为160g/L,碳酸氢铵溶液浓度为260g/L。通过碳酸氢铵流量调节pH,控制反应pH在6.5-8.0之间,反应温度为45℃±2℃。 反应3.5h后停止加料,得到含有碳酸钴的浆料;碳酸氢铵和硝酸钴中钴离子的摩尔比为3.5:1;(2)将含有碳酸钴的浆料输送至微孔过滤器进行固液分离,压强为0.18Mpa,得到母液和滤饼,母液进入回收系统,滤饼卸料至漏斗式下料槽,然后用纯水冲洗至带搅拌浆的浆化槽中,浆化30min后得到浆化后的物料,将浆化后的物料通过泵输送至带搅拌浆的浆洗槽中进行恒温水洗,启动搅拌,搅拌速度为100rpm,保持洗涤温度为25℃,洗涤6h。重复进行上述固液分离与浆化洗涤2次,洗涤结束后物料经板框压滤机压干,得到碳酸钴沉淀。将碳酸钴沉淀转移至闪蒸干燥机,进料口温度为200℃,出料口温度为85℃,使得干燥后粉末水分含量<3%,干燥后物料经过气流破碎、筛分后得到碳酸钴前驱体;(3)将碳酸钴前驱体送至还原工段的多温区钢带式还原炉,还原炉中依次设置三个温区,分别为进炉区、中间区和出炉区,进炉区温度为250℃,中间温区温度为350℃,出炉区温度250℃,在进炉区停留时间为2h,在中间温区的停留时间为1h,在出炉区中的停留时间为1h。钢带速度为160mm/min,还原后所得钴粉依次经气流破碎机、筛分机、混料包装等设备后得到超细钴粉,超细钴粉的Fsss粒度为0.59μm。实施例4制得的碳酸钴前驱体的D50粒径为3.4μm,松装密度为0.48g/cm3,比表面积为65m2/g;碳酸钴前驱体中Ca、Na、Fe、Mg含量小于16ppm,Cl含量小于76ppm,杂质含量具体参见表4,从表4中可以看出,本实施例4制得的碳酸钴前驱体杂质含量小,纯度高。表4实施例4制得的碳酸钴前驱体中的杂质含量NiCuFeCaMgMnZnPb0.0009%0.0002%0.0005%0.0010%0.0003%0.0002%0.0001%0.0001%CdNaAlSiCrSAsCl0.0001%0.0011%0.0002%0.0002%0.0002%0.0015%0.0001%0.0065%实施例5一种低成本制备超细钴粉的方法,包括以下步骤:(1)向合成反应釜中加入纯水(反应底液)至搅拌桨处,升高反应釜温度 至70℃后,启动搅拌桨,控制转速为100rpm,同时并流加入氯化钴溶液和碳铵溶液,其中氯化钴溶液中钴离子浓度为135g/L,碳酸铵溶液浓度为260g/L。通过碳酸铵流量调节pH,控制反应pH在7.1-8.3之间,反应温度为70℃±2℃。反应0.5h后停止加料,得到含有碳酸钴的浆料;碳酸铵和氯化钴中钴离子的摩尔比为2.8:1;(2)将含有碳酸钴的浆料输送至板框压滤机进行固液分离,压强为19Mpa,得到母液和滤饼,母液进入回收系统,滤饼卸料至漏斗式下料槽,然后用纯水冲洗至带搅拌浆的浆化槽中,浆化40min后得到浆化后的物料,将浆化后的物料通过泵输送至带搅拌浆的浆洗槽中进行恒温水洗,启动搅拌,搅拌速度为200rpm,保持洗涤温度为55℃,洗涤3.0h。重复进行上述固液分离与浆化洗涤1次,洗涤结束后物料经板框压滤机压干,得到碳酸钴沉淀;将碳酸钴沉淀转移至闪蒸干燥机,进料口温度为210℃,出料口温度为92℃,使得干燥后粉末水分含量<3%,干燥后物料经过气流破碎、筛分后得到碳酸钴前驱体;(3)将碳酸钴前驱体送至还原工段的多温区管式还原炉,还原炉中依次设置两个温区,分别为低温区和高温区,低温区的温度为200℃,停留时间为4h,高温区的温度350℃,停留时间为4h。还原后所得钴粉依次经气流破碎机、筛分机、混料包装等设备后得到超细钴粉,超细钴粉的Fsss粒度为0.96μm。实施例5制得的碳酸钴前驱体的D50粒径为4.4μm,松装密度为0.49g/cm3,比表面积为52m2/g;碳酸钴前驱体中Ca、Na、Fe、Mg含量小于18ppm,Cl含量小于65ppm,杂质含量具体参见表5。从表5中可以看出,本实施例5制得的碳酸钴前驱体杂质含量小,纯度高。表5实施例5制得的碳酸钴前驱体中的杂质含量NiCuFeCaMgMnZnPb0.0012%0.0001%0.0007%0.0013%0.0005%0.0001%0.0001%0.0001%CdNaAlSiCrSAsCl0.0001%0.0016%0.0001%0.0002%0.0003%0.0017%0.0001%0.060%本发明采用液相沉淀-一步还原法制备超细钴粉,通过优化工艺、简化过程,多途径缩减生产成本,最终实现了低成本条件下规模化生产超细钴粉的目的。以上所述是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也视为本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1