一种ZrC/SiC复相陶瓷前驱体及其制备方法与流程
本发明属于复相材料领域,具体地说,涉及一种zrc/sic复相陶瓷前驱体及其制备方法。
背景技术:
碳化物陶瓷具有高熔点,高强度、高硬度的特点,并且拥有良好的力学性能、化学稳定性和耐氧化性。其中以zr、hf、ta、nb的碳化物性能更为突出,集中了大量的研究。碳化物在有氧环境中会转化为相应致密的氧化物层附着在基体表面阻止氧气的进一步侵蚀和气流冲刷,可以达到对基体在极端环境中的有效保护。然而碳化物具有陶瓷固有脆性大的缺点,需要对其韧性进行改性。碳纤维作为唯一可耐3000℃高温的纤维增强体,可通过缠绕编织等手段制备预制件,结合碳化物陶瓷制备陶瓷基复合材料,达到高强、高韧、耐高温、抗氧化的目的。
sic或zrc陶瓷作为传统的碳化物陶瓷材料应用广泛,sic陶瓷抗氧化温度上限为1700℃,高于此温度时会发生主动氧化形成气态sio导致材料表面烧蚀率急剧增加;且环境压力的降低,水汽的侵蚀都会加剧氧化速率;zrc陶瓷高温有氧环境下产物为zro2,zro2具有更高的熔点(2700℃),较低的氧扩散系数和热导率,可以有效的阻止外界氧和水汽对材料的进一步侵蚀,但是zrc陶瓷存在抗热震性能差,热膨胀系数的的问题,高温环境下会对材料本身造成缺陷和裂纹,使其力学性能下降。研究表明,掺杂一定量sic的zrc复相陶瓷基复合材料可以有效的弥补zrc材料的缺陷,并具有更高的抗氧化耐温等级。
目前制备纤维增强陶瓷基复合材料的方法主要有前驱体浸渍裂解法(pip),化学气相沉积法(cvi),反应熔渗法(rmi)和泥浆法(si),其中采用pip法制备的陶瓷基复合材料具有以下优点:①可通过前驱体分子的设计实现对复合材料陶瓷基体组成、微结构和性能的控制;②工艺简单、成型温度低、裂解温度低;③对纤维的机械损伤和热损伤小,对纤维增强体的适应性好;④能获得成分均匀、纯度高的单组元或多组元的陶瓷基体;⑤可制备大型形状复杂的近尺寸的复合材料零部件。然而目前,工业化制备碳化锆(zrc)的主要方法是固体碳和金属氧化物粉末进行碳热还原反应,这种方法需要的反应温度较高,通常在1900~2300℃,生产周期较长,且制备的zrc粉体粒径比较大,烧结活性差,与c/c和含硅前驱体复合时,难以混匀,易与炭材料存在明显界面,产生应力集中,造成产品开裂而影响材料的烧蚀性能。因此制备与硅源稳定相容的zrc前驱体变得至关重要,吸引了该领域的许多关注和研究。溶胶-凝胶法是制备zrc陶瓷粉体的主要方法,但是在作为pip法的前驱体使用时,存在有效浓度低、稳定性差、不易储存的缺点,且高温陶瓷产率低,使得制备周期长,成本高,不适用于制备大尺寸陶瓷基复合材料构件;另外该方法同硅源的相容性较差,较难实现复相陶瓷的制备。聚合物前驱体法通过含锆小分子聚合形成zr-c分子主链,或zr-o分子主链聚合物复配碳源的方法,获得稳定的zrc陶瓷前驱体,稳定的液相前驱体同多种硅源相容性良好,可设计性强。
中国专利cn105218099a公开了一种非极性碳化锆液相前驱体的制备方法,涉及将锆醇盐在乙酰丙酮的配位保护下进行可控水解制备非极性聚锆氧烷,随后与碳源二乙烯基苯复配制成。该体系在实际使用过程中存在以下问题:(1)由于反应程度不够,聚锆氧烷分子中残留的zr-or及zr-oh的含量相对较高,导致碳化锆前驱体稳定性不足,在实际使用过程中与空气中的水接触易发生凝胶,贮存期只有1个月。一般复合材料浸渍需要至少数十轮次,周期长达数月,碳化锆前驱体稳定性不足会导致浸渍重复性和稳定性下降,同时增加了前驱体的损耗和复合材料制备的成本。(2)使用的碳源二乙烯基苯为小分子,与聚锆氧烷不发生反应,因此在二者共固化时,二乙烯基苯易挥发导致碳源流失,为了保证碳源充足,需要在密闭的高压容器中固化。随之带来以下劣势:一方面复合材料制件的尺寸通常较大,因此需要的对应尺寸的高压反应容器,大大提高了制造成本;另一方面,固化反应在高的压力下存在易爆等安全风险,从而限制了碳化锆前驱体的工程化应用。
有鉴于此,特提出本发明。
技术实现要素:
本发明要解决的技术问题在于克服现有技术的不足,提供一种zrc/sic复相陶瓷前驱体及其制备方法,采用改性酚醛作为碳源,并且在制备方法中采用高温预聚的方式制备非极性锆源,并在制备zrc过程中使非极性锆源与改性酚醛在一定温度下反应,有效克服了zrc前驱体稳定性不足,易与空气中的水接触易发生凝胶的技术问题。
为解决上述技术问题,本发明采用技术方案的基本构思是:
本发明提供了一种zrc/sic复相陶瓷前驱体,所述前驱体以非极性聚锆氧烷为锆源,改性酚醛为碳源,聚碳硅烷为硅源反应制备而成;所述改性酚醛中的部分羟基被醚化,且含有碳碳不饱和官能团。
上述方案中,改性酚醛分子链中的羟基被醚化,使不饱和官能团与氧原子相连,而所述不饱和官能团为烯基和/或炔基,具有较好的反应活性,可在复相材料的形成过程中,使改性酚醛中的烯键和/或炔键与聚锆氧烷和聚碳硅烷之间发生交联反应,进一步将碳源固定在复相材料中,使得zr、si和c均匀分布在复相陶瓷前驱体中,提高了前驱体陶瓷的产率。
本发明的进一步方案为:所述改性酚醛中羟基的醚化率为80~99%,优选为85~95%。
上述方案中,改性酚醛中的部分羟基被醚化,其醚化率较高,即代表碳源中可与聚锆氧烷中分子链交联的碳碳不饱和基团含量较高,提高了交联密度,避免碳源在后续高温处理中发生流失。而调低优选醚化率的最大值,则是考虑使碳源中少量未经醚化的酚羟基与聚锆氧烷反应以进一步去除聚锆氧烷中的活性基团,提高前驱体的稳定性。
本发明的进一步方案为:所述复相陶瓷前驱体中,锆元素与碳元素的摩尔比为1:1~3,优选为1:1~2;锆元素与硅元素的摩尔比为1:0.1~15,优选为1:0.3~6。
本发明的进一步方案为:所述非极性聚锆氧烷中zr-or的摩尔含量为0~3%,r为氢基和/或烷基。
本发明的进一步方案为:所述复相陶瓷前驱体在25℃下放置12个月后,粘度的变化率不大于10%。
本发明还提供了一种如上所述的zrc/sic复相陶瓷前驱体的制备方法,所述制备方法在在对锆醇盐进行配位保护和水解缩聚后,进行高温预聚制得非极性聚锆氧烷;之后将制得的非极性聚锆氧烷与改性酚醛混合在一定温度下反应后制得zrc前驱体。
上述制备方法中,在非极性聚锆氧烷制备步骤中增加了高温预聚步骤,此步骤使得原方法中大部分活性的zr-or和zr-oh基团在高温下缩合,减少前驱体树脂的活性基团,增加其使用过程中的稳定性。此外,增加了非极性聚锆氧烷与改性酚醛碳源进行反应的步骤,由于碳源中的的部分羟基被醚化,且含有碳碳不饱和基团,可使得碳源中少量未经醚化的酚羟基与聚锆氧烷的反应,进一步去除前驱体溶液中的活性基团;与此同时,碳源通过化学反应与聚锆氧烷分子链相交联,避免后续高温处理过程中碳源的流失;而碳碳不饱和键的引入则增加了碳源的交联密度,提高其碳保留率,从而提高了陶瓷产率。
根据上述制备方法,制备非极性聚锆氧烷包括如下步骤:
(1)配位保护:在25~100℃的温度下,将一定量的络合剂加入到锆醇盐的一元醇溶液中,反应0.5~5h;
(2)水解缩聚:在25~100℃的温度下,在步骤(1)反应后的溶液中再加入一定量的水和一元醇的混合液,进行水解缩聚;
(3)高温预聚:对步骤(2)水解缩聚后的溶液加热,使其在100~180℃温度下预聚0.5~5h,之后加入非极性溶剂得到非极性聚锆氧烷;
其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:0.5~6:0.5~2:6~20;优选的,步骤(1)和(2)中的反应温度为60~95℃,步骤(3)中的预聚温度为120~150℃。
根据上述制备方法,制备zrc前驱体包括将非极性聚锆氧烷与改性酚醛混合在30~130℃下反应0.5~5h;优选的,非极性聚锆氧烷与改性酚醛混合在50~80℃下反应2~4h。
根据上述制备方法,所述制备方法还包括将聚碳硅烷在非极性溶剂中溶解并搅拌0.5~6h制得sic前驱体溶液,并将sic前驱体溶液与所述zrc前驱体按一定比例混合后在30~130℃下反应0.5~5h后进行固化;优选的,sic前驱体溶液与所述zrc前驱体按一定比例混合后在50~80℃下反应2~4h。
上述制备方法中,sic前驱体与zrc前驱体在一定温度下进行反应固化可进一步增加碳源与硅源的交联程度。
根据上述制备方法,所述固化包括在110~300℃条件下进行4~24h的常压固化,之后在1500~1800℃的惰性气氛下裂解制得复相陶瓷前驱体;所述固化过程包括:在120℃下固化2~3h,之后在160℃下固化2~3h,之后在180℃下固化2~3h,之后在220℃下固化2~3h,最后在250℃下固化2h制得;或者,在120℃下固化1~2h,之后在140℃下固化1~2h,之后在160℃下固化1~2h,之后在180℃下固化1~2h,之后在200℃下固化1~2h,之后在220℃下固化1~2h,最后在250℃下固化2~4h制得。
上述制备方法中,所述裂解温度优选为1550~1700℃。
根据上述制备方法,所述络合剂为乙酰丙酮和/或乙酰乙酸乙酯。
根据上述制备方法,所述的一元醇为异丙醇,正丙醇,正丁醇,乙二醇甲醚,乙二醇乙醚中的一种或几种。
根据上述制备方法,所述的非极性溶剂为正己烷,正庚烷,甲苯,二甲苯中的一种或几种。
根据上述制备方法,所述制备方法具体包括如下步骤:
(1)在25~100℃的温度下,将一定量的络合剂加入到锆醇盐的一元醇溶液中,反应0.5~5h;之后在25~100℃的温度下,将反应后的溶液中再加入一定量的水和一元醇的混合液,进行水解缩聚;其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:0.5~6:0.5~2:6~20;
(2)对步骤(1)中水解缩聚后的溶液中加入一定量的非极性溶剂,加热使其在100~180℃温度下预聚0.5~5h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷以一定比例与改性酚醛树脂混合并在30~130℃下反应0.5~5h,得到zrc前驱体;
(4)将聚碳硅烷溶解于非极性溶剂中,搅拌0.5~6h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按一定比例混合后搅拌1~6h混合,并在30~130℃下反应0.5~5h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在110~300℃条件下进行4~24h的常压固化,之后在1500~1800℃的惰性气氛下裂解制得复相陶瓷前驱体。
采用上述技术方案后,本发明与现有技术相比具有以下有益效果:
1.本发明采用锆醇盐络合水解的简单反应制备出非极性聚锆氧烷,通过增加高温预聚以及和碳源反应的手段减少前驱体中的活性基团,极大的增加了前驱体的稳定性,室温保存1年后粘度变化不超过10%;
2.本发明选用的碳源为部分羟基被醚化的酚醛树脂,具有较高的碳保留率,且具有结构稳定的特点,提高了前驱体陶瓷的产率;
3.使用本发明提供的制备方法制得的前驱体化学性质稳定,陶瓷产率高,粘度可调范围广,可溶于多种有机溶剂,且具备树脂的特性,加工性优良,克服了传统无机法工艺性差的缺点;同时,硅源聚碳硅烷pcs的加入比例可根据需求任意调节;适用于前驱体浸渍裂解,等离子喷涂等制备工艺,在制备涂层及陶瓷纤维方面也具有潜在的应用。
4.本发明的制备方法中,热解温度较低,前驱体在1600℃即转化为zrc/sic复相陶瓷;低温能够降低工艺过程的能耗,减小对基体的高温损伤及制件中的热应力,减少烧结中的变形从而提高了成品中的合格率等,同时制备的陶瓷晶粒较小,颗粒尺寸均匀,可以防止涂层的高温性能下降。
下面结合附图对本发明的具体实施方式作进一步详细的描述。
附图说明
附图作为本发明的一部分,用来提供对本发明的进一步的理解,本发明的示意性实施例及其说明用于解释本发明,但不构成对本发明的不当限定。显然,下面描述中的附图仅仅是一些实施例,对于本领域普通技术人员来说,在不付出创造性劳动的前提下,还可以根据这些附图获得其他附图。在附图中:
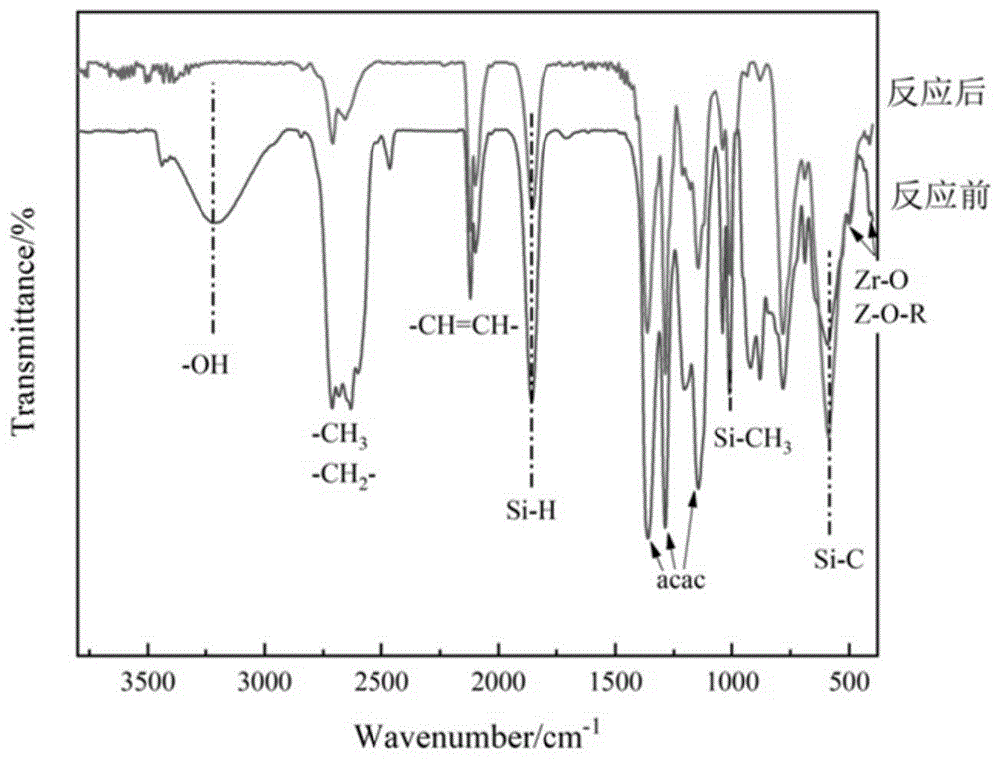
图1是本发明前驱体共混反应前以及反应后的红外对比图;
图2是本发明zrc/sic复相陶瓷的xrd图;
图3是本发明zrc/sic复相陶瓷的sem图。
需要说明的是,这些附图和文字描述并不旨在以任何方式限制本发明的构思范围,而是通过参考特定实施例为本领域技术人员说明本发明的概念。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对实施例中的技术方案进行清楚、完整地描述,以下实施例用于说明本发明,但不用来限制本发明的范围。
实施例1
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在25℃的温度下,将1mol络合剂乙酰丙酮加入到含有1mol锆醇盐和5mol正丙醇的锆醇盐正丙醇溶液中,反应2h;之后在25℃的温度下,将反应后的溶液中再加入1mol水和1mol正丙醇,进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:1:1:6;
(2)在步骤(1)中水解缩聚后的溶液中加入7mol二甲苯,加热使其在125℃温度下预聚1h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为97%的炔丙基酚醛以锆元素与碳元素的摩尔比为1:1混合,并在50℃下反应2h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌3h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:0.61混合后搅拌3h混合,并在50℃下反应2h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃、140℃、160℃、180℃、200℃、220℃、250℃各保温2h使之固化,然后置于石墨炉中,惰性气氛加热至1600℃,得到zrc/sic复相陶瓷。
实施例2
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在40℃的温度下,将0.8mol络合剂乙酰乙酸乙酯加入到含有1mol锆醇盐和5mol正丁醇的锆醇盐正丁醇溶液中,反应2h;之后在40℃的温度下,将反应后的溶液中再加入1mol水和2mol正丁醇,进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:0.8:1:7;
(2)在步骤(1)中水解缩聚后的溶液中加入7mol加入二甲苯,加热使其在125℃温度下预聚1h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为82%的炔丙基酚醛以锆元素与碳元素的物质的量比为1:1.2混合,并在50℃下反应2h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌3h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:0.65混合后搅拌3h混合,并在50℃下反应2h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃、140℃、160℃、180℃、200℃、220℃、250℃各保温2h使之固化,然后置于石墨炉中,惰性气氛加热至1600℃,得到zrc/sic复相陶瓷。
实施例3
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在25℃的温度下,将1mol络合剂乙酰丙酮加入到含有1mol锆醇盐和5mol异丙醇的的锆醇盐异丙醇溶液中,反应2h;之后在25℃的温度下,将反应后的溶液中再加入1mol水和2mol异丙醇,进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:1:1:7;
(2)在步骤(1)中水解缩聚后的溶液中加入7mol二甲苯,加热使其在125℃温度下预聚1h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为82%的炔丙基酚醛以锆元素与碳元素的摩尔比为1:1混合,并在50℃下反应2h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌3h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:0.63混合后搅拌3h混合,并在50℃下反应2h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃、140℃、160℃、180℃、200℃、220℃、250℃各保温2h使之固化,然后置于石墨炉中,惰性气氛加热至1600℃,得到zrc/sic复相陶瓷。
实施例4
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在25℃的温度下,将1mol络合剂乙酰丙酮加入到含有1mol锆醇盐和5mol乙二醇甲醚的的锆醇盐乙二醇甲醚溶液中,反应2h;之后在40℃的温度下,将反应后的溶液中再加入1mol水和2mol乙二醇甲醚,进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:1:1:7;
(2)在步骤(1)中水解缩聚后的溶液中加入7mol二甲苯,加热使其在125℃温度下预聚1h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为82%的炔丙基酚醛以锆元素与碳元素的质量比为1:1.2混合,并在50℃下反应2h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌3h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:0.71混合后搅拌3h混合,并在50℃下反应2h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃、140℃、160℃、180℃、200℃、220℃、250℃各保温2h使之固化,然后置于石墨炉中,惰性气氛加热至1600℃,得到zrc/sic复相陶瓷。
实施例5
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在100℃的温度下,将0.5mol络合剂乙酰丙酮加入到含有1mol锆醇盐和16mol正丙醇和正丁醇混合溶液(摩尔比1:1)的锆醇盐一元醇溶液中,反应0.5h;之后在95℃的温度下,将反应后的溶液中再加入0.5mol水和4mol正丙醇和正丁醇混合溶液(摩尔比1:1),进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:0.5:0.5:20;
(2)在步骤(1)中水解缩聚后的溶液中加入20mol正己烷,加热使其在120℃温度下预聚1.5h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为99%的炔丙基酚醛以锆元素与碳元素的质量比为1:1.8混合,并在80℃下反应0.5h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌3.5h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:6混合后搅拌4h混合,并在80℃下反应0.5h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃下固化1h,之后在140℃下固化1h,之后在160℃下固化2h,之后在180℃下固化2h,之后在200℃下固化2h,之后在220℃下固化2h,最后在250℃下固化4h,然后置于石墨炉中,惰性气氛加热至1500℃,得到zrc/sic复相陶瓷。
实施例6
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在95℃的温度下,将2mol络合剂乙酰乙酸乙酯加入到含有1mol锆醇盐和5mol乙二醇乙醚的锆醇盐乙二醇乙醚溶液中,反应1h;之后在100℃的温度下,将反应后的溶液中再加入2mol水和1mol乙二醇乙醚,进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:2:2:6;
(2)在步骤(1)中水解缩聚后的溶液中加入6mol正庚烷,加热使其在150℃温度下预聚2h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为95%的炔丙基酚醛以锆元素与碳元素的质量比为1:2混合,并在100℃下反应0.8h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌2h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:0.3混合后搅拌2h混合,并在100℃下反应0.8h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃下固化2h,之后在160℃下固化2h,之后在180℃下固化2h,之后在220℃下固化2h,最后在250℃下固化2h,然后置于石墨炉中,惰性气氛加热至1800℃,得到zrc/sic复相陶瓷。
实施例7
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在60℃的温度下,将3mol络合剂乙酰乙酸乙酯和3mol络合剂乙酰丙酮加入到含有1mol锆醇盐和5mol异丙醇的锆醇盐异丙醇溶液中,反应2h;之后在60℃的温度下,将反应后的溶液中再加入1.5mol水和5mol异丙醇,进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:6:1.5:10;
(2)在步骤(1)中水解缩聚后的溶液中加入10mol甲苯,加热使其在100℃温度下预聚5h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为85%的炔丙基酚醛以锆元素与碳元素的质量比为1:2.5混合,并在30℃下反应4h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌4h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:0.1混合后搅拌1h混合,并在30℃下反应4h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃下固化3h,之后在160℃下固化3h,之后在180℃下固化3h,之后在220℃下固化3h,最后在250℃下固化2h,然后置于石墨炉中,惰性气氛加热至1700℃,得到zrc/sic复相陶瓷。
实施例8
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在75℃的温度下,将3mol络合剂乙酰乙酸乙酯加入到含有1mol锆醇盐和5mol乙二醇甲醚的锆醇盐乙二醇甲醚溶液中,反应3h;之后在75℃的温度下,将反应后的溶液中再加入1.5mol水和10mol乙二醇甲醚,进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:3:1.5:15;
(2)在步骤(1)中水解缩聚后的溶液中加入10mol正己烷和5mol正庚烷,加热使其在180℃温度下预聚0.5h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为80%的炔丙基酚醛以锆元素与碳元素的质量比为1:3混合,并在130℃下反应5h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌6h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:15混合后搅拌6h混合,并在130℃下反应5h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃下固化1h,之后在140℃下固化1h,之后在160℃下固化1h,之后在180℃下固化2h,之后在200℃下固化2h,之后在220℃下固化2h,最后在250℃下固化3h,然后置于石墨炉中,惰性气氛加热至1600℃,得到zrc/sic复相陶瓷。
实施例9
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在40℃的温度下,将1mol络合剂乙酰乙酸乙酯加入到含有1mol锆醇盐和5mol正丁醇的锆醇盐正丁醇溶液中,反应4h;之后在40℃的温度下,将反应后的溶液中再加入1mol水和7mol正丁醇,进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:1:1:12;
(2)在步骤(1)中水解缩聚后的溶液中加入12mol二甲苯,加热使其在125℃温度下预聚2h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为88%的炔丙基酚醛以锆元素与碳元素的质量比为1:1.2混合,并在70℃下反应1h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌1h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:3混合后搅拌1.8h混合,并在70℃下反应1h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃下固化2h,之后在160℃下固化2h,之后在180℃下固化2h,之后在220℃下固化2h,最后在250℃下固化2h,然后置于石墨炉中,惰性气氛加热至1600℃,得到zrc/sic复相陶瓷。
实施例10
本实施例中,采用如下方法制备zrc/sic复相陶瓷前驱体:
(1)在25℃的温度下,将0.8mol络合剂乙酰乙酸乙酯加入到含有1mol锆醇盐和5mol正丙醇的锆醇盐正丙醇溶液中,反应5h;之后在25℃的温度下,将反应后的溶液中再加入1mol水和2mol正丙醇,进行水解缩聚,其中,锆醇盐,络合剂,水和一元醇的摩尔比为1:0.8:1:7;
(2)在步骤(1)中水解缩聚后的溶液中加入3mol甲苯和4mol二甲苯,加热使其在125℃温度下预聚3h,得到非极性聚锆氧烷;
(3)将步骤(2)制得的非极性局锆氧烷和醚化率为93%的炔丙基酚醛以锆元素与碳元素的质量比为1:1混合,并在60℃下反应0.5h,得到zrc前驱体;
(4)将聚碳硅烷溶解于二甲苯中,搅拌0.5h得到sic前驱体;
(5)将步骤(3)和(4)制得的zrc前驱体和sic前驱体按锆元素与硅元素的摩尔比1:1混合后搅拌1.4h混合,并在60℃下反应0.5h后得到复相前驱体;
(6)将步骤(5)制得的复相前驱体在120℃、140℃、160℃、180℃、200℃、220℃、250℃各保温2h使之固化,然后置于石墨炉中,惰性气氛加热至1600℃,得到zrc/sic复相陶瓷。
对比例1
在实施例1的基础上,选择醚化率为79%的改性酚醛作为碳源,其他条件同实施例1,进行复相相陶瓷前驱体的制备,
对比例2
在实施例1的基础上,选择醚化率为100%的改性酚醛作为碳源,其他条件同实施例1,进行复相相陶瓷前驱体的制备。
对比例3
在实施例1的基础上,去掉步骤(2),其他实施方式同实施例1。
对比例4
在实施例1的基础上,去掉步骤(3)中非极性聚锆氧烷和改性酚醛在50℃下反应2h的过程,其他实施方式同实施例1。
实验例1
对实施例1~10和对比例1~4制得的zrc/sic复相陶瓷前驱体进行粘度变化的测试,其结果如下表所示:
由上表可知,根据本申请的实施例所制得的zrc/sic复相陶瓷前驱体在室温储存12个月后的粘度变化小于10%,而对比例1和2虽然也采用了实施例中的方法,但其粘度变化超出10%的范围,分析是由于碳源酚醛树脂中羟基的醚化程度过低或过高导致的,而对比例3和4中生产的前驱体在放置后粘度变化极大,则是由于缺少了高温预聚以及非极性聚锆氧烷与碳源的反应步骤。由此可见,本申请在制备过程中增加了高温预聚步骤以及将非极性聚锆氧烷与碳源进行反应的方法减少了前驱体中的活性基团,极大的增加了前驱体的储存稳定性。
实验例2
对实施例1~10和对比例1~4制得的zrc/sic复相陶瓷前驱体的陶瓷产率进行测试,结果如下表:
由上表可知,本发明的实施例制得的zrc/sic复相陶瓷前驱体的陶瓷产率要高于对比例,其中,对比例1中改性酚醛羟基的醚化率较低,即碳源中可与聚锆氧烷中分子链交联的碳碳不饱和基团含量较低,无法防止碳源在后续高温处理中发生流失,而对比例2中改性酚醛羟基的醚化率较高,使碳源中不存在未经醚化的酚羟基,因此无法与聚锆氧烷反应以进一步去除聚锆氧烷中的活性基团,从而降低了稳定性,影响了产率。而对比例3和4由于缺少了高温预聚和非极性聚锆氧烷与碳源的反应步骤,无法有效减少前驱体溶液中的活性基团,而碳源也无法与聚锆氧烷分子链形成交联,使碳源在后续高温处理过程中流失,从而降低了陶瓷生产率。
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!