无甲醛的酚醛树脂、下游产物及其合成与用途的制作方法
本发明涉及包含由例如葡萄糖和酚类化合物原位产生的5-羟甲基糠醛(HMF)的反应产物的树脂组合物、此类树脂的合成以及下游用途和产物。
背景
酚醛(PF)树脂是在涂料、粘合剂、铸塑、工程材料、日用品等中具有广泛应用的第一个商业化合成树脂。近年来甲醛致癌效应的发现(Zhan等人,2009)以及减少挥发性有机化合物(VOC)更严格的环境法规已经引发了对新型PF制造的需求,以减少生产和使用期间的甲醛排放,例如,或者减少最终生产阶段的废气。
因此制造商正寻求比常规聚合物(例如PF)更加绿色和更环境友好的替代物(Netravali和Chabba,2003)。生物质正日益成为燃料和化学品的重要原料(Zakzeski等人,2010)。木质素与纤维素和半纤维素一起构成了木质纤维素生物质的三种主要组分。木质素是三种单体的聚合物:愈创木基(G)、紫丁香基(syringyl)和对羟苯基丙烷(p-H)-型(Tejado等人,2007)。在其结构中,木质素的酚基特别令人关注,并且在酚醛树脂中作为原油基苯酚的替代已经引起了研究人员的兴趣(Effendi、Gerhauser和Bridgwater,2008)。
例如,已经用木质素和腰果酚替代苯酚,但是甲醛继续引起环境问题并且美国规定了其暴露水平(Kowatsch,2010;Hahnenstein等人,1994)。尽管经济性相对较差,研究者报道一种方法用更昂贵的树脂代替PF树脂(Kurple,1989)。
已经在合成木质素改性的酚醛(LPF)树脂中对使用木质素作为苯酚的替代作出了大量努力,但是将木质素直接并入PF合成已经是一个挑战,因为粗木质素具有比苯酚更少与醛反应的反应位点(Wang等人,2009)。通常为获得更多反应性官能团而实施木质素改性,其包括酚化(Alonso等人,2005)、羟甲基化(Alonso等人,2004)、脱甲基化(Ferhan等人,2013)和热解聚/液化(Cheng等人,2012;Cheng等人,2013)。发现最终的树脂性能非常依赖于木质素的化学和物理性质。木质素的直接酚化由于其简单易行被广泛应用于酚醛树脂(Alonso等人,2005)。
葡萄糖是纤维素、半纤维素和淀粉的主要结构单元,并且在自然界中是最丰富的可再生固定碳源。在化石资源接近预计消耗的情况下,葡萄糖在某些化学转变后可以是燃料(生物乙醇和生物丁醇,二甲基呋喃等)和其它化学品的未来碳源。已经在水、有机溶剂、氯盐和离子液体中表现了葡萄糖至HMF(平台化合物)的转变(Yan等人,2009;Zhao等人,2007;Li等人,2009;Binder和Raines,2009)。
通常将六亚甲基四胺(HMTA)(氨和甲醛的缩合产物)用于线型酚醛树脂的固化。由于HMTA在固化和应用中分解形成氨和甲醛,其使用也是受限的(Nielsen等人,1979;Richmond等人,1948)。亚甲基桥在线型酚醛合成期间形成于酚的苯环之间并且在线型酚醛树脂的酚环上保留至少一个邻位或者对位,线型酚醛的一般结构为(Knop和Pilato,1985):
通过使用较高的温度,进一步形成其它亚甲基桥,但是该方法的应用受限于HMTA含有的有害空气污染物(Lytle、Bertsch和McKinley,1998)。因此,开发绿色固化剂作为HMTA的替代物用于线型酚醛树脂固化也已经受到了日益增长的研究关注。
Simitzis等人(1996)产生了用HMTA与以下组分之一的混合物固化线型酚醛树脂:来自压榨的橄榄和分离油的残余物、硫酸盐木质素(KL)、羟甲基化硫酸盐木质素(KLH)和纤维素(CEL)。发现固化反应的活化能(Ea)和反应速率常数(k)为HMTA<HMTA/KLH<生物质<KL<CEL。其表明尽管Ea和k随着不同的固化剂而变化,但是反应级数(n)实际上相同(n=1)。然而,该研究未提出这些新型固化剂交联的机制(Simitzis等人,1996)。
已有报道将具有以下结构的2,6-二(羟甲基)-对甲酚(a)、3,3',5,5'-四(羟甲基)-4,4'-异亚丙基双酚(b)和2,6-双(2-羟基-3-羟甲基-5-甲基苄基)-4-甲酚(c):
用作线型酚醛树脂的固化剂。获得了与用HMTA固化的那些相比具有较高的物理机械特性的硬聚合物(Sergeev等人,1995)。木质素具有与以上固化剂结构类似的甲氧基化苯基丙烷结构(Zakzeski等人,2010),因此当用作固化线型酚醛PF树脂的HMTA替代物时,应遵循与以上固化剂类似的交联机制(Grenier-Loustalot等人,1996)。
通过用各种有机溶剂处理木材或者甘蔗渣获得的有机溶剂型木质素(OL)通常硫含量较低并且具有高纯度(Sarkanen等人,1981)。已有将OL作为纤维素乙醇过程中的副产物进行商业生产。硫酸盐木质素(KL)是木质纤维素材料进行硫酸盐化学制浆的副产物,其中采用高pH和大量的氢氧化钠水溶液和硫化钠在423-453K的温度下进行约2h以溶解木质素。工业上产生大量KL(每年约7000万吨),但是目前主要在纸浆厂/造纸厂的回收锅炉中作为低价值的燃料用于产生热能/电能。
使用PF树脂作为聚合物基体的纤维增强复合材料(FRC)是线型酚醛PF树脂的典型应用。由于其高强度、高刚度和良好的耐腐蚀性,使用PF树脂的FRC已经在风车叶片、船、航空航天、汽车、民用基础设施、运动以及娱乐产品中得到普及。用成本竞争力强的绿色组分产生的生物复合材料更有前景。在过去数十年,生物复合材料(如具有生物基聚合物基体材料的玻璃纤维增强复合材料)在汽车和装饰市场的用途中显著增长(Shibata等人,2008;Suharty等人,2008)。
最常用于PF线型酚醛的固化剂仍然是六亚甲基四胺(HMTA)。线型酚醛与HMTA或者多聚甲醛之间的固化条件、反应机理和动力学参数已经吸引了许多研究关注。例如,将快速定量13C NMR光谱应用于表征聚合度、数均分子量和未反应的邻酚环和对酚环的数量(Ottenbourgs等人,1995)。通过使用固态13C NMR讨论线型酚醛树脂和多聚甲醛的固化行为(Ottenbourgs等人,1998;Bryson等人,1983)。该技术表现出可定量确定甲醛/苯酚比率和固化转化程度。然而,发现多聚甲醛无法完全固化线型酚醛。Zhang等人也使用13C NMR和15N NMR技术研究了固化时线型酚醛树脂和HMTA的化学过程(Zhang等人,1997;Zhang等人,1998;Lim等人,1999)。尤其注意在固化过程期间形成作为反应中间体的苄胺和苯并噁嗪。形成亚甲基键合以在较低温度下以对位-对位键合连接线型酚醛分子,然而它们比邻位连接的中间体热稳定性更差。
固化参数和条件对于酚醛材料的性能至关重要。最常见的分析之一是通过差示扫描量热法(DSC)进行的。已经报道了约144kJ/mol的活化能和反应常数(De Medeiro等人,2003)。通过自加速效应和三阶唯象方程描述由流变力学光谱记录的固化反应(Markovic等人,2001)。王等人还评估了分子量和分子量分布对HMTA固化的线型酚醛的固化动力学以及热、流变学和机械性能的影响(Wan等人,2011)。他们报道了具有较低分子量的线型酚醛树脂表现出较高的反应热和反应性、在加热时更快的分解速率、在850℃下较低的炭残余物,并且复合材料呈现出较高的弯曲强度。
概述
本文发现有可能使己糖产生的HMF与酚类化合物聚合以产生可固化树脂。原位产生HMF并且发现以一锅法与酚类化合物反应。
优选由葡萄糖(特别是D-葡萄糖)原位产生HMF。可使用其它己糖(通常比葡萄糖更昂贵):阿洛糖、阿卓糖、甘露糖、古洛糖、艾杜糖、半乳糖、果糖和塔罗糖,包括任何前述的混合物,包括与葡萄糖的混合物,以及包括任何前述或者所有前述的D-立体异构体。
HMF的对应反应物为酚类化合物。在本发明的上下文中,“酚类化合物”为包含羟基取代的苯基环的化合物,其中所述环的邻位碳和对位碳(相对于含有羟基的碳)中的至少一个是未取代的(即,含有氢)。因此此类碳原子可用于反应以形成如本文所述的酚醛树脂。此类酚类化合物包括苯酚自身(C6H5OH)、生物酚(例如,酚化的解聚木质素)。因此产物树脂可以是例如苯酚-HMF(PHMF)树脂或者生物酚-HMF(BPHMF)树脂。
产生的树脂是可固化的,因此是可以交联的,以产生下游产品(例如粘合剂)。发现可能使用木质素产生的树脂固化,例如不使用固化剂HMTA,尽管HMTA也表现出对本发明的树脂是有用的。树脂可用作生产复合材料。
在酚类化合物(树脂由其制备)为生物酚(生物酚-HMF,BPHMF)的实施方案中,Mw优选为约2500g/mol至约4000g/mol。“生物酚”意指来自如在木质素、树皮、木材和作物残余物等所发现的木质纤维素生物质的酚类化合物或者腰果酚。根据使用差示折光计如由凝胶渗透色谱测量的线性聚苯乙烯标准品的Mw来表达“Mw”。
将本文所示例的PHMF树脂在铬氯化物催化剂存在下使用生物基交联剂OL和KL进行热交联。PHMF树脂和木质素的热固化表明在约120℃下开始交联并且通过放热的热量释放进行表征。通过DSC扫描显示玻璃态聚合物(特征在于133℃的Tg)。热重量测试表明失重峰在Ti~230℃开始并且在Tmax~400℃最大。将用OL/KL固化的PHMF树脂用于产生包含高百分比的可再生原料的生物复合材料。因此木质素(OL/KL)固化的PHMF树脂作为HMTA固化的PF树脂系统的替代在用于具有满意性能的生物基热固性材料方面表现出巨大的潜力。
在一个方面,本发明因此是制备可交联的酚醛树脂的方法。所述方法包括步骤:
(i)在酚类化合物以及促进酚类化合物的芳香族环的碳原子与HMF的甲酰基的碳之间形成共价键的催化剂存在下,将己糖转化为5-羟甲基糠醛(HMF),以形成树脂。
在一个方面,如在以下所述的实施例中,催化剂可以是Friedel-Crafts烷基化催化剂。催化剂可包括YCl2、YCl3和季铵盐的混合物,其中Y为Cr或者Cu或者Zn。季铵盐可以是例如四乙基氯化铵(TEAC)或者四甲基氯化铵。
在一个方面,步骤(i)的催化剂包含路易斯酸。
可选择催化剂以原位催化己糖转化为HMF以及形成树脂。
优选的己糖为葡萄糖(包括D-葡萄糖)。
步骤(i)的酚类化合物可以是未取代的苯酚。酚类化合物可以是取代的苯酚(例如腰果酚)。酚类化合物可以是从例如木质纤维素生物质(例如热解油、液化生物质、木质素、解聚木质素或者液化木质素中的一种或者多种)获得的一种或者多种生物酚。
步骤(i)优选包括将包含己糖、苯酚和催化剂的混合物加热至足以形成液体混合物的温度。所述温度通常为至少80℃、或者至少90℃、或者至少100℃、或者至少110℃、或者至少120℃,因此较高的温度还促进HMF形成。
在实施方案中,步骤(i)的己糖:酚类化合物的酚环的摩尔比为0.5:1至3:1。
在实施方案中,己糖:酚环的摩尔比为至少1,并且步骤(i)的混合物包括水。在包括水的情况下,其通常以不多于混合物重量的20%的量存在,但是其可不多于混合物重量的15%、或者不多于混合物重量的10%、或者不多于混合物重量的9%、或者不多于混合物重量的8%、或者不多于混合物重量的7%、或者不多于混合物重量的6%。在实施方案中,水以以下的量存在:混合物重量的至少2%、或者混合物重量的至少2%、或者混合物重量的至少3%、或者混合物重量的至少4%、或者混合物重量的至少5%、或者混合物重量的至少6%。
在实施方案中,将步骤(i)进行以下时间段:1小时至12小时、或者1小时至10小时、或者1小时至8小时、或者1小时至6小时、或者2小时至10小时、或者3小时至10小时、或者4小时至10小时。
本发明的实施方案还可包括从步骤(i)形成的树脂中去除未反应的糖的步骤(ii),通常,在步骤(i)之后。步骤(ii)可包括水洗。
本发明还包括产生包含多个呋喃基环和苯基环的反应产物的方法,邻近的呋喃基环和苯基环通过-CH2-或者-CHOH-基团彼此共价连接。所述方法包括:
(a)在苯酚以及促进苯酚与HMF之间形成所述-CH2-基团和所述-CHOH-基团的催化剂存在下,将己糖转化为5-羟甲基糠醛(HMF)。
以上结合基于步骤(i)的方法描述了该方法的其它更详细的方面。
在其它实施方案中,本发明包括制备树脂的可固化的反应混合物,所述混合物包含:(a)己糖;(b)苯酚或者生物酚;以及(c)催化剂,三种组分(a)、(b)和(c)如以上和本文其它地方所述。
本发明包括根据任何前述方法实施方案制备的酚醛树脂。
本发明包括为酚类化合物(例如,苯酚或者生物酚)与5-羟甲基糠醛的反应产物的聚合物树脂。
在实施方案中,此类树脂为具有与酚部分的环碳原子(即并入树脂的苯酚分子的芳香族环)共价连接的HMF的甲酰基的碳原子的反应产物。在实施方案中,反应产物包括与酚部分的环状芳香族碳原子(特别是相对于酚基的羟基位于邻位和对位的碳原子)共价连接的HMF的羟甲基的碳原子。
通常,树脂具有约500g/mol至约5000g/mol的重均分子量(Mw)。在酚为未取代的苯酚(苯酚-HMF树脂)的实施方案中,优选的树脂具有以下范围的Mw:约500g/mol至约4000g/mol、约500g/mol至约3000g/mol、约500g/mol至约2000g/mol、约500g/mol至约1500g/mol、约500g/mol至约1000g/mol、约600g/mol至约3000g/mol、约600g/mol至约2500g/mol、约600g/mol至约2000g/mol、约600g/mol至约1500g/mol或者约600g/mol至约1500g/mol,或者约600g/mol、约700g/mol、约800g/mol、约900g/mol、约1000g/mol、约1100g/mol、约1200g/mol、约1300g/mol、约1400g/mol或者约1500g/mol。
在实施方案中,本发明的树脂具有约2至约20的多分散指数(Mw/Mn),更优选为约2至约15、约2至约10、约2至约5,或者约2、约3、约4或者约5。
在实施方案中,酚的芳香族环和呋喃基环与糖的比率为约0.5至约3,更优选为约0.5至约1.5。
本发明的另一方面为制备聚合物树脂组合物的方法,所述方法包括步骤:
(A)混合如本文所述的树脂和交联剂。
在实施方案中,交联剂为具有至少两个烷羟基的有机分子。羟基可以是伯羟基。交联剂的实例为木质素。
交联剂相对于树脂的重量可为约10%至约30%,更优选为约15%至约25%。
交联剂可以是HMTA。
因此本发明还包括含有本发明的树脂和交联剂的树脂组合物。此外,交联剂可以是具有至少两个烷羟基的有机交联剂,以允许所述试剂参加与树脂的酚部分的芳香族环碳的交联反应。
本发明还包括热固化树脂组合物的方法,其涉及通过将树脂组合物加热至组合物变得固化使以上所述的树脂组合物热固化。加热树脂组合物可包括将组合物加热至约100℃至约200℃的温度,更优选为约120℃至约180℃。可将树脂组合物加热约5分钟至约120分钟的时间,更优选为约30分钟至约60分钟。
本发明还包括从以上所述的固化方法的实施方案获得的固化组合物。
还可将本发明的固化组合物描述成包括交联聚合物,其中所述聚合物包含HMF和苯酚或者生物酚的单体单元,并且聚合物的分子通过与多羟基化合物的反应彼此交联。多羟基化合物可以是木质素。
固化的组合物可以是例如粘合剂。
在整个说明书中讨论了本发明的其它实施方案。关于本发明的一个方面所讨论的任何实施方案也适用于本发明的其它方面,反之亦然。将详述实施例中的实施方案理解为适用于本发明的所有方面的本发明的实施方案。
当在权利要求和/或说明书中与术语“包含/包括”结合使用时,词语“一个/一种(a)”或者“一个/一种(an)”的使用可意指“一个/一种(one)”,但是其还与“一个或者多个/一种或者多种”、“至少一个/至少一种”和“一个或者多于一个/一种或者多于一种”的含义一致。
将术语“约”用于表示数值包括用于确定数值的装置或者方法的误差的标准偏差。
将权利要求中的术语“或者”的使用用于意指“和/或”,除非明确指明仅指替代项或者替代项是相互排斥的。
如本申请所使用,词语“包含”、“具有”、“包括”或者“含有”以及任何它们的形式都是包括式的或者开放式的,并且不排除其它的、未提到的要素或者方法步骤。
根据以下详细描述,本发明的其它目的、特征和优点将变得显而易见。然而,应当理解,仅以示例的方式给出了详细描述和说明本发明的具体实施方案的具体实施例,因为根据这种详细说明,本发明的主旨和范围内的各种变化和修改对于本领域技术人员将变得显而易见。附图简述
现将通过实施例、参考附图描述实施方案,其中:
图1为由葡萄糖和苯酚获得的PHMF树脂的FTIR光谱;
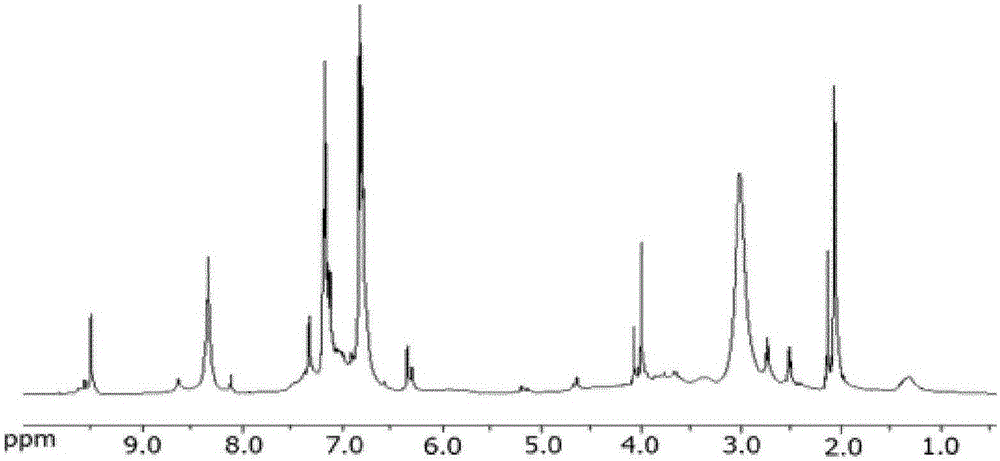
图2为由葡萄糖和苯酚获得的PHMF树脂的1H-NMR光谱;
图3为由葡萄糖和苯酚获得的PHMF树脂的13C-NMR光谱;
图4为由试剂HMF和苯酚合成的PHMF树脂的13C-NMR光谱;
图5表示DHL(最上面的曲线)、PHMF(中间曲线)和BPHMF(下面的曲线)树脂的分子量分布;
图6表示BPHMF树脂(下部迹线)、DHL(上部迹线)和PDHL(中间迹线)的FTIR光谱;
图7表示PHMF、OL、具有HMTA的PHMF和具有OL的PHMF的DSC曲线;
图8表示PHMF(底部迹线)和具有1,4-双-(羟甲基)苯的固化PHMF(上部迹线)的FTIR光谱;
图9表示PHMF(下图)和具有1,4-双-(羟甲基)苯的固化PHMF(上图)的13C NMR光谱;
图10表示用(a)OL、(b)KL固化的PHMF树脂的热稳定性及其与(c)HMTA固化的PHMF树脂的热稳定性对比;
图11为由玻璃纤维布增强并且用OL(上图)和HMTA(下图)固化的PHMF树脂制备的复合材料的照片;
图12表示用(a)OL、(b)KL和(c)HMTA固化的PHMF树脂复合材料的DMA图:将储能模量(E',迹线在各图的右上方开始)和tanδ(迹线在各图的左下方开始)对温度作图;
图13为用于根据ASTM 638的抗拉强度测试的复合材料试样的照片,用HMTA固化的PHMF(左图)和用HMTA固化的线型PF酚醛(右图);
图14表示在氮气气氛中具有不同量的HMTA(10wt%–20wt%)固化PHMF树脂的TG和DTG图;
图15表示在空气气氛中具有不同量的HMTA(10wt%–20wt%)固化PHMF树脂的TG和DTG图;
图16表示用15wt%HMTA固化的BPHMF树脂(a)和PHMF树脂(b)的DSC图;
图17表示由TGA测定的固化的BPHMF树脂在氮气(实线)和空气(虚线)中的热稳定性和分解速率;以及
图18表示(a)BPHMF-玻璃纤维复合材料与(b)PHMF–玻璃纤维复合材料(b)对比的动态力学分析。
详述
已经在水、有机溶剂和离子液体中实现了葡萄糖至5-羟甲基糠醛(HMF)的转变(Yan等人,2009;Zhao等人,2007;Li等人,2009;Binder和Raines,2009)。在许多金属之间,已经发现Zr和Cr是葡萄糖转化为HMF的有效催化剂。在SO4/ZrO2和SO4/ZrO2-Al2O3催化剂存在下,可将葡萄糖在水溶液中转化为具有48%收率的HMF(Yan等人,2009)。Zhao等人(Zhao等人,2007)发现铬氯化物(CrCl2和CrCl3)连同烷基-咪唑氯化物离子液体以多至69%的收率催化葡萄糖转化为HMF。Li等人(2009)使用Zhao的催化剂体系在微波辐射下获得了葡萄糖至HMF的91%转化率。Binder和Raines(2009)发现极性非质子有机溶剂中的季铵卤化盐和碱金属卤(Cl,Br)化盐可在铬氯化物催化的葡萄糖至HMF的转化中代替离子液体作为共催化剂。获得高达80%的收率。
已知糠醛与苯酚在碱性(Wu和Fu,2006)条件和酸性(Long等人,2008)条件下反应以形成酚醛聚合物。在此,发现可能使HMF与苯酚或者生物酚反应(HMF已经原位产生),即在苯酚或者生物酚存在下,HMF与其反应以产生酚醛树脂。将葡萄糖用作原位产生HMF的来源储备。
PHMF树脂合成
可用离子液体四乙基氯化铵实现葡萄糖至HMF的转化(Zhao等人,2007;Yuan等人,2011)。先前的工作表明CrCl2和CrCl3催化葡萄糖经过果糖中间体转化为HMF,但是CrCl2比CrCl3得到更高的HMF收率。表1和表2中的实验结果表明,在所使用的反应体系中(120℃,在含水或者不含水的CrCl2/CrCl3/TEAC催化剂的情况下),葡萄糖转化率为90%-100%,同时检测到非常低浓度的游离HMF(通常<1.5wt%),表明在形成HMF后经由苯酚-HMF树脂化原位消耗HMF。还通过苯酚的相当大的转化率(根据苯酚/葡萄糖摩尔比,依照表1为36%-63%或者表2为68%-92%)和所得聚合物材料–PHMF树脂的大分子量(700g/mol-900g/mol的Mw)来证明苯酚-HMF树脂化反应。
表1
缩写:Ph=苯酚,Glu=葡萄糖,C1=CrCl2,C2=CrCl3,C3=TEAC。
*PHMF树脂的全部PDI(多分散指数,Mw/Mn)为3-4。
a使用试剂级HMF。
表2
催化剂浓度:CrCl2/CrCl3/TEAC=0.02/0.01/0.03(M/M/M)。
结果也表明,使用CrCl2/TEAC或者CrCl3/TEAC(在表1中第6和第7项)催化剂体系,苯酚和葡萄糖的转化率比用CrCl2/CrCl3/TEAC催化剂的转化率低得多,特别是在CrCl2/TEAC的情况下。这有可能是因为CrCl2(尽管在形成HMF中更具活性)比CrCl3酸性更弱,而苯酚与HMF之间的树脂化/缩合反应需要路易斯酸催化剂。
先前(Yuan等人,2011;Moubarik等人,2009)已经表示120℃是适于将葡萄糖转化为HMF的温度,并且这是本发明所使用的温度。
表1中所示的结果表示3小时后在固定的催化剂浓度下,将葡萄糖/苯酚摩尔比从0.6增加至1.5导致苯酚转化率从36%稳定增加至63%,并且葡萄糖的转化率超过90%。当葡萄糖/苯酚比率增加超过1.5时,发现最初的磁力搅拌变得困难,大概是由于葡萄糖的高熔点以及其在苯酚中的低溶解度。为了使葡萄糖/苯酚比率高于1.5,添加水以促进葡萄糖溶解。为了维持含水反应介质的反应温度(120℃),因此使用压力反应器。由于水是葡萄糖转化为HMF以及苯酚与HMF的缩合反应的副产物,因此将水添加至反应体系对于树脂合成不是有利的。因此有水存在的反应(表2)比无水的实验(表1)进行更长时间。对比表1和表2的结果,可见在1.5:1至2:1的葡萄糖/苯酚摩尔比时,在添加水的压力反应器(表2)中的苯酚转化率(68-92%)与较低的葡萄糖/苯酚比率时的转化率相比要高得多。这表明在较高的葡萄糖/苯酚摩尔比下的反应有利于苯酚转化为PHMF树脂。也正如表1和表2所示,树脂的分子量还随着葡萄糖/苯酚比率的增加而增加。
由表2所示的结果表示了反应时间对苯酚转化率和PHMF形成反应的积极作用。随着反应时间从5h增加至8h,苯酚转化率和葡萄糖转化率分别从69%增加至92%以及从80%增加至99%,伴随着PHMF产物的Mw增加。表1中的第1-4项表示在3小时后,大多数葡萄糖都被转化。对比表1中的第2项和第5项,对于再反应2小时,苯酚转化率的增加比葡萄糖转化率的增加更多。这可能是因为聚合物链主要以HMF终止,如方案1所采用的形成苯酚-HMF树脂的可能反应机理所示。
方案1
合成苯酚-HMF树脂的反应机理:
在CrCl2、CrCl3和TEAC存在下,可将葡萄糖异构化为果糖,然后将果糖脱水,失去三个水分子以形成HMF(Zhao等人,2007;Binder和Raines,2009;Yuan等人,2011)。在路易斯酸(CrCl3)存在下,苯酚的对位和邻位的富电子碳可与HMF的亲电子醛基进行亲核加成。在路易斯酸存在下,HMF中的羟甲基还可通过Friedel-Crafts烷基化机理与相对于苯酚的羟基位于对位和邻位的苯酚碳反应。最终产物为具有支链的与线型酚醛树脂的结构类似的树脂,但是具有由呋喃环替代的一些苯环以及由羟基亚甲基键合替代的一些亚甲基键合。由于呋喃也是富电子芳香族环,因此还可发生呋喃环与HMF中的醛基或者羟甲基之间的反应。这可能有助于高的葡萄糖消耗。
已经观察到(Yuan等人,2011;Girisuta、Janssen和Heeres,2006;Huber、Iborra和Corma,2006)适量水的存在可提高葡萄糖转化为HMF,但是大量的水促进HMF分解成乙酰丙酸并且降低HMF收率。在本系统中,新形成的HMF在其形成后与苯酚快速反应,其明显地驱动脱水反应向前以维持低的HMF浓度以及减弱其转化为副产物的趋势。这由GC-MS分析确认为未检测到乙酰丙酸。因此这些实验结果表示了原位一锅法反应的益处。已知在酸性条件下(Hu等人,2011;Dee和Bell,2011),可将葡萄糖转化为腐黑物(脱水葡萄糖的脱水聚合物)、HMF和HMF的降解产物。因此,存在形成的树脂为PHMF和腐黑物的混合物的可能性,但是腐黑物的存在可能不会显著影响PHMF的有效性,只要其可并入固化产物。发现最终的树脂不可溶于水,但是可溶于大多数有机溶剂,包括丙酮、四氢呋喃和醇类。这表明树脂产物不是葡萄糖的低聚物,而是高度脱水的聚合物产物。可通过将反应混合物溶于丙酮来纯化PHMF树脂,然后将其沉淀于水/甲醇中以去除催化剂和未反应的葡萄糖和苯酚。对于苯酚/葡萄糖=1:2,8h反应,纯化后的收率为57%(产物重量除以进料,理论收率为75%)。由于催化剂是无毒的并且在促进树脂固化反应中也具有活性,未反应的苯酚可在固化阶段进行反应,实际上,不需要进一步纯化。
图1所示的合成树脂的IR光谱显示了在芳香族环的1400-1600cm-1区域特征的吸收带,即在可归于PHMF树脂的苯酚和呋喃环结构的1592cm-1、1505cm-1和1450cm-1的碳-碳伸缩振动。参见方案1的途径。在1230cm-1和1000cm-1的吸收分别表明存在共轭和非共轭的C-O伸缩。在748cm-1的吸收可归于芳香族C-H键的平面外弯曲。在3275cm-1、2910cm-1和1702cm-1的吸收可分别归于OH、亚甲基(-CH2-)和C=O(醛)伸缩,这是HMF中的醛和羟甲基与苯酚对位和邻位反应位点的缩合反应以形成PHMF-CH(OH)-和-CH2-键合的证据,如方案1所示。
在图2所示的PHMF树脂的质子NMR光谱中,除了水的峰(2.8-3.1ppm)和丙酮的峰(d6-丙酮2.0ppm)以外,大多数峰可归于与芳香族碳原子键合的氢原子(6-8ppm),即PHMF树脂中的苯酚环和HMF环的质子。在9.5ppm的峰是来自并入的HMF的醛基的质子。在8.3ppm的峰是由于具有氢键合的苯酚环的羟基质子。在4.0ppm的峰可归于亚甲基质子。
还使用13C-NMR表征PHMF树脂的结构。参见图3。可将峰指定如下:醛碳,178ppm;与呋喃环的氧相邻的碳,162ppm;羟基取代的苯酚碳,156和157ppm;与呋喃环的氧和醛基相邻的碳,152ppm;在苯酚环上的OH连接的碳的间位的碳,129ppm;在呋喃环上与氧间位的碳,119ppm;在苯酚环上的OH连接的碳的邻位的碳,115ppm;在呋喃环上与氧和CHO间位的碳,110ppm;亚甲基和次甲基,52ppm、56ppm;以及溶剂,d6-二甲基亚砜,40ppm。可将剩余的未经确认的峰归于葡萄糖聚合物的碳。来自苯酚和葡萄糖的PHMF树脂的13C-NMR光谱与由苯酚和试剂级纯的HMF合成的PHMF非常相似。参见图4。
元素分析(C,H,O)表明在苯酚/葡萄糖比率为1:1.5的情况下,纯化的PHMF树脂所具有的C、H、O含量(wt.%)为66.2、5.5、27.0。来自试剂HMF的PHMF所具有的C、H、O含量为70.7、4.8、24.3,与由交替的苯酚-HMF单元组成的PHMF树脂引起的C、H、O含量(71.3wt.%、5.0wt.%和23.8wt.%)类似。来自苯酚-葡萄糖的PHMF树脂的O含量比苯酚-HMF树脂的O含量高约11%,可能是由于葡萄糖与苯酚的比率较高。
BPHMF树脂合成
将从酶水解过程的残余物中提取的木质素称为酶水解木质素(HL)(Jin等人,2010)。酸水解木质素(商购的木材的酸糖化过程的副产物)是木质纤维素残余物的一部分(Dizhbite等人,1999)。木质素、纤维素和其它碳水化合物是水解残余物的主要组分。其它类型的木质素(例如,碱溶性硫酸盐木质素、水溶性木质素磺酸盐、有机溶剂型木质素)也可用于合成生物酚HMF(BPHMF)树脂。按照PHMF的类似合成方法,用酚化的解聚HL(PDHL)合成BPHMF树脂。在如实验部分所述的条件下(14.10g酚化的DHL(包含50wt%苯酚和50wt%DHL)、13.5g(0.075mol)葡萄糖以及3g水和总共0.3g催化剂在120℃下反应8h),合成的BPHMF树脂的毛收率为85%。可通过增加反应底物中的葡萄糖水平来进一步提高BPHMF树脂的收率。
DHL、PDHL和BPHMF树脂的表征
用于本工作的原始HL来自硬木材的酶水解,并且其具有重均分子量Mw>20000g/mol。图5和表3呈现了从BPHMF树脂获得的近似分子量和分布及其与DHL和PDHL的对比。
表3
DHL和PDHL的GPC图表明DHL和PDHL具有类似的分子量和分布,这与酚化过程中的低的苯酚转化率(4%)一致。甚至在如此低的苯酚转化率情况下,由于DHL和纯苯酚的分子量存在较大差别,平均三分之二的DHL分子为酚化的苯酚。BPHMF的GPC图表现出比DHL和PDHL更宽的重量分布和较大的多分散性值。更有趣的是,BPHMF的大多数组分表现出比PDHL更低的停留时间,如GPC图的峰所示(图5)。在树脂化过程中与葡萄糖衍生的HMF缩合时,PDHL的分子量和PDI增加了三倍,表明树脂化反应进行良好,并且BPHMF树脂具有宽的分子量分布(PDI=8.34)以及大的Mw(9030g/mol)和Mn(1082g/mol)。根据对PHMF树脂的结构分析和相关参考(Yuan等人,2014),可通过方案1提出反应机理。
方案1
由生物酚和原位产生的HMF合成BPHMF树脂的反应机理
图6表示BPHMF树脂的红外光谱。BPHMF树脂具有与PF树脂类似的化学结构。在3400cm-1至3387cm-1存在典型的羟基吸收,并且在2980cm-1至2875cm-1存在C-H伸缩。碳-碳伸缩和芳香族振动的强烈吸收在1592cm-1,表示苯酚、木质素和HMF的大量芳香族化合物。芳香族环振动在1510cm-1和1450cm-1,H-O弯曲在1355cm-1,甲氧基在1260cm-1,芳香族环与氧伸缩在约1215cm-1,愈创木基带(C-O)在1015cm-1,以及芳香族环与氢的平面外弯曲在748cm-1。因为剩余另外的醛基,由于BPHMF中的羧酸而存在1663cm-1处的CHO(羰基)伸缩。
3.2固化PHMF树脂
DSC测量(图7)表示可使用有机溶剂型木质素(OL,20wt.%)(视为绿色固化剂)固化苯酚/葡萄糖比率=1:2树脂,在约142℃下表现出明显的放热峰。相反,PHMF树脂和OL的DSC曲线各自都没有观察到明显的放热行为。固化反应是在CrCl3催化下木质素中的羟烷基与PHMF中的芳香族环(苯酚和呋喃环)之间的典型的Fridel-Craft烷基化反应(Fadel等人,1987)。正如所料,PHMF树脂是可用HMTA固化的,其在139℃表现出明显的固化行为,与PHMF和OL以及PF和HMTA之间的固化行为类似(De Medeiros等人,2003)。热固性(体型)酚醛树脂和线型PF树脂的固化可释放甲醛。对于热固性酚醛树脂(resole),因为各苯环连接两个至三个羟甲基,当固化时,首先在不同苯环的两个羟甲基之间形成CH2-O-CH2醚键,然后进一步加热醚键将释放甲醛并且在两个苯环之间形成更稳定的亚甲基键合。对于线型PF树脂的固化,尽管它是在苯环之间仅有亚甲基键合的、直链或者轻度支化的聚合物,它的固化通常需要六亚甲基四胺(HMTA)作为固化剂,其在交联期间通常伴随着甲醛的形成。本发明的PHMF树脂具有与线型酚醛类似的结构,它也可通过HMTA固化,这将释放甲醛。为了避免释放有毒化学品,使用绿色固化剂(例如OL或者KL)是更加环境友好的。
据作者所知,还未曾开发出OL/KL与线型酚醛之间的固化反应。PHMF树脂和OL/KL的化学结构是复杂的,并且如果烷基化是机理,存在的羟基亚甲基将明显干扰结构分析。在该条件下,1,4-双-(羟甲基)苯是表现OL/KL用于研究这些反应以阐明PHMF树脂和木质素的固化机理的理想模型化合物。
通过FTIR光谱(图8)对整个固化过程的监测显示,1,4-双-(羟甲基)苯的羟基在固化后几乎消失了。参与固化过程的特征官能团的演变使得确认出现了CH2基团的不对称伸缩振动带(在2978cm-1观察)。
方案2
提出的固化反应机理
图9中的碳(13C)NMR光谱表明了PHMF树脂用1,4-双-(羟甲基)苯固化前与固化后相比的关键区别。应当注意,对应于PHMF树脂中的对位的未取代的芳香族碳的峰(119ppm)消失了,表明一些基团与该对位的酚碳连接。同时在固化后形成取代的对位酚碳(139ppm)表明来自模型化合物的羟基与PHMF反应。此外,还在72ppm和64ppm检测到了归属于另外的羟基亚甲基官能团的峰。对于这些发现的一种可能解释为与通过铬氯化物的催化效应的PHMF和1,4-双-(羟甲基)苯的烷基化过程有关(Elavarasa等人,2011)。基于以上分析,方案2中提出并且描述了PHMF与OL/KL之间的固化机制。
PHMF树脂的下游处理的表征(玻璃化转变、固化和热稳定性测试)
已经制备和表征PHMF树脂,进行实验,所述实验确立使用树脂作为其它材料的前体的可行性。在第一个实施例中,使用天然聚合物有机溶剂型木质素(20wt.%)或者HMTA固化树脂。
玻璃化转变温度(Tg)确立了良好使用树脂的温度,因此其被认为是聚合物材料和复合材料的重要参数。已经发展了测定聚合物体系中的Tg的许多方法(Cai和Lin,2005)。分别用OL和KL以10%、20%、30%和40%的四种重量百分比固化纯的树脂。研究了所有固化树脂的玻璃化转变温度。如表4所示,Tg为110℃至135℃,在20wt%交联剂时具有最大值。明显地,OL/KL的添加显著提高了PHMF树脂的使用温度。同时,OL/KL的低比率(10%)对于交联是不足的,但是30%wt和40%wt的OL/KL是过量的,因此导致较小的Tg,Tg从30%至40%持续减少就是很好的例子。最佳的固化剂比率确定为树脂的20%。应当注意,用OL/KL固化的PHMF树脂具有与那些PF酚醛相同的Tg,其被报道为134℃(Pérez等人,2011)。
表4
进行TGA实验以检测用OL/KL/HMTA固化的PHMF聚合物的热重量行为和热性能。图10表示固化的PHMF树脂在加热时的TGA热失重谱图。从TGA数据来看,确定了固化的PHMF树脂在700℃下的热稳定性系数并且示于表5中,所述热稳定性系数包括起始分解温度(Ti)、50%失重的温度(T50%)、最大分解速率的温度(Tmax)和残余质量。结果表示PHMF聚合物在高至约222℃(Ti)下是热稳定的,并且对于OL(397℃)和KL(407℃)固化的PHMF具有高的Tmax,比用HMTA固化的那些几乎高100℃。
表5
此外,固化的PHMF树脂在250℃至500℃表现出强烈的热分解阶段。Lee等人在PF树脂中(Lee等人,2003)和Tejadoa在木质素取代的木质素-苯酚-甲醛树脂中(Tejado等人,2007)也观察到了这类行为。通常认为热分解是通过无规断链的热降解,其由于大量的烷基而涉及单元间键合的破碎,当降解一些其它芳香族环/低聚物时,将单体酚释放进入气相。由于HMTA在加热时被分解成甲醛和氨,在已经消耗反应位点后可形成具有小分子量的一些固化的树脂。相对于用较大分子量的OL固化的那些树脂,这些具有较小网络的树脂可在较低温度下分解。然而,由于OL结构中受限的反应位点的数目,后一种树脂可导致低的交联密度,因此PHMF和HMTA体系在700℃下表现出略高的残余物。因此,OL/KL具有总体上与用HMTA固化的那些相当的热稳定性。
PHMF树脂的下游产物
将葡萄糖基PHMF树脂用作发展复合材料的热固性基体的结构单元。用在适度地高温下固化的可热聚合的PHMF树脂通过敞模以及手工层叠(layup)注入纤维来制备基于天然组分的生物复合材料。
图11表示通过热固化由PHMF树脂浸渍的玻璃纤维获得的生物复合材料。天然原料在PHMF树脂中的最终含量大于83%,在负载有50%重量的玻璃纤维的组合物中达到大于41%。
将PHMF-玻璃纤维复合材料的动态力学性能随温度变化进行测量。除了玻璃化转变温度(Tg)值的信息以外,也可获得其它三种重要的参数用于全面理解复合材料的粘弹行为,即储能模量(E′)、损耗模量(E″)和δ的正切(tanδ),它们通过以下表达式进行相互关联(intercorrelated):
研究了OL/KL对复合材料试样的机械性能的作用,并且在图12中阐明了DMA图,其中储能模量(E')和tanδ对温度作图。代表固化树脂的刚度的储能模量与载荷循环期间储存的能量成比例。通过tanδ的峰值温度确定本工作中的固化样品的Tg。众所周知,在相同的固化剂和组分的情况下,热固性树脂的不同Tg值反映单体的不同结构和交联密度。Tanδ峰的宽度用较宽的峰反映聚合物网络异质性,所述较宽的峰表示更多多相聚合物。通过应用最佳量的木质素或者HMTA,Tg值为约267℃(OL)和280℃(HMTA)。这意味着,与HMTA相对比,来自木质素的羟基亚甲基对网络中的片段移动性提供具有类似空间限制的刚性键,因此导致相近的Tg。尽管用KL固化的PHMF树脂表现出较小的Tg(220℃),但是它们明显比腰果酚-甲醛酚醛树脂高~130℃(da Silva Santos等人,2010)。此外,需要更多的工作以便改善树脂的机械性能,同时改善其玻璃化转变温度以及进一步降低固化温度。
由PHMF树脂以及15wt%HMTA(玻璃纤维与树脂+固化剂的质量比=1/1)在120℃持续30min、150℃持续30min以及180℃持续1h的固化程序下制备玻璃纤维增强的复合材料。根据ASTM 638,热压的复合材料形成哑铃状,示例于图13中。表6对比了玻璃纤维-PHMF/PF复合材料的抗拉强度。基于抗拉强度的结果,玻璃纤维-PHMF复合材料与玻璃纤维-PF复合材料相当。
表6
完全固化的玻璃纤维增强的PHMF树脂复合材料的机械性能
在表7中可见HMTA的添加量对完全固化的玻璃纤维增强的PHMF复合材料的抗拉性能的作用。实验测量表示随着HMTA含量增加,断裂强度(即,抗拉强度)和破坏应力的值显著较高。例如,PHH10(表示具有约10wt%HMTA的PHMF树脂基体)试样具有分别为101Mpa和4.85%的抗拉强度值和断裂伸长率。随着HMTA添加量的增加,PHH15试样和PHH20试样分别具有109MPa和114Mpa的较高抗拉强度值,表明较高的HMTA添加量在树脂分子之间产生更多交联。与PHH10试样相比,PHH15试样和PHH20试样也具有较高的破坏应力,分别为5.55%和6.9%。然而,发现杨氏模量随着HMTA添加量的增加而单调地从21GPa降低至16Gpa,意味着增加固化剂的量降低了复合材料的刚度。
表7
尽管PHMF基复合材料的拉伸断裂强度不是非常高,但是依然比一些PF基复合材料或者其它类型的树脂基复合材料更好。例如,玻璃/聚酯复合材料表现出70MPa的最大拉伸断裂强度和700MPa的模量(Mouritz等人,1997)。酚醛片状模塑复合材料(SMC)具有96MPa的拉伸断裂强度(Pilato,2010),并且长玻璃纤维增强的PF树脂模塑化合物的抗拉强度为115MPa,这与PHMF基复合材料在此获得的值非常接近。George等人综述了天然纤维增强的塑料复合材料,特别是关于它们最佳的机械性能(George、Sreekala和Thomas,2001),其中这些生物复合材料具有35MPa的最大抗拉强度。此外,在ASTM D638标准测试下列出了一些复合材料的强度,例如玻璃增强的聚酯和尼龙分别具有143MPa和162MPa的抗拉强度。
对于完全固化的玻璃纤维增强的PHMF和PF树脂复合材料的矩形梁的3点弯曲测试,由方程(2)计算弯曲模量:
由方程(3)定义弯曲强度:
其中w(b)和h(d)是梁的宽度(mm)和高度(mm),L为支撑跨度的长度(mm),d为由于在断裂点施加载荷F(N)的挠度。
表8
由表8所示的3点弯曲测试结果证明,增加的HMTA添加量导致较高的破坏应力但是使最大载荷从PHH10的227N降低至PHH20的193N。因此,增加HMTA添加量使抗弯模数或者弯曲模量从PHH10的8.7GPa大幅度地降低至PHH20的2.5GPa。这种结果可能归因于交联网络在导致较高交联密度的较高HMTA添加量下的较大片段移动性。相反,增加的HMTA含量导致弯曲强度从PHH10的145MPa显著下降至PHH20的82MPa。这种结果是可预料的,因为高度交联的聚合物(例如PHH20)将是更加易碎的。在较高水平的固化剂情况下,可以以苯并噁嗪环的形式产生过量的交联,导致增加固化复合材料的交联密度和片段移动性,因此影响弯曲性能(Anseth等人,1995;Zhu等人,2004)。当与其它常见工业复合材料的弯曲性能相对比时,PHMF-基复合材料的表现令人满意。例如,报道酚醛SMC具有158MPa弯曲强度和8.2GPa弯曲模量(Pilato,2010),处于PHH10的数值的类似范围中。研究用于汽车应用的天然纤维和玻璃纤维增强的聚合物复合材料具有与PHMF基复合材料的那些类似(差别小于15%)的弯曲性能(Holbery和Houston,2006)。玻璃纤维增强的不饱和的聚酯表现出80MPa的弯曲强度和6.0GPa的模量,其低于或者类似于PHMF基复合材料的弯曲强度和模量。
HMTA固化的PHMF树脂的热稳定性
为了检测HMTA添加量对固化的PHMF树脂的热稳定性的作用,收集和分析了氮气气氛和空气气氛下的TGA数据。图14表示了不同用量的HMTA(10-20wt.%)固化的PHMF树脂对温度的失重和分解。表9概述了由TG/DTG图产生的特征。
在氮气气氛中,当HMTA添加量从10wt.%增加至15wt.%时,起始温度T5(5%失重)从297℃增加至315℃,并且在20wt.%HMTA时未发生变化。随着HMTA添加量的增加而改善的PHMF的热稳定性可能是由于基体的较大交联,其可从图14中的TG/DTG图中推断:分解速率(DTG)峰的高度在约320℃下随着HMTA添加量的增加而降低。该峰可能是由于存在于HMTA固化的PHMF体系中的醚基的降解。所有树脂在455℃表现出第二个分解峰,其可对应于脂肪族基团的降解。对于PHH10、PHH15和PHH20,在800℃获得的残炭率分别高达59%、61%和63%,这也表明随着HMTA添加量的增加,PHMF树脂的交联密度越高,可能是由于在加热时增加形成PHMF树脂中的苯并噁嗪环。
表9
a在5%失重的温度
图15突出了PHH10、PHH15和PHH20在空气中的失重和分解速率。表9也概述了由TG/DTG图产生的特征。通常,起始失重和最大失重速率的温度转移至较高的温度,并且转移的程度随着HMTA添加量的增加而增加。类似的现象已在文献中进行了报道,其中高的耐热性归因于中间体固化成苯并噁嗪型结构(Hatfield和Maciel,1987)。与N2中分别为~320℃和~450℃的温度相比,HMTA固化的PHMF树脂在空气中的第一分解温度和第二分解温度(主要的)分别为~350℃和~650℃。树脂的最显著的分解发生在~650℃,其中芳香族结构通过燃烧遭到破坏并且炭在800℃下完全被氧化。
BPHMF树脂的固化行为
由于BPHMF树脂包含极其少的反应位点,可需要其它固化剂(例如环氧树脂、木质素和HMTA)以实现树脂的交联和固化。此外,随着温度增加,也可在木质素和树脂之间发生自固化,因为在木质素中存在可有助于交联的额外的官能团(例如羟基亚甲基和羟基)。可能的反应可以是树脂中的芳香族基与木质素中的羟基亚甲基之间的烷基化反应。图16表现了制备的BPHMF在10℃/min的加热速率下于氮气中与PHMF树脂相对比的DSC测量结果,两者都使用HMTA作为固化剂。DSC曲线表明两种树脂的固化反应在120℃开始,在约150℃出现峰并且在200℃之后结束,这对于许多生物酚醛树脂是常见的现象(Cheng等人,2012;Cheng等人,2013)。
BPHMF需要比PHMF更长的时间用于完成固化过程。可通过木质素衍生的生物酚的低反应性以及如之前在树脂与木质素官能团之间解释的自固化机制来解释这种现象。
热稳定性
还使用TGA-DTG研究了合成的BPHMF树脂的热稳定性。如图17所示,BPHMF树脂在300℃之后于空气或者氮气气氛中开始分解,并且该温度比PHMF树脂(图14和图15的PHH15)的高50℃,表明BPHMF树脂的热稳定性比PHMF树脂的更佳。表10表示BPHMF树脂和PHMF树脂在氮气中加热时的热稳定性对比。BPHMF的最大分解温度比PHMF的高60℃,意味着由于存在木质素衍生的生物酚而显著增强了耐热性。此外,BPHMF树脂比PHMF树脂产生更高的碳残余物(在800℃下约60wt%),比PHMF树脂的碳残余物几乎高10%(表10)。这可能是由于木质素结构中的醚键及其高芳香族部分的较慢分解。有趣地,BPHMF树脂在空气中表现出持久的热稳定性,如由500℃下较高的重量残余(70%-80%)所示,在500℃之上时树脂在空气中急剧地分解。
表10
动态力学分析
将BPHMF-玻璃纤维复合材料的动态力学性能作为温度函数进行测量。图18表示BPHMF-玻璃纤维复合材料与PHMF–玻璃纤维复合材料对比的动态力学分析。通常储能模量的值随着温度的增加呈下降趋势。然而,储能模量并不是在整个温度范围内都等量下降,其中其值的显著下降发生在190℃-300℃,这对应于BPHMF玻璃纤维复合材料和PHMF玻璃纤维复合材料的玻璃化转变温度范围。在190℃以下,温度区的特征为玻璃态行为,其中材料的刚度与其余温度区相比为其最大值。如图18所示,储能模量在100℃下随着温度的增加存在轻微下降,这主要是由于不完全固化。当温度增加超过Tg时,复合材料样品将进行玻璃态(高刚性)区向软橡胶态平坦区的转变。
Tanδ用作聚合物材料的弹性相与黏滞相之间的平衡指示器(Hassan等人,2011)。由损耗角正切图获得的阻尼峰对应于玻璃化转变温度Tg。对比BPHMF玻璃纤维复合材料(图18(a))与PHMF玻璃纤维复合材料(图18(b))之间的阻尼峰,当与PHMF聚合物基体的玻璃化转变温度(243℃)对比时,将DHL并入聚合物基体(BPHMF)导致其玻璃化转变温度Tg(272℃)的增加。
使用由HMTA完全固化的BPHMF树脂的玻璃纤维增强塑料的机械性能
表11给出了编织的玻璃纤维布-PHMF树脂FRC复合材料的抗拉强度与BPHMF FRC的抗拉强度的对比,两者都用15wt%的HMTA固化。BPHMF复合材料具有约89MPa的抗拉强度,比PHMF复合材料的抗拉强度低20%,但是两个值实际上与商购的酚类FRC的抗拉强度相当。
表11
因此,本发明提供由酚化的解聚水解木质素与原位衍生自葡萄糖的HMF在催化剂存在下的反应获得的无甲醛的生物酚HMF树脂(BPHMF)。BPHMF树脂的毛收率可为85重量%。通过FTIR的结构分析表示树脂化是成功的并且获得的BPHMF树脂的分子量(重均重量)为约9030g/mol,与PDHL的2107g/mol的Mw相比,表明在合成期间PDHL与HMF大量缩聚。BPHMF仅具有2800g/mol的数均分子量。发现用HMTA固化的BPHMF树脂在氮气或者空气中于高至300℃下是热稳定的。与PHMF树脂相比,BPHMF树脂需要较高的温度用于固化,但是其具有较大的储能模量和Tg,因此其具有更好的热力学性能。
用不同量的HMTA固化剂(约10wt.%至20wt.%)固化的玻璃纤维增强的PHMF树脂进行热分析、物理分析和机械分析。通常,发现通过增加HMTA的量增强了复合材料的抗拉性能、热稳定性、储能模量、交联密度和玻璃化转变温度。然而,弯曲性能、流变学测试和耐化学品性测试表明15wt.%HMTA刚好足以在酚醛树脂中形成饱和的三维交联。使用太多HMTA可形成来自HMTA的不稳定末端基团,这降低了树脂的整体耐化学品性并且弱化固化过程。结果表明使用PHMF树脂以聚合物基体并入作为纤维增强复合材料的一部分的可行性。即使在使用HMTA作为固化剂时,也可获得无甲醛的产物。
实验
材料
从Sigma-Aldrich获得试剂纯苯酚(99.0%)、CrCl2(95.0%)、CrCl3·6H2O(98.0%)、D-葡萄糖(99.5%)、四乙基氯化铵水合物(99.0%)和5-羟甲基糠醛(99.0%)。从Caledon实验室或者Fisher Scientific获得四氢呋喃(THF,HPLC级)、二甲基亚砜(DMSO-d6)和0.005M H2SO4HPLC级水。由Lignol Canada供应用作固化剂的有机溶剂型木质素。所有试剂未经进一步处理/纯化进行使用。
甲醛(大约37%)来自Anachemia,Montreal,QC,并且按原样使用。由FPInnovations供应水解木质素,其为来自用于生物产物的其硬木材分馏过程的副产物(或者称为“TMP-生物过程”)(Yuan等人,2011)。HL包含由残余的纤维素和碳水化物平衡的>50-60wt%木质素。分子量被认为>20000g/mol,但是由于在溶剂中的不溶性,其是不可测量的。用于本工作中的溶剂为蒸馏水、丙酮(Fisher Scientific,Fair Lawn,NJ)和氢氧化钠溶液(大约50%,Ricca Chemical Co.,Arlington,TX),所有都按原样使用。
从Sigma-Aldrich获得六亚甲基四胺(HMTA)并且用作BPHMF树脂的固化剂。BGF玻璃纤维布购买自Freeman,Ohio。
使用其他单独的专利申请的解聚过程,在低的操作压力下(<150psi)于150-300℃在溶剂中操作30min-120min获得解聚的水解木质素(DHL)。该过程产生高收率的解聚HL(DHL)(70wt%)以及~10wt%的SR。发现该过程对于非常高分子量(Mw>20000g/mole)的HL解聚/液化为具有更低分子量(1000-2000g/mol)的DHL而言,是非常有成本效益和高效率的。虽然对于合成反应是使用低分子量的木质素更为有利的,但是对于本发明来说解聚过程的本身并不重要,本发明需要的是具有重均分子量<5000g/mol的解聚/液化木质素作为制备BPHMF树脂的生物酚原料。
酚化的DHL(PDHL)在本工作中通过以下制备:等量DHL和苯酚以及2%硫酸和溶剂丙酮,加热至120℃进行反应3h,然后将反应器冷却至室温。通过旋转蒸发和真空干燥去除溶剂。
PHMF树脂的合成
首先在配备有冷凝器以及中间颈部的氮气出口、分别在两侧颈部的氮气入口和温度计的100mL三颈反应器中于常压下进行苯酚-HMF(PHMF)树脂的合成。在典型试验中,用氮气吹扫反应器,然后添加9.41g(0.100mol)苯酚、16.20g(0.090mol)葡萄糖、0.0610g CrCl2(在反应混合物中约0.02M)、0.0570g CrCl3·6H2O(0.01M)和0.1640g(0.06M)四乙基氯化铵(TEAC)。将反应器浸于预热至120℃的油浴中并且用磁力搅拌器在氮气气氛下搅拌3至8小时。为了对比,也使用试剂级HMF以1:0.9的HMF/苯酚比率在类似于以上所述的那些条件下合成PHMF树脂。在100mL ACE玻璃压力反应器中以葡萄糖与苯酚的较高比率进行树脂合成。通常,将7.05g苯酚(0.075mol)、27.0g(0.15mol)葡萄糖、6.00g水和0.100g CrCl2(0.02M)、0.0940g CrCl3·6H2O(0.01M)和0.300g(0.06M)TEAC添加至反应器。用氮气通过橡胶隔片排空和吹扫反应器,然后用Teflon塞子密封。将反应器置于预热至120℃的油浴中并且用磁力搅拌器搅拌5小时。在冷却后,用80/20(v/v)甲醇/水稀释反应混合物以形成均一溶液。然后收取该溶液的样品并且用HPLC溶剂进一步稀释用于葡萄糖、苯酚和HMF分析。首先使用旋转蒸发仪去除溶剂来纯化产物(以去除未反应的葡萄糖和苯酚),然后将剩余的材料溶于丙酮,并且将PHMF沉淀于90/10(v/v)水/甲醇中。在真空干燥后,沉淀过程产生半固态的黑色PHMF产物。通过GPC、FTIR、NMR和DSC来表征PHMF产物的分子量、结构和固化性能。
固化过程:通过将设计量的OL/KL均匀添加至树脂中并且在120℃下加热1小时、在150℃下加热1小时以及然后在180℃下后固化1小时经由烷基化反应制备OL/KL固化的PHMF树脂。在聚合之前,将树脂和固化剂溶于丙酮以形成均匀的混合物并且去除丙酮。作为对比,根据出版物中常用的量用15wt%HMTA固化PHMF(Mwaikambo和Ansell,2001)。
BPHMF树脂的合成
将酚化的DHL用作原料以合成BPHMF树脂。本文表明了合成具有50%苯酚取代水平的程序。在BPHMF树脂的典型合成试验中,将14.10g酚化的DHL(包含50wt%苯酚和50wt%DHL)、13.5g(0.075mol)葡萄糖、3g水和总的0.3g催化剂(与PHMF树脂合成所用的催化剂相同)装入有Teflon塞子的100mL玻璃压力反应器。将反应器置于预热的120℃油浴中并且用磁力搅拌器搅拌。由于反应对温度敏感,在过程中严格控制温度。因此,可通过将温度维持在所期望的水平来降低副产物。在反应8小时后,将反应在水浴中冷却至室温来停止。通过旋转蒸发和真空干燥使产物干燥。在所得的BPHMF树脂上进行GC-MS测试以便计算苯酚至树脂的转化率。确定苯酚转化率为约60%,并且在本工作中通过蒸汽蒸馏回收未反应的游离苯酚(然而,实际上,并不需要回收未反应的苯酚,这是因为可用固化剂在树脂固化阶段固化未反应的苯酚)。此外,还可通过增加树脂化过程中的葡萄糖添加量进一步改善苯酚转化率。
原料和产物表征
使用各种技术表征原料和产物的化学结构/热/机械性能,所述技术包括凝胶渗透色谱(GPC)、傅里叶变换红外光谱(FTIR)、气相色谱-质谱(GC-MS)、差示扫描量热仪(DSC)、热重分析(TGA)和动态力学分析(DMA)。
用具有智能ITR/ATR配件的Nicolet 6700傅里叶变换红外光谱在500cm-1至4000cm-1扫描获得FTIR光谱。
通过使用配备有Varian 5mm三重共振间接检测HCX探针的Varian Inova 600NMR(在298K扫描16-32次)光谱仪来获得1H NMR和13C NMR(核磁共振)光谱。使用2s循环延时、3.6s采集时间、45度顶尖角(pw=4.8us)和0ppm至14ppm(sw=9000.9Hz)的谱宽。选择d6-DMSO作为液体样品的溶剂和固态13C交叉极化魔角旋转(CPMAS)的细粉末。
用Waters Breeze仪器(具有折射率和紫外检测器的1525二元泵)进行HPLC(高效液相色谱法)分析。通过使用Bio-Rad Aminex HPX-87H柱和HPLC级0.005M H2SO4水作为流动相(流速为0.6mL/min)分析葡萄糖、苯酚和HMF含量。
使用THF作为流动相以1mL/min的流速使用聚苯乙烯标准品用于校正,在Waters Styrylgel HR1凝胶渗透色谱(GPC,1525二元泵,设置在270nm的UV检测器,在40℃的Waters Styragel HR1柱)上测量分子量。
用差示扫描量热仪(DSC,Mettler-Toledo,Switzerland)在50mL/min N2下以10℃/min的加热速率在40℃至250℃下于坩埚中评估树脂的热固化性能。
使用TGA 2050(热重分析仪,TA Instruments)测量未固化和固化的PHMF树脂的热重行为。将约10mg样品置于铂盘并且以10℃/min的升温速率在50mL min-1的N2气氛中加热至700℃。
使用动态力学分析(DMA)测量固化复合材料的热机械性能。使用Netzsch 242C DMA通过三点弯曲测试具有20×10×1mm3概略尺寸的矩形样品。在具有5μm偏差的1Hz的驱动频率下测试样品,同时以5℃min-1的扫描速率将温度从30℃升温至250℃。
例如,以下详细说明了表征BPHMF树脂的原料和产物的程序。通过凝胶渗透色谱(GPC,Waters Styrylgel HR1)使用THF作为洗脱液以及线型聚苯乙烯作为标准品用于校正测量DHL酯化之前和之后以及BPHMF树脂的分子量分布曲线。使用FTIR(Perkin-Elmer Spectrum Two IR Spectrometers)分析BPHMF树脂的官能团结构。如先前所述,将树脂中的其余游离苯酚浓度在连接有5977A MSD的Agilent 7890B GC上通过GC-MS使用30m×0.5mm×0.25μm DB-5柱用以下温度程序进行检测:在50℃的起始温度下保持1min,然后以30℃min-1升温至280℃的最终温度保持1min。用差示扫描量热仪(DSC,Mettler-Toledo,Schwerzenbach,Switzerland)在50mL/min N2下以10℃/min的升温速率在50℃至250℃下于密封的坩埚中评估树脂的热固化性能。通过热重分析(TGA)用5-10mg热预固化树脂(在120℃下进行30min、150℃下进行30min以及180℃下进行1h)在Perkin-Elmer热分析系统上用Pyris 1型TGA部件检测BPHMF树脂的热稳定性。在氮气中以20ml/min的流速测量样品;升温速率为10℃/min。使用动态力学分析仪(DMA Q800,TA Instruments)以5℃/min表征固化的BPHMF复合材料的动态力学性能(例如玻璃化转变温度,Tg)。通过将BPHMF和HMTA的均匀溶液以1/1比率(w/w)涂覆于玻璃纤维上并且保持在室温下直到溶剂去除来制备复合材料。通过在Carver液压型热压机中的热压以与树脂固化相同的程序在35MPa(5000psi)载荷下固化样品。
复合材料的制备
通过将成分在50℃下溶于90%丙酮和10%水(v/v)的混合物中制备HMTA和PHMF(或者BPHMF)线型酚醛基体的混合物。用100:10(PHH10)、100:15(PHH15)和100:20(PHH20)重量比的PHMF树脂(PH)和HMTA(H)产生三种制剂。将树脂混合物以1:1的重量比涂覆于玻璃纤维并且在室温下保持24小时以使溶剂蒸发。通过Carver液压型热压机中的热压在将载荷逐渐增加多至35MPa(5000psi)于120℃下固化30min、于150℃下再固化30min并且然后于180℃下进一步固化60min来固化干燥的树脂。在180℃下热处理5h时,将完全固化的复合板切成如ASTM标准所需的样品。考虑到热压过程中不可避免的树脂损失,完全固化的复合材料包含约40%的树脂,并由马弗炉中燃烧复合材料所确认。为了便于对比,如上所述制备用HMTA固化的常规PF和玻璃纤维增强的PF复合材料。PF树脂(PF)和HMTA(H)的重量比为100:15(PFH15)。
机械性能测试
使用Admet 7000通用试验机根据ASTM D 638测量拉伸性能。使用长度为180mm且握持部分的宽度为10mm的哑铃状试样。十字头速度为10mm/min并且总的延伸范围为25mm。施加拉伸应力直到样品失效并且将失效前的最大施加应力记录为抗拉强度。获得应力-应变关系并且其斜率确定杨氏模量。各测试重复进行五次。
按照ASTM D790-M93使用相同的通用试验机确定复合材料试样的弯曲模量和弯曲强度。用50mm的支撑跨度在三点弯曲装置上进行尺寸为127mm×13mm×3mm的矩形试样并且以1.2mm/min的十字头速度负载至各试样的中心直到失效。在失效前将弯曲模量和弯曲断裂强度记录为最大施加应力并且以载荷-位移曲线的斜率获得弯曲模量。
参考文献
Alonso,M.V.,Oliet,M.,Pérez,J.M.,Rodrlguez,F.,Echeverrla,J.,2004.Determination of curing kinetic parameters of lignin–phenol–formaldehyde resol resins by several dynamic differential scanning calorimetry methods,Thermochim.Acta.419,161-167.
Alonso,M.V.,Oliet,M.,Rodrlguez,F.,Garcla,J.,Gilarranz,M.,Rodrlguez,J.,2005.Modification of ammonium lignosulfonate by phenolation for use in phenolic resins,Bioresour.Technol.96,1013-1018.
Anseth,K.S.,Kline,L.M.,Walker,T.A.,Anderson,K.J.,Bowman,C.N.,1995.Reaction kinetics and volume relaxation during polymerizations of multiethylene glycol dimethacrylates,Macromolecules.28,2491-2499.
Binder,J.B.,Raines,R.T.,2009.Simple chemical transformation of lignocellulosic biomass into furans for fuels and chemicals,J.Am.Chem.Soc.131,1979-1985.
Bryson,R.L.,Hatfield,G.R.,Early,T.A.,Palmer,A.R.,Maciel,G.E.,1983.Carbon-13NMR studies of solid phenolic resins using cross polarization and magic-angle spinning,Macromolecules.16,1669-1672.
Cai,S.X.,Lin,C.H.,2005.Flame‐retardant epoxy resins with high glass‐transition temperatures from a novel trifunctional curing agent:Dopotriol,Journal of Polymer Science Part A:Polymer Chemistry.43,2862-2873.
Cheng,S.,Wilks,C.,Yuan,Z.,Leitch,M.,Xu,C.C.,2012.Hydrothermal degradation of alkali lignin to bio-phenolic compounds in sub/supercritical ethanol and water–ethanol co-solvent,Polym.Degrad.Stab.97,839-848.
Cheng,S.,Yuan,Z.,Leitchb,M.,Anderson,M.,Xu,C.C.,2013.Highly efficient de-polymerization of organosolv lignin using a catalytic hydrothermal process and production of phenolic resins/adhesives with the depolymerized lignin as a substitute for phenol at a high substitution ratio,Ind.Crop.Prod.44,315-322.
da Silva Santos,R.,de Souza,A.A.,De Paoli,M.,de Souza,Cleide Maria Leite,2010.Cardanol-formaldehyde thermoset composites reinforced with buriti fibers:Preparation and characterization,Composites Part A:Applied Science and Manufacturing.41,1123-1129.
De Medeiros,E.S.,Agnelli,J.A.M.,Joseph,K.,De Carvalho,L.H.,Mattoso,L.H.C.,2003.Curing behavior of a novolac‐type phenolic resin analyzed by differential scanning calorimetry,J Appl Polym Sci.90,1678-1682.
Dee,S.J.,Bell,A.T.,2011.A Study of the Acid‐Catalyzed Hydrolysis of Cellulose Dissolved in Ionic Liquids and the Factors Influencing the Dehydration of Glucose and the Formation of Humins,ChemSusChem.4,1166-1173.
Dizhbite,T.,Zakis,G.,Kizima,A.,Lazareva,E.,Rossinskaya,G.,Jurkjane,V.,Telysheva,G.,Viesturs,U.,1999.Lignin—a useful bioresource for the production of sorption-active materials,Bioresour.Technol.67,221-228.
Effendi,A.,Gerhauser,H.,Bridgwater,A.V.,2008.Production of renewable phenolic resins by thermochemical conversion of biomass:A review,Renewable and Sustainable Energy Reviews.12,2092-2116.
Elavarasan,P.,Kondamudi,K.,Upadhyayula,S.,2011.Kinetics of phenol alkylation with tert-butyl alcohol using sulfonic acid functional ionic liquid catalysts,Chem.Eng.J.166,340-347.
Fadel,A.,Yefsah,R.,Salaun,J.,1987.Anhydrous ferric chloride dispersed on silica gel.IV[1–3]:A catalyst for alkylation of aromatic compounds in dry medium,Reactive Polymers,Ion Exchangers,Sorbents.6,93-97.
Ferhan,M.,Yan,N.,Sain,M.,2013.A New Method for Demethylation of Lignin from Woody Biomass using Biophysical Methods,J.Chem.Eng.Process.Technol.4,160.
George,J.,Sreekala,M.S.,Thomas,S.,2001.A review on interface modification and characterization of natural fiber reinforced plastic composites,Polym.Eng.Sci.41,1471-1485.
Girisuta,B.,Janssen,L.,Heeres,H.,2006.Green chemicals:A kinetic study on the conversion of glucose to levulinic acid,Chem.Eng.Res.Design.84,339-349.
Grenier-Loustalot,M.F.,Larroque,S.,Grenier,P.,Bedel,D.,1996.Phenolic resins:4.Self-condensation of methylolphenols in formaldehyde-free media,Polymer.37,955-964.
Hahnenstein,I.,Hasse,H.,Kreiter,C.G.,Maurer,G.,1994.1H-and13C-NMR-Spectroscopic Study of Chemical Equilibria in Solutions of Formaldehyde in Water,Deuterium Oxide,and Methanol,Ind Eng Chem Res.33,1022-1029.
Hassan,A.,Rahman,N.A.,Yahya,R.,2011.Extrusion and injection-molding of glass fiber/MAPP/polypropylene:effect of coupling agent on DSC,DMA and mechanical properties,J.Reinf.Plast.Compos.30,1223-1232.
Hatfield,G.R.,Maciel,G.E.,1987.Solid-state NMR study of the hexamethylenetetramine curing of phenolic resins,Macromolecules.20,608-615.
Holbery,J.,Houston,D.,2006.Natural-fiber-reinforced polymer composites in automotive applications,JOM.58,80-86.
Hu,X.,Lievens,C.,Larcher,A.,Li,C.,2011.Reaction pathways of glucose during esterification:Effects of reaction parameters on the formation of humin type polymers,Bioresour.Technol.102,10104-10113.
Huber,G.W.,Iborra,S.,Corma,A.,2006.Synthesis of transportation fuels from biomass:chemistry,catalysts,and engineering,Chem.Rev.106,4044-4098.
Jin,Y.,Cheng,X.,Zheng,Z.,2010.Preparation and characterization of phenol–formaldehyde adhesives modified with enzymatic hydrolysis lignin,Bioresour.Technol.101,2046-2048.
Knop,A.,Pilato,L.A.,1985.Phenolic resins:chemistry,applications,and performance:future directions.Springer-Verlag Berlin.
Kowatsch,S.,2010.Formaldehyde,Anonymous Springer,第25-40页。
Kurple,K.R.,1989.Foundry resins.
Lee,Y.K.,Kim,D.J.,Kim,H.J.,Hwang,T.S.,Rafailovich,M.,Sokolov,J.,2003.Activation energy and curing behavior of resol‐and novolac‐type phenolic resins by differential scanning calorimetry and thermogravimetric analysis,J Appl Polym Sci.89,2589-2596.
Li,C.,Zhang,Z.,Zhao,Z.K.,2009.Direct conversion of glucose and cellulose to 5-hydroxymethylfurfural in ionic liquid under microwave irradiation,Tetrahedron Lett.50,5403-5405.
Lim,A.S.C.,Solomon,D.H.,Zhang,X.,1999.Chemistry of novolac resins.X.Polymerization studies of HMTA and strategically synthesized model compounds,J.Appl.Polym.Sci.37,1347-1355.
Long,D.H.,Zhang,J.,Yang,J.H.,Hu,Z.J.,Li,T.Q.,Cheng,G.,Zhang,R.,Ling,L.C.,2008.Preparation and microstructure control of carbon aerogels produced using m-cresol mediated sol-gel polymerization of phenol and furfural,New Carbon Materials.23,165-170.
Lytle,C.,Bertsch,W.,McKinley,M.,1998.Determination of novolac resin thermal decomposition products by pyrolysis-gas chromatography-mass spectrometry,J.Anal.Appl.Pyrolysis.45,121-131.
Markovic,S.,Dunjic,B.,Zlatanic,A.,Djonlagic,J.,2001.Dynamic mechanical analysis study of the curing of phenol‐formaldehyde novolac resins,J.Appl.Polym.Sci.81,1902-1913.
Moubarik,A.,Pizzi,A.,Allal,A.,Charrier,F.,Charrier,B.,2009.Cornstarch and tannin in phenol–formaldehyde resins for plywood production,Industrial Crops and Products.30,188-193.
Mouritz,A.P.,Leong,K.H.,Herszberg,I.,1997.A review of the effect of stitching on the in-plane mechanical properties of fibre-reinforced polymer composites,Compos.Part A-Appl.S.28,979-991.
Mwaikambo,L.,Ansell,M.,2001.Cure characteristics of alkali catalysed cashew nut shell liquid-formaldehyde resin,J.Mater.Sci.36,3693-3698.
Netravali,A.N.,Chabba,S.,2003.Composites get greener,Materials today.6,22-29.
Nielsen,A.T.,Moore,D.W.,Ogan,M.D.,Atkins,R.L.,1979.Structure and chemistry of the aldehyde ammonias.3.Formaldehyde-ammonia reaction.1,3,5-Hexahydrotriazine,J.Org.Chem.44,1678-1684.
Ottenbourgs,B.,Adriaensens,P.,Carleer,R.,Vanderzande,D.,Gelan,J.,1998.Quantitative carbon-13 solid-state nmr and FT-Raman spectroscopy in novolac resins,Polymer.39,5293-5300.
Ottenbourgs,B.T.,Adriaensens,P.J.,Reekmans,B.J.,Carleer,R.A.,Vanderzande,D.J.,Gelan,J.M.,1995.Development and optimization of fast quantitative carbon-13 NMR characterization methods of novolac resins,Ind.Eng.Chem.Res.34,1364-1370.
Pérez,J.,Rodríguez,F.,Alonso,M.,Oliet,M.,2011.Time–temperature–transformation cure diagrams of phenol–formaldehyde and lignin–phenol–formaldehyde novolac resins,J Appl Polym Sci.119,2275-2282.
Pilato,L.,2010.Phenolic resins:A century of progress.Springer Verlag.
Richmond,H.,Myers,G.,Wright,G.F.,1948.The Reaction between Formaldehyde and Ammonia,J.Am.Chem.Soc.70,3659-3664.
Sarkanen,S.,Teller,D.C.,Hall,J.,McCarthy,J.L.,1981.Lignin.18.Associative effects among organosolv lignin components,Macromolecules.14,426-434.
Sergeev,V.,Shitikov,V.,Nechaev,A.,Chizhova,N.,KUDRYAVSTEVA,N.,1995.Hydroxymethyl derivatives of phenols as curing agents for novolacs,Polymer science.Series B.37,273-276.
Shibata,S.,Cao,Y.,Fukumoto,I.,2008.Flexural modulus of the unidirectional and random composites made from biodegradable resin and bamboo and kenaf fibres,Compos.Part A-Appl.S.39,640-646.
Simitzis,J.,Karagiannis,K.,Zoumpoulakis,L.,1996.Influence of biomass on the curing of novolac-composites,European polymer journal.32,857-863.
Suharty,N.S.,Wirjosentono,B.,Firdaus,M.,Handayani,D.S.,Sholikhah,J.,Maharani,Y.A.,2008.Synthesis of degradable bio-composites based on recycle polypropylene filled with bamboo powder using a reactive process,J.Physi.Sci.19,105-115.
Tejado,A.,Pena,C.,Labidi,J.,Echeverria,J.,Mondragon,I.,2007.Physico-chemical characterization of lignins from different sources for use in phenol–formaldehyde resin synthesis,Bioresour.Technol.98,1655-1663.
Wu,D.,Fu,R.,2006.Synthesis of organic and carbon aerogels from phenol–furfural by two-step polymerization,Microporous and mesoporous materials.96,115-120.
Wan,J.,Wang,S.,Li,C.,Zhou,D.,Chen,J.,Liu,Z.,Yu,L.,Fan,H.,Li,B.G.,2011.Effect of molecular weight and molecular weight distribution on cure reaction of novolac with hexamethylenetetramine and properties of related composites,Thermochim.Acta.530,32-41.
Wang,M.,Leitch,M.,Xu,C.,2009.Synthesis of phenol–formaldehyde resol resins using organosolv pine lignins,Eur.Polym.J.45,3380-3388.
Yan,H.,Yang,Y.,Tong,D.,Xiang,X.,Hu,C.,2009.Catalytic conversion of glucose to 5-hydroxymethylfurfural over SO42-/ZrO2and SO42-/ZrO2–Al2O3solid acid catalysts,Catalysis Communications.10,1558-1563.
Yuan,Z.,Browne,T.C.,Zhang,X.,2011.Biomass fractionation process for bioproducts,美国专利US20110143411A1.
Yuan,Z.,Xu,C.C.,Cheng,S.,Leitch,M.,2011.Catalytic conversion of glucose to 5-hydroxymethyl furfural using inexpensive co-catalysts and solvents,Carbohydr.Res.346,2019-2023.
Yuan,Z.,Zhang,Y.,Xu,C.,2014.Synthesis and Thermomechanical Property Study of Novolac Phenol-Hydroxymethyl Furfural(PHMF)Resin,RSC Adv.4,31829-31835.
Zakzeski,J.,Bruijnincx,P.C.A.,Jongerius,A.L.,Weckhuysen,B.M.,2010.The catalytic valorization of lignin for the production of renewable chemicals,Chem.Rev.110,3552.
Zhang,L.,Steinmaus,C.,Eastmond,D.A.,Xin,X.K.,Smith,M.T.,2009.Formaldehyde exposure and leukemia:a new meta-analysis and potential mechanisms,Mutation Research/Reviews in Mutation Research.681,150-168.
Zhang,X.,Potter,A.C.,Solomon,D.H.,1998.The chemistry of novolac resins:Part 7.Reactions of para-hydroxybenzylamineintermediates,Polymer.39,1957-1966.
Zhang,X.,Looney,M.G.,Solomon,D.H.,Whittaker,A.K.,1997.The chemistry of novolac resins:3.13C and 15N n.m.r.studies of curing with hexamethylenetetramine,Polymer.38,5835-5848.
Zhao,H.,Holladay,J.E.,Brown,H.,Zhang,Z.C.,2007.Metal chlorides in ionic liquid solvents convert sugars to5-hydroxymethylfurfural,Science.316,1597-1600.
Zhu,J.,Chandrashekhara,K.,Flanigan,V.,Kapila,S.,2004.Curing and mechanical characterization of a soy‐based epoxy resin system,J.Appl.Polym.Sci.91,3513-3518.
- 还没有人留言评论。精彩留言会获得点赞!