一种生产莽草酸的重组菌株及其制备方法与应用与流程
本发明涉及基因工程重组菌领域,具体涉及一种生产莽草酸的重组菌株及其制备方法与应用。
背景技术:
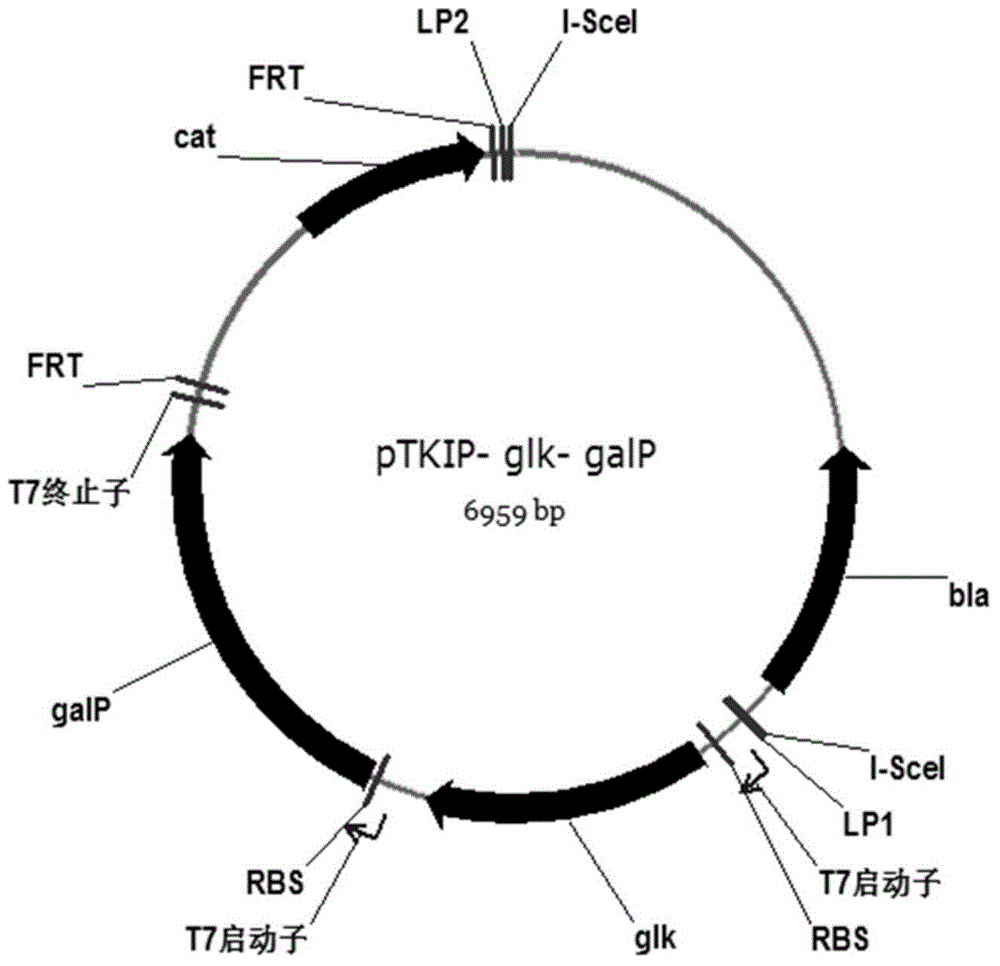
:莽草酸(Shikimicacid),化学名为[3R-(3α,4α,5β)]-3,4,5-三羟基-1-环己烯-1-羧酸。随着H5N1、H1N1和H7N9亚型流感病毒的连续爆发,罗氏公司的专利药“达菲”(磷酸奥司他韦,Oseltamivir)成为世界各国争相贮备的首选抗流感病毒药物。作为“达菲”合成的关键手性起始原料,莽草酸也引起人们的广泛关注。获得莽草酸的途径主要有三方面:植物提取、化学合成和微生物合成。植物提取的来源主要是木兰科植物八角,但利用八角提取莽草酸,生产工艺复杂,得率较低,原料易受季节气候等因素限制,难以满足大规模生产所需,而且大量砍伐植物对环境也造成巨大危害。化学合成则由于产率不高,对环境造成污染等缺点受到限制。因此,利用微生物来生产莽草酸具有极广阔的应用前景,其中利用大肠杆菌合成莽草酸成为近些年的研究热点。目前大多研究在强化关键基因时都利用质粒表达载体进行过表达,然而质粒在传代的过程中不稳定,需要添加抗生素维持其稳定性,所添加的抗生素也会污染环境,导致耐药菌的产生。另外基因表达需添加诱导剂或转变温度进行诱导,其中常见诱导剂IPTG价格昂贵,且对细胞及人体有一定的毒性,而工业生产中大型发酵罐转变温度较难实现,因此通过质粒表达基因的重组菌株不适于工业化生产。近年来通过定点整合的方式将关键基因直接整合到大肠杆菌的染色体上进行强化表达的策略取得了成功,所得到的重组菌株能够稳定表达所强化的基因,且无需抗生素及诱导剂的添加。该方法已在微生物代谢产物的合成中取得了重要进展,也成为微生物合成植物来源化合 物的研究热点及主要趋势。目前对于利用染色体整合的方式进行莽草酸的微生物合成报道很少,已有的一篇文献报道了通过染色体整合的方式获得生产莽草酸的重组菌株,采用了化学诱导染色体进化技术提高了莽草酸合成途径的相关基因在染色体上的拷贝数,之后得到的菌株通过摇瓶发酵莽草酸产量为3.12g/L,但未进行发酵罐培养(Cui,Y.Y.,Ling,C.,Zhang,Y.Y.,Huang,J.,Liu,J.Z.2014.ProductionofshikimicacidfromEscherichiacolithroughchemicallyinduciblechromosomalevolutionandcofactormetabolicengineering.MicrobCellFact,13,21.)。该方法操作步骤较繁琐,还涉及到噬菌体的整合与化学诱导,其原理是将噬菌体先整合到染色体上,再将基因整合到噬菌体整合位点上。而噬菌体整合位点有限且随机,因此具有很大的不确定性和不可重复性,并且获得的菌株莽草酸发酵产量仍然不够理想,不利于工业生产。技术实现要素:本发明所要解决的技术问题是为了克服现有的微生物法合成莽草酸的生产过程中需添加抗生素和诱导剂并且产量不够理想、不利于工业生产的问题,提供了一种改进的重组莽草酸生产菌株及其制备方法。该改进的重组莽草酸生产菌株发酵后所得的莽草酸产量较高,生产莽草酸时无需抗生素及诱导剂的添加,降低了生产成本,简化了生产工艺。本发明技术方案之一:一种重组大肠杆菌菌株,其自身不包含下述基因:aroK、aroL、tyrR与磷酸烯醇式丙酮酸-糖磷酸转移酶系统操纵子(ptsHIcrr);其操纵子ptsHIcrr位点与基因tyrR位点包含了下述基因:aroG、aroB、tktA、aroE、glk、galP和ppsA,所述基因aroG、aroB、tktA、aroE、glk、galP和ppsA转录表达由大肠杆菌内具有活性的启动子驱动,由大肠杆菌内具有活性的终止子终止。本发明中,所述重组大肠杆菌菌株出发菌株可以为本领域常规的能生产莽草酸的大肠杆菌菌株,较佳地为E.coliJW0379-1菌株。所述重组大肠杆菌 菌株出发菌株较佳地通过基因敲除的方法剔除基因aroK、aroL、tyrR与操纵子ptsHIcrr。所述基因敲除的方法可以为本领域常规的基因敲除的方法,较佳地为通过在所述重组大肠杆菌菌株的出发菌株内表达重组系统进行敲除,所述重组系统较佳地为FLP/FRT和/或Red重组系统。所述基因aroG、aroB、tktA、aroE、glk、galP和ppsA为本领域常规所指的基因,可以通过合成获得,也可以通过从生物体内分离或PCR扩增得到,较佳地为通过从生物体内PCR扩增得到。所述基因aroG、aroB、tktA、aroE、glk、galP和ppsA可以为任何生物来源,只要其具有与大肠杆菌的aroG、aroB、tktA、aroE、glk、galP和ppsA基因相同的功能即可,较佳地来自大肠杆菌,更佳地来自E.coliJW0379-1菌株。所述基因aroG、aroB、tktA、aroE、glk、galP和ppsA可以以任意的顺序被整合到所述ptsHIcrr位点与所述tyrR位点,只要在所述ptsHIcrr位点整合有选自所述基因aroG、aroB、tktA、aroE、glk、galP和ppsA中的任意1个、2个、3个、4个、5个或6个,同时在所述tyrR位点整合有剩下的基因即可。较佳地,所述基因aroG、aroB、tktA、aroE、glk和galP位于所述ptsHIcrr位点,所述基因ppsA位于所述tyrR位点,更佳地,所述基因aroG、aroB、tktA、aroE、glk和galP按该先后顺序排列位于所述ptsHIcrr位点,所述基因ppsA位于所述tyrR位点。所述大肠杆菌内具有活性的启动子为本领域常规所指的能够在大肠杆菌体内驱动转录的启动子,较佳地为选自PT7、PT5、Ptac、Ptrc、Plac、PBAD和Ptrp中的一种,更佳地为PT7,所述大肠杆菌内具有活性的终止子可以为本领域常规所指的能够在大肠杆菌内终止转录的序列,较佳地为T7终止子。本发明中,所述重组大肠杆菌菌株较佳地还可以包括含有所述基因aroG、aroB、tktA和aroE的载体,所述载体可以为本领域常规所指的载体,包括原核载体和真核载体,较佳地可以为原核表达载体如pET-28a、pETDuet-1或pRSFDuet-1,更佳地为pETDuet-1。本发明技术方案之二:一种制备如前所述重组大肠杆菌菌株的方法,其包括以下步骤:从所述重组大肠杆菌出发菌株中敲除基因aroK、aroL、tyrR 和操纵子ptsHIcrr,在ptsHIcrr位点与tyrR位点整合基因aroG、aroB、tktA、aroE、glk、galP和ppsA,所述基因aroG、aroB、tktA、aroE、glk、galP和ppsA转录表达由大肠杆菌内具有活性的启动子驱动,由大肠杆菌内具有活性的终止子终止。所述方法中的所述敲除基因与所述整合基因的步骤可以为任意的顺序,只要所述大肠杆菌出发菌株中所述基因aroK、aroL、tyrR和操纵子ptsHIcrr被敲除,所述基因aroG、aroB、tktA、aroE、glk、galP和ppsA被整合到所述ptsHIcrr位点与所述tyrR位点即可。所述基因aroG、aroB、tktA、aroE、glk、galP和ppsA可以以任意的顺序被整合到所述ptsHIcrr位点与所述tyrR位点,只要在所述ptsHIcrr位点整合有选自所述基因aroG、aroB、tktA、aroE、glk、galP和ppsA中的任意1个、2个、3个、4个、5个或6个,同时在所述tyrR位点整合有剩下的基因即可。较佳地,所述基因aroG、aroB、tktA、aroE、glk和galP可以按任意顺序被整合到所述ptsHIcrr位点,所述基因ppsA被整合到所述tyrR位点,更佳地,所述基因aroG、aroB、tktA、aroE、glk和galP按该顺序被整合到所述ptsHIcrr位点,所述基因ppsA被整合到所述tyrR位点。较佳地,所述方法包括如下步骤:(1)从所述重组大肠杆菌出发菌株中敲除aroL与aroK基因,获得敲除菌株(△aroLaroK);(2)从步骤(1)所述敲除菌株(△aroLaroK)中敲除ptsHIcrr操纵子,得敲除菌株(△aroLaroK,△ptsHIcrr);(3)将大肠杆菌内具有活性的启动子和终止子连同所述基因aroG、aroB、tktA和aroE整合到步骤(2)所述敲除菌株(△aroLaroK,△ptsHIcrr)的所述ptsHIcrr位点,得整合菌株(△aroLaroK,△ptsHIcrr::GBAE);(4)将大肠杆菌内具有活性的启动子和终止子连同所述基因glk和galP整合到步骤(3)所述整合菌株(△aroLaroK,△ptsHIcrr::GBAE)的所述ptsHIcrr位点,得整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP)。(5)从步骤(4)中所述整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP) 中敲除tyrR基因,得整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR)。(6)将大肠杆菌内具有活性的启动子连同所述基因ppsA整合到步骤(5)所述整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR)的所述tyrR位点,得整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA)。更佳地,所述方法还可以包括步骤(7),所述步骤(7)为:将含有所述基因aroG、aroB、tktA和aroE的载体转化到步骤(6)所述整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA)中,得整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA)/pGBAE。步骤(7)中,所述含有所述基因aroG、aroB、tktA和aroE的载体可以为本领域常规的所指的载体,包括原核载体和真核载体,较佳地可以为原核表达载体如pET-28a、pETDuet-1或pRSFDuet-1,更佳地为pETDuet-1。步骤(1)中,所述重组大肠杆菌出发菌株为能生产莽草酸并且aroL基因被敲除的大肠杆菌菌株,较佳地为E.coliJW0379-1菌株。所述敲除可以指本领域常规所指的敲除基因的方法,较佳地为通过在所述重组大肠杆菌菌株的出发菌株内表达重组系统进行敲除。所述重组系统可以为本领域常规用到的重组系统,较佳地为FLP/FRT和/或Red重组系统。所述敲除较佳地通过下述步骤进行:用FLP/FRT重组系统在敲除所述大肠杆菌出发菌株中自带的抗生素抗性基因后,再在其中表达RED重组酶,将aroK基因被所述抗生素抗性基因替换的片段转化进入所述大肠杆菌出发菌株进行同源重组,获得敲除菌株(△aroLaroK)。所述aroK基因被所述抗生素抗性基因替换的片段较佳地为被neo抗性基因替换的片段,所述aroK基因被neo抗性基因替换的片段可以从生物体内PCR扩增得到或化学合成得到,较佳地为从生物体内PCR扩增得到,更佳地为从大肠杆菌中PCR扩增得到,最佳地为从E.coliJW5947-1菌株中PCR扩增得到。所述转化可以为本领域常规的转化质粒载体的操作,较佳地为化转或电转,更佳地为电转。所述步骤(1)较佳地还可以包括:将T7噬菌体基因组整合到所述敲除菌株(△aroLaroK)上,得敲除菌株(△aroLaroK,DE3)。所述整合可以根据本领域常规的操作进行, 较佳地使用λDE3溶源化试剂盒进行。步骤(2)中,所述敲除较佳地通过下述步骤进行:将两端含有与操纵子ptsHIcrr两端50bp同源的序列的四环素抗性基因tetA片段转化入步骤(1)所述敲除菌株(△aroLaroK)进行同源重组,在平板上挑选具有四环素抗性的菌落检测,即得敲除了ptsHIcrr操纵子的敲除菌株(△aroLaroK,△ptsHIcrr)。所述转化可以为本领域常规的转化质粒载体的操作,较佳地为化转或电转,更佳地为电转。步骤(3)中,所述整合可以指本领域常规所指的整合基因到基因组上的方法,较佳地为通过表达重组酶系统将含有所述基因aroG、aroB、tktA和aroE的打靶载体整合到基因组上。所述打靶载体可以为本领域常规的载体,其含有所述基因aroG、aroB、tktA和aroE、含有所述基因大肠杆菌内具有活性的启动子如PT7、PT5、Ptac、Ptrc、Plac、PBAD或Ptrp和大肠杆菌内具有活性的终止子,较佳地含有所述基因aroG、aroB、tktA和aroE、T7启动子和T7终止子,更佳地为打靶载体pTKIP-GBAE。所述重组酶系统可以为本领域常规的重组酶系统,较佳地为RED重组酶。所述基因aroG、aroB、tktA和aroE为本领域常规所指的基因,可以通过合成获得,也可以通过从生物体内分离或PCR扩增得到,较佳地为通过从生物体内PCR扩增得到。所述基因aroG、aroB、tktA和aroE可以为任何生物来源,只要其具有与大肠杆菌的aroG、aroB、tktA和aroE基因相同的功能即可,较佳地来自大肠杆菌,更佳地来自E.coliBW25113菌株。所述整合较佳地通过下述步骤进行:将RED重组酶表达载体与所述基因aroG、aroB、tktA和aroE的打靶载体pTKIP-GBAE转化进入步骤(2)所述敲除菌株(△aroLaroK,△ptsHIcrr),挑选无氨苄青霉素抗性和四环素抗性而有氯霉素抗性的阳性转化子。所述转化可以为本领域常规的转化质粒载体的操作,较佳地为化转或电转,更佳地为电转。步骤(4)中,所述整合可以指本领域常规所指的整合基因到基因组上的方法,较佳地为通过表达重组酶系统将含有所述基因glk和galP的打靶载 体整合到基因组上。所述打靶载体可以为本领域常规的载体,含有所述基因glk和galP、含有大肠杆菌内具有活性的启动子,如PT7、PT5、Ptac、Ptrc、Plac、PBAD或Ptrp和大肠杆菌内具有活性的终止子,较佳地含有所述基因glk和galP、T7启动子和T7终止子,更佳地为打靶载体pTKIP-glk-galP。所述重组酶系统可以为本领域常规的重组酶系统,较佳地为Red重组酶。所述基因glk和galP为本领域常规所指的基因,可以通过合成获得,也可以通过从生物体内分离或PCR扩增得到,较佳地为通过从生物体内PCR扩增得到。所述基因glk和galP可以为任何生物来源,只要其具有与大肠杆菌的glk和galP基因相同的功能即可,较佳地来自大肠杆菌,更佳地来自E.coliBW25113菌株。所述整合较佳地通过下述步骤进行:将RED重组酶表达载体转化入步骤(3)所述整合菌株(△aroLaroK,△ptsHIcrr::GBAE),扩增出两端含有与步骤(3)所述重组菌株(△aroLaroK,△ptsHIcrr::GBAE)上的基因cat两端50bp同源的序列的四环素抗性基因tetA片段,将其转化入步骤(3)所述表达RED重组酶的整合菌株(△aroLaroK,△ptsHIcrr::GBAE)中进行同源重组,挑选具有四环素抗性的菌落,得到用基因tetA替换掉基因cat的菌株,再转化表达RED重组酶与含有所述基因glk与galP的打靶载体pTKIP-glk-galP进行同源重组,挑取无氨苄青霉素抗性和四环素抗性而有氯霉素抗性的菌落进行菌落PCR验证,在经过验证的菌株中转化表达FLP/FRT重组系统的表达载体,删除抗性基因neo。所述转化可以为本领域常规的转化质粒载体的操作,较佳地为化转或电转,更佳地为电转。步骤(5)中,所述敲除较佳地包括下述步骤:将含有与步骤(4)所述整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP)上的基因tyrR两端50bp同源的序列的四环素抗性基因tetA片段转化入步骤(4)所述整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP)中进行同源重组,挑选具有四环素抗性的阳性转化子。所述转化可以为本领域常规的转化质粒载体的操作,较佳地为化转或电转,更佳地为电转。步骤(6)中,所述整合可以指本领域常规所指的整合基因到基因组上 的方法,较佳地为通过表达重组酶系统将含有所述基因ppsA的打靶载体整合到基因组上。所述打靶载体可以为本领域常规的载体,含有所述基因ppsA、大肠杆菌内具有活性的启动子,如PT7、PT5、Ptac、Ptrc、Plac、PBAD或Ptrp中的一种和大肠杆菌内具有活性的终止子,较佳地含有所述基因ppsA、T7启动子和T7终止子,更佳地为打靶载体pTKIP-ppsA。所述重组酶系统可以为本领域常规的重组酶系统,较佳地为Red重组酶。所述基因ppsA为本领域常规所指的基因,可以通过合成获得,也可以通过从生物体内分离或PCR扩增得到,较佳地为通过从生物体内PCR扩增得到。所述基因ppsA可以为任何生物来源,只要其具有与大肠杆菌的ppsA基因相同的功能即可,较佳地来自大肠杆菌,更佳地来自E.coliBW25113菌株。所述整合较佳地包括下述步骤:将RED重组酶表达载体和含有所述基因ppsA的打靶载体转化入步骤(5)所述整合菌株(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR)进行同源重组,挑取无氨苄青霉素抗性和四环素抗性而有氯霉素抗性的菌落检测。所述转化可以为本领域常规的转化质粒载体的操作,较佳地为化转或电转,更佳地为电转。本发明技术方案之三:一种制备莽草酸的方法,其包括以下步骤:发酵培养前述重组大肠杆菌菌株(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA),从培养液获得;所述发酵培养的培养基中葡萄糖与甘油的质量比为1∶5~5∶1。所述发酵培养可以为本领域常规的操作,包括发酵罐发酵培养和摇瓶发酵培养。所述摇瓶发酵培养较佳地包括下述步骤:挑取单菌落接种于20mLLB培养基中,37℃过夜培养,然后按5%(v/v)接种量接种至20mL发酵培养基中,在30℃、220rpm条件下进行发酵培养,48h后取样。所述发酵罐培养较佳地包括下述步骤:挑取单菌落接种于20mLLB培养基中,37℃过夜培养,然后按1%(v/v)接种量接种至LB培养基中,37℃培养8h,接着按5%(v/v)接种量接种至5L发酵罐中进行发酵。发酵罐的培养基装量为2.5L,培养温度为30℃,空气通量为4~6L/min,溶氧浓度为5%~70%,较佳地为10%~50%,更佳地为30%,发酵液pH为7.0。所述pH较佳地通 过添加NH4OH或20%H3PO4维持在7.0。发酵6h时一次性加入5g/L甘油、10g/L酵母浸出粉FM888及胰蛋白胨5g/LFP318,8h开始流加400g/L葡萄糖溶液使罐内葡萄糖含量维持在5~15g/L。所述发酵培养基组成可以为本领域常规的培养基,包括摇瓶发酵培养基与发酵罐发酵培养基。所述摇瓶发酵培养基较佳地包括:20g/L葡萄糖、10g/L酵母浸出粉FM888、5g/L胰蛋白胨FP318、2g/L柠檬酸、0.5g/L谷氨酸钠、3g/LK2HPO4,1g/LKH2PO4、1g/LNaCl、3g/LMgSO4,灭菌前调节pH至7.0;所述发酵罐发酵培养基较佳地包括总浓度为20g/L葡萄糖和甘油、20g/L酵母浸出粉FM888、10g/L胰蛋白胨FP318,2g/L柠檬酸、0.5g/L谷氨酸钠、3g/LK2HPO4,1g/LKH2PO4、1g/LNaCl、3g/LMgSO4,灭菌前调节pH为7.0,所述葡萄糖和甘油的质量比为1:5~5:1,较佳地为2:3~3:2,更佳地为1:1。在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。本发明所用试剂和原料均市售可得。本发明的积极进步效果在于:本发明提供的改进的莽草酸生产菌株及其制备方法通过定点整合的方法将莽草酸合成途径的关键基因aroG、aroB、tktA、aroE、glk、galP和ppsA整合到大肠杆菌染色体上的磷酸烯醇式丙酮酸-糖磷酸转移酶系统操纵子(ptsHIcrr)位点与酪氨酸调控基因tyrR位点上,具有很高的确定性和重复性。根据该方法所制备的重组菌株莽草酸的发酵产量较高,生产莽草酸时无需抗生素及诱导剂的添加,降低生产成本,简化生产工艺。附图说明图1:质粒pTKIP-GBAE的结构示意图。图2:质粒pTKIP-glk-galP的结构示意图。图3:质粒pTKIP-ppsA的结构示意图。图4:基因aroG,aroB,tktA,aroE以及glk,galP定点整合到染色体基因ptsHIcrr位点过程示意图。图5:基因ppsA定点整合到染色体基因tyrR位点过程示意图。图6:重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA)的PCR验证图。数字1、2、3分别代表菌株BW25113,BW25113(△ptsHIcrr::GBAEKP,△tyrR::ppsA)和BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA)。组I和组II用于验证aroL和aroK基因双敲除;组III、组IV、组V和组VI用于验证基因aroG,aroB,tktA,aroE,glk和galP定点整合入染色体基因ptsHIcrr位点;组VII和组VIII用于验证基因ppsA定点整合入染色体基因tyrR位点。图7:莽草酸生产菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA)/pGBAE在5L发酵罐发酵过程中,菌株生长情况(OD600)和莽草酸产量随时间的变化情况。具体实施方式下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。下述实施例中所用的菌株有大肠杆菌BW25113(F-,Δ(araD-araB)567,ΔlacZ4787(::rrnB-3),λ-,rph-1,Δ(rhaD-rhaB)568,hsdR514)、JW0379-1(F-,Δ(araD-araB)567,ΔlacZ4787(::rrnB-3),ΔaroL753::kan,λ-,rph-1,Δ(rhaD-rhaB)568,hsdR514)和JW5947-1(F-,Δ(araD-araB)567,ΔlacZ4787(::rrnB-3),λ-,ΔaroK725::kan,rph-1,Δ(rhaD-rhaB)568,hsdR514),均从ColiGeneticStockCenter(NewHaven,USA)购买获得,具体信息参见文献(BabaT,AraT,HasegawaM,etal.ConstructionofEscherichiacoliK-12in-frame,single-geneknockoutmutants:theKeiocollection.MolSystBiol2006.2:1-11)。下述实施例中所用到的质粒有表达载体pETDuet-1(购自Novagen,产品编号71146),抗性基因消除质粒pCP20(购自ColiGeneticStockCenter),基因定点整合工具质粒pTKRED(购自Addgene,产品编号41062)、pTKIP-cat(购自Addgene,产品编号41065)、pTKS/CS(购自Addgene,产品编号41063),具体信息及基因定点整合原理参考文献(Kuhlman,T.E.,Cox,E.C.Site-specificchromosomalintegrationoflargesyntheticconstructs.NucleicAcidsRes.2010Apr;38(6):e92.Epub2010Jan4);大肠杆菌λDE3化试剂盒购自Novagen公司。实施例中用到的基础培养基:LB(Luria-Bertani)培养基:酵母提取物5g/L,胰蛋白胨10g/L,NaCl10g/L;LB固体培养基另加10g/L琼脂粉。RDM(EZRichDefinedMedium)培养基:购自Teknova,Inc.。酵母浸出粉FM888和胰蛋白胨FP318均购自安琪酵母公司。以下实施例中所采用的分子生物学实验技术包括PCR扩增、质粒提取、质粒转化、DNA片段连接、酶切、凝胶电泳、感受态制备等都采用常规方法,具体可参见《分子克隆实验指南》(第三版)(SambrookJ,RussellDW,JanssenK,ArgentineJ.黄培堂等译,2002,北京:科学出版社)。实施例中所用到的主要仪器:色谱柱CarbomixH-NP10(10μm,7.8×300mm),购自苏州赛分科技有限公司。以下实施例中所用到的引物(由上海捷瑞生物工程有限公司合成)见表1。表1(划线序列为酶切位点,粗体序列为用于同源重组的50bp同源序列。)实施例1打靶质粒pTKIP-GBAE的构建(1)所需基因均以菌株BW25113的染色体为模板扩增得到。用引物G1(SEQIDNO:1)和G2(SEQIDNO:2)扩增得到基因aroG,PCR扩增条件如下:PCR体系(20μL):模板10ng,10μM引物各1μL,2×pfu酶PCR反应混合液10μL,添加ddH2O至20μL。扩增程序:94℃预变性5min;94℃变性30s,68℃退火30s(每个循环减0.5℃),72℃延伸1min/kb,共35个循环;72℃再延伸10min。将扩增到的基因aroG和质粒pETDuet-1用限制性内切酶BamHI酶切后连接得到质粒pETDuet-G;用引物B1(SEQIDNO:3)和B2(SEQIDNO:4)扩增得到基因aroB,将基因aroB和质粒pETDuet-1用限制性内切酶BamHI和HindIII酶切后连接得到质粒pETDuet-B;用引物A1(SEQIDNO:5)和A2(SEQIDNO:6)扩增得到基因tktA,将基因tktA和质粒pETDuet-1用限制性内切酶BglII和EcoRV酶切后连接得到质粒pETDuet-A;用引物E1(SEQIDNO:7)和E2(SEQIDNO:8)扩增得到基因aroE,将基因aroE和质粒pETDuet-1用限制性内切酶BamHI和HindIII酶切后连接得到质粒pETDuet-E;(2)将质粒pETDuet-B上的基因aroB连同其rbs序列用引物GB1(SEQIDNO:9)和GB2(SEQIDNO:10)扩增得到,扩增条件同前述,用限制性内切酶EcoRI和PstI酶切后连接到质粒pETDuet-G上得到质粒pETDuet-GB;将质粒pETDuet-E上的基因aroE连同其rbs序列用引物AE1(SEQIDNO:11)和AE2(SEQIDNO:12)扩增得到,扩增条件同前述,用限制性内切酶EcoRV和AvrII酶切后连接到质粒pETDuet-A上得到质粒pETDuet-AE;将质粒pETDuet-AE用限制性内切酶BglII和AvrII酶切后的片段连接到pETDuet-GB相应位点得到质粒pETDuet-GBAE;(3)将质粒pETDuet-GBAE上的基因aroG、aroB、tktA和aroE连同各自的rbs序列以及T7启动子终止子用引物Duet-F(SEQIDNO:13)和Duet-R(SEQIDNO:14)扩增得到,扩增条件同前述,用限制性内切酶ApaI和NheI酶切后连接到质粒pTKIP-cat上得到质粒pTKIP-GBAE,其结构如图1所示。实施例2打靶质粒pTKIP-glk-galP的构建(1)以菌株BW25113为模板,用引物glk1(SEQIDNO:15)和glk2(SEQIDNO:16)扩增得到基因glk片段,扩增条件同前述,将其和质粒pETDuet-1用限制性内切酶EcoRI和PstI酶切后连接得到质粒pETDuet-glk; 用引物galP1(SEQIDNO:17)和galP2(SEQIDNO:18)扩增得到基因galP片段,扩增条件同前述,将其和质粒pETDuet-glk用限制性内切酶BglII和XhoI酶切后连接得到质粒pETDuet-glk-galP;(2)将质粒pETDuet-glk-galP上的基因glk和galP连同各自的rbs序列以及T7启动子终止子用引物Duet-F(SEQIDNO:13)和Duet-R(SEQIDNO:14)扩增得到,扩增条件同前述,用限制性内切酶ApaI和NheI酶切后连接到质粒pTKIP-cat上得到质粒pTKIP-glk-galP,其结构如图2所示。实施例3打靶质粒pTKIP-ppsA的构建(1)以菌株BW25113为模板,用引物ppsA1(SEQIDNO:19)和ppsA2(SEQIDNO:20)扩增得到基因ppsA片段,扩增条件同前述,将其和质粒pETDuet-1用限制性内切酶EcoRI和XhoI酶切后连接得到质粒pETDuet-ppsA;(2)将质粒pETDuet-ppsA上的基因ppsA连同其rbs序列以及T7启动子终止子用引物Duet-F(SEQIDNO:13)和Duet-R(SEQIDNO:14)扩增得到,扩增条件同前述,用限制性内切酶ApaI和NheI酶切后连接到质粒pTKIP-cat上得到质粒pTKIP-ppsA,其结构如图3所示。实施例4整合菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)的构建1、构建aroL和aroK双基因敲除菌株BW25113(△aroLaroK,DE3)(1)将质粒pCP20通过电转化的方法转入菌株E.coliJW0379-1,电转化条件为:使用0.2cm电转化杯,电压2.5KV,时间5ms。在添加100mg/L氨苄青霉素抗性的LB平板上挑选阳性转化子,于LB液体培养基中37℃培养过夜,使质粒pCP20表达FLP酶,特异性识别染色体上的FRT位点进行卡那霉素抗性基因neo的去除,再升温至42℃培养2h,以去除质粒pCP20,在无抗性LB平板上划线分离单菌落,通过菌落PCR验证挑选出删除卡那 霉素抗性基因neo的菌株;(2)将质粒pTKRED电转化入(1)得到的菌株,电转化条件同前述,挑选阳性转化子于30℃培养过夜,按照1%(v/v)的比例接种于50mL的LB液体培养基内培养,待培养液OD600≈0.3时,加入终浓度为2mM的IPTG诱导重组酶,继续培养至OD600≈0.6时制备电转化感受态细胞;所述电转化感受态细胞通过下述方法制得:将菌液放于4℃冰箱内预冷10min后,转入冰上预冷的50mL离心管中。在4500rpm,4℃条件下离心10min。弃上清,加入20mL预冷的无菌水重悬菌体,相同条件下离心。弃上清,加入15mL预冷的10%甘油重悬菌体。离心后弃上清,加入适当10%甘油(约600μL)混匀菌体,分装于1.5mL离心管中(100μL/管)备用。(3)以菌株E.coliJW5947-1为模板,用引物K1(SEQIDNO:23)和K2(SEQIDNO:24)进行PCR扩增,扩增条件同前述,得到aroK基因被neo基因替换的片段(>100ng),将其电转化入(2)中制备的新鲜感受态细胞中进行同源重组,电转化条件同前述,37℃培养2h后涂布加入30mg/L卡那霉素的LB平板,37℃过夜,挑选菌落进行PCR验证,得到aroK基因被neo基因替换的菌株;(4)同(1),将pCP20电转化到步骤(3)所得菌株,电转化条件同前述,删除抗性基因neo,用引物L1(SEQIDNO:21)/L2(SEQIDNO:22)和K1(SEQIDNO:23)/K2(SEQIDNO:24)进行菌落PCR验证,结果如图6所示,得到双基因敲除菌株BW25113(△aroLaroK)。(5)使用Novagen公司的λDE3溶源化试剂盒对所得双基因敲除菌株BW25113(△aroLaroK)进行λDE3化,得双基因敲除菌株(△aroLaroK,DE3)。2、敲除葡萄糖转运PTS系统(ptsHIcrr)(1)将质粒pTKRED电转化入菌株BW25113(△aroLaroK,DE3),电转化条件同前述,制备电转化感受态细胞,制备方法同前述;(2)将质粒pTKS/CS线性化后做为模板,用引物PTS-CS1(SEQIDNO: 25)/PTS-CS2(SEQIDNO:26)扩增得到含有基因ptsHIcrr两端50bp同源序列的四环素抗性基因tetA片段,扩增条件同前述,将其电转化入(1)制备的感受态细胞中进行同源重组,电转化条件同前述,在平板上挑选具有四环素抗性的菌落进行菌落PCR验证,得到敲除了葡萄糖转运PTS系统(ptsHIcrr)的菌株BW25113(△aroLaroK,△ptsHIcrr,DE3)。3、构建基因整合菌株BW25113(△aroLaroK,△ptsHIcrr::GBAE,DE3)(1)将质粒pTKRED和pTKIP-GBAE电转化入菌株BW25113(△aroLaroK,△ptsHIcrr,DE3),挑选阳性转化子在RDM培养基(加0.5%甘油)中活化,按2%(v/v)接种量接种至20mLRDM培养基(加0.5%甘油)中,30℃培养至OD600≈0.6时加入2mMIPTG和0.2%(w/v)L-阿拉伯糖诱导过夜。次日加入34mg/L氯霉素继续培养6h,稀释菌液至105×,取100μL涂布添加了氯霉素的LB平板;(2)挑取无氨苄青霉素抗性和四环素抗性而有氯霉素抗性的菌落进行菌落PCR验证并送测序,得到基因整合菌株BW25113(△aroLaroK,△ptsHIcrr::GBAE,DE3)。4、构建基因整合菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,DE3)(1)将质粒pTKRED电转化入菌株BW25113(△aroLaroK,△ptsHIcrr::GBAE,DE3),电转化条件同前述,制备电转化感受态细胞,制备方法同前述;(2)用引物CM-CS1(SEQIDNO:27)/PTS-CS2(SEQIDNO:26)扩增得到含有菌株BW25113(△aroLaroK,△ptsHIcrr::GBAE,DE3)上的基因cat两端50bp同源序列的四环素抗性基因tetA片段,扩增条件同前述,将其电转化入(1)制备的感受态细胞中进行同源重组,电转化条件同前述,在平板上挑选具有四环素抗性的菌落进行菌落PCR验证,即得到用基因tetA替换掉基因cat的菌株;(3)将质粒pTKRED和pTKIP-glk-galP电转化入(2)得菌株进行同源重组,电转化条件同前述,挑取无氨苄青霉素抗性和四环素抗性而有氯霉 素抗性的菌落进行菌落PCR验证;(4)将pCP20电转化到(3)所得菌株,电转化条件同前述,删除抗性基因neo,用引物PTS-up(SEQIDNO:28)/PTS-down(SEQIDNO:29)等进行菌落PCR验证后(图6)送测序,得到基因整合菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,DE3)。1-4的流程如图4所示。5、敲除酪氨酸调控基因tyrR(1)将质粒pTKRED电转化入菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,DE3),电转化条件同前述,制备感受态细胞,制备方法同前述;(2)用引物tyrR-CS1(SEQIDNO:30)/tyrR-CS2(SEQIDNO:31)扩增得到含有菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,DE3)上的基因tyrR两端50bp同源序列的四环素抗性基因tetA片段,扩增条件同前述,将其电转化入(1)制备的感受态细胞中进行同源重组,电转化条件同前述,在平板上挑选具有四环素抗性的菌落进行菌落PCR验证,即得到基因tyrR敲除菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR,DE3)。6、构建重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)将质粒pTKRED和pTKIP-ppsA电转化入菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR,DE3)进行同源重组,电转化条件同前述,挑取无氨苄青霉素抗性和四环素抗性而有氯霉素抗性的菌落,用引物tyrR-up(SEQIDNO:32)、tyrR-down(SEQIDNO:33)和ppsA-M1(SEQIDNO:34)等进行菌落PCR验证(图6)并送测序,得到基因整合菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)。5-6的过程如图5所示。实施例5重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)/pGBAE的构建将质粒pETDuet-GBAE电转化入实施例4所得的重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3),电转化条件同前述,即得莽草酸生产菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)/pGBAE。实施例6重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)/pGBAE摇瓶发酵生产莽草酸摇瓶发酵培养基组成为:葡萄糖20g/L、酵母浸出粉FM88810g/L、胰蛋白胨FP3185g/L、柠檬酸2g/L、谷氨酸钠0.5g/L、K2HPO43g/L,KH2PO41g/L、NaCl1g/L、MgSO43g/L,灭菌前调节pH为7.0。(1)挑取单菌落接种于装有20mLLB培养基的三角瓶中,37℃过夜培养,然后按5%(v/v)接种量接种至装有20mL发酵培养基的250mL三角瓶中,在30℃、220rpm条件下进行发酵培养,48h后取样。(2)莽草酸含量的测定:取样1mL,12000rpm离心5分钟取上清通过0.45μm滤膜过滤后进行莽草酸含量的测定(HPLC):,柱温55℃,紫外检测器波长设定214nm,进样量10μL,流动相为0.25mMH2SO4,流速为0.6mL/min。检测发现莽草酸产量为4.14g/L。实施例7重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)/pGBAE发酵罐发酵生产莽草酸发酵罐培养时发酵培养基组成为:葡萄糖10g/L、甘油10g/L、酵母浸出粉FM88820g/L、胰蛋白胨FP31810g/L,其余同摇瓶培养基配方。发酵6h时一次性加入甘油5g/L、酵母浸出粉FM88810g/L及胰蛋白胨FP3185g/L,8h开始流加400g/L的葡萄糖溶液使罐内葡萄糖含量维持在5~15g/L。发酵过程中定时取样测定菌株生长情况(OD600)和莽草酸生产情况。(1)挑取单菌落接种于装有20mLLB培养基的三角瓶中,37℃过夜培养,然后按1%(v/v)接种量接种至装有LB培养基的三角瓶中,37℃培养 8h,接着按5%(v/v)接种量接种至5L发酵罐中进行发酵。发酵罐的培养基装量为2.5L,培养温度为30℃,空气通量为4~6L/min,溶氧控制在30%,通过添加NH4OH或20%H3PO4维持发酵液pH为7.0。(2)莽草酸含量的测定:取样1mL,12000rpm离心5分钟取上清通过0.45μm滤膜过滤后进行莽草酸含量的测定(HPLC):,柱温55℃,紫外检测器波长设定214nm,进样量10μL,流动相为0.25mMH2SO4,流速为0.6mL/min。(3)以发酵培养时间对OD600和莽草酸含量作图,结果如图7所示,15h后菌株生长进行稳定期,从10h开始莽草酸开始大量合成,培养至48h,莽草酸产量最高达到27.4g/L。实施例8重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)摇瓶发酵生产莽草酸发酵培养基、发酵操作步骤及检测方法同实施例6,检测结果重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)莽草酸摇瓶发酵产量为2.83g/L。实施例9-12用碳源构成比例不同的培养基在发酵罐中发酵培养重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)/pGBAE生产莽草酸菌株、发酵操作步骤及检测方法同实施例7,发酵培养基除葡萄糖和甘油的比例不同外其余组分同实施例7,结果见表1。表1实施例碳源(共20g/L)莽草酸产量(g/L)实施例9葡萄糖+甘油(1:5)23.4实施例10葡萄糖+甘油(5:1)22.6实施例11葡萄糖+甘油(2:3)26.5实施例12葡萄糖+甘油(3:2)26.3对比例1-3中间菌株BW25113(ΔaroL/aroK,ΔptsHIcrr::GBAE,DE3)、BW25113(ΔaroL/aroK,ΔptsHIcrr::GBAEKP,DE3)和BW25113(ΔaroL/aroK,ΔptsHIcrr::GBAEKP,ΔtyrR,DE3)摇瓶发酵生产莽草酸发酵培养基、发酵操作步骤及检测方法同实施例6,检测结果见表3。表2表2结果显示,对比例1-3的中间菌株莽草酸摇瓶发酵产量均显著低于BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)菌株的产量。对比例4-5用碳源构成全部为葡萄糖或全部为甘油的培养基在发酵罐中发酵培养重组菌株BW25113(△aroLaroK,△ptsHIcrr::GBAEKP,△tyrR::ppsA,DE3)/pGBAE生产莽草酸菌株、发酵操作步骤及检测方法同实施例7,发酵培养基除碳源不同外其余组分同实施例7,结果见表3。表3对比例碳源(20g/L)莽草酸产量(g/L)对比例4葡萄糖20.3对比例5甘油8表3结果显示,当发酵罐发酵所用的培养基碳源全部为葡萄糖时,发酵产量与实施例9~12相比明显下降;当碳源全部为甘油时产量急剧降低。综上所述,本发明所得到的重组菌株在发酵过程中无需添加抗生素,不用添加诱导剂进行诱导,发酵工艺简单,能够有效生产莽草酸,可应用于发酵生产莽草酸的产业化中,且对生产其它代谢产物的大肠杆菌工程菌株的构建具有一定的指导意义。应理解,在阅读了本发明的上述内容之后,本领域技术人员可以对本发明的相关条件作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1