聚合物共混物组合物及制备方法与流程
本发明是在以由美国能源部授予的基本合同号DE-AC05-00OR22725的政府支持下完成的。政府在本发明中具有某些权利。
发明领域
本发明总体上涉及聚合物共混物(polymer blend)和复合组合物(composite composition),并且更具体地,涉及这样的在如抗拉强度和韧性这样的性质方面具有有用特性的组合物。
发明背景
当今可用的大量结构材料要么以良好的机械(即抗拉)强度为特征,要么以良好的伸长率(韧性)为特征,但是通常不具有导致鲁棒(robust)或坚固(rugged)(即,既坚固且坚韧)的材料的这两种特性的组合。具有这样的改善的物理特性的材料在大量应用中将是有用的和有利的,所述应用包括在关键结构应用和耐冲击应用,其中遇到高负荷或突然的机械应力。在这样的应用中,具有高抗拉强度但低韧性的材料由于它们的脆性而易于失效。具有高抗拉强度连同改进的韧性的材料大大地较不易于发生这样的失效。
发明概述
本公开内容涉及可用作用于大量应用的工业塑料树脂和结构材料的高性能聚合物共混物和它们的复合物。本文中描述的聚合物共混物的特征在于有益的机械性质(例如,高强度和韧性)的组合,这使得它们特别可用在其中遇到高负荷或机械应力的关键结构应用中。在许多实施方案中,本文中描述的聚合物共混物是热塑性的,这有利地向它们提供足够程度的可模制性(成型性,moldability)、弹性、可再循环性(可回收性,recyclability)和/或延展性,以将它们模制成各种各样的有用形状。
在一些实施方案中,聚合物共混物材料包括:(i)第一聚合物,所述第一聚合物含有其中至少一个氢原子结合至选自氧、氮和硫的杂原子的氢键供给基团(hydrogen bond donating group),(例如,羟基、胺、酰胺、硫醇、羧基、磺酸和膦酸基团);或者第一聚合物的阴离子形式(阴离子形式的第一聚合物,anionic version of the first polymer),其中至少一部分的结合至杂原子的氢原子不存在(absent)并且被至少一个电子对代替;(ii)第二聚合物,所述第二聚合物含有氢键接受基团(hydrogen bond accepting group),如选自腈、卤素和醚官能团的那些;和(iii)至少一种改性剂,所述改性剂选自碳粒子(carbon particle)、含醚的聚合物和路易斯酸化合物(Lewis acid compound)。在聚合物共混物材料中,如果第二聚合物含有醚官能团,则所述至少一种改性剂选自碳粒子和路易斯酸化合物。通常,所述路易斯酸化合物在性质上是非聚合物的。
本公开内容还涉及用于制备上述聚合物共混物材料或复合物中的任一种的方法。在特别的实施方案中,所述方法包括将包含组分(i)、(ii)和(iii)的混合物均匀地共混。所述方法可以还包括模制过程,其可以包括本领域已知的成形(shaping)、加热和/或压制(pressing)过程中的任一个,以制备所述聚合物共混物材料的成形制品。
附图简述
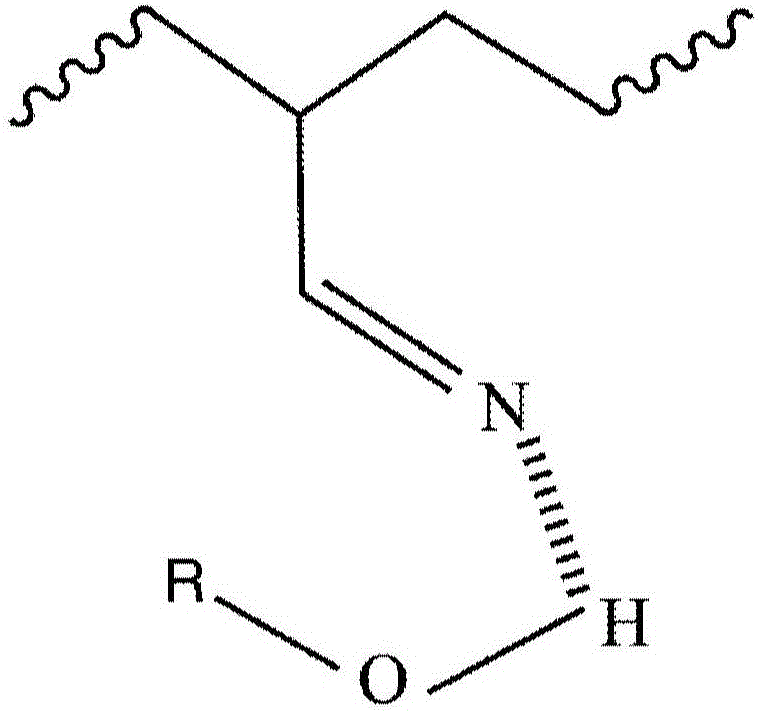
图1A、1B:显示氢键合(图1A)和配价(dative)键合(图1B)位点的代表性化学结构。
图2:对于含有炭黑、硼酸并且在过氧化二异丙苯的存在下交联的软木木质素-天然橡胶和软木木质素-丁腈橡胶共混物的代表性动态力学储存模量(dynamic mechanical storage modulus)和损耗角正切谱(loss tangent spectra)。
图3A(上图、下图)、3B(上图、下图):对于图3A:多至1000%的应变的硬木(HW)木质素-PEO共混物的代表性拉伸应力-应变分布图(图3A,上图),连同多至60%的应变轴的放大部分(图3A,下图)。对于图3B:在有和没有结合硼酸(BA)的情况下,多至1000%的应变的软木(SW)木质素-PEO共混物的代表性拉伸应力-应变分布图,以及仅对于PEO的对照曲线(图3B,上图),连同多至200%的应变轴的放大部分(图3B,下图)。
图4A、4B:在有或没有没有DCP的情况下,具有PEO和/或BA的硬木(HW)木质素-NBR共混物(图4A)和具有PEO和/或BA的软木(SW)木质素-NBR共混物(图4B)的代表性拉伸应力-应变分布图,以及在有或没有DCP的情况下,对于仅含有木质素和NBR的共混物的对照曲线(其中“NBR”指的是丁腈橡胶,且“DCP”指的是过氧化二异丙苯)。
图5A(上图、下图)、5B(上图、下图):对于图5A:多至600%的应变的具有PEO和/或BA的软木(SW)木质素-NBR-CB-DCP共混物的代表性拉伸应力-应变数据(图5A,上图),连同多至100%的应变轴的放大部分(图5A,下图)。对于图5B:多至1250%的应变的具有PEO和/或BA的硬木(HW)木质素-NBR-CB-DCP共混物的代表性拉伸应力-应变数据(图5B,上图),连同多至200%的应变轴的放大部分(图5B,下图)。在每个图中还包括了对于仅含有木质素-NBR-CB-DCP的共混物的对照曲线,以及在有或没有DCP的情况下,对于仅含有木质素和NBR的对照曲线。
图6A、6B:分别是对于基于软木(图6A)和硬木(图6B)木质素的组合物,对于木质素//NBR/CB/BA/DCP/PEO的共混物在可变的PEO负载(如表8中提供的)下的代表性应力-应变分布图。
发明详述
在第一方面,本公开内容涉及一种聚合物共混物材料(聚合物共混材料,polymer blend material),其包含:(i)第一聚合物,所述第一聚合物含有氢键供给基团,如选自羟基、胺、硫醇、羧基、磺酸和膦酸基团或它们的组合的那些;(ii)第二聚合物,所述第二聚合物含有氢键接受基团,如选自腈、卤素和醚官能团或它们的组合的那些;和(iii)至少一种改性剂,所述改性剂选自碳粒子、含醚聚合物和路易斯酸化合物。所述氢键供给基团含有至少一个结合至杂原子(即,不同于碳,如氧、硫或氮)的氢原子,使得该氢原子可以参与与氢键接受基团的氢键合相互作用。氢键接受基团通常不含有结合至杂原子的氢原子。如本文所使用的,术语“聚合物”通常指的是具有至少或大于5、10、15、20、30、40或50个连接的单体单元的分子,并且可以是均聚物或共聚物,其中共聚物可以是,例如,嵌段、无规、交替、接枝或支化的共聚物。
在第一实施方案中,含有氢键供给基团的第一聚合物与含有氢键接受基团的第二聚合物经由在第一聚合物的氢原子与第二聚合物的氢接收基团之间的氢键相互作用接合(engage)。对于含羟基的第一聚合物和含腈的第二聚合物的情形,图1A中描绘了代表性的氢键相互作用。在第二实施方案中,含有氢键供给基团的第一聚合物与含有氢键接受基团的第二聚合物经由配价键合相互作用接合。配价键,也称为偶极键,是由于分子的富电子区域(即,在一个或多个原子上的一个或多个电子对,如由路易斯碱提供的)与另一个分子的贫电子区域之间的相互作用所致形成的共价键。同样对于含羟基的第一聚合物和含腈的第二聚合物的情形,图1B中描绘了代表性的配价键相互作用。值得注意的是,对条件的某些调节可以促使第一聚合物与第二聚合物要么通过氢键要么通过配价键相互作用而相互作用。可以改变相互作用类型的一种具体条件是pH。例如,第一聚合物中的羧酸和酚基团可以部分地或完全地通过与碱(例如,金属氢氧化物,如氢氧化钠)相互作用而去质子化,在此情况下,去质子化的羧酸(即,羧酸盐)或去质子化的苯酚(即,酚盐)通过配价键合相互作用部分地或排他地与第二聚合物接合。因此,出于前述考虑,在一些实施方案中,术语“氢键供给基团”可以是指不具有能够在氢键合相互作用中接合的氢原子的基团,而是包含阴离子基团,所述阴离子基团具有一个或多个能够在配价键合相互作用中接合的电子对。在其他实施方案中,可以包含质子酸(例如,强或弱的无机或有机酸,如HCl或乙酸),以确保占优势的氢键合相互作用。一些官能团(例如,醇或胺)可以交替地或同时地通过氢键合和/或配价键合相互作用而相互作用。
在一个实施方案中,术语“第一聚合物”和“第二聚合物”指的是单独(分开,separate)的化合物。在含有选自第一和第二聚合物两者的官能团的单一聚合物(例如,丙烯腈-乙烯醇共聚物,如在美国专利3,388,199中描述的)的情况下,可以将该单一聚合物看作第一或第二聚合物(即,组分(i)或(ii)),但是不充当第一和第二聚合物两者。特别地,因为经常使用PAN-丙烯酸共聚物代替纯的PAN聚合物用于类似应用中,所以本公开内容通常将PAN-丙烯酸聚合物视为属于第一或第二聚合物而不充当第一和第二聚合物两者,尽管本公开内容可以包括PAN-丙烯酸充当第一和第二聚合物两者的可能性。在一些实施方案中,第二聚合物不含有上述任一种氢键供给基团的一个或多个,或完全不含有任何氢键供给基团,和/或第一聚合物不含有上述任一种氢键接受基团的一个或多个,或完全不含有任何氢键接受基团。在另一个实施方案中,含有选自第一和第二聚合物两者的官能团的单一聚合物可以充当组分(i)和(ii)两者。在该后一种实施方案中,聚合物共混物可以仅包含两种组分,即,与组分(iii)的改性剂组合的含有选自第一和第二聚合物两者的官能团的单一聚合物(例如,丙烯腈-乙烯醇共聚物)。因此,聚合物共混物包括单独的第一和第二聚合物(即,单独的组分i和ii)的可能性,或充当组分i和ii两者的单一聚合物的可能性,其中,在聚合物共混物中,要么两种单独的聚合物要么单一的聚合物与至少一种改性剂(组分iii)组合。在聚合物不具有重叠的组分(i)和(ii)的官能团(即,第一聚合物不具有任何对于第二聚合物来说可接受的氢键接受基团,或第二聚合物不具有任何氢键供给基团)的情况下,聚合物共混物不包括将组分(iii)在不存在第二聚合物的情况下与第一聚合物组合的可能性,或将组分(iii)在没有第一聚合物的情况下与第二聚合物组合的可能性。,氢键供给基团和氢键接受基团两者都需要存在于要么单独的聚合物中要么单一聚合物中,以正确地充当组分(i)和(ii)。
(i)和/或(ii)的聚合物可以独立地具有宽范围的重均分子量(Mw)的任一个,如精确地、大约、至少、高于、多至或少于,例如,10,000,000g/mol,5,000,000g/mol,1,000,000g/mol,500,000g/mol,400,000g/mol,300,000g/mol,200,000g/mol,100,000g/mol,50,000g/mol,10,000g/mol,5,000g/mol,2,500g/mol,2,000g/mol,1,500g/mol,1,000g/mol,500g/mol或250g/mol,或者在由前述示例值中的任意两个所界定的范围内。聚合物也可以独立地具有宽范围的数均分子量Mn的任一个,其中n可以对应于上文对于Mw提供的数中的任一个,以及,例如5、10、20、50、100或200,并且其中Mn可以对应于上文提供的Mw值中的任一个或其中的范围。
含有羟基(OH)基团的聚合物可以具有结合至脂族(即,非芳族,例如,烷基或烯基)或芳族基团的羟基。在一些实施方案中,羟基至少部分地或仅结合至脂族基团,而在其他实施方案中,羟基至少部分地或仅结合至芳族(例如,苯基)基团,而仍然在其他实施方案中,羟基至少部分地或仅结合至脂族和芳族基团两者。含羟基的聚合物的一些实例包括聚乙烯醇、多糖(例如,纤维素、半纤维素、淀粉、葡聚糖、几丁质、壳聚糖和果胶)、含羟基的乙烯基加成聚合物(例如,聚(丙烯酸2-羟乙酯))、含羟基的聚酰亚胺和含苯酚的聚合物,如木质素、丹宁酸、聚(乙烯基苯酚)、聚(苯乙烯-共-烯丙基醇)、酚醛树脂、酚醛清漆和甲阶段酚醛树脂(resole)。在一些实施方案中,含羟基的聚合物仅含有羟基官能团,如附接到烃主链,而在其他实施方案中,含羟基的聚合物包括不同于羟基的官能团,如醚基团、羧基基团或氨基基团。以上描述的所有含羟基的聚合物都是本领域熟知的。在一个实施方案中,如果含羟基的聚合物不含有氢键接受基团(或仅含有羟基官能团),则它可以仅起到第一聚合物(组分i)的作用。在另一个实施方案中,如果含羟基的聚合物含有氢键接受基团(例如,腈或醚基团),则它也可以(即,另外)起到第二聚合物(组分ii)的作用。
在特别的实施方案中,含羟基的聚合物是木质素。木质素可以是广泛多种自然中存在的或本领域已知的木质素组合物中的任一种。如本领域已知的,自然中存在的木质素组合物通常不是均匀的。木质素是无规聚合物,其在植物物种之间显示明显的组成上的变化。许多其他条件,如环境条件、年龄和加工的方法,都影响木质素组成。木质素主要在三种醇单元即对香豆基醇(p-coumaryl alcohol)、愈创木基醇和芥子醇的比率上有区别。香豆基醇、愈创木基醇和芥子醇的聚合作用分别形成木质素聚合物的对羟基苯基(H)、愈创木基(G)和丁香基(S)组分。前体木质素可以具有广泛多种的H、G和S组分的相对重量%(wt%)的任一种。如在一些种子中观察到的,木质素也可以由咖啡醇(caffeyl alcohol)单元组成,例如,Chen等,PNAS,109(5),1772-1777(2012)。例如,前体木质素可以含有(对于每种组分来说独立地)至少、多至或少于1重量%、2重量%、5重量%、10重量%、20重量%、30重量%、40重量%、50重量%、60重量%、70重量%、80重量%或90重量%或其范围的咖啡醇、H、G和S组分中的任一种。通常,每种醇组分的重量%的和是100%,或者如果考虑其他次要组分,为至少98%。不同的木材和植物来源(例如,硬木、软木、柳枝稷和甘蔗渣)在它们的木质素组成上经常是大大不同的。
除了木质素的天然变化之外,还存在基于木质素被加工的方式的在组成上的变化。例如,前体木质素可以是Kraft木质素、亚硫酸木质素(即,木质素磺酸盐)或无硫木质素。如本领域已知的,Kraft木质素指的是由Kraft过程得到的木质素。在Kraft过程中,将氢氧化钠和硫化钠的组合(称为“白液”)与在生物质中存在的木质素反应,以形成暗色的带有硫醇基的木质素。Kraft木质素通常是具有高浓度的酚基团的水和溶剂不可溶的材料。通常可以在碱性水溶液中使它们可溶。还如本领域已知的,亚硫酸木质素指的是由亚硫酸盐过程得到的木质素。在亚硫酸盐过程中,将亚硫酸盐或亚硫酸氢盐(取决于pH)连同抗衡粒子与木质素反应,以形成带有磺酸酯(SO3H)基团的木质素。该磺酸酯基团赋予亚硫酸木质素相当大程度的水溶性。存在若干类型的本领域已知的无硫木质素,包括从生物质转化技术(如在乙醇生产中使用的那些)、溶剂制浆(即,“有机溶剂(organosolv)”过程)和碱法制浆获得的木质素。特别地,有机溶剂木质素是通过从木质纤维来源(如切碎的木头)溶剂萃取随后沉淀而获得的。由于在制备有机溶剂木质素中使用的明显较温和的条件(即,相对于Kraft和亚硫酸盐过程),有机溶剂木质素通常更纯、较少降解,并且通常比Kraft和亚硫酸盐木质素具有更窄的分子量分布。也可以将这些木质素热除去挥发成分,以制备具有较少脂族羟基的变体,以及具有升高的软化点的分子重构的形式。前述木质素类型中的任意一种或多种可以使用(或排除)作为本文所述的方法中的组分用于制备聚合物共混物。
木质素也可以是具有特定的或优化的H、G和S组分的比率的木质素的改造形式。可以通过例如引起木质素中的化学结构和生物质中的总木质素含量变化的本领域已知的转基因和重组DNA方法来改造木质素(例如,F.Chen等,Nature Biotechnology,25(7),第759-761页(2007)和A.M.Anterola等,Phytochemistry,61,第221-294页(2002))。木质素的改造特别涉及改变木质素的G和S组分的比率(D.Guo等,The Plant Cell,13,第73-88页,(2001年1月)。特别地,木材制浆动力学研究显示,S/G比率的增加明显增强木质素移除的速率(L.Li等,Proceedings of The National Academy of Sciences of The United States of America,100(8),第4939-4944页(2003))。S单元变得与两种木质醇(lignol)单体共价连接;另一方面,G单元连接到三个其他单元。这样,增加的G含量(降低S/G比率)通常产生具有更多C-C键合的高度支化的木质素结构。相反,增加的S含量通常导致更多的β-芳基醚(β-O-4)连接,其容易在化学脱木质素化期间(例如,如在Kraft制浆过程中)断裂(与C-C键相比)。已经证实,减少木质素含量和改变S/G比率提高可生物转化能力和脱木质素化脱木质素化。因此,对于脱木质素化可以使用较不苛刻和损害性的条件(即,与使用强酸或碱的当前实践相比),这将提供更好适用于更高附加价值的应用的更加改进的木质素,所述应用包括韧性聚合物共混物的制造、碳材料制备(例如碳纤维、碳粉、活性炭、微孔和介孔碳)以及芳烃原料的热解或催化制备。
已经研究了留下高木质素含量残余物的实验室规模的生物质发酵(S.D.Brown等,Applied Biochemistry and Biotechnology,137,第663-674页(2007))。这些残余物将含有具有取决于生物质来源(例如,木材物种、草和秸秆)变化的分子结构的木质素。从这些高品质木质素生产附加价值的产品会大大改进生物精炼厂的总体运行成本。已经提出了多种化学路径以从木质素获得附加价值的产品(J.E.Holladay等,来自生物质的顶级附加值化学品:第二卷-对来自生物精炼厂木质素的潜在候选者的筛选结果(Top Value-Added Chemicals from Biomass:Volume II-Results of Screening for Potential Candidates from Biorefinery Lignin),DOE Report,PNNL-16983(2007年10月))。
在一些实施方案中,木质素可以是交联的木质素,其是可熔融加工的或经得起熔融加工。“交联的”意指木质素在木质素结构中的苯环碳原子之间含有亚甲基(即,-CH2-)和/或亚乙基(即,-CH2CH2-)连接(即连接基团)。“可熔融加工的”意指可以将交联的木质素熔融或在特定的玻璃化转变温度开始转化成熔融的、高度粘稠的或橡胶状的状态。随后可以更容易地加工熔融的或高度粘稠的木质素,例如通过混合、模制、施加在表面上或溶解在溶剂中。
分离的木质素可以具有为至少300、500、1,000、5,000或10,000g/mol的数均或重均分子量(即,分别为Mn或Mw)。在不同的实施方案中,木质素可以交联至以下程度:它具有精确地、大约、至少或大于例如以下值的数均或重均分子量:10,000g/mol、25,000g/mol、50,000g/mol、75,000g/mol、100,000g/mol、125,000g/mol、150,000g/mol、175,000g/mol或200,000g/mol,或在由前述示例值中的任何两个所界定的范围内的分子量。
交联的木质素的玻璃化转变温度(Tg)通常高于室温(典型地,15、20、25或30℃)。在不同的实施方案中,木质素(得自生物质的分离的木质素,或它的交联的衍生物)具有精确地、大约、至少或大于以下值的玻璃化转变温度:40℃、50℃、60℃、70℃、80℃、90℃、100℃、105℃、110℃、115℃、120℃、125℃、130℃、140℃、150℃、160℃、170℃、180℃、190℃、200℃、210℃、220℃、230℃、240℃或250℃,或在由前述值中的任何两个界定的范围内的Tg。
木质素(处于从生物质分离的原始形式,或它的交联的衍生物)优选基本上可溶于极性有机溶剂或碱性水溶液中。如本文所使用的,术语“基本上可溶”通常表明至少1、2、5、10、20、30、40、50或60克的木质素完全溶解在1公升(100mL)的极性有机溶剂或碱性水溶液中。在其他实施方案中,溶解性表示为溶液中木质素的重量%。在特别的实施方案中,木质素具有足够的溶解性,以制备在极性有机溶剂或碱性水溶液中至少5重量%、10重量%、15重量%、20重量%、30重量%、40重量%或50重量%的溶液。极性有机溶剂可以是非质子的或质子的。极性非质子溶剂的一些实例包括有机醚(例如,二乙醚、四氢呋喃和二烷)、腈(例如,乙腈、丙腈)、亚砜(例如,二甲亚砜)、酰胺(例如,二甲基甲酰胺、N,N-二甲基乙酰胺)、有机氯化物(例如,二氯甲烷、氯仿、1,1,-三氯乙烷)、酮(例如,丙酮、2-丁酮)和二烷基碳酸酯(例如,碳酸乙二酯、碳酸二甲酯、碳酸二乙酯)。极性有机质子溶剂的一些实例包括醇(例如,甲醇、乙醇、异丙醇、正丁醇、叔丁醇、戊醇、己醇、辛醇等)、二醇(例如,乙二醇、二甘醇、三甘醇)和质子胺(例如,乙二胺、乙醇胺、二乙醇胺和三乙醇胺)。碱性水溶液可以是具有至少(或高于)8、9、10、11、12或13的pH的任何含水溶液。碱化溶质(alkalizing solute)可以是,例如,碱氢氧化物(例如,NaOH或KOH)、氨或氢氧化铵。也可以使用任何这些溶剂的组合。在一些实施方案中,可以排除一种或多种种类或特定类型的溶剂。
含有其他氢键供给基团如胺(-NH2或-NHR,其中R是烃)、酰胺(-C(O)NH2或-C(O)NHR,其中R是烃)、硫醇(-SH)、羧基(-COOH)、磺酸(-SO3H)、磺酰胺(-SO2NH2)和膦酸(-PO3H2)基团的聚合物可以具有结合至脂族(即,非芳族,例如,烷基或烯基)或芳族基团的氢键供给基团,如以上对于含羟基聚合物描述的。含胺的聚合物的一些实例包括聚苯胺、聚(乙烯基苯胺)、聚乙烯基胺、聚醚胺和含氨基的聚膦腈。含酰胺的聚合物的实例包括聚丙烯酰胺和聚酰胺(例如,尼龙)。含硫醇的聚合物的一些实例包括聚(乙烯硫醇)、硫醇化壳聚糖(例如,壳聚糖-巯基丁胺(thiobuylamidine))和聚对巯基苯乙烯。含羧基的聚合物的一些实例包括聚丙烯酸、聚甲基丙烯酸、聚(4-乙烯基苯甲酸)、聚马来酸、聚富马酸、聚天冬氨酸和聚谷氨酸。含磺酸的聚合物的一些实例包括聚(乙烯基磺酸)、聚(乙烯基苯甲酸磺酸)(poly(vinylbenzoic sulfonic acid))、聚(2-丙烯酰酰胺基-2-甲基丙烷磺酸)、磺酸化的聚烯烃(例如,美国申请公开号2013/0084455和2013/0214442,将其内容通过引用结合于此)和其他这样的聚合物,其公开在例如美国专利3,230,201和8,445,141中,将其内容通过引用结合于此。含磺酰胺的聚合物的一些实例包括pH-敏感的聚合物和这类的凝胶(例如,通过聚合4-氨基-N-[4,6-二甲基-2-嘧啶基]苯磺酰胺),如在例如,美国专利6,103,865;S.Kang等,Macromolecular Symposia,第172卷,第1期,第149-156页,2001年7月;和S.Y.Park等,Macromolecular Rapid Communications,第20卷,第5期,第269-273页,1999年5月中进一步描述的,将其内容通过引用结合于此。含膦酸的聚合物的一些实例包括通过乙烯基膦酸、亚乙烯基二膦酸、异丙烯基膦酸和2-丙烯酰胺基-2-甲基丙烷膦酸的加成聚合衍生的那些,如在例如美国专利5,534,235和8,637,174中描述的,将其内容通过引用结合于此。所有以上描述的聚合物都是本领域熟知的。在一个实施方案中,含有氢键供给基团的聚合物,如果它不含有氢键接受基团(或仅含有一种或多种类型的氢键供给基团),可以仅起到第一聚合物(组分i)的作用。在另一个实施方案中,含有氢键供给基团的聚合物,如果它含有氢键接受基团(例如,腈或醚基),也(即,另外)可以起到第二聚合物(组分ii)的作用。
如在组分(ii)中提供的,含有腈基团的聚合物可以具有结合至脂族(即,非芳族,例如,烷基或烯基)或芳族基团的腈基团,如以上对于含羟基的聚合物描述的。含腈的聚合物的一些实例包括聚丙烯腈(PAN)、丁腈橡胶(NBR)、丙烯腈丁二烯苯乙烯(ABS)、苯乙烯丙烯腈(SAN)、含有在离子传导材料中的芳族腈基团的聚合物(例如,美国申请公开号2006/0258836)和其他含腈的聚合物,如在E.N.Zil’berman等,Russian Chemical Reviews,第55卷,第1号,1986中描述的那些,或例如在L.Sheng等,Journal of Polymer Science,Part A: Polymer Chemistry,第52卷,第1期,第21-29页,2004年1月中描述的聚(亚芳基醚醚腈),将其内容通过引用结合于此。还有其他含腈的聚合物包括也如本领域熟知的,聚氰基丙烯酸烷基酯,如聚(乙基-2-氰基丙烯酸酯)或聚氰基丙烯酸丁酯,以及本领域熟知的,作为结构粘合剂的任何氰基丙烯酸酯的聚合衍生物。
在特别的实施方案中,含腈的聚合物是PAN或其衍生物或共聚物。在一些实施方案中,含PAN的聚合物是均聚的PAN。在其他实施方案中,含PAN的聚合物是PAN与至少一种非PAN节段或嵌段的共聚物。在这样的共聚物中的PAN可以为主要量(即,大于50摩尔%)、次要量(即,少于50摩尔%)或等量。共聚物可以是,例如,嵌段、无规、交替或接枝的共聚物。非PAN共聚物单元典型地是从本领域已知的用于制备这样的聚合物的任何不饱和(通常为烯烃)单体前体衍生的加成聚合物。在特别的实施方案中,非PAN共聚物单元衍生自不饱和羧酸酯前体分子、不饱和酰胺前体分子或它们的组合。不饱和羧酸酯前体分子通常含有至少一个碳-碳双键和羧酸或羧酸酯基团,其中烯烃基团经常结合至羧酸或羧酸酯基团的羰基碳原子。不饱和羧酸酯前体分子的一些实例包括:丙烯酸甲酯、丙烯酸乙酯、丙烯酸丙酯、丙烯酸丁酯、甲基丙烯酸甲酯、(丙烯酸2-羟乙基酯)、乙酸乙烯酯、丙烯酸、甲基丙烯酸和衣康酸。不饱和酰胺前体分子通常含有至少一个碳-碳双键和酰胺基(其可以是N-取代的或N,N-二取代的),其中烯烃基团经常结合至酰胺基的羰基碳原子。不饱和酰胺前体分子的一些实例包括丙烯酰胺、甲基丙烯酰胺、其N-烷基衍生物和其N,N-二烷基衍生物。
如在组分(ii)中提供的,含有卤素的聚合物可以具有结合至脂族(即,非芳族,例如,烷基或烯基)或芳族基团的卤原子,如以上对于含羟基聚合物描述的。卤原子可以是,例如,氟、氯和溴原子。氟化的聚合物的一些实例包括聚(氟乙烯)、聚(偏二氟乙烯)、聚(四氟乙烯)、氟化的乙烯-丙烯共聚物、聚(乙烯四氟乙烯)、聚(全氟磺酸)和氟弹性体。氯化的聚合物的一些实例包括聚(氯乙烯)、聚偏二氯乙烯、乙烯-氯三氟乙烯共聚物、聚氯丁二烯、卤化的丁基橡胶、氯化的聚乙烯、氯磺酸化的聚乙烯、氯化的聚丙烯、氯化的乙烯-丙烯共聚物和氯化的聚氯乙烯。溴化的聚合物的一些实例包括聚(溴乙烯),和本领域已知的溴化的阻燃剂,如溴化环氧树脂、聚(溴化的丙烯酸酯)、溴化的聚碳酸酯和溴化的多元醇。
如在组分(ii)中提供的,含有醚官能团的聚合物在本文中意指在范围相当于如在组分(iii)中提供的一类“含醚的聚合物”。含醚的聚合物可以是,例如,聚亚烷基氧化物(聚环氧烷烃,polyalkylene oxide)(即,聚乙二醇)或其共聚物。聚亚烷基氧化物的一些实例包括聚环氧乙烷、聚环氧丙烷、聚环氧丁烷和它们的共聚物或与乙烯、丙烯或烯丙基缩水甘油醚的共聚物。含醚的聚合物也可以是,例如,聚乙烯基氰基乙基醚,如在例如美国专利2,341,553中描述的,将其内容通过引用结合于此。含醚的聚合物也可以是,例如,PVA的醚化形式,如聚(乙烯基甲基醚),其可以对应于CAS号9003-09-2。含醚的聚合物也可以是,例如,苯基醚聚合物,其可以是聚苯基醚(PPE)或聚亚苯醚(polyphenylene oxide,PPO)。含醚的聚合物也可以包括环醚基团,如环氧化物或缩水甘油基,或如在例如美国专利4,260,702中进一步描述的,将其内容通过引用结合于此。环醚聚合物可以是环酸酐改性的聚乙烯醇缩醛,如在美国专利6,555,617中进一步描述的,或环状或螺环状聚缩醛醚,如在例如A.G.Pemba等,Polym.Chem.,5,3214-3221(2014)中进一步描述的,将其内容通过引用结合于此。在一些实施方案中,环状或非环状醚基团足以与第一聚合物的氢键供给基团反应以形成与第一聚合物的共价键。在其他实施方案中,环状或非环状醚基团不是足以与第一聚合物的氢键供给基团反应以形成与第一聚合物的共价键。在还是其他的实施方案中,含醚的聚合物可以是环状的或非环状的含硫醚聚合物,如聚苯基硫醚或聚亚苯硫醚。
在第一组实施方案中,含羟基的聚合物与含腈的聚合物、含醚的聚合物或含卤素的聚合物混合或组合。在第二组实施方案中,含胺的聚合物与含腈的聚合物、含醚的聚合物或含卤素的聚合物混合或组合。在第三组实施方案中,含酰胺的聚合物与含腈的聚合物、含醚的聚合物或含卤素的聚合物混合或组合。在第四组实施方案中,含硫醇的聚合物与含腈的聚合物、含醚的聚合物或含卤素的聚合物混合或组合。在第五组实施方案中,含羧基的聚合物与含腈的聚合物、含醚的聚合物或含卤素的聚合物混合或组合。在第六组实施方案中,含磺酸的聚合物与含腈的聚合物、含醚的聚合物或含卤素的聚合物混合或组合。在第七组实施方案中,含膦酸的聚合物与含腈的聚合物、含醚的聚合物或含卤素的聚合物混合或组合。对于本发明的目的,上述聚合物的组合中的任一种起到聚合物组分(i)和(ii)的混合物的作用,其随后与至少一种改性剂(组分iii)混合,以形成聚合物共混物材料。
聚合物组分(i)和/或(ii)中的任一种,或组分(iii)的含醚的聚合物,可以具有任何合适的玻璃化转变温度(Tg),如精确地、大约、至少、高于、多至或少于,例如,-120℃、-100℃、-50℃、0℃、10℃、20℃、30℃、40℃、50℃、60℃、70℃、80℃、90℃、100℃、110℃、120℃、130℃、140℃、150℃、160℃、170℃、180℃、190℃或200℃,或由前述示例值中的任何两个界定的范围内的Tg。所得的聚合物共混物也可以具有选自上文提供的示例值中的任何一个或在由以上示例值的任何两个界定的范围内的Tg。
聚合物组分(i)和(ii)可以以任何合适的按重量计的量(即,作为重量%)存在。在不同的实施方案中,聚合物组分(i)或(ii)的量相对于在最终共混物材料中的组分的总重量独立地为,精确地、大约、至少、高于、多至或少于,例如,1重量%、2重量%、5重量%、10重量%、15重量%、20重量%、25重量%、30重量%、40重量%、50重量%、60重量%、70重量%、80重量%、90重量%、95重量%、98重量%或99重量%,或者为在由前述值中的任何两个界定的范围内的量,其中术语“大约”通常表明距离指示的值不大于±10%、±5%或±1%。可以按组分(i)和(ii)之间的重量比陈述前述量中的任一个。例如,如果组分(i)和(ii)各自的量为相对于共混物材料中的组分的总重量为约50重量%,则(i)和(ii)为约1∶1的重量比。组分(i)和(ii)的组合重量可以为,例如,至少或高于最终共混物材料重量的30、40、50、60、70、80、90 95、98或99重量%,或在其中的范围内。通常,组分(i)、(ii)和(iii)的组合重量构成了聚合物共混物的重量的主体(bulk),即,组分(i)、(ii)和(iii)的组合重量典型地为至少或高于最终共混物材料重量的50、60、70、80、90 95、98或99重量%。
聚合物组分(i)和(ii)与至少一种选自碳粒子、含醚的聚合物和路易斯酸化合物的改性剂(组分iii)混合,条件是,如果第二聚合物(组分ii)或充当组分(i)和(ii)的单一聚合物含有醚官能团,则该至少一种改性剂(组分iii)选自碳粒子和路易斯酸化合物中的一种或两者(即,不是含醚的聚合物)。在组分(i)的聚合物除氢键供给基团之外还含有醚基团的情况下,则如果组分(ii)不包括含醚的聚合物,组分(iii)可以是含醚的聚合物;其中,如果组分(iii)是含醚的聚合物,则它是与组分(i)中的含醚聚合物不相同的,并且优选不含有氢键供给基团。改性剂起到有利地改变聚合物共混物材料的物理性质的作用,通常是通过改进聚合物共混物材料的极限拉伸(即,韧性)特性。在第一实施方案中,仅选择碳粒子作为改性剂(即,排除含醚的聚合物和路易斯酸化合物)。在第二实施方案中,仅选择含醚的聚合物作为改性剂(即,排除碳粒子和路易斯酸化合物)。在该后一种实施方案中,组分(ii)的聚合物选自不同于含醚的聚合物,而组分(i)的聚合物可以包括或可以不包括醚基团连同氢键供给基团。在第三实施方案中,仅选择路易斯酸化合物作为改性剂(即,排除碳粒子和含醚聚合物)。在第四实施方案中,选择碳粒子与含醚的聚合物的组合作为改性剂。在第五实施方案中,选择碳粒子与路易斯酸化合物的组合作为改性剂。在第六实施方案中,选择含醚的聚合物与路易斯酸化合物的组合作为改性剂。在第七实施方案中,选择碳粒子、含醚的聚合物与路易斯酸化合物的组合作为改性剂。
碳粒子可以是至少部分或全部由元素碳构成的现有技术中已知的碳粒子中的任何一种,并且可以是导电的,半导电的或不导电的。碳粒子可以是纳米粒子(例如,至少1、2、5或10nm,且多至20、50、100、200或500nm)、微粒子(例如,至少1、2、5或10μm,且多至20、50、100、200或500μm)或大粒子(例如,高于500μm或至少或多至1、2、5、10、20、50或100mm)。碳粒子的一些实例包括炭黑(“CB”)、碳洋葱(carbon onion)(“CO”)、球形富勒烯(例如,巴基球(buckminsterfullerene),即,C60,以及任何更小或更大的巴基球如C20或C70),管状富勒烯(例如,单壁的、双壁的或多壁的碳纳米管)、碳纳米金刚石和碳纳米芽(nanobud),它们全部具有本领域熟知的组成和物理及电学性质。如本领域已知的,完全石墨化的碳纳米金刚石可以认为是碳洋葱。
在一些实施方案中,碳粒子排他地由碳制成,而在其他实施方案中,碳粒子可以包括一定量的非碳非氢(即杂-掺杂剂)元素中的一种或组合,如氮、氧、硫、硼、硅、磷,或金属,如碱金属(例如,锂)、碱土金属、过渡金属、主族金属(例如,Al、Ga或In)、或稀土金属。二元碳组合物的一些实例包括碳化硅(SiC)和碳化钨(WC)。杂元素的量可以是小量(例如,多至0.1、0.5、1、2或5重量%或摩尔%)或更显著的量(例如,大约、至少或多至10、15、20、25、30、40或50重量%或摩尔%)。在一些实施方案中,从碳粒子中排除碳粒子的具体提及的种类或具体类型中的任何一种或多种,或杂-掺杂剂元素的具体提及的种类或具体类型中的任何一种或多种。
在一些实施方案中,碳粒子可以是本领域已知的高强度碳纤维组合物中的任一种。如本领域已知的,碳纤维具有长于其宽度大小的长度大小。碳纤维组合物的一些实例包括通过热解聚丙烯腈(PAN)、黏胶纤维(viscose)、人造纤维(rayon)、沥青(pitch)、木质素、聚烯烃制备的那些,以及气相生长的碳纳米纤维、单壁的和多壁的碳纳米管,这些中的任何一种可以是或可以不是杂原子掺杂的,如利用氮、硼、氧、硫或磷掺杂的。碳粒子也可以是二维碳材料,如石墨烯、氧化石墨烯、或石墨烯纳米带,它们可以衍生自例如天然石墨、碳纤维、碳纳米纤维、单壁碳纳米管和多壁碳纳米管。碳纤维典型地具有高的抗拉强度,如至少500、1000、2000、3000、5000、7,000、10,000或20,000MPa,具有通常为大约钢或更高的坚硬程度(例如,100-1000GPa)。
路易斯酸化合物可以是具有路易斯酸特性(即由于电子缺乏而是强烈地亲电性的)的本领域已知的化合物中的任一种。路易斯酸化合物的一些实例包括含硼化合物(例如,硼酸、硼酸盐、硼酸酯、硼烷和卤化硼,如BF3、BCl3和BBr3)、含铝化合物(例如,氢氧化铝、铝酸盐、铝酸酯和卤化铝,如AlF3、AlCl3和AlBr3)和含锡化合物(例如,锡酸、锡酯(例如,乙酸锡(II)或2-乙基己酸锡(II))、锡醇盐(例如,乙醇锡(IV))和卤化锡,如SnF4、SnCl4、SnBr4和SnI4)。路易斯酸化合物优选在以下程度不是可逆地与两种聚合物组分(i)和(ii)或组合物的其他组分反应,即当相对于没有路易斯酸化合物时聚合物共混物材料在它的物理性质方面没有被改进的程度。
聚合物共混物材料可以进一步包括或可以不进一步包括金属氧化物组合物作为附加的改性剂。金属氧化物组合物的金属可以是,例如,碱金属、碱土金属、主族金属、过渡金属或镧系金属。碱金属氧化物的一些实例包括Li2O、Na2O、K2O和Rb2O。碱土金属氧化物组合物的一些实例包括BeO、MgO、CaO和SrO。主族金属氧化物组合物的一些实例包括B2O3、Ga2O3、SnO、SnO2、PbO、PbO2、Sb2O3、Sb2O5和Bi2O3。过渡金属氧化物组合物的一些实例包括Sc2O3、TiO2、Cr2O3、Fe2O3、Fe3O4、FeO、Co2O3、Ni2O3、CuO、Cu2O、ZnO、Y2O3、ZrO2、NbO2、Nb2O5、RuO2、PdO、Ag2O、CdO、HfO2、Ta2O5、WO2和PtO2。镧系金属氧化物组合物的一些实例包括La2O3、Ce2O3和CeO2。在一些实施方案中,从聚合物共混物中排除前述金属氧化物的一个或多个种类或具体类型。
相对于组分(i)、(ii)和(iii)的重量之和或相对于最终聚合物共混物的重量,改性剂的量(即,重量百分比,或“重量%”)可以是任何合适的量,但是典型地不大于约10、15、20、25或30重量%。在不同的实施方案中,改性剂的量可以为精确地、大约、至少、高于、多至或少于,例如,1重量%、2重量%、3重量%、4重量%、5重量%、10重量%、12重量%、15重量%、20重量%、25重量%或30重量%,或者在由前述值中的任何两个界定的范围内的量。
当组合物不含溶剂或基本上未溶剂化时,含有至少组分(i)、(ii)和(iii)的聚合物共混物材料优选具有至少或高于1MPa的抗拉强度,并且更优选至少或高于10、15、20或30MPa。在不同的实施方案中,聚合物共混物材料可以表现出至少或高于1MPa、2、MPa、3MPa、4MPa、5MPa、10MPa、12MPa、15MPa、20MPa、25MPa、30MPa、35MPa、40MPa、45MPa、50MPa、60MPa、70MPa、80MPa、90MPa、100MPa、200MPa、300MPa、400MPa、500MPa、600MPa、700MPa、800MPa、900MPa或1000MPa的抗拉强度,或在由前述示例值中的任何两个界定的范围内的抗拉强度。
含有至少组分(i)、(ii)和(iii)的聚合物共混物材料优选具有至少或高于5%、且更优选至少或高于10%、20%、30%、40%或50%的伸长率。在不同的实施方案中,聚合物共混物材料可以表现出至少或高于5%、10%、15%、20%、30%、40%、50%、60%、70%、80%、90%、100%、110%、120%、150%、180%、200%、250%、300%、400%或500%的伸长率,或在由前述示例值中的任何两个界定的范围内的伸长率。在一些实施方案中,聚合物共混物表现出至少或高于1000%、1500%、2000%或甚至2500%的伸长率。
在另一方面,本公开内容涉及用于制备以上描述的聚合物共混物材料的方法。在该方法中,将组分(i)、(ii)和(iii)混合并均匀地共混,以形成所述聚合物共混物材料。可以以液体形式(如果可应用的话)、溶液形式或粒子形式包括组分(i)、(ii)和/或(iii)中的任一个。在粒子的情况下,所述粒子可以独立地是纳米粒子(例如,至少1、2、5或10nm,且多至20、50、100、200或500nm)、微粒子(例如,至少1、2、5或10μm,且多至20、50、100、200或500μm)、或大粒子(例如,高于500μm或至少或多至1、2、5、25、50或100mm)。典型地,如果以粒子形式提供组分(i)-(iii)中的任一个,则通过适当的加热将聚合物粒子熔融或软化,以允许聚合物的均匀共混和粒子的均匀分散。可以通过本领域已知的任何方法将所述组分均匀共混,以获得固体、半固体、凝胶、浆料或液体混合物的均匀共混物。可应用的共混过程的一些实例包括简单或高速混合、复合(compounding)、挤出或球磨,所有这些是本领域熟知的。
通过“均匀地共混”意指:在大(例如,毫米)尺度下,至少不存在组分(i)和(ii)的可分辨区域,尽管可以存在或可以不存在组分(iii)的可分辨的区域。相(iii)组分的一些保持为固体相,处于单质状态(例如,碳粒子)或以晶体薄片相(例如,聚环氧乙烷)。换言之,均匀的共混物至少对于组分(i)和(ii)来说具有改性的或增容的相结构(不必需是单一相结构,但是经常具有与单独相相关的保持的但移动了的Tg)。改性的相结构通常表明在不损失每个组分特性的情况下在微米尺度或接近分子水平的同质一体化。不同于组分(i)、(ii)或(iii)的组分可以以均匀的或非均匀的形式存在。在附加的非均匀组分的情况下,当前描述的具有组分(i)、(ii)和(iii)的聚合物共混物可以认为是其中结合了附加的非均匀组分的“均匀基质”。优选地,全部组分保持它们的阶段性身份,并且组分良好地以纳米尺度分散。在该情况下,组分(i)可以提供刚性或高Tg相;组分(ii)可以提供挠性、弹性和低Tg;且组分(iii)可以提供一定程度的相(i)和(ii)之间的相互作用中的协同,从而起到界面附着促进剂和/或材料性能增强剂的作用。在一些实施方案中,如果组分(i)和(ii)形成完全可混溶的相,它们将表现出单一的Tg。
在一些实施方案中,被共混的混合物还包括自由基交联剂。自由基交联剂是任何在共混过程期间和/或随后在调节(conditioning)过程、活化过程、固化过程和/或成形过程期间产生自由基以实现组分(i)和/或(ii)的交联的物质。典型地,自由基交联剂在热或照射暴露下分解,以形成反应性自由基。自由基交联剂可以是,例如,本领域已知的任何自由基聚合引发剂。在特别的实施方案中,自由基交联剂是有机过氧化物化合物。有机过氧化物化合物的一些实例包括过氧化二异丙苯(DCP)、叔丁基过氧化物、苯甲酰基过氧化物、甲基乙基酮过氧化物和丙酮过氧化物。自由基交联剂可以备选地是无机过氧化物化合物,如过二硫酸盐。自由基交联剂也可以选自或可以不选自非过氧化物自由基产生化合物,如偶氮化合物(例如,AIBN或ABCN)或卤素(例如,Br2或I2)。在一些实施方案中,自由基交联可以通过物理手段如通过将材料暴露至产生用于组分交联的自由基的电子束(例如,Stelescu等,The Scientific World Journal,684047,2014)或紫外(UV)照射(例如,Naskar等,Carbon,43(5)1065-1072,2005)来完成。烃聚合物通过暴露至电子束照射产生自由基。在一些实施方案中,为了促进UV交联,聚合物共混物还可以用当暴露至UV照射时产生自由基的丙烯酸酯和/或共轭酮(二苯甲酮衍生物)添加剂改性。
典型地,使聚合物共混物材料经过成形过程,以制备聚合物共混物的所需形状。成形过程可以包括,例如,模制(例如,倾倒,注入或压缩模塑)、挤出、熔纺、熔体压制或冲压,所有这些都是本领域熟知的。
在仍然其他的方面,本发明涉及含有上文描述的聚合物共混物的制品。该制品典型地是其中期望一定程度的韧性连同高机械强度的制品。共混物可以进一步用例如连续的碳、陶瓷或金属纤维增强,以制备复合材料部件。制品可以用作或包含在任何有用的组件中,如结构支撑体、汽车的内部或外部、家具、工具或器具,或高强度片材或板材。在一些实施方案中,聚合物共混物可以制备并用作涂层或膜,如保护膜。可以通过例如将共混物熔融或将共混物的组分溶解在合适的溶剂中,随后将该液体施加到合适的基板上并随后移除溶剂,来将聚合物共混物作为涂层或膜。如果聚合物共混物具有适当地相当程度的弹性,则聚合物共混物也可以起到粘结剂、粘合剂或分散剂的作用。
以下为了举例说明的目的提出了实施例,以描述本发明的某些具体实施方案。然而,本发明的范围不以任何方式受本文提出的实施例的限制。
实施例1
具有金属氧化物作为改性剂的聚合物共混物的制备(比较)
第1部分:在CW Brabender 3-片式混合机中的材料的反应合成
混合运行1:在表1的第一栏中示出了混合组合物。将混合机在设置到60℃的温度预热。将马达设置为60rpm的速度,并且将运行时间设置为9分钟。在混合机到达所需温度后,开始该混合运行,其首先超过随后冷却回降至60℃。随后加入丁腈橡胶样品,并且允许橡胶软化两分钟,之后立即加入ZnO。将混合物共混两分钟,随后立即加入1pphr(份/百份橡胶)DCP加载。允许混合物混合五分钟。随后将样品回收并储存在室温,直至第二次混合运行。
混合运行2:将温度设置到140℃,速度设置在60rpm,并且运行时间设置为6分钟。在混合机到达所需温度后,开始该混合运行,其首先超过并随后冷却回降至140℃。然后,加入软木Kraft木质素样品,并且允许木质素熔融1分钟,之后加入来自混合运行1的橡胶样品。将混合物运行五分钟,并将样品回收并储存在室温,直至模制。
第2部分:树脂样品模制成片材
将经混合的样品材料插入到压缩模具中,以制备模制的片材。将模具的板加热到150℃(302°F)。在达到温度后,将板紧紧加压(但没有登记的(registered)压力)达10秒,并随后释放。然后,将板加压至5公吨的压力达10秒,并释放。最后,将板加压至5公吨的压力并保持10分钟。在10分钟时,将加热板用冷却水冷却。冷却进行十分钟,随后释放剩余的压力并取回样品片材。将该片材小心地储存直至使用它来切出哑铃状体。结果示于下表1中。
表1
根据表1中的数据,所研究的制剂可以用木质素替代60%的丁腈橡胶,其中在保持>500%的断裂伸长率的同时,在抗拉强度和模量方面的改善很小。这些结果是令人鼓舞的,但是该制剂是有缺欠的,特别是在抗拉强度(即,峰值应力)方面。
实施例2
具有硼酸作为改性剂的聚合物共混物的制备
第1部分:在CW Brabender 3-片式混合机中的材料的反应合成
在表2的第一栏中示出了混合组合物。在此实验中,避免混合运行1,计划将DCP加载加倍,假设50%的DCP将到达木质素相,其将充当自由基清除剂并通过将反应猝灭结合至橡胶。
将混合机在设置到140℃的温度预热。将马达速度设置在60rpm。在混合机到达所需温度后,开始混合运行,其首先超过并随后冷却回降至140℃。然后,加入软木Kraft木质素样品,并且允许木质素剪切两分钟,之后逐渐加入原丁腈橡胶。将混合物共混6分钟,之后加入3重量%的硼酸(相对于总的橡胶+木质素质量)。随后将混合物共混4分钟,之后加入2phr的DCP。随后将混合物共混10分钟,并且将样品回收并储存在室温,直至模制。
第2部分:树脂样品模制成片材
将混合的样品材料插入到压缩模具中,以制备模制的片材。将模具的板加热到185℃(365°F)。在达到温度后,将板紧紧加压(但没有登记的压力)达10秒,并随后释放。然后,将板加压至5公吨的压力达10秒,并释放。最后,将板加压至5公吨的压力并保持10分钟。在10分钟时,将加热板用冷却水冷却。冷却进行十分钟,随后释放剩余的压力并取回样品片。将片材小心地储存直至使用它以切出哑铃状体。对于来自相同材料的另一个模制样本,将模制时间保持至30分钟,随后开始冷却。两个样品的结果提供在下表2中。
表2
以上结果显示,使用硼酸代替ZnO将抗拉强度提高了两倍多。此外,极限伸长率进一步下降至300-400%的水平。结果还显示,较长的模制时间改善了性质。增加交联或者退火也有助于增强性质。结果是令人鼓舞的,但是进行了努力以进一步改进制剂,特别是在保持良好伸长率的同时的抗拉强度方面。
实施例3
具有硼酸和炭黑作为改性剂的聚合物共混物的制备
第1部分:在CW Brabender 3-片式混合机中的材料的反应合成
遵循实施例1和2的方案。在此实验中,进一步增加DCP加载,连同加入20pphr炭黑。而且,在这组实验中,改变橡胶材料的类型,以更好地理解橡胶类型对共混物的机械性质的影响。在不同的制剂中,除了丁腈橡胶或丙烯腈-丁二烯橡胶(NBR),使用了其他橡胶,如天然橡胶(NR)、丁苯橡胶(SBR)、丁二烯橡胶(BR)和溴化的异丁烯对甲基-苯乙烯三元共聚物(ExxproTM-3433)。
混合运行1:将混合机在设置到140℃的温度预热。将马达速度设置在30rpm。在混合机到达所需温度后,开始混合运行,其首先超过并随后冷却回降至140℃。然后,加入橡胶,并且允许橡胶剪切两分钟,之后逐渐加入炭黑。将混合物共混7分钟,之后停止混合,并将橡胶混合物移出并冷却。
混合运行2:将混合机在设置到140℃的温度预热。随后将木质素加入,并允许熔融并剪切两分钟,之后向经剪切的木质素中加入橡胶黑(black)混合物。将混合物再共混六分钟。然后,加入3重量%的硼酸(相对于总的橡胶+木质素质量)。随后将混合物共混4分钟,之后加入4.2phr的DCP。随后将混合物共混10分钟,并且将样品回收并储存在室温,直至模制。在另一个具有丁腈橡胶的样品中,使用4.8pphr DCP。
第2部分:树脂样品模制成片材
将经混合的样品材料插入到压缩模具中,以制备模制的片材。将模具的板加热到185℃(374°F)。在达到温度后,将板紧紧加压(但没有登记的压力)达10秒,并随后释放。然后,将板加压至5公吨的压力达10秒,并释放。最后,将板加压至5公吨的压力并保持30分钟。在30分钟时,将加热板用冷却水冷却。冷却进行十分钟,随后释放剩余的压力并取回样品片。将片材小心地储存直至使用它以制成犬骨状体。结果提供在下表3中。
表3
以上结果显示,炭黑的使用将共混物的抗拉强度从4MPa提高到6.4MPa(提高60%),其中极限伸长率在200-300%水平。橡胶组成的变化表明和任何其他橡胶相比,与NBR更高程度的相容性。如果可以优化DCP加载,则溴丁基橡胶或SBR可以是良好的,但是BR和NR与木质素熔融体大大地不相容。基于此结果,可以假定,可以通过其他添加剂的选择进一步改善NBR和木质素相之间的潜在界面相互作用。对于含有NR和NBR的代表性木质素共混物的动态力学储存模量和损耗角正切谱示于图2中。储存模量值随着温度增高而下降。在玻璃化转变温度。储存模量的下降是高的。损耗角正切(tan(δ))值(其是损耗与储存模量的比率)显示在Tg的最大值。从图2明显的是,含有NR/木质素的共混物具有与橡胶(-56℃)和木质素(148℃)相相关的两个独特的Tg。然而,对于NBR/木质素共混物,木质素Tg在损耗角正切谱中看起来不显著。位移的NBR Tg看起来在0℃,并且由于均匀的木质素-橡胶改性相所致,肩部看起来在60-130℃温度范围。NBR/木质素共混物在超过100℃不显示储存模量的下降。含有ExxproTM-3433(溴丁基橡胶)的共混物显示比含有NR、SBR或BR的共混物稍微更好的性质。
实施例4
其中结合有硼酸的木质素/PEO共混物
使用两种类型的木质素:硬木有机溶剂(HW)和软木(SW)Kraft木质素。将60g的木质素与40g聚环氧乙烷(PEO,分子量5,000,000)混合。在所述制剂的两种中,向木质素-PEO共混物加入10g的硼酸。组合物示于下表4中。如下制备制剂:将半大小的Brabender Intelli-Torque Palsti-Corder预热至140℃,并且将木质素加入至混合室。硬木木质素变成熔融的流体,而软木保持颗粒状粉末。在以50rpm剪切两分钟后,加入PEO。当包括硼酸时,将它在四分钟的总混合时间后加入。在总共12分钟的总混合时间后,移出熔融的混合物。将该材料在190℃在Teflon片中间压缩模制。按照ASTM D882方法,在装备有5N测压元件的MTS Alliance RT/5装置中,以0.5英寸/分钟应变速率进行拉伸测试。使用冲模从模制的片材切割用于拉伸测试的哑铃状样本(ASTM D-638-5-1MP)。组合物的机械性质和热学特性总结在下表4中。
表4.木质素/PEO共混物的组成和性质
*Tg数据来自第二加热循环,以10℃/分钟扫描速率,且熔融峰来自第一加热循环,以10℃/分钟扫描速率,在DSC运行中。
木质素通过与聚环氧乙烷混合形成可混溶的共混物。然而,共混物保持非常脆,具有大量导致单一相玻璃化转变温度(Tg)的氢键合。硼酸的结合降低了共混物的Tg,并且允许延展性的很小改善。拉伸应力-应变曲线图示于图3A和3B中,其显示了在有和没有结合硼酸(BA)的情况下,硬木(HW)木质素-PEO共混物(图3A)和软木(SW)木质素-PEO(图3B)的代表性拉伸应力-应变分布图,以及对于仅PEO的对照曲线。
从这些结果,清楚的是,木质素与PEO强烈相互作用,并且可混溶的混合物具有比纯PEO更高的屈服应力。软木木质素(在性质上是更刚性的)显示比在PEO基质中的硬木木质素更高的屈服应力。BA的结合降低了PEO/木质素相互作用(氢键合),因为硼酸促进与木质素中的一些羟基的缩合。在软木木质素/PEO共混物中,屈服应力的减少是主要的,这表明与硬木木质素和硼酸的反应相比,硼酸和软木木质素之间出乎意料的更强的反应。在所有含有PEO的木质素共混物中,软木木质素、PEO和硼酸混合物也显示高的断裂伸长率(120%)。
实施例5
增韧的木质素-丁腈橡胶共混物和不同添加剂的效果
将硬木(HW)和软木(SW)木质素与丙烯腈-丁二烯橡胶(NBR)混合。将共混物的橡胶相通过有机过氧化物交联。在一些组合物中,用炭黑强化橡胶相,并且将木质素与硼酸复合(complexed)。在一些情况下,进一步用PEO对组合物改性,目的是增强如在实施例4中观察到的屈服应力。组合物示于下表5中。如下制备制剂:将半大小的Brabender Intelli-Torque Palsti-Corder预热至140℃。将橡胶捏合2分钟,随后加入炭黑。将橡胶和炭黑混合5分钟,随后从混合机移出。向保持在140℃的混合室加入木质素。硬木木质素变成熔融的流体,但是软木木质素保持颗粒状粉末。在以50rpm剪切两分钟后,加入PEO。在将木质素与PEO混合三分钟后,加入(预混合的)加载了炭黑的橡胶,并且再混合六分钟。随后加入硼酸,接着再混合四分钟。在该时间点,加入过氧化二异丙苯,并且混合直至达到均匀的扭力。在其中漏过了具体添加剂加载的一些组合物中,它们遵循相同的程序,但没有加入这样的成分,并且混合次数或多或少是类似的。将熔融混合的材料在它是热的时候从混合室移出。
表5.木质素/NBR共混物的组成和性质
*Tg数据来自第二加热循环,以10℃/分钟扫描速率,来自对于软橡胶相(1)和硬木质素相(2)的DSC运行;NA=未分析的。
将熔融混合的制剂在190℃、在Teflon片中间、在9吨压力下压缩模制,并随后在压力下冷却。按照ASTM D882方法,在装备有5N测压元件的MTS Alliance RT/5装置中,以0.5英寸/分钟应变速率进行拉伸测试。使用冲模从模制的片材切割用于拉伸测试的哑铃状样本(ASTM D-638-5-1MP)。在以10℃/分钟扫描的差示扫描量热计中对模制的样本进行热学分析。
没有CB加载的二元、三元、四元和五元共混物
研究了表5中所示的组合物,以理解在存在或不存在PEO这两种情况下,DCP在木质素/NBR共混物中的作用。性质总结在表5中。结果显示,SW/NBR共混物比HW/NBR共混物更坚固。这可能是由于SW木质素分子的较高刚性度导致的。NBR通过过氧化二异丙苯的交联增强了共混物的性质。NBR/HW/PEO共混物表现出PEO的屈服应力特性。如从可见的屈服应力明显的,PEO可能保持为独立的或排除的相(在NBR/HW/PEO中)。然而,这样的屈服应力在SW/NBR/PEO混合物中并不是清楚地可见。从该表格数据,SW/NBR共混物通过PEO的增塑和软化也是明显的。PEO增塑作用在DCP-固化的SW/NBR共混物中是主要的。在DCP-交联的HW/NBR中,这样的PEO相的增塑作用不是主要的。
接下来,通过将硼酸结合在SW和HW的木质素/NBR/PEO/DCP共混物中,研究五元共混物。由PEO、BA、DCP组成的HW和SW组合物两者不像简单的用DCP交联的木质素/NBR共混物那样坚固或坚韧。PEO和BA的存在导致了对所有组合物的增塑作用,除了PEO在HW组合物中保持相分离(表现出屈服应力)的事实之外。代表性的应力-应变分布图示于图4A和4B中,其显示了具有PEO和/或DCP的硬木(HW)木质素-NBR共混物(图4A)和具有PEO和/或DCP的软木(SW)木质素-NBR共混物(图4B)的代表性拉伸应力-应变分布图,以及对于仅含有木质素和NBR的共混物的对照曲线。
具有CB加载的二元、三元四元和五元共混物
在下表6和7中分别示出了制剂和性质。对于基于软木和硬木木质素的组合物,分别在图5A和5B中显示了代表性的拉伸应力-应变数据。从图5A明显的是,尽管SW/NBR/PEO共混物不表现出PEO相的屈服应力(图4B),但硼酸和炭黑的同时存在导致了SW/NBR共混物中对于PEO相的屈服应力的显著改善。备选地,从图5B明显的是,尽管HW/NBR/PEO共混物表现出更适当的与PEO相有关的屈服应力(图4A),但硼酸和炭黑的同时存在导致与在HW/NBR共混物中的PEO相有关的屈服应力的显著降低。因此,取决于成分,木质素/NBR共混物(HW或SW共混物)可以通过控制相的相互作用来调整以具有所需的力学性质。
表6.木质素/NBR共混物的组成
表7.木质素/NBR共混物的性质
*Tg数据来自第二加热循环,以10℃/分钟扫描速率,在DSC运行中。
如在表7中观察到的,NBR/SW二元共混物比NBR/HW二元共混物更坚固。这可能是由于SW木质素分子的较高刚性度导致的。NBR通过过氧化二异丙苯的交联增强了共混物的性质。在上文没有CB加载的实施例中,研究了在有或没有用DCP交联的情况下的木质素/NBR的二元、三元和四元共混物以及在PEO存在下的类似组合物。在这些组合物的一些中,橡胶相通过结合炭黑的的强化进一步使共混物增韧。同样地,在所有这些共混物中,SW木质素共混物保持更坚固。然而,在SW/NBR/CB/DCP共混物中,硼酸的结合稍微降低了性质。前述具有SW木质素的结果与将硼酸结合到HW/NBR/CB/DCP共混物中出人意料地相反,其形成更坚固和更坚韧的共混物。这表明硼酸在DCP交联的NBR/HW共混物中的增容作用,这可能是通过HW木质素和NBR通过硼酸的共交联,由此形成具有改善的界面的共连续形貌所介导。
在实施例4中,结果表明,通过硼酸的结合,在软木木质素和PEO共混物中的屈服应力降低。在没有CB加载的上述实施例中,结果表明,通过硼酸的加入,SW/NBR三元或五元共混物中的强度降低,而HW/NBR三元或五元共混物保持不受影响(图4A和4B)。NBR/木质素共混物中的PEO的存在也不改善强度。PEO/硼酸的组合也不增强强度。炭黑的存在增强NBR/木质素的强度,但是在PEO或硼酸的存在下,在HW/NBR共混物中的强度增强是主要的。炭黑/PEO/硼酸共混物在NBR/SW组合中显示协同效果,得到抗拉强度的显著增强。然而,这样的组合对于NBR/HW木质素共混物来说通常是有害的。这种出乎意料的结果可能是由于HW木质素与SW木质素之间的结构差异导致的。
SW/NBR/CB/BA/DCP共混物的PEO增容作用相对HW/NBR/CB/BA/DCP共混物的PEO增塑作用
具有变化的PEO加载的木质素/NBR/CB/BA/DCP制剂的组成和性质分别在以下显示在表8和9中。
表8.对于SW和HW两者的木质素/NBR共混物的组成
表9.木质素/NBR共混物的性质
*Tg数据得自第二加热循环,以10℃/分钟扫描速率,在DSC运行中。
如所示的,在HW/NBR/CB/BA/DCP共混物中结合PEO降低了抗拉强度,而在SW/NBR/CB/BA/DCP共混物中的PEO增加抗拉强度。对于处于变化的PEO加载(如表8中提供的)下的木质素//NBR/CB/BA/DCP的共混物,分别对于基于软木(图6A)和硬木(图6B)木质素的组合物,代表性的应力-应变分布图示于图5A和5B中。从图6A,清楚的是,在含有SW的共混物中,PEO的屈服应力变得非常高,并且它随着PEO加载的增大而增加,伴以极限伸长率的减小。出人意料的是,PEO的结合增加了含有HW的共混物的相对柔软基质的延展性(图6B),并且PEO屈服强度明显低(小于在含有SW的共混物中观察到的值的三分之一)。
尽管已经显示并描述了本发明当前考虑的优选实施方案,但本领域技术人员可以进行保持在由所附权利要求限定的本发明的范围内的各种改变和改进。
- 还没有人留言评论。精彩留言会获得点赞!