一种亚氧化钛歧化分解制备特定比例导电TiO负载黑色氧化钛的方法与流程
本发明涉及一种一步法负载特定比例的导电tio来提高黑色氧化钛的导电能力的方法,具体涉及一种亚氧化钛歧化分解制备特定比例导电tio负载黑色氧化钛的方法,属于合成制备领域。且,该导电tio负载黑色氧化钛,突破黑色氧化钛的半导体导电限制,可应用于电催化、光催化等领域。
背景技术:
在1972年,fujishima和honda(nature,1972,238:37-38)首次提出tio2在光解水中的应用。tio2的禁带宽度较宽,金红石相3.0ev,锐钛矿相3.2ev,是一种宽禁带半导体材料,只能吸收并利用波长380nm以下太阳光,这部分能量其实只占太阳光总体的5%。
为提高材料的光吸收效率,增强对能量更高的可见光和近红外光的吸收,研究人员开展了一系列研究。目前主要有两种趋势,一种是形成一种核壳结构,吸收光谱在可见光及红外区有一“肩高”;另一种是进行体相掺杂,使得吸收光谱向可见光区整体推移。目前人们通过缺陷调控,可以获得不同颜色的二氧化钛,可使tio2对光的吸收拓展到可见光区,使催化活性显著提高,颜色也由白色变为黄色、灰色、红色、蓝色直至黑色。
当光学吸收边接近1000nm时,禁带宽度由3.0ev降低至1.3ev,tio2也就变成黑色。2011年,陈小波等人(science,2011,331:746-750)用高压纯h2(20.0bar)处理tio2,在200℃保温5天,制备出黑色tio2,在400-800nm有一肩高状的吸收,带尾延伸到1100nm,禁带宽度达到1.5ev。2013年,黄富强研究员、汪宙博士(advancedfunctionalmaterials,2013,23:5444–5450)采用氢等离子注入的方法,制备出黑色的tio2-xhx,hrtem测试表明其为内核结晶,外壳非晶的结构,在太阳光谱能量的吸收高达83%,可见光区和红外区达到77%。相同条件下,p25仅利用了5%能量,高压h2处理的也只利用了30%。除了采用还原性气体外,黄富强研究员、汪宙博士(rscenergy&environmentalscience,2013,6,3007)从al-ti合金获得灵感,设计了双温区管式炉使tio2变为黑色的方法,一端采用800℃熔融铝获得低的氧分压,另一端300-500℃下保温促使tio2形成更多的氧空位。黄富强研究员、林天全等人(rscenergy&environmentalscience,2014,7,967)在此基础上实现了阴离子重掺杂取代氧位,研究了不同离子取代后对太阳光利用效率作用大小为:h<i<s<n,在光催化方向有较大的应用前景。除了非接触式al还原外,也可以换用al、mg、zn等活泼金属接触式地还原tio2,达到还原加掺杂的目的。这些研究发展了离子注入、高压h2还原、活泼金属双温区法以及梯度掺杂等技术,提供了氧化钛光吸收增强的新方案。
亚氧化钛是一种黑度非常好的活性电极材料,能够保护电池的两极,也可用作双极性电池材料和led黑色矩阵材料。常见的亚氧化钛有tio、ti2o3、ti3o5以及以ti4o7为代表的magneli相。不同于缺陷态的黑色氧化钛,这些亚氧化钛由于ti-o元素的重新分布,均有各自的晶体结构,在一定化学比下是可以稳定存在的。因此,可以通过设计ti-o比例,以获得相应的纯物质。需要指出的,这些低价的氧化钛,化学式均符合通式tino2n-1,可以看作是tio和tio2的组合。但从其xrd图谱中,并不会显露出这两相的谱线。因此,在结构上,亚氧化钛是独立的。
黑色氧化钛的导电性,相对于金红石及锐钛矿相,导电性有所提高,但仍属于半导体范畴。在电催化及光电化学方面,半导体性质的黑色氧化钛,不利于电子的转移。一般来说,要提高这类材料的导电性,需要加入炭黑或乙炔黑等碳材料,来辅导电子转移。
技术实现要素:
为此,本发明提供了一种亚氧化钛歧化分解制备特定比例导电tio负载黑色氧化钛的方法,将亚氧化钛和卤化铵混合后,在500~700℃下进行热处理,使亚氧化钛发生歧化分解,得到导电tio负载黑色氧化钛;所述亚氧化钛的化学式为tino2n-1,n≥2。需要说明是,以下所说的比例,均是按物质的量进行计算的,为摩尔比。
在本公开中,将常温稳定的亚氧化钛,置于卤化铵气氛使其发生歧化反应生成tio和tio2双相结构(参见图8)。从xrd图谱,可标定产物为立方相的tio和金红石相的tio2(例如,参见图3)。卤化铵可以辅助亚氧化钛发生歧化分解反应,中间价态向两边散开。这归因于卤化铵产生的气氛抑制了归中反应,同时形成了特定核壳结构。而且,低价氧化钛tio具有良好的导电性,接近金属,可以用来改善黑色氧化钛的导电性,而不引入其他杂质元素。
较佳地,所述亚氧化钛选自ti2o3、ti3o5和ti4o7中至少一种。不同的亚氧化钛tino2n-1为起始原料,所制备的tio负载黑色氧化钛产物,满足摩尔比为tio:tio2=1:(n-1)。也就是说,当本发明的方法所选取的亚氧化钛为ti2o3时,所制备的导电tio负载黑色氧化钛为摩尔比tio:tio2=1:1的歧化产物;当本发明的方法所选取的亚氧化钛为ti3o5时,所制备的导电tio负载黑色氧化钛为摩尔比tio:tio2=1:2的歧化产物;当本发明的方法所选取的亚氧化钛为ti4o7时,所制备的导电tio负载黑色氧化钛为摩尔比tio:tio2=1:3的歧化产物。
关于所述亚氧化钛,可利用还原剂还原二氧化钛以制得。按照所需制备的亚氧化钛的化学式,将按摩尔比略过量的还原剂和二氧化钛混合,置于真空容器中,然后在800~1100℃下保温4~10小时,得到所述亚氧化钛。适当提高还原剂的比例可以确保生成物的纯度。适度过量2~10mol%为优选。可选地,称取摩尔比(还原剂:二氧化钛)略适度大于1:1的物料,可合纯一氧化钛。称取摩尔比(还原剂:二氧化钛)略适度大于1:3的物料,可合纯三氧化二钛。称取摩尔比(还原剂:二氧化钛)略适度大于1:5的物料,可合纯五氧化三钛。称取摩尔比(还原剂:二氧化钛)略适度大于1:1的物料,可合纯七氧化四钛。可选地,合成纯的ti2o3、ti3o5是在800~850℃;合成纯的ti4o7是在820~880℃,合成纯的tio是在1000~1100℃。合成温度可以适当降低,并相应增加保温时间。
在所述亚氧化钛的制备中,所述还原剂优选为钛粉、镁粉、铁粉、铝粉、c、h2中的至少一种;所制得的亚氧化钛为纯相,并且其本身是干燥的。较佳的,选用钛粉,免除后续洗涤过程、干燥过程和热处理过程。
在本发明的导电tio负载黑色氧化钛的制备方法,较佳地,所述卤化铵选自氟化铵、氯化铵、溴化铵、碘化铵中的至少一种。
较佳地,所述卤化铵nh4x(其中x为f离子、氯离子、碘离子、溴离子等)和亚氧化钛的摩尔比为(0.1~1):1,更优选为(0.5~1):1(按比值的百分数记作50~100mol%)。当卤化铵和亚氧化钛的摩尔比小于0.5,亚氧化钛会部分残留。
较佳地,所述热处理的气氛为真空气氛、或惰性气氛;所述惰性气氛为氩气气氛。
较佳地,所述热处理的时间为0.5~8小时。
另一方面,本发明还提供了一种根据上述的方法制备的导电tio负载黑色氧化钛,所述tio负载黑色氧化钛具有核壳结构,其核为黑色的氧化钛,壳为导电的tio。所得,导电tio负载黑色氧化钛中tio和黑色tio2的摩尔比为1:(n-1)。
有益效果:
本发明首次发现了一个亚氧化钛的歧化反应,生成物为特定比例的立方相tio和金红石相tio2,这两相都是高温稳定相。而且,卤化铵(例如nh4cl)高温分解的氢卤酸在这个歧化反应起主要作用,刻蚀氧化物为钛的氯化物,结合中间产物水,生成相应的氧化物。本发明进一步测试了所得产物的导电性,接近金属,而主体为黑色的氧化钛。
附图说明
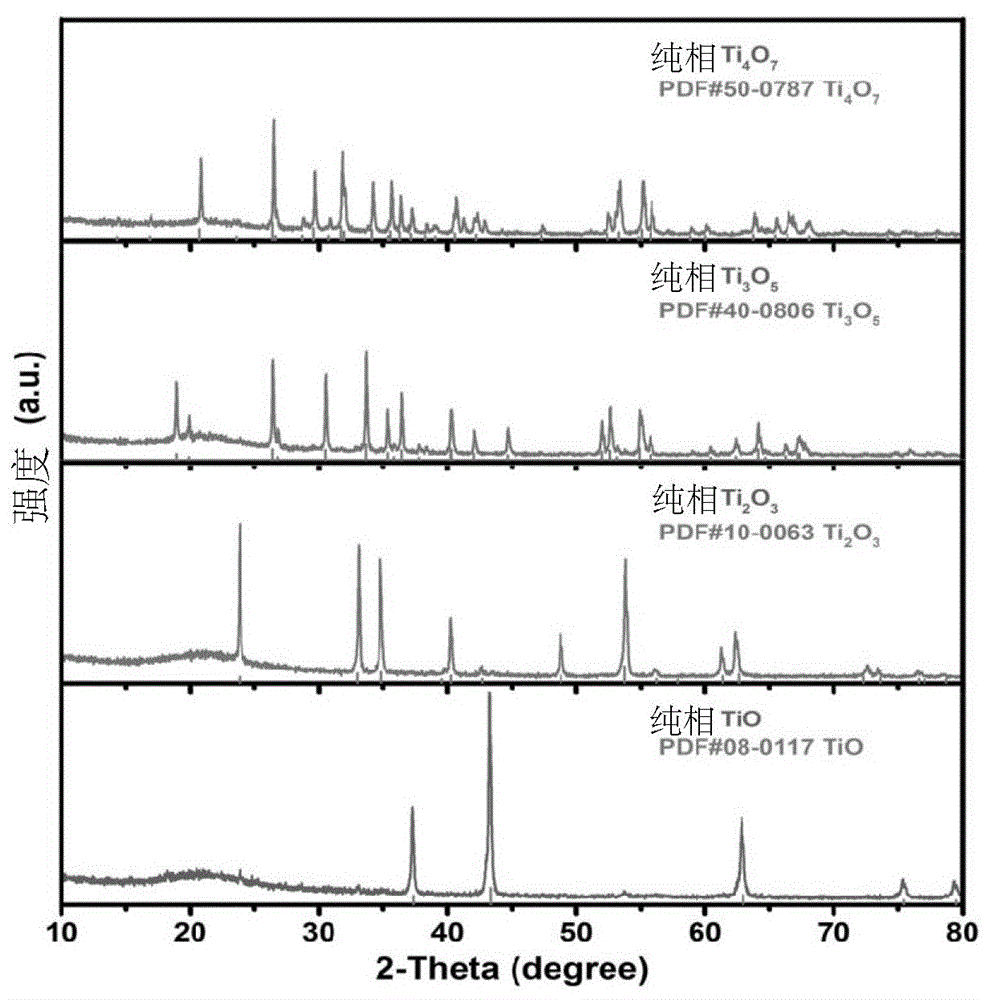
图1为实施例1-4中分别制备的亚氧化钛纯相的xrd图谱;
图2为实施例2-4中反应前的亚氧化钛和氯化铵原料xrd图谱;
图3为实施例2-4中反应后不同亚氧化钛对应的生成物xrd图谱(nh4cl:tino2n-1=0.5:1);
图4为tio与rutile(金红石相)含量关系的标准曲线;
图5为不同亚氧化钛歧化产物的含量标定图;
图6为实施例2中ti2o3歧化产物的tem表征图;
图7为实施例3中ti3o5歧化产物压片后测的i-v测试图;
图8为本专利中歧化反应所得产物的组成示意图;
图9为补充的对比样的xrd实验结果(以ti4o7为例);
图10为补充的反应时间的xrd实验结果(以ti4o7为例)。
具体实施方式
以下通过下述实施方式进一步说明本发明,应理解,下述实施方式仅用于说明本发明,而非限制本发明。
在本公开中,首次利用亚氧化钛的歧化分解制备特定比例的导电tio负载黑色氧化钛,实现了一步法合成特定摩尔比例的tio和tio2的目的。而且,导电tio的存在,可提高黑色tio2的导电性。
以下示例性地说明本发明提供的亚氧化钛歧化分解制备特定比例导电tio负载黑色氧化钛的方法。
亚氧化钛的制备。通过混合特定比例的还原剂(例如,金属钛粉等)与白色金红石相tio2,于封闭真空容器中,经800~1100℃高温反应获得化学式为tio、ti2o3、ti3o5、ti4o7的亚氧化钛作为原料。具体来说,根据计算的配料比例,称取相应量的金属钛粉和二氧化钛粉体,玛瑙烟研钵中混合均匀,置于玻璃管中。采用真空封管的方法,制成封闭容器。然后在马弗炉中,于800-1100℃保温4-10h,获得理想的原料。应注意,在还原剂的选择上还有很多,例如镁粉、铁粉、铝粉、c或还原性气体(氢气等)等。为减少杂质的存在以及避免洗涤过程,因此优选选用以商品化的钛粉及二氧化钛作为原料,来合成原料。如若通过其他金属或c、h2反应获得,还需要后续进行洗涤干燥以除去水;并且产物纯相与否,不影响后续与卤化铵晶体的反应。根据计算的钛粉和二氧化钛比例,适当提高钛粉的比例(还原剂及二氧化钛的比例需依据化学反应式,且还原剂适当过量2-10mol%),于封闭容器中,可制备出相应的纯相亚氧化钛。作为一个示例,本公开方法合纯的ti2o3和ti3o5是在800℃,合纯的ti4o7是在850℃,合纯的tio是在1100℃。温度可以适当降低,相应增加保温时间即可。此外,为达到缩短反应时间、提高生成物纯度、控制生成物尺寸的目的,需确保反应物干燥且选用的原料为微米纳米尺寸。
在可选的实施方式中,采用真空玻璃管作为封闭容器,真空度低于0.1pa。按摩尔比称取一定量钛粉和二氧化钛,为确保生成物的纯度,适当提高还原剂钛粉的比例,适度过量2-10mol%。可选地,称取1份钛粉和1份二氧化钛,可合纯一氧化钛;称取1份钛粉和3份二氧化钛,可合纯三氧化二钛;称取1份钛粉和5份二氧化钛,可合纯五氧化三钛;称取1份钛粉和7份二氧化钛,可合纯七氧化四钛。
将亚氧化钛和卤化铵混合后,在500~700℃下进行热处理一定时间,得到导电tio负载黑色氧化钛。其中,卤化铵选自氟化铵、氯化铵、碘化铵中的至少一种。卤化铵和亚氧化钛的摩尔比选用nh4x:tino2n-1=(0.1~1):1,优选为nh4x:tino2n-1=(0.5~1):1。热处理的气氛可为真空气氛、或惰性气氛;所述惰性气氛为氩气气氛。热处理的时间可为0.5~8小时(见附图10)。本发明制备的歧化产物即使不经过洗涤,该试样的残余量仍在xrd检测限内。最后可用去离子水洗涤产物,可去除试样中残留的卤化铵。作为一个详细的示例,在玛瑙研钵中研碎摩尔比为2:1的亚氧化钛(ti2o3、ti3o5、ti4o7)和氯化铵混合物,于封闭真空管中,经500~700℃高温处理一段时间,即可获得亚稳态的tio和tio2生成物,且摩尔比分别为1:1、1:2、1:3。应注意,选用的真空容器(例如,真空管等),能耐受400~1100℃温度范围;选用尽可能低的真空度,便于计算获取纯的亚氧化钛应盈留的还原剂比例。
进一步优选,选取卤化铵中的氯化铵实施本发明。一是因为氟化铵产生的氢氟酸在高温会严重腐蚀管壁,会破坏密封容器的真空度;二是因为溴化铵和碘化铵,副产物会有颜色较深的单质溴和碘出现,造成洗涤步骤必不可少;三是氯化铵的分解产物均为无色物质,凝结的氯化铵粉体较轻,可用扇风的方式有效去除,可省去洗涤步骤。以氯化铵为亚氧化钛歧化反应的催化剂,混合不同比例的氯化铵和亚氧化钛,可以获得不同歧化程度的产物;当氯化铵比例较高时,可确保歧化反应完全。需要注意的是,氯化铵的量需要控制,以免分解的气体让封闭容器爆破。
应注意,对于热处理采用的升温速率、降温速率不影响歧化产物的生成。其中,降温过程可选用淬火、空冷或随炉冷却等。
此外,本发明为促进该反应的实际应用,实验补充了升温制度对这一歧化过程的影响。结果表明,升温速率快慢不影响反应;降温过程,不论是淬火、空气冷却还是随炉冷却,同样不影响歧化反应(附图10中,即快放快出的产物,在不同时间下的实验结果)。如ti4o7,反应温度在600℃,反应时间在30分钟,即可歧化完全(附图10)。
下面进一步例举实施例以详细说明本发明。同样应理解,以下实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,本领域的技术人员根据本发明的上述内容作出的一些非本质的改进和调整均属于本发明的保护范围。下述示例具体的工艺参数等也仅是合适范围中的一个示例,即本领域技术人员可以通过本文的说明做合适的范围内选择,而并非要限定于下文示例的具体数值。
实施例1:合成纯相tio,建立标准曲线
称取1g二氧化钛和0.6g钛粉,于研钵中研磨均匀,置于玻璃管中。待真空封管后,置于马弗炉中,120min升温到1100℃,保温6h。经xrd测试,随炉冷却样为单斜相tio,淬火样为立方相tio。分别称取设置质量比例的立方相tio与rutile原料,混合均匀,测试xrd,读取tio2(110)与tio(200)的衍射峰面积比值,并建立物质的量比值间的关系。混合样分别编号为s1(0.09gtio2+0.01gtio),s2(0.08gtio2+0.02gtio),s3(0.07gtio2+0.03gtio),s4(0.06gtio2+0.04gtio)和s5(0.05gtio2+0.05gtio)。建立的标准曲线,如图4所示。
实施例2:合成纯相ti2o3,并开展歧化反应
称取1g二氧化钛和0.21-0.3g钛粉,于研钵中研磨均匀,置于玻璃管中。待真空封管后,置于马弗炉中,120min升温到800℃,保温6h,随炉冷却后取出。经xrd测试,原料为纯相ti2o3。分别称取该纯相ti2o30.1g和氯化铵0.0372g(100mol%)、0.0277g(75mol%)、0.0186g(50mol%)、0.0093g(25mol%)、0.0037g(10mol%),研钵中研磨均匀,置于玻璃管中。待真空封管后,置于马弗炉中,120min升温到600℃,保温6h,随炉冷却后取出。生成物经xrd测试及标准曲线标定,确定各生成物的含量。本实施例中,当氯化铵和纯相tio和的摩尔比为50mol%以上时,只检测到tio与tio2共存的情形,如图3。
实施例3:合成纯相ti3o5,并开展歧化反应
称取1g二氧化钛和0.12-0.15g钛粉,于研钵中研磨均匀,置于玻璃管中。待真空封管后,置于马弗炉中,120min升温到800℃,保温6h,随炉冷却后取出。经xrd测试,原料为纯相ti3o5。分别称取该纯相ti3o50.1g和氯化铵0.0238g(100mol%)、0.0179g(75mol%)、0.0119g(50mol%)、0.0060g(25mol%)、0.0024g(10mol%),研钵中研磨均匀,置于玻璃管中。待真空封管后,置于马弗炉中,120min升温到600℃,保温6h,随炉冷却后取出。生成物经xrd测试及标准曲线标定,确定各生成物的含量。本实施例中,当氯化铵和纯相tio和的摩尔比为50mol%以上时,只检测到tio与tio2共存的情形,如图3。
实施例4:合成纯相ti4o7,并开展歧化反应
称取1g二氧化钛和0.085-0.095g钛粉,于研钵中研磨均匀,置于玻璃管中。待真空封管后,置于马弗炉中,120min升温到850℃,保温6h,随炉冷却后取出。经xrd测试,原料为纯相ti4o7。分别称取该纯相ti4o70.2g和氯化铵0.0352g(100mol%)、0.0264g(75mol%)、0.0176g(50mol%)、0.0088g(25mol%)、0.0035g(10mol%),研钵中研磨均匀,置于玻璃管中。待真空封管后,置于马弗炉中,120min升温到600℃,保温6h,随炉冷却后取出。生成物经xrd测试及标准曲线标定,确定各生成物的含量。本实施例中,当氯化铵和纯相tio和的摩尔比为50mol%以上时,只检测到tio与tio2共存的情形,如图3所示。
对比示例
a)将0.1g原料ti4o7置于玻璃管中,加入mgcl2·6h2o(摩尔比mgcl2·6h2o:ti4o7=0.5:1,记作50mol%mgcl2·6h2o);待真空封管后,置于马弗炉中,120min升温到600℃,保温6h,随炉冷却后取出;
b)将0.1g原料ti4o7置于玻璃管中,加入fe(oh)3(摩尔比fe(oh)3:ti4o7=0.5:1,记作50mol%fe(oh)3);待真空封管后,置于马弗炉中,120min升温到600℃,保温6h,随炉冷却后取出;
c)将1g原料ti4o7置于管式炉中,在大流量氨气气氛下,120min升温到600℃,保温6h,随炉冷却后取出;
d)将0.1g原料ti4o7置于玻璃管中,加入nacl(摩尔比nacl:ti4o7=1:1,记作100mol%nacl);待真空封管后,置于马弗炉中,120min升温到800℃,保温6h,随炉冷却后取出;e)将0.1g原料ti4o7置于玻璃管中,加入kcl(摩尔比kcl:ti4o7=1:1,记作100mol%kcl);待真空封管后,置于马弗炉中,120min升温到800℃,保温6h,随炉冷却后取出;
f)从xrd结果(图9)可以看出,对比样a和对比样b,分解产生的水汽,将亚氧化钛全部氧化为rutile相的tio2,并未有tio生成;对比样c,亚氧化钛在高浓度的nh3气中,主相仍为ti4o7原料,部分转化为ruitle相的tio2,并也未检测出有tio生成;对比样d和对比样e,反应温度即使上升到800℃,也未有反应发生。
为弄清反应的机理,分析了反应过程可能的物质。a)考虑到氯化铵在高温分解反应为氨气和氯化氢,猜想是否因为有还原性氨气。对此,在管式炉中,通入大流量氨气,结果并未出现tio的峰位。b)又考虑到有氯化氢气体,会刻蚀亚氧化钛,同时生成水蒸汽,因此补充做了水的反应,xrd发现全部转化为rutile相。c)考虑到ticl4在600℃为气态,很可能氯离子趋向于结合钛。实验补充了kcl和nacl与ti4o7的反应,达到熔融状态,也并未反应,因此,排除氯离子的作用,焦点集中在氯化氢上。d)为证明氢氯酸对亚氧化钛起刻蚀,促进了这一歧化过程,选用了其他卤化铵与ti4o7进一步实施本发明。采用氟化铵、碘化铵进行上述相同实验,结果相同。根据上述实验可知,ti4o7可在卤化铵作用下,生成立方相tio和金红石相tio2。同样,ti2o3、ti3o5可在卤化铵作用下,生成立方相tio和金红石相tio2。
图3为实施例2-4中反应后不同亚氧化钛对应的生成物xrd图谱,从图中可知,产物由tio和tio2两相构成。分析产物中两相的峰面积,可以看到不同反应物对应的产物的含量是不一样的;
图4为tio与rutile(金红石相)物相含量与xrd峰面积比值关系的标准曲线,从图中可知,根据xrd特定晶面的衍射峰面积比值与物质的量的比值线性相关,r=0.99。其中特定晶面分别选取tio(200)和tio2(110)晶面;
图5为不同亚氧化钛歧化产物的含量标定图,从图中可以看出理论标定值与实验测定值一致。利用图4的标准曲线,标定了ti4o7生成的生成物比例为tio:tio2=1:3。这一结果,同样在ti2o3和ti3o5中得到体现。例如,ti2o3生成的生成物比例为tio:tio2=1:1,ti3o5生成的生成物比例为tio:tio2=1:2;
图6为实施例2中ti2o3歧化产物(纯相ti2o30.1g和氯化铵0.0186g(50mol%))的tem表征图,从图中可知产物为核壳结构;从saed图可以看出,内核为rutile相,外壳由rutile和tio(壳层尺寸小于光斑尺寸,因此为两相);
图7为实施例3中ti3o5歧化产物(纯相ti3o50.1g和氯化铵0.0119g(50mol%)压片后测的i-v测试图,从图中可知电阻6.42ω,为导体,区别于半导体的黑色氧化钛;
图8为本专利的歧化所得产物的组成示意图,从图中可知在卤化铵(nh4x)作用下,亚氧化钛(tino2n-1)最终分解为特定比例的tio和tio2,并且产物具有核壳结构;
图9为对比样的xrd图,由图可知,这些对比样均未将ti4o7转变成tio和tio2两相,即没有发生歧化。其中50mol%mgcl2·6h2o和50mol%fe(oh)3加热过程生成水,排除了水作为歧化反应发生的可能;nh3中气氛处理的试样未发生歧化,排除了卤化铵分解产生的nh3气作为反应主因的可能;100mol%nacl和100mol%kcl,不会使ti4o7发生反应,排除了氯离子(cl-)的作用。因此,是卤化铵(nh4x)分解产生的hx,使歧化反应发生。
- 还没有人留言评论。精彩留言会获得点赞!