一种抗肿瘤活性化合物的制备方法与流程
本发明属于天然药物领域,特别是涉及一种抗肿瘤活性化合物的制备方法。
背景技术:
云南藤黄(GarciniayunnanensisHu)是我国的特有植物,分布于云南省西南部,生于丘陵、坡地的杂木林中,海拔1,300米至1,600米。研究表明其主要成分oblongifolinC和guttiferoneK具有良好的药理活性。WafaaHamed等(J.Nat.Prod.2006,69:774-777)从Garciniaoblongifolia的树皮(500g)中分离得到化合物oblongifolinC(110mg,得率:0.022%),其采用乙酸乙酯浸泡提取,两次硅胶柱层析分离,以庚烷-乙酸乙酯混合溶液进行洗脱,采用制备液相以乙腈-水-0.1%甲酸混合溶液洗脱进行纯化,制得。GangXu等(J.Agric.FoodChem.2008,56(23):11144-11150)从Garciniayunnanensis的果皮(9.0kg)中分离得到化合物oblongifolinC(9.375g换算所得产量,得率:0.10%),其采用丙酮浸泡提取,提取物经娃胶柱层析分离,三氯甲烷、乙酸乙酯、丙酮依次洗脱,三氯甲烷洗脱部位再进行硅胶柱层析分离,正己烷-丙酮混合溶液进行梯度洗脱,洗脱液再采用制备液相经反相C18柱纯化制得。Sheng-XiongHuang等(J.Nat.Prod.2009,72:130-135)从Garciniaoblongifolia的树皮(5.4kg)中分离得到化合物oblongifolinC(140mg换算所得产量,得率:0.003%),其采用丙酮浸泡提取,提取物水混悬后以三氯甲烷萃取,三氯甲烷萃取部位用硅胶柱层析分离,以正己烷-丙酮混合溶液进行梯度洗脱,洗脱液再采用制备液相经反相C18柱以甲醇-水混合溶液进行梯度洗脱,再经过凝胶LH-20柱以甲醇进行洗脱,再采用制备液相以含0.3%甲酸的乙腈-0.3%甲酸的水混合溶液为流动相进行洗脱,纯化,制得。ShugengCao等(J.Nat.Prod.2007,70:686-688)从Rheediacalcicola的果实(130g)中分离得到化合物guttiferoneK(1.32g换算所得产量,得率:1.02%),其采用乙醇渗滤提取,回收溶剂后提取物用甲醇-水(9:1)混悬,以正己烷萃取。甲醇-水层稀释为70%甲醇溶液,再用二氯甲烷萃取。二氯甲烷层回收有机溶剂后,残渣用甲醇复溶,经C18固相萃取柱,后采用制备液相以甲醇-水(85:15)洗脱,制得。MilenaMasullo等(J.Agric.FoodChem.2008,56(13):5205-5210)从Garciniacambogia的果实(200g)中分离得到化合物guttiferoneK(1.26g换算所得产量,得率:0.63%),其采用乙醇浸 泡提取20天,回收有机溶剂后,提取物以水混悬,用二乙醚萃取,二乙醚萃取物再采用半制备高效液相经反相ODS色谱柱分离纯化,以含0.1%三氟乙酸的水-含0.1%三氟乙酸的乙腈溶液为洗脱液洗脱,纯化,制得。GangXu等(J.Agric.FoodChem.2008,56(23):11144-11150)从Garciniayunnanensis的果皮(9.0kg)中分离得到化合物guttiferoneK(5.625g换算所得产量,得率:0.06%),其采用丙酮浸泡提取,提取物经硅胶柱层析分离,三氯甲烷、乙酸乙酯、丙酮依次洗脱,氯仿洗脱部位再进行硅胶柱层析分离,正己烷-丙酮混合溶液进行梯度洗脱,洗脱液再经过硅胶柱层析分离,以石油醚-丙酮混合溶液进行梯度洗脱,后再采用制备液相经反相C18柱,以乙腈-0.1%乙酸水为流动相洗脱,纯化,制得。Huiyang等(J.Agric.FoodChem.2010,58(8):4749-4755)从GarciniaIivingstonei果肉(2.8kg)中分离得到化合物guttiferoneK(21.15mg换算所得产量,得率:0.00076%),其采用甲醇浸泡提取,回收有机溶剂后用水混悬,依次用乙酸乙酯、正丁醇萃取,乙酸乙酯萃取部位经两次凝胶SephadexLH-20柱层析分离,用甲醇洗脱,后再采用制备液相进行纯化制得。GiovannaR.Almanza等(Nat.Prod.Comm.2011,6(9):1269-124)从Rheediaacuinata的茎皮(1500g)中分离得到化合物guttiferoneK(446mg,得率:0.03%),其采用石油醚浸泡提取后,药渣再经二氯甲烷浸泡提取,二氯甲烷提取物再经减压液相柱色谱(VLC)纯化,以二氯甲烷-石油醚及甲醇-二氯甲烷为流动相依次进行梯度洗脱,洗脱液在经多次柱层析,后经重结晶纯化制得。之前本发明人申请的专利(徐宏喜等,抗肿瘤活性化合物的制备方法,申请号:201310410325.2,公开号:103626644A)从GarciniayunnanensisHu果皮(9.0kg)中分离得到guttiferoneK(5.625g换算所得产量,得率:0.06%),采用甲醇、乙醇、丙酮或丁酮浸泡提取,提取物经硅胶柱层析分离,三氯甲烷洗脱,再经MCI柱或大孔树脂柱层析分离,以乙醇-水的混合溶液进行梯度洗脱,洗脱液经中压ODS柱柱层析分离,以乙醇-水的混合溶液进行梯度洗脱,后采用半制备高效液相经反相ODS色谱柱分离纯化,以等比例的甲醇与乙腈的混合溶液再与0.1%甲酸水的混合溶液进行梯度洗脱,纯化制得。
技术实现要素:
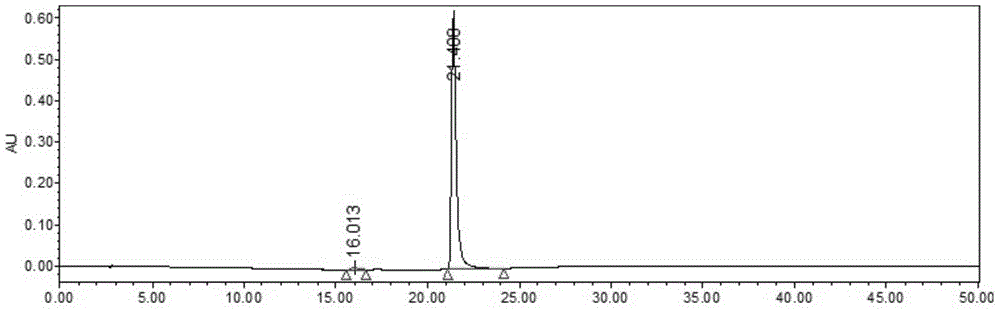
本发明的目的旨在提供一种利用pH区带高速逆流色谱法快速大量制备两种来源于云南藤黄的抗肿瘤化合物的方法,其弥补了现有技术中存在的固态支持物或载体的不可逆吸附、损耗和变性等缺点,使分离回收率提高,且耗时短、操作简单、得率高。具体地说,本发明的第一方面是提供了一种式(Ⅰ)化合物的制备方法,所述方法包括 如下步骤:(a)将云南藤黄果实、枝或叶粉末用60%以上乙醇、甲醇或丙酮等溶剂室温渗漉提取或冷凝回流提取,减压浓缩提取液至无有机溶剂味,得浸膏A;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)作为溶剂系统,上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相,对浸膏C采用高速逆流色谱分离,待所有馏分洗脱结束后,再用等量的浸膏C经上相溶解后进样,通过高速逆流色谱分离,紫外检测器检测,采用Waterse2695高效液相色谱仪在如下色谱条件下检测分析:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水,梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集合并含有目标化合物的流份,减压、浓缩,干燥。在一优选例中,所述步骤(a)为:将云南藤黄果实粉末用95%乙醇室温渗漉提取,减压浓缩95%乙醇提取液至无醇味,得浸膏A。本发明第二方面是提供了式(Ⅱ)化合物的制备方法,所述方法包括如下步骤:(a)将云南藤黄果实、枝或叶粉末用60%以上乙醇、甲醇或丙酮等溶剂室温渗漉提取或冷凝回流提取,减压浓缩提取液至无有机溶剂味,得浸膏A;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)作为溶剂系统,上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相,对浸膏C采用高速逆流色谱分离,待所有馏分洗脱结束后,再用等量的浸膏C经上相溶解后进样,通过高速逆流色谱分离,紫外检测器检测,采用Waterse2695高效液相色谱仪在如下色谱条件下检测分析:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水,梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集合并含有目标化合物的流份,减压、浓缩,得粗品D;(e)以正己烷:叔丁基甲醚:95%乙醇:水(7:3:7.5:2.5,v/v/v/v)作为溶剂系统,上相加入0.1%乙酸作为固定相,下相加入0.08%三乙胺作为流动相,对粗品D采用高速逆流色谱分离,紫外检测器检测,采用Waterse2695高效液相色谱仪检测分析,色谱条件同(d),收集合并含有目标化合物的流份;(f)对上述(e)所得含有目标化合物的流份,采用与(e)相同的高速逆流色谱分离条件纯化,紫外检测器检测,采用Waterse2695高效液相色谱仪检测分析,色谱条件同(d),收集合并含有目标化合物的流份,减压、浓缩、干燥。在一优选例中,所述步骤(a)为:将云南藤黄果实粉末用95%乙醇室温渗漉提取,减压浓缩95%乙醇提取液至无醇味,得浸膏A。本发明各个方面的细节将在随后的章节中得以详尽描述。通过下文以及权利要求的描述,本发明的特点、目的和优势将更为明显。附图说明图1是本发明实施例1粗品D的HSCCC-UV检测图谱;图2是本发明实施例1的HPLC-UV检测图谱;图3是本发明实施例1的HPLC-UV检测图谱;图4是本发明实施例1的HPLC-UV检测图谱;图5是本发明实施例5的HSCCC-UV检测图谱;图6是本发明实施例5的HSCCC-UV检测图谱;图7是本发明实施例5的HPLC-UV检测图谱;图8是本发明实施例5的HPLC-UV检测图谱;图9是本发明实施例5的HPLC-UV检测图谱。具体实施方式本发明所述的,可用本发明的方法快速、大量制备的抗肿瘤化合物,是从云南藤黄果实中提取获得的,其具有如下化学结构式:简而言之,本发明的方法是将分离的溶剂系统置于分液漏斗中充分摇匀后静置分层,上相加入保留酸作为固定相,下相加入洗脱碱作为流动相。先用固定相充满整个柱子,上相溶液溶解浸膏C进样,再泵入下相洗脱,待流动相开始流出色谱柱时,收集分离物,将不同成分分别收集,待所有成分洗脱结束后,再将等量的浸膏C经上相溶解后进样,通过逆流色谱分离,紫外检测器检测,高效液相检测分析,收集合并含有目标化合物的流份,减压浓缩, 分离得到纯度﹥98%的化合物oblongifolinC及粗品D。粗品D再经过两次pH区带逆流色谱的纯化后,得到纯度﹥99%的化合物guttiferoneK。本发明首次采用连续pH曲带高速逆流色谱进样洗脱模式(即连续两次进样),通过增大了进样量,可从354.9g云南藤黄果实中快速大量制备抗肿瘤活性化合物oblongifolinC(8.37g换算所得产量,得率:2.36%)和guttiferoneK(7.65g换算所得产量,得率:2.16%)。由此可见,本发明的方法的得率要远高于现有技术中已有方法的得率。而且,其还弥补了现有方法存在的过程繁琐,耗时长且伴随有样品损失等诸多缺点。下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。除非另外说明,否则所有的百分数、比率、比例、或份数按重量计。除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。本发明提到的上述特征,或实施例提到的特征可以任意组合。本专利说明书所揭示的所有特征可与任何组合物形式并用,说明书中所揭示的各个特征,可以任何可提供相同、均等或相似目的的替代性特征取代。因此除有特别说明,所揭示的特征仅为均等或相似特征的一般性例子。制备抗肿瘤化合物oblongifolinC实施例1(a)将云南藤黄果实粉末359.4g(2014年6月采自云南省德宏州,植物样本由上海中医药大学张红梅博士鉴定,并留样于上海中医药大学中药创新实验室,标号为CMED-0477)用30L95%乙醇渗漉提取,合并95%乙醇提取液,减压浓缩至无醇味得浸膏A206.9g;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,上相8L,下相24L对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B62.9g;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C31.0g;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)对浸膏C采用高速逆流色谱分离:按体积比将溶剂系统至于分液漏斗中,摇匀静置分层一段时间后,将上下相 分开,向上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相。采用TBE-300C型高速逆流色谱仪,柱体积300mL,进样圈20mL,TBP5002泵,TBP-2000UV/2000D紫外检测器,DC-0506低温恒温槽,BSZ-100自动接收器,梅特勒-托利PH计。先用固定相充满整个柱子,上相溶液溶解上述浸膏C2.0g进样,调整主机转速为850rpm,柱温25℃,检测波长351nm,以2mL/min的流速泵入流动相,每3分钟每管收集馏分,待所有馏分洗脱结束后,等量的浸膏C2.0g经上相溶解后进样,通过高速逆流色谱分离,紫外检测器检测,HSCCC-UV图谱如图1所示,采用Waterse2695高效液相色谱仪检测分析,色谱条件为:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水;梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集含有目标化合物的流份(185-250min,475-534min),减压浓缩,得纯度﹥98%的化合物oblongifolinC1.080g。化合物oblongifolinC的HPLC-UV色谱图及相对峰面积数据如下:UV245nm(图2)峰号保留时间面积相对峰面积%高度积分类型116.013879800.745848bb221.4081177542099.26607558bb表1UV280nm(图3)峰号保留时间面积相对峰面积%高度积分类型18.081516820.711949bb216.018651970.904631bb321.409711661998.38352502bb表2UV351nm(图4)峰号保留时间面积相对峰面积%高度积分类型12.78179790.092666bb221.409882106499.89462576bb324.55521650.02404bb表3化合物oblongifolinC的结构鉴定数据如下:EI-MS:670[M]+;HRESI-MS:693.4147[M+Na]+;[α]20D(c=0.21,CHCl3):+14.5;UV(EtOH):λmax(logε)230(sh),282(3.93),356(sh)nm;IR(CHCl3):νmax3535,3338,2927,1727,1646,1611,1523,1442,1383,1290,1215,1114cm-1;H1-NMR(600MHzinCD3OD+0.1%TFA-d)及C13-NMR(150MHzinCD3OD+0.1%TFA-d)数据见表4所示。表4化合物oblongifolinC的H1-NMR、C13-NMR数据实施例2(a)将云南藤黄果实粉末300.6g用30L60%乙醇冷凝回流提取,合并60%乙醇提取液,减压浓缩至无醇味得浸膏A186.2g;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,上相8L,下相24L对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B52.4g;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C25.8g;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)对浸膏C采用高速逆流色谱分离:按体积比将溶剂系统至于分液漏斗中,摇匀静置分层一段时间后,将上下相分开,向上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相。采用TBE-300C型高速逆流色谱仪,柱体积300mL,进样圈20mL,TBP5002泵,TBP-2000UV/2000D紫外检测器,DC-0506低温恒温槽,BSZ-100自动接收器,梅特勒-托利PH计。先用固定相充满整个柱子,上相溶液溶解上述浸膏C2.0g进样,调整主机转速为850rpm,柱温25℃,检测波长351nm,以2mL/min的流速泵入流动相,每3分钟每管收集馏分,待所有馏分洗脱结束后,等量的浸膏C2.0g经上相溶解后进样,通过高速逆流色谱分离,紫外检测器检测,采用Waterse2695高效液相色谱仪检测分析,色谱条件为:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水;梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集含有目标化合物的流份(185-250min,475-534min),减压浓缩,得纯度﹥98%的化合物oblongifolinC1.012g。实施例3(a)将云南藤黄果实粉末320.2g用30L甲醇渗漉提取,合并甲醇提取液,减压浓缩至 无醇味得浸膏A183.7g;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,上相8L,下相24L对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B55.8g;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C26.4g;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)对浸膏C采用高速逆流色谱分离:按体积比将溶剂系统至于分液漏斗中,摇匀静置分层一段时间后,将上下相分开,向上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相。采用TBE-300C型高速逆流色谱仪,柱体积300mL,进样圈20mL,TBP5002泵,TBP-2000UV/2000D紫外检测器,DC-0506低温恒温槽,BSZ-100自动接收器,梅特勒-托利PH计。先用固定相充满整个柱子,上相溶液溶解上述浸膏C2.0g进样,调整主机转速为850rpm,柱温25℃,检测波长351nm,以2mL/min的流速泵入流动相,每3分钟每管收集馏分,待所有馏分洗脱结束后,等量的浸膏C2.0g经上相溶解后进样,通过高速逆流色谱分离,紫外检测器检测,采用Waterse2695高效液相色谱仪检测分析,色谱条件为:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水;梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集含有目标化合物的流份(185-250min,475-534min),减压浓缩,得纯度﹥98%的化合物oblongifolinC1.107g。实施例4(a)将云南藤黄果实粉末350.2g用30L丙酮冷凝回流提取,合并丙酮提取液,减压浓缩至无酮味得浸膏A198.7g;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,上相8L,下相24L对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B60.8g;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C30.2g;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)对浸膏C采用高速逆流色谱分离:按体积比将溶剂系统至于分液漏斗中,摇匀静置分层一段时间后,将上下相分开,向上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相。采用TBE-300C型高速逆流色谱仪,柱体积300mL,进样圈20mL,TBP5002泵,TBP-2000UV/2000D紫外检测器,DC-0506低温恒温槽,BSZ-100自动接收器,梅特勒-托利PH计。先用固定相充满整 个柱子,上相溶液溶解上述浸膏C2.0g进样,调整主机转速为850rpm,柱温25℃,检测波长351nm,以2mL/min的流速泵入流动相,每3分钟每管收集馏分,待所有馏分洗脱结束后,等量的浸膏C2.0g经上相溶解后进样,通过高速逆流色谱分离,紫外检测器检测,采用Waterse2695高效液相色谱仪检测分析,色谱条件为:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水;梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集含有目标化合物的流份(185-250min,475-534min),减压浓缩,得纯度﹥98%的化合物oblongifolinC1.004g。制备抗肿瘤化合物guttiferoneK实施例5(a)将云南藤黄果实粉末359.4g用30L95%乙醇渗漉提取,合并95%乙醇提取液,减压浓缩至无醇味得浸膏A206.9g;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,上相8L,下相24L对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B62.9g;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C31.0g;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)对浸膏C采用高速逆流色谱分离:按体积比将溶剂系统至于分液漏斗中,摇匀静置分层一段时间后,将上下相分开,向上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相。采用TBE-300C型高速逆流色谱仪,柱体积300mL,进样圈20mL,TBP5002泵,TBP-2000UV/2000D紫外检测器,DC-0506低温恒温槽,BSZ-100自动接收器,梅特勒-托利PH计。先用固定相充满整个柱子,上相溶液溶解上述浸膏C2.0g进样,调整主机转速为850rpm,柱温25℃,检测波长351nm,以2mL/min的流速泵入流动相,每3分钟每管收集馏分,待所有馏分洗脱结束后,等量的浸膏C2.0g经上相溶解后进样,通过逆流色谱分离,紫外检测器检测,HSCCC-UV图谱如图1所示,采用Waterse2695高效液相色谱仪检测分析,色谱条件:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水;梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集含有所述化合物的流份(135-170min,420-460min),减压浓缩,得粗品D1.376g。(e)以正己烷:叔丁基甲醚:95%乙醇:水(7:3:7.5:2.5,v/v/v/v)作为溶剂系统,上 相加入0.1%乙酸作为固定相,下相加入0.08%三乙胺作为流动相,对上述粗品D1.376g采用高速逆流色谱分离,色谱分离检测条件与(d)所示相同,HSCCC-UV图谱如图5所示,收集含有目标化合物的流份(90-170min),减压浓缩,得目标化合物流份1.051g。(f)1.051g含有目标化合物的流份,采用上述(e)相同的逆流色谱分离检测条件纯化,HSCCC-UV图谱如图6所示,收集含有目标化合物的流份,减压浓缩,得纯度﹥99%的化合物guttiferoneK978mg。化合物guttiferoneK的HPLC-UV色谱图及相对峰面积数据如下:UV245nm(图7)峰号保留时间面积相对峰面积%高度积分类型18.432543190.265021bb29.715414210.202490bb313.0212067526199.101264857bb414.081911500.446653bb表5UV280nm(图8)峰号保留时间面积相对峰面积%高度积分类型18.439358410.282460bb29.733242820.192055bb312.657121520.091835bb413.0221270743599.03771752bb514.103518360.403787bb表6UV351nm(图9)峰号保留时间面积相对峰面积%高度积分类型16.25672940.051069bb213.0221538610499.50943752bb314.068693580.455239bb表7化合物guttiferoneK的结构鉴定数据如下:HRFABMS:603.3712[M+H]+;[α]23D(c=0.35,CHCl3):-2;UV(MeOH):λmax(logε)241(sh),255(4.54),325(4.14)nm;IR(film):νmax3348,2968,2916,2857,1728,1670,1640,1602,1542,1519,1440,1376,1289,1191,1116cm-1;H1-NMR(500MHzinCD3OD)及C13-NMR(125MHzinCD3OD)数据见表8所示。表8化合物guttiferoneK的H1-NMR、C13-NMR数据实施例6(a)将云南藤黄果实粉末300.6g用30L60%乙醇冷凝回流提取,合并60%乙醇提取液,减压浓缩至无醇味得浸膏A186.2g;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,上相8L,下相24L对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B52.4g;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C25.8g;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)对浸膏C采用高速逆流色谱分离:按体积比将溶剂系统至于分液漏斗中,摇匀静置分层一段时间后,将上下相分开,向上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相。采用TBE-300C型高速逆流色谱仪,柱体积300mL,进样圈20mL,TBP5002泵,TBP-2000UV/2000D紫外检测器,DC-0506低温恒温槽,BSZ-100自动接收器,梅特勒-托利PH计。先用固定相充满整个柱子,上相溶液溶解上述浸膏C2.0g进样,调整主机转速为850rpm,柱温25℃,检测波长351nm,以2mL/min的流速泵入流动相,每3分钟每管收集馏分,待所有馏分洗脱结束后,等量的浸膏C2.0g经上相溶解后进样,通过逆流色谱分离,紫外检测器检测,HSCCC-UV图谱如图1所示,采用Waterse2695高效液相色谱仪检测分析,色谱条件:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水;梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集含有所述化合物的流份(135-170min,420-460min),减压浓缩,得粗品D1.405g。(e)以正己烷:叔丁基甲醚:95%乙醇:水(7:3:7.5:2.5,v/v/v/v)作为溶剂系统,上相加入0.1%乙酸作为固定相,下相加入0.08%三乙胺作为流动相,对上述粗品D1.405g采用高速逆流色谱分离,色谱分离检测条件与(d)所示相同,收集含有目标化合物的流份(90-170min),减压浓缩,得目标化合物流份1.138g。(f)1.138g含有目标化合物的流份,采用上述(e)相同的逆流色谱分离检测条件纯化,收集含有目标化合物的流份,减压浓缩,得纯度﹥99%的化合物guttiferoneK1.056mg。实施例7(a)将云南藤黄果实粉末320.2g用30L甲醇渗漉提取,合并甲醇提取液,减压浓缩至无醇味得浸膏A183.7g;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,上相8L,下相24L对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B55.8g;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C26.4g;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)对浸膏C采用高速逆流色谱分离:按体积比将溶剂系统至于分液漏斗中,摇匀静置分层一段时间后,将上下相分开,向上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相。采用TBE-300C型高速逆流色谱仪,柱体积300mL,进样圈20mL,TBP5002泵,TBP-2000UV/2000D紫外检测器,DC-0506低温恒温槽,BSZ-100自动接收器,梅特勒-托利PH计。先用固定相充满整个柱子,上相溶液溶解上述浸膏C2.0g进样,调整主机转速为850rpm,柱温25℃,检测波长351nm,以2mL/min的流速泵入流动相,每3分钟每管收集馏分,待所有馏分洗脱结束后,等量的浸膏C2.0g经上相溶解后进样,通过逆流色谱分离,紫外检测器检测,HSCCC-UV图谱如图1所示,采用Waterse2695高效液相色谱仪检测分析,色谱条件:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水;梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集含有所述化合物的流份(135-170min,420-460min),减压浓缩,得粗品D1.320g。(e)以正己烷:叔丁基甲醚:95%乙醇:水(7:3:7.5:2.5,v/v/v/v)作为溶剂系统,上相加入0.1%乙酸作为固定相,下相加入0.08%三乙胺作为流动相,对上述粗品D1.320g采用高速逆流色谱分离,色谱分离检测条件与(d)所示相同,收集含有目标化合物的流份(90-170min),减压浓缩,得目标化合物流份1.024g。(f)1.024g含有目标化合物的流份,采用上述(e)相同的逆流色谱分离检测条件纯化,收集含有目标化合物的流份,减压浓缩,得纯度﹥99%的化合物guttiferoneK986mg。实施例8(a)将云南藤黄果实粉末350.2g用30L丙酮冷凝回流提取,合并丙酮提取液,减压浓缩至无醇味得浸膏A198.7g;(b)以正己烷:乙酸乙酯:95%乙醇:水(5:5:5:5,v/v/v/v)作为溶剂系统,上相8L,下相24L对浸膏A进行萃取,收集上相有机相浓缩,得浸膏B60.8g;(c)对浸膏B进行SephadexLH20凝胶柱柱层析分离:以氯仿:甲醇(1:1,v/v)的混合溶液进行洗脱,薄层层析检测,收集含有目标化合物的流份,减压浓缩得浸膏C30.2g;(d)以正己烷:乙酸乙酯:95%乙醇:水(9.75:0.25:8:2,v/v/v/v)对浸膏C采用高速逆流色谱分离:按体积比将溶剂系统至于分液漏斗中,摇匀静置分层一段时间后,将上下相分开,向上相加入0.1%乙酸作为固定相,下相加入0.025%三乙胺作为流动相。采用TBE-300C型高速逆流色谱仪,柱体积300mL,进样圈20mL,TBP5002泵,TBP-2000UV/2000D紫外检测器,DC-0506低温恒温槽,BSZ-100自动接收器,梅特勒-托利PH计。先用固定相充满整个柱子,上相溶液溶解上述浸膏C2.0g进样,调整主机转速为850rpm,柱温25℃,检测波长351nm,以2mL/min的流速泵入流动相,每3分钟每管收集馏分,待所有馏分洗脱结束后,等量的浸膏C2.0g经上相溶解后进样,通过逆流色谱分离,紫外检测器检测,HSCCC-UV图谱如图1所示,采用Waterse2695高效液相色谱仪检测分析,色谱条件:色谱柱:AgilentZORBAXSB-C18column,5μm,4.6I.D.×250mm,流动相:C:乙腈,D:0.1%甲酸水;梯度洗脱程序:0-10min,C:D=90:10→95:5,10-20min,C:D=95:5→100:0,20-25min,C:D=100:0→90:10,25-40min,C:D=90:10→90:10,流速:1.0mL/min,检测波长:351nm,收集含有所述化合物的流份(135-170min,420-460min),减压浓缩,得粗品D1.413g。(e)以正己烷:叔丁基甲醚:95%乙醇:水(7:3:7.5:2.5,v/v/v/v)作为溶剂系统,上相加入0.1%乙酸作为固定相,下相加入0.08%三乙胺作为流动相,对上述粗品D1.413g采用高速逆流色谱分离,色谱分离检测条件与(d)所示相同,收集含有目标化合物的流份(90-170min),减压浓缩,得目标化合物流份1.162g。(f)1.162g含有目标化合物的流份,采用上述(e)相同的逆流色谱分离检测条件纯化,收集含有目标化合物的流份,减压浓缩,得纯度﹥99%的化合物guttiferoneK1.061mg。本发明所涉及的多个方面已做如上阐述。然而,应理解的是,在不偏离本发明精神之前提下,本领域专业人员可对其进行等同改变和修饰,所述改变和修饰同样落入本申请所附权利要求的覆盖范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1