用于制备石墨烯纳米带的邻三联苯的制作方法
石墨烯由二维碳层组成且具有许多突出的特性。其不仅比钻石更硬,极其耐撕裂且不透气,而且其也为优异的电和热导体。由于这些突出的特性,石墨烯在物理学、材料科学和化学中已受到相当大的关注。基于石墨烯的晶体管被视为目前使用的硅组分的潜在后继者。然而,因为石墨烯为半金属,其与硅相比缺乏电子带隙且因此不具有对电子应用至关重要的开关能力。
石墨烯纳米带(通常缩写为GNR)为来源于石墨烯晶格的具有超薄宽度的石墨烯条带。其为基于新型石墨烯的电子装置的有前景的构建单元。除导电锯齿形边缘(ZGNR)与主要半导体扶手椅边缘石墨烯纳米带(AGNR)之间的最重要区别以外,GNR几何形状的更一般的变化允许经由一维(ID)量子限制进行间隙调谐。一般而言,带宽度增加导致带隙整体降低,其中AGNR的迭加振荡特征最大化。
制备GNR的标准“自上而下(top-down)”方法,如石墨烯晶格的光刻图案化和碳纳米管的解链(unzipping)(例如描述于US 2010/0047154和US 2011/0097258中),仅得到不同GNR的混合物。另外,宽度低于10nm的纳米带的比例相当低或甚至为零。然而,对于高效率电子装置,石墨烯纳米带的宽度需要受到精确控制且优选低于10nm,且其边缘需要为光滑的,因为即使微小偏离于理想边缘形状也会严重降低电子特性。
由于此类“自上而下”方法的固有限制,实现结构上限定明确的GNR仍是困难的。通过溶液介导的脱氢环化反应(例如J.Wu,L.Gherghel,D.Watson,J.Li,Z.Wang,C.D.Simpson,U.Kolb,K.Mullen,Macromolecules 2003,36,7082-7089;L.Dossel,L.Gherghel,X.Feng,K.Mullen,Angew.Chem.Int.Ed.2011,50,2540-2543;Y.Fogel,L.Zhi,A.Rouhanipour,D.Andrienko,H.J.Rader,K.Mullen,Macromolecules 2009,42,6878-6884;以及A.Narita等人,Nature Chemistry 2014,6,126-132)和表面辅助脱氢环化反应(例如J.Cai等人,Nature 2010,470-473;S.Blankenburg等人,ACS Nano 2012,6,2020;S.Linden等人,Phys.Rev.Lett.2012,108,216801)的“自下而上(bottom-up)”化学合成方法最近已显现为用于合成GNR的有前景的途径。
与“自上而下”方法相比,基于溶液介导或表面辅助的脱氢环化反应的“自下而上”化学合成方法提供了通过使特制三维聚亚苯基前体反应来制成限定明确且均匀的GNR的机会。这些基于聚亚苯基的聚合物前体由结构可在现代合成化学能力内定制的小分子构建。
然而,所有这些“自下而上”方法迄今为止仅允许制备微量的石墨烯纳米带。此外,所得石墨烯纳米带经常由于其主链中的统计学上排布的“扭结”而没有清楚限定,或仅具有低分子量。
因此,本发明的一个目的是提供新的制备石墨烯纳米带的方法。本发明的又一目的是提供用于制备石墨烯纳米带的适合的低聚亚苯基单体和适合的聚合物前体。
所述问题通过通式(I)的邻三联苯解决:
其中
R1、R2、R3和R4独立地选自下组:H;CN;NO2;和饱和、不饱和或芳族C1-C40烃基,其可被F、Cl、OH、NH2、CN和/或NO2 1至5取代,且其中一个或多个-CH2-基团可被-O-、-NH-、-S-、-C(=O)O-、-OC(=O)-和/或-C(=O)-替代;和
X与Y相同或不同,且选自下组:F、Cl、Br、I、OTf(三氟甲磺酸酯基)。
优选地,R1、R2、R3和R4独立地选自下组:H、未被取代的C1-C40烷基基团和未被取代的C1-C40烷氧基基团。
更优选地,R1和R2独立地选自下组:H、未被取代的C1-C20烷基基团和未被取代的C1-C20烷氧基基团;且R3和R4为H。
在本申请的一个实施方案中,R1和R2为H。
在本发明的上下文中,表述“C1-C40烃基”包括所有种类的由碳原子和氢原子组成的基团。实例为直链或支化C1-C40烷基、直链或支化C2-C40烯基、直链或支化C2-C40炔基和C6-C40芳基。
当可能时,C1-C40烷基基团可为直链或支化的。实例为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、2-戊基、3-戊基、2,2-二甲基丙基、1,1,3,3-四甲基戊基、正己基、1-甲基己基、1,1,3,3,5,5-六甲基己基、正庚基、异庚基、1,1,3,3-四甲基丁基、1-甲基庚基、3-甲基庚基、正辛基、1,1,3,3-四甲基丁基和2-乙基己基、壬基、癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基、二十烷基、二十一烷基、二十二烷基、二十三烷基、二十四烷基、二十五烷基、二十六烷基、二十七烷基、二十八烷基、二十九烷基、三十烷基、三十一烷基、三十二烷基、三十三烷基、三十四烷基、三十五烷基、三十六烷基、三十七烷基、三十八烷基、三十九烷基和四十烷基。
C2-C40烯基基团为直链或支化烯基基团,例如乙烯基、烯丙基、甲代烯丙基、异丙烯基、2-丁烯基、3-丁烯基、异丁烯基、正戊-2,4-二烯基、3-甲基-丁-2-烯基、正辛-2-烯基、正十二-2-烯基、异十二烯基、正十二-2-烯基和正十八-4-烯基。
C2-40炔基基团为直链或支化的。实例为乙炔基、1-丙炔-3-基、1-丁炔-4-基、1-戊炔-5-基、2-甲基-3-丁炔-2-基、1,4-戊二炔-3-基、1,3-戊二炔-5-基、1-己炔-6-基、顺-3-甲基-2-戊烯-4-炔-1-基、反-3-甲基-2-戊烯-4-炔-1-基、1,3-己二炔-5-基、1-辛炔-8-基、1-壬炔-9-基、1-癸炔-10-基和1-二十四炔-24-基。
C6-C40芳基基团的实例为苯基、萘基、联苯基、三联苯基、芘基、芴基、菲基、蒽基、并四苯基(tetracyl)、并五苯基(pentacyl)或并六苯基(hexacyl)。
C1-C40烷氧基为直链或支化烷氧基,例如甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、戊氧基、异戊氧基、叔戊氧基、庚氧基、辛氧基、异辛氧基、壬氧基、癸氧基、十一烷氧基、十二烷氧基、十四烷氧基、十五烷氧基、十六烷氧基、十七烷氧基、十八烷氧基、十九烷氧基、二十烷氧基、二十一烷氧基、二十二烷氧基、二十三烷氧基、二十四烷氧基、二十五烷氧基、二十六烷氧基、二十七烷氧基、二十八烷氧基、二十九烷氧基、三十烷氧基、三十一烷氧基、三十二烷氧基、三十三烷氧基、三十四烷氧基、三十五烷氧基、三十六烷氧基、三十七烷氧基、三十八烷氧基、三十九烷氧基和四十烷氧基。
本发明的问题进一步通过使用通式(I)的邻三联苯制备石墨烯纳米带来解决。
因此,本发明的另一方面为一种制备石墨烯纳米带的方法,其包括以下步骤:
(a)使通式(I)的邻三联苯聚合,形成具有通式(II)重复单元的聚合物前体
其中R1、R2、R3和R4如上文所定义;和
(b)使所述聚合物前体脱氢环化,形成具有通式(III)重复单元的石墨烯纳米带
其中R1、R2、R3和R4如上文所定义。
在本发明的一个优选实施方案中,(a)聚合在溶液中进行。例如,具有通式(II)重复单元的聚合物前体可通过在二甲基甲酰胺(DMF)中或在甲苯与DMF的混合物中的Yamamoto-缩聚获得(T.Yamamoto,Progr.Polym.Sci.1992,17,1153-1205;T.Yamamoto,Bull.Chem.Soc.Jpn.1999,72,621-638;T.Yamamoto,T.Kohara,A.Yamamoto,Bull.Chem.Soc.Jpn.1981,54,1720-1726。)。用于Yamamoto-缩聚的适合催化剂可由双(环辛二烯)镍(0)、1,5-环辛二烯和2,2'-联吡啶的化学计算量混合物例如在甲苯与DMF的混合物中制备。取决于具体取代基R1和R2,聚缩合反应在50℃至110℃温度下,优选在70℃至90℃温度下进行。通过小心地将反应混合物滴入稀甲醇盐酸中实现Yamamoto-缩聚反应的淬灭和镍基团的分解。通常,形成白色沉淀,其可通过过滤来收集。其他适合的聚缩合反应依赖于例如Ullmann型偶联和Glaser型偶联。通过适合的共聚单体,邻三联苯也可应用于例如Suzuki-Miyaura型偶联、Negishi型偶联、Stille型偶联和Kumada型偶联。
在本发明的一个实施方案中,(b)脱氢环化在溶液中进行。例如,具有通式(III)重复单元的石墨烯纳米带的制备可使用路易斯酸,如氯化铁(FeCl3)、氯化钼(MoCl5)或三氟甲磺酸铜(Cu(OTf)2)在二氯甲烷与硝基甲烷的混合物中进行。或者,石墨烯纳米带的制备可使用苯基碘(III)双(三氟乙酸盐)(PIFA)和BF3醚合物在无水二氯甲烷中进行。已知当PIFA经路易斯酸活化时,其容易与宽范围的基底反应,以优异收率得到联芳基产物(Takada,T.;Arisawa,M.;Gyoten,M.;Hamada,R.;Tohma,H.;Kita,Y.J.Org.Chem.1998,63,7698-7706)。此外,其可应用于三亚苯基的合成(King,B.T.;Kroulik,J.;Robertson,C.R.;Rempala,P.;Hilton,C.L.;Korinek,J.D.;Gortari,L.M.J.Org.Chem.2007,72,2279-2288)和六迫六苯并蒄(HBC)衍生物的合成(Rempala,P.;Kroulik,J.;King,B.T.J.Org.Chem.2006,71,5067-5081)。重要的是,通过此程序排除了在施用氯化铁时经常观测到的不希望的氯化。此类类型的脱氢环化反应的适当变化可在文章“Cyclodehydrogenation in the Synthesis of Graphene-Type Molecules”(M.Kivala,D.Wu,X.Feng,C.Li,K.Mullen,Materials Science and Technology 2013,373-420)及其中所引用的文献中找到。
一般而言,通过在溶液中进行脱氢环化获得的石墨烯纳米带的分子量在1,000g/mol至1,000,000g/mol,优选20,000g/mol至200,000g/mol范围内。
在本发明的另一个优选实施方案中,(a)聚合和(b)脱氢环化在惰性表面上进行。因此,具有通式(III)重复单元的石墨烯纳米带通过在高真空条件下在此表面上直接生长来制备。藉此,首先使通式(I)的邻三联苯在升高的温度下聚合,形成具有通式(II)重复单元的聚合物前体,其接着在进一步升高的温度下反应形成具有通式(III)重复单元的石墨烯纳米带。
使用超高真空(UHV)条件的表面辅助自下而上方法已描述于J.Cai等人,Nature 466,第470-473页(2010)中和此后的少数出版物(S.Blankenburg等人,ACS Nano 2012,6,2020;S.Linden等人,Phys.Rev.Lett.2012,108,216801)中。或者,可使用WO 2014/045148 A1中所公开的表面辅助的自下而上方法。此方法具有不需要施加超高真空的优点。
在本发明的上下文中,表述“惰性表面”包括使通式(I)的邻三联苯和/或具有通式(II)重复单元的聚合物前体吸附/沉积,且随后聚合和/或脱氢环化而不与所述化合物自身不可逆地反应的所有种类的固体基底的表面。“惰性表面”可优选地充当聚合和/或脱氢环化反应的催化剂。惰性表面可为金属表面,如Au、Ag、Cu、Al、W、Ni、Pt或Pd表面,优选为Au和/或Ag表面。该表面也可为金属氧化物表面,如氧化硅、氮氧化硅、硅酸铪、氮化硅酸铪、硅酸锆、二氧化铪和二氧化锆或氧化铝、氧化铜、氧化铁。该表面也可由半导体材料,如硅、锗、砷化镓、碳化硅和二硫化钼制成。该表面也可为诸如氮化硼、氯化钠或方解石的材料。该表面可为导电、半导电或绝缘的。
在表面上的沉积可通过真空沉积(升华)方法、基于溶液的方法(如旋涂、喷涂、浸涂、印刷、电喷雾沉积)或激光诱导的解吸或转移方法进行。沉积方法也可为直接的表面至表面转移。优选地,沉积通过真空沉积方法进行。优选地,其为真空升华方法。
取决于上文所描述的表面辅助方法,反应步骤(a)和(b)中所施加的压力通常低于10-5毫巴,经常低于10-5毫巴。
优选地,通过热活化诱导步骤(a)中的聚合。然而,也可使用诱导聚合的任何其他能量输入,如辐射。活化温度取决于所用表面和通式(I)的邻三联苯的取代模式。通常,温度在100℃至300℃范围内。
任选地,步骤(a)可重复一次或数次,然后在步骤(b)中进行部分或完全脱氢环化。
如上文所示,本发明的方法的步骤(b)包括将具有通式(II)重复单元的聚合物前体至少部分地,优选完全地脱氢环化,形成具有通式(III)重复单元的石墨烯纳米带。脱氢环化反应通常在200℃至500℃范围内的温度下进行。
优选地,表面辅助方法在方法步骤(a)与(b)之间不包括任何中间步骤。步骤(a)和(b)可直接彼此跟随和/或重迭。
一般而言,通过在表面上直接生长获得的具有通式(III)重复单元的石墨烯纳米带的分子量在2,000g/mol至1,000,000g/mol,优选4,000g/mol至100,000g/mol范围内。
可通过扫描隧道显微镜(STM)技术有效研究共价键合的二维分子阵列(array)。表面受限共价键形成的实例涉及Ullmann偶联、酰亚胺化、卟啉的交联和杂环碳烯与多胺的低聚。用于在表面上直接生长石墨烯纳米带和石墨烯网状物的化学驱动程序就在最近已由Müllen组(MPI-P Mainz,Germany)和Fasel组(EMPA Dübendorf,Switzerland)建立(Bieri,M.;Treier,M.;Cai,J.;K.;Ruffieux,P.;O.,P.;Kastler,M.;Rieger,R.;Feng,X.;Müllen,K.;Fasel,R.;Chem.Commun.2009,45,6919;Bieri,M.;Nguyen,M.T.;O.;Cai,J.;Treier,M.;K.;Ruffieux,P.;Pignedoli,C.A.;Passerone,D.;Kastler,M.;Müllen,K.;Fasel,R.;J.Am.Chem.Soc.2010,132,16669;Treier,M.;Pignedoli,C.A.;Laino,T.;Rieger,R.;Müllen,K.;Passerone,D.;Fasel,R.Nature Chemistry 2011,3,61;Cai,J.;Ruffieux,P.;Jaafar,R.;Bieri,M.;Braun,T.;Blankenburg,S.;Muoth,M.;Seitsonen,A.P.;Saleh,M.;Feng,X.;Müllen,K.;Fasel,R.Nature 2010,466,470-473。)。在不受理论束缚的情况下,可由这些研究推断金属表面上的纳米带形成经由自由基路径进行。在经由超高真空(UHV)升华(10-11毫巴至10-5毫巴,优选10-10毫巴至10-7毫巴)将官能化单体沉积在表面上之后,脱卤被认为发生在通过退火至100℃至200℃的热活化之后。这产生双自由基物质,其扩散在表面上且彼此偶联,导致碳-碳键形成。这些自由基加成反应在中间热水平(100℃至300℃,优选150℃至220℃)下进行且为随后在更高温度(200℃至500℃,优选380℃至420℃)下的脱氢环化的前提条件。仅当在第一阶段期间形成足够分子量的聚合物质时,随后才将进行分子的完全石墨化,其中避免了材料由表面热解吸。
对于UHV表面辅助聚合和脱氢环化,需要刚性和平面度足够高的官能单体,这些官能单体辅助金属基底上的平面定向。此外,该方法允许石墨烯纳米带的拓扑定制,因为它们的形状通过前体单体的官能性图案和几何形状确定。单体设计中不需要对烷基链加溶,因为此表面受限程序中不涉及基于溶剂的方法。
本申请的又一方面为一种用于制备石墨烯纳米带的聚合物前体,其具有通式(II)重复单元:
其中R1、R2、R3和R4如上文所定义。
本申请的另一方面为具有通式(III)重复单元的石墨烯纳米带:
其中R1、R2、R3和R4如上文所定义。
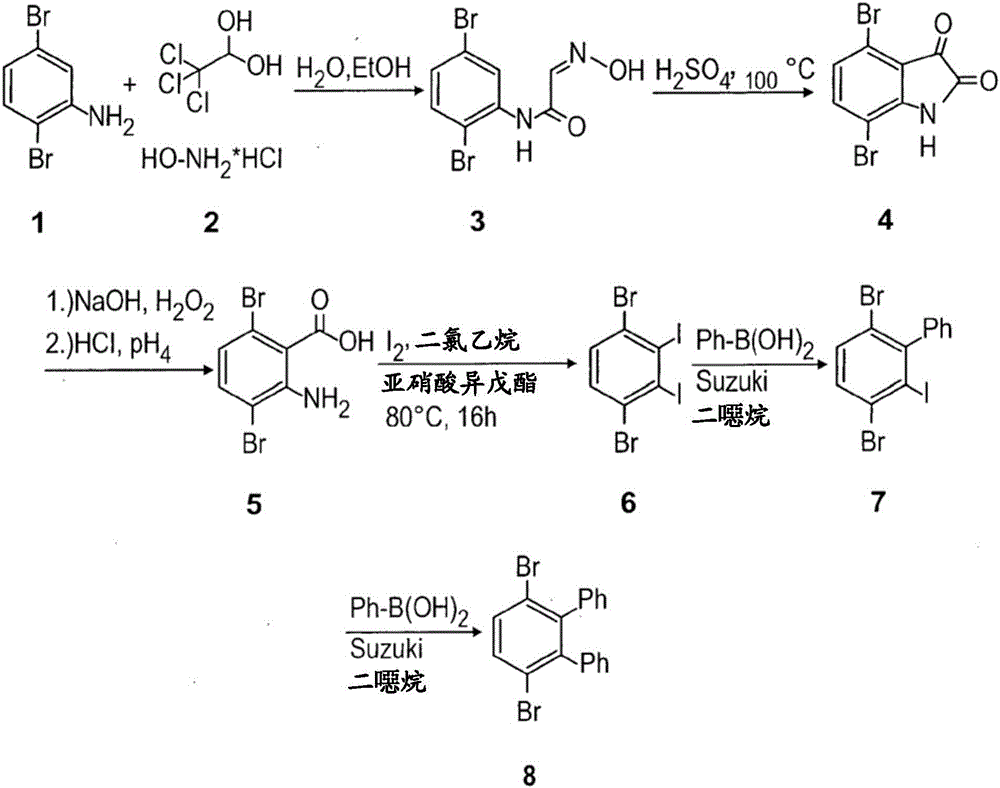
通式(I)的邻三联苯可根据以下所示的流程1至3合成。所用的反应条件和溶剂纯粹为说明性的;当然也可使用其他条件和溶剂且可易于由本领域技术人员确定。作为合成通式(I)的邻三联苯的起始材料,使用市售2,5-二卤苯胺1(流程1)。在反应序列的第一步骤中,使2,5-二卤苯胺1与水合氯醛2和盐酸羟胺在碱性条件下反应,形成(2,5-二卤苯基)-2-(羟亚胺基)乙酰胺3。
接着,使(2,5-二卤苯基)-2-(羟亚胺基)乙酰胺3在高温下经受硫酸,产生4,7-二卤吲哚啉-2,3-二酮4。
流程1
向4,7-二卤吲哚啉-2,3-二酮4和氢氧化钠在水中的溶液中添加过氧化氢水溶液,将反应混合物加热至50℃(流程2)。在冷却和酸处理之后,获得2-氨基-3,6-二卤苯甲酸5,随后使其与碘和亚硝酸异戊酯反应,产生1,4-二溴-2,3-二碘苯6。
流程2
接着,使1,4-二溴-2,3-二碘苯6进行相继的两次Suzuki偶联反应(流程3)。1,4-二溴-2,3-二碘苯6与硼酸9的第一Suzuki偶联反应可例如在催化量的四(三苯基膦)钯(0)(Pd(PPh3)4)和碱(如碳酸钠)存在下在二噁烷中在高温下进行。由此获得的单偶联联苯(IV)可进行第二Suzuki反应。通式(I)的邻三联苯可例如通过将单偶联联苯(IV)、芳基硼酸10、钯(0)催化剂和碱的反应混合物在二噁烷中加热至100℃数天来合成。在纯化之后,可使通式(I)的邻三联苯进行聚合。
流程3
利用本文所公开的石墨烯纳米带的各种制造制品,包括电子装置、光学装置和光电装置,如场效应晶体管(例如薄膜晶体管)、光伏装置、有机发光二极管(OLED)、互补金属氧化物半导体(CMOS)、互补倒相器、D触发器、整流器和环形振荡器,也在本发明的范围内,制造这些制品的方法也在本发明的范围内。
因此,本发明的另一方面为具有如上文所定义的通式(III)重复单元的石墨烯纳米带在电子、光学或光电装置中的用途。优选地,装置为有机场效应晶体管装置、有机光伏装置或有机发光二极管。
因此,本发明进一步提供制备程序限定明确的电子带隙的半导体材料的方法,电子带隙可通过选择分子前体而为特定应用进行定制。所述方法可包括:制备包含本文所公开的溶解或分散于液体介质(如溶剂或溶剂混合物)中的一种或多种本发明化合物的组合物;将该组合物沉积在基底上以提供半导体材料前体;和处理(例如加热)该半导体前体以提供包含本文所公开的一种或多种化合物的半导体材料(例如薄膜半导体)。在各种实施方案中,液体介质可为有机溶剂、无机溶剂(如水)或其组合。在一些实施方案中,组合物可进一步包含一种或多种独立地选自以下的添加剂:清洁剂、分散剂、粘合剂、相容剂、固化剂、引发剂、保湿剂、消泡剂、润湿剂、pH调节剂、杀生物剂和抑菌剂。例如,可包含表面活性剂和/或聚合物(例如聚苯乙烯、聚乙烯、聚-α-甲基苯乙烯、聚异丁烯、聚丙烯、聚甲基丙烯酸甲酯及其类似物)作为分散剂、粘合剂、相容剂和/或消泡剂。在一些实施方案中,沉积步骤可通过印刷,包括喷墨印刷和各种接触印刷技术(例如网版印刷、凹版印刷、胶印、移印、平版印刷、柔版印刷和微接触印刷)进行。在其他实施方案中,沉积步骤可通过旋涂、滴铸、区域浇铸、浸涂、刮涂、喷涂或真空过滤进行。
本发明进一步提供制造制品,如本文所描述的各种装置,其包括具有本发明的半导体材料的复合材料和基底组件和/或介电组件。基底组件可选自掺杂硅、氧化铟锡(ITO)、ITO涂布的玻璃、ITO涂布的聚酰亚胺或其他塑料、铝或单独或涂布在聚合物或其他基底上的其他金属、掺杂聚噻吩及其类似物。介电组件可由以下制备:无机介电材料,如各种氧化物(例如SiO2、Al2O3、HfO2);有机介电材料,如各种聚合物材料(例如聚碳酸酯、聚酯、聚苯乙烯、聚卤乙烯、聚丙烯酸酯);和自组装超晶格/自组装纳米介电(SAS/SAND)材料(例如描述于Yoon,M-H.等人,PNAS,102(13):4678-4682(2005)中);以及混杂有机/无机介电材料(例如描述于US2007/0181961A1中)。复合材料也可包含一个或多个电触点。用于源电极、漏电极与栅电极的适合材料包括金属(例如Au、Al、Ni、Cu)、透明导电氧化物(例如ITO、IZO、ZITO、GZO、GIO、GITO)和导电聚合物(例如聚(3,4-乙二氧基噻吩)聚(苯乙烯磺酸盐)(PEDOT:PSS)、聚苯胺(PANI)、聚吡咯(PPy))。一种或多种本文所描述的复合材料可包含各种有机电子、光学和光电装置内,如有机薄膜晶体管(OTFT),具体而言,有机场效应晶体管(OFET),以及传感器、电容器、单极电路、互补电路(例如倒相器电路)及其类似装置。
因此,本发明的又一方面为一种包含薄膜半导体的电子、光学或光电装置,所述薄膜半导体包含具有如上文所定义的通式(III)重复单元的石墨烯纳米带。优选地,装置为有机场效应晶体管装置、有机光伏装置或有机发光二极管。
本发明的石墨烯纳米带在其中有用的其他制造制品为光伏装置或太阳能电池。本发明化合物可呈现宽的光吸收和/或极正向偏移的还原电位,使得它们对此类应用而言是合意的。因此,本文所描述的化合物可在光伏设计中用作n型半导体,所述光伏设计包括形成p-n结的相邻p型半导体材料。化合物可呈薄膜半导体形式,其可沉积在基底上,形成复合材料。在此类装置中采用本发明化合物在本领域技术人员的知识范围内。
因此,本发明的另一方面涉及制造包括本发明的半导体材料的有机场效应晶体管的方法。本发明的半导体材料可用于制造各种类型的有机场效应晶体管,包括:顶栅顶接触电容器结构、顶栅底接触电容器结构、底栅顶接触电容器结构和底栅底接触电容器结构。
在某些实施方案中,可使用SiO2作为电介质,以顶接触几何形状用本发明石墨烯纳米带在掺杂硅基底上制造OTFT装置。在特定实施方案中,可在室温下或在升高的温度下沉积包括至少一种本发明化合物的活性半导体层。在其他实施方案中,可通过如本文所描述的旋涂或印刷来施加包括至少一种本发明化合物的活性半导体层。对于顶接触装置,可使用遮蔽罩、电子束光刻和剥离技术或在本领域技术人员的知识范围内的其他适合的结构化方法将金属触点图案化在膜顶部。
通过以下实施例更详细地说明本发明。
实施例
图1至图7显示:
图1:3',6'-二溴-1,1':2',1"-三联苯8(邻三联苯(I),其中R1=R2=R3=R4=H,且X=Y=Br)的合成途径。
图2:1,4-二溴-2,3-二碘苯6的1H NMR(300MHz,CD2Cl2)。
图3:1,4-二溴-2,3-二碘苯6的13C NMR(75MHz,CD2Cl2)。
图4:3',6'-二溴-1,1':2',1"-三联苯8的1H NMR(300MHz,CD2Cl2)。
图5:3',6'-二溴-1,1':2',1"-三联苯8的13C NMR(75MHz,CD2Cl2)。
图6:在Au表面上聚合和脱氢环化之后由3',6'-二溴-1,1':2',1"-三联苯8获得的9-AGNR的STM图像。
图7:显示STM图像与AGNR结构的化学模型叠加的放大。
实施例1制备(2,5-二卤苯基)-2-(羟亚胺基)乙酰胺3
如S.-J.Garden,J.-C.Torres,A.-A.Ferreira,R.-B.Silva,A.-C.Pinto,Tetrahedron Lett.1997,38,1501中所描述合成(2,5-二卤苯基)-2-(羟亚胺基)乙酰胺3。因此,在1L圆底烧瓶中放入10g(39.85mmol)2,5-二卤苯胺1、7.91g(47.82mmol)水合氯醛、4.15g(59.78mmol)盐酸羟胺和48g硫酸钠。添加300mL乙醇和300mL水并将反应混合物在80℃下搅拌12h。在冷却至室温之后,将沉淀物过滤,用乙酸乙酯与己烷(1:10)的混合物洗涤并在真空下干燥,以72%的收率获得白色固体状(2,5-二卤苯基)-2-(羟亚胺基)乙酰胺3。
1H-NMR:(300MHz,DMSO):δ=12.54(s,1H),9.51(s,1H),8.15(d,1H),7.6(m,2H),7.34(dd,1H)ppm
13C-NMR:(300MHz,DMSO):δ=160.45,143.10,136.73,134.18,129.15,126.50,120.58,114.96ppm。
实施例2制备4,7-二卤吲哚啉-2,3-二酮4
如S.-J.Garden等人,Tetrahedron Lett.1997,38,1501中所描述,将浓硫酸(45mL)在250mL圆底烧瓶中加热至50℃。添加干燥的(2,5-二卤苯基)-2-(羟亚胺基)乙酰胺3(5g,15.6mmol)并将反应混合物加热至100℃30min。将所得紫色混合物冷却至室温并倾入冰水(300mL)中,沉淀出浅橙色固体4,7-二卤吲哚啉-2,3-二酮4。将沉淀物过滤并在真空中干燥,以56%的收率获得4。
1H-NMR:(300MHz,DMSO):δ=11.43(s,1H),7.66(d,1H),7.17(d,1H)ppm
13C-NMR:(300MHz,DMSO):δ=181.08,158.94,151.06,140.64,127.86,118.36,103.68ppm。
实施例3制备2-氨基-3,6-二卤苯甲酸5
根据出版物:V.Lisowski,M.Robba,S.Rault,J.Org.Chem.2000,65,4193中所描述的合成程序合成2-氨基-3,6-二卤苯甲酸5。因此,将4,7-二卤吲哚啉-2,3-二酮4(3g,10mmol)溶解于50mL 5%氢氧化钠中并加热至50℃。逐滴添加30%过氧化氢(50mL)并将所得混合物在50℃下搅拌额外30min。在冷却至室温之后,将溶液过滤并用1M盐酸酸化至pH 4。将米色沉淀物过滤并在真空中干燥,以65%的收率获得2-氨基-3,6-二卤苯甲酸5。
1H-NMR:(300MHz,DMSO):δ=13.73(b s,1H),7.38(d,1H),6.79(d,1H),5.58(b s,1H)ppm
13C-NMR:(300MHz,DMSO):δ=167.32,144.12,134.32,121.09,118.96,107.86ppm。
实施例4制备1,4-二溴-2,3-二碘苯6
根据文章:O.S.Miljanic,K.P.C.Vollhardt,G.D.Whitener Synlett2003,29-34中公布的程序合成1,4-二溴-2,3-二碘苯6。向碘(2.58g,10.17mmol)和亚硝酸异戊酯(1.64mL,12.21mmol)在200mL 1,2-二氯乙烷中的搅拌和回流溶液中逐滴添加2-氨基-3,6-二卤苯甲酸5在15mL二噁烷中的溶液。将所得混合物回流1h,冷却至室温,过滤并用5%硫代硫酸钠水溶液洗涤滤液。有机相经硫酸镁干燥,并蒸发溶剂。通过用己烷进行快速柱层析纯化所得残余物,以60%的收率获得无色针状1,4-二溴-2,3-二碘苯6。光谱数据与文献值一致。
1H-NMR:(300MHz,CD2Cl2):δ=7.45(s,2H)ppm
13C-NMR:(300MHz,CD2Cl2):δ=133.25,128.09,117.52ppm。
实施例5制备3',6'-二溴-1,1':2',1"-三联苯8
将1,4-二溴-2,3-二碘苯6(250mg,0.5mmol)和苯基硼酸(65.63mg,0.5mmol)溶解于10mL二噁烷中并添加1mL 2M碳酸钠水溶液。使氩气鼓泡通过溶液45min,接着添加四(三苯基膦)钯(0)(60mg,0.1mol%)。使氩气鼓泡通过溶液额外15min并将反应混合物在80℃下搅拌2天。在冷却至室温之后,用水/二氯甲烷萃取溶液,有机相经硫酸镁干燥,并蒸发溶剂。通过柱层析(PE:DCM 9:1)纯化粗混合物,以60%的收率获得单偶联产物7。
将第二碘在类似Suzuki偶联反应中与额外当量的苯基硼酸偶联。将溶液在100℃下在氩气下搅拌3天。通过柱层析(PE:DCM 9:1)纯化粗反应混合物,以10%的收率获得3',6'-二溴-1,1':2',1"-三联苯8。可由乙醇再结晶无色固体。
1H-NMR:(300MHz,CD2Cl2):δ=7.49(s,2H),7.12-7.05(m,6H),6.93-6.90(m,4H)ppm
13C-NMR:(300MHz,CD2Cl2):δ=144.24,140.56,133.14,130.23,127.85,127.45,123.63ppm
FD-MS:m/z=388.0。
实施例6表面辅助制备石墨烯纳米带
将Au(111)单晶(Surface Preparation Laboratory,Netherlands)用作用于生长N=9扶手椅石墨烯纳米带(9-AGNR)的基底。首先通过重复氩离子轰击和退火至480℃的循环来清洁基底,然后冷却至室温进行沉积。通过以的速率进行升华将3',6'-1,1':2',1"-三联苯8沉积至干净表面上。然后将Au(111)基底在175℃下后退火10min以诱导聚合并在400℃下后退火10min以形成GNR。将来自Omicron Nanotechnology GmbH,Germany的低温STM(LT-STM)用于表征9-AGNR样品的形态。模型与STM图像之间的一致性证明可在Au(111)表面上由3',6'-二溴-1,1':2',1"-三联苯8合成9-AGNR(图6)。
- 还没有人留言评论。精彩留言会获得点赞!
- 过渡金属与石墨烯的复合纳米线及其制备方法与流程
- 一种氧化石墨烯表面原位生长二氧化硅的纳米杂化填料及其制备方法与流程
- GO增强CNT覆膜砂的智能混凝土无线传感器的制造方法与工艺
- 一种基于纳米纤维素晶须MoS2/石墨烯复合对电极材料及其制备方法和应用与流程
- 聚醚砜树脂/石墨烯纳米片多孔纳米复合材料、制备方法及其用途与流程
- 一种改性石墨烯与纳米纤维素复合温敏材料的制备方法与制造工艺
- 石墨烯掺杂改性纳米钙钛矿型La1‑xSrxMnO3复合材料及其制备方法和应用与制造工艺
- 一种在二氧化锰电极表面对苯胺进行原位氧化的方法与制造工艺
- 一种氧化石墨烯/银纳米颗粒/金字塔形硅三维拉曼增强基底及制备方法和应用与制造工艺
- 纳米羧基化石墨烯复合涤纶导电织物的制备方法与制造工艺
- 一种氧化石墨烯表面原位生长二氧化硅的纳米杂化填料及其制备方法与流程
- 一种基于纳米纤维素晶须MoS2/石墨烯复合对电极材料及其制备方法和应用与流程
- 一种改性石墨烯与纳米纤维素复合温敏材料的制备方法与制造工艺
- 一种石墨烯生产设备的制造方法与工艺
- 石墨烯掺杂改性纳米钙钛矿型La1‑xSrxMnO3复合材料及其制备方法和应用与制造工艺
- 一种氧化石墨烯/银纳米颗粒/金字塔形硅三维拉曼增强基底及制备方法和应用与制造工艺
- 纳米羧基化石墨烯复合涤纶导电织物的制备方法与制造工艺
- 硅烷纳米石墨烯陶化膜及其制备方法与制造工艺
- 纳米氧化石墨烯在促进小鼠胚胎干细胞培养和自我更新中的应用的制造方法与工艺
- 一种纳米石墨烯改性蓝藻基复合生物塑料及其制备方法与制造工艺
- 一种基于纳米纤维素晶须MoS2/石墨烯复合对电极材料及其制备方法和应用与流程
- 一种改性石墨烯与纳米纤维素复合温敏材料的制备方法与制造工艺
- 石墨烯掺杂改性纳米钙钛矿型La1‑xSrxMnO3复合材料及其制备方法和应用与制造工艺
- 一种多层石墨烯纳米碳管三维碳材料填充纳米硅复合材料及其制备方法与制造工艺
- 一种氧化石墨烯/银纳米颗粒/金字塔形硅三维拉曼增强基底及制备方法和应用与制造工艺
- 一种碳纳米管和石墨烯协同增强铝基复合材料及制备方法与制造工艺
- 纳米氧化石墨烯在促进小鼠胚胎干细胞培养和自我更新中的应用的制造方法与工艺
- 一种碳纳米管和石墨烯复配润滑油的制备方法与制造工艺
- 一种纳米石墨烯改性蓝藻基复合生物塑料及其制备方法与制造工艺
- 用于制备石墨烯纳米带的邻三联苯的制造方法与工艺