一种尺寸可控过渡金属氧化物超细纳米颗粒及其快速制备方法和应用
本发明属于纳米材料制备技术领域,具体涉及一种尺寸可控过渡金属氧化物超细纳米颗粒及其快速制备方法和应用。
背景技术:
金属氧化物纳米材料作为最具有市场应用潜力的新兴材料之一,其潜在的重要性毋庸置疑。其中,过渡金属氧化物纳米材料由于其独特的小尺寸和高比表面积等性能而受到科研人员的广泛关注。例如,ni基、co基的氧化物纳米材料在超级电容器、电化学催化剂、二次可充电离子电池等电化学储能领域方面获得了广泛的应用;fe等金属纳米材料具有优秀的局域表面等离子共振特性,在电化学、生物学、医学、电子学等领域都有广泛的应用。
近年来,20nm以下的超细氧化物纳米颗粒吸引了越来越多的关注,原因在于超细纳米颗粒的小尺寸效应使得其表面的电子态和键态与其固体颗粒内部有所不同,导致其过渡金属具有多种价态,将有利于电荷的存储与转移;同时,表面元素配位环境的改变会导致其表面活性位点的增加,改善其电化学反应动力学,从而提高它们在储能,特别是电催化领域的应用能力。
然而,过渡金属氧化物纳米颗粒,特别是20nm以下超细纳米颗粒的制备虽已见诸报道,但是到目前为止,大多采用的是有机溶液合成法。例如,2019年4月26日公开的公开号为cn109678118a的专利,公开了一种金属氧化物纳米颗粒及金属纳米颗粒的制备方法,将金属的乙酰丙酮盐溶于三氯甲烷等有机溶剂作为浸渍液,浸渍水溶性无机盐,干燥后高温(500~1000℃)煅烧或还原,得到仅小于100nm金属氧化物纳米颗粒;2020年6月23日公开的公开号为cn111320193a的专利,公开了金属氧化物纳米颗粒及金属纳米颗粒的制备方法,利用含过渡金属元素的硝酸盐和乙二醇甲醚等有机溶液之间的反应形成凝胶,再经过高温(600~1000℃)煅烧获得10~40nm的氧化物颗粒。上述这些有机溶液合成方法不仅实验方法较为复杂,需要煅烧温度高,消耗过大,而且合成周期偏长,有机物溶液还容易造成环境的污染。
而一些水系体系制备的过渡金属氧化物纳米颗粒则粒径较大,2016年9月14日公开的公开号为cn105940145a的专利,公开了过渡金属氧化物颗粒及其制备方法,采用电化学辅助制备的氧化锰颗粒为微米级,粒径较大。
2017年6月20日公开的公开号cn106865625a的专利公开了一种由过渡金属氧化物纳米粒子构成的纳米片及其制备方法,添加模板有增加了制备步骤和消耗成本。
因此,发展快速、简易、低消耗、高效、产物粒径小的过渡金属氧化物纳米颗粒制备方法依然充满挑战。
技术实现要素:
本发明的目的在于提供一种尺寸可控过渡金属氧化物超细纳米颗粒,粒径3~15nm,且粒子分布均匀。
本发明另一目的在于提供一种尺寸可控过渡金属氧化物超细纳米颗粒的快速制备方法,利用高浓度金属盐的水系快速成核-沉淀法,简单快速的制备了超细的过渡金属氧化物纳米颗粒,在该纳米颗粒中,大小均一,通过温度的选择控制纳米颗粒的平均直径在3~15nm。一般来说,温度越高粒径越大,这是由于在高温下纳米颗粒极易团聚。本方法操作简单,周期短,合成效率高,重复性好,且对环境绿色无污染,对多种过渡金属氧化物均有效。
本发明还有一个目的在于提供一种尺寸可控过渡金属氧化物超细纳米颗粒的应用,用于超级电容器、二次离子电池电极材料或高效电化学催化领域。
本发明具体技术方案如下:
一种尺寸可控过渡金属氧化物超细纳米颗粒的快速制备方法,包括以下步骤:
1)将可溶性过渡金属硫酸盐溶于水中,形成溶液a;
2)将碱性材料溶于naclo或kclo溶液中,形成溶液b;
3)将b溶液和a溶液滴加混合,搅拌后,离心分离,沉淀物洗涤,干燥;
4)步骤3)产物进行热处理,得到尺寸可控过渡金属氧化物超细纳米颗粒。
步骤1)中,溶液a中过渡金属硫酸盐浓度为1.6~2.0mol/l;硝酸盐对于此反应很剧烈,有一定危险,氯化物效果达不到本申请所述的效果,本发明选择过渡金属硫酸盐,进行本发明反应,不仅反应安全可控,而且产品性能优异。
步骤1)中,所述过渡金属硫酸盐选自硫酸镍、硫酸铁、硫酸亚铁、硫酸钴或硫酸锰中的一种或多种。
步骤2)中所述naclo溶液或kclo溶液质量浓度5~8%;naclo或kclo溶液提供沉淀材料,clo-和硫酸根离子由于团簇的占位较大,他们之间会形成较大的配位团簇,使得金属离子不易于团聚,同时clo-与金属离子优先结合形成的强配位中间体会形成保护层阻碍晶体扩大生长,减缓纳米材料生长速率;
步骤2)中溶液b中碱性材料的浓度为3~4.5mol/l。
步骤2)中所述的碱性材料选自碱性金属氢氧化物,为氢氧化钠naoh或氢氧化钾koh或氢氧化锂lioh。
步骤3)中所述b溶液和a溶液滴加混合是指将a溶液滴加到b溶液中,或者将b溶液加入到a溶液;滴加时间为15-20分钟;滴加混合的作用是促进快速成核,防止团聚。
步骤3)所述搅拌,时间为90~150分钟,为常温搅拌,搅拌是促进反应充分和防止团聚,始终搅拌,搅拌速度200-400转每分钟,搅拌不能太慢,搅拌太慢容易导致团聚。
步骤3)中,所述沉淀物洗涤是指用去离子水洗涤若干次,直到上清液ph为中性。
步骤4)中所述热处理是指200~400℃条件下热处理1-3h;
步骤4)中所述热处理是指在马弗炉中热处理,空气气氛下即可。
步骤4)中所述尺寸可控过渡金属氧化物超细纳米颗粒为nio、fe3o4、fe2o3、co3o4获mn3o4中的一种或多种。
本发明提供的一种尺寸可控过渡金属氧化物超细纳米颗粒,采用上述方法制备得到,颗粒大小均匀、分散性好,粒径可以控制在3~15nm。
本发明提供的一种尺寸可控过渡金属氧化物超细纳米颗粒的应用,用于超级电容器、二次离子电池电极材料或高效电化学催化领域。具体为:作为锂离子电池负极材料、钠离子电池负极材料或电催化析氧oer材料、电催化析氢her材料的应用。
本发明先制备高浓度金属盐和沉淀剂,混合反应,过渡金属离子成核,clo-离子诱发强氧化反应,形成含有羟基基团插层的过渡金属氧化物纳米层,该产物不同于普通金属氯化物和naoh反应形成的金属氢氧化物沉淀,主要区别是羟基的掺入可以进一步的隔断金属氧化物,防止其团聚。热处理发生除去含氧羟基基团的分解反应,最终形成小颗粒的氧化物;本发明利用高浓度金属盐的水系快速成核-沉淀法,采用可溶性过渡金属硫酸盐为原料,碱性金属氢氧化物和naclo或kclo为沉淀剂,简单快速的制备了尺寸可控的过渡金属氧化物超细纳米颗粒。不仅避免了有机体系的繁琐步骤和高温处理,还解决了水系体系中制备的尺寸大且不可控的缺点。本发明提供的尺寸可控过渡金属氧化物超细纳米颗粒大小均匀、分散性好,粒径可以控制在3~15nm。本发明产品颗粒小,能利用完全和提高活性位点,容量、倍率性能也就越好。这种超细纳米颗粒在超级电容器、二次离子电池电极材料和高效电化学催化领域能等方面发挥重要作用。本发明制备的一种尺寸可控的过渡金属氧化物超细纳米颗粒,该方法通过温度的控制即可获得粒径不同的纳米颗粒,粒径3~15nm,粒径小且粒子分布均匀。本发明的制备方法操作简单,周期短,合成效率高,重复性好,且对环境绿色无污染,易于推广。
附图说明
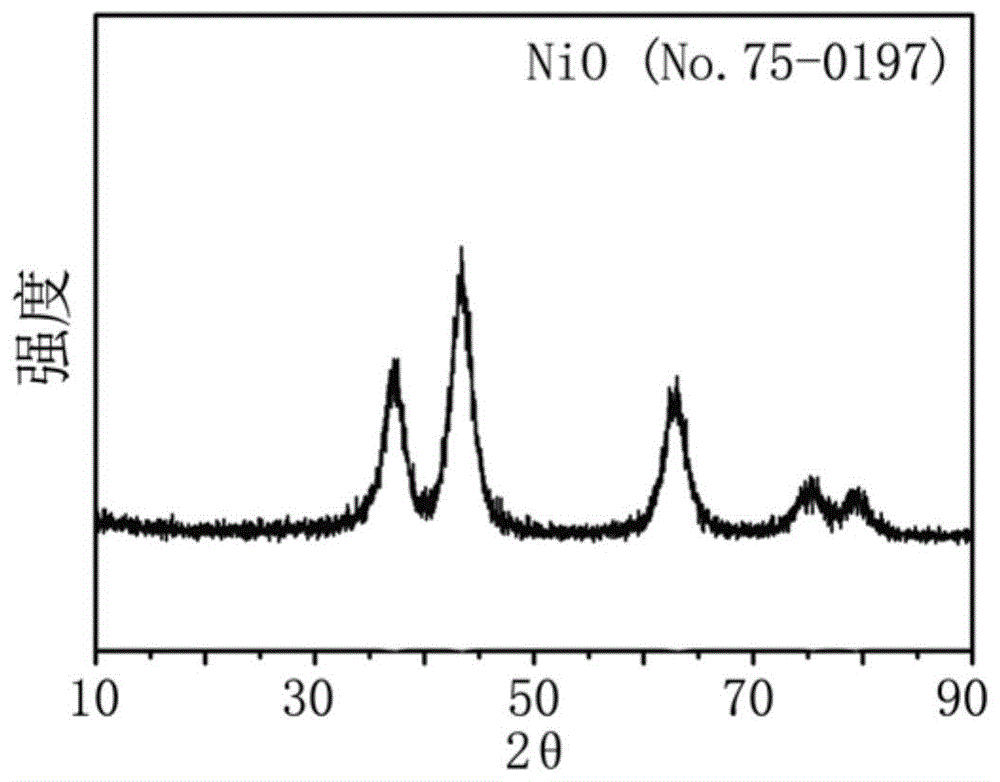
图1为实施例1所制得的nio纳米颗粒的x射线粉末衍射图;
图2为实施例1所制得的nio纳米颗粒的tem图;
图3为实施例1所制得的nio纳米颗粒的粒径分布图;
图4为实施例1所制得的超细nio纳米颗粒作为超级电容器电极材料在koh溶液中的恒电流充放电曲线;
图5为对比例1所制得的nio纳米颗粒sem图(图中a)以及其作为超级电容器电极材料在koh溶液中的恒电流充放电曲线(图中b);
图6为实施例2所制得的nio纳米颗粒的tem图;
图7为实施例3所制得的超细co3o4纳米颗粒的x射线粉末衍射图;
图8为实施例3所制得的超细co3o4纳米颗粒的tem图;
图9为实施例3所制得的超细co3o4纳米颗粒的电催化析氧的lsv测试曲线;
图10为对比例2所制得的co3o4颗粒sem图(图中a)以及其电催化析氧的lsv测试曲线(图中b);
图11为实施例4所制得的mn3o4纳米颗纳米颗粒的x射线粉末衍射图;
图12为实施例4所制得的mn3o4纳米颗纳米颗粒的tem图;
图13为实施例5所制得的mn3o4纳米颗纳米颗粒的tem图;
图14为对比例3所制备的mn3o4纳米颗纳米颗粒的sem图;
图15为实施例6所制得的fe2o3纳米颗粒的x射线粉末衍射图;
图16为实施例6所制得的fe2o3纳米颗粒的tem图;
图17为实施例6所制得的fe2o3纳米颗粒的电催化析氧的lsv测试曲线;
图18为对比例4所制得的fe2o3颗粒sem图(图中a)以及其电催化析氧的lsv测试曲线(图中b)。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
下述实施例中所用的试验材料和试剂等,如无特殊说明,均可从商业途径获得。
实施例中未注明具体技术或条件者,均可以按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。
实施例1
一种尺寸可控nio超细纳米颗粒的快速制备方法,包括以下步骤:
1)称取15.5g的六水硫酸镍溶于35ml去离子水中,形成1.69mol/l的a溶液;
2)称取5.1g的naoh溶于36ml5%的naclo形成b溶液,naoh的浓度为3.5mol/l;
3)将b溶液缓慢的滴加到a溶液中,匀速搅拌90分钟;离心分离,并用去离子水洗涤多次,直到上清液ph为中性,50℃干燥后,放到马弗炉中300℃热处理2小时,得到nio纳米颗粒。
通过x射线粉末衍射xrd对最终产物的样品进行表征,如图1所示,产物主要成分为pdf卡片号为75-0197的nio。
通过透射电子显微镜tem对产物的形貌和尺寸进行表征,从图2和图3可以看出所获得的nio颗粒大小均一,其平均尺寸约为5nm。
一种尺寸可控过渡金属氧化物超细纳米颗粒的应用,用于超级电容器;具体为:对本实施例1获得的超细nio纳米颗粒进行超级电容器的电化学性能测试:
以nio:活性碳:ptfe质量比为8:1:1的比例混合,均匀的涂覆在泡沫镍集流体上,以hg/hgo和铂丝分别作为参比电极和对电极,在3mol/l的koh电解液中测试nio电极的恒电流充放电性能。如图4所示,在1a/g的电流密度下,本实施例制备的超细nio纳米颗粒可以获得高达1936f/g的比容量,这源于小的纳米颗粒有利于质子插层反应的充分进行。
对比例1
该对比例用于观测原料中naclo盐对于nio纳米颗粒粒径的影响,具体制备步骤采用实施例1中的技术方案,区别在于步骤2):称取5.1g的naoh溶于36ml的去离子水中形成b溶液,naoh的浓度为3.5mol/l;。
通过扫描电子显微镜sem对产物的形貌和尺寸进行表征,从图5中a可以看出所获得的超细nio颗粒的尺寸范围为30-60nm,明显大于实施例1中的nio尺寸。
继续对本对比例1获得的nio纳米颗粒进行超级电容器的电化学性能测试:
以nio:活性碳:ptfe质量比为8:1:1的比例混合,均匀的涂覆在泡沫镍集流体上,以hg/hgo和铂丝分别作为参比电极和对电极,在3mol/l的koh电解液中测试nio电极的恒电流充放电性能。如图5中b所示,在1a/g的电流密度下,本对比例制备的nio大纳米颗粒仅获得1326f/g的比容量,这是由于大的颗粒不利于质子插层反应的充分进行。
实施例2
该实施例观测不同的热处理温度对于nio纳米颗粒粒径的影响,具体制备步骤采用实施例1中的技术方案,区别在于步骤3)中放入马弗炉中的加热温度为400℃。
通过透射电子显微镜tem对产物的形貌和尺寸进行表征,从图6可以看出所获得的超细nio颗粒大小均一,其粒径的平均尺寸为10nm。
实施例3
一种co3o4超细纳米颗粒的快速制备方法,包括以下步骤:
1)称取18.5g的六水硫酸钴溶于35ml去离子水中,形成2.00mol/l的a溶液;
2)称取7.5g的koh溶于35ml8%的naclo形成b溶液,koh的浓度为3.8mol/l;
3)将b溶液缓慢的滴加到a溶液中,匀速搅拌120分钟,离心分离,并用去离子水洗涤多次,直到上清液ph为中性,50℃干燥后,放到马弗炉中300℃热处理2小时,得到黑色co3o4纳米颗粒。
通过x射线粉末衍射xrd对最终产物的样品进行表征,如图7所示,产物成分为pdf卡片号为42-1467的co3o4材料。
通过透射电子显微镜tem对产物的形貌和尺寸进行表征,从图8可以看出所获得的超细co3o4颗粒大小均一,平均粒径为4nm。
一种超细co3o4纳米颗粒在电化学催化析氧oer中的应用,具体为:对本实施例获得的超细co3o4纳米颗粒进行电化学催化析氧oer的性能测试:
以co3o4:乙炔黑:萘酚质量比为8:1:1的比例混合,均匀的涂覆在旋转圆盘电极上,以hg/hgo和铂丝分别作为参比电极和对电极,设置旋转圆盘电极的转数为1600rpm/min,在1mol/l的koh电解液中测试co3o4电极的lsv曲线。如图9所示,在10ma/cm2的电流密度时,本实施例3所制备的超细co3o4纳米颗粒的电势为1.59v,即过电位仅为360mv。
对比例2
该对比例源于文献中制备co3o4的普通方法,用于验证本发明工艺对于纳米材料粒径和电化学性能的影响,对比样具体制备步骤为:
1)4.76g六水氯化钴和1.42g表面活性剂聚氧乙烯-聚氧丙烯醚嵌段共聚物f127溶于30ml乙醇中。
2)上述溶液100℃热回流8小时,在得到凝胶产物后,40℃下老化2天,再40℃下老化10天。
3)上述2)产物150℃下加热15分钟,再400℃下热处理3小时。
通过扫描电子显微镜sem对产物的形貌和尺寸进行表征,从图10中a可以看出所按照文献获得的co3o4颗粒的尺寸范围为300-500nm,明显大于实施例3中的co3o4尺寸。
继续对本对比例获得的co3o4纳米材料进行电化学催化析氧oer的性能测试:
以co3o4:乙炔黑:萘酚质量比为8:1:1的比例混合,均匀的涂覆在旋转圆盘电极上,以hg/hgo和铂丝分别作为参比电极和对电极,设置旋转圆盘电极的转数为1600rpm/min,在1mol/l的koh电解液中测试co3o4电极的lsv曲线。如图10中b所示,在10ma/cm2的电流密度时,本对比例2所制备的co3o4纳米材料的电势为1.63v,即过电位高达400mv,远高于实施例3的超细co3o4纳米颗粒。
实施例4
一种超细mn3o4纳米颗粒的快速制备方法,包括以下步骤:
1)称取10.2g的硫酸锰溶于35ml去离子水中,形成1.93mol/l的a溶液;
2)称取5.1g的naoh溶于36ml8%的naclo形成b溶液,naoh的浓度为3.5mol/l;
3)将b溶液缓慢的滴加到a溶液中,匀速搅拌100分钟,离心分离,并用去离子水洗涤多次,直到上清液ph为中性,50℃干燥后,放到马弗炉中300℃热处理2小时,得到黑色mn3o4纳米颗粒。
通过x射线粉末衍射xrd对最终产物的样品进行表征,如图11所示,产物成分为pdf卡片号为24-0734的mn3o4。
通过透射电子显微镜tem对产物的形貌和尺寸进行表征,从图12可以看出所获得的超细mn3o4颗粒的大小均一,平均粒径为5nm。
实施例5
该实施例观测不同的热处理温度对于mn3o4纳米颗粒粒径的影响,具体制备步骤采用实施例4中的技术方案,区别在于步骤3)中放入马弗炉中的加热温度为400℃。
通过透射电子显微镜tem对产物的形貌和尺寸进行表征,从图13可以看出所获得的mn3o4纳米颗粒的大小均一,其粒径的平均尺寸为15nm。
对比例3
该对比例用于观测原料中naclo盐对于mn3o4纳米颗粒粒径的影响,具体制备步骤采用实施例4中的技术方案,区别在于步骤2):称取5.1g的naoh溶于36ml的去离子水中形成b溶液,naoh的浓度为3.5mol/l。
通过扫描电子显微镜sem对产物的形貌和尺寸进行表征,从图14可以看出所获得的超细nio颗粒的尺寸范围为60-200nm,明显大于实施例4中的mn3o4尺寸。
实施例6
一种超细fe2o3纳米颗粒的制备方法,包括以下步骤:
1)称取18.5g的七水硫酸亚铁溶于35ml去离子水中,形成1.90mol/l的a溶液;
2)称取5.1g的naoh溶于36ml5%的naclo形成b溶液,naoh的浓度为3.5mol/l;
3)将b溶液缓慢的滴加到a溶液中,匀速搅拌90分钟,离心分离,并用去离子水洗涤多次,直到上清液ph为中性,50℃干燥后,放到马弗炉中300℃热处理2小时,得到fe2o3纳米颗粒。
通过x射线粉末衍射xrd对最终产物的样品进行表征,如图15所示,产物成分为pdf卡片号为33-0664的fe2o3材料。
通过透射电子显微镜tem对产物的形貌和尺寸进行表征,从图16可以看出所获得的fe2o3纳米颗粒的大小均一,平均粒径为10nm。
一种超细fe2o3纳米颗粒在电化学催化析氧oer中的应用,具体为:对本实施例所制备的fe2o3纳米颗粒进行电化学催化析氧oer的性能测试:
以fe2o3:乙炔黑:萘酚质量比为8:1:1的比例混合,均匀的涂覆在旋转圆盘电极上,以hg/hgo和铂丝分别作为参比电极和对电极,设置旋转圆盘电极的转数为1600rpm/min,在1mol/l的koh电解液中测试fe2o3电极的lsv曲线。如图17所示,在10ma/cm2的电流密度时,本实施例6所制备的超细fe2o3纳米颗粒的电势为1.62v,即过电位仅为390mv。
对比例4
该对比例用于观测原料浓度对于fe2o3纳米颗粒粒径的影响,具体制备步骤采用实施例6中的技术方案,区别在于步骤1):称取1.85g的七水硫酸亚铁溶于35ml去离子水中,形成0.19mol/l的a溶液;2)称取0.51g的naoh溶于36ml5%的naclo形成b溶液,naoh的浓度为0.35mol/l。
通过扫描电子显微镜sem对产物的形貌和尺寸进行表征,从图18中a可以看出所按照文献获得的fe2o3颗粒的尺寸范围为50-250nm,明显大于实施例6中的fe2o3尺寸。
继续对本对比例获得的co3o4纳米材料进行电化学催化析氧oer的性能测试:
以fe2o3:乙炔黑:萘酚质量比为8:1:1的比例混合,均匀的涂覆在旋转圆盘电极上,以hg/hgo和铂丝分别作为参比电极和对电极,设置旋转圆盘电极的转数为1600rpm/min,在1mol/l的koh电解液中测试fe2o3电极的lsv曲线。如图18中b所示,在10ma/cm2的电流密度时,本对比例2所制备的fe2o3纳米材料的电势为1.68v,即过电位高达450mv,远高于实施例6的超细fe2o3纳米颗粒,这是由于大的块状颗粒暴露的活性位点数较少导致的。
- 还没有人留言评论。精彩留言会获得点赞!