用于锂离子电池的阴极活性材料的制作方法
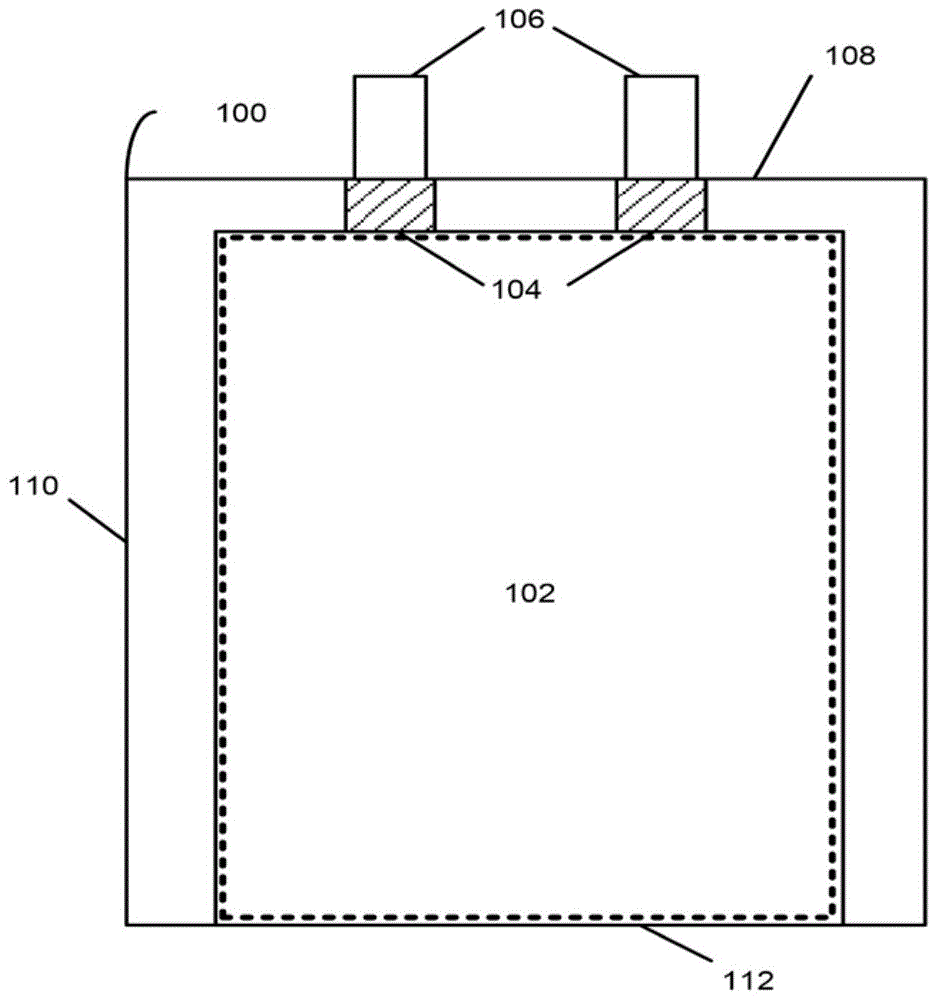
本申请是国际提交日为2017年3月14日(优先权日2016年3月14日)、于2018年9月进入中国国家阶段、中国国家申请号为201780014650.3、发明名称为“用于锂离子电池的阴极活性材料”的发明专利申请的分案申请。相关申请的交叉引用根据35u.s.c.§119(e),本申请要求以下专利的权益:美国临时专利申请序列号62/307,956,名称为“surface-modifiedcathodeactivematerialsforlithium-ionbatteries”,提交于2016年3月14日;以及美国专利申请序列号62/307,964,名称为“cathodeactivematerialsforlithium-ionbatteries”,提交于2016年3月14日。每个专利申请的内容全文以引用方式并入本文。本公开整体涉及电池,并且更具体地涉及用于锂离子电池的阴极活性材料。美国政府许可权本发明基于wfo提案no.85f59在美国政府的支持下进行。本发明基于appleinc.与argonnenationallaboratory(隶属于美国能源部)之间的crada1500801进行。美国政府拥有本发明的某些权利。
背景技术:
:常用的可再充电电池类型是锂电池,诸如锂离子或锂聚合物电池。由于电池供电设备变得越来越小以及更强大,给这些设备供电的电池需要在更小的体积中存储更多的能量。因此,可通过能够改善装置中电池的体积能量密度的机制来促进电池驱动装置的使用。可在锂离子电池的阴极活性材料中使用锂过渡金属氧化物。这些化合物可包括锂钴氧化物或其衍生物。这些化合物可为粉末形式。技术实现要素:在第一方面,本公开涉及根据式(iii)的化合物:liαco1-xmxalγoδ(iii)其中m为b、na、mn、ni、mg、ti、ca、v、cr、fe、cu、zn、al、sc、y、ga、zr、mo、ru或它们的组合,0.95≤α≤1.10,0<x<0.50,0≤γ≤0.05,并且1.95≤δ≤2.60。在一些方面,m为mn、ni或它们的组合,0.95≤α≤1.10,0<x<0.50,0≤γ≤0.05,并且1.95≤δ≤2.60。在另一方面,本公开涉及包含颗粒的粉末。颗粒包含根据式(iii)的化合物。在另一方面,本公开涉及包含具有芯和涂层的颗粒的粉末。涂层设置在芯的至少一部分上。芯包含选自式(i)、式(iia)、式(iib)和式(iii)化合物的化合物:liαmoδ(i)(x)[li2m1o3]·(1-x)[lim2o2](iia)(x)[li2m1o3]·(1-x)[li1-ym2o2](iib)liαco1-xmxalγoδ(iii)其中,当化合物为式(i)时,m选自co、mn、ni以及它们的组合,0.95≤α≤2,并且1.95≤δ≤3;当化合物为式(iia)时,0≤x≤1,m1选自ti、mn、zr、mo、ru以及它们的组合,并且m2选自b、na、mg、ti、ca、v、cr、mn、fe、co、ni、cu、zn、al、sc、y、ga、zr、mo、ru以及它们的组合;当所述化合物为式(iib)时,0≤x≤1,0≤y≤1,m1选自ti、mn、zr、mo、ru以及它们的组合,并且m2选自b、na、mg、ti、ca、v、cr、mn、fe、co、ni、cu、zn、al、sc、y、ga、zr、mo、ru以及它们的组合;并且当所述化合物为式(iii)时,0.95≤α≤1.10,m选自b、na、mn、ni、mg、ti、ca、v、cr、fe、cu、zn、al、sc、y、ga、zr、mo、ru以及它们的组合,0<x<0.50,0≤γ≤0.05,并且1.95≤δ≤2.60。在一些方面,化合物为式(i),m选自co、mn、ni以及它们的组合,1≤α≤2,并且2≤δ≤3。在一些方面,化合物为式(iia),0≤x≤1,m1选自ti、mn、zr、mo、ru以及它们的组合,并且m2选自b、na、mg、ti、ca、v、cr、mn、fe、co、ni、cu、zn、al、sc、y、ga、zr、mo、ru以及它们的组合。在一些方面,化合物为式(iia),并且0<x≤0.10。在一些方面,化合物为式(iib),0≤x≤1,0≤y≤1,m1选自ti、mn、zr、mo、ru以及它们的组合,并且m2选自b、na、mg、ti、ca、v、cr、mn、fe、co、ni、cu、zn、al、sc、y、ga、zr、mo、ru以及它们的组合。在一些方面,化合物为式(iii),并且0<x≤0.10。在一些方面,化合物为式(iii),0.95≤α≤1.10,m选自mn、ni以及它们的组合,0<x<0.50,0≤γ≤0.05,并且1.95≤δ≤2.60。涂层包含氧化物材料、氟化物材料或它们的组合。在一些方面,芯包含根据式(iia)的化合物。在一些方面,芯包含根据式(iib)的化合物。在一些方面,芯包含根据式(iii)的化合物。在其它变型中,0.001≤γ≤0.03。在一个方面,本公开涉及由式(iv)表示的化合物:liαco1-xmnxoδ(iv)其中0.95≤α≤1.10,0≤x≤0.10,并且1.90≤δ≤2.20。在另一方面,0<x≤0.10。在另一方面,本公开涉及由式(iv)表示的化合物,其中0.98≤δα≤1.01。在另一方面,本公开涉及由式(iv)表示的化合物,其中1.00≤α≤1.05。在另一方面,0≤x≤0.10。在另一方面,本公开涉及由式(iv)表示的化合物,其中0.95≤α≤1.05并且0.02≤x≤0.05。在另一方面,本公开涉及由式(iv)表示的化合物,其中1.01≤α≤1.05并且0.02≤x≤0.05。在另一方面,本公开涉及由式(iv)表示的化合物,其中0.95≤α≤1.05并且x=0.04。在另一方面,本公开涉及由式(iv)表示的化合物,其中1.01≤α≤1.05并且x=0.04。在另一方面,本公开涉及由式(iv)表示的化合物,其中0.98≤α≤1.01并且x=0.03。在另一方面,化合物具有式(va)或式(vb)的结构:(x)[li2mno3]·(1-x)[licoo2](va)(x)[li2mno3]·(1-x)[li(1-y)co(1-y)mnyo2](vb)其中0≤x≤0.10,并且任选地0≤y≤0.10。在一些变型中,0<y≤0.10。在一些方面,本公开涉及包含颗粒的粉末,其中颗粒包含由式(iv)表示的化合物:liαco1-xmnxoδ。在各个方面,0.95≤α≤1.10,0≤x≤0.10,并且1.90≤δ≤2.20。在各个方面,0.95≤α≤1.10,0<x≤0.10,并且1.90≤δ≤2.20。在一些实施方案中,颗粒的至少一部分具有平滑表面。在这些实施方案的各种情况下,颗粒的至少一部分具有等于或大于2.2g/cm3的振实密度。在这些实施方案的各种情况下,颗粒的至少一部分具有平滑表面和等于或大于2.2g/cm3的振实密度。在另一方面,本公开涉及包含本文所述的粉末的阴极活性材料。在另一方面,本公开涉及具有设置在集流体上的阴极活性材料的阴极。在另一方面,本公开涉及电池单元,该电池单元包括具有阳极集流体和设置在阳极集流体上的阳极活性材料的阳极以及阴极。在另一方面,本公开涉及包括由电池组驱动的一组部件的便携式电子设备。在另一方面,本公开涉及制备本文所述粉末的方法。首先,通过将前体溶解于溶剂中以形成前体溶液来制备前体溶液(例如,铝盐和/或氟化物盐前体)。将前体溶液加入到颗粒粉末中以形成湿法浸渍的粉末。将湿法浸渍的粉末加热至高温以形成具有本文所述组合物的颗粒。在另一方面,将多种前体(例如,铝盐和氟化物盐)溶解于第一溶剂和第二溶剂中,以分别形成第一溶液和第二溶液。然后将第一溶液和第二溶液混合以制备前体溶液,然后将其添加到如本文所述的颗粒中。在另一方面,本公开涉及通过干混方法制备颗粒。纳米晶体材料的颗粒与包含式(iv)的化合物的颗粒混合。纳米晶体材料颗粒和包含式(iv)的颗粒经受压缩力、剪切力或它们的组合。纳米晶体材料颗粒结合到粉末颗粒的表面上,从而形成粉末颗粒上的涂层。在另一方面,本公开涉及由式(vii)或式(viii)表示的化合物:liαco1-x-ymymnxoδ(vii)liαco1-x-yalymnxoδ(viii)当化合物由式(vii)表示时,在各个方面,m为选自b、na、mg、ti、ca、v、cr、fe、ni、cu、zn、al、sc、y、ga、zr、mo和ru的至少一种元素。在一些变型中,0.95≤α≤1.30,0<x≤0.30,0≤y≤0.10,并且1.98≤δ≤2.04。在各个方面,化合物为具有晶体结构的单相化合物。在另外的方面,α>1+x并且/或者α<1+x。在另外的方面,本公开涉及包含颗粒的粉末。颗粒的平均直径可为至少5μm。在一些方面,颗粒的平均直径可为至少20μm。在一些方面,颗粒可包含次级颗粒,每个次级颗粒包含多个烧结在一起的初级颗粒。在一些变型中,平均次级颗粒的至少30%由单个初级颗粒形成。在另外的方面,本公开涉及包含化合物或粉末的阴极活性材料。在另外的方面,本公开涉及电池单元,该电池单元包括阳极和包含阴极活性材料的阴极。在一些方面,电池单元可具有大于或等于750wh/kg的第一循环放电能量。在一些方面,电池单元在10个充电放电循环之后可具有大于或等于70%的能量保留率。在一些方面,电池单元在52个放电循环之后可具有至少65%的能量电容保留率。附图说明通过以下结合附图的详细描述,将容易理解本公开,其中类似的参考标号指代类似的结构元件,并且其中:图1为根据例示性实施方案的电池单元的俯视图;图2为根据例示性实施方案的电池单元的一组层的侧视图;图3a-图3c为根据例示性实施方案的一系列扫描电子显微图,这些扫描电子显微图分别示出了基料粉末、涂覆有0.1重量%alf3的基料粉末,以及涂覆有0.1重量%al2o3的粉末;图4为根据例示性实施方案表示三个硬币半电池(每个引入有单个阴极活性材料)在第一个充电和放电循环期间的性能的数据的曲线图;图5为根据例示性实施方案表示图4的四个硬币半电池在速率测试中电容随增加的循环而变化的数据的曲线图;图6为根据例示性实施方案表示图4的四个硬币半电池在寿命测试中电容随增加的循环而变化的数据的曲线图;图7为根据例示性实施方案表示图4的四个硬币半电池在速率测试中能量密度随增加的循环而变化的数据的曲线图;图8为根据例示性实施方案表示图4的四个硬币半电池在寿命测试中能量密度随增加的循环而变化的数据的曲线图;图9为对应于速率测试中的图4的四个硬币半电池中每一个的充电放电特征的数据的曲线图;图10为对应于寿命测试中的图4的四个硬币半电池中每一个的充电放电特征的数据的曲线图;图11为表示速率测试中的图4的四个硬币半电池各自的dq/dv特征的数据的曲线图;以及图12为表示寿命测试中的图4的四个硬币半电池各自的dq/dv特征的数据的曲线图。图13为根据例示性实施方案表示摩尔比对包含liαco0.96mn0.04oδ的阴极活性材料的电容和效率的影响的数据的曲线图;图14为根据例示性实施方案表示摩尔比(由α表示)和mn含量(由x表示)对liαco0.96mn0.04oδc轴晶格参数的影响的数据的曲线图;图15为根据例示性实施方案表示liαco0.96mn0.04o2的c轴晶格参数随摩尔比的变化的数据的曲线图;图16为根据例示性实施方案的一组扫描电镜显微图,示出了在900℃、1000℃、1050℃和1100℃下制备的粉末;图17为根据例示性实施方案的一组扫描电镜显微图,示出了温度和摩尔比对颗粒形态的影响α;图18为根据例示性实施方案表示混合比对包含liαco0.96mn0.04oδ的阴极活性材料的电容和效率的影响的数据的曲线图;图19为根据例示性实施方案的一组扫描电镜显微图,示出了包含liαco1-xmnxoδ且通过共沉淀方法制备的阴极活性材料;图20为根据例示性实施方案的通过共沉淀方法制备且包含liαco0.96mn0.04oδ、δliαco0.90mn0.10oδ和liαco0.84mn0.16oδ(具有不同α值)的阴极活性材料的x射线衍射图;图21为根据例示性实施方案的通过共沉淀方法制备且包含liαco0.78mn0.22oδδ和liαco0.72mn0.28oδ(具有不同α值)的阴极活性材料的x射线衍射图;图22为根据例示性实施方案的一组扫描电镜显微图,示出了包含liαco1-xmnxoδ且通过凝胶方法制备的阴极活性材料;图23为根据例示性实施方案的通过凝胶方法制备且包含li1.131co0.90mn0.10o2、li1.198co0.84mn0.16o2、li1.241co0.78mn0.22o2和li1.301co0.72mn0.28o2的阴极活性材料的x射线衍射图;图24为根据例示性实施方案的包含licoo2、li1.05co0.96mn0.04o2、li1.05co0.93mn0.07o2、li1.110co0.90mn0.10o2和li1.19co0.72mn0.28o2的阴极活性材料的微分电容曲线图;图25为根据例示性实施方案的包含li1.05co0.96mn0.04o2、li1.05co0.93mn0.07o2、li1.110co0.90mn0.10o2和li1.19co0.72mn0.28o2的阴极活性材料的电压特征图;图26为根据例示性实施方案的随着mn被取代、co1-xmnx和锂比率[li]/[co1-xmnx]而变化的放电能量密度的等值线图;图27为根据例示性实施方案的随着mn被取代、co1-xmnx和锂比率[li]/[co1-xmnx]而变化的能量保留率的等值线图;图28为根据例示性实施方案的包含liαco0.99-yalymn0.01oδ、liαco0.98-yalymn0.02oδ、liαco0.97-yalymn0.03oδ和liαco0.96-yalymn0.04oδ的阴极活性材料的微分电容曲线图;图29为根据例示性实施方案的包含li_co0.97alymn0.03oδ、li_co0.97alymn0.03oδ、li_co0.97alymn0.03oδ和li_co0.97alymn0.03oδ的阴极活性材料的微分电容曲线图;图30为根据例示性实施方案的包含li_co0.97mn0.03oδ、li_co0.97mn0.03oδ和li_co0.97mn0.03oδ的阴极活性材料的放电能量与循环计数的曲线图;图31为根据例示性实施方案的包含liαco0.98-yalymn0.02oδ、liαco0.97-yalymn0.03oδ和liαco0.96-yalymn0.04oδ的阴极活性材料的放电能量与循环计数的曲线图;图32为根据例示性实施方案的liαco0.97mn0.03oδ和liαco0.97mn0.03oδ的核磁共振图案;图33为根据例示性实施方案的包含li_co0.97-yalymn0.03oδ(添加了0.077、0.159和0.760重量%的al2o3)的阴极活性材料的放电能量与循环计数的曲线图;图34a为根据例示性实施方案的通过焙烧前体制备且包含liαco1-x-yalymnxoδ的阴极活性材料的颗粒的扫描电镜显微图;图34b为根据例示性实施方案的图34a的阴极活性材料的扫描电镜显微图,但其中前体在更高烧结温度下焙烧;图35a为根据例示性实施方案的通过在1050℃下焙烧前体制备且包含liαco1-x-yalymnxoδ的阴极活性材料的颗粒的粒度分布;图35b为根据例示性实施方案的通过在1085℃下焙烧前体制备且包含liαco1-x-yalymnxoδ的阴极活性材料的颗粒的粒度分布;图36为根据例示性实施方案的包含li.01co1-x-yalymnxoδ的阴极活性材料的表面积变化和能量保留率与煅烧温度的曲线图;图37为根据例示性实施方案的包含li1.01co1-x-yalymnxoδ的阴极活性材料的初始放电电容和库仑效率与煅烧温度的曲线图;图38为根据例示性实施方案的包含licoo2、liαco0.99mn0.01oδ、liαco0.98mn0.02oδ、liαco0.97mn0.03oδ、liαco0.96mn0.04oδ和liαco0.93mn0.07oδ的阴极活性材料的热流与煅烧温度的曲线图;图39为根据例示性实施方案的包含αlicoo2、liαco0.98mn0.02oδ、liαco0.97mn0.03oδ、liαco0.96mn0.04oδ和liαco0.93mn0.07oδ的阴极活性材料的c轴晶格参数与锂含量的曲线图;图40为根据例示性实施方案的包含licoo2、liαco0.96mn0.04oδ和liαco0.93mn0.07oδ的阴极活性材料的拉曼光谱的曲线图;图41为根据例示性实施方案的包含αliαco0.96mn0.04oδ和liαco0.93mn0.07oδ的阴极活性材料的52个循环之后的放电电容与锂含量的曲线图;图42为根据例示性实施方案的包含li1.025co0.96-yalymn0.04oδ和li1.00co0.93-yalymn0.07oδ的阴极活性材料的微分电容曲线图;以及图43为根据例示性实施方案的包含αliαco0.96mn0.04oδ的阴极活性材料的第一循环充电电容、第一循环放电电容和第一循环库仑效率与锂含量的数据图。具体实施方式给出以下描述是为了使本领域的任何技术人员能够制定并使用实施方案,并且以下描述是在特定应用及其要求的情境中提供的。对于本领域的技术人员而言,对本发明所公开的实施方案的各种修改将是显而易见的,并且可以将本文所定义的一般原理应用于其他实施方案和应用而不脱离本公开的实质和范围。应当理解,以下描述不旨在将实施方案限制于一个优选实施方案。相反,其旨在涵盖可被包括在由所附权利要求限定的所述实施方案的实质和范围内的另选形式、修改形式和等同形式。因此,本公开不限于所示的实施方案,但要符合根据本文公开的原理和特征的最广泛的范围。如本文所用,除非另外指明,否则用于阴极活性材料的所有组合物代表已制备材料(即“原始”材料)的组合物。这些组合物的材料未暴露于另外的工艺,诸如分别在锂离子电池充电和放电期间脱锂化和锂化。可在商业锂离子电池的阴极活性材料中使用锂钴氧化物。这些化合物通常包含锂钴氧化物或其衍生物。此类阴极活性材料的性能可通过提高其电容、工作电压和重量电极密度来增强。颗粒的形态也可影响阴极活性材料的性能。颗粒可包括初级颗粒和次级颗粒。初级颗粒和次级颗粒的粒度分布、形状和孔隙率可影响锂钴氧化物电极的密度。次级颗粒由较小初级颗粒的附聚物构成,通常也被称为晶粒。可对形状和密度的次级颗粒特性进行控制。可使用提供增大的电容、工作电压和重量电极密度的化合物和颗粒来改善电池的性能。本文的公开内容解决了这些和其它需求。图1示出了根据一个实施方案的电池单元100的俯视图。电池单元100可对应于用于给消费、医疗、航空、国防和/或运输应用中使用的设备供电的锂离子或锂聚合物电池单元。电池单元100包括具有多个层的叠堆102,所述多个层包括具有阴极活性涂层的阴极、隔板和具有阳极活性涂层的阳极。更具体地讲,叠堆102可包括一条阴极活性材料(例如涂覆有锂化合物的铝箔)和一条阳极活性材料(例如涂覆有碳的铜箔)。叠堆102还包括设置在一条阴极活性材料与一条阳极活性材料之间的一条隔板材料(例如导电聚合物电解质)。阴极、阳极和隔板层可以平面构型保持平坦或可被卷绕成卷绕构型(例如,“凝胶卷”)。电池单元可包封在柔性袋中。返回图1,在电池单元100的组装期间,叠堆102包封在柔性袋中。叠堆102可为平面或卷绕构型,但其它构型也是可以的。柔性袋通过沿折叠线112折叠柔性片材而形成。例如,柔性片材可由具有聚合物膜(例如聚丙烯)的铝制成。在折叠柔性片材之后,例如可以通过沿着侧面密封部110和沿着平台密封部108施加热来密封柔性片材。柔性袋的厚度可小于120微米以改善电池单元100的封装效率、电池单元100的密度或这两者。叠堆102还包括耦合到阴极和阳极的一组导电引片106。导电引片106可延伸穿过袋中的密封部(例如,使用密封带104形成的密封部)以为电池单元100提供端子。然后,导电引片106可用于将电池单元100与一个或多个其它电池单元电耦合以形成电池组。电池可组合在任何构型的电池组中。例如,电池组可以串联、并联或串并联构型耦合电池单元来形成。这样耦合的单元可封装在硬质壳体中以完成电池组,或可嵌入便携式电子设备(诸如膝上型计算机、平板电脑、移动电话、个人数字助理(pda)、数码相机和/或便携式媒体播放器)的外壳内。图2示出了根据本发明所公开的实施方案的电池单元(如,图1的电池单元100)的一组层的侧视图。该组层可包括阴极集流体202、阴极活性涂层204、隔板206、阳极活性涂层208和阳极集流体210。阴极集流体202和阴极活性涂层204可形成电池单元的阴极,阳极集流体210和阳极活性涂层208可形成电池单元的阳极。为了形成电池单元,可将该组层以平面构型堆叠,或堆叠然后卷绕成卷绕构型。如上所述,阴极集流体202可为铝箔,阴极活性涂层204可为锂化合物,阳极集流体210可为铜箔,阳极活性涂层208可为碳,隔板206可包含导电聚合物电解质。应当理解,本文所述的阴极活性材料可与本领域已知的任何电池单元或其部件结合使用。例如,除卷绕电池单元之外,层可被堆叠和/或用于形成其它类型的电池单元结构,诸如双单元结构。所有此类电池单元结构在本领域中是已知的。本发明公开了表面改性和结构稳定的锂钴氧化物材料。该材料可用作锂离子电池中的阴极活性材料。在各个方面,过渡金属氧化物为锂钴氧化物的变型。在一个方面,本公开涉及式(i)的化合物:liαmoδ(i)其中m为co、mn、ni或它们的任何组合,0.95≤α≤2并且2≤δ≤3。在一些变型中,1≤α≤2。在一些变型中,1.20≤αδδ在一些变型中,1.40≤αδδ在一些变型中,1.60≤αδδ在一些变型中,1.80≤αδδ在一些变型中,α≤1.8。在一些变型中,α≤1.6。在一些变型中,α≤1.4。在一些变型中,α≤1.2。在一些变型中,α≤1.8。此外,在一些变型中,2.2≤δ。在一些变型中,2.4≤δ。在一些变型中,2.6≤δ。在一些变型中,2.8≤δ。在一些变型中,δ≤2.8。在一些变型中,δ≤2.6。在一些变型中,δ≤2.4。在一些变型中,δ≤2.2。应当理解,α和δ的边界可以如上的任何变型进行组合。在各个方面,本公开涉及式(iia)的化合物:(x)[li2m1o3]·(1-x)[lim2o2](iia)其中m1为具有+4的平均氧化态(即,四价)的一个或多个阳离子,m2为具有+3的平均氧化态(即,三价)的一个或多个阳离子,并且0≤x≤1。在一些变型中,m1选自ti、mn、zr、mo、ru以及它们的组合。在特定变型中,m1为mn。在一些变型中,m2选自b、na、mg、ti、ca、v、cr、mn、fe、co、ni、cu、zn、al、sc、y、ga、zr、ru、mo以及它们的组合。在一些变型中,m2选自mg、ca、v、cr、mn、fe、co、ni、cu、zn、al、sc、y、ga、zr以及它们的组合。在特定变型中,m2为co。应当理解,在过量过渡金属氧化物的表示中,m=[m2]1-x[m1]x,α=1+x,并且δ=2+x。在一些变型中,α≤1.2。在一些变型中,α≤1.4。在一些变型中,α≤1.6。在一些变型中,α≤1.8。在一些变型中,1.2≤α。在一些变型中,1.4≤α。在一些变型中,1.6≤α。在一些变型中,1.8≤α。应当理解,x的边界可以如上的任何变型进行组合。对于本文所公开的实施方案,钴是主要过渡金属组分,其耐受高电压以及用于锂离子电池中所采用阴极活性材料的高体积能量密度。在各个方面,本公开涉及式(iib)的化合物:(x)[li2m1o3]·(1-x)[li1-ym2o2](iib)其中m1为具有+4的平均氧化态(即,四价)的一个或多个阳离子,m2为一个或多个阳离子,0≤x≤1,并且0≤y≤1。在一些变型中,m1选自ti、mn、zr、mo、ru以及它们的组合。在特定变型中,m1为mn。在一些变型中,m2选自b、na、mg、ti、ca、v、cr、mn、fe、co、ni、cu、zn、al、sc、y、ga、zr、ru、mo以及它们的组合。在一些变型中,m2选自mg、ti、ca、v、cr、mn、fe、co、ni、cu、zn、al、sc、y、ga、zr以及它们的组合。在一些变型中,m2选自mg、ca、v、cr、mn、fe、co、ni、cu、zn、al、sc、y、ga、zr以及它们的组合。在特定变型中,m2为co和mn。应当理解,在过量过渡金属氧化物的表示中,m=[m2]1-x[m1]x,α=1+x-y+xy,并且δ=2+x。在一些变型中,α≤0.98。在一些变型中,α≤1.0。在一些变型中,α≤1.1。在一些变型中,α≤1.2。在一些变型中,0.95≤α。在一些变型中,1.3≤α。在一些变型中,1.6≤α。在一些变型中,1.8≤α。应当理解,α的边界可以如上的任何变型进行组合。对于本文所公开的实施方案,钴是主要过渡金属组分,其耐受高电压以及用于锂离子电池中所采用阴极活性材料的高体积能量密度。在一些变型中,本公开涉及由式(iii)表示的化合物:liαco1-xmxalγoδ(iii)其中m选自b、na、mn、ni、mg、ti、ca、v、cr、fe、cu、zn、al、sc、y、ga、zr、mo、ru或它们的一种组合或它们的任何组合;0.95≤α≤1.10;0<x<0.50;0≤γ≤0.05;并且1.95≤δ≤2.60。在一些变型中,m为mn、ni或它们的任何组合。在一些变型中,m为mn、ni或它们的组合,0.95≤α≤1.10,0<x<0.50,0≤γ≤0.05,并且1.95≤δ≤2.60。在一些变型中,0.01≤γ≤0.03。在一些变型中,0.001≤γ≤0.005。在一些变型中,0.002≤γ≤0.004。在一些变型中,γ为0.003。在一些变型中,0.02≤γ≤0.03。在式(iii)的此类变型中,其中γ≠0(即,存在铝),颗粒内的铝分布可为均匀的或可被偏置成邻近颗粒的表面。其它铝分布也是可能的。在一些变型中,al为至少500ppm。在一些变型中,al为至少750ppm。在一些变型中,al为至少900ppm。在一些变型中,al小于或等于2000ppm。在一些变型中,al小于或等于1500ppm。在一些变型中,al小于或等于1250ppm。在一些变型中,al为约1000ppm。在式(iii)的另外变型中,1.02≤α≤1.05并且0.02≤x≤0.05。在式(iii)的另外变型中,1.03≤α≤1.05并且x=0.04。应当理解,如上所述的组分可为任何组合。式(i)、(iia)、(iib)和(iii)的各种化合物可包含mn4+。不希望受限于任何理论或作用模式,掺入的mn4+可提高氧化物在高电压充电(例如,4.5v)下的稳定性,并且在过渡通过4.1至4.3v区域时(即,在充电和放电期间)还可帮助保持的晶体结构(即,α-nafeo2结构)。在一些实施方案中,本公开涉及包含颗粒的粉末。在一些方面,颗粒可包含式(iii)和新式的化合物。在一些变型中,颗粒包含具有式(i)、式(iia)、式(iib)或式(iii)的化合物的芯和设置在芯的至少一部分上的涂层。在另外的变型中,颗粒包含具有式(iii)的化合物的芯和设置在芯的至少一部分上的涂层。应当理解,粉末可用作阴极活性材料的一部分或全部。本文所述的颗粒可用于电池中的阴极活性材料。此类阴极活性材料可耐受等于或高于常规材料(即,相对于li/li+氧化还原对)的电压且无电容衰减。电容衰减将降低电池性能,原因可能为阴极活性材料的结构不稳定性、电解质在高电压下的副反应、表面不稳定性、阴极活性材料溶解于电解质中或它们的一些组合。在颗粒包含式(iii)的化合物并且包含铝的变型中,铝可被称为掺杂剂。此类铝掺杂剂可均匀地分布在整个颗粒中,或者沿颗粒的表面定位。在另外的变型中,颗粒包含芯和涂层。涂层可为氧化物材料、氟化物材料或它们的组合。在一些变型中,涂层可为与芯的表面接触的材料层或沿芯的表面形成的反应层。涂层可包含氧化物材料(例如,zro2、al2o3等)、氟化物材料(例如,alf3)或它们的组合(例如,aloxfy)。在一些实施方案中,氧化物材料包含选自al、co、li、zr、mg、ti、zn、mn、b、si、ga和bi的至少一种元素。在这些实施方案中,氧化物材料可包括含氧阴离子,诸如磷酸根(例如,alpo4、co3(po4)2、licopo4等)。在一些实施方案中,氟化物材料包含选自al、co、mn、ni、li、ca、zr、mg、ti和na的至少一种元素。涂层可包含选自alf3、al2o3、alpo4、co3(po4)2、licopo4和zro2的一种或多种组合物。涂层可为本领域已知的任何量。在一些变型中,涂层的量可小于或等于总颗粒的7重量%。在一些变型中,涂层的量可小于或等于总颗粒的5重量%。在一些变型中,涂层的量可小于或等于总颗粒的0.8重量%。在一些变型中,涂层的量可小于或等于总颗粒的0.6重量%。在一些变型中,涂层的量可小于或等于总颗粒的0.4重量%。在一些变型中,涂层的量可小于或等于总颗粒的0.3重量%。在一些变型中,涂层的量可小于或等于总颗粒的0.2重量%。在一些变型中,涂层的量可小于或等于总颗粒的0.1重量%。在各个方面,该量可选择为使得阴极活性材料的电容不受负面影响。涂层可包含多层涂层材料。涂层也可为连续涂层或不连续涂层。不连续涂层的非限制性示例包括具有空隙或裂缝的涂层以及由其间具有间隙的颗粒形成的涂层。其它类型的不连续涂层也是可能的。包含本文所述颗粒的粉末可用作锂离子电池中的阴极活性材料。此类阴极活性材料可耐受等于或高于常规材料(即,相对于li/li+氧化还原对)的电压且无电容衰减。电容衰减将降低电池性能,原因可能为阴极活性材料的结构不稳定性、电解质在高电压下的副反应、表面不稳定性、阴极活性材料溶解于电解质中或它们的一些组合。在各个方面,如本文所述的化合物和/或颗粒在作为阴极活性材料掺入到电池中时,使得锂离子电池可在高电压下充电且无电容衰减。不希望受限于特定机制或作用模式,化合物可在充电至较高电压期间阻碍或延缓来自α-nafeo2晶体结构的结构偏差。在各个方面,如本文所述的化合物在作为阴极活性材料掺入到电池中时,使得锂离子电池具有大于4.2v的电压且无电容衰减。在另外的方面,此类电池具有大于4.3v的电压。在各个方面,锂钴氧化物材料涉及大于4.4v的电压。在各个方面,锂钴氧化物材料涉及大于4.5v的电压。具有包含本发明所公开的颗粒的阴极活性材料的电池可在高电压下表现出改善的电池性能。例如,颗粒在高电压(例如4.5v)下的循环中提供增大的电池电容。在某些变型中,电池的衰减速率在4.5v的充电电势下小于0.7mah/g/循环。在一些变型中,电池的衰减速率在4.5v的充电电势下小于0.6mah/g/循环。在一些变型中,电池的衰减速率在4.5v的充电电势下小于0.5mah/g/循环。在一些另外的变型中,包含本发明所公开的颗粒的阴极活性材料的电池在每个循环中损失较少的每单位质量的能量。在一些实施方案中,当在4.5v的电势下操作时,此类电池在每个循环中损失小于4wh/kg。本公开还涉及制备本发明所公开的化合物和颗粒的方法。在颗粒或芯包含式(vi)的化合物的实施方案中,铝可通过表面改性方法引入。例如但不限于,可将颗粒涂覆包含铝的涂层并随后加热。热能可促进颗粒与涂层之间的反应,从而将铝注入到芯中(例如,掺杂)。在另一个非限制性示例中,可将颗粒暴露于包含铝的溶液中。颗粒与溶液之间的化学反应可形成包含铝的表面反应层。可随后将颗粒加热(例如,以将铝从表面反应层扩散到颗粒中,将表面反应层转化为涂层等)。在另一个非限制性示例中,颗粒可与包含铝的颗粒接触,例如在研磨期间。机械能产生压缩力、剪切力或它们的组合,以将铝颗粒熔合到颗粒中(例如,将al2o3纳米粒子粘合到颗粒上)。这些表面改性方法可使得芯实现0<γ≤0.03之间的铝含量。其它表面改性方法也是可能的。在各种实施方案中,包括化合物和/或粉末的电池的性能可增大电池电容和/或减少满充电池随时间推移的可用电力损耗。本公开还涉及通过湿加工或干加工来改性颗粒表面的方法。在另一方面,本公开涉及制备颗粒的方法。制备包含溶解于溶剂中的第一量的前体的前体溶液。粉末包含根据式(i)、(iia)、(iib)或(iii)的化合物。将前体溶液加入到粉末中以形成湿法浸渍的粉末。将湿法浸渍的粉末加热至高温。在另一方面,将第一前体溶解到溶剂的第一部分中以形成第一溶液。将第一溶液加入到粉末中以形成部分湿法浸渍的粉末。将第二前体溶解于溶剂的第二部分中以形成第二溶液。将第二溶液加入到部分湿法浸渍的粉末中以制备湿法浸渍的粉末。随后在高温下加热湿法浸渍的粉末。湿法浸渍涉及将溶剂加入粉末颗粒中,直至颗粒表现出湿稠度(例如,类似于糊剂)。在各个方面,可选择溶剂的量,使得当将前体溶液添加到粉末中时,如此制得的湿法浸渍的粉末表现出湿稠度。该方法还包括在高温下加热湿法浸渍的粉末。应当理解,溶剂的量可通过如下方式确定:选择已知量的粉末,逐步添加溶剂,直至粉末的所有颗粒看起来润湿但不流动(即,表现出湿稠度)。当采用该方法时,溶剂与粉末的比例(例如,每克粉末的溶剂克数)可按需缩放以适应不同量的粉末。随后可选择至少一种前体的浓度以将所需量的材料施用到颗粒的表面上。该方法的代表性变型参考实施例1、3、4、5和6进行描述。在一些实施方案中,加热湿法浸渍的粉末包括干燥湿法浸渍的粉末。在一些实施方案中,至少一种前体包含铝(例如,参见实施例1)。在一些实施方案中,制备前体溶液包括将第一前体溶解于溶剂的第一部分中以形成第一溶液,以及将第二前体溶解于溶剂的第二部分中以形成第二溶液。溶剂的第一部分和溶剂的第二部分总计可对应于溶剂的量。在这些实施方案中,制备前体溶液还包括将第一溶液与第二溶液混合,从而形成前体溶液。在一些情况下,第一前体包含铝,第二前体包含磷酸盐(例如,参见实施例3)。在其它情况下,第一前体包含钴,第二前体包含磷酸盐(例如,参见实施例4)。在其它情况下,第一前体包含铝,第二前体包含锂(例如,参见实施例5)。在一些实施方案中,制备前体溶液包括将第一前体溶解于溶剂的第一部分中以形成第一溶液,将第二前体溶解于溶剂的第二部分中以形成第二溶液,以及将第三前体溶解于溶剂的第三部分中以形成第三溶液。溶剂的第一部分、溶剂的第二部分和溶剂的第三部分总计对应于溶剂的量。在此类实施方案中,制备前体溶液还包括将第一溶液、第二溶液和第三溶液混合,从而形成前体溶液。在一些实施方案中,第一前体、第二前体和第三前体中的至少一者包含锂。在一些实施方案中,第一前体包含钴,第二前体包含锂,并且第三前体包含磷酸盐(例如,参见实施例6)。在一些实施方案中,将第一溶液加入到粉末中包括干燥部分湿法浸渍的粉末。在一些实施方案中,加热湿法浸渍的粉末包括干燥湿法浸渍的粉末。在一些实施方案中,第一前体包含铝,第二前体包含氟(例如,参见实施例2)。根据一个例示性实施方案,用于改性颗粒表面的方法包括搅拌颗粒悬浮液。颗粒包含式(iii)的化合物。该方法还涉及在搅拌的同时向颗粒悬浮液中添加一种或多种前体。在一些实施方案中,该方法还包括在添加前体溶液之后过滤颗粒。该方法的代表性变型参考实施例7至9进行描述。在一些实施方案中,该方法包括在添加至少一种前体之后过滤颗粒并且将经过滤的颗粒加热至高温。在一些实施方案中,前体溶液可包含铝或钴(例如,参见实施例8)。金属前体的非限制示例包括:铝前体,例如al(no3)3;以及钴前体,例如co(no3)3。根据一个例示性实施方案,用于改性粉末中颗粒表面的方法包括将纳米晶体材料颗粒与粉末颗粒共混。粉末颗粒可包含根据式(i)、(iia)或(iii)的化合物,如本文所述。颗粒可与纳米晶体材料混合并且同时共混,和/或经受压缩力、剪切力或它们的组合。此类力可诱导纳米晶体材料颗粒结合到粉末颗粒的表面上。纳米晶体材料颗粒和粉末颗粒可以一定比例共混,使得纳米晶体材料颗粒在粉末颗粒上形成预定量的涂层。该方法的代表性变型参考实施例10至12进行描述。在一些实施方案中,纳米晶体材料颗粒可包含氧化铝材料、氟化铝材料或它们的组合。在一些变型中,本公开涉及由式(iv)表示的化合物:liαco1-xmnxoδ(iv)其中0.95≤α≤1.10,0≤x≤0.10,并且1.90≤δ≤2.20。在一些变型中,0.98≤α≤1.01。在式(iv)的一些变型中,0.98≤α≤1.01并且x=0.03。在式(iv)的一些变型中,1.00≤α≤1.05。在一些变型中,0<x≤0.10。在另外的变型中,本公开涉及由式(iv)表示的化合物,其中0.95≤α≤1.05并且0.02≤x≤0.05。在另一方面,本公开涉及由式(iv)表示的化合物,其中0.95≤α≤1.05并且x=0.04。在一些变型中,x=0.03。在式(iv)的另外变型中,1.01≤α≤1.05并且0.02≤x≤0.05。在式(iv)的另外变型中,1.01≤α≤1.05并且x=0.04。在式(iv)的一些变型中,1.00<α≤1.10。在式(iv)的其它变型中,1.00<α≤1.05。在另一方面,本公开涉及由式(iv)表示的化合物,其中0.98≤α≤1.01,x=0.03,并且δ=2。应当理解,α表示锂含量与总过渡金属含量(即co和mn的总含量)的摩尔比。在各个方面,增大锂含量可增大电容、改善稳定性、增大包含化合物的颗粒的重量密度、增大颗粒密度和/或增大阴极活性材料的颗粒强度。在各个方面,减小锂含量可增大电容、改善稳定性、增大包含化合物的颗粒的重量密度、增大颗粒密度和/或增大阴极活性材料的颗粒强度。在一些变型中,式(iv)的化合物可表示为两相的固溶体,即li2mno3和licoo2的固溶体。在这些变型中,化合物可根据式(va)进行描述:(x)[li2mno3]·(1-x)[licoo2](va)其中mn为具有+4的平均氧化态(即,四价)的阳离子,co为具有+3的平均氧化态(即,三价)的阳离子。式(va)的更紧凑标记如下所示:li1+xco1-xmnxo2+x(vi)在式(vi)中,x包括mn和co两者。由于mn和co之间存在不同的价,这样包含mn可能影响化合物的锂含量和氧含量。在式(iv)中,'x'的组合物可为0≤x≤0.10。在一些变型中,0<x≤0.10。在此类变型中,锂含量在式(vi)中可为1至1.10,氧含量可为2至2.10。然而,本文所公开的化合物具有可独立于x而变化的锂含量和氧含量。例如但不限于,由于本领域的技术人员有意选择的合成条件,锂含量和氧含量可能在化学计量值方面有所不同。因此,式(v)和(vi)中的下标并非意在限制式(iv),即,α不一定等于1+x,并且δ不一定等于2+x。应当理解,由式(iv)表示的化合物的锂含量和氧含量可相对于式(vi)的化学计量值为欠化学计量或过化学计量。在一些变型中,式(iv)的化合物可表示为两相的固溶体,即li2mno3和licoo2的固溶体。在这些变型中,化合物可根据式(vb)进行描述:(x)[li2mno3]·(1-x)[li(1-y)co(1-y)mnyo2](vb)其中mn为具有+4的平均氧化态(即,四价)的阳离子,co为具有+3的平均氧化态(即,三价)的阳离子。本公开还涉及包含本文所述化合物的粉末。在各个方面,本公开涉及一种粉末,该粉末包含具有liαco1-xmnxoδ的颗粒,其中0.95≤α≤1.10,0≤x≤0.10,并且1.90≤δ≤2.20。在一些变型中,0<y≤0.10。粉末可用作阴极活性材料的一部分或全部(即,阴极活性材料包含粉末)。在一些实施方案中,0.98≤α≤1.01并且x=0.03。在一些实施方案中,1.00≤α≤1.05。在另外的实施方案中,1.01≤α≤1.05并且0.02≤x≤0.05。在另外的实施方案中,1.01≤α≤1.05并且x=0.04。在一些实施方案中,1.00<α≤1.10。在其它实施方案中,1.00<α≤1.05。在一些变型中,式(iv)的化合物可表示为两相的固溶体,即li2mno3和licoo2的固溶体。在这些变型中,化合物可根据式(va)或式(vb)进行描述,其中mn为具有+4的平均氧化态(即,四价)的阳离子,co为具有+3的平均氧化态(即,三价)的阳离子。另选地,更紧凑的标记表示为式(vc):li1+x-y-xyco(1-x)*(1-y)mn(x+y-x*y)o2+x(vc)在式(vc)中,x可包括mn和co两者。不希望受限于特定机制或作用模式,由于mn和co之间存在不同的价,包含mn可能影响化合物的锂含量和氧含量。在式(vc)中,'x'和'y'的组合至少为零且小于或等于0.10。在一些变型中,'x'和'y'的组合可大于零且小于或等于0.10。在此类变型中,对于其它式,锂含量可为本文所述的任何范围。在一些变型中,锂可为0.9至1.10。在一些变型中,氧含量可为2至2.10。应当理解,就本文提出的所有式而言,本文所公开的化合物具有可独立于x和y而变化的锂含量和氧含量。化合物和粉末可为锂离子电池的阴极活性材料,如本文所述。这些阴极活性材料分别在锂离子电池充电和放电期间通过释放和储存锂离子来帮助储能装置。不希望受限于特定机制或作用模式,当粉末用作阴极活性材料时,粉末的特性可提供改善的电池性能。包含本文所述化合物的粉末在先前已知的化合物中具有增大的振实密度。包含这些粉末作为阴极活性材料的电池具有增大的体积能量密度。在一些情况下,具有此类阴极活性材料的电池具有大于130mah/g的比电容。在一些情况下,比电容大于140mah/g。在一些情况下,比电容大于150mah/g。在一些情况下,比电容大于160mah/g。在一些情况下,比电容大于170mah/g。增大阴极活性材料中的初始锂含量(例如,已制备的锂含量)可增大体积能量密度和/或循环寿命。阴极活性材料可表现出较高的振实密度和/或改善的颗粒强度。在一些情况下,阴极活性材料可表现出等于或大于2.1g/cm3的振实密度。在一些情况下,阴极活性材料可表现出等于或大于2.2g/cm3的振实密度。在一些情况下,阴极活性材料可表现出等于或大于2.3g/cm3的振实密度。在一些情况下,阴极活性材料可表现出大于或等于2.4g/cm3的振实密度。在一些变型中,本公开涉及用于锂离子电池的阴极活性材料,该阴极活性材料包含具有mn4+的四价金属的锂钴氧化物。在这些材料中,三价co离子co3+可用作提供电容的宿主。不希望受限于任何理论或作用模式,掺入mn4+可提高锂钴氧化物在高电压充电下的稳定性,并且在过渡通过4.1至4.6v区域时(即,在充电和放电期间)还可帮助保持的晶体结构。在一些情况下,电压充电可等于或大于4.4v。在一些情况下,电压充电可等于或大于4.5v。在一些情况下,电压充电可等于或大于4.6v。mn的程度可影响阴极活性材料所需的额外锂量。例如但不限于,具有92%的co和8%的mn的锂钴氧化物的组合物可对应于6摩尔%至10摩尔%的过量单位的锂含量。然而,一般来讲,锂含量的范围可基于锰取代的程度而变化,并且参照图13所示,本领域的技术人员可使用经验数据来确定锂含量范围。图13呈现了根据例示性实施方案表示摩尔比对包含liαco0.96mn0.04oδ(即,x=0.04)的阴极活性材料的电容(即,第一循环放电电容)和效率的影响的数据的曲线图。在横坐标上指示的摩尔比对应于α,这是锂含量对总过渡金属含量的摩尔比(即,在图13中被描述为“li/tm比”)。为了确定摩尔比,使用电感耦合等离子体光发射光谱测定(icpoes)来化学表征阴极活性材料,如本领域技术人员已知的那样。电容和效率分别列于纵坐标的左侧和右侧。参照图13,第一曲线300表示电容随摩尔比的变化,拟合于第一组数据点302。第二曲线304表示效率随摩尔比的变化,拟合于第二组数据点306。对于该特定组合物(即x=0.04),第一曲线300在对应于0.98≤α≤1.05的范围内表现出最大值。然而,应当理解,范围限制可响应于x的变化而改变,x表示阴极材料中mn的量。一般来讲,本领域的技术人员可改变α和x以实现所需的电容和效率组合。在某些变型中,本文所述的化合物可允许过量的锂存储。在图13中,显示增加的锂(即,0.98<α≤1.05)可增大阴极材料相对于单位(即,α=1.00)的电容。这种增大的电容允许本领域的技术人员能够将具体(增大的)电容与期望的效率相匹配。此类增大的电容是非预期的。在各个方面,额外的锂通常可预期用作阴极材料中的污染物,从而劣化其性能。实际上,当过量的锂超过阈值(例如,在图13中超过α~1.05)时,电容和效率两者均降低。在各个方面,本文所述的化合物提供比已知化合物增大的阴极活性材料电容和稳定性。图14呈现了根据例示性实施方案表示摩尔比(由α表示)和mn含量(由x表示)对式(iv)化合物(即,liαco0.96mn0.04oδ)的c轴晶格参数的影响的数据的曲线图。组合物中的氧可对应于δ=2,但来自该化学计量的变型也是可能的(即,在1.90≤δ≤2.20内)。使用x射线衍射技术生成数据,如本领域技术人员已知的那样。licoo2的衍射图案的(003)峰的特征是根据x和α偏移。对于给定的x(例如,0.04或4摩尔%),c晶格随α增大而收缩(即,当α从1.0062增大至1.0925)。不希望受限于任何理论或作用模式,据信由于存在mn4+,过量的锂将移动到liαco0.96mn0.04oδ过渡金属层中。这种移动增大了材料的锂电容和稳定性。图15呈现了根据例示性实施方案表示liαco0.96mn0.04oδ的c轴晶格参数随摩尔比的变化的数据的曲线图。(摩尔比α在横坐标上指示为“icpli/tm”)。组合物中的氧可对应于δ=2,但来自该化学计量的变型也是可能的(即,在1.90≤δ≤2.20内)。为了确定α,使用icpoes来化学表征阴极活性材料,如本领域技术人员已知的那样。c轴晶格参数的变化沿循s形曲线。s形曲线趋于在α的极值处变成渐近线(即,图15所示的α的最低值和最高值)。本公开还涉及制备包含liαco1-xmnxoδ的粉末的方法。该方法包括将锂源与前体颗粒混合以产生反应物,然后将反应物加热至高温的步骤。可煅烧锂离子阴极材料的前体以获得正极材料。然而,很难控制颗粒烧结过程以产生高密度颗粒并且同时产生高电容材料。由于高温下的高蒸发速率,现有技术的程序添加了至多10重量%的额外锂的可变量。前体颗粒包含含有co和mn的过渡金属氢氧根离子。锂源的非限制性示例包括氢氧化锂(即,lioh)和碳酸锂(即,li2co3)。然而,其它锂源也是可能的。该方法还包括将反应物加热至等于或高于800℃的温度。在这些方法中,0≤x≤0.10并且1.90≤δ≤2.20。在一些变型中,0<x≤0.10。选择锂源与前体颗粒之比,使得0.95≤α≤1.10。由于加热期间锂的蒸发损失,该比率可等于或大于摩尔比α。在一些实施方案中,温度介于800至1200℃之间。在一些方面,加热温度为约800至约1000℃。在其它方面,加热温度为约1000至约1100℃。在其它方面,加热温度为约1100至约1200℃。在其它方面,加热温度为约900至约1000℃。在其它方面,加热温度为约800至约900℃。不希望受限于任何机制或作用模式,前述范围内的温度使得粉末颗粒表现出适合锂离子电池应用的足够重量密度和强度。这些范围也对应于改善的电容和第一循环效率。同样,不希望受限于任何机制或作用模式,应当理解,高重量密度能够增大阴极活性材料的能量密度。颗粒强度可改善电池制造期间的有效处理和电池工作期间的循环稳定性。在各个方面,摩尔比(即,α)、温度和对应的加热周期可控制颗粒的烧结和压实。图16呈现了根据例示性实施方案的一组扫描电镜显微图,示出了在900℃、1000℃、1050℃和1100℃下制备的粉末。温度从左到右升高,放大率从上到下增大。粉末颗粒的组合物对应于α=1.04并且x=0.04。在900℃和1000℃下,粉末颗粒保持前体颗粒的片状结构。粉末分别表现出2.0和2.1g/cm3的振实密度。然而,在1050℃和1100℃下,粉末中单个颗粒的形态改变为平滑的(即,平滑表面)。这种平滑形态显示出部分熔融。对应的振实密度增大至2.3至2.4g/cm3。这些粉末表现出更大的强度,因为晶粒(即初级颗粒)已生长并且更好地结合在一起。此外,晶粒的数量减少,并且其中存在较少的晶界。在这些方法中,额外的锂含量控制前体颗粒的烧结程度。当包含作为阴极活性材料时,此类额外的锂含量还控制粉末的电容。应当理解,应存在足够的锂,以与前体颗粒反应和烧结,但不具有使前体颗粒过度烧结的风险。过度烧结可产生固体料团。即使可避免固体料团,过量的锂可降低阴极活性材料的电容和效率。颗粒密度和强度随摩尔比(即,α)增大而增大。图17呈现了根据例示性实施方案的一组扫描电镜显微图,示出了温度和摩尔比(α)对颗粒形态的影响。粉末的组合物(即其中的颗粒)对应于x=0.04,并且α从1.00渐进地增大至1.04、至1.06、至1.08以及至1.10(即,图17中从左到右)。示出了两个温度,即1050℃和1100℃。组合物中的氧可对应于δ=2,但来自该化学计量的变型也是可能的(即,在1.90≤δ≤2.20内)。加热温度可对粉末的表面形态和增大的振实密度产生影响。在图17所示的实施方案中,在1050℃和小于1.02的α值下,次级颗粒在煅烧后自由流动。然而,初级晶粒没有很好的结合强度。然而,在较大的摩尔比下,单个晶粒更好地融合在一起。在1100℃下,次级颗粒具有平滑表面和良好结合的晶粒。随着摩尔比增大(即,在1100℃下),次级颗粒开始结合在一起,从而形成颗粒的刚性烧结料团。该刚性烧结料团可通过研磨而破碎。在一些实施方案中,1.00≤α≤1.05。在该方法的其它实施方案中,1.00<α≤1.10。在另外的实施方案中,1.00<α≤1.05。在一些实施方案中,1.01≤α≤1.05并且0.02≤x≤0.05。在另外的实施方案中,1.01≤α≤1.05并且x=0.04。图18呈现了根据例示性实施方案表示混合比对包含liαco0.96mn0.04oδ的阴极活性材料的电容和效率的影响的数据的曲线图。混合比对应于锂源与前体颗粒混合的比。阴极活性材料包括图17所示并且在1050℃下煅烧的粉末。组合物中的氧可对应于δ=2,但来自该化学计量的变型也是可能的(即,在1.90≤δ≤2.20内)。在图18中,横坐标示出了混合比从1.00增大至1.10。在混合比为1.04(对应于通过icp-oes测量的α=1.01)时,阴极活性材料具有192mah/g的最大放电电容(即,从4.5v放电至2.75v期间)。对于混合比1.06、1.08和1.10(即,分别为通过icp-oes测量的α=1.02、1.03和1.04),也可获得约190mah/g的高放电值。在一些变型中,本公开涉及由式(vii)表示的化合物:liαco1-x-ymymnxoδ(vii)其中0.95≤α≤1.30,0<x≤0.30,0≤y≤0.10,1.98≤δ≤2.04,并且m为选自b、na、mg、ti、ca、v、cr、fe、co、ni、cu、zn、al、sc、y、ga、zr、ru和mo的至少一种元素。式(vii)的化合物为单相。该化合物可具有三角形晶体结构。在另一些变型中,0.98≤α≤1.16并且0<x≤0.16。在一些变型中,0.98≤α≤1.16,0<x≤0.16,0<y≤0.05,1.98≤δ≤2.04在一些变型中,本公开涉及由式(viii)表示的化合物:liαco1-x-yalymnxoδ(viii)其中0.95≤α≤1.30,0<x≤0.30,0≤y≤0.10,并且1.98≤δ≤2.04。在一些变型中,0.96≤α≤1.04,0<x≤0.10,0≤y≤0.10,并且1.98≤δ≤2.04。在一些变型中,对于由式(viii)表示的化合物,0.98≤α≤1.01,0.02≤x≤0.04,0≤y≤0.03,并且1.98≤δ≤2.04。式(viii)的化合物为单相。该化合物可具有三角形晶体结构。在一些情况下,对于由式(vii)和(viii)表示的化合物,α>1+x。在其它情况下,α<1+x。因此,式(vii)和(viii)中的α可偏离α=1+x,从而可能与li2mno3和(1-x)lico1-ymyo2之间的固溶体相关联。该固溶体可由xli2mno3·(1-x)lico1-ymyo2和xli2mno3·(1-x)li1-yco1-ymyo2表示,或者以紧凑标记由li1+xco1-x-y+xym(1-x)*ymnxo2+x或li1+x-y+xyco1-x-y+xym(1-x)*ymnxo2+x表示。如上所述,各种化合物不包括第二相,诸如具有不同晶体结构的第二相。应当理解,li2mno3为具有单斜c2/m晶体结构的“岩盐”相。因此,基于li2mno3和lico1-ymyo2之间的固溶体的阴极活性材料具有表现出单斜c2/m晶体结构的“岩盐”相的部分。除与lico1-ymyo2相关的任何相之外,还出现该“岩盐”相,从而使固溶体为双相(或多相)。相比之下,由式(vii)和(viii)表示的阴极活性材料及其变型为单相并且仅具有三角形晶体结构。不希望受限于任何特定机构或作用方式,将锰掺入式(vii)和(viii)的化合物中,稳定其晶体结构,但m的其它成分也可能有助于稳定。化合物在其中均匀分布有mn的晶体结构中包括co的亚晶格。另选地,在一些变型中,锰的簇(例如,对、三重态等)出现在co的亚晶格中并且均匀分布于其中。簇可例如通过核磁共振(nmr)检测,如本文所述。化合物中存在mn可限制在电池工作(例如,充电、放电等)期间从晶体结构发生相变。存在mn还可改善化合物在较高电压(例如,等于或大于4.0伏的电压)下的氧化稳定性。在一些变型中,x对应于mn对co的取代程度。mn取代的程度可与在阴极活性材料中使用时化合物的稳定性相关。在各个方面,mn对co的取代可大于或等于取代下限。另选地,mn对co的取代可等于或小于取代限。在一些变型中,x为至少0.001。在一些变型中,x为至少0.01。在一些变型中,x为至少0.02。在一些变型中,x为至少0.03。在一些变型中,x为至少0.04。在一些变型中,x为至少0.05。在一些变型中,x为至少0.06。在一些变型中,x为至少0.07。在一些变型中,x为至少0.08。在一些变型中,x为至少0.09。在一些变型中,x为至少0.10。在一些变型中,x为至少0.12。在一些变型中,x为至少0.14。在一些变型中,x为至少0.16。在一些变型中,x为至少0.18。在一些变型中,x为至少0.20。在一些变型中,x为至少0.22。在一些变型中,x为至少0.24。在一些变型中,x为至少0.26。在一些变型中,x为至少0.28。在一些变型中,x小于或等于取代上限。在一些变型中,x小于或等于0.30。在一些变型中,x小于或等于0.28。在一些变型中,x小于或等于0.26。在一些变型中,x小于或等于0.24。在一些变型中,x小于或等于0.22。在一些变型中,x小于或等于0.20。在一些变型中,x小于或等于0.18。在一些变型中,x小于或等于0.16。在一些变型中,x小于或等于0.14。在一些变型中,x小于或等于0.12。在一些变型中,x小于或等于0.10。在一些变型中,x小于或等于0.09。在一些变型中,x小于或等于0.08。在一些变型中,x小于或等于0.07。在一些变型中,x小于或等于0.06。在一些变型中,x小于或等于0.05。在一些变型中,x小于或等于0.04。在一些变型中,x小于或等于0.03。应当理解,在任何组合中,取代下限和上限可以上述任何变型组合以限定x的范围。例如但不限于,x可在0.001至0.01的范围内。x可在0.02至0.05的范围内(即0.02≤x≤0.07)。在另一个非限制性示例中,x可在0.24至0.28的范围内(即0.06≤x≤0.10)。在另一个非限制性示例中,x可在0.24至0.28的范围内(即0.22≤x≤0.28)。上限和下限的其它组合也是可能的。此外,不希望受限于任何特定机制或作用模式,可在式(vii)和(viii)的化合物中选择一定含量的锂以稳定其晶体结构并提高电池性能。锂的含量可选择性地补充对co的取代程度(即,经由mn、m或铝取代)。例如但不限于,α可被选择为约1+x。相对于α=1,此选择可提高晶体结构在锂化和去锂化期间的稳定性(参见图29)。在另一个非限制性示例中,αδ可被选择为大于1+x以容纳除mn之外的取代(即,m和y)。这些取代物可使得化合物具有改善的放电能量(参见图33)。在又一个示例中,锂含量可根据α<1+x来选择以增强电池性能。如图30所示,li1.003co0.97mn0.03o2比li1.014co0.97mn0.03o2保持更高的重复循环后放电能量,但后者具有更高的α值。在一些变型中,0.98≤li/me≤1.01。应当理解,α对应于锂与co及其取代物(即,对于式(vii),为m和mn,对于式(viii),为m、mn和铝)之比。对于式(vii),该比率可描述为[li]/[co1-x-ymymnx]。对于式(vii),该比率可描述为[li]/[co1-x-yalymnx]。对于y=0的化合物,α对应于锂与co和mn之比,即[li]/[co1-xmnx]。后一比率可被称为锂与过渡金属之比(即,li/tm)。在一些变型中,这些化合物保持单相并且具有三角形晶体结构。在一些变型中,α可等于或大于下限。在一些变型中,α为至少0.95。在一些变型中,α为至少0.98。在一些变型中,α为至少1.00。在一些变型中,α大于1.00。在一些变型中,α为至少1.02。在一些变型中,α为至少1.04。在一些变型中,α为至少1.06。在一些变型中,α为至少1.08。在一些变型中,α为至少1.10。在一些变型中,α为至少1.12。在一些变型中,α为至少1.14。在一些变型中,α为至少1.16。在一些变型中,α为至少1.20。在一些变型中,α为至少1.22。在一些变型中,α为至少1.24。在一些变型中,α为至少1.26。在一些变型中,α为至少1.28。类似地,α可小于或等于下限。在一些变型中,α小于或等于1.30。在一些变型中,α小于或等于1.28。在一些变型中,α小于或等于1.26。在一些变型中,α小于或等于1.24。在一些变型中,α小于或等于1.22。在一些变型中,α小于或等于1.20。在一些变型中,α小于或等于1.16。在一些变型中,α小于或等于1.14。在一些变型中,α小于或等于1.12。在一些变型中,α小于或等于1.10。在一些变型中,α小于或等于1.08。在一些变型中,α小于或等于1.06。在一些变型中,α小于或等于1.04。在一些变型中,α小于或等于1.02。在一些变型中,α小于或等于1.00。在一些变型中,α小于1.00。在一些变型中,α小于或等于0.98。在这些变型中,化合物也保持单相并且具有三角形晶体结构。应当理解,α取代下限和上限可以上述任何变型组合以限定范围。例如但不限于,α可在0.95至1.00的范围内(即,0.95≤α≤1.00)。在另一个非限制性示例中,α可在1.00至1.06的范围内(即,1.00≤α≤1.08)。在另一个非限制性示例中,α可在1.22至1.28的范围内(即,1.22≤α≤1.28)。上限和下限的其它组合也是可能的。还应当理解,x是上下限以及α的上下限可以上述任何变型组合以限定x和α的范围组合。例如但不限于,x≥0.03并且0.98≤α≤1.10。在另一个非限制性示例中,0.02≤x≤0.10并且0.95≤α≤1.12。在另一个非限制性示例中,0.04≤x≤0.12并且0.95≤α≤1.16。其它范围组合也是可能的。在一些变型中,α接近1+x。在这些变型中,α可能接近1+x并且公差不大于5%。公差可对应于(1–t)*(1+x)≤α≤(1+x)*(1+t),其中t≤0.05。在一些变型中,公差小于或等于±5.0%。在一些情况下,公差小于或等于±4.5%。在一些情况下,公差小于或等于±4.0%。在一些情况下,公差小于或等于±3.5%。在一些情况下,公差小于或等于±3.0%。在一些情况下,公差小于或等于±2.5%。在一些情况下,公差小于或等于±2.0%。在一些情况下,公差小于或等于±1.5%。在一些情况下,公差小于或等于±1.0%。在一些情况下,公差为至少±1.0%。在一些情况下,公差为至少±0.5%。在一些情况下,公差为至少±1.0%。在一些情况下,公差为至少±1.5%。%。在一些情况下,公差为至少±2.0%。在一些情况下,公差为至少±2.5%。在一些变型中,公差为至少±3.0%。在一些情况下,公差为至少±3.5%。在一些情况下,公差为至少±4.0%。在一些情况下,公差为至少±4.5%。应当理解,当α接近1+x时,对应的化合物保持单相特征并且其中不含li2mno3。此外,化合物在充电和放电期间可表现出改善的相变抗性,以及改善的放电能量。此类化合物的非限制性示例包括li1.050co0.96mn0.04o2、li1.074co0.96mn0.04o2、li1.197co0.78mn0.22o2和li1.247co0.72mn0.28o2。在一些变型中,化合物选自li1.050co0.96mn0.04o2、li1.074co0.96mn0.04o2、li1.081co0.96mn0.04o2、li1.089co0.96mn0.04o2、li1.050co0.93mn0.07o2、li1.065co0.90mn0.10o2、li1.100co0.90mn0.10o2、li1.110co0.90mn0.10o2、li1.158co0.90mn0.10o2、li0.975co0.84mn0.16o2、li1.050co0.84mn0.16o2、li1.114co0.84mn0.16o2、li1.197co0.78mn0.22o2、li1.190co0.72mn0.28o2和li1.247co0.72mn0.28o2。在这些化合物中,锰取代钴且不导致形成li2mno3,即,化合物为单相并且具有三角形晶体结构。在一些变型中,化合物为li0.991mn0.03co0.97o2。在一些变型中,化合物为li0.985mn0.03co0.97o2。本公开还涉及包含本文所述化合物的粉末。在各个方面,本公开涉及包括含有上述任何化合物的颗粒的粉末。粉末可用作阴极活性材料的一部分或全部(即,阴极活性材料包含粉末)。化合物和粉末可为锂离子电池的阴极活性材料,如本文所述。这些阴极活性材料分别在锂离子电池充电和放电期间通过释放和储存锂离子来帮助储能装置。不希望受限于特定机制或作用模式,化合物可在电池单元的充电和放电期间改善阴极活性材料的体积能量密度、能量保留率和/或循环能力。化合物可改善阴极活性材料的热稳定性。在一些变型中,颗粒具有大于或等于第一下限的平均粒径。在一些变型中,颗粒具有至少5μm的平均直径。在一些变型中,颗粒具有至少10μm的平均直径。在一些变型中,颗粒具有至少15μm的平均直径。在一些变型中,颗粒具有至少20μm的平均直径。在一些变型中,颗粒具有至少25μm的平均直径。在一些变型中,颗粒具有小于或等于第一上限的平均粒径。在一些变型中,颗粒具有小于或等于30μm的平均直径。在一些变型中,颗粒具有小于或等于25μm的平均直径。在一些变型中,颗粒具有小于或等于20μm的平均直径。在一些变型中,颗粒具有小于或等于15μm的平均直径。在一些变型中,颗粒具有小于或等于10μm的平均直径。在一些变型中,颗粒具有小于或等于5μm的平均直径。应当理解,第一下限和第一上限可以上述任何变型组合以限定平均粒径的第一范围。例如但不限于,平均粒径可在10μm至20μm的范围内。在另一个非限制性示例中,平均粒径可在20μm至25μm的范围内。其它范围也是可能的。具有前述平均粒径的颗粒,无论其通过第一下限、第一上限还是二者(即,第一范围)来表征,都可根据共沉淀方法进行处理。在一些变型中,颗粒具有大于或等于第二下限的平均粒径。在一些变型中,颗粒具有至少200nm的平均直径。在一些变型中,颗粒具有至少300nm的平均直径。在一些变型中,颗粒具有至少400nm的平均直径。在一些变型中,颗粒具有至少500nm的平均直径。在一些变型中,颗粒具有至少600nm的平均直径。在一些变型中,颗粒具有至少700nm的平均直径。在一些变型中,颗粒具有小于或等于第二上限的平均粒径。在一些变型中,颗粒具有小于或等于800nm的平均直径。在一些变型中,颗粒具有小于或等于700nm的平均直径。在一些变型中,颗粒具有小于或等于600nm的平均直径。在一些变型中,颗粒具有小于或等于500nm的平均直径。在一些变型中,颗粒具有小于或等于400nm的平均直径。在一些变型中,颗粒具有小于或等于300nm的平均直径。应当理解,第二下限和第二上限可以上述任何变型组合以限定平均粒径的第二范围。例如但不限于,平均粒径可在300nm至500nm的范围内。在另一个非限制性示例中,平均粒径可在400nm至800nm的范围内。其他范围也是可能的。具有前述平均粒径的颗粒,无论其通过第二下限、第二上限还是二者(即,第二范围)来表征,都可根据溶胶-凝胶方法进行处理。在一些变型中,颗粒为由附聚的初级颗粒形成的次级颗粒。附聚的初级颗粒可被烧结在一起。在一些情况下,次级颗粒具有大于或等于下限的平均粒径。下限的非限制性示例包括15μm、20μm和25μm。在某些情况下,次级颗粒具有小于或等于上限的平均粒径。上限的非限制性示例包括30μm、25μm和20μm。应当理解,下限和上限可以上述任何变型组合以限定平均粒径的范围。例如但不限于,平均粒径可在15μm至20μm的范围内。在另一个非限制性示例中,平均粒径可在20μm至25μm的范围内。其它范围也是可能的。在一些变型中,单个初级颗粒占据由对应的次级颗粒所占据体积的一定百分比。在一些情况下,该百分比大于或等于下限。在一些变型中,单个初级颗粒占据由对应的次级颗粒所占据体积的至少30%。在一些变型中,单个初级颗粒占据由对应的次级颗粒所占据体积的至少35%。在一些变型中,单个初级颗粒占据由对应的次级颗粒所占据体积的至少40%。在一些变型中,单个初级颗粒占据由对应的次级颗粒所占据体积的至少45%。在一些变型中,单个初级颗粒占据由对应的次级颗粒所占据体积的至少50%。在一些变型中,单个初级颗粒占据由对应的次级颗粒所占据体积的至少55%。在一些变型中,单个初级颗粒占据由对应的次级颗粒所占据体积的至少60%。在一些变型中,单个初级颗粒占据由对应的次级颗粒所占据体积的至少65%。在一些变型中,单个初级颗粒占据小于或等于由对应的次级颗粒所占据体积的70%。在一些变型中,单个初级颗粒占据小于或等于由对应的次级颗粒所占据体积的65%。在一些变型中,单个初级颗粒占据小于或等于由对应的次级颗粒所占据体积的60%。在一些变型中,单个初级颗粒占据小于或等于由对应的次级颗粒所占据体积的55%。在一些变型中,单个初级颗粒占据小于或等于由对应的次级颗粒所占据体积的50%。在一些变型中,单个初级颗粒占据小于或等于由对应的次级颗粒所占据体积的45%。在一些变型中,单个初级颗粒占据小于或等于由对应的次级颗粒所占据体积的40%。在一些变型中,单个初级颗粒占据小于或等于由对应的次级颗粒所占据体积的35%。应当理解,下限和上限可以上述任何变型组合以限定百分比的范围。例如但不限于,百分比可在30%至50%的范围内。然而,其它范围也是可能的。如本文所述,可通过使用较高的烧结温度来形成较大粒度和由单个初级颗粒占据的次级颗粒的百分比。不希望受限于特定机制或作用模式,在一些情况下,颗粒不容易破裂,从而可提供比常规颗粒增强的稳定性。在化合物中包括mn和/或铝来取代co、改变锂的量和/或包括al2o3涂层均可降低或有可能降低不稳定的相变。不希望受限于特定机制或作用模式,另外的元素也可在较高的电池截止电压上限下为化合物提供更大的氧化稳定性。在一些变型中,对于至少4.4v和li0/li+的情况,化合物、颗粒和/或阴极活性材料可具有更大的稳定性。在一些变型中,颗粒具有增大的颗粒强度。当颗粒用于阴极活性材料时,增大的颗粒强度导致增大的能量保留率。在一些变型中,阴极活性材料中mn的增加量提供改善的电池稳定性。在一些变型中,增加的mn量升高分解的起始温度。在一些变型中,增加的mn量可导致化合物在分解温度下减少热释放量。在一些变型中,阴极活性材料具有至少700wh/kg的第一循环放电能量。在一些变型中,阴极活性材料具有至少725wh/kg的第一循环放电能量。在一些变型中,阴极活性材料具有至少750wh/kg的第一循环放电能量。在一些变型中,阴极活性材料具有至少775wh/kg的第一循环放电能量。在一些变型中,阴极活性材料具有至少800wh/kg的第一循环放电能量。在一些变型中,阴极活性材料具有至少825wh/kg的第一循环放电能量。在一些变型中,阴极活性材料具有至少850wh/kg的第一循环放电能量。在一些变型中,阴极活性材料具有至少875wh/kg的第一循环放电能量。在一些变型中,阴极活性材料具有至少180mah/g的第一循环放电电容。在一些变型中,阴极活性材料具有至少185mah/g的第一循环放电电容。在一些变型中,阴极活性材料具有至少190mah/g的第一循环放电电容。在一些变型中,阴极活性材料具有至少195mah/g的第一循环放电电容。在一些变型中,阴极活性材料具有至少200mah/g的第一循环放电电容。在一些变型中,阴极活性材料具有至少205mah/g的第一循环放电电容。在一些变型中,阴极活性材料具有至少210mah/g的第一循环放电电容。在一些变型中,阴极活性材料具有至少215mah/g的第一循环放电电容。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少65%的能量电容保留率。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少67%的能量电容保留率。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少69%的能量电容保留率。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少71%的能量电容保留率。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少73%的能量电容保留率。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少75%的能量电容保留率。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少77%的能量电容保留率。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少79%的能量电容保留率。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少81%的能量电容保留率。在一些变型中,阴极活性材料在52个充电放电循环之后具有至少83%的能量电容保留率。化合物、粉末和阴极活性材料可用于如本文所述的电池。材料可用于电子设备。本文的电子设备可指本领域已知的任何电子设备,包括便携式电子设备。例如,电子设备可为电话诸如移动电话和座机电话,或任何通信设备诸如智能电话(包括例如iphonetm),以及电子邮件发送/接收设备。电子设备还可为娱乐设备,包括便携式dvd播放器、常规dvd播放器、蓝光影碟播放器、视频游戏控制器、音乐播放器诸如便携式音乐播放器(例如,)。电子设备可为显示器的一部分,显示器诸如数字显示器、电视显示器、电子书阅读器、便携式web浏览器(例如,)、手表(例如,applewatch)或计算机监视器。电子设备还可为提供控制的设备的一部分,诸如控制图像、视频、声音的流(例如,apple),或者其可为电子设备的远程控制。此外,电子设备可为计算机或其附件的一部分,诸如硬盘塔外壳或保护套、膝上型计算机外壳、膝上型计算机键盘、膝上型计算机触控板、台式计算机键盘、鼠标和扬声器。电池和电池组还可应用于诸如手表或时钟等设备。由电池或电池组驱动的部件可包括但不限于微处理器、计算机可读存储介质、输入和/或输出设备,诸如键盘、触控板、触摸屏、鼠标、扬声器等。实施例以下实施例仅用于说明目的。对于本领域的技术人员将显而易见的是,可在不脱离本公开的范围的情况下对材料和方法实施多种修改。实施例1—湿法浸渍以形成al2o3涂层边搅拌边将去离子水(millipore超纯水,18mω·cm)逐滴加入10g基料粉末(即,10gli1.04co0.96mn0.04o2.04基料粉末)中。当粉末被润湿但仍然松散时,停止搅拌。(在粉末形成湿软或粘性团块之前,停止添加去离子水。)然后计算比率r,该比率等于所添加水的量(重量或体积)除以基料粉末的量。将粉末润湿至合适湿度所需的水量取决于所用基料粉末的表面积。一般来讲,较大的表面积需要更多的水。接着,选择一定量(例如,重量)的基料粉末。铝盐前体(例如,硝酸铝九水合物)的量被确定为会对应于期望量基料粉末(例如,0.1重量%)上al2o3涂层的期望水平。然后使用比率(即,用r乘以基料粉末的重量)来计算去离子水的量。表1给出了基料粉末、溶剂(即,去离子水)和铝盐前体的类型和量。然后测量表1中给出的量,包括其量为预定的去离子水的量。将硝酸铝九水合物溶于去离子水中以形成澄清溶液。边搅拌边将澄清溶液以液滴形式加至玻璃容器中的基料粉末。澄清溶液添加完毕后,继续搅拌基料粉末几分钟以确保充分混合。形成润湿的松散粉末。将润湿的松散粉末于80℃的烘箱中干燥过夜。然后将干燥粉末转移到al2o3坩埚中,并在120℃下热处理2小时。该热处理后,在停滞空气中于500℃下进行后续热处理4小时。将经热处理的粉末通过325目筛网。偶尔需要用研钵和研杵进行轻度研磨,以破坏经热处理的粉末的凝聚部分。表1.用于10gli1.04co0.96mn0.04o2上0.1重量%al2o3涂层的材料材料试剂/材料用量基料粉末li1.04co0.96mn0.04o210g溶剂(用于溶解涂层前体)去离子水~0.9–1ml或g涂层前体al(no3)3·9h2o0.075g实施例2—湿法浸渍以形成alf3涂层根据相对于实施例1所述的程序确定比率r。接着,选择一定量(例如,重量)的基料粉末。铝盐前体(例如,硝酸铝九水合物)和氟化盐前体的量被确定为会对应于期望量基料粉末(例如,0.1重量%)上alf3涂层的水平。为确保反应完全,alf3中氟化盐前体的量为氟化物化学计算量的两倍(即,所选择的al与f的摩尔比为1:6)。然后使用比率(即,用r乘以基料粉末的重量)来计算所需去离子水的量。将硝酸铝九水合物溶于第一部分去离子水中以形成第一澄清溶液。将氟化铵溶于第二部分去离子水中以形成第二澄清溶液。将基料粉末转移到玻璃容器中,并向其中以液滴形式快速加入第一澄清溶液(即,以“淹没”基料粉末)。将基料粉末搅拌2分钟,并在105℃下干燥以得到粉饼。将粉饼破碎成松散粉末(例如,用研钵和研杵),然后转移到新鲜的玻璃容器中。轻轻地敲拍新鲜的玻璃容器以将松散粉末装在其中。边搅拌边将第二澄清溶液快速添加至装于玻璃容器中的粉末(即,与第一澄清溶液类似)。将该混合物搅拌2分钟,然后于105℃下干燥。将干燥粉末转移至铝烤箱,在流动的氮气下于120℃热处理2小时。然后在400℃下将经热处理的粉末加热5小时,得到经热处理的粉饼。经热处理的粉饼易于解体,对其进行轻度研磨,并通过325目筛网进行筛分。实施例3—湿法浸渍以形成alpo4涂层将预定量的基料粉末(即,li1.04co0.96mn0.04o2)称量到玻璃容器中。基于所称的基料粉末量计算期望的alpo4涂层量(例如,5重量%)所需的铝前体和磷酸盐前体的量。所用的铝前体包括各种铝盐,诸如硝酸铝、乙酸铝或可溶于水或醇的其它铝盐。所用的磷酸盐前体为磷酸二氢铵(nh4h2po4)、磷酸氢二铵[(nh4)2hpo4]或这两者的组合。将al与p的摩尔比保持在0.9和1.1之间。将铝前体和磷酸盐前体分别溶解于少量水或醇中以形成溶液。然后将两种溶液混合在一起。通过改变磷酸铵盐的比率来调节混合溶液的ph以防止沉淀。用玻璃棒或刮刀搅拌基料粉末,同时将混合溶液逐滴加至基料粉末上。溶液的体积使得基料粉末被初步润湿并且充分混合(即表现出湿稠度)。于50-80℃干燥后,将经干燥的基料粉末在停滞空气中于700℃下干燥5h。实施例4—湿法浸渍以形成co3(po4)2涂层将预定量的基料粉末(即,li1.04co0.96mn0.04o2)称量到玻璃容器中。基于称量的基料粉末量来计算期望的co3(po4)2涂层量(例如,5重量%)所需的钴前体和磷酸盐前体的量。所用的钴前体包括各种钴盐,诸如硝酸钴、乙酸钴,或可溶于水或醇的其它钴盐。所用的磷酸盐前体为磷酸二氢铵(nh4h2po4)、磷酸氢二铵[(nh4)2hpo4]或这两者的组合。将co与p的摩尔比保持在1.4和1.6之间。将钴前体和磷酸盐前体分别溶于少量水或醇中以形成溶液。然后将两种溶液混合在一起。通过改变磷酸铵盐的比率来调节混合溶液的ph以防止沉淀。用玻璃棒或刮刀搅拌基料粉末,同时将混合溶液逐滴加至基料粉末上。所添加溶液的体积使得基料粉末被初步润湿并且充分混合(即表现出湿稠度)。于50-80℃干燥后,将经干燥的基料粉末在停滞空气中于700℃下干燥5h。实施例5—湿法浸渍以形成li-al2o3涂层将预定量的基料粉末(即,li1.04co0.96mn0.04o2)称量到玻璃烧杯中。基于称量的基料粉末量来计算期望的涂层量(例如,0.5重量%)所需的铝前体的量。铝前体包括各种铝盐,诸如硝酸铝、乙酸铝或可溶于水或醇的其它铝盐。将铝前体溶于少量的水或醇中以形成第一澄清溶液。使用介于0.25和1.05之间的li与al摩尔比计算期望的锂前体量。所用的锂前体为氢氧化锂、硝酸锂、乙酸锂或可溶于水或醇的其它锂盐。将所需量的锂前体溶解于少量的水或醇中以形成第二澄清溶液。将第一澄清溶液和第二澄清溶液混合在一起。然后边搅拌边将混合溶液逐滴加至基料粉末。所添加溶液的体积使得基料粉末被初步润湿,但不湿软(即,表现出湿稠度)。于50-80℃干燥后,将经干燥的基料粉末在停滞空气中于500℃下干燥4h。也可改变第一澄清溶液(即,铝溶液)的ph以改善涂层特性,诸如涂层密度和均匀度。实施例6—湿法浸渍以形成li-co3(po4)2涂层将预定量的基料粉末(即,li1.04co0.96mn0.04o2)称量到玻璃烧杯中。基于称量的基料粉末量来计算期望的涂层量(例如,0.5重量%)所需的钴前体、磷酸盐前体和锂前体的量。钴前体包括各种钴盐,诸如硝酸钴、乙酸钴,或可溶于水或醇的其它钴盐。所用的磷酸盐前体为磷酸二氢铵(nh4h2po4)、磷酸氢二铵[(nh4)2hpo4]、磷酸锂或它们的组合。将co与p的摩尔比保持在1.4和1.6之间。使用介于0.3和1.05之间的li与co摩尔比计算期望的锂前体量。所用的锂前体为氢氧化锂、硝酸锂、乙酸锂或可溶于水或醇的其它锂盐。将钴前体、磷酸盐前体和锂前体分别溶于少量的水或醇中以形成对应的澄清溶液。然后将三种溶液混合在一起。然后边搅拌边将混合溶液逐滴加至基料粉末。所添加溶液的体积使得基料粉末被初步润湿,但不湿软(即,表现出湿稠度)。于50-80℃干燥后,将经干燥的基料粉末在停滞空气中于700℃下热处理5h。实施例7—悬浮处理以形成al2o3涂层将al(no3)3水性溶液与基料粉末(li1.02co0.96mn0.04o2)悬浮液混合,然后将其泵入搅拌槽反应器。在搅拌的同时,通过反馈泵使用氨溶液将反应ph值保持在9.3。将悬浮液搅拌2h,过滤、干燥,并且在400℃下于空气中煅烧5h。实施例8—悬浮处理以形成co3(po4)2涂层将第一co(no3)3水性溶液和第二磷酸二氢铵水性溶液泵入搅拌槽反应器中的基料粉末(li1.02co0.96mn0.04o2)悬浮液中。将合并体积搅拌2h,过滤并干燥。将干燥粉末在700℃下于空气中煅烧5h。实施例9—悬浮处理以形成alpo4涂层将第一al(no3)3水性溶液和第二磷酸二氢铵溶液泵入搅拌槽反应器中的基料粉末(li1.02co0.96mn0.04o2)悬浮液中。将合并体积搅拌2h,过滤并干燥。将干燥粉末在700℃下于空气中煅烧5h。实施例10—干法处理以形成al2o3涂层将预定量的基料粉末(li1.02co0.96mn0.04o2)称量并倒入干式涂布机(nobilta,nob-130,细川密克朗公司(hosokawamicronltd))中。接着,根据预定基料粉末(例如,0.5重量%)上所需的涂层量称量al2o3纳米晶体粉末。将称量的al2o3纳米晶体粉末倒入干式涂布机中。干式涂布机包括高速旋转混合器,该高速旋转混合器通过机械过程将al2o3纳米晶体粉末的颗粒粘结至基料粉末中的颗粒(即,沿着其表面)。对于0.5重量%的涂层,将2.5gal2o3纳米晶体粉末与500g基料粉末充分混合。将速度控制在4000rpm。5分钟后,形成涂覆有al2o3的基料粉末。实施例11—干法处理以形成alf3涂层将预定量的基料粉末(li1.02co0.96mn0.04o2)称量并倒入干式涂布机(nobilta,nob-130,细川密克朗公司(hosokawamicronltd))中。接着,根据预定基料粉末(例如,0.1重量%)上所需的涂层量称量alf3纳米晶体粉末。将称量的alf3纳米晶体粉末倒入干式涂布机中。对于0.1重量%的涂层,将0.5galf3与500g基料粉末充分混合。将速度控制在4000rpm。5分钟后,形成涂覆有alf3的基料粉末。实施例12—干法处理以形成al2o3和alf3的涂层将预定量的基料粉末(li1.02co0.96mn0.04o2)称量并倒入干式涂布机(nobilta,nob-130,细川密克朗公司(hosokawamicronltd))中。接着,根据预定基料粉末(例如,0.1重量%)上所需的涂层量称量al2o3纳米晶体粉末和alf3纳米晶体粉末。将称量的纳米晶体粉末倒入干式涂布机中。对于0.1重量%的涂层,将0.25gal2o3和0.05galf3与500g基料粉末充分混合。将速度控制在4000rpm。5分钟后,形成涂覆有al2o3和alf3的基料粉末。实施例13—粉末表征通过扫描电镜(sem)、电感耦合等离子体发射光谱(icp-oes)和maccor测试仪评估某些涂覆粉末的形态、组成和电化学性能。图3a-图3c示出了根据例示性实施方案的一系列扫描电子显微图,这些扫描电子显微图分别示出了基料粉末、涂覆有0.1重量%alf3的基料粉末,以及涂覆有0.1重量%al2o3的粉末。基料粉末对应于包含li1.02co0.96mn0.04o2的颗粒。湿法浸渍之前和之后的粉末仅显示出细小差别。与未涂覆的粉末(即图3a)相比,涂覆粉末(即图3b和图3c)的表面显示出如污渍或凸块指示的毛绒和粗糙。用icp-oes测定涂覆在基料粉末样品上的al2o3和alf3的量。表2和表3分别示出了al2o3和alf3湿法浸渍基料粉末的结果。目标涂覆水平与测量值的比较结果表明测量值与其针对≥0.2重量%的涂覆水平的对应目标值匹配得非常好。表2.涂覆有al2o3的基料粉末和未涂覆基料粉末的icp-oes结果表3.涂覆有alf3的基料粉末和未涂覆基料粉末的icp-oes结果实施例14对2032硬币半电池进行电化学测试,所述硬币半电池具有大约15mg/cm2的阴极活性材料负载。2032硬币半电池使用的电解质包含以ec:emc为溶剂的1.2mlipf6,其中ec:emc按重量计的比率为3:7。将电池置于maccor系列2000测试仪上,并于室温下以恒电流模式循环,采用的电压窗口为4.5v至2.75v。在每个电压窗口下进行形成、速率和循环的一系列电化学测试。在形成测试期间,恒定电流(0.2c)在充电过程期间被施加到电池,接着恒定电压充电直到电流等于或小于0.05c。随后,电池以恒定电流(0.2c)放电直到放电结束。电池充电和放电重复三次。在速率测试期间,对于所有速率测试,将充电速率固定到0.7c,然后恒定电压充电,直到电流等于或小于0.05c。施加五种不同的放电速率0.1c、0.2c、0.5c、1c和2c,直到电池完全放电为止。针对每个速率进行三次循环。最后,进行50次循环来调查循环寿命。施加与速率测试的充电条件相同的充电条件。放电速率针对所有循环被固定到0.5c。此处我们示出基料粉末li1.04co0.96mn0.04o2和两种执行得最多的涂覆样品(li1.04co0.96mn0.04o2-al2o30.05重量%和li1.04co0.96mn0.04o2-alf30.1重量%)的循环数据。图4示出了根据例示性实施方案表示三个硬币半电池(每个引入有单个阴极活性材料)在第一个充电和放电循环期间的性能的数据的曲线图。针对三个硬币半电池中每个硬币半电池的单个阴极活性材料分别对应于基料粉末li1.04co0.96mn0.04o2(即,“hw168”)、涂覆有0.05重量%al2o3的li1.04co0.96mn0.04o2(即,“hw168-al2o30.05重量%)以及涂覆有0.1重量%alf3的li1.04co0.96mn0.04o2(即,“hw168-alf30.1重量%)。三个硬币半电池的性能通过以下两个条柱来表征:最左侧的条柱指示第一循环充电电容,并且最右侧的条柱指示第一循环放电电容。在图4中,阴极活性材料中存在涂层轻微降低了第一循环充电电容和放电电容,因为相对于未涂覆的变型,涂覆有al2o3和涂覆有alf3的变型均显示出降低了4mah/g电容。该值高于针对此类少量涂层的预期值。电容的此类降低可归因于涂覆期间锂的损失,如由表3和表4中的icp-oes数据所指示。然而,图4所示的性能(其为初始性能)不代表后续充电和放电循环中硬币半电池的性能。图5和图6示出了根据例示性实施方案表示图4的三个硬币半电池的电容随增加的循环而变化的数据的曲线图。图7和图26示出了根据例示性实施方案表示图4的三个硬币半电池的能量密度随增加的循环而变化的数据的曲线图。图5和图7对应于速率测试,并且图6和图8对应于寿命测试。图5显示,一直到1c速率时,涂层的存在并不影响引入li1.04co0.96mn0.04o2、涂覆有al2o3的li1.04co0.96mn0.04o2以及涂覆有alf3的li1.04co0.96mn0.04o2的硬币半电池的性能。一直到1c(即,对于c/10、c/5、c/2和1c)的速率时,它们的电容均类似。对应于涂覆有al2o3的li1.04co0.96mn0.04o2的硬币半电池显示在2c的速率时,性能(相对)降低。图6示出了寿命测试,在该图中更清晰地突出了涂层的有益效果。对应于经涂覆的li1.04co0.96mn0.04o2变型的硬币半电池显示出相对于未经涂覆的li1.04co0.96mn0.04o2具有改善的电容。与经涂覆的li1.04co0.96mn0.04o2相关的硬币半电池仅损失了4-5mah/g。引入未经涂覆的li1.04co0.96mn0.04o2的硬币半电池开始时具有较低的电容,即相对于引入经涂覆的li1.04co0.96mn0.04o2那些而言具有较低的电容,因为该硬币半电池在老化测试和速率测试后已循环了19次。此类预老化相比于未预老化的那些(即,利用经涂覆的li1.04co0.96mn0.04o2的那些)导致更快的劣化。经过26次寿命循环测试之后,损失了15mah/g以上的电容。在图7和图8中观察到与图5和图6所述类似的能量密度趋势。然而,与未经涂覆的li1.04co0.96mn0.04o2相关的硬币半电池开始时的能量密度低于两个经涂覆的样品的硬币半电池的能量密度(图7和图8)。涂覆有alf3的样品的能量密度高于涂覆有al2o3的样品的能量密度。图9和图10示出了表示图4的三个硬币半电池中的每一个的充电放电概况的数据的曲线图。图9示出了速率测试,并且图10示出了寿命测试。这些图证实了本文所公开的涂层的优点。相对于引入基料粉末li1.04co0.96mn0.04o2的硬币半电池,引入涂覆有al2o3和涂覆有alf3的li1.04co0.96mn0.04o2的硬币半电池的曲线形状变化较不明显。实际上,对应于未涂覆的li1.04co0.96mn0.04o2的变型随着循环次数的增加显示出更快的衰减。图11和图12示出了表示图4的三个硬币半电池中的每一个的dq/dv概况的数据的曲线图。图11示出了速率测试,并且图12示出了寿命测试。每个图中的曲线每5次循环针对速率测试和寿命测试进行绘制。大约3.85v处的降低峰表征逐渐进行的循环期间,每个硬币半电池中阴极活性材料的结构稳定性。阴极活性材料的劣化(即,结构不稳定性)反映在降低峰朝更低电压偏移和增宽上。在速率测试期间,对应于基料粉末li1.04co0.96mn0.04o2的硬币半电池显示出最高的劣化,降低峰距3.85v具有最快的偏移且峰增宽较多。相比之下,对应于其涂覆变型的硬币半电池显示出较慢的偏移和较少的增宽。速率测试期间的峰偏移和增宽相较于寿命测试期间更不明显。这种行为由锂离子随充电速率/放电速率的渐进式增加而来回循环所致。在寿命测试期间,引入基线licoo2的硬币半电池显示出进一步的劣化,而引入li1.04co0.96mn0.04o2及其涂覆变型的那些几乎没有显示出劣化。实施例15—通过共沉淀方法得到的阴极活性材料用蒸馏水填充3.5升搅拌槽反应器,并将其加热至60℃。边以1100rpm的速率搅拌蒸馏水,边向槽反应器中引入氮气气流。分别将硫酸锰和硫酸钴溶于蒸馏水中,以制备总浓度为2.0m和预定摩尔比(即,[mn]:[co])的第一水性溶液。比率包括代表性示例,诸如[mn]:[co]=0.00:1.00、0.04:0.96、0.07:0.93、0.10:0.90、0.16:0.84以及0.28:0.72。将第一水性溶液以100ml/h的流动速率连续滴入槽反应器的蒸馏水中,以制备合并的水性溶液。使用联接到泵的ph控制器通过添加氢氧化钠和氨的第二水性溶液将合并水性溶液的ph固定至11.5。经过300小时的运行时间,颗粒在合并水性溶液中成核并生长,从而形成最终的前体颗粒。在175℃下洗涤、过滤并干燥最终前体颗粒12h。最终的前体颗粒用于形成组合物limnxco1-xo2的阴极活性材料,其中x和1-x对应于预定摩尔比,即[mn]:[co]=[x]:[1-x]。用li2co3粉末和最终前体颗粒的粉末进行固态反应。用不同摩尔比的li2co3和最终前体颗粒得到[li]:[mnxco1-x]比率有所不同的阴极活性材料(即,锂与总过渡金属含量的比率)。在轨道式搅拌器中共混li2co3与最终前体颗粒的粉末以制备混合粉末。共混后,将混合粉末转移到氧化铝托盘中并在流动空气中于700℃下加热10小时。烘箱的斜坡速率为每分钟5℃。在700℃下加热后,将已反应的混合粉末置于烘箱中通过自然热量损失冷却至环境温度。将所得的中间粉末用研钵和研杵研磨、筛分并在流动空气中于1050℃下再烧制15小时。斜坡速率为每分钟5℃,并且在烧制后,将所得的烧结粉末置于烘箱中通过自然热量损失冷却至环境温度。将烧结粉末破碎,用研钵和研杵研磨并筛分以制备阴极活性材料。使用brukerd8通过粉末x射线衍射来表征阴极活性材料的样品(参见图20和图21)。由上述共沉淀方法制备的阴极活性材料的代表性示例包括licoo2、li0.987co0.96mn0.04o2、li1.050co0.96mn0.04o2、li1.074co0.96mn0.04o2、li1.081co0.96mn0.04o2、li1.089co0.96mn0.04o2、li0.981co0.93mn0.07o2、li1.050co0.93mn0.07o2、li0.984co0.90mn0.10o2、li1.065co0.90mn0.10o2、li1.100co0.90mn0.10o2、li1.110co0.90mn0.10o2、li1.158co0.90mn0.10o2、li0.975co0.84mn0.16o2、li1.050co0.84mn0.16o2、li1.114co0.84mn0.16o2、li0.994co0.78mn0.22o2、li1.100co0.78mn0.22o2、li1.197co0.78mn0.22o2、li0.973co0.72mn0.28o2、li1.087co0.72mn0.28o2、li1.190co0.72mn0.28o2和li1.247co0.72mn0.28o2。图19示出了根据上述共沉淀方法制备的阴极活性材料的扫描电子显微图。显微图表示由致密烧结的初级颗粒形成的次级颗粒。此类致密烧结的次级颗粒典型地用于由共沉淀方法制备的阴极活性材料。阴极活性材料的组合物对应于li0.96co0.93mn0.07o2、li0.98co0.93mn0.07o2和li1.00co0.93mn0.07o2。图20示出了由以下的组合物表示的阴极活性材料的x射线粉末衍射图案:li1.074co0.96mn0.04o2、li1.081co0.96mn0.04o2、li1.089co0.96mn0.04o2、li1.065co0.90mn0.10o2、li1.110co0.90mn0.10o2、li1.158co0.90mn0.10o2、li0.975co0.84mn0.16o2、li1.050co0.84mn0.16o2和li1.114co0.84mn0.16o2。根据上述共沉淀方法制备这些阴极活性材料。在图20中,衍射图案组由底部到顶部排列,底部和顶部分别对应于锰含量的增加,即,从0.04增加至0.10增加至0.16。然而,在每个群中,锂与过渡金属之比(即,[li]/[mnxco1-x])从底部到顶部以三的增量减少。参考条柱指示预期的li2mno3、co3o4和limno2的峰,分别以粉色、灰色和蓝色示出。图20中的阴极活性材料大多为单相。在2θ=20°附近没有峰,表明在阴极活性材料中不存在li2mno3。此外,尽管用更多的mn取代co,但每个阴极活性材料的晶体结构(如由空间群表示)依然为然而,对于较低的[li]/[mnxco1-x]值,即,li0.975co0.84mn0.16o2,出现m3o4金属氧化物相(例如,co3o4)化学计量。图21示出了由以下的组合物表示的阴极活性材料的x射线粉末衍射图案:li0.994co0.78mn0.22o2、li1.100co0.78mn0.22o2、li1.197co0.78mn0.22o2、li0.973co0.72mn0.28o2、li1.087co0.72mn0.28o2、li1.19co0.72mn0.28o2和li1.247co0.72mn0.28o2根据上述共沉淀方法制备这些阴极活性材料。在图3中,衍射图案组由底部到顶部排列,底部和顶部分别对应于锰含量的增加,即从0.22增加至0.26。然而,在每个群中,锂与过渡金属之比(即,[li]/[mnxco1-x])从底部到顶部以三的增量减少。示出的参考条柱指示预期的li2mno3和co3o4的峰。在图21中,mn取代co的程度高于图20。然而,每个阴极活性材料的晶体结构(如由空间群所示)为li0.994co0.78mn0.22o2、li1.10co0.78mn0.22o2、li0.973co0.72mn0.28o2以及li1.087co0.72mn0.28o2的衍射图案在2θ=20°附近显示出峰。此类峰指示在这些阴极活性材料中存在小比例的li2mno3。然而,在由[li]/[mnxco1-x]的值接近1+x,即li1.197co0.78mn0.22o2与li1.247co0.72mn0.28o2的组合物表示的阴极活性材料中不存在峰。后面这些阴极活性材料为单相。度量1+x对应于xli2mno3·(1-x)licoo2(即,li1+xco1-xmnxo2+x)的理想的固体-溶液化学计量。实施例16—通过溶胶凝胶法得到的阴极活性材料以预定摩尔比(即,[mn]:[co])且总共2mol制备乙酸锰和乙酸钴的第一水性溶液。如下所述,预定比率包括代表性示例,诸如[mn]:[co]=0.10:0.90、0.16:0.84、0.22:0.78以及0.28:0.72。将1.0m的第二柠檬酸水性溶液加至第一水性溶液,并通过磁力搅拌混合,以制备合并溶液。将合并的溶液加热至80℃以形成凝胶,随后将其在80℃下保持6小时。然后将凝胶转移到箱式炉中并在350℃下煅烧4小时。冷却后,将所得的饼用研钵和研杵研磨、筛分并在流动空气中于900℃下烧制12小时。斜坡速率为每分钟5℃,并且在烧制后,将所得的阴极活性材料置于烘箱中通过自然热量损失冷却至环境温度。使用brukerd8通过粉末x射线衍射来表征阴极活性材料的样品(参见图23)。由上述溶胶凝胶法制备的阴极活性材料的代表性示例包括li1.131co0.90mn0.10o2、li1.198co0.84mn0.16o2、li1.241co0.78mn0.22o2和li1.301co0.72mn0.28o2。图22示出了根据上述溶胶凝胶法制备的阴极活性材料的扫描电子显微图。显微图指示细小颗粒的片状聚集体的尺寸小于1μm。此类细小颗粒形态是溶胶凝胶法制备的阴极活性材料的典型形态。阴极活性材料的组合物对应于li1.1co0.1al0.01mg0.01mn0.89o2,并且为li1.28co0.258al0.02mg0.02co0.68o2。图23示出了由以下的组合物表示的阴极活性材料的x射线粉末衍射图案:li1.131co0.90mn0.10o2、li1.198co0.84mn0.16o2、li1.241co0.78mn0.22o2和li1.301co0.72mn0.28o2。根据上述溶胶凝胶法制备这些阴极活性材料。阴极活性材料为单相。每个阴极活性材料的晶体结构(如由空间群所示)为在2θ=20附近没有峰,表明这些阴极活性材料中不存在li2mno3。在2θ=65附近的峰分离表明良好结晶的分层结构。实施例17—电池性能图24示出了由以下的组合物表示的阴极活性材料的微分电容曲线:licoo2、li1.05co0.96mn0.04o2、li1.05co0.93mn0.07o2、li1.110co0.90mn0.10o2和li1.19co0.72mn0.28o2根据上述共沉淀方法制备这些阴极活性材料。在第一充电放电循环期间,以c/5的速率对2032硬币半电池进行微分电容测量。在图24中,纵坐标表示dq/dv的量度,并且横坐标表示电化学势或电压的量度。licoo2不可逆的相变峰出现在约4.45v的电势处。但是在用mn取代co后,相变偏移至约4.55v的电势,峰强度降低。此类行为表明取代co(例如,用mn取代co)可产生高电压稳定性的阴极活性材料。图25示出了由以下的组合物表示的阴极活性材料的电压分布曲线:li1.05co0.96mn0.04o2、li1.05co0.93mn0.07o2、li1.110co0.90mn0.10o2和li1.19co0.72mn0.28o2。根据上述共沉淀方法制备这些阴极活性材料,然后将其装到2032硬币半电池中。电压分布对应于充电放电速率为c/10、电压窗口为2.75–4.6v的第一充电放电循环。在图25中,纵坐标表示硬币半电池的电化学势(即,v)的量度,并且横坐标表示积聚电容的量度(即,mah/g)。对于所有组合物而言,实现了高比电容(即,>150mah/g)以及高平均电压(即,>3.7v)。对于li1.19co0.72mn0.28o2阴极活性材料而言,第一充电曲线中大约4.5v处的平台期表明材料中存在对li2mno3类相的激活过程。实施例18—调整电池性能应当理解,因素诸如取代co(例如,co1-x-ymymnx)和锂与co之比及其取代(即,[li]/[co1-x-ymymnx])影响阴极活性材料中存在的相。如由图20-图21和图23所证实的那样,可选择此类因素以制备单相的阴极活性材料(例如,晶体结构)。此外,如图24和图25所证实的那样,也可以选择此类因素以提升电池性能(例如,增加电压稳定性)。图26示出了放电能量密度的等值曲线图,该等值曲线图随取代(即,co1-xmnx)和锂的比率(即,[li]/[co1-xmnx])而变化。放电能量密度对应于第一循环期间并且在c/10的充电放电率下获得的2032硬币半电池的测量结果。等值曲线图通过样品测量和预测建模的组合生成。2032硬币半电池使用根据上述溶胶凝胶方法制备的阴极活性材料(其中0≤x≤0.28)。在图26中,存在两个区域,这两个区域表明高能量密度(即,>700wh/kg):[1]第一区域,mn含量高达约12%(即,x≤0.12)并且比率高达约1.15(即,[li]/[co1-xmnx]≤1.15),以及[2]第二区域,mn含量高于约25%(即,x>0.25),并且比率高于约1.25(即,[li]/[co1-xmnx]>1.25)。图27示出能量保留率的等值曲线图,该等值曲线图随取代(即,co1-xmnx)和锂的比率(即,[li]/[co1-xmnx)而变化。能量保留率对应于10次循环后并且在c/3的充电放电率下获得的2032硬币半电池的测量结果。等值曲线图通过样品测量和预测建模的组合生成。2032硬币半电池使用根据上述溶胶凝胶方法制备的阴极活性材料(其中0≤x≤0.28)。类似于图26中,图27中存在两个区域,这两个区域表明高能量密度(即,>700wh/kg):[1]第一区域,mn含量高达约12%(即,x≤0.12)并且比率高达约1.15(即,[li]/[co1-xmnx]≤1.15),以及[2]第二区域,mn含量高于约25%(即,x>0.25),并且比率高于约1.25(即,[li]/[co1-xmnx]>1.25)。实施例19将liαco1-x-yalymnxoδ的四个样品制成硬币电池,采用li金属阳极,并在2.75-4.5v下以c/5的充电/放电率循环。四个样品对应于x=0.01,0.02、0.03和0.04,其中α=1.0;y的范围为0.001-0.003;并且δ为约2.0。图28和图29示出微分电容相对于电化学电势的导数(即,dq/dv与v)的曲线图,该曲线图示出mn和li含量对电池单元性能的影响。在licoo2阴极的充电和放电期间,由六边形相到单斜晶相的相变发生在4.0-4.3v之间(参见图29)。x=0.01的mn取代也发生相变(参见图28)。相变导致晶格的体积膨胀,晶格的体积膨胀可促成电极的电容衰减。在此种情况下,以大于x=0.01的比例的mn取代co减轻了该相变,如图28中针对x=0.02、0.03和0.04的组合物所示。相变还取决于化合物中的li含量。当考虑如在liαco0.97mn0.03o2中的mn取代时,如果α≥1.0则可减轻相变。图29示出,随着li含量从0.977增加至1.014,dq/dv曲线中介于4.0-4.3v之间的特征性相变峰降低至α=1.014的平整线。另一方面,li过量的有益效果受其对体积能量密度和保留的影响限制。图30示出在介于2.75-4.5v、速率为c/5的循环期间,liαco0.97mn0.03o2中li含量对放电能量的影响。化学计量组合物,α=1.003,显示出754wh/g的最大能量,并且在25个循环中,损失了该能量的约8%。富含li的样品(即,α=1.014)具有相似的最大能量,但是损失该能量的10%。亚化学计量组合物(即,α<1.0)显示出更低的能量。这种行为以及采用2%mn(即,x=0.02)的重复测试表明在其中α为约1.00的锂含量下获得了最佳能量和能量保留率。材料中每种金属的成分值由高精度icp-oes分析确定,特别是锂含量的测量(即,α)。类似的电化学测量(未示出)也示出了铝含量对减弱六边形相至单斜晶相相变的影响。由于铝被取代为另外的liαco1-x-yalymnxoδ固定组合物,相变得到抑制。可能存在出现体积能量、能量保留率和相变抑制的a、x和y。从该检查中确定以≥3%(即,x≥0.03)的总量对mn、al和li、额外或其它可被取代成该结构的元素进行的任何取代将减弱在4.0-4.3v期间六边形到单斜晶体的转变。似乎额外的元素也与它们氧化态的总和相关,即mn4+、al3+和li+加在一起应达到某水平以防止相变。这是因为已经观察到,要防止相变,对于给定的mn含量需要不同的li化学计量。实施例20如图31所示,确定的最佳mn含量(即,x)介于x=0.02和x=0.04之间,该含量使得mn取代的licoo2在2.75-4.5v、c/5的速率的循环中实现最大的体积能量密度和能量保留率。在2.75v至4.5v以及c/5的速率下对包含x=0.02、0.03和0.04的三种组成并包含相似的li和al含量的电池单元进行循环。对应于x=0.03的组合物尽管相较于x=0.02显示出稍微较小的初始能量,但显示出最高的能量保留率。实施例21—核磁共振固态6li核磁共振(nmr)测量识别出liαco1-xmnxoδ中存在mn-mn聚集。该聚集最终将导致形成li2mno3,因为mn和li含量增加超出富含li的组合物的相限。不希望受限于特定机制或运作模式,mn聚集稳定阴极结构,这可向本文所述的材料提供如任何电化学测试中所示的高电压稳定性。但是hr-xrd和nmr未示出在该研究中考虑的组合物中形成有任何li2mno3。在hr-xrd和nmr光谱中均不存在对应于li2mno3的新的相峰。图32示出了x=0.03和x=0.04的mn取代licoo2的比较结果。对指定共振态的nmr量化显示x=0.04时锰的聚集是x=0.03时锰聚集的两倍,而不是如预期那样有25%的增加。虽然mn取代被示为稳定licoo2晶体结构,但较大的mn聚集体趋于将li引入过渡金属(tm)层,使得在较高电压(4.5v)下时,li从晶体结构中被引出,如果过渡金属层中的li落入锂层中,过渡金属层中形成的空缺减弱晶体结构的稳定性。实施例22—添加铝制备三种组合物li1.01co0.97-yalymn0.03oδ的阴极活性材料,分别将li和mn的含量固定至1.01和0.03,而al含量变化为0.077重量%、0.159重量%和0.760重量%。以2.75v-4.5v、c/5的速率循环,在半电池中对阴极活性材料进行测试。图33示出了由于al取代增加,因此放电能量减少。然而,随着al的增加,能量保留率有所提升,最多0.76重量%的al取代在25次循环后表现出最好的放电能量。实施例23—颗粒形态为实现稳定和高能量密度,可在足够的温度下将liαco1-x-yalymnxoδ组合物的阴极活性材料处理足够的时间,使得次级颗粒包含致密的单颗粒(即,初级颗粒)。这些致密的单颗粒可赋予高强度以在电极制造和电池单元组装期间耐受压延过程。图34a-图34b和图35a-图35b示出了最佳处理以实现高强度颗粒的影响。在足够的温度下处理前体粉末足够的时间后,可进一步烧结在图34a中所见的多颗粒结构以获得如图34b中更大且强度更大的颗粒,该颗粒更加难以碎裂。该改善的强度在图35a-图35b中示出。由于烧结所致的部分互连,在1050℃(图35a)下煅烧的前体粉末的尺寸分布从18生长至22μ。当将粉末压成丸状并且颗粒在压力下碎裂时(即,模拟电极层合物压延),颗粒尺寸分布由于颗粒-颗粒键(即,初级颗粒之间)断裂而减少至双峰分布,并且颗粒破裂成更小的初级颗粒。然而,当在相同处理时间下温度上升至1085℃时,在处理期间烧结在一起的较大的单颗粒破裂成初始的前体尺寸,但不会进一步破裂。这种强度防止形成新表面,诸如不由al2o3涂层保护并且经受与电极的相互作用的表面。实施例24—能量保留率煅烧温度不仅影响颗粒强度,还影响作为电极的阴极活性材料的能量保留率。随着煅烧温度增加,能量保留率也增加至介于1075-1080℃(图36)之间的最大值。压实在渐增的温度下煅烧的粉末后表面积的变化随着颗粒强度的增加而稳定,并且由于压碎颗粒,因此没有新表面露出。图36示出阴极活性材料的强度(稳定的表面积变化)与能量保留率之间的相关性。在图36中,阴极活性材料具有其中锂与过渡金属之比(li/tm)为1.01(即,α=1.01且y=0)的组合物。阴极材料的第一循环的放电电容和库伦效率也与煅烧温度相关。图37示出了这一关系。初始的放电电容以及初始能量随着材料的煅烧温度从1050℃增加至1092℃而有效地降低。库伦效率是在第一放电期间插入回阴极的li的量的量度,显示从未来充电/放电循环消除的li的部分。最大效率在1080℃处发生,而有关颗粒强度的电容则最佳为1070-1080℃。煅烧温度的这种敏感性是所提出的本发明的新颖点的一部分,因为其被示出为影响颗粒强度、能量保留率、li含量、循环性、材料的电容以及能量。实施例25—热稳定性li离子电池材料的能量增高,随之而来的是意外能量释放的风险增大,例如,热可导致电池单元着火。对充电和暴露至电极的阴极活性材料的差示扫描量热测定(dsc)有助于确定热失效的风险。图38示出了mn=1-7摩尔%(即,x=0.01-0.07)的五个mn取代licoo2组合物的dsc测量结果。相比于商用lco(即,licoo2),mn1%组合物(即,x=0.01)示出在形成更多热量的放热反应中具有较低的起始温度。然而,随着mn含量的增加,反应的起始温度升高,并且释放的总热量降低直到mn7%。mn=4%之后,起始温度升高,但反应的热量释放开始再次增加。表4列举了这些值。基于dsc测量结果,最佳mn取代介于mn3-4摩尔%之间(即,0.03≤x≤0.04)。表4.dsc测量结果汇总实施例26—晶格参数对材料性能至关重要的li含量(即,α)与晶体结构的c-晶格参数的变化相关联,该晶格参数的变化如图39所示。根据mn含量(随着mn增加,c-晶格参数也增加),li含量将随着li的增加而降低c-晶格值。还呈现出来的是随着mn含量增加,材料在阴极活性材料中容纳过量锂同时维持晶体结构的能力增加。lico(1-x)mno2系统的该相图提供在不形成二次相的情况下最佳li添加的图。实施例27—拉曼光谱分层的licoo2和mn取代的licoo2(即,x=0.04和0.07)的拉曼光谱在图40中示出。利用785nm光子激发获得拉曼光谱。根据因子群分析,分层的具有晶体结构的licoo2预计显示出两个拉曼活性模式,即,一个在大约596cm-1处,其为a1g对称性(缘于沿c轴线的对称氧振动),并且另一个在大约486cm-1处,其为eg对称性(缘于在a/b晶面中的两个简并对称氧振动)。随着将mn加入结构中,新的拉曼散射特征出现在高于596cm-1带以及低于486cm-1带的频率处。这种新的散射缘于mn-o键伸缩振动,该伸缩振动与形成在整个过渡金属层中的各种mn-co-li占位相关联。由这些新的mn-o振动引起的键的相对强度随着mn取代的增加而增加。320cm-1附近的带来自caf2窗口,拉曼光谱通过该窗口获得。实施例28—氧化稳定性图41中示出7%mn取代(即,x=0.07)与4%mn取代(即,x=0.04)相比的氧化稳定性结果。将半电池从开路值充电至4.65v,然后在连续的循环运行中于4.0v和4.65v之间循环。第52次循环为介于2.75v至4.65v之间进行的全循环。在图41中,第52次循环时,放电电容相对于锂与过渡金属之比(即,α,其中y=0)的曲线图显示最佳电容出现在li含量(α)接近一处。在这些循环条件下,相比于x=0.04,x=0.07的取代在52次循环后保持较高的电容,这表明在这些条件下其具有更好的电容保留率。在图42中,绘制了引入li1.00co0.93-yalymn0.07oδ阴极活性材料的半电池的dq/dv以及引入有li1.025co0.96-yalymn0.04oδ阴极活性材料的半电池的dq/dv。图42示出了发生在大约4.53v处的高电压下,x=0.07样品的降低过程。然而,x=0.04样品的另一个降低峰在4.47v处是明显的,这表明可能的不稳定性或另外的相变。此类不稳定性或相变可导致循环时的结构化转变。注意,该降低过程不存在各自的充电峰,这可表明不可逆性,并且因此表明当循环至4.65v的较高电压时的内部电容保留,如图41所讨论。实施例29—电容和库仑效率第一循环的电容示于图43中,其中第一循环充电和放电值以及所计算的库伦效率作为锂与过渡金属比率(即,α,其中y=0)的函数绘制。库仑效率在近似化学计量值时具有更好的改善。α值越大,越多的li可从阴极活性材料中引出。然而,相反,li含量越高,所得到的放电电容越低。因此,研究了最佳α。实施例30—x射线吸收根据整个串联的所有化合物的x射线吸收(xas)结果(此处未示出),对于mn取代的licoo2样品中所有的α值,co处于+3氧化态,并且mn处于+4氧化态。因此,计算出的化学计量是li0.983co0.914mn0.069o2,并且li在li层中是不足够的。将li阳离子从li层中移出以满足过渡金属层导致li0.966[li0.017co0.914mn0.069]o2。(注意,括号中li、co和mn的值总计为一。)出于本
发明内容的目的,我们将该材料进一步区分为在li层中具有约5%li(不充足)并且在tm层中具有一些少量的li以与mn结合,从而形成mn-li-mn结构域以替代co-co-co结构域,该mn-li-mn结构域具有电荷补偿,是所得晶格limn2稳定性的关键。然而,这些单元的尺寸如由密度泛函理论(dft)总能计算(未示出)所建议那样受限,并且示出在模式中具有比在上述循环条件下x=0.04更大的稳定性。在上述描述中,为了解释的目的,所使用的特定命名提供对所述实施方案的彻底理解。然而,对于本领域的技术人员而言将显而易见的是,实践所述实施方案不需要这些具体细节。因此,出于举例说明和描述的目的,呈现了对本文所述的具体实施方案的前述描述。它们并非旨在是穷举性的或将实施方案限制到所公开的精确形式。对于本领域的普通技术人员而言将显而易见的是,根据上述教导内容,许多修改和变型是可能的。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1