磁性复合颗粒及其制备方法和应用与流程
本发明属于纳米材料领域,涉及一种磁性复合颗粒及其制备方法和应用,具体涉及一种核壳卫星结构的磁性复合颗粒(AgMNP–AuNRs)及其制备方法和应用。
背景技术:
由于纳米级粗糙的金属表面对吸附在上面的探针分子具有巨大的拉曼增强效应,因此表面增强拉曼光谱(SERS)是一种非常重要的、可以应用到小分子痕量检测的振动光谱分析技术。基于它的高灵敏以及指纹识别特性,SERS已经在很多领域得到了应用。SERS能够得到应用,甚至未来可以在临床诊断领域发挥作用的关键在于高性能SERS增强基底的可控制备。
在尺度为1~5nm的纳间隙处会形成SERS“热点”,处于SERS“热点”区域的探针分子的拉曼信号会被强烈的放大。目前,虽然已经有许多带有“热点”结构的SERS增强基底被报道,但是大多数已经被报道的SERS“热点”结构是零维,一维或者二维材料,SERS“热点”的密度和数量都比较有限。
在过去的几年间,将磁性内核和贵金属纳米颗粒复合在一起形成核壳卫星结构纳米复合颗粒来构筑SERS基底的方法,引起广泛的关注和研究。研究人员发现,这类基于磁性内核和贵金属纳米颗粒的复合物,相比传统的胶体类SERS基底具有一些独特的优势。第一,它们非常适合检测溶解在水中或者是乙醇中的化学物质,因为它们的超顺磁性保证了它们可以在外加磁场的作用下,对目标化学物质做到很好的富集,而不用经过离心过程,可以避免离心损失,甚至是离心导致的沉降。第二,在外加磁场的作用下,这类磁性基底可以团聚在一起,就会形成颗粒间的“热点”,从而进一步的增强SERS信号。在过去的十几年间有许多研究人员研究了这类核壳卫星结构的磁性复合颗粒的构筑方法。比如,有的学者通过化学键(Au-S)直接连接Fe3O4和金纳米颗粒来构筑Fe3O4–AuNPs磁性复合物。有的学者通过静电方法,利用带正电的Fe3O4来吸附带负电的金纳米颗粒来直接构筑这类Fe3O4–AuNPs磁性复合物。有的学者通过静电方法,利用PEI修饰的Fe3O4(带正电)来吸附PVP修饰的金纳米棒(带负电)构筑Fe3O4–AuNRs磁性复合物。但是,这类核壳卫星结构SERS增强基底的特点就是,磁性内核都没有SERS增强能力,只是充当磁核,SERS增强由外围连接的贵金属纳米颗粒保证,SERS增强效果有限。
技术实现要素:
本发明要解决的技术问题是克服现有技术的不足,提供一种具有很好的磁响应能力和SERS性能的磁性复合颗粒,还提供一种制备时间短、工艺简单、通用性强、适用性广的磁性复合颗粒的制备方法,并相应提供上述磁性复合颗粒作为SERS增强基底的检测应用。
为解决上述技术问题,本发明采用以下技术方案:
一种磁性复合颗粒,所述磁性复合颗粒为核壳卫星结构,所述磁性复合颗粒包括银壳磁珠和金纳米棒,所述银壳磁珠为磁性复合颗粒的磁性内核,所述银壳磁珠表面通过聚(2-乙烯吡啶)与所述金纳米棒结合;所述银壳磁珠是由锰铁氧体和连续包被在锰铁氧体上的银壳组成。
上述的磁性复合颗粒中,优选的,所述磁性复合颗粒的磁饱和值为10emu/g~35emu/g;和/或,所述银壳磁珠的磁饱和值为40emu/g~50emu/g,所述锰铁氧体的粒径为100nm~300nm,所述银壳的厚度为15nm~30nm。
作为一个总的发明构思,本申请还提供一种磁性复合颗粒的制备方法,包括以下步骤:
(1)将银壳磁珠加入到聚(2-乙烯吡啶)溶液中,经超声处理后,在银壳磁珠表面形成聚(2-乙烯吡啶)多聚物层,用磁铁富集,得到聚(2-乙烯吡啶)修饰的银壳磁珠;
(2)制备金纳米棒溶液;
(3)将步骤(1)所得聚(2-乙烯吡啶)修饰的银壳磁珠与步骤(2)制备的金纳米棒溶液进行混合,保证金纳米棒过量,经超声反应后,通过磁富集,得到磁性复合颗粒。
上述的磁性复合颗粒的制备方法中,优选的,所述步骤(1)中,所述聚(2-乙烯吡啶)溶液中聚(2-乙烯吡啶)的浓度为2mg/mL~10mg/mL,所述聚(2-乙烯吡啶)溶液的溶剂为乙醇;和/或,所述银壳磁珠与所述聚(2-乙烯吡啶)溶液的比值为5mg~20mg∶2mL~6mL;和/或,所述超声处理的时间为30min~60min。
上述的磁性复合颗粒的制备方法中,优选的,所述步骤(1)中,所述银壳磁珠的制备方法包括以下步骤:
(1.1)将锰铁氧体加入到聚醚酰亚胺溶液中,超声,在锰铁氧体表面形成带正电的聚醚酰亚胺多聚物层,用磁铁富集,得到聚醚酰亚胺修饰的锰铁氧体;
(1.2)将聚醚酰亚胺修饰的锰铁氧体加入到带负电的金种子溶液中,超声,使所述聚醚酰亚胺修饰的锰铁氧体的表面吸附上带负电的金种子,然后用磁铁富集,得到中间产物;
(1.3)将步骤(1.2)所得的中间产物超声分散于聚乙烯吡咯烷酮溶液中,加入硝酸银溶液,超声混合,再加入甲醛溶液和氨水溶液进行超声反应,最后用磁铁富集,得到银壳磁珠。
上述的磁性复合颗粒的制备方法中,优选的,所述步骤(1.1)中,所述聚醚酰亚胺溶液中聚醚酰亚胺的浓度为2mg/mL~5mg/mL;所述锰铁氧体与所述聚醚酰亚胺溶液的比值为0.1g∶50mL;所述超声的时间为10min~30min;
和/或,所述步骤(1.2)中,所述金种子溶液主要由氯金酸溶液、柠檬酸钠溶液和硼氢化钠溶液加入水中搅拌制备得到;
和/或,所述步骤(1.3)中,所述聚乙烯吡咯烷酮溶液中聚乙烯吡咯烷酮的浓度为1.0mg/mL~1.5mg/mL;所述硝酸银溶液中硝酸银的浓度为2mg/mL~4mg/mL;所述甲醛溶液中甲醛的浓度为36wt%~38wt%;所述氨水溶液中氨水的浓度为25wt%~28wt%;所述中间产物、聚乙烯吡咯烷酮溶液、硝酸银溶液、甲醛溶液和氨水溶液的比值为10mg∶200mL∶1mL∶150μL∶100μL~500μL。
上述的磁性复合颗粒的制备方法中,优选的,所述步骤(2)中,所述金纳米棒溶液的制备方法包括以下步骤:
(2.1)将氯金酸溶液加入到十六烷基三甲基溴化铵溶液中,在恒温搅拌条件下,加入硼氢化钠溶液,经反应后,停止搅拌,继续恒温老化,得到金种子溶液;
(2.2)将氯金酸溶液加入到十六烷基三甲基溴化铵溶液中,然后加入硝酸银溶液和盐酸溶液,再加入抗坏血酸溶液,最后加入金种子溶液,在恒温水浴下进行生长,得到初步的金纳米棒溶液;
(2.3)将初步的金纳米棒溶液进行离心处理,然后用水重悬,得到金纳米棒溶液。
上述的磁性复合颗粒的制备方法中,优选的,所述步骤(2.1)中,所述氯金酸溶液的浓度为0.5mM,所述十六烷基三甲基溴化铵溶液的浓度为0.2M,所述硼氢化钠溶液的浓度为0.01M,所述氯金酸溶液、十六烷基三甲基溴化铵溶液、硼氢化钠溶液的体积比为2.5mL∶2.5mL∶0.3mL;所述恒温搅拌的温度为29℃,所述反应的时间为1min,所述恒温老化的温度为29℃,所述恒温老化的时间为1h~2h;
和/或,所述步骤(2.2)中,所述氯金酸溶液的浓度为1mM,所述十六烷基三甲基溴化铵溶液的浓度为0.2M,所述硝酸银溶液的浓度为0.01M,所述盐酸溶液的浓度为2M,所述抗坏血酸溶液的浓度为0.1M;所述氯金酸溶液、十六烷基三甲基溴化铵溶液、硝酸银溶液、盐酸溶液、抗坏血酸溶液、金种子溶液的体积比为20mL∶20mL∶0.4mL∶0.38mL∶0.32mL∶140μL;所述恒温水浴的温度为29℃,所述生长的时间为10h~16h;
和/或,所述步骤(2.3)中,所述初步的金纳米棒溶液的离心次数为3次~5次,离心转速为7500rpm~8500rpm,离心时间为6min~7min。
上述的磁性复合颗粒的制备方法中,优选的,所述步骤(3)中,所述超声反应的时间为30min~60min。
作为一个总的发明构思,本申请还提供一种上述的磁性复合颗粒或上述的制备方法制得的磁性复合颗粒在作为表面增强拉曼散射增强基底中的应用。
本发明的步骤(1.1)中,锰铁氧体的制备过程如下:
将含铁的前驱体和含锰的前驱体溶解于溶剂中,加入表面活性剂,然后油浴加热至表面活性剂溶解,停止加热,加入静电稳定剂并搅拌,再进行溶剂热合成反应,冷却后,用磁铁富集反应产物,得到锰铁氧体;
所述含铁的前驱体为FeCl3·6H2O,所述含锰的前驱体为MnCl2·4H2O,所述溶剂为乙二醇,所述表面活性剂为聚乙烯吡咯烷酮,所述静电稳定剂为醋酸钠;所述FeCl3·6H2O、MnCl2·4H2O、乙二醇、聚乙烯吡咯烷酮和醋酸钠的比值为360mg∶131.94mg∶20mL∶2g∶1.5g;
更优选的,所述油浴加热的温度为110℃~130℃,加热时间为10min~20min;所述溶剂热合成反应的温度为200℃~210℃,反应时间为10h~16h。
本发明的步骤(1.2)中,金种子溶液制备时,更优选的,所述氯金酸溶液中氯金酸浓度为1wt%;所述柠檬酸钠溶液中柠檬酸钠浓度为1wt%;所述硼氢化钠溶液中硼氢化钠的浓度为0.1M;所述氯金酸溶液、柠檬酸钠溶液、硼氢化钠溶液和水的体积比为3.4∶2.94∶12∶400。
本发明的磁性复合颗粒的应用中,更优选的,所述应用包括磁性复合颗粒在检测生物化学物质中的应用,所述生物化学物质能与金或银表面通过化学键结合,包括以下步骤:
将所述磁性复合颗粒加入到含生物化学物质的甲醇溶液中超声反应,用磁铁富集,得到反应后的磁性复合颗粒;将反应后的磁性复合颗粒重悬于乙醇中,点至硅片上进行光谱测试,得到拉曼测试信号,利用所述拉曼测试信号对所述生物化学物质的浓度进行定性或定量检测。
本发明中,磁性复合颗粒可写作AgMNP-AuNRs,银壳磁珠为AgMNPs,金纳米棒为AuNRs。
本发明中,浓度M即为mol/L,mM即为mmol/L。
本发明为了进一步提高SERS增强效果,提出了一种新型的三维(3D)核壳卫星结构的磁性复合颗粒,以银壳磁珠为磁性内核,以金纳米棒为卫星纳米颗粒构筑了三维核壳卫星结构磁性复合颗粒。这种结构的内核和卫星之间形成纳间隙,构成高密度的SERS“热点”,与磁引诱导致的团聚形成的颗粒间的“热点”一起进一步增强目标分子的拉曼信号,显著增强该区域目标分子的拉曼信号。本发明磁性内核和卫星颗粒都有SERS增强能力,在银壳磁珠和卫星金纳米棒之间形成了高密度的“热点”结构。本发明制备的磁性复合颗粒非常适用于生化小分子的高灵敏SERS检测。
与现有技术相比,本发明的优点在于:
(1)本发明的磁性复合颗粒具有核壳卫星结构,磁性内核为银壳磁珠(AgMNPs),卫星纳米颗粒为金纳米棒(AuNRs),具有很好的磁响应能力以及SERS性能。磁响应能力由超顺磁性的内核保证,SERS性能由银壳磁珠和金纳米棒共同保证。
(2)本发明的磁性复合颗粒通过共价键法构筑,利用P2VP层连接银壳磁珠和金纳米棒,这种结构的内核和卫星之间形成纳间隙,纳间隙尺寸在1nm以下,构成高密度的SERS“热点”,可以显著增强该区域目标分子的拉曼信号。
(3)本发明的磁性复合颗粒的制备方法的关键在于P2VP分子上有许多吡啶基团,这些吡啶基团可以通过氮原子提供孤对电子与金属表面形成共价键。因此,只需要P2VP这一种功能分子就可以实现核壳卫星结构的AgMNP-AuNRs的构筑。当P2VP修饰到银壳磁珠表面之后,还有许多空余的吡啶基团可以继续连接金纳米棒。
(4)本发明的磁性复合颗粒的制备方法,磁性内核是具有超顺磁性、具有良好SERS性能的银壳磁珠,并且银壳磁珠的制备是以锰铁氧体为内核通过超声辅助种子生长法实现的。且可以根据需要,通过控制硝酸银溶液的加入量,在不同粒径的锰铁氧体上包被任意厚度的银壳。因而本发明的制备方法通用性强、适应性广。
(5)本发明的磁性复合颗粒的应用,该磁性复合颗粒用作生化传感器对生物化学物质进行检测时,尤其是对含有巯基或者二硫键这类可以与金或银表面形成化学键的物质时,可以通过磁富集对样品进行浓缩,检测灵敏度高,操作方便,而且有望为人体激素、病原体、残留化学毒素等生物化学物质的检测提供便利,尤其是在低浓度生物化学物质SERS检测上,提供快速、实用、高灵敏的SERS增强基底。
综上,本申请通过共价键连接(P2VP)的方法,实现了核壳卫星结构的磁性复合颗粒的可控制备,反应简单,操作方便,制备的磁性复合颗粒,具有很好的SERS活性以及磁响应能力。在生物样品分离以及生物医学检测应用等领域都有很好的应用前景。
附图说明
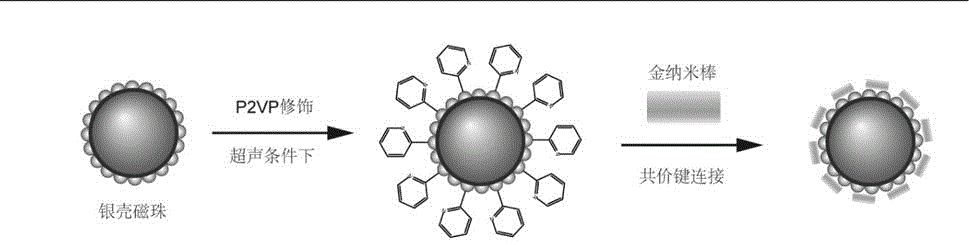
图1为本发明的磁性复合颗粒(AgMNP-AuNRs)的制备方法示意图。
图2为本发明实施例1制得的MnFe2O4、MnFe2O4@PEI-auseeds和银壳磁珠的透射电子图,其中,(a)图为MnFe2O4;(b)图为MnFe2O4@PEI-auseeds;(c)图为银壳磁珠。
图3为本发明实施例1制得的MnFe2O4、MnFe2O4@PEI-auseeds和银壳磁珠的XRD图谱。
图4为本发明实施例1制得的MnFe2O4、MnFe2O4@PEI-auseeds和银壳磁珠的磁响应曲线。
图5为本发明实施例1制得的银壳磁珠的扫描电镜图。
图6为本发明实施例1制得的金纳米棒的透射电子显微镜照片。
图7为本发明实施例1制得的金纳米棒的紫外-可见吸收光谱图。
图8为本发明实施例1制得的磁性复合颗粒(AgMNP-AuNRs)的扫描电子显微镜照片(a和b),透射电子显微镜照片(c)以及高分辨率透射电子显微镜照片(d)。
图9为本发明实施例1制得的磁性复合颗粒(AgMNP-AuNRs)的磁滞曲线图。
图10为本发明实施例1制得的磁性复合颗粒(AgMNP-AuNRs)用于检测Thiram的检测流程图。
图11为本发明实施例1制得的磁性复合颗粒(AgMNP-AuNRs)检测Thiram的光谱图。
图12为本发明实施例1制得的磁性复合颗粒(AgMNP-AuNRs)检测Thiram的强度-浓度校正曲线图。
具体实施方式
以下结合说明书附图和具体优选的实施例对本发明作进一步描述,但并不因此而限制本发明的保护范围。
以下实施例中所采用的材料和仪器均为市售。
实施例1:
一种本发明的磁性复合颗粒(具体为AgMNP-AuNRs),该磁性复合颗粒为核壳卫星结构,包括银壳磁珠和金纳米棒,银壳磁珠为磁性复合颗粒的磁性内核,银壳磁珠表面通过聚(2-乙烯吡啶)与金纳米棒结合,银壳磁珠是由锰铁氧体和连续包被在锰铁氧体上的银壳组成。
本实施例中,所述磁性复合颗粒的磁饱和值为31.1emu/g,磁性内核为超顺磁性的银壳磁珠,粒径为360nm,其中锰铁氧体的粒径为300nm,银壳的厚度为30nm,银壳磁珠的磁饱和值为46.4emu/g。银壳磁珠外围的金纳米棒尺寸为72×18nm,LSPR共振峰为840nm。
一种上述本实施例的磁性复合颗粒(AgMNP-AuNRs)的制备方法,如图1所示,包括以下步骤:
(1)制备银壳磁珠
(1.1)制备单分散超顺磁性的锰铁氧体(MnFe2O4)
将(4/3mmol, 360 mg)FeCl3·6H2O和(2/3mmol, 131.94 mg)MnCl2·4H2O通过磁力搅拌溶解到20mL乙二醇(EG)中,加入2g聚乙烯吡咯烷酮(PVP),转移到120℃的油浴中,加热10min,直到溶液呈透明状,停止加热;再加入1.5g醋酸钠,转移到磁力搅拌器上,搅拌30min,得到比较均匀的混合溶液后转移到50mL的聚四氟乙烯反应釜中。置入提前加热到200℃的烘箱中,反应10h后取出,冷却至室温,用磁铁富集反应产物,将反应产物用乙醇清洗3~5次,再用水清洗3~5次。在60℃的真空烘箱中烘干,得到粒径为300nm的锰铁氧体(MnFe2O4),备用。
本步骤中,利用乙二醇作为溶剂的溶剂热法制备超顺磁性的锰铁氧体,作为银壳磁珠的磁性内核。制备过程中,三氯化铁和氯化锰被用作前驱体,而EG既充当溶剂又充当还原剂。聚乙烯吡咯烷酮作为表面活性剂,醋酸钠被用作静电稳定剂,防止颗粒团聚。
(1.2)制备PEI修饰的锰铁氧体(MnFe2O4@PEI)
称取0.1g锰铁氧体加入到50mL聚醚酰亚胺(PEI)水溶液中,其中,聚醚酰亚胺的浓度为5mg/mL,超声20min,利用静电结合作用,在MnFe2O4表面形成一层厚度为2nm的带正电的PEI多聚物层,后用磁铁富集清洗,得到聚醚酰亚胺修饰的锰铁氧体(MnFe2O4@PEI)。
通过超声时间可以控制PEI多聚物层的厚度,PEI多聚物层的厚度优选为1nm~3nm,不宜过厚,否则会影响产物的磁响应特性,也会影响产物的表面形貌。
(1.3)制备金种子粒径为4nm的带负电的金种子溶液
在500mL蓝盖瓶中加入400mL水,并开始以950rpm的转速进行磁力搅拌;先后加入3.4mL浓度为1wt%的HAuCl4溶液和2.94mL浓度为1wt%的柠檬酸钠溶液;再加入新制的12mL浓度为0.1M的硼氢化钠溶液,室温搅拌4h后,得到金种子粒径为4nm的带负电的金种子溶液,备用。
(1.4)制备吸附金种子的聚醚酰亚胺修饰的锰铁氧体(MnFe2O4@PEI-auseeds)
将0.1g MnFe2O4@PEI 加入到200mL步骤(1.3)所得的带负电的金种子溶液中超声1h,用磁铁富集清洗后,得到中间产物MnFe2O4@PEI-auseeds。该MnFe2O4@PEI-auseeds可以长期存放(1~3个月)。
4 nm金种子的量优选为200mL~300mL,只要保证金种子过量即可。
(1.5)制备银壳磁珠
将步骤(1.4)所得的中间产物超声分散于聚乙烯吡咯烷酮溶液中,加入硝酸银溶液,超声混合,再加入甲醛溶液和氨水溶液进行超声反应,最后用磁铁富集,得到银壳磁珠。具体过程如下:
利用超声辅助种子生长法在MnFe2O4@PEI-auseeds上包被银壳,将10mg中间产物MnFe2O4@PEI-auseeds加入到200mL浓度为1.5mg/mL的聚乙烯吡咯烷酮水溶液中,超声2min;加入1mL硝酸银溶液(2mg/mL),超声2min;加入150μL浓度为36-38wt%的甲醛溶液,再加入300μL浓度为25-28wt%的氨水溶液超声1min后,用磁铁富集得到银壳磁珠。
(2)银壳磁珠P2VP修饰
在超声条件下,将20mg聚(2-乙烯吡啶)(P2VP)溶解到4mL乙醇中,形成5mg/mL的P2VP溶液,然后加入10mg银壳磁珠,超声处理1h,再利用磁富集的手段,用乙醇清洗5次,去除过量的P2VP,即得到P2VP修饰的银壳磁珠。
(3)金纳米棒的制备
(3.1)将2.5mL、浓度为0.5mM的氯金酸溶液加入到2.5mL、0.2M的十六烷基三甲基溴化铵(CTAB)溶液中,在500rpm、29℃的恒温磁力搅拌条件下,向上述溶液中加入300μL、0.01M新配置的NaBH4溶液,1min后,溶液呈棕黄色,关闭磁力搅拌,将得到的种子液在29℃下继续老化1h,得到金种子溶液,备用;
(3.2)将20mL浓度为1mM的氯金酸溶液加入到20mL、0.2M十六烷基三甲基溴化铵溶液中,轻微摇匀后,向溶液中加入0.4mL、0.01M的硝酸银溶液,轻微摇匀后,加入0.38mL、2M盐酸溶液,再加入0.32mL、0.1M抗坏血酸(AA)溶液后,轻微摇匀,溶液呈无色,最后加入140μL上述步骤(3.1)得到的金种子溶液后轻微摇匀,置于预先设置到29℃的恒温水浴中,生长12h,得到初步的金纳米棒溶液;
(3.3)将初步的金纳米棒溶液离心3次,离心速度为7500rpm,每次的离心时间为6min,尽可能的去除金纳米棒表面的十六烷基三甲基溴化铵,然后用水重悬(可以重悬到10 mL去离子水中,得到4倍浓缩的金纳米棒溶液,即4×AuNRs),备用。
(4)核壳卫星结构的磁性复合颗粒(AgMNP-AuNRs)的制备
将步骤(2)所得P2VP修饰过的银壳磁珠与步骤(3)所得的经过三次离心处理的金纳米棒进行混合,保证金纳米棒过量,超声反应30min,两者通过共价键结合,形成核壳卫星结构的AgMNP-AuNRs磁性复合颗粒。然后,通过磁富集的手段,用去离子水清洗三次,去除过量的金纳米棒,得到核壳卫星结构的AgMNP-AuNRs磁性复合颗粒。
图2为本实施例步骤(1.1)制得的MnFe2O4、步骤(1.4)制得的MnFe2O4@PEI-auseeds和步骤(1.5)制得的银壳磁珠的透射电子图。其中,(a)图为MnFe2O4的透射电子图,由图可见,所得的锰铁氧体粒径均一,单分散性好;(b)图为MnFe2O4@PEI-auseeds的透射电子图,(c)图为银壳磁珠的透射电子图,由图可见,纳米级粗糙的银壳连续地包被在磁性内核上,此结构能赋予银壳磁珠优异的SERS性能。图3为本实施例步骤(1.1)制得的MnFe2O4、步骤(1.4)制得的MnFe2O4@PEI-auseeds和步骤(1.5)制得的银壳磁珠的XRD图谱,其中,(a)为MnFe2O4;(b)为MnFe2O4@PEI-auseeds;(c)为银壳磁珠。从图3中的曲线a可以清楚地观察到合成的MnFe2O4的衍射峰(图中的实心方框)在29.6,35.1,42.5,52.6,56.1和61.6度,分别对应于MnFe2O4的(220),(311),(400),(422),(511)和(440)晶面。金种子被吸附到MnFe2O4上之后,在38.2度处(图中的实心圆形)出现了一个新的对应于金的(111)晶面的X射线衍射峰(图3的曲线b)。当连续的银壳形成之后,我们在2θ值38.1,44.3,64.4,77.5,和81.5处又观察到了五个新的衍射峰(图3的曲线c),分别对应于银的(111),(200),(220),(311)和(222)晶面(实心倒三角)。
图4为本实施例步骤(1.1)制得的MnFe2O4、步骤(1.4)制得的MnFe2O4@PEI-auseeds和步骤(1.5)制得的银壳磁珠的磁响应曲线,其中,(a)为MnFe2O4;(b)为MnFe2O4@PEI-auseeds;(c)为银壳磁珠。首先,图4中没有出现磁滞回线,也就说明了制备得到的所有磁性纳米颗粒都是超顺磁性的。而这种超顺磁性的特性可以防止纳米颗粒的团聚,也能够使得这些磁性纳米颗粒在没有外加磁场作用时,很好的分散以及重悬到溶液中。这种特性也方便了它们在生物样品分离以及检测方面的应用。如图4所示,制备得到的银壳磁珠的磁饱和值大约是46.4emu/g(曲线c)。
本实施例所制得的银壳磁珠的扫描电镜照片如图5所示,由图可见,所制备的银壳磁珠外壳比较粗糙并且连续,是由彼此相邻的较大的银颗粒构成的。
图6和图7分别为本实施例步骤(3)制得的金纳米棒的透射电子显微镜照片和紫外-可见吸收光谱。从图6可以看出,制备得到的金纳米棒尺寸比较均一,大颗粒较少,长径比大约为4∶1。从图7可以看出,制备得到的金纳米棒的SPR共振峰大约为840nm。
图8为本实施例步骤(4)制得的核壳卫星结构的磁性复合颗粒(AgMNP-AuNRs)的扫描电子显微镜照片(a和b),透射电子显微镜照片(c)以及高分辨率透射电子显微镜照片(d)。从图中可以看出金纳米棒是被成功连接在银壳磁珠表面,在两者连接处几乎观察不到CTAB和P2VP分子层,说明两者之间可以形成间隙为1nm左右的纳间隙。
图9为本实施例步骤(4)制得的磁性复合颗粒(AgMNP-AuNRs)的磁滞曲线。从图中可以看出,在连接金纳米棒之后,磁饱和值由46.4emu/g降到了31.1emu/g。磁饱和值的逐步降低也说明了非磁性材料的成功包覆,这也是和电镜表征的结果是相互印证的。
一种上述本实施例的磁性复合颗粒(AgMNP-AuNRs)作为SERS增强基底在对农残福美双(Thiram)检测中的应用,如图10所示,包括以下步骤:
(1)将本实施例制得的AgMNP-AuNRs分散在乙醇中,得到浓度为10mg/mL的AgMNP-AuNRs分散液;将6个不同浓度(10-6 M、10-7 M、10-8 M、10-9 M、10-10 M和10-11 M)的1mL福美双的甲醇溶液分别加入到6个1.5mL的离心管中,然后每个离心管中加入5μL上述的磁性复合颗粒分散液,置入超声中反应1h。
(2)用磁铁富集反应后的AgMNP-AuNRs,并用乙醇清洗3~5次后,重悬到5μL乙醇中,用移液枪将重悬的反应后的AgMNP-AuNRs点到硅片上,晾干后进行光谱测试,利用得到的拉曼测试信号即可对待测样品中的福美双浓度进行定性或定量检测。
所得的测试结果如图11和图12所示,图11为拉曼光谱图,图12为强度-浓度校正曲线图。由图11可见,福美双的主峰(1383 cm-1)在10-10M浓度以上都清晰可见。由此可见,制备得到的磁性复合颗粒对福美双的检测限是10-10M。图12说明福美双的浓度和SERS强度之间存在比较好的校正曲线关系,也就是说该磁性复合颗粒检测福美双的动态范围为10-6M~10-10M。
以上所述仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例。凡属于本发明思路下的技术方案均属于本发明的保护范围。应该指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下的改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!