制备5‑氟色醇的方法与流程
已知色醇衍生物是合成药物活性物质的结构单元。5-氟色醇[2-(5-氟-1H-吲哚-3-基)-乙醇]例如描述为在被取代的螺环己烷化合物的合成中的结构单元,被取代的螺环己烷化合物尤其自WO 2004/043967和WO 2008/040481已知,例如(1r,4r)-6’-氟-N,N-二甲基-4-苯基-4’,9’-二氢-3’H-螺[环己烷-1,1’-吡喃并-[3,4b]-吲哚]-4-胺和(1r,4r)-6’-氟-N-甲基-4-苯基-4’,9’-二氢-3’H-螺[环己烷-1,1’-吡喃并-[3,4b]-吲哚]-4-胺。这些化合物用作混合型NOP-/μ-阿片-受体激动剂且在治疗例如急性疼痛、内脏疼痛、神经疼痛、癌症疼痛和慢性疼痛的疼痛方面得到特别关注。
在本领域中已知色醇的多种合成路线。例如,已知使用靛红(isatine)作为原料经由多步合成来合成色醇。该路线需要使用例如LiAlH4的强还原剂,这使得这些反应难以从实验室扩大到工业规模。这类合成例如由C.A. Demerson等人在J.Med.Chem. 1976, 19, 391-395中描述。还参见WO 2008009415、WO 2007/124903、WO 2008101659和US 2006/0166947A1。
合成色醇的另一路线使用费歇尔-吲哚反应以使芳基肼和醛在例如强无机酸如H2SO4的酸、固定型非均相布朗斯特酸性催化剂如蒙脱石K10或路易斯酸如ZnCl2存在下环化。参看例如K. R. Campos等人,Org.Lett., 第6卷;第1期,2004, 79-82;J. Soubhye等人,J.Med.Chem.2010, 53, 8747-8759;P.K. Singh等人,Tetrahedron Letters 49 (2008) 3335-3340;WO 03/099824 A1;B. McKittrick等人,J.Hetereocycli cChem, 27, 2151 (1990);US 2006/0166947和R. E. Mewshaw等人,J.Med.Chem.2004, 47, 3823-3842。这些反应(就明确描述的那些而言)在使用与水混溶的有机共溶剂以及水的均相水性溶剂中进行。所使用的共溶剂例如为THF、二甲基乙酰胺、乙腈、DMF或二噁烷。根据P.K. Singh等人(参见上文),在纯水性体系中且使用蒙脱石K10作为非均相催化剂,色醇反应产物和烯醇醚原料形成高浓度的有机层。据说这导致形成不需要的副产物,且建议使用某些极性的与水混溶的共溶剂,以避免形成非均相溶剂体系且由此降低不需要的副产物的形成。在所有上述合成路线中,如果完全离析,色醇则经由例如柱色谱或快速色谱法的色谱法纯化,使得这些方法不适合在较大的工业规模上使用。
在使用费歇尔-吲哚环化反应来合成色醇时遇到的问题之一是形成不需要的副产物。在这些反应中观察到的一个副反应导致经由最初形成的色醇与未反应的烯醇醚如二氢吡喃或二氢呋喃反应形成三元醇化合物,例如:
。
对于7-乙基色醇(在非甾族消炎药依托度酸的制备中的中间体)的合成,已知使用2,3-二氢呋喃和乙基苯基肼盐酸盐的两步费歇尔-吲哚反应合成,其中,在第一步骤中,形成相应苯腙,随后使其进一步反应以给出产物。该产物在若干纯化步骤之后经由自环己烷中结晶来离析。据描述改进的选择性和增加的产率可借助于下列措施实现:a) 控制在腙的初始形成期间的pH值(弱酸性),b) 在腙形成的最后加入NaHSO3以去除过量的4-羟基丁醛,c) 在费歇尔环化步骤期间缓慢加入H2SO4,d) 用乙醇和甲苯稀释费歇尔反应体系以通过从水相中原位萃取所形成的产物来降低分子间相互反应的发生,和e) 调节在反应之后的pH值以达到约8的pH值。据说所述反应提供具有高于80%的纯度的7-乙基色醇的粗产物并以高于60%的产率产生纯化产物。(参见Y. Lü等人,Journal of Chemical Engineering of Chinese Universities, 第1期,第24卷,2010,第127-131页。)
经由费歇尔-吲哚反应合成5-氟色醇由P.K. Singh等人(参见上文)、K. R. Campos等人(参见上文)和J. Soubhye等人(参见上文)描述。例如如由K. R. Campos描述的典型反应流程表示如下:
。
反应流程1:苯肼和烯醇醚的酸催化费歇尔-吲哚环化以得到色醇。
在费歇尔-吲哚环化反应期间所有这些程序使用N,N-二甲基乙酰胺(DMA或DMAC)作为与水混溶的有机共溶剂。据描述反应产物经由色谱法自粗产物离析。
5-氟色醇具有约60℃的相对较低的熔点。因此,化合物结晶的容易度强烈地取决于其纯度。残留副产物和溶剂阻碍结晶且5-氟色醇常常只能作为油性的高度粘性的流体离析。在设法使合成扩大到工业规模时这也会产生问题,因为与固体结晶产物相比,油性流体在工业规模中操作难得多。在上述引文中描述为选择用于合成5-氟色醇的水混溶共溶剂的DMAC在反应之后难以去除且如果DMAC例如仅经由蒸馏去除,而不是在粗产物经由色谱法纯化期间去除,则在产物中见到相当大量的残留溶剂。另外,DMAC具有毒性且对环境是危险的且因此应该在药品的工业合成中避免。由于其本征毒性,在药用产物中仅容许非常少量的残留DMAC,且使其从其合成期间使用的活性剂或中间体产物中去除需要另外的高成本且耗时的纯化步骤。
因此,需要5-氟色醇的合成,其为简单、成本有效的,其可以数千克的规模进行且其以相对较高的纯度和足够的产率提供产物。
因此,本发明的目的在于提供至少部分地克服现有技术合成的缺点的合成。该目的经由如本文限定的本发明实现。
本发明提供基于费歇尔-吲哚环化反应的方法,这允许可以数千克的规模进行的5-氟色醇的单步合成。已经意外地发现,根据本发明的方法产生相对较高纯度的5-氟色醇,其可在不使用色谱法的情况下经由简单沉淀/结晶离析。此外,已经意外地发现,根据本发明的方法,在费歇尔-吲哚反应的[3+3]环化之前从反应介质中去除过量的醛不是必需的。此外,利用根据本发明的方法,可避免使用认为对健康和环境危险的溶剂。
另一方面,本发明提供使用经由根据本发明的第一方面的方法得到的5-氟色醇制备(1r,4r)-6’-氟-N,N-二甲基-4-苯基-4’,9’-二氢-3’H-螺[环己烷-1,1’-吡喃并-[3,4b]-吲哚]-4-胺的方法。
制备5-氟色醇的本发明方法包括以下步骤:
a) 提供包含4-氟苯基-肼、活化剂、水和选自四氢呋喃、2-甲基四氢呋喃和乙酸异丙酯的至少一种非质子有机溶剂的混合物;
b) 向混合物中加入至多约1.1当量的2,3-二氢呋喃在选自四氢呋喃、2-甲基四氢呋喃和乙酸异丙酯的至少一种非质子有机溶剂中的溶液以与4-氟苯基肼反应,从而给出5-氟色醇,其中在步骤a)和b)中提供的非质子溶剂和水以相对于彼此的比率提供,使得在添加步骤b)之前或期间形成包含液体有机相和液体水相的非均相反应混合物;
c) 使有机相与水相分离;
d) 使有机相与至少一种无机盐的水溶液接触以形成包含液体有机相和液体水相的非均相混合物;
e) 使有机相与非均相混合物的水相分离;和
f) 经由沉淀步骤从有机相中离析5-氟色醇。
已经意外地发现,当如上所述在非均相反应介质中进行4-氟苯基肼、2,3-二氢呋喃和活化剂的费歇尔-吲哚型反应时,副产物的产生显著降低,这继而使得能够在不使用昂贵的色谱法或不使用环境危险且有毒的溶剂如DMAC的情况下从反应混合物中离析高产率的具有相当高纯度的结晶5-氟色醇。
在根据本发明的方法的实施方案中,在步骤a)中形成的混合物为包含液体有机相和液体水相的非均相混合物。
本文中用以描述非均相反应介质和在所述方法期间形成的任何其他非均相混合物的性质的术语“水性液相”和“有机液相”中“水性液相”的意义应理解为其中主要溶剂为水、但另外的溶剂(特别是有机溶液)也可溶解于水相中的液相。反过来,“有机液相”为其中主要溶剂为有机溶剂、但一些水也可溶解于有机相中的相。本领域技术人员意识到,当使包含有机溶剂的有机相与水接触时,水将在某种程度上溶解于有机溶剂中且有机溶剂将在某种程度上溶解于水中,直至达到相平衡的状态,这取决于每种溶剂在彼此中的相对溶解度。在这方面,术语“水混溶的”有机溶剂用以描述在文献中描述为以任何比率溶解于水中的那些溶剂,例如乙腈、DMF、DMAC、二噁烷和THF。与水仅在有机溶剂:水的特定比率内形成均相混合物的有机溶剂在本文中分别描述为与水部分混溶或与水不混溶。
与在关于制备5-氟色醇的上文提到的现有技术方法中的教导相反,在这些方法中费歇尔-吲哚环化都在包含单一液相的反应介质中进行,在根据本发明的方法中,费歇尔-吲哚环化以如下方式进行,其中反应直接从添加步骤b)开始在非均相反应介质中发生或者这一非均相反应介质在包含2,3-二氢呋喃的溶液的添加过程中形成。非均相反应介质包含液体水相和液体有机相,其中所述有机相包含选自四氢呋喃、2-甲基四氢呋喃和乙酸异丙酯的至少一种非质子有机溶剂。所述有机相还可包含上文列出的非质子有机溶剂中的一种或多种的混合物。尽管不想受理论约束,但还是认为在该反应介质中,在费歇尔-吲哚环化中在水相中形成的5-氟色醇迅速移动到有机相中且因此不再进行与在水相中存在的活化的2,3-二氢呋喃的副反应。另外,认为有机溶剂在水中的相应溶解度或者水在有机溶剂中的相应溶解度提供给反应物在非均相反应介质的两个液相中的条件,这产生相应反应物在两相中的分布平衡,这增加了反应对于形成目标反应产物,即5-氟色醇的选择性。因此,与用于制备5-氟色醇的现有技术方法相反,现有技术方法通过使用例如DMAC的与水高度混溶的极性有机溶剂作为共溶剂连同以足以形成单一均相流体相的量的水一起设法避免非均相反应介质的存在,而根据本发明的方法使用非均相反应介质来控制不需要的副产物的出现。发现,当使用非极性的与水不混溶的有机非质子溶剂(例如甲苯或苯,烷烃,例如戊烷、己烷、庚烷或环己烷;和卤代烷烃,例如二氯乙烷)时,不需要的副产物以使其在没有经由色谱法先前纯化的情况下不可能离析以结晶形式的反应产物的程度形成。再次不想受理论限制,但还是认为,当使用这类与水不混溶的非质子有机溶剂时,5-氟色醇在这些有机溶剂中的溶解度不够高,不足以有效地从水相中移除5-氟色醇。在用其他极性的与水部分混溶或与水混溶的溶剂如乙醇、正丙醇、异丙醇、2-甲基-1-丙醇、正丁醇、乙酸或二噁烷的情况下,不可能形成非均相反应混合物和/或在反应期间以抑制5-氟色醇经由简单沉淀/结晶离析的量形成副产物。再次不想受理论限制,但还是认为,对于这些溶剂,反应物(包括活化剂)、水和非质子溶剂在反应介质的相应相中的分配无法使反应具有充分选择性,从而产生足够纯度的目标5-氟色醇,使得其能够经由沉淀/结晶容易地离析。类似地,使用例如乙酸正丁酯、乙酸异丁酯、乙酸叔丁酯和甲基叔丁基醚的溶剂无法给出令人满意的结果。意外地发现,在费歇尔-吲哚环化所需要的反应条件下,乙酸异丙酯表现出足够的化学稳定性且不经历脱酯过程,而乙酸乙酯和乙酸叔丁酯倾向于该反应,使得这些溶剂已经出于该原因而不适合在根据本发明的方法中使用。
另外,发现当如由Y. Lü等人(参见上文)关于7-乙基色醇的合成所述(即在包含水、乙醇和甲苯作为溶剂的非均相反应介质中)进行反应时,该反应也缺乏选择性且使得5-氟色醇不可能容易地离析。意外地发现,在该文献中作为水混溶性溶剂描述的四氢呋喃也可用于根据本发明的方法中,原因是其水混溶性以可形成包含水性液相和有机液相的非均相反应介质的方式受反应介质中反应物存在的影响。此外,发现通过使用上述溶剂(这些溶剂在环境压力下的沸点全部远低于DMAC的沸点),附着到粗产物的任何残留溶剂可例如经由在减压和/或高温下干燥而相对容易地去除。这在5-氟色醇待经沉淀/结晶离析和/或纯化时特别有帮助。
另外,发现副产物的出现通过将2,3-二氢呋喃相对于4-氟苯基肼的量限制到1.1当量而降低。再次,尽管不想受理论限制,但还是认为,在2,3-二氢呋喃的相对量较高的情况下,5-氟色醇与2,3-二氢呋喃的副反应的反应速率增加到对反应的选择性有害的程度且这样形成的量的副产物使得很难接着离析足够纯度的产物。优选在反应中使用这两种反应物中的每一种的理论上需要的相对量,即等摩尔量的4-氟苯基肼和2,3-二氢呋喃。如果4-氟苯基肼相对于2,3-二氢呋喃以过量使用,优选以相对于4-氟苯基肼而言不低于0.9当量的比率使用,因为显而易见,相对于更昂贵试剂4-氟苯基肼盐酸盐而言的产率将显著减小。因此,在根据本发明的方法的其他实施方案中,将2,3-二氢呋喃相对于4-氟肼盐酸盐的比率选择在约0.9-1.1当量的范围内。
有机反应产物的离析和纯化常经由溶剂萃取进行,例如将液体水相加到含有粗产物的液体有机相中以形成细分散的非均相混合物且随后将混合物相分离成液体水相和液体有机相,由此产物在这两相之一中富集,随后将其与另一者中离析并进一步处理。该纯化步骤的先决条件是所形成的混合物可再次迅速分离并且不形成稳定乳液。这对于大规模/工业规模工艺特别重要,因为已知相分离所需要的时间随批量大小而增加。意外地发现,当使用至少一种无机盐的水溶液来洗涤有机相时,粗产物可通过萃取从反应介质中离析并含有目标化合物的有机相而高效地离析并纯化。使用水性盐溶液的萃取将大量有机副产物移入水相中,但几乎没有移除产物。另外,发现在使用至少一种无机盐的水溶液来洗涤有机相时,而不是在使用纯水作为初始洗涤步骤中的水相时,在洗涤步骤中形成的细分散的非均相混合物再次迅速分离。如果需要的话,水可作为随后洗涤中的洗涤液使用。与如在现有技术中所述在反应介质中使用DMAC不同,容易地实现从粗产物中去除残留有机溶剂,这使得去除共溶剂所需要的洗涤步骤的数目减少,由此减少在合成中产生的废料的量并降低工艺所需的时间,这两者都促进工艺成本的进一步降低。
本领域技术人员意识到,在本发明中用以形成5-氟色醇的费歇尔-吲哚环化反应为在文献中描述为在腙形成和随后的[3+3]-重排下肼与醛的反应的反应。因此,本领域技术人员清晰可见,a) 在反应中使用的试剂2,3-二氢呋喃表示必须经由水解(参见以下反应流程2)活化以与4-氟苯基肼反应的受保护的醛和b) 这类水解需要活化剂。
反应流程2:2,3-二氢呋喃的活化
这类活化剂可例如为有机或无机布朗斯特酸,即有机或无机质子酸;非均相布朗斯特酸催化剂或无机路易斯酸或其混合物。
在根据本发明的方法的一个实施方案中,所述活化剂为至少一种有机或无机质子酸或非均相布朗斯特酸催化剂。
在本发明方法的又一实施方案中,相对于4-氟苯基肼,所述活化剂为至少等摩尔量的有机或无机质子酸,该有机或无机质子酸具有在-6.5至+10范围内的pKa值。优选所述酸选自H2SO4、HCl、NH4HSO4、(NH4)2SO4、乙酸、NH4Cl、H3PO4、(NH4)H2PO4、HClO4或(NH4)ClO4。作为活化剂,还可使用这些酸的混合物。在本发明方法的另一实施方案中,所述活化剂选自HCl、H2SO4、H3PO4、乙酸和NH4Cl或其混合物。在另一实施方案中,所述活化剂为HCl。在另一实施方案中,所述活化剂为H2SO4。已经发现,当使用HCl或H2SO4作为活化剂时,完成费歇尔-吲哚环化所需要的反应时间可减至最少,特别是在所述酸以相对于4-氟苯基肼的量而言过量使用时,4-氟苯基肼本身充当布朗斯特碱。如果使用较弱酸作为活化剂,则发现在指定反应温度下完成反应所需的反应时间较久,但与使用较强酸相比较反应的选择性可增加。在本发明的其他实施方案中,所述酸选自NH4Cl、NH4HSO4和(NH4)2SO4。根据费歇尔-吲哚环化所用的活化剂、批量大小和反应温度,反应时间通常在2-48小时范围内。出于经济原因,当然优选使用使得反应时间相对较短(例如在2-24小时范围内)的反应条件。
在本发明的另一实施方案中,所述活化剂为至少一种有机或无机质子酸,相对于4-氟苯基肼的量,其中所述一种或多种质子酸在反应介质中以超过至少1当量的量存在。在本发明方法的另一实施方案中,所述活化剂为质子酸,其中所述质子酸以其与4-氟苯基肼的加成盐的形式提供在反应介质中。因此,在本发明方法的其他实施方案中,所述活化剂为HCl或H2SO4,这些可通过将盐4-氟苯基肼盐酸盐或H2SO4与4-氟苯基肼的加成盐加到反应混合物中而提供在反应介质中。如果将活性剂以其与4-氟苯基肼的加成盐的形式加到反应介质中,则优选将另外量的活性剂加到反应混合物中,使得活性剂以相对于4-氟苯基肼的量而言过量存在。如果例如在反应介质中使用4-氟苯基-肼盐酸盐,可将另外的HCl或另一活化剂加到反应介质中,即,另外量的质子有机或无机酸,例如H2SO4、NH4HSO4、(NH4)2SO4、乙酸、NH4Cl、H3PO4、(NH4)H2PO4、HClO4或(NH4)ClO4。在本发明方法的其他实施方案中,另外质子酸的量选择为相对于4-氟苯基肼的量而言为0.1-2,具体地为0.5-1.5且甚至更具体地为1.0摩尔当量。
4-氟苯基肼通常以其与HCl的加成盐的形式市售购得,即作为4-氟苯基肼盐酸盐,使得这成为在本发明方法中使用的第一选择。如果不同的加成盐将通过使4-氟苯基肼盐酸盐例如经由用NaOH水溶液中和而转化成其游离碱且随后加入质子酸来产生加成盐而待使用,则必须小心操作以确保加入足够量的质子酸以补偿会中和部分质子酸的任何残留NaOH的存在。结果,在转化的产物中将存在小于等摩尔量的活化剂。这将以有害方式影响随后的环化反应,将反应速率降低到将不允许经由沉淀/结晶离析5-氟色醇的程度。
在本发明方法的另一实施方案中,所述活化剂作为4-氟苯基肼与例如HCl或H2SO4的第一酸和选自NH4Cl、NH4HSO4或(NH4)2SO4的第二较弱酸的酸加成盐的组合存在于非均相反应介质中。已经发现,与仅使用酸加成盐相比较,在费歇尔-吲哚环化期间在反应介质中第二较弱酸的额外存在进一步加速反应且在反应介质的水相中产生pH < 7、优选pH < 4的pH值。除了在反应介质中作为酸加成盐的一部分存在的酸之外的第二较弱酸的使用提供相对于4-氟苯基肼的摩尔量而言过量的酸。费歇尔-吲哚环化的速度可受相对于4-氟苯基肼而言摩尔过量的酸进一步影响。当使用4-氟苯基肼和第二较弱酸的酸加成盐的组合时,例如4-氟苯基肼盐酸盐和NH4Cl、NH4HSO4或(NH4)2SO4,第二弱酸可例如以相对于4-氟苯基肼而言0.5-2、优选0.5-1.5摩尔当量的量使用。例如,在本发明的一个实施方案中,所述酸加成盐4-氟苯基肼盐酸盐与另外量的NH4Cl组合使用。特别地,第二酸NH4Cl可以相对于4-氟苯基肼的量而言0.5-1.5、优选1摩尔当量的量存在。
在本发明的又一实施方案中,所述活化剂为催化量的非均相布朗斯特酸催化剂。在本发明方法的其他实施方案中,所述活化剂为催化量的非均相布朗斯特酸催化剂,所述非均相布朗斯特酸催化剂选自Amberlyst-15、Amberlite-120、Indion-130、蒙脱石K10和沸石HY,且优选选自蒙脱石K10和Amberlyst-15。
在根据本发明方法的一些实施方案中,在所述反应介质中使用的非质子有机溶剂选自乙酸异丙酯和2-甲基四氢呋喃或这两者的混合物,且优选为2-甲基四氢呋喃。发现,当在反应介质中使用2-甲基四氢呋喃时,反应选择性地进行以产生足够纯度的粗产物,从而实现产物的容易纯化和离析。
在根据本发明的方法的其他实施方案中,水和非质子有机溶剂在反应介质中以约1:3至约3:1、优选约2:3至约3:2的体积比存在。另外,发现,特别是在这些边界内,所述反应在高选择性下产生5-氟色醇。
在根据本发明的方法的其他实施方案中,在步骤(b)中的反应在至少50℃、更优选至少55℃、最优选至少约60℃的温度下进行。已经发现,在约至少60℃的反应温度下,4-氟苯基肼或其盐在反应介质中的溶解度显著增加,其与高温一起使得反应迅速且受控。因此,在本发明的优选实施方案中,反应温度在60℃-80℃、特别是65℃-75℃范围内。在一个优选的实施方案中,所述反应温度为约70℃。
2,3-二氢呋喃加到非均相反应混合物中的速度不是关键性的。应该以使得反应受控且使完成反应所需的总反应时间减至最少的速度加入。通常,添加时间应该选择在约15分钟至约2小时的范围内,且常选择为1小时。
所述反应可在环境压力下或在高压下使用适合在这样的高压下使用的反应容器进行。
此外发现,如果相分离步骤(c)和/或(e)在高温下进行,则这些步骤所需的时间可显著降低。因此,在本发明方法的又一实施方案中,相分离步骤(c)和/或相分离步骤(e)在高温下,优选在约40-60℃的温度下进行。
类似地,发现如果洗涤步骤(d)也在高温下进行,则该步骤的效率可增加。因此,在根据本发明的方法的另一实施方案中,洗涤步骤(d)在高温下,优选在约40-60℃下优选使用NaCl水溶液进行。
在根据本发明的方法的另一实施方案中,所有洗涤和相分离步骤都在约40-60℃的高温下进行。
在根据本发明的方法的又一实施方案中,离析步骤(f)包括以下步骤:
(f.1.) 从在步骤(e)中得到的有机相去除非质子有机溶剂;
(f.2.) 向在步骤(f.1.)中得到的残留物中加入水或至少一种无机盐的水溶液和在20℃下具有低于5g/l的水溶解度的有机溶剂以形成包含水相和有机相的非均相混合物;
(f.3.) 从在步骤(f.2.)中得到的非均相混合物中分离有机相;
(f.4.) 使5-氟色醇从在步骤(f.3.)中得到的有机相中沉淀。
很显然,对于从在步骤(f.2.)中得到的非均相混合物中分离有机相,必须进行所述混合物的水相和有机相的相分离,即不许非均相混合物形成稳定的乳液,不然有机相的离析和水相的去除将不可能。
在步骤(f.1.)中的非质子有机溶剂可例如通过蒸发,特别是在减压和/或高温下蒸发而去除。为了促进5-氟色醇从有机相中沉淀/结晶,部分有机溶剂可经由蒸发去除和/或可将5-氟色醇在所述有机溶剂中的溶液冷却到例如低于约10℃、例如约6℃或更低的温度。
在本发明方法的其他实施方案中,在步骤(f.2.)中加入的有机溶剂为甲苯或苯,优选为甲苯。
此外,在根据本发明的方法的其他实施方案中,在步骤(f.2.)中非均相混合物的形成和/或在步骤(f.3.)中相的分离在高温下、优选在约40-60℃的温度下进行。
在根据本发明的方法的又一实施方案中,所述方法包括以下步骤:
a.) 形成包含4-氟苯基肼、以相对于4-氟苯基肼至少等摩尔的量的至少一种质子有机或无机酸、水和选自四氢呋喃、2-甲基四氢呋喃和乙酸异丙酯的至少一种非质子有机溶剂,其中水和非质子有机溶剂在反应介质中以约1:3至约3:1、优选约2:3至约3:2的体积比存在;
b) 在至少50℃、优选至少60℃的温度下向所述混合物中加入相对于4-氟苯基肼的量而言约0.9-1.1当量的2,3-二氢呋喃在选自四氢呋喃、2-甲基四氢呋喃和乙酸异丙酯的至少一种非质子有机溶剂中的溶液,以与4-氟苯基肼反应,以给出5-氟色醇,其中在步骤a)和b)中提供的非质子溶剂和水以相对于彼此的比率提供,使得在添加步骤b)之前或期间形成包含液体有机相和液体水相的非均相反应混合物;
c) 使液体有机相与液体水相分离;
d) 在约40-约60℃的高温下使液体有机相与至少一种无机盐的水溶液接触以形成包含有机液相和水性液相的非均相混合物;
e) 将所述非均相混合物分离成液体有机相和液体水相并离析液体有机相;
f.1.) 从在步骤(e)中得到的有机相中去除非质子有机溶剂;
f.2.) 向在步骤(f.1.)中得到的残留物中加入水或至少一种无机盐的水溶液和在20℃下具有低于5g/l的水溶解度的有机溶剂以形成包含液体水相和液体有机相的非均相混合物;
f.3.) 从在步骤(f.2.)中得到的非均相混合物中分离有机相;
f.4.) 使5-氟色醇从在步骤(f.3.)中得到的有机相中沉淀。
再次,应该很明显,为了从非均相混合物中分离液体有机相,将混合物相分离成其水相和有机相必须发生,即,不许混合物是这两相的稳定乳液。
优选在步骤(f.2.)中加入的有机溶剂选自甲苯和苯且优选为甲苯。
优选所述活化剂选自有机质子酸和无机质子酸,所述有机质子酸和无机质子酸选自H2SO4、HCl、NH4HSO4、(NH4)2SO4、乙酸、NH4Cl、H3PO4、(NH4)H2PO4、HClO4或(NH4)ClO4或其混合物,特别是选自H2SO4和HCl或其混合物。优选所述质子酸以相对于在反应中使用的4-氟苯基肼的量而言过量存在。
优选在步骤a)中使用的非质子有机溶剂为2-甲基四氢呋喃或乙酸异丙酯或所述两种溶剂的混合物。
优选在步骤a)中形成的混合物为包含液体有机相和液体水相的非均相混合物。
优选,在步骤a)中,相对于4-氟苯基肼而言,将至少1当量的选自HCl、HBr、H2SO4、H3PO4和HClO4的第一酸和另外量的选自NH4Cl、NH4HSO4、(NH4)2SO4、(NH4)H2PO4、(NH4)2HPO4和(NH4)3PO4的第二酸用作活性剂。优选第一酸选自HCl和H2SO4且第二酸选自NH4Cl或(NH4)2SO4。第二酸可例如以相对于4-氟苯基肼的量而言0.5-2、优选0.5-1.5摩尔当量的量使用。在本发明的一些实施方案中,第二酸以相对于4-氟苯基肼而言等摩尔量使用。例如,在本发明的一个实施方案中,所述酸加成盐4-氟苯基肼盐酸盐与另外量的NH4Cl组合使用。特别地,第二酸NH4Cl可以相对于4-氟苯基肼的量而言0.5-1.5、优选1摩尔当量的量存在。优选在根据本发明的方法中的所有洗涤和相分离步骤在高温下,特别在约40℃-60℃的温度下进行。
在本发明的另一方面,提供制备任选以其生理学上可接受的酸加成盐的形式的根据式(I)的化合物的方法,
(I),
其包括以下步骤:
(a) 通过上述发明方法提供5-氟色醇,和
(b) 使所述5-氟色醇在酸催化的Oxa-Pictet-Spengler反应中与在每种情况下任选以酸加成盐的形式的根据式(II)的化合物反应以形成根据式(I)的化合物,
(II)
其中根据式(II)的化合物的基团R2和R3共同表示=O,或者与连接它们的碳原子一起形成选自和的环状部分,
其中R4和R5在每种情况下彼此独立地代表选自H和CH3的0、1、2、3或4个取代基。
附图
图1a-1c示出在以下实施例3中描述的样品1-3的1H-NMR光谱。
实施例
下面的实施例进一步说明本发明,但不应该视为限制其范围。
缩写:
DMAC:二甲基乙酰胺
THF:四氢呋喃
RT:反应温度
n.d.:未测定
Ta:添加2,3-二氢呋喃(dihydrotetrafuran)的温度
tad:2,3-二氢呋喃的添加时间
trct.:在加入2,3-二氢呋喃之后的反应时间
DMF:二甲基甲酰胺
MTBE:甲基叔丁基醚
2-MeTHF:2-甲基四氢呋喃
比较实施例1:
以下反应程序基于K. R. Campos等人在Org.Lett., 第6卷,第1期,2004, 79-82(参见上面)中描述的程序发展。
将30g 4-氟苯基肼盐酸盐(185mmol)悬浮在150ml二甲基乙酰胺(DMAC)和300ml H2SO4 (4%水溶液)中。在搅拌的同时将悬浮液加热到75-80℃,由此使肼溶解。经30-60分钟的时间将12.9g 2,3-二氢呋喃(185mmol)在150ml DMAC中的溶液逐滴加到反应混合物中且将反应混合物在80-85℃下搅拌2小时。随后将反应混合物冷却到周围温度且加入600ml乙酸乙酯。在5-10分钟的大力搅拌之后,用分液漏斗分离各相。将有机相离析并分别使用250ml氯化钠水溶液(5%)洗涤4次。有机溶剂经由在减压下且在高温(45-60℃,160-9mbar)下蒸发而从洗涤的有机相中去除。在20-25℃下将残留物溶解在为约其4倍重量的甲苯中且将溶液在周围温度下搅拌1小时。如果尚未结晶,则将晶种加到溶液中。将溶液冷却到0-3℃并保持在该温度下2-3小时,同时继续搅拌所述溶液。沉淀的晶体经由过滤离析并用10-20ml冷甲苯(0-3℃)洗涤。离析的固体在45℃下在减压(50-60mbar)下干燥且产物以约25%产率得到作为具有60±1℃的熔点的蜡样几乎白色的晶体。
尽管该程序产生以足够纯度的5-氟色醇以允许产物经由结晶纯化并离析,但是产物仅以相对较低的产率得到。另外,必须用水洗涤有机相四次以去除DMAC。在没有该重复的洗涤程序的情况下,不可能使产物结晶。
比较实施例2:
基于如上文比较实施例1中描述的程序,尝试基于Y. Lü等人在Journal of Chemical Engineering of Chinese Universities, 第1期,第24卷,2010,第127-131页(参见上文)中的公开内容来调试该程序,即,通过使用水/乙醇/甲苯的非均相溶剂混合物代替水和DMAC的均相溶剂混合物。
将5g 4-氟苯基肼盐酸盐(30.75mmol)连同3.5g氯化铵(65.43mmol)与12.5ml甲苯、12.5ml乙醇和46.5ml水混合。将混合物加热到70℃且在连续搅拌下在该温度下经30分钟的时间逐滴加入由2.16g 2,3-二氢呋喃(30.75mmol)在12ml甲苯和12ml乙醇中的第二混合物。在搅拌的同时将反应混合物保持在70℃下过夜且在17小时之后冷却到55℃。加入5% NaCl在水(33.75ml)中的溶液且在大力搅拌的同时将混合物加热到55℃。让有机相和水相在50℃下分离。将有机相离析且将溶剂在减压(< 10mbar)下且在高温(65℃)下去除。取出粗产物的样品且5-氟色醇和若干副产物(包括“副产物A”)(参见下文)的含量经由HPLC-分析测定。尝试使5-氟色醇从粗产物在甲苯中的溶液中结晶,但未成功。
使用以5:1和1:5的体积比的甲苯和乙醇的混合物作为费歇尔-吲哚环化反应的有机溶剂类似地重复实验。这些实验中没有一个可能使5-氟色醇从粗产物在甲苯中的溶液中结晶。尽管不想受理论限制,但还是据信这归因于在反应中形成的大量副产物,在不使用色谱法的情况下这些副产物难以从产物中分离。一种这样的副产物例如为下列化合物,其在下文中将称为“副产物A”
“副产物A”
表1列出如在粗产物中测定的5-氟色醇和“副产物A”的相应纯度/含量。
表1:
比较实施例3:
类似地重复根据比较实施例的程序,但DMAC被不同的有机溶剂替换。对通用程序的另外改变连同分析结果在下文表2中的标题“注解”中列出:
表2:
实施例1:
出于一般实验室安全考虑,以下反应程序在N2气氛下进行。使500g 4-氟苯基肼盐酸盐(3.08mol)悬浮在2.5升 2-甲基四氢呋喃和氯化铵的水溶液[350g氯化铵(6.54mol)在4650ml水中]的混合物中。将悬浮液加热到70℃,由此使4-氟苯基肼完全溶解于由水溶液和有机溶剂形成的非均相反应介质中。经1小时的时间,在搅拌的同时将溶解于2.4升2-甲基四氢呋喃中的215.5g 2,3-二氢呋喃(3.08mol)加到非均相反应介质中,且在完成添加之后,将反应混合物在70℃下再搅拌16-24小时。随后将混合物冷却到50℃,且在不搅拌下15分钟之后使得相分离,在维持温度在50℃下的同时离析有机相和水相。向有机相中加入由溶解于3375ml水中的180g氯化钠组成的洗涤液且将混合物在50℃下大力搅拌10分钟。停止搅拌且使得有机相在15分钟期间从水相中分离且随后离析,这全部在维持温度在50℃下的同时进行。有机溶剂经由在55℃在减压(< 10mbar)下从有机相中去除。将所得残留物在50℃的温度下用甲苯(4050ml)和水(1250ml)的混合物萃取,在维持在50℃下的同时离析有机相且包含在所述相中的1400ml甲苯经由在不超过65℃的温度下且在减压(150mbar)下蒸发而去除。在继续搅拌的同时,将剩余有机溶液谨慎地冷却到5℃以使5-氟色醇以结晶形式沉淀。在10-15℃的温度下,将晶种加到溶液中。在搅拌的同时将溶液再在5℃下维持16-24小时。随后,沉淀物经由过滤离析且用冷却的甲苯(+3℃,总计500ml)洗涤几次。所得固体在干燥箱中在35℃下干燥过夜,以约65%产率提供为黄色至白色固体的5-氟色醇(熔点:62℃;纯度(HPLC>98%)。
还对以下溶剂进行了经由使结晶产物从根据上述程序得到的粗产物的相应溶液中沉淀进行的纯化/离析试验:庚烷、环己烷、乙醚、二异丙醚、甲基叔丁基醚、丙酮、甲基乙基酮、乙酸乙酯、甲醇、乙醇、异丙醇和苯。另外,对下列混合物进行了试验:乙酸乙酯:庚烷(1:1);异丙醇:庚烷(1:1);和丙酮:庚烷(1:1)。
除了苯以外,发现这些溶剂或溶剂混合物无一不适合经由如上文关于甲苯所述的沉淀/结晶进行的纯化步骤。
实施例2:
出于一般实验室安全考虑,以下反应在N2气氛下进行。使12g 4-氟苯基肼盐酸盐(73.81mol)悬浮在30ml 乙酸异丙酯和30ml 2-甲基四氢呋喃与氯化铵的水溶液[8.4g氯化铵(157mol)在111.6ml水中]的混合物中。将悬浮液加热到70℃。经25分钟的时间,在搅拌的同时将溶解于28.8ml 2-甲基四氢呋喃中的5.17g 2,3-二氢呋喃(73.8mmol)加到非均相反应介质中。在添加完成之后,将反应混合物在70℃下再搅拌16小时。随后将混合物冷却到55-50℃并停止搅拌。在停止搅拌之后15分钟,在维持温度在50℃下的同时,分离有机相和水相。向有机相中加入由溶解于81ml水中的4.32g氯化钠组成的洗涤液且将混合物在50℃下大力搅拌10分钟。停止搅拌且使有机相在15分钟期间与水相分离且随后离析,这全部在维持温度在50℃下的同时进行。有机溶剂在55℃下在减压(< 50mbar)下从有机相中去除。将所得残留物在50℃的温度下用甲苯(40.5ml)和水(12.5ml)的混合物萃取,且在维持在50℃下的同时离析有机相。包含在所述相中的10ml甲苯随后经由在高温下且在减压下蒸发而去除。在继续搅拌的同时,将剩余有机溶液冷却到约5℃以使5-氟色醇以结晶形式沉淀。为了帮助沉淀,将晶种加到溶液中。在搅拌的同时将溶液维持在约5℃下过夜。随后,沉淀物经由过滤离析且用冷却的甲苯(+3℃,总共12ml)洗涤几次。所得固体在干燥箱中在45℃下干燥过夜并以约42.3%产率(5.6g)和98.56%的纯度(HPLC)提供结晶5-氟色醇。
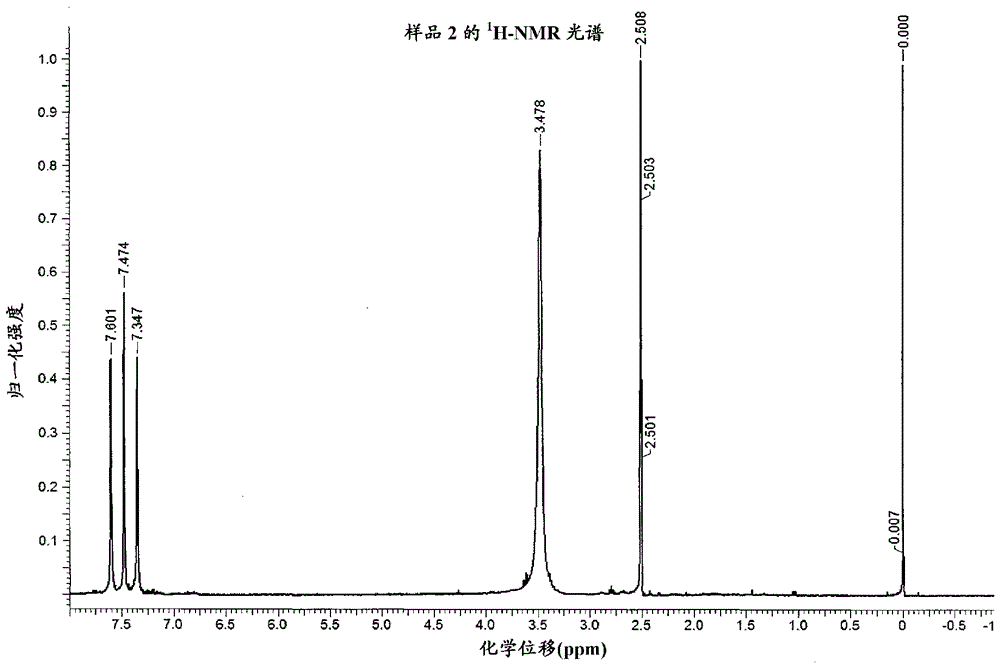
实施例3
出于一般实验室安全考虑,以下反应在N2气氛下进行。使5g 4-氟苯基肼盐酸盐(30.75mol)悬浮在25ml 2-甲基四氢呋喃和氯化铵水溶液[3.5g氯化铵(65.4mol)在46.5ml水中]的混合物中。将悬浮液加热到70℃。经12分钟的时间,在搅拌的同时将溶解于24ml 2-甲基四氢呋喃中的2.15g 2,3-二氢呋喃(30.75mmol)加到非均相反应介质中。在添加完成之后,将反应混合物在70℃下再搅拌16小时。随后将混合物冷却到55℃并停止搅拌。在停止搅拌之后15分钟,在维持温度在55℃下的同时,分离有机相和水相。取出水相的样品进行NMR分析(样品1)。向有机相中加入由溶解于33.7ml水中的1.8g氯化钠组成的洗涤液且将混合物在55℃下大力搅拌10分钟。停止搅拌且使有机相在15分钟期间与水相分离。再次,取出水相的样品进行NMR分析(样品2)。随后离析有机相,这全部在维持温度在55℃下的同时进行。有机溶剂在55℃下在减压(< 50mbar)下从有机相中去除。将所得残留物在50℃的温度下用甲苯(40.5ml)和水(12.5ml)的混合物萃取。再次,取出水相的样品进行NMR分析(样品3)且随后在维持在50℃的同时离析有机相。将溶剂从有机相中去除,产生准备随后结晶的粗产物的残留物。取出所述残留物的样品以便分析其HPLC-纯度。粗产物具有94.4%的HPLC-纯度,证实所述方法以代表性方式进行。
在所述合成期间取得的不同样品的NMR-光谱示于图1中。意外地的是,从这些样品得到的1H-NMR光谱证实在样品1和2中基本没有见到产物。这从缺乏5-氟色醇特征性的1H-峰而显而易见。在样品1和2的光谱中,在对于结合到芳环的质子特有的区域中见到的仅有信号为可指派给未反应的4-氟苯基肼的信号。样品2的NMR-光谱还示出用氯化钠水溶液的洗涤步骤不仅从有机相中选择性地去除4-氟苯基肼,而且去除其他有机副产物。可指派给产物的1H-NMR峰在从样品3得到的光谱中首次见到,样品3从所述方法中的一个阶段取得,其中2-甲基四氢呋喃已被去除并用甲苯替换。因此,使用溶剂2-甲基四氢呋喃使得能够从反应混合物中有效离析并纯化5-氟色醇。
实施例4
a) 以小规模类似地进行如实施例1中所述的反应,不同之处在于使用1.15当量的2,3-四氢呋喃(2.48g;35.37mmol)代替等摩尔量。这使得产物的总产率显著降到30.8%,产物作为橙色/褐色粘稠固体离析,且在40℃下在减压(75-150mbar)下干燥过夜之后熔融。认为这表明在产物中存在显著量的副产物/杂质。这通过测定物质的HPLC-纯度证实,发现纯度仅为93.46%。
b) 以小规模类似地进行如在实施例1中所述的反应,不同之处在于使用0.95当量的2,3-四氢呋喃(4.38g;62.51mmol)代替等摩尔量。该变化以令人满意的产率(55%)和令人满意的纯度(98.7%)生成产物。
实施例5
以较小规模类似地进行如在实施例1中所述的反应(30.72mmol 4-氟苯基肼),不同之处在于,在第一实验中,在加入2,3-二氢呋喃之后在非均相反应介质中存在的水与2-甲基四氢呋喃的比率为约1:3 (24.5ml 2-MeTHF/71.5ml H2O;实施例5.1),在第二实验中,该比率为约1:5 (16.5ml 2-MeTHF/81.5ml H2O;实施例5.2),且在第三实验中,该率为约5:1 (83ml 2-MeTHF/15ml H2O;实施例5.3),[代替如在实施例1中使用的以约1:1的比率的2-MeTHF和水(4.9l 2-MeTHF:4.65l H2O)]。
在第一实验(实施例5.1.)中,产物的总产率显著降低(25.4%)且再次,经由结晶得到的固体在40℃下在减压下干燥过夜时熔融。发现产物的HPLC-纯度为93.49%。
在第二实验(实施例5.2.)中,在环化反应之后的相分离不完全且所述实验因此在该阶段终止。
在第三实验(实施例5.3.)中,水的量不足以将所有氯化铵溶解于反应介质中,这使得在环化反应之后的相分离困难。用甲苯萃取是不可能的,因为在反应的该阶段得到的粗产物(根据HPLC-纯度分析含有约3%的副产物A)不溶于甲苯中。所述实验因此在该阶段终止。
实施例6
出于一般实验室安全考虑,以下反应在N2气氛下进行。使11.27g 4-氟苯基肼盐酸盐(69.32mmol)悬浮在56ml四氢呋喃、113ml硫酸水溶液(4%)的混合物中并将混合物加热到63℃,由此形成橙色溶液。经66分钟的时间且在63-65℃的温度下,在搅拌的同时,将溶解于56ml四氢呋喃中的4.85g 2,3-二氢呋喃(69.32mmol)加到所述溶液中。在添加过程期间,溶液首先变得不透明,随后形成包含液体有机相和液体水相的非均质两相体系。将非均相反应介质在65℃下另外搅拌2小时,随后冷却到10℃。冷却的非均相反应介质用225ml乙酸异丙酯萃取且将所得有机相与水相分离并用90ml去离子水洗涤两次。有机溶剂经由在减压(< 1mbar)下在45℃下蒸发而去除。将这样得到的残留物称重(10.2g)且加入其重量的2.6倍的甲苯。将所得溶液在5-8℃下搅拌约16小时,其中在搅拌2小时之后将晶种加到溶液中。沉淀的固体经由过滤离析并用8-10ml的冷却的甲苯(5-8℃)洗涤两次。将所得固体在45℃下在减压(75-100mbar)下干燥22小时,产生具有99.67%的HPLC-纯度的3.0g产物(25%产率)。
实施例7
出于一般实验室安全考虑,以下反应在N2气氛下进行。将20g 4-氟苯基肼盐酸盐(123mmol)悬浮在100ml四氢呋喃与14g NH4Cl在186ml水中的水溶液的混合物中。将混合物加热到65℃。在该温度下,盐酸肼并未全部溶解且混合物形成非均相体系,在混合物中的部分THF形成液体有机相。经50分钟的时间且在65℃的温度下,在搅拌的同时,将溶解于96ml四氢呋喃中的8.6g 2,3-二氢呋喃(123mmol)加到非均相反应介质中。将非均相反应介质在70℃下另外搅拌16小时,随后冷却到50℃。在该温度下,使有机相与水相分离。将氯化钠的水溶液(7.2g NaCl在135ml水中)加到有机相中并将混合物在50℃下大力搅拌15分钟。搅拌停止且在10分钟之后,使有机相与有机相分离。有机溶剂经由在45℃下在减压(< 10bar)下蒸发而从有机相中去除。向这样得到的残留物中加入160ml甲苯和50ml水且将所得混合物大力搅拌10分钟。在搅拌中止之后,混合物形成三相体系。将固相离析且让剩余的两个液相再分离12分钟。将有机相分离且56ml甲苯经由蒸发(55℃/90mbar)去除,以由56ml甲苯替换。再次,经由蒸发(55℃/90mbar)去除56ml甲苯。随后将所得溶液冷却到5℃,加入晶种且随后在5-8℃下将溶液搅拌约16小时。沉淀的固体经由过滤离析并用很少的甲苯洗涤。将固体在30℃和减压(75-100bar)干燥两天,产生7.8g (35.4%)的具有98.76%的HPLC-纯度的黄色产物。
实施例8
为了测试不同非质子酸的用途,使用如下文在表3中所述的试剂进行如在实施例2中所述的方法。试剂和溶剂的量基于11.27g 4-氟苯基肼盐酸盐(69.3mmol)的量计算。与实施例2中所述的程序的不相符之处在下表中指出。
表3:
实施例9
在该实施例中,证明非4-氟苯基肼盐酸盐的加成盐可用于根据本发明的方法中:
220g 4-氟苯基肼盐酸盐与1100ml 2-MeTHF和440ml H2O一起提供。为了释放肼,加入32%NaOH水溶液,同时冷却。在中和反应之后,将含有4-氟苯基肼的有机相与水相分离。随后,将94.84g 2,3-二氢呋喃加到分离的有机相中且将所得混合物缓慢(5.5小时)加到已经加热到70℃的440g NH4HSO4在2046ml水和1056ml 2-MeTHF中的溶液中。在搅拌下将反应保持在该温度下历时另外16.5小时。
将反应混合物冷却到53℃并让各相分离。随后去除水相并加入氯化钠溶液(79.2g NaCl在1485ml水中)。将所得混合物在50℃的温度下大力搅拌,随后再次让各相分离并去除水相。溶剂经由在55℃和减压(<10mbar)下蒸发而从剩余有机相中去除。在55℃下加入1782ml甲苯和550ml水以溶解残留物。在相分离之后,经由在65℃和减压(150mbar)下蒸发而去除650ml甲苯。将剩余溶液冷却到低于60℃,加入晶种并使5-氟色醇从溶液中沉淀出。产物经由过滤分离并用少量冷甲苯洗涤几次。剩余结晶固体在干燥箱中在45℃下且在减压下干燥过夜。5-氟色醇以58%的产率得到。产物具有99.5%的HPLC-纯度。
实施例10
在该实施例中,将市售蒙脱石K10作为非均相布朗斯特酸催化剂使用。
将10g (61.5mmol)的4-氟苯基肼盐酸盐悬浮在50ml 2-甲基四氢呋喃中。将蒙脱石K10 (7g)在93ml水中的悬浮液加到该悬浮液中。在大力搅拌下将混合物加热到72℃。接着,经50分钟的时间,将4.3g (61.5mmol)的2,3-二氢呋喃在50ml 2-甲基四氢呋喃中的溶液加到该混合物中。在继续搅拌的同时将反应混合物维持在72℃下过夜(约16小时)。随后,混合物的不溶性组分经由经玻璃料(孔径3)过滤而去除。使有机相与水相彼此分离且分离的有机相通过加入3.6g氯化钠在68ml水中的溶液并在50℃下搅拌混合物10分钟来洗涤。再次,将有机相和水相彼此分离且溶剂在60℃和< 10mbar的减少的压力下去除。向残留物中加入80ml甲苯和25ml水且将混合物在50℃下搅拌10分钟。使水相和有机相再次彼此分离且有机溶剂在减压(< 10mbar)下且在60℃下从有机相中去除。将残留物溶解于28ml甲苯中并冷却到5-8℃过夜。为了便于结晶,加入5-氟色醇的晶种。将沉淀物滤出并在40℃下且在75-100bar的减少的压力下干燥2天。得到6.2g (56%)/99.5%纯度(HPLC)的5-氟色醇。
- 还没有人留言评论。精彩留言会获得点赞!