一种制备亚胺的方法与流程
本发明属于制备亚胺领域,具体涉及到一种由醇类化合物和芳香硝基化合物为底物,在无碱助剂条件下通过转氢偶联反应制备亚胺的方法。
背景技术:
亚胺类化合物是非常重要的一类有机合成中间体和重要的氮源,因其具有独特的抑菌、杀菌、抗肿瘤、抗病毒的生物活性以及良好的配位化学性能,广泛应用于医药、农药等领域。
制备亚胺的方法已有报道。CN 101781230 B公布了以胺和酮为原料,以碘为催化剂,在共沸脱水剂存在下加热回流,制备亚胺产物。最近有相关文献报道,以醇与伯胺为原料,钌、铱等过渡金属配合物作为催化剂,均相催化脱水生成相应的亚胺(Angew.Chem.Int.Ed.2010,49,1468.)。但胺的来源是硝基化合物,若能直接从硝基化合物制备亚胺则不必分离出中间产物胺,生产工艺就会变得简单。CN 103058805 A报道了在过渡金属负载型固体催化剂作用下,以芳香醇和硝基化合物为原料,合成相应的N-亚苄基苯胺和N-苄基苯胺化合物。但产物的选择性不够高。Cui等报道了以Au/Ag–Mo nano-rods为催化剂,以芳香醇和硝基化合物为原料,合成相应的亚胺(Chem.Commun.,2012,48,9391.),但反应过程中添加了碳酸钾,碱会腐蚀化工设备。
因此,已报道的方法路线中大多采用胺类,作为N源与醇、醛或者酮在催化剂存在的条件下合成亚胺,或用硝基化合物与醇在加入碱的情况下合成亚胺,因此开发出高原子经济性合成路线,从更廉价易得的硝基苯为原料,不加碱的合成方法,催化剂可以多次循环利用具有重要的研究意义。
技术实现要素:
针对上述问题,本发明提供一种制备亚胺类化合物的方法,该方法在无碱助剂条件下,通过转氢偶联反应制备亚胺类化合物。
本发明反应式为:
本发明采用的技术方案为:
在反应釜中,芳香硝基化合物和醇类化合物为底物,加入溶剂和硫化钼基催化剂,通入惰性气体置换合成釜内部的气氛,密闭,搅拌,加热反应,反应完成后,离心分离催化剂,蒸干溶剂,通过柱分离提纯得到产物亚胺类化合物。
所述芳香硝基化合物中的芳香环为连有一个或两个以上取代基的苯 环、稠环或杂环;取代基选自H,F,Cl,Br,I,CH3,OCH3,NH2,NO2,CHO,Ph中的一个或两个以上,取代基相同或不同;
所述醇类化合物分子式为Rx-(C6H5-X)-CH2OH,x=1~5的整数,其中,R=H,F,Cl,Br,I,CH3,OCH3,NH2,NO2,CHO,Ph,x代表取代基的数目,当x>1时,R为相同取代基或不同取代基;或者,醇类化合物分子式为CH3(CH2)y-OH,其中,y=0-10。
芳香硝基化合物中所述稠环为萘环,蒽环;所述杂环为吡啶环,噻吩环,呋喃环,咪唑环。
所述醇类化合物和芳香硝基化合物的摩尔比为10:1~0.5:1;
醇类化合物于反应体系中的浓度为0.01~2mol·L-1,硝基化合物于反应体系中的浓度为0.005-1mol·L-1。
所述硫化钼基催化剂选自二硫化钼,三硫化钼,金属掺杂的二硫化钼中一种或两种以上;
所述金属掺杂的二硫化钼,其中,金属选自Au、Pd、Pt、Rh、Ru、Ag、Al、Fe、Co、Cu、Zn、Bi、Pb中一种或两种以上,金属优选为Pt、Pd、Rh、Ag中的一种或两种以上,金属与钼的摩尔比范围为1:100-1:2;金属占金属掺杂的二硫化钼总质量的数值大于0小于等于16wt%;Mo占金属掺杂的二硫化钼总质量的范围34-60wt%。
所述金属掺杂的二硫化钼的制备方法为:
将金属盐前驱体与钼酸铵前驱体溶解于水中,Ar保护下,加入硫源混合,将混合液体转移到水热釜中,100-600℃水热处理1-10h,用饱和氢氧化钠溶液于20-100℃处理1-20h,水离心洗涤,80-150℃干燥。
所述硫源为硫化铵、硫氢化铵、硫化钠或二硫化碳中的一种或几种。
其中金属前驱体盐为:
H2PdCl4,H2PtCl6,RhCl3,RuCl3,AgNO3,AlCl3,FeCl3,Co(NO3)2,Cu(NO3)2,Zn(NO3)2,BiCl3,Pb(NO3)2盐中的一种或两种以上。
更好的催化活性催化剂的制备为:
将金属盐前驱体与钼酸铵前驱体溶解于水中,Ar保护下加入硫化铵、或二硫化碳中的一种或两种,将液体转移到水热釜中。200-400℃水热处理4-8h,用0.2M-饱和的氢氧化钠溶液于60-80℃处理3-15h,水离心洗涤,80-150℃干燥。Pt、Pd、Rh的合金表现出了更好的催化活性。
本方法所述溶剂为甲苯,二甲苯,三甲苯,乙苯,氯苯,1,4-二氧六环,乙腈中一种或两种以上;优选溶剂为甲苯,二甲苯,氯苯,乙腈。
催化剂的加入量为底物总质量的0.1~10wt%,所述惰性气氛为氩气或氮气。
反应温度为80℃~200℃,反应时间为2~24h;
反应结束后离心去除催化剂,并用乙醇洗涤催化剂三次,将洗涤液与反应液合并;
选择15:1石油醚和乙酸乙酯柱分离产物,浓缩纯化后得到亚胺产物。
本发明的优点和有益效果为:
本发明涉及一种由廉价易得的醇类化合物与芳香硝基化合物为底物偶联制备亚胺的方法,在不同的多相硫化钼基催化剂催化作用下,能得到高的转化率以及亚胺选择性,且该发法为原子经济性反应,反应中不需要加碱添加剂。
附图说明
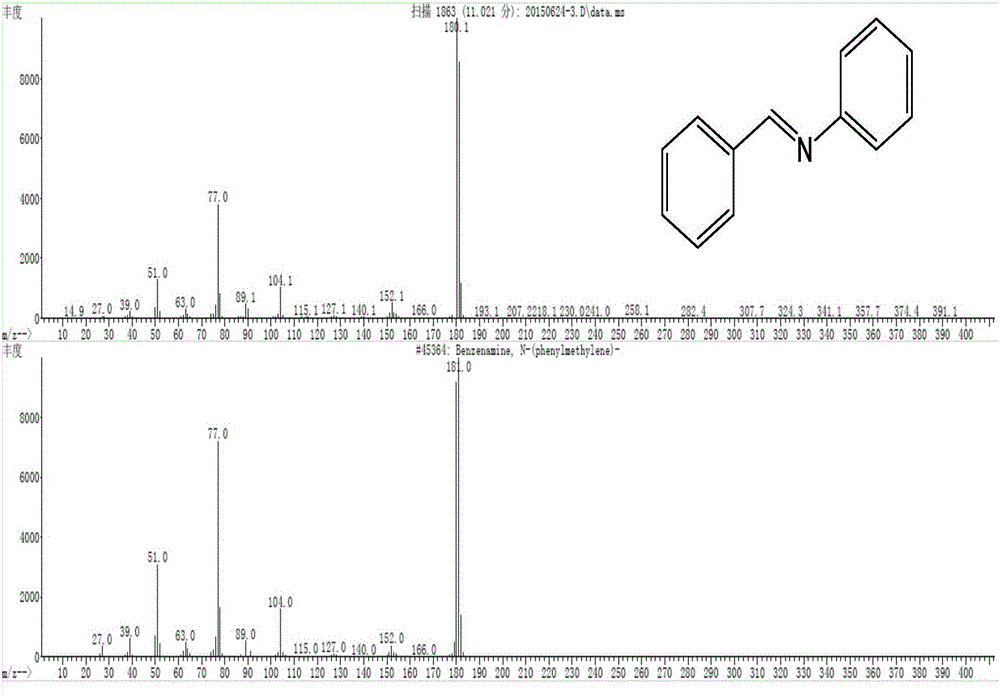
图1为亚胺合成实施例1制备得到的N-苄烯基苯胺的质谱图。
具体实施方式
为了对本发明进行进一步详细说明,下面给出几个具体实施案例,但本发明不限于这些实施例。
一、硫化钼基催化剂的制备方法
制备步骤如下:
将金属盐前驱体与钼酸铵前驱体溶解于水中,Ar保护下加入硫化铵、硫氢化铵、硫化钠或二硫化碳,将液体转移到水热釜中。100-600℃水热处理1-10h。用0.2M-饱和的氢氧化钠溶液于20-100℃处理1-20h。水离心洗涤,80-150℃干燥。
实施例1
将1mL 0.1M的HAuCl4与10mmol的钼酸铵溶解于20mL水中,加入10mL浓度16-20%的硫化铵水溶液,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
实施例2
将1mL 0.1M的HAuCl4与10mmol的钼酸铵溶解于20mL水中,加入10mL浓度16-20%的硫化铵水溶液,Ar保护条件下将液体转移入水热釜中600℃处理1h,用0.2M氢氧化钠溶液于100℃处理20h。水离心洗涤,80℃干燥。
实施例3
将1mL 0.1M的HAuCl4与10mmol的钼酸铵溶解于20mL水中,加入10mL浓度16-20%的硫化铵水溶液,Ar保护条件下将液体转移入水热釜中100℃处理10h,用饱和氢氧化钠溶液于80℃处理4h。水离心洗涤,150℃干燥。
实施例4
将10mL 0.1M的HAuCl4与4mmol的钼酸铵溶解于20mL水中,加入10mL浓度16-20%的硫化铵水溶液,Ar保护条件下将液体转移入水热釜中100℃处理10h,用1M氢氧化钠溶液于20℃处理8h。水离心洗涤,80℃干燥。
实施例5
将1mL 0.1M的H2PtCl6与10mmol的钼酸铵溶解于20mL水中,加入10mL浓度16-20%的硫化铵水溶液,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
实施例6
将1mL 0.1M的HAuCl4、1mL 0.1M的AgNO3与10mmol的钼酸铵溶解于20mL水中,加入10mL浓度16-20%的硫化铵水溶液,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
实施例7
将1mL 0.1M的H2PdCl4、0.1M的Cu(NO3)2与10mmol的钼酸铵溶解于20mL水中,加入10mL浓度16-20%的硫化铵水溶液,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
实施例8
将1mL 0.1M的RhCl3、0.1M的RuCl3与10mmol的钼酸铵溶解于20mL水中,加入5mL浓度16-20%的硫化铵水溶液与5mL CS2,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
实施例9
将1mL 0.1M的AlCl3、0.1M的Ni(NO3)2与10mmol浓度16-20%的钼酸铵水溶液溶解于20mL水中,加入10mL CS2,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
实施例10
将1mL 0.1M的FeCl3、0.1M的Ni(NO3)2与10mmol浓度16-20%的钼酸铵水溶液溶解于20mL水中,加入10mL CS2,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
实施例11
将1mL 0.1M的BiCl3与10mmol浓度16-20%的钼酸铵水溶液溶解于20mL水中,加入10mL CS2,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
实施例12
将1mL 0.1M的Zn(NO3)2与10mmol浓度16-20%的钼酸铵水溶液溶解于20mL水中,加入10mL CS2,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
实施例13
将10mmol浓度16-20%的钼酸铵水溶液溶解于20mL水中,加入10mL浓度16-20%的硫氢化铵,Ar保护条件下将液体转移入水热釜中100℃处理10h,用0.2M氢氧化钠溶液于20℃处理1h。水离心洗涤,80℃干燥。
二、亚胺合成
实施例1:N-苄烯基苯胺制备
在25mL耐压釜中,加入苯甲醇(1.62g,15mmol)、硝基苯(615mg,5mmol)、催化剂Pt/MoS2(50mg)(其中Pt质量百分含量为2%)、甲苯5.5mL用于搅拌的磁子,氩气置换反应体系后,反应釜置加热至150℃,搅拌反应24小时。气相色谱分析硝基苯转化率>99%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
转氢偶联反应机理:
实施例2:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于反应釜加热至180℃,搅拌反应15小时。气相色谱分析硝基苯转化率>99%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例3:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于反应釜加热至200℃,搅拌反应2小时。气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例4:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为PdPt/MoS2,气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例5:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为AgPt/MoS2,气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性 >90%。
实施例6:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为Fe/MoS2,气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例7:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为Pb/MoS2,气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例8:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为RuAg/MoS2,气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例9:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为CoAl/MoS2,气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例10:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为MoS2,气相色谱分析硝基苯转化率>55%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>99%。
实施例11:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为MoS3,气相色谱分析硝基苯转化率>85%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例12:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为Au/MoS2,气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例13:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为AuAg/MoS2,气相色谱分析硝基苯转化率>90%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性 >90%。
实施例14:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为Zn/MoS2,气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例15:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于苯甲醇(5.4g,50mmol)、硝基苯(615mg,5mmol)。气相色谱分析硝基苯转化率>99%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>99%。
实施例16:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于苯甲醇(270mg,
2.5mmol)、硝基苯(615mg,5mmol)。气相色谱分析硝基苯转化率>60%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>96%。
实施例17:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为反应后回收的催化剂即:反应完后,过滤,离心,用乙醇洗涤5次,干燥。气相色谱分析硝基苯转化率>99%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>99%。
实施例18:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为反应2次后回收的催化剂即:反应完后,过滤,离心,用乙醇洗涤5次,干燥。气相色谱分析硝基苯转化率>96%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>99%。
实施例19:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为反应3次后回收的催化剂即:反应完后,过滤,离心,用乙醇洗涤5次,干燥。气相色谱分析硝基苯转化率>96%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>98%。
实施例20:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于催化剂为反应4次后回收的催化剂即:反应完后,过滤,离心,用乙醇洗涤5次,干燥。气相色谱分析硝基苯转化率>95%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>96%。
实施例21:对甲基N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于所用醇为对甲基苯甲醇 (6.1g,50mmol)。气相色谱分析硝基苯转化率>99%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到对甲基N-苄烯基苯胺,选择性>99%。
实施例22:对氯N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于所用醇为对氯苯甲醇
(7.1g,50mmol)。气相色谱分析硝基苯转化率>99%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到对氯N-苄烯基苯胺,选择性>99%。
实施例23:对N-丁烯苯胺制备
反应步骤同实施例1,与实施例1不同之处在于所用醇为正丁醇(3.7g,50mmol)。气相色谱分析硝基苯转化率>80%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-丁烯苯胺,选择性>99%。
实施例24:N-苄烯基对氯苯胺制备
反应步骤同实施例1,与实施例1不同之处在于硝基化合物为4-硝基氯苯(787mg,5mmol)。气相色谱分析硝基苯转化率>99%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到对氯N-苄烯对氯苯胺,选择性>99%。
实施例25:N-苄烯基苯胺制备
反应步骤同实施例1,加入苯甲醇1mmol,溶剂为10mL二甲苯。气相色谱分析硝基苯转化率>59%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>90%。
实施例26:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于加入硝基苯为0.02mmol溶剂为甲苯和氯苯个1mL。气相色谱分析硝基苯转化率>89%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>97%。
实施例27:N-苄烯基苯胺制备
反应步骤同实施例1,与实施例1不同之处在于溶剂为2.5mL二甲苯。气相色谱分析硝基苯转化率>99%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-苄烯基苯胺,选择性>99%。
实施例28:N-(噻吩-2-亚甲基)-苯胺制备
反应步骤同实施例1,与实施例1不同之处在于2-噻吩甲醇(2.8g,
25mmol)。气相色谱分析硝基苯转化率>80%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-(噻吩-2-亚甲基)-苯胺,选择性>89%。
实施例29:N-(吡咯-3-亚甲基)-苯胺制备
反应步骤同实施例1,与实施例1不同之处在于所用醇为吡咯烷-3-甲醇(2.5g,25mmol)。气相色谱分析硝基苯转化率>79%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-(吡咯-3-亚甲基)-苯胺,选择性>89%。
实施例30:N-(2-萘基-1亚甲基)-苯胺制备
反应步骤同实施例1,与实施例1不同之处在于所用醇1-萘乙醇(4.3g,25mmol)。气相色谱分析硝基苯转化率>88%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到N-(2-萘基-1亚甲基)-苯胺,选择性>80%。
实施例31:3-亚苄基氨基吡啶制备
反应步骤同实施例1,与实施例1不同之处在于所用硝基化合物为3-硝基吡啶(0.86g,5mmol)。气相色谱分析硝基苯转化率>78%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到3-亚苄基氨基吡啶,选择性>90%。
实施例32:2-亚苄基氨基呋喃制备
反应步骤同实施例1,与实施例1不同之处在于所用硝基化合物为2-硝基呋喃(0.56g,5mmol)。气相色谱分析硝基苯转化率>78%,反应混合物离心除去催化剂,回收的催化剂循环使用。产物经高分辨质谱测定确认,得到3-亚苄基氨基吡啶,选择性>90%。
- 还没有人留言评论。精彩留言会获得点赞!