钛酸盐化合物、碱金属钛酸盐化合物和制备其的方法、以及使用钛酸盐化合物和碱金属钛酸盐化合物作为活性材料的蓄电装置与流程
本发明涉及钛酸化合物、碱金属钛酸盐化合物、以及用于制备这些的方法。本发明还涉及含有钛酸化合物和/或碱金属钛酸盐化合物的电极和蓄电装置。
背景技术:
以便携式装置作为主要应用的锂离子蓄电池的用途持续扩大,并且最近也已经对于大尺寸蓄电系统、移动体等新领域研究了锂离子蓄电池的应用。对于这些应用来说,使用具有高Li吸留(occlusion)和释放电势的负极活性材料的锂离子蓄电池吸引了注意,因为它们具有可归因于它们的高电势的高安全性。另一方面,锂离子蓄电池具有能量密度变低的问题。因此,进行了对于保持高电势且同时具有大容量的钛酸化合物的开发(例如,非专利文献1,和专利文献1和2)。
非专利文献1公开了多个类型的钛酸化合物;并且应理解,在这些中,H2Ti12O25具有低的第一-循环充电和放电效率以及低的在充电和放电循环进程中涉及的容量减少,并且有前途作为电极活性材料。然而,锂脱嵌容量(deintercalation capacity)为约200mAh/g,并且要求进一步的容量增加。
本发明的发明人在专利文献1和2中公开了作为电极活性材料的钛酸化合物及其制备方法。然而,锂脱嵌容量在第二循环中最高为约210mAh/g,并且要求进一步的容量增强。
引用清单
非专利文献
非专利文献1:Akimoto等,Journal of The Electrochemical Society,158(5),A546-A549(2011)
专利文献
专利文献1:JP 2008-255000 A
专利文献2:JP 2010-254482 A
发明概述
技术问题
本发明的一个目的是提供一种钛酸化合物,其可以解决如上所述的当前问题,并且当用其作为蓄电装置的电极活性材料时,其可以提供容量能够得到更大增强并且充电和放电循环性质、倍率性质(rate property)等出色的蓄电装置。
解决问题的方案
本发明的发明人在研究改进含有钛酸化合物的电极活性材料的放电容量(Li脱嵌容量)的过程中,认为使得活性材料的粒径小是有效的。然而,已经搞清的是,当使得活性材料的平均粒径小时,尽管初始Li嵌入容量变高,但Li脱嵌容量的改善比那小;也就是说,充电和放电效率降低,并且在充电和放电循环中涉及的Li脱嵌容量明显降低,表明该活性材料作为电极活性材料是不足的。随后作为对解决方法的彻底研究的结果,发现了,通过使得钛酸化合物具有在特定范围内的比表面积(SSA)(不是通过仅仅使得平均粒径小,而是通过减少超细粒子的数量且同时使得平均粒径小),并且通过使得钛酸化合物含有等于或大于特定量的量的具有在特定范围内的长轴直径的粒子,可以获得钛酸化合物,当该钛酸化合物用作电极活性材料时,其能够使得Li脱嵌容量高且充电和放电效率高,并且能够减小在充电和放电循环中涉及的Li脱嵌容量的降低速率;并且这样的钛酸化合物的倍率性质也是出色的。此发现导致完成了本发明。
也就是说,本发明是:
(1)一种钛酸化合物,所述钛酸化合物具有通过使用氮吸附的BET单点法测得的为10至30m2/g的比表面积,并且具有各向异性的形状,
其中所述钛酸化合物中,基于粒子的数量的60%以上的的粒子具有在0.1<L≤0.9μm范围内的通过电子显微术测得的长轴直径L。
(2)根据(1)所述的钛酸化合物,其中所述钛酸化合物中,基于粒子的数量的60%以上的粒子具有在1.0<L/S≤4.5范围内的长径比L/S,所述长径比L/S由通过电子显微术得到的各个粒子的长轴直径L和短轴直径S算得。
(3)根据(1)或(2)所述的钛酸化合物,所述钛酸化合物在使用CuKα辐射作为辐射源的粉末X射线衍射图案中具有至少在2θ=14.0°、24.8°、28.7°、43.5°、44.5°和48.6°的位置(对每种情况有±0.5°的误差)的峰,其中在取在2θ=14.0°(有±0.5°的误差)处的峰的强度为100的情况下,除了在2θ=14.0°处的峰之外,在10.0°≤2θ≤20.0°之间没有观察到具有20以上的强度的峰。
(4)根据(1)至(3)中任一项所述的钛酸化合物,所述钛酸化合物具有0.05以下的比率h2/h1,
其中
在电压V-dQ/dV曲线中,h1是在介于1.5V至1.7V之间的电压V下的dQ/dV的最大值,且
h2是在介于1.8V至2.0V之间的电压V下的dQ/dV的最大值,
其中所述电压V-dQ/dV曲线是通过将在硬币型蓄电池的Li脱嵌侧获得的电压V-容量Q曲线的容量Q相对于V求微分而确定的,
其中所述蓄电池使用所述钛酸化合物作为工作电极以及使用金属Li作为对电极。
(5)根据(1)至(4)中任一项所述的钛酸化合物,其中所述钛酸化合物的硫元素含量按SO3计为0.1至0.5质量%。
(6)根据(1)至(5)中任一项所述的钛酸化合物,其中所述粒子包含由通式H2Ti12O25表示的化合物作为主组分。
(7)一种碱金属钛酸盐化合物,所述碱金属钛酸盐化合物具有通过使用氮吸附的BET单点法测得的为5至15m2/g的比表面积,并且具有各向异性的形状,
其中所述碱金属钛酸盐化合物中,基于粒子的数量的60%以上的粒子具有在0.1<L≤0.9μm范围内的通过电子显微术测得的长轴直径L。
(8)根据(7)所述的碱金属钛酸盐化合物,其中所述碱金属钛酸盐化合 物中,基于粒子的数量的60%以上的粒子具有在1.0<L/S≤4.5范围内的长径比L/S,所述长径比L/S由通过电子显微术得到的各个粒子的长轴直径L和短轴直径S算得。
(9)根据(7)或(8)所述的碱金属钛酸盐化合物,其中所述粒子包含由通式Na2Ti3O7表示的化合物作为主组分。
(10)一种用于制备根据以上(7)至(9)中任一项的碱金属钛酸盐化合物的方法,所述方法包括研磨碱金属钛酸盐化合物直至其比表面积变成10m2/g以上的步骤,以及将所得的研磨的材料退火的步骤。
(11)根据(10)所述的用于制备碱金属钛酸盐化合物的方法,其中所述研磨以湿法研磨进行。
(12)根据(11)所述的用于制备碱金属钛酸盐化合物的方法,所述方法还包括,在所述湿法研磨步骤后,在没有过滤分离的情况下将所述碱金属钛酸盐化合物和分散介质干燥的步骤。
(13)根据(12)所述的用于制备碱金属钛酸盐化合物的方法,其中所述干燥通过喷雾干燥器进行。
(14)根据(10)至(13)中任一项所述的用于制备碱金属钛酸盐化合物的方法,其中进行所述退火,直至将所述碱金属钛酸盐化合物的比表面积在所述退火后降低至在所述退火前其比表面积的20至80%。
(15)根据权利要求(10)至(14)中任一项所述的用于制备碱金属钛酸盐化合物的方法,所述方法包括烧制混合物从而制备出具有10m2/g以下的比表面积的碱金属钛酸盐化合物的步骤,所述混合物至少含有具有按SO3计的硫元素含量为0.1至1.0质量%的氧化钛、和碱金属化合物。
(16)根据(15)所述的用于制备碱金属钛酸盐化合物的方法,其中所述氧化钛具有通过使用氮吸附的BET单点法测得的为80至350m2/g的比表面积。
(17)一种用于制备钛酸化合物的方法,所述方法包括:将通过根据(10)至(16)中任一项的制备方法获得的碱金属钛酸盐化合物与酸性水溶液接触从而用质子取代所述碱金属钛酸盐化合物中的碱金属阳离子的至少一部分的步骤。
(18)一种用于制备钛酸化合物的方法,所述方法还包括:加热通过 根据(17)的制备方法获得的质子取代的钛酸化合物的步骤。
(19)根据(18)所述的用于制备钛酸化合物的方法,其中在所述加热步骤中的加热温度为150至350℃。
(20)根据(10)至(19)中任一项所述的用于制备钛酸化合物的方法,其中所述碱金属是钠。
(21)一种电极活性材料,所述电极活性材料包含根据(1)至(9)中任一项所述的钛酸化合物和/或碱金属钛酸盐化合物。
(22)一种蓄电装置,所述蓄电装置包含根据(21)所述的电极活性材料。
本发明的有益效果
当使用根据本发明的钛酸化合物作为蓄电装置的电极活性材料时,可以获得与常规蓄电装置相比具有较高容量、较高充电和放电效率、减小的在充电和放电循环中涉及的Li脱嵌容量的降低速率、和较好倍率性质的蓄电装置。
附图简述
[图1]图1是图示根据本发明的蓄电装置的一个实例的示意图。
[图2]图2是实施例1的扫描电子显微镜照片。
[图3]图3是实施例1的粉末X射线衍射图案。
[图4]图4是比较例1的扫描电子显微镜照片。
[图5]图5是比较例2的扫描电子显微镜照片。
[图6]图6是比较例2的粉末X射线衍射图案。
[图7]图7是比较例4的扫描电子显微镜照片。
[图8]图8是实施例1至3和比较例1和4的长轴直径的累积相对频率分布。
[图9]图9是实施例1至3和比较例1和4的长径比的累积相对频率分布。
[图10]图10是实施例1和比较例2的充电和放电曲线。
[图11]图11是实施例1和比较例2的dQ/dV对V的曲线。
实施方案描述
本发明的技术构成以及它的作用和效果如下。
本发明是一种钛酸化合物,其中,其粒子具有通过使用氮吸附的BET单点法测得的为10至30m2/g的比表面积,并且具有各向异性的形状,其中所述钛酸化合物基于粒子的数量包含60%以上的具有在0.1<L≤0.9μm范围内的通过电子显微术测得的长轴直径L的粒子。
根据本发明的钛酸化合物是其晶格由Ti、H和O构成并且与具有结晶水和吸附水的二氧化钛明显不同的化合物。
根据本发明的钛酸化合物优选具有以下组成式。
HxTiyOz (1)
其中x/y为0.06至4.05;且z/y为1.95至4.05。
满足式(1)的化合物具体包括由通式HTiO2、HTi2O4、H2TiO3、H2Ti3O7、H2Ti4O9、H2Ti5O11、H2Ti6O13、H2Ti8O17、H2Ti12O25、H2Ti18O37、H4TiO4和H4Ti5O12表示的钛酸化合物。通过粉末X射线衍射测量的峰位置确认这些化合物的存在。
在这些中,优选的是在粉末X射线衍射测量(使用CuKα辐射)中至少在14.0°、24.8°、28.7°、43.5°、44.5°和48.6°的位置(对每种情况有±0.5°的误差)显示明显有区别的峰的钛酸化合物;并且更优选的是,在取在2θ=14.0°(有±0.5°的误差)处的峰的强度为100的情况下,除了在2θ=14.0°处的峰之外,钛酸化合物在10.0°≤2θ≤20.0°之间没有被观察到具有20以上的强度的峰。显示这样的X射线衍射图案的钛酸化合物包括由通式H2Ti12O25表示的钛酸化合物。
在本发明中,钛酸化合物可以不仅是具有化学计量的组成的钛酸化合物,也甚至可以是其中一些元素缺欠或过量的非化学计量的组成的钛酸化合物,只要是由上述通式表示的即可。而且,其他元素可以被取代成氢、钛和氧的一部分,并且可以存在于空隙位置中。这样的元素的实例包括碱金属元素,如锂、钠、钾和铯;并且其含量优选为按钛酸化合物中的碱金属氧化物计为0.4质量%以下。可以例如通过X射线荧光分析计算该含量。
而且,在本发明的范围内,包括具有源自其他晶体结构的粉末X射线衍射峰的钛酸化合物,也就是说,具有除了作为主相的上述钛酸化合物之 外的副相的钛酸化合物。在具有副相的情况下,在取主相的主峰的强度为100的情况下,归属于副相的主峰的强度优选30以下,且更优选10以下,且再更优选观察不到副相的主峰,即,钛酸化合物是单相。副相的实例包括锐钛矿型、金红石型和青铜型氧化钛。还可以存在多个类型的钛酸化合物相。
根据本发明的钛酸化合物具有通过使用氮吸附的BET单点法测得的为10至30m2/g的比表面积。测量可以通过一般的使用氮吸附的BET单点法进行,其中在用液氮冷却样品管的同时使得氮气被吸附在样品上。
而且,根据本发明的钛酸化合物具有各向异性的形状。各向异性的形状指的是如板状、针状、棒状、柱状、纺锤状或纤维状的形状。在其中通过多个一次粒子的聚集而形成二次粒子的情况下,所述形状指的是一次粒子的形状。可以通过电子显微镜照片检查一次粒子的形状。但是,钛酸化合物的粒子不是必需全部都具有各向异性的形状,而是可以一部分粒子含有具有各向同性的形状或不确定的形状。
而且,根据本发明的钛酸化合物基于粒子的数量包含60%以上的具有在0.1<L≤0.9μm范围内的通过电子显微术测得的长轴直径L的粒子。
通过电子显微术确定长轴直径L的分布按如下进行。首先,通过扫描电子显微镜取得10,000X照片,并且放大该照片,使得在放大比例尺上1cm对应于0.5μm。将在照片上的粒子的形状(即,投影图像)近似为粒子内切于其中的矩形或正方形;随机选择至少100个其短边为1mm以上的一次粒子,并且测量所选的粒子的长边和短边。随后,通过对所得的长边和短边的测量值除以放大倍数,确定各粒子的长轴直径L和短轴直径S。将这样确定的长轴直径L落在0.1μm的各级间隔中(每级的上限值被包含在该级内)的粒子的数量计数,并且除以粒子的总数,从而确定按相对于L的粒子数量计的累积相对频率分布。基于这样获得的按相对于L的粒子数量计的累积相对频率分布,通过从L=0.9μm处的累积率(%)减去L=0.1μm处的累积率(%),可以算得0.1<L≤0.9μm的粒子按粒子计的占有率(%)。
为了达到本发明的益处,必须的是,使得钛酸化合物具有在特定范围内的比表面积,并且使得钛酸化合物含有等于或大于特定量的量的具有在 特定范围内的长轴直径的粒子。当比表面积超过30m2/g时,大概因为具有0.1μm以下粒径的超细粒子变得过多,所以尽管第一-循环Li嵌入容量变得明显地高,但是Li脱嵌容量的改进小于它(充电和放电效率降低),并且进一步地,在充电和放电循环的进程中涉及的Li脱嵌容量明显降低。另一方面,当比表面积小于10m2/g时,实现不了Li脱嵌容量和倍率性质的改进。而且,即使比表面积在10至30m2/g的范围内,当基于粒子的数量具有0.1<L≤0.9μm的长轴直径L的粒子占少于60%的粒子时,因为这样的状态使得超细粒子和粗粒子各以大的量含有,所以充电和放电效率降低并且实现不了倍率性质的改进,并且在充电和放电循环中的Li脱嵌容量保持低下。基于粒子的数量包含60%以上的具有在0.1<L≤0.9μm范围内的长轴直径的粒子表示长轴直径比较均等、并且可以高度平衡Li脱嵌容量、循环性质和倍率性质的这样一种状态。
优选使得比表面积在10至25m2/g的范围内,且更优选使得其在12至25m2/g的范围内。具有在0.1<L≤0.9μm范围内的长轴直径L的粒子按粒子的数量计优选占65%以上,更优选70%以上。具有0.1<L≤0.6μm的长轴直径L的粒子按粒子的数量计优选占35%以上,且更优选50%以上。
根据本发明的钛酸化合物优选基于粒子的数量包含60%以上的具有在1.0<L/S≤4.5范围内的长径比L/S的粒子,所述长径比L/S由通过电子显微术得到的各个粒子的长轴直径L和短轴直径S算得。当钛酸化合物粒子具有各向异性性时,观察到(尽管不清楚原因)Li脱嵌容量很可能变高。另一方面,当长径比变得过高时,观察到倍率性质的降低,并且当制造电极时,难以使充填密度变高。通过使得基于粒子的数量的60%以上的粒子中含有具有在1.0<L/S≤4.5的范围内的长径比L/S的粒子,可以同时满足高Li脱嵌容量和高电极充填密度,并且变得容易达到最佳的比表面积和长轴直径。
通过电子显微术确定长径比L/S的分布按如下进行。由通过上述方法测定的各粒子的长轴直径L和短轴直径S计算各粒子的L/S。将这样确定的L/S落在0.5的各级间隔中(每级的上限值被包含在该级内)的粒子的数量计数,并且除以粒子的总数,从而确定按相对于L/S的粒子数量计的累积相对频率分布。基于这样获得的按相对于L/S的粒子数量计的累积相对 频率分布,通过从L/S=4.5处的累积率(%)减去L/S=1.0处的累积率(%),可以算得1.0<L/S≤4.5的粒子按粒子计的占有率(%)。
钛酸化合物基于粒子的数量包含优选65%以上的具有在1.0<L/S≤4.5范围内的长径比L/S的粒子,且更优选其70%以上。而且,钛酸化合物包含优选55%以上的具有在1.5<L/S≤4.0范围内的长径比L/S的粒子,且更优选其60%以上。
根据本发明的钛酸化合物可以含有硫元素,且可以使得它的量按后述的换算计为0.1至0.5质量%。当使得钛酸化合物含有硫元素时,因为钛酸化合物的一次粒子容易地呈现各向异性的形状(板状、棒状、方柱状、针状),所以可以增强Li脱嵌容量。当硫的量小于0.1质量%,对于一次粒子来说变得难以呈现各向异性的形状;并且当超过0.5质量%时,Li脱嵌容量变得易于逆向减少。
作为通过X射线荧光分析测得的钛酸化合物中硫的质量%的按SO3计的值,确定硫元素的含量。
进一步优选的是,根据本发明的钛酸化合物是具有0.05以下的h2/h1比率的钛酸化合物,其中在电压V和dQ/dV曲线中,h1是在介于1.5V至1.7V之间的电压V下的dQ/dV的最大值,且h2是在介于1.8V至2.0V之间的电压V下的dQ/dV的最大值,所述电压V和dQ/dV曲线是将在使用钛酸化合物作为工作电极的活性材料且使用金属Li作为对电极的硬币型蓄电池的Li脱嵌侧上的电压V-容量Q曲线相对于V求微分而确定的。
电压V和dQ/dV的曲线按如下确定。首先,如在后述的实施例1中所述,使用钛酸化合物作为工作电极并且使用金属锂作为对电极来制造硬币型蓄电池。将硬币蓄电池充电(Li嵌入)至1V,并且随后以0.1C放电(Li脱嵌)至3V。此时,以5mV的电压变化量的间隔和/或以120秒的间隔获得Li脱嵌侧上的电压V-容量Q的数据。基于这样获得的数据,绘制V-Q曲线。
随后,通过单纯移动平均法,分别将所得的电压V和容量Q的数据平滑化。具体地,在以时间序列排列的(2n+1)个数据(n是任选的,但是可以为2)中,用所述(2n+1)个数据的平均值替换中间的一个第(n+1)个数据。
随后,按如下确定通过将在这些平滑化的数据中第i个点上的Qi相对 于V求微分获得的值。也就是说,确定相对于V的通过该点和该点的前后两点共三点(Vi-1,Qi-1)、(Vi,Qi)和(Vi+1,Qi+1)的二次函数;并且将该二次函数相对于V求微分,并且代入V=Vi,从而获得微分值。为了确定通过三点的二次函数,如果使用拉格朗日插值公式,计算是容易的(参考文献:Hideyo Nagashima,″数值计算法(NUMERICAL METHOD)(改订2版)″(Maki Shoten Publishing Co.)(日文)。
当在上述条件下绘制它的在Li脱嵌侧的微分曲线时,钛酸化合物具有至少两个在1.5至1.7V之间的峰,但是在一些情况下也在1.8至2.0V之间观察到峰。那么,如在本发明中的,当使得各个电势范围内的最大值的关系如上所述时,使得钛酸化合物的Li脱嵌容量高,倍率性质特别是循环性质出色。使得h2/h1为0.02以下是更优选的。已知的是,当存在一定量以上的副相如氧化钛和非晶态相时,在1.8V至2.0V之间出现最大值h2。
而且,根据本发明的钛酸化合物优选其结晶度高。具体地,在使用CuKα辐射作为辐射源的粉末X射线衍射图案中,优选的是在2θ=14.0°(误差:±0.5°)处的峰强度I2与在2θ=24.8°(误差:±0.5°)的最大峰强度I1的峰强度比率I2/I1为1.5以上;且2至5是更优选的。这说明归属于2θ=24.8°的晶面明显发展;并且这样的钛酸化合物,变得Li脱嵌容量高并且显示出色的循环性质(尽管不清楚原因)。
在本发明中,按如下进行粉末X射线衍射测量。通过使用CuKα辐射作为它的线源,并且设定扫描速度在5°/分钟,测量2θ=5至70°的角度范围的衍射。为了计算峰强度比率,使用通过从峰强度的测量值中减去背景强度而获得的值。通过使用拟合方法进行背景的消除(进行简单的峰搜索,并且除去峰部分,并且随后,通过对剩余的数据赋予拟合操作而获得多项式)。
根据本发明的钛酸化合物可以具有由一次粒子的聚集形成的二次粒子的形状,和由一次粒子和/或二次粒子的进一步聚集形成的聚集体的形状。本发明中的二次粒子是处于一次粒子本身相互牢固结合的状态的二次粒子,并且不是通过由粒子间相互作用如范德华力或通过机械压紧导致的内聚力所形成的二次粒子,而是不容易通过通常的工业操作(如混合、粉碎、 过滤、水洗、运输、称重、装袋和堆垛)崩塌的二次粒子,并且大部分作为二次粒子保持。尽管一次粒子具有各向异性的形状,但对二次粒子的形状没有特别的限制,并且可以使用具有各种形状的二次粒子。二次粒子的平均粒径(通过激光散射法得到的中值直径)优选在1至50μm的范围内。如上所述,对二次粒子的形状也没有特别的限制,并且可以使用具有各种形状的二次粒子,但是球形的二次粒子是优选的,因为增强了可流动性。相反,与二次粒子不同,聚集体通过上述工业操作崩塌。如二次粒子一样,对聚集体的形状没有特别的限制,并且可以使用各种形状的聚集体。
此外,本发明是一种碱金属钛酸盐化合物,所述碱金属钛酸盐化合物具有通过使用氮吸附的BET单点法测得的为5至15m2/g的比表面积,并且具有各向异性的形状,其中所述碱金属钛酸盐化合物基于粒子的数量包含60%以上的具有在0.1<L≤0.9μm范围内的通过电子显微术测得的长轴直径L的粒子。比表面积、粒子形状和长轴直径分布可以通过如上所述的方法确定。
根据本发明的碱金属钛酸盐化合物可以用作电极活性材料,并且可以进一步用作钛酸化合物的原料。特别是,当碱金属钛酸盐化合物用作钛酸化合物的原料时,碱金属钛酸盐化合物适合用于根据本发明的钛酸化合物的制备。
碱金属钛酸盐化合物优选具有以下组成式:
MxTiyOz (2)
其中M是选自由碱金属元素组成的组中的一种或两种碱金属元素;x/y为0.05至2.50,且z/y为1.50至3.50;在M是两种元素的情况下,x指两种元素的总数。
满足式(2)的化合物更具体地包括显示MTiO2、MTi2O4、M2TiO3、M2Ti3O7、M2Ti4O9、M2Ti5O11、M2Ti6O13、M2Ti8O17、M2Ti12O25、M2Ti18O37和M4Ti5O12的X射线衍射图案的化合物(M在该式中是选自由钠、钾、铷和铯组成的组中的一种或两种)。
该化合物更优选包括显示以下的与众不同的X射线衍射图案的化合物:钛酸钠化合物如NaTiO2、NaTi2O4、Na2TiO3、Na2Ti6O13、Na2Ti3O7和Na4Ti5O12,钛酸钾化合物如K2TiO3、K2Ti4O9、K2Ti6O13和K2Ti8O17,以及 钛酸铯化合物如Cs2Ti5O11。特别地,Na2Ti3O7是优选的。
在本说明书中,显示MTiO2等的X射线衍射图案的碱金属钛酸盐化合物不仅是具有MTiO2等的化学计量的组成的碱金属钛酸盐化合物;而且是在那范围内包括甚至是其一些元素是缺欠或过量的并且其具有非化学计量的组成的碱金属钛酸盐化合物,只要碱金属钛酸盐化合物显示MTiO2等的化合物的与众不同的X射线衍射图案即可。
例如,显示Na2Ti3O7的X射线衍射图案的钛酸钠化合物含有除了化学计量的组成的Na2Ti3O7之外,还含有不具有Na2Ti3O7的化学计量的组成但是在粉末X射线衍射测量(使用CuKα辐射)中显示在10.5°、15.8°、25.7°、28.4°、29.9°、31.9°、34.2°、43.9°、47.8°、50.2°和66.9°的2θ位置处(其任一个的误差为±0.5°)的Na2Ti3O7的与众不同的峰的钛酸钠化合物。
而且,在本发明的范围内包括具有源自其他晶体结构的峰的碱金属钛酸盐化合物,即,具有除主相之外的副相的碱金属钛酸盐化合物。在具有副相的情况下,在取主相的主峰的强度为100的情况下,归属于副相的主峰的强度优选为30以下,且更优选为10以下,且再更优选地,钛酸盐化合物是不含有副相的单相。
根据本发明的碱金属钛酸盐化合物优选基于粒子的数量包含60%以上的具有在1.0<L/S≤4.5范围内的长径比L/S的粒子,所述长径比L/S由通过电子显微术得到的各个粒子的长轴直径L和短轴直径S算得。这样的碱金属钛酸盐化合物尤其适合作为用于根据本发明的钛酸化合物的制备的原料。可以通过上述方法确定长径比的分布。碱金属钛酸盐化合物基于粒子的数量优选包含65%以上具有在1.0<L/S≤4.5范围内的长径比L/S的粒子,且更优选70%以上。而且,碱金属钛酸盐化合物基于粒子的数量优选包含55%以上具有在1.5<L/S≤4.0范围内的长径比L/S的粒子,且更优选60%以上。
此外,本发明是一种用于制备碱金属钛酸盐化合物的方法,所述方法包括研磨碱金属钛酸盐化合物直至其比表面积变成10m2/g以上的步骤(步骤1),以及将所得的研磨的材料退火的步骤(步骤2)。通过对碱金属钛酸盐化合物进行根据本发明的方法,可以获得上述碱金属钛酸盐化合物,即,其具有通过使用氮吸附的BET单点法测得的为5至15m2/g的比表面积, 并且具有各向异性的形状,其中所述碱金属钛酸盐化合物基于粒子的数量包含60%以上的具有在0.1<L≤0.9μm范围内的通过电子显微术测得的长轴直径L的粒子。
可以通过上述方法简单地制备根据本发明的碱金属钛酸盐化合物。
被供应用于研磨的碱金属钛酸盐化合物(在下文中,在某些情况下称为“研磨前体”)含有上述碱金属钛酸盐化合物作为主相,且可以含有副相。在具有副相的情况下,且主相的主峰的强度被看作是100的情况下,归属于副相的主峰的强度优选为50以下,且更优选为30以下,且再更优选地,碱金属钛酸盐化合物是不含有副相的单相。
在本发明中,制备步骤包括研磨碱金属钛酸盐化合物(研磨前体)直至其比表面积变成10m2/g以上的步骤(作为步骤1),以及将所得的研磨的材料退火的步骤(作为步骤2)。对于碱金属钛酸盐化合物的合成来说,因为通常需要在高温烧制原料混合物,所以导致了粒子本身的粒子生长和烧结,并且获得了提供许多粗粒子和具有小的比表面积的碱金属钛酸盐化合物。因此,通过使用碱金属钛酸盐化合物作为原料制得的钛酸化合物提供许多粗粒子,并且变得具有小的比表面积。那么,通过进行本发明的步骤1,可以减少粗粒子的数量,并且可以使得比表面积大。然而,当要最终制得的钛酸化合物用作电极活性材料时,在仅仅进行步骤1的情况下,由于在研磨的材料中含有许多超细粒子并且由于碱金属钛酸盐化合物的结晶度的降低和副相的形成,因而初始充电和放电效率以及循环性质下降。那么,通过进行步骤2,因为反之超细粒子被吸附在其他粒子中并消失并且结晶性回复,所以不导致过多的粒子本身的粒子生长和烧结,可以制备具有适合于钛酸化合物的制备的比表面积并具有均匀的粒径分布的碱金属钛酸盐化合物。
如果进行研磨,则充分进行到碱金属钛酸盐化合物的比表面积变成10m2/g以上,并且优选进行研磨直到比表面积变成13m2/g以上。如果通过设定研磨条件并进行一次或多次研磨,则使比表面积达到目标比表面积,是足够的。因为当研磨进行到此范围时可以达到本发明的益处,所以对比表面积没有特别的上限,但是因为研磨需要能量,所以如果使比表面积为30m2/g以下就足够了。通过上述使用氮吸附的BET单点法测量比表面积。 可以使用中值直径作为研磨的量度的指标。可以使此时的中值直径为例如1.0μm以下,且优选使其为0.6μm以下。优选的是,确定与上述比表面积的相关性,并且基于该相关性建立目标中值直径。
对于研磨,可以使用公知的研磨机器。可以在干燥状态下进行研磨,例如,通过使用冲击磨如锤式粉碎机、销棒粉碎机(pin mill)、或离心磨,磨碎机如双轴式轮碾机或滚筒辗粉机,压缩破碎机如片式破碎机(flake crusher)、滚碎机或颚式粉碎机,或空气流动型磨如喷射磨机;或者可以在湿润状态下进行研磨,例如,通过使用砂磨、球磨、DYNO-MILL等。从高效研磨的观点出发,优选使用湿法研磨,或者如果干法研磨,则使用磨碎机,并且湿法研磨是尤其优选的。
对用于湿法研磨的分散介质没有特别的限制,并且可以使用公知的材料。分散介质的实例包括极性溶剂,如水、乙醇和乙二醇。此外,对于研磨来说,可以使用公知的介质,其实例包括氧化锆、二氧化钛、锆石和氧化铝。为了浆液的粘度调整,在喷雾干燥中的造粒困难的情况下,并且为了使得粒径的控制容易,可以通过加入有机粘合剂进行研磨。所用的有机添加剂的实例包括(1)乙烯基系化合物(聚乙烯醇、聚乙烯基吡咯烷酮、等),(2)纤维素系化合物(羟乙基纤维素、羧甲基纤维素、甲基纤维素、乙基纤维素等),(3)蛋白质系化合物(明胶、阿拉伯树胶、酪蛋白、酪蛋白酸钠、酪蛋白酸铵等),(4)丙烯酸系化合物(聚丙烯酸钠、聚丙烯酸铵等),(5)天然聚合物化合物(淀粉、糊精、琼脂、海藻酸钠等),和(6)合成的聚合物化合物(聚乙二醇等);并且可以使用选自其中的至少一种。在这些中,不含有无机组分如钠的化合物是更优选的,因为它们容易通过干燥、退火或加热而分解或挥发。
在其中步骤1通过湿法研磨进行的情况下,在湿法研磨步骤后,优选在不将碱金属钛酸盐化合物从分散介质中过滤分离的情况下进行干燥;并且特别是在使用水作为分散介质的情况下,本制备方法是优选的。包含Na2Ti3O7的碱金属钛酸盐化合物通常具有高的离子可交换性,并且容易释放碱金属。本制备方法是优选的,这是因为:如果释放碱金属,则所得的组成偏离用作材料的碱金属钛酸盐化合物的组成;并且当随后进行步骤2的退火时,作为结果形成副相,并且当将最终制备的钛酸化合物用作电极 活性材料时,降低Li脱嵌容量和循环性质。干燥方法的实例包括减压干燥、蒸发至干燥、冷冻干燥和喷雾干燥;在这些中,喷雾干燥是工业上优选的。
如果进行喷雾干燥,则根据浆液的性质和加工能力,所用的喷雾干燥器可以适当地选自盘型、压力喷嘴型、双流喷嘴型、三流喷嘴型、四流喷嘴型等。例如通过调整浆液中的固体含量浓度,或者在盘型的情况下通过调整盘的转动频率,或者在压力喷嘴型、双流喷嘴型、三流喷嘴型、四流喷嘴型等的情况下通过调整喷雾压力和喷嘴直径,或者以其他方式,来控制喷射的液滴的大小,从而可以进行对二次粒子的直径的控制。作为双流喷嘴型,例如,可以使用Ohkawara Kakoki Co.,Ltd.制造的Twin-Jet Nozzle;并且作为三流喷嘴型和四流喷嘴型,可以使用例如Fujisaki Electric Co.,Ltd.制造的Trispire Nozzle and Micro Mist Spray Dryer。关于干燥温度,优选的是,使得入口端口温度在150至250℃的范围内,且使得出口端口温度在70至120℃的范围内。在其中浆液的粘度低并且造粒困难的情况下,或者为了使得粒径的控制更容易,可以使用有机粘合剂。所用的有机粘合剂的实例包括(1)乙烯基系化合物(聚乙烯醇、聚乙烯基吡咯烷酮等),(2)纤维素系化合物(羟乙基纤维素、羧甲基纤维素、甲基纤维素、乙基纤维素等),(3)蛋白质系化合物(明胶、阿拉伯树胶、酪蛋白、酪蛋白酸钠、酪蛋白酸铵等),(4)丙烯酸系化合物(聚丙烯酸钠、聚丙烯酸铵等),(5)天然聚合物化合物(淀粉、糊精、琼脂、海藻酸钠等),和(6)合成的聚合物化合物(聚乙二醇等);并且可以使用选自其中的至少一种。在这些中,不含有无机组分如钠的化合物是更优选的,应为它们容易通过干燥、退火或加热而分解或挥发。
可以例如通过将研磨的材料放在加热炉中,将研磨的材料加热至预定的温度,保持温度一定时间,并且冷却加热的材料,来进行对研磨的材料的退火(步骤2)。作为加热炉,可以使用公知的加热设备,例如,流化炉,固定炉、旋转窑或隧道窑。在退火中的气氛可以根据目的任选地建立,并且可以使得该气氛为,例如,非氧化性气氛如氮气或氩气,还原性气氛如氢气或一氧化碳气体,或氧化性气氛如空气或氧气。
优选的是,进行退火,直至将所述碱金属钛酸盐化合物的比表面积相 对于其在所述研磨后比表面积的20至80%。当降低比率低于此范围时,超细粒子在其他粒子中的吸附和结晶度改善不足;并且当高于此范围时,导致粒子本身的粒子生长和烧结,并且作为结果减少研磨的效果。其进一步优选的范围是25至70%。尤其优选的是,使得退火后碱金属钛酸盐化合物的比表面积在5至15m2/g的范围内。为了达到此范围,退火温度适当地在400至800℃的范围内。其进一步优选的范围是450至750℃。为了促进反应和抑制产物的烧结,可以重复进行两次以上退火。可以适当地设定退火时间,但是如果退火温度在上述温度范围内,则约1至10小时是合适的。可以适当地建立升温速率和冷却速率。在退火后,当需要时,可以对碱金属钛酸盐化合物进行压碎步骤。
通过烧制混合物获得上述研磨前体,所述混合物至少含有氧化钛和碱金属化合物,并且上述研磨前体优选是通过使用具有优选0.1至1.0质量%、更优选0.2至1.0质量%的按SO3计的硫元素含量的氧化钛制备的前体。当使得氧化钛含有在上述范围内的硫元素时,因为最终获得的钛酸化合物的一次粒子变得容易形成各向异性的形状,所以可以增强Li脱嵌容量。相反,当该含量低于0.2质量%,尤其是低于0.1质量%时,一次粒子可能难以形成各向异性的形状;并且当超过1.0质量%时,因为硫元素与Na反应并且制得其他相如Na2SO4,并且难以获得单相的碱金属钛酸盐化合物如Na2Ti3O7,所以Li脱嵌容量变得易于逆向减少。可以类似于上述钛酸化合物中的硫元素含量的测量,通过X射线荧光分析确定硫元素含量。而且,如果使得此处要制备的碱金属钛酸盐化合物的比表面积为10m2/g以下,则因为容易发展研磨步骤(步骤1)和退火步骤(步骤2)的组合的效果,优选连续地进行这些步骤。
氧化钛包括氧化钛类,如TiO、Ti4O7、Ti3O5、Ti2O3和TiO2,由TiO(OH)2、TiO2·xH2O(x是任意的)等表示的氧化钛水合物或含水的氧化钛。作为氧化钛水合物或含水的氧化钛,可以使用由TiO(OH)2或TiO2·H2O表示的偏钛酸,由TiO2·2H2O表示的正钛酸,它们的混合物等。氧化钛包括晶态氧化钛和非晶态氧化钛,并且在晶态氧化钛的情况下,可以使用金红石型、锐钛矿型或板钛矿型氧化钛或它们的混合晶体,或它们的混合物。
优选的是,具有通过使用氮吸附的BET单点法测得的为80至350m2/g 的比表面积。如果使用具有在此范围内的比表面积的氧化钛,是优选的,因为增强了在后续的烧制中氧化钛与碱金属化合物的反应性,并且可以减小在研磨前体中除了碱金属钛酸盐化合物之外的副相的主峰的强度。
对于碱金属化合物没有特别的限制,只要是含有碱金属的化合物(碱金属化合物)即可。例如,在其中碱金属是Na的情况下,碱金属化合物包括盐如Na2CO3和NaNO3,氢氧化物如NaOH,以及氧化物如Na2O和Na2O2。而且,在其中碱金属是K的情况下,碱金属化合物包括盐如K2CO3和KNO3,氢氧化物如KOH,以及氧化物如K2O和K2O2。在这些中,从成本、工艺中的可操作性和对潮解的抑制的观点出发,优选使用钠化合物。
可以使用任选的方法进行混合。其实例包括将碱金属化合物和氧化钛在干燥状态下或在湿润状态下混合的方法。可以通过搅拌并混合两者,例如,通过使用干法磨机如流体能量磨或冲击磨,高速搅拌器如Henschel混合器或高速混合器,混合器如样品混合器等,进行两者的干法混合。可以例如通过将两种化合物分散成浆液并经过湿法研磨机如砂磨、球磨、罐磨或DYNO-MILL,进行湿法混合。此时,可以加热浆液。根据具体情况,可以在混合后将浆液通过喷雾干燥器进行喷雾干燥。如果混合通过磨机进行或通过喷雾干燥进行,是优选的,因为增强了在后续的烧制中氧化钛与碱金属化合物的反应性。
碱金属化合物与氧化钛的共混比率可以与目标碱金属钛酸盐化合物的组成一致。例如,在制备Na2Ti3O7的情况下,将两者共混,使得Na/Ti变成摩尔比为0.67至0.72。此处,优选的是,碱金属化合物在共混量上比其由碱金属钛酸盐化合物的化学计量比算得的共混量稍大,例如,大1至6mol%。
随后,将至少含有氧化钛和碱金属化合物的混合物烧制,并使其反应,从而获得研磨前体。例如,通过将原料放在加热炉中,加热至预定温度,并且保持温度一定时间,来进行烧制。所用的加热炉和气氛可以与在上述退火步骤中的加热炉和气氛相同。
烧制温度优选在700至1,000℃的范围内,这使得容易获得具有高主相比率的研磨前体。当烧制温度低于该温度范围时,碱金属钛酸盐化合物的制备反应几乎不进行;并且当高于该温度范围时,易于引起产物本身的 坚固烧结。进一步优选的温度范围是750至900℃。为了促进反应和抑制产物的烧结,可以将烧制重复进行两次以上。可以适当地设置烧制时间,并且约1至100小时是合适的。也可以适当地建立升温速率和冷却速率。冷却可以通常是自然冷却(在炉中的自发冷却)或缓慢冷却。在此,在制备碱金属钛酸盐化合物的温度,因为粒子生长是时不可避免的,所以形成微米量级的粗粒子。
此外,本发明是一种用于制备钛酸化合物的方法,所述方法包括将通过上述制备方法获得的碱金属钛酸盐化合物(其具有通过使用氮吸附的BET单点法测得的为5至15m2/g的比表面积,并且具有各向异性的形状,其中所述碱金属钛酸盐化合物基于粒子的数量包含60%以上的具有在0.1<L≤0.9μm范围内的通过电子显微术测得的长轴直径L的粒子)与酸性水溶液接触从而用质子取代所述碱金属钛酸盐化合物中的碱金属阳离子的至少一部分的步骤(步骤3);并且该方法可以获得属于碱金属钛酸盐化合物的质子取代物的钛酸化合物(在下文中,也称为“质子取代物”)。质子取代物可以用作电极活性材料,或者可以用作后述将通过加热步骤获得的钛酸化合物的原料。
该方法的具体实例包括以下方法:其中制备含有分散在分散介质中的碱金属钛酸盐化合物的分散液,并且向分散液中加入酸性水溶液。作为分散介质,可以使用例如水。作为酸性水溶液,可以使用其中酸性化合物溶解在水中的酸性水溶液。
酸性化合物包括无机酸如盐酸、硫酸、硝酸和氢氟酸,以及它们的混合物。当使用这些时,反应容易进行,并且如果这些是盐酸或硫酸,是优选的,因为可以在工业上有优势地进行反应。
对酸性化合物的量和浓度没有特别的限制,但优选的是,该量是等于或大于在碱金属钛酸盐化合物中所含的碱金属的反应等效重量的量,并且使得游离酸的浓度为2N以下。对反应温度没有特别的限制。但优选的是,在低于100℃的范围内的温度下进行反应,在这样的温度时,待制备的质子取代物的结构几乎不变化。加工时间为1小时至7天,优选2小时至1天。而且,为了缩短加工时间,溶液可以适当地用新鲜的溶液替换。
在本步骤中,优选的是,尽可能减少在质子取代物中的碱金属的含量; 并且优选的是,使碱金属钛酸盐化合物与酸性化合物反应,以使得在与酸性化合物的反应的步骤中获得的质子取代物中的碱金属(M)的含量变成按M的氧化物计为1.0质量%以下。反应具体包括(1)使与酸性化合物的反应的温度为40℃以上,(2)重复进行与酸性化合物的反应两次以上,和(3)在三价钛离子的存在下与酸性化合物反应,并且可以通过组合这些方法中的两种以上来进行反应。在方法(1)中,优选使反应温度如上所述低于100℃。方法(3)具体包括:其中向酸性化合物或它的溶液中加入可溶性三价钛化合物如三氯化钛的方法,和其中将可溶性四价钛化合物如硫酸氧钛或四氯化钛还原从而导致存在三价钛离子的方法。酸性化合物或它的溶液中的三价钛离子浓度优选在0.01至1质量%的范围内。
根据本发明的制备方法可以使在质子取代物中的碱金属含量为1.0质量%以下,并且进一步为0.5质量%以下,并且可以进一步明显缩短步骤3来所需的时间。这可能是由于通过根据本发明的制备方法得到的碱金属钛酸盐化合物大概具有高结晶度并且几乎不具有粗粒子导致的。因为可以以这样的方式减少质子取代物中的碱金属含量,所以在后续加热步骤中对组成的控制变得容易,并且容易获得蓄电池性质出色的活性材料。
当需要时,将获得的质子取代形式清洁并固液分离,并随后干燥。清洁可以使用水、酸性水溶液等。固液分离可以使用公知的过滤方法。干燥也可以使用公知的干燥方法,但是因为结构取决于温度而发生变化,所以适宜地建立干燥温度。
质子取代物的具体实例包括H2Ti3O7、H2Ti4O9和H2Ti5O11。优选地使得其比表面积为13至35m2/g。
此外,本发明是一种用于制备钛酸化合物的方法,所述方法还包括加热在上述步骤3中获得的质子取代物的步骤(步骤4)。当加热质子取代物时,在质子取代物的构成元素中的氢原子和氧原子的一部分从晶格中消除,从而引起晶格的重排,并且将消除的氧和氢作为水释放,从而获得钛酸化合物。例如,通过将质子取代物放在加热炉中,将质子取代物加热至预定温度,并且保持温度一定时间,来进行加热。所用的加热炉和气氛可以与在上述退火步骤中的加热炉和气氛相同。
根据质子取代物的种类和目标钛酸化合物的种类,适当地建立加热温 度。例如,在其中通过使用H2Ti3O7作为质子取代物合成作为钛酸化合物的H2Ti12O25的情况下,可以伴随H和O的消除获得目标钛酸盐化合物H2Ti12O25。在这种情况下,加热温度在150℃至350℃的范围内,并且优选在250℃至350℃的范围内。常规上,在通过使用H2Ti3O7作为质子取代物合成作为钛酸化合物的H2Ti12O25的情况下,如在专利文献1中描述的,合适的加热温度在200℃至270℃的范围内,并且考虑到加工时间、变量等的工业方面,实践上需要在约260℃进行加热。然而,通过使用由采用根据本发明的方法制备的碱金属钛酸盐化合物作为原料,可以扩展加热温度的可接受范围,并且允许放松制备条件控制,这导致工业上的益处。
而且,在通过使用H2Ti4O9作为质子取代物合成作为钛酸化合物的H2Ti12O25的情况下,优选在250至650℃的范围内、更优选在300至400℃的范围内的温度下进行加热。在其中通过使用H2Ti5O11作为质子取代物合成作为钛酸化合物的H2Ti12O2的情况下,优选在200至600℃的范围内、更优选在350至450℃的范围内的温度下进行加热。
加热时间通常为0.5至100小时,且优选1至30小时;并且加热温度越高,可以使加热时间越短。
这样获得的钛酸化合物使得粒子几乎不含有超细粒子,并且具有比较均匀的粒径和特定的比表面积。从而,当使用钛酸化合物作为电极活性材料时,获得在Li脱嵌容量高、充电和放电效率高、能够减小在充电和放电循环中涉及的Li脱嵌容量的降低速率、并且倍率性质出色的钛酸化合物。这样的钛酸化合物不能够仅简单地通过研磨钛酸化合物以制造其细粒子而获得。
根据本发明的钛酸化合物和碱金属钛酸盐化合物的Li脱嵌容量、充电和放电效率、循环性质和倍率性质中任一项都是出色的。因此,使用含有这样的化合物作为电极活性材料的电极作为构成部件的蓄电装置是具有高容量、能够进行锂离子等的可逆的嵌入和脱嵌反应、并且保证高可靠性的蓄电装置。
根据本发明的蓄电装置具体包括锂二次电池、钠二次电池、镁二次电池、钙二次电池和电容器,它们是由含有根据本发明的钛酸化合物作为它们的电极活性材料的电极、对电极、隔离体和电解液构成的。
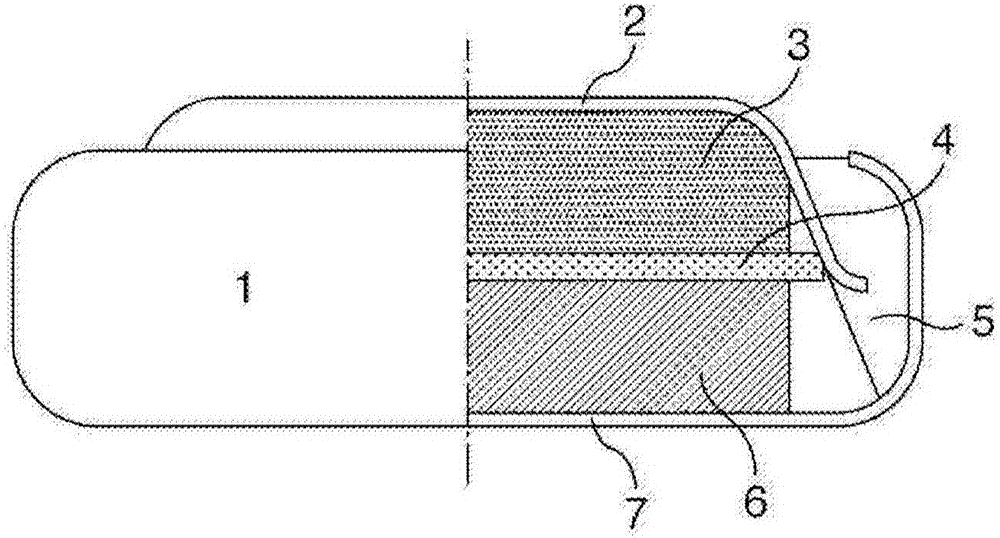
也就是说,除了使用根据本发明的钛酸化合物和/或碱金属钛酸盐化合物作为电极材料之外,可以原样使用公知的锂二次电池、钠二次电池、镁二次电池、钙二次电池和电容器的蓄电池元件;并且蓄电池可以是硬币型、纽扣型、圆柱型、层压体型、全固体型等中的任何类型。图1是图示了一个实例的示意图,其中将作为根据本发明的蓄电装置的一个实例的锂二次蓄电池用于硬币型锂二次蓄电池。硬币型蓄电池1由负极端子2、负极3、(电解质或隔离体+电解液)4、绝缘包装5、正极6和正极罐7构成。
当需要时,将含有根据本发明的钛酸化合物和/或碱金属钛酸盐化合物的活性材料与导电剂、粘合剂等共混,从而制备电极混合物,其压接在集流体上,从而制造电极。作为集流体,可以优选使用铜网、不锈钢网、铝网、铜箔、铝箔等。作为导电剂,可以优选使用乙炔黑、Ketjen Black等。作为粘合剂,可以优选使用聚四氟乙烯、聚偏二氟乙烯等。
对在电极混合物中的含有钛酸化合物和/或碱金属钛酸盐化合物的活性材料、导电剂、粘合剂等的共混物没有特别的限制,但是如果共混物使得导电剂为1至30质量%(优选5至25质量%);粘合剂为0至30质量%(优选3至10质量%);且余量为含有根据本发明的钛酸化合物和/或碱金属钛酸盐化合物的活性材料,通常是足够的。活性材料可以含有除了钛酸化合物和/或碱金属钛酸盐化合物之外公知的活性材料,但优选的是,钛酸化合物和/或碱金属钛酸盐化合物占电极容量50%以上,且80%以上是更优选的。
在根据本发明的蓄电装置中,在锂二次电池中,作为上述电极的对电极,可以使用公知的起正极作用并且能够吸留和释放锂的活性材料。作为这样的活性材料,可以使用各种类型的氧化物和硫化物;并且可以使用,例如,二氧化锰(MnO2)、氧化铁、氧化铜、氧化镍、锂锰复合氧化物(例如,LixMn2O4or LixMnO2)、锂镍复合氧化物(例如,LixNiO2)、锂钴复合氧化物(LixCoO2)、锂镍钴复合氧化物(例如,LixNi1-yCoyO2)、锂锰钴复合氧化物(LixMnyCo1-yO2)、锂镍锰钴复合氧化物(LixNiyMnzCo1-y-zO2)、具有尖晶石结构的锂锰镍复合氧化物(LixMn2-yNiyO4)、具有橄榄石结构的锂磷氧化物(LixFePO4、LixFe1-yMnyPO4、LixCoPO4、LixMnPO4、等等)和锂硅氧化物(Li2xFeSiO4、等等)、硫酸铁(Fe2(SO4)3)、钒氧化物(例如,V2O5)、 和由xLi2MO3·(1-x)LiM′O2表示的固溶型复合氧化物(M和M′各是相同的或不同的一种或两种以上的金属)。可以将这些混合并使用。在此,在上文中,x、y和z各自优选在0至1的范围内。而且,作为正极活性材料,可以使用有机材料和无机材料,包括导电的聚合物材料如聚苯胺和聚吡咯、二硫化物系聚合物材料、硫(S)和氟化碳。
而且,在根据本发明的蓄电装置中,在锂二次电池中,作为上述电极的对电极,可以使用公知的起负极作用并且能够吸留和释放锂的活性材料,如金属锂、锂合金、碳系材料如石墨和MCMB(中间相碳微球)。
在根据本发明的蓄电装置中,在钠二次电池中,作为上述电极的对电极,可以使用公知的起正极作用并且能够吸留和释放钠的活性材料,如钠过渡金属复合氧化物如钠铁复合氧化物、钠铬复合氧化物、钠锰复合氧化物和钠镍复合氧化物。
而且,在根据本发明的蓄电装置中,在钠二次电池中,作为上述电极的对电极,可以使用公知的起负极作用并且能够吸留和释放钠的活性材料,如金属钠、钠合金和碳系材料如石墨。
在根据本发明的蓄电装置中,在镁二次电池和钙二次电池中,作为上述电极的对电极,可以使用公知的起正极作用并且能够吸留和释放镁和钙的活性材料,如钠过渡金属复合氧化物如镁过渡金属复合氧化物和钙过渡金属复合氧化物。
而且,在根据本发明的蓄电装置中,在镁二次电池和钙二次电池中,作为上述电极的对电极,可以使用公知的起负极作用并且能够吸留和释放镁和钙的活性材料,如金属镁、镁合金、金属钙、钙合金和碳系材料如石墨。
而且,在根据本发明的蓄电装置中,在电容器中,可以制造不对称电容器,所述不对称电容器使用碳材料如石墨作为它们的上述电极的对电极。
而且,在根据本发明的蓄电装置中,所用的隔离体、蓄电池容器等可以是公知的蓄电池元件。
而且,在根据本发明的蓄电装置中,对于非水性电解质来说,可以使用其中电解质溶解在非水性有机溶剂中的液体非水性电解质(非水性电解液),其中聚合物材料含有非水性溶剂和电解质的聚合物凝胶状电解质,锂 离子传导性聚合物固体电解质或无机固体电解质等。
非水性有机溶剂起到参与锂蓄电池的电化学反应的离子可以在其中迁移的介质的作用。作为这样的非水性有机溶剂,可以使用例如碳酸酯系、酯系、醚系、酮系、或其他疏质子溶剂,或醇系溶剂。
作为碳酸酯系溶剂,可以使用碳酸二甲酯(DMC)、碳酸二乙酯(DEC)、碳酸二丙酯(DPC)、碳酸甲基丙基酯(MPC)、碳酸乙基丙基酯(EPC)、碳酸乙基甲基酯(EMC)、碳酸乙二酯(EC)、碳酸丙二酯(PC)、碳酸丁二酯(BC)等。
作为酯系溶剂,可以使用乙酸甲酯、乙酸乙酯、乙酸正丙酯、乙酸二甲酯、丙酸甲酯、丙酸乙酯、γ-丁内酯(GBL)、δ-癸内酯、戊内酯、甲羟戊酸内酯、己内酯等。
作为醚系溶剂,可以使用二丁醚、四甘醇二甲醚、二甘醇二甲醚、二甲氧基乙烷、2-甲基四氢呋喃、四氢呋喃等。
作为酮系溶剂,可以使用环己酮等。
作为醇系溶剂,可以使用乙醇、异丙醇等。
作为其他疏质子溶剂,可以使用腈如R-CN(R是C2-C20直链或支链或环状结构烃基,并且可以含有双键、芳环或醚键),酰胺如二甲基甲酰胺,二氧戊环如1,3-二氧戊环,环丁砜等。
非水性有机溶剂可以由单一物质构成,或可以是两种以上溶剂的混合物。在非水性有机溶剂是两种以上溶剂的混合物的情况下,根据蓄电池性能适当调整两种以上溶剂的混合比率,并且例如,可以使用环状碳酸酯如EC和PC,或主要由环状碳酸酯及具有低于环状碳酸酯粘度的粘度的非水性溶剂构成的混合溶剂,等等。
作为电解质,可以使用碱盐;并且优选使用锂盐。锂盐的实例包括六氟磷酸锂(LiPF6)、四氟硼酸锂(LiBF4)、六氟砷酸锂(LiAsF6)、高氯酸锂(LiClO4)、锂双三氟甲磺酰亚胺(LiN(CF3SO2)2、LiTSFI)和三氟偏磺酸锂(LiCF3SO3)。这些可以单独使用或以两种以上的混合物使用。
非水性溶剂中的电解质的浓度优选为0.5至2.5mol/l。当浓度为0.5mol/l以上时,可以降低电解质的电阻,并且可以提高充电和放电性质。另一方面,当在2.5mol/l以下时,可以抑制电解质的熔点和粘度的升高, 并且可以使电解质在通常温度下是液体。
液体非水性电解质(非水性电解液)可以还含有能够改善锂蓄电池的低温性质等的添加剂。作为添加剂,可以使用例如碳酸酯系物质、亚硫酸乙二酯(ES)、二腈化合物和丙烷磺内酯(PS)。
可以例如从由以下各项组成的组中选择碳酸酯系物质:碳酸亚乙烯酯(VC),具有一个以上选自由卤素(例如,-F、-Cl、-Br、-I等)、氰基(CN)和硝基(-NO2)组成的组中的取代基的碳酸亚乙烯酯衍生物,和具有一个以上选自由卤素(例如,-F、-Cl、-Br、-I等)、氰基(CN)和硝基(-NO2)组成的组中的取代基的碳酸亚乙酯衍生物。
添加剂可以是单独一种物质,或可以是两种以上物质的混合物。具体地,电解液可以还含有一种以上选自由以下各项组成的组中的添加剂:碳酸亚乙烯酯(VC)、碳酸氟代亚乙酯(FEC)、亚硫酸亚乙酯(ES)、丁二腈(SCN)和丙烷磺内酯(PS)。
电解液优选含有作为溶剂的碳酸乙二酯(EC)和作为电解质的锂盐。电解液优选含有选自碳酸亚乙烯酯(VC)、亚硫酸乙二酯(ES)、丁二腈(SCN)和丙烷磺内酯(PS)的至少一种作为添加剂。据推测这些溶剂和添加剂具有在负极的钛酸化合物上形成膜的作用,并且提高抑制在高温环境中气体逸出的效果。
相对于非水性有机溶剂和电解质的100质量份总量,添加剂的含量优选为10质量份以下,且更优选0.1至10质量份。当含量在此范围内时,可以改善蓄电池的温度性质。添加剂的含量再更优选为1至5质量份。
对于构成聚合物凝胶状电解质的聚合物材料来说,可以使用公知的材料。可以使用例如单体的聚合物,如聚丙烯腈、聚丙烯酸酯、聚偏二氟乙烯(PVdF)和聚环氧乙烷(PEO),以及与其他单体的共聚物。
对于聚合物固体电解质的聚合物材料来说,可以使用公知的材料。可以使用例如单体的聚合物,比如聚丙烯腈、聚偏二氟乙烯(PVdF)和聚环氧乙烷(PEO),以及与其他单体的共聚物。
作为无机固体电解质,可以使用公知的材料。例如,可以使用含有锂的陶瓷材料。适当地使用Li3N玻璃和Li3PO4-Li2S-SiS2玻璃。
实施例
在下文中,将给出实施例,并且将使本发明的特征更加清楚。本发明不限于这些实施例。
下面将描述各样品的物理性质的测量方法。
(比表面积的测量)
采用比表面积分析仪(Monosorb MS-22,由Quantachrome Instruments制造),通过使用氮吸附的BET单点法,测量样品的比表面积。
(X射线衍射术)
通过装备有高速一维探测器D/teX Ultra的粉末X射线衍射仪Ultima IV(两者均由Rigaku Corp.制造)测量样品的粉末X射线衍射。通过使用Cu-Kα作为X射线源,在5至70°的2θ角度和5°/分钟的扫描速度进行测量。通过与PDF卡片或公知文献比较,进行化合物的鉴定。峰强度使用从测量后的数据通过背景消除(拟合方法:进行单峰搜索,并且除去峰部分,并且随后,通过对剩余的数据赋予拟合操作获得多项式)获得的值。通过从X射线衍射图阅读背景消除后在2θ=14.0°处的峰强度I1和在2θ=24.8°处的峰强度I2,来确定峰强度比I2/I1。
(电子显微术)
通过扫描电子显微镜(SEM)(S-4800,由Hitachi High-Technologies Corp.制造),在10,000X的视野下观察样品粒子,打印图像使得1em相当于0.5μm,并且随机选择并测量100个其短边为1mm以上的粒子,来确定样品粒子的长轴直径L和短轴直径S。从结果确定长径比L/S。从这些数据,生成按L和L/S的粒子数计的累积相对频率分布。也通过使用扫描电子显微镜检查样品的形状。
(组成的分析)
通过使用波长分散X射线荧光分析仪(RIX-2100,由Rigaku Corp.制造),测量样品的硫和钠的浓度。通过从样品中的S和Na的量计算SO3和 Na2O的质量,并将所述质量除以样品的质量,来确定硫和钠的含量。
(中值直径)
通过激光衍射散射法测量中值直径。具体地,通过使用粒径分布分析仪(LA-950,由HORIBA Ltd.制造),测量中值直径。作为分散介质,使用纯水;并且将折射率设置在2.5。
实施例1
将2,000g的锐钛矿型二氧化钛(比表面积SSA:90m2/g,硫元素含量:按SO3计0.3质量%,由Ishihara Sangyo Kaisha Ltd.制造)和820g的碳酸钠通过使用Henschel混合机(MITSUI HENSCHEL FM20C/I,由Mitsui Mining Co.,Ltd.制造)以1,800rpm混合10min。将混合物中的2,400g充入烧箱中,并通过使用电炉在空气中在800℃的温度烧制6小时,从而获得研磨前体(样品A1)。样品A1的比表面积为8.2m2/g,并且通过粉末X射线衍射测量确认,样品A1是具有良好结晶度的Na2Ti3O7的单相。
(步骤1)
将1,000g所得的样品A1加入至4,000g纯水中,从而制备具有20质量%的固体含量的浆液。通过使用填有80%的直径为0.5mm的锆珠的湿法磨机(型号:MULTI LAB,由Shinmaru Enterprises Corp.制造),在10m/sec的盘圆周速度和120ml/分钟的浆液进料量的条件下,研磨浆料。在研磨后,样品的中值直径为0.31μm。通过使用喷雾干燥器(型号:L-8i,由Ohkawara Kakouki Co.,Ltd.制造),在入口端口温度为190℃且出口端口温度为90℃的条件下,将浆液喷雾干燥,从而获得样品A2。样品A2的比表面积为21.0m2/g。
(步骤2)
将所得的样品A2在电炉中在700℃的空气中退火5小时,从而获得样品A3。样品A3的比表面积为8.2m2/g,并且由于退火导致的比表面积的减少率为61%。而且,通过粉末X射线衍射确认,样品A3是具有良好 结晶度的Na2Ti3O7的单相。
通过扫描电子显微镜检查样品A3的粒子形状,并且其为棒形。而且,作为通过上述方法确定粒子的长轴直径、短轴直径和长径比的结果,关于长轴直径L,0.1<L≤0.9μm的粒子的比例为85%,且0.1<L≤0.6μm的粒子的比例为53%。关于长径比,1<L/S≤4.5的粒子的比例为83%,且1.5<L/S≤4.0的粒子的比例为68%。
(步骤3)
将1,000g的此样品A3浸入其中将563g的70%硫酸加入到3,437g的纯水形成的水溶液中,使其在60℃在搅拌下反应5小时,随后过滤并用水洗涤,并在120℃干燥。干燥的粉末中的830g浸入其中将60g的70%硫酸加入到3,260g的纯水形成的水溶液中,使其在70℃在搅拌下反应5小时,随后过滤并用水洗涤,并在120℃干燥12小时,从而获得质子取代物(样品A4)。样品A4的比表面积为16.9m2/g。
通过X射线荧光分析对所得的样品A4分析它的化学组成,其被测定按Na2O计为0.087质量%的钠,得到99.9%的Na移除率,并合理给出接近完全质子交换的H2Ti3O7的化学式。进一步通过粉末X射线衍射清楚的是,样品A4是具有良好结晶度的H2Ti3O7的单相。
(步骤4)
将780g所得的样品A4在电炉中在260℃的空气中加热脱水15小时,从而获得钛酸化合物(样品A5)。样品A5的比表面积为16.1m2/g。
样品A5的扫描电子显微镜照片示于图2中。样品A5的粒子形状是保持作为起始原料的样品A3的形状的棒形。而且,作为通过上述方法确定粒子的长轴直径、短轴直径和长径比的结果,关于长轴直径L,0.1<L≤0.9μm的粒子的比例为85%,且0.1<L≤0.6μm的粒子的比例为53%。关于长径比,1.0<L/S≤4.5的粒子的比例为83%,且1.5<L/S≤4.0的粒子的比例为68%。在此,关于数均值(100个粒子的平均值),长轴直径L为0.68μm;短轴直径S为0.21μm;且长径比L/S为3.24。
样品A5的使用CuKα辐射的粉末X射线衍射图示于图3中。所得的 样品A5至少具有在2θ=14.0°、24.8°、28.7°、43.5°、44.5°和48.6°位置的峰(其中任一个的误差为±0.5°),并且在2θ=10至20°之间具有一个峰,这如在过去的报告中所见,表明是H2Ti12O25衍射图案特征。在2θ=24.8°(误差:±0.5°)处的峰强度I2与在2θ=14.0°(误差:±0.5°)处的峰强度I1的峰强度比I2/I1为2.95。而且,在取2θ=14.0°(误差:±0.5°)处的峰的强度为100的情况下,除了在2θ=14.0°处的峰之外,在10.0°≤2θ≤20.0°之间没有观察到具有20以上的强度的峰。
通过X射线荧光分析分析样品A5的化学组合,并且按SO3计的硫元素含量为0.27质量%。而且,按Na2O计的钠含量为0.092质量%。
实施例2
将样品A1用作研磨前体,并且湿法研磨,直至通过加强步骤1的研磨条件使得中值直径变成0.24μm,并在与实施例1中相同的条件下喷雾干燥,从而获得样品B2。样品B2的比表面积为24.3m2/g。
随后,在与实施例1中相同的条件下对样品B2进行退火(步骤2),从而获得样品B3。样品B3的比表面积为8.1m2/g,并且由于退火导致的比表面积的减少率为67%。通过扫描电子显微镜检查样品B3的粒子形状,并且为棒形。而且,关于长轴直径,0.1<L≤0.9μm的粒子的比例为81%,且0.1<L≤0.6μm的粒子的比例为64%。关于长径比,1.0<L/S≤4.5的粒子的比例为75%,且1.5<L/S≤4.0的粒子的比例为66%。通过粉末X射线衍射进一步确认,样品B3是具有良好结晶度的Na2Ti3O7的单相。
随后,在与实施例1中相同的条件下对样品B3进行质子取代(步骤3),从而获得质子取代物(样品B4)。样品B4的比表面积为18.0m2/g。进一步通过粉末X射线衍射清楚的是,样品B4是具有良好结晶度的H2Ti3O7的单相。
随后,在与实施例1中相同的条件下对样品B4进行加热(步骤4),从而获得钛酸化合物(样品B5)。样品B5的比表面积为16.4m2/g。
作为通过扫描电子显微镜观察样品B5的结果,样品B5的粒子形状是保持作为起始原料的样品B3的形状的棒形。而且,关于长轴直径,0.1<L≤0.9μm的粒子的比例为81%,且0.1<L≤0.6μm的粒子的比例为64%。 关于长径比,1.0<L/S≤4.5的粒子的比例为75%,且1.5<L/S≤4.0的粒子的比例为66%。在此,关于数均值(100个粒子的平均值),长轴直径L为0.62μm;短轴直径S为0.20μm;且长径比L/S为3.46。
作为样品B5的粉末X射线衍射的结果,样品B5至少具有在2θ=14.0°、24.8°、28.7°、43.5°、44.5°和48.6°位置的峰(其中任一个的误差为±0.5°),并且在2θ=10至20°之间具有一个峰,这如在过去的报告中所见,表明是H2Ti12O25衍射图案特征。而且,在2θ=24.8°(误差:±0.5°)处的峰强度I2与在2θ=14.0°(误差:±0.5°)处的峰强度I1的峰强度比I2/I1为3.48。而且,在取2θ=14.0°(误差:±0.5°)处的峰的强度为100的情况下,除了在2θ=14.0°处的峰之外,在10.0°≤2θ≤20.0°之间没有观察到具有20以上的强度的峰。
通过X射线荧光分析分析样品B5的化学组合,并且按SO3计的硫元素含量为0.28质量%。而且,按Na2O计的钠含量为0.059质量%。
实施例3
将样品A1用作研磨前体,并且湿法研磨,直至通过减缓步骤1的研磨条件使得中值直径变成0.53μm,并在与实施例1中相同的条件下喷雾干燥,从而获得样品C2。样品C2的比表面积为16.0m2/g。
随后,在与实施例1中相同的条件下对样品C2进行退火(步骤2),从而获得样品C3。样品C3的比表面积为7.0m2/g,并且由于退火导致的比表面积的减少率为56%。通过扫描电子显微镜检查样品C3的粒子形状,并且为棒形。而且,关于长轴直径,0.1<L≤0.9μm的粒子的比例为69%,且0.1<L≤0.6μm的粒子的比例为36%。关于长径比,1<L/S≤4.5的粒子的比例为67%,且1.5<L/S≤4.0的粒子的比例为59%。还通过粉末X射线衍射确认,样品C3是具有良好结晶度的Na2Ti3O7的单相。
随后,在与实施例1中相同的条件下对样品C3进行质子取代(步骤3),从而获得质子取代物(样品C4)。样品C4的比表面积为14.2m2/g。进一步通过粉末X射线衍射清楚的是,样品C4是具有良好结晶度的H2Ti3O7的单相。
随后,在与实施例1中相同的条件下对样品C4进行加热(步骤4),从 而获得钛酸化合物(样品C5)。样品C5的比表面积为12.9m2/g。
作为通过扫描电子显微镜观察样品C5的结果,样品C5的粒子形状是保持作为起始原料的样品C3的形状的棒形。而且,关于长轴直径,0.1<L≤0.9μm的粒子的比例为69%,且0.1<L≤0.6μm的粒子的比例为36%。关于长径比,1.0<L/S≤4.5的粒子的比例为67%,且1.5<L/S≤4.0的粒子的比例为59%。在此,关于数均值(100个粒子的平均值),长轴直径L为0.86μm;短轴直径S为0.22μm;且长径比L/S为4.23。
作为样品C5的粉末X射线衍射的结果,样品C5至少具有在2θ=14.0°、24.8°、28.7°、43.5°、44.5°和48.6°位置的峰(其中任一个的误差为±0.5°),并且在2θ=10至20°之间具有一个峰,表明是H2Ti12O25衍射图案特征,这如在过去的报告中所见。而且,在2θ=24.8°(误差:±0.5°)处的峰强度I2与在2θ=14.0°(误差:±0.5°)处的峰强度I1的峰强度比I2/I1为2.82。而且,在取2θ=14.0°(误差:±0.5°)处的峰的强度为100的情况下,除了在2θ=14.0°处的峰之外,在10.0°≤2θ≤20.0°之间没有观察到具有20以上的强度的峰。
通过X射线荧光分析分析样品C5的化学组合,并且按SO3计的硫元素含量为0.20质量%。而且,按Na2O计的钠含量为0.12质量%。
实施例4
如在实施例1中获得钛酸化合物(样品D5),不同之处在于将步骤4的加热温度变更至350℃。作为样品D5的粉末X射线衍射的结果,样品D5至少具有在2θ=14.0°、24.8°、28.7°、43.5°、44.5°和48.6°位置的峰(其中任一个的误差为±0.5°),并且在2θ=10至20°之间具有一个峰,表明是H2Ti12O25衍射图案特征,这如在过去的报告中所见。而且,在取2θ=14.0°(误差:±0.5°)处的峰的强度为100的情况下,除了在2θ=14.0°处的峰之外,在10.0°≤2θ≤20.0°之间没有观察到具有20以上的强度的峰。由此,变得清楚的是通过使用采用根据本发明的方法制得的碱金属钛酸盐化合物作为原料,可以扩大在步骤4中用于加热的可接受的温度范围。
比较例1
通过使用样品A1作为研磨前体,不进行步骤1(研磨)和步骤2(退火),在与实施例1中相同的条件下进行质子取代(步骤3),来获得质子取代物(样品E4)。样品E4的比表面积为16.7m2/g。随后,在与实施例1中相同的条件下对样品E4进行加热(步骤4),从而获得钛酸化合物(样品E5)。样品E5的比表面积为14.9m2/g。
样品E5的扫描电子显微镜照片示于图4中。粒子主要含有棒形粒子,并且存在许多粗粒子。而且,关于粒子的长轴直径,0.1<L≤0.9μm的粒子的比例为31%,且0.1<L≤0.6μm的粒子的比例为10%。关于长径比,1.0<L/S≤4.5的粒子的比例为51%,且1.5<L/S≤4.0的粒子的比例为43%。在此,关于数均值,长轴直径L为1.31μm;短轴直径S为0.30μm;且长径比L/S为4.88。
作为样品E5的粉末X射线衍射的结果,样品E5至少具有接近在2θ=14.0°、24.8°、28.7°、43.5°、44.5°和48.6°位置的峰,并且在2θ=10至20°之间具有一个峰,表明是H2Ti12O25衍射图案特征,这如在过去的报告中所见。而且,在2θ=24.8°处的峰强度I2与在2θ=14.0°处的峰强度I1的峰强度比I2/I1为1.49。
通过X射线荧光分析分析样品E5的化学组成,并且按SO3计的硫元素含量为0.24质量%。而且,按Na2O计的钠含量为0.31质量%。
比较例2
采用通过使用样品A1作为研磨前体并进行如在实施例1中的湿法研磨(步骤1)获得的上述样品A2。通过不进行退火(步骤2),并且在与实施例1中相同的条件下对样品A2进行质子取代(步骤3),获得质子取代物(样品F4)。样品F4的比表面积为61.9m2/g。随后,在与实施例1中相同的条件下对样品F4进行加热(步骤4),从而获得钛酸化合物(样品F5)。样品F5的比表面积为46.8m2/g。
样品F5的扫描电子显微镜照片示于图5中。发现了,样品F5的粒子主要是棒形粒子和比较小的、各向同性的角状粒子,但是在这些粒子表面上存在超细粒子。
样品F5的使用CuKα辐射的粉末X射线衍射图案示于图6中。所得 的样品F5至少具有接近在2θ=14.0°、24.8°、28.7°、43.5°、44.5°和48.6°位置的峰,并且在2θ=10至20°之间具有一个峰,这如在过去的报告中所见,表明是H2Ti12O25衍射图案特征。而且,在接近2θ=24.8°处的峰强度I2与在2θ=14.0°处的峰强度I1的峰强度比I2/I1为2.65。
通过X射线荧光分析分析样品F5的化学组成,并且按SO3计的硫元素含量为0.46质量%。而且,按Na2O计的钠含量为0.063质量%。
比较例3
使用通过采用样品A1作为研磨前体并进行如在实施例2中的湿法研磨(步骤1)获得的上述样品B2。通过不进行退火(步骤2),并且在与实施例1中相同的条件下对样品B2进行质子取代(步骤3),获得质子取代物(样品G4)。样品G4的比表面积为81.5m2/g。随后,在与实施例1中相同的条件下对样品G4进行加热(步骤4),从而获得钛酸化合物(样品G5)。样品G5的比表面积为61.4m2/g。
作为通过扫描电子显微镜观察样品G5的结果,发现了,样品G5的粒子主要是棒形粒子和比较小的、各向同性的角状粒子,但是在这些粒子表面上存在超细粒子。还观察到,存在的超细粒子的量比在样品F5中大。
作为样品G5的粉末X射线衍射的结果,样品G5至少具有接近在2θ=14.0°、24.8°、28.7°、43.5°、44.5°和48.6°位置的峰,并且在2θ=10至20°之间具有一个峰,这如在过去的报告中所见,表明是H2Ti12O25衍射图案特征。而且,在接近2θ=24.8°处的峰强度I2与在2θ=14.0°处的峰强度I1的峰强度比I2/I1为3.44。
通过X射线荧光分析分析样品G5的化学组成,并且按SO3计的硫元素含量为0.79质量%。而且,按Na2O计的钠含量为0.091质量%。
比较例4
将2,000g的金红石型二氧化钛(SSA:6.2m2/g,硫元素含量:按SO3计0.0质量%,由Ishihara Sangyo Kaisha Ltd.制造)和820g的碳酸钠通过使用Henschel混合机(MITSUI HENSCHEL FM20C/I,由Mitsui Mining Co.,Ltd.制造)以1,800rpm混合10min。将混合物中的2,400g充入烧箱中,并 通过使用电炉在800℃温度的空气中烧制6小时,从而获得研磨前体(样品H1)。样品H1的比表面积为1.2m2/g,并且通过粉末X射线衍射测量确认,样品H1是具有良好结晶度的Na2Ti3O7的单相。
如在比较例1中获得钛酸化合物(样品H5),不同之处在于使用样品H1作为研磨前体。样品H5的比表面积为5.6m2/g。
作为通过扫描电子显微镜观察样品H5的结果(图7),粒子中的许多是板状粒子,并且存在许多粗粒子。而且,关于粒子的长轴直径,0.1<L≤0.9μm的粒子的比例为1%,且0.1<L≤0.6μm的粒子的比例为0%。关于长径比,1.0<L/S≤4.5的粒子的比例为94%,且1.5<L/S≤4.0的粒子的比例为73%。
作为样品H5的粉末X射线衍射的结果,样品H5至少具有接近在2θ=14.0°、24.8°、28.7°、43.5°、44.5°和48.6°位置的峰,并且在2θ=10至20°之间具有一个峰,这如在过去的报告中所见,表明是H2Ti12O25衍射图案特征。而且,在接近2θ=24.8°处的峰强度I2与在2θ=14.0°处的峰强度I1的峰强度比I2/I1为1.65。
通过X射线荧光分析分析样品H5的化学组成,并且按SO3计的硫元素含量为0.30质量%。而且,按Na2O计的钠含量为0.26质量%。
在表1中示出了实施例和比较例的样品的比表面积和具有0.1<L≤0.9μm的长轴直径L的钛酸化合物的粒子的比例(%)。
[表1]
注:比表面积的单位是m2/g
对于实施例1至3和比较例1和4(样品A5、B5、C5、E5和H5)来说,关于长轴直径L和长径比L/S的按粒子数计的累积相对频率分布分别示于图8和图9中。变得清楚的是,进行了研磨(步骤1)和退火(步骤2)的样品A5、B5和C5具有比没有进行研磨(步骤1)和退火(步骤2)的样品E5和H5更小的长轴直径,并且具有中等的长径比。
蓄电池性质评价1:Li脱嵌容量、充电和放电效率以及循环性质的评价
通过使用样品A5至C5以及E5和H5作为它们的电极活性材料,来制备锂二次电池,并且评价它们的充电和放电性质。将描述蓄电池的形式和测量条件。
将上述各样品、作为导电剂的乙炔黑粉末、和作为粘合剂的聚四氟乙烯树脂以5∶4∶1的质量比混合,在研钵中捏合在一起,拉伸成片形式,并且成形为直径10mm的圆,从而制造出粒料形式。调整厚度,使得粒料的质量变成约10mg。将此粒料夹在两个以10mm直径切出的铝网之间,并在9MPa加压,从而制造工作电极。
将工作电极在220℃的温度真空干燥4小时,并且在手套箱中在氩气氛下在露点-60℃的温度以下,作为工作电极组装在可密封的硬币型评价电池中。作为评价电池,使用一种其材料是不锈钢(SUS316)并且具有20mm外径和3.2mm高度的评价电池。作为它的对电极,使用成形为直径为12mm的圆的厚度为0.5mm的金属锂。作为它的非水性电解液,使用碳酸亚乙酯和碳酸二甲酯(体积比为1∶2)的混合溶液,其中以1mol/l的浓度溶解有LiPF6。
将工作电极放在评价电池的下罐上;在其上放置作为隔离体的多孔聚丙烯膜;并且将非水性电解液滴在其上。进一步,放置对电极、厚度为1mm的用于调整厚度的间隔体和弹簧(各是SUS316的);并且在其上覆盖具有聚丙烯衬垫的上罐,并且将外周缘部分填缝并密封。
通过设置电压范围为1.0至3.0V,充电和放电电流为0.11mA,并室温下以恒定电流进行11次充电和放电的循环,来进行充电和放电容量的测量。图10显示作为典型实例的实施例1和比较例2的第一-循环充电和放电曲线。
取此时的第一循环的Li脱嵌容量为初始容量。
而且,取其与第一循环的Li嵌入容量的比率,即(第一-循环Li脱嵌容量/第一-循环Li嵌入容量)×100,作为充电和放电效率。可以说,此值越高,充电和放电效率越高。
从第12次循环起,通过将充电和放电电流设置在0.22mA,在室温以恒定电流进行59次充电和放电的循环,并评价循环性质。这样总共进行了70次循环,并且从第70次循环的Li脱嵌容量,取(第70次循环Li脱嵌容量/第一-循环Li脱嵌容量)×100作为循环性质。此值越高,循环性质越好。
蓄电池性质评价2:V-dQ/dV
按如下确定上述的微分曲线V-dQ/dV。将评价电池充电(Li嵌入)至1V,并且随后以0.1C放电(Li脱嵌)至3V。此时,以5mV的电压变化量的间隔和/或以120秒的间隔获得Li脱嵌侧上的电压V-容量Q的数据。基于这样获得的数据,绘制V-Q曲线。通过使用第二循环的Li脱嵌曲线,并且在计算微分值之前,通过单纯移动平均法,分别将所得的电压V和容量Q的数据平滑化。具体地,在以时间序列排列的5个数据中,用5个数据的平均值替换中间的第三个数据。对所有数据进行此过程,并绘制平滑化的V-Q曲线。
随后,计算微分值。按如下确定通过将在这些平滑化的数据中第i个点上的Qi相对于V求微分获得的值。也就是说,确定相对于V的通过该点和该点的前后两点共三点(Vi-1,Qi-1)、(Vi,Qi)和(Vi+1,Qi+1)的二次函数;并且将该二次函数相对于V求微分,并且代入V=Vi,从而获得微分值。为了确定通过三点的二次函数,使用拉格朗日插值公式。图11示出了作为典型实例的实施例1和比较例2的V-dQ/dV曲线。
随后,读取1.5至1.7V之间的电压的dQ/dV的最大值h1和1.8至2.0 V之间的电压的dQ/dV的最大值h2,并计算比率h2/h1。
蓄电池性质评价3:倍率性质(Li嵌入侧)
通过使用样品A5至C5以及E5和H5作为它们的电极活性材料,来制备锂二次电池,并且评价该锂二次电池的充电和放电性质。将描述蓄电池的形式和测量条件。
将上述各样品、作为导电剂的乙炔黑粉末、和作为粘合剂的聚偏二氟乙烯(PVdF)树脂的N-甲基-2-吡咯烷酮(NMP)溶液混合,从而变成按固体含量计的质量比为83∶10∶7;并且进一步添加NMP,使得固体含量变成35重量%。通过行星式混合机(Awatorirentaro ARE-310,由Thinky Corp.制造)将混合物捏合,从而制造糊料。将制得的糊料涂敷在铝箔上,在80℃干燥20min,冲成12mm的圆,并在10MPa加压,从而制造电极。调整涂敷量(涂敷厚度),使得冲成12mm的电极上的活性材料量变成9mg。
将此工作电极在120℃真空干燥4小时,并且随后在手套箱中在氩气氛下在露点-60℃的温度以下,作为正极组装在可密封的硬币型评价电池中。作为评价电池,使用一种其材料是不锈钢(SUS316)并且具有20mm外径和3.2mm高度的评价电池。作为它的负极,使用成形为14mm直径的圆形且厚度为0.5mm的金属锂。作为它的非水性电解液,使用碳酸亚乙酯和碳酸二甲酯(体积比为1∶2)的混合溶液,其中以1mol/1的浓度溶解有LiPF6。
将工作电极放在评价电池的下罐上;在其上放置作为隔离体的多孔聚丙烯膜;并且将非水性电解液滴在其上。进一步,在其上放置负极、厚度为1mm的用于调整厚度的间隔体和弹簧(各是SUS316的);并且在其上覆盖具有聚丙烯衬垫的上罐,并且将外周缘部分填缝并密封。
通过设置电压范围为1.0至3.0V,将放电(Li脱嵌)电流固定在0.33mA,设置充电(Li嵌入)电流在0.33或8.25mA,在室温在恒定电流下进行充电和放电容量的测量。在此,从当电流值为0.33mA时的容量和当电流值为8.25mA时的容量出发,取(在8.25mA的Li嵌入容量/在0.33mA的Li嵌入容量)×100作为倍率性质。此值越高,倍率性质越好。
将上述蓄电池性质评价的结果集体地示于表2中。清楚的是,尽管比 表面积大于30m2/g的比较例2和3的初始容量高,但充电和放电效率低,且循环性质也低下。而且,h2/h1也高于0.05。由此,暗示产生了副相。其中具有0.1<L≤0.9um的长轴直径的粒子的比例低于60%的比较例1也具有低初始容量。而且,清楚的是,任何实施例都具有比任何比较例更高的倍率性质。由上文变得清楚的是,当使用根据本发明的钛酸化合物作为蓄电装置的电极活性材料时,可以获得与常规蓄电装置相比容量更高、充电和放电效率更高、在充电和放电循环中涉及的Li脱嵌容量的降低速率减小更多、且倍率性质更好的蓄电装置。
[表2]
工业实用性
根据本发明,能够提供钛酸化合物和/或碱金属钛酸盐化合物,当它们用作蓄电装置的电极活性材料时,可以提供容量能够更加得到增强并且充电和放电循环性质和倍率性质出色的蓄电装置。
附体标记说明
1:硬币型蓄电池
2:负极端子
3:负极
4:电解质,或隔离体+电解液
5:绝缘包装
6:正极
7:正极罐
- 还没有人留言评论。精彩留言会获得点赞!