共聚聚硅氮烷、其制造方法以及包含其的组合物以及使用了其的二氧化硅质膜的形成方法与流程
本发明涉及一种新型共聚聚硅氮烷,特别涉及一种可用于在低温制造二氧化硅质膜的新型共聚聚硅氮烷。另外,本发明也涉及该共聚聚硅氮烷的制造方法、包含该共聚聚硅氮烷的组合物、以及使用了该组合物的二氧化硅质膜的形成方法。
背景技术:
已知晓,可使用聚硅氮烷、特别是可溶于有机溶剂的全氢聚硅氮烷作为氮化硅前体(例如参照专利文献1)。另外近年来,作为用于形成半导体装置等电子设备中的层间绝缘膜的方法,已知晓通过涂布包含聚硅氮烷的涂布组合物,在其后利用焙烧将聚硅氮烷转化为二氧化硅质膜的方法(例如参照专利文献2~5)。
全氢聚硅氮烷具有由-SiH2-NH-表示的重复单元,在专利文献1中,作为合成方法,记载了使二卤代硅烷与碱的加合物与氨进行反应的方法。
此外,作为聚硅氮烷的制造方法,人们提出了:(a)使胺与SiCl4、SiH2Cl2等硅卤化物进行反应的方法,(b)使用可进行脱氢化的KH等碱金属氢化物催化剂将硅氮烷制成聚硅氮烷的方法,(c)使用过渡金属络合物催化剂,利用硅烷化合物与胺类化合物的脱氢反应而合成硅氮烷的方法,(d)使用CF4SO3H这样的酸催化剂使得氨基硅烷与氨进行胺交换的方法,(e)利用大量的氨或胺将氨基硅烷进行胺交换的方法,(f)使得多氨基硅烷化合物与多氢化含氮化合物在碱性催化剂的存在下进行胺交换反应的方法等各种方法(例如参照专利文献6)。
关于聚硅氮烷的二氧化硅质转化膜,除了用作前述的层间绝缘膜之外,也用作半导体装置等电子设备中的钝化膜、保护膜、平整化膜等各种膜,但是对于这样的电子设备中的膜,人们要求其具有绝缘性、膜的平整性、对于酸/碱、溶剂等的耐受性、以及高的阻隔性等特性。利用使用含有聚硅氮烷的涂布液形成二氧化硅质膜的方法,从而可形成满足这样的特性的膜,但是在全氢聚硅氮烷方面,无法通过在低温进行焙烧而获得具有耐受电压特性和对溶剂的耐受性的二氧化硅质膜。由此,为了将全氢聚硅氮烷转化为具有充分的绝缘性和/或耐受电压特性、对溶剂的耐受性的二氧化硅质膜,在高温下与水蒸气接触的工序是必需的。
现有技术文献
专利文献
专利文献1:日本特公昭63-16325号公报
专利文献2:日本特开2011-54898号公报
专利文献3:日本特开2005-45230号公报
专利文献4:日本特开平9-31333号公报
专利文献5:日本特开平9-275135公报
专利文献6:WO97/24391
技术实现要素:
发明想要解决的课题
本发明基于上述那样的情形而完成,目的在于提供一种共聚聚硅氮烷,其可利用在低温的焙烧工序而获得具有耐受电压特性和对溶剂的耐受性的二氧化硅质膜。
另外,本发明的另一目的在于提供前述共聚聚硅氮烷的制造方法、包含前述共聚聚硅氮烷的组合物、使用了该组合物的,在低温的焙烧工序后形成具有耐受电压特性和对溶剂的耐受性的二氧化硅质膜的方法。
用于解决问题的方案
本发明人等进行了深入研究,结果发现如下见解,基于此见解而完成了本发明:通过并用在溶液中使二卤代硅烷化合物与伯胺类化合物进行反应的胺解(aminolysis)、以及与氨进行反应的氨解(ammonolysis)从而获得共聚聚硅氮烷化合物,将由此获得了的共聚聚硅氮烷化合物在300℃以下的焙烧温度下,转化为具有比历来公知的无机聚硅氮烷优异的耐受电压特性和对溶剂的耐受性的二氧化硅质膜。
即,本发明涉及以下所示的共聚聚硅氮烷、该共聚聚硅氮烷的制造方法、包含前述共聚聚硅氮烷的组合物、以及使用该组合物制造二氧化硅质膜的方法。
(1)一种共聚聚硅氮烷,其至少具有由下述通式(I)表示的重复单元、以及由下述通式(II)表示的重复单元,NR3/SiH1,2(SiH1,2表示SiH1与SiH2的合计量)为0.005~0.3。
通式(I):
(式中,R1、R2各自独立地表示氢原子、烃基、含烃基的甲硅烷基、含烃基的氨基、或者烃氧基,R3表示烷基、烯基、烷氧基、环烷基、芳基、或者烷基甲硅烷基。)
通式(II):
(式中,R1、R2与上述同样定义。)
(2)一种共聚聚硅氮烷,其为上述(1)所述的共聚聚硅氮烷,其特征在于,通式(I)以及通式(II)的R1以及R2的任一个都是氢原子。
(3)一种共聚聚硅氮烷,其为上述(1)或(2)所述的共聚聚硅氮烷,其特征在于,聚苯乙烯换算重均分子量为700~4,000。
(4)根据上述(1)所述的共聚聚硅氮烷的制造方法,其特征在于,使得由通式(III)表示的二卤代硅烷化合物与由式(IV)表示的伯胺类化合物进行反应,然后进一步与氨进行反应,
(式中,R1、R2各自独立地表示氢原子、烃基、含烃基的甲硅烷基、含烃基的氨基、或者烃氧基,X表示F、Cl、Br、或者I。)
R3-NH2(IV)
(式中R3表示烷基、烯基、烷氧基、环烷基、芳基、或者烷基甲硅烷基)。
(5)一种共聚聚硅氮烷的制造方法,其为上述(4)所述的共聚聚硅氮烷的制造方法,其特征在于,前述伯胺类化合物相对于二卤代硅烷化合物而言按摩尔比计以0.1~3.0使用,氨相对于前述二卤代硅烷化合物而言按摩尔比计以0.2~3.1使用。
(6)一种共聚聚硅氮烷的制造方法,其为上述(4)或者(5)所述的共聚聚硅氮烷的制造方法,其特征在于,反应在溶剂中进行,在与前述伯胺的反应之前,使得二卤代硅烷化合物与碱进行反应而形成二卤代硅烷化合物的加合物,其后使该二卤代硅烷化合物的加合物与前述伯胺类化合物反应。
(7)一种共聚聚硅氮烷的制造方法,其为上述(4)~(6)中任一项所述的共聚聚硅氮烷的制造方法,其特征在于,通式(III)的R1以及R2均为氢原子,X为氯原子。
(8)一种涂布组合物,其特征在于,含有上述(1)~(3)中任一项所述的共聚聚硅氮烷。
(9)一种二氧化硅质膜的形成方法,其特征在于,将上述(8)所述的涂布组合物涂布于基板,进行干燥,然后在氧化气氛中在300℃以下加热。
发明的效果
使用了以往的全氢聚硅氮烷的二氧化硅质膜制造方法中,必须进行高温焙烧,在低温时无法获得充分的耐受电压特性、对溶剂的耐受性等。然而通过使用本发明的共聚聚硅氮烷,可利用300℃以下的低温焙烧而形成具有耐受电压特性和对溶剂的耐受性的二氧化硅质膜。因此,也可适用于制造耐热性不充分的半导体元件的层间绝缘膜等。
附图说明
图1所示为由实施例2获得的共聚聚硅氮烷的FT-IR谱图。
图2所示为由实施例2获得的共聚聚硅氮烷的1H NMR谱图。
图3所示为由实施例3获得的共聚聚硅氮烷的FT-IR谱图。
图4所示为由实施例3获得的共聚聚硅氮烷的1H NMR谱图。
具体实施方式
如前述的那样,本发明的共聚聚硅氮烷至少具有由通式(I):-Si(R1)(R2)-NR3-表示的重复单元、以及由通式(II):-Si(R1)(R2)-NH-表示的重复单元(式中,R1、R2各自独立地表示氢原子、烃基、含烃基的甲硅烷基、含烃基的氨基、或者烃氧基,R3表示烷基、烯基、烷氧基、环烷基、芳基、或者烷基甲硅烷基),NR3/SiH1,2比为0.005~0.3的共聚聚硅氮烷。
关于上述本发明的共聚聚硅氮烷,例如,可通过并用在溶剂中使二卤代硅烷化合物与伯胺类化合物进行反应的胺解、以及与氨进行反应的氨解从而调制。在此过程中,通过进行使用了伯胺类化合物的胺解,然后进行使用了氨的氨解,从而获得在聚合物中含有在氮原子上被氢原子以外的基团取代的骨架的共聚聚硅氮烷。
作为前述溶剂,如果是可将前述二卤代硅烷化合物、前述伯胺类化合物、形成出的共聚聚硅氮烷溶解,另外不与共聚聚硅氮烷制造时使用的化合物、中间产物、最终产物进行反应的溶剂,则可以是任一种溶剂,但作为优选者而列举出:例如,(a)芳香族化合物、例如、苯、甲苯、二甲苯、乙苯、二乙苯、三甲苯、三乙苯等,(b)饱和烃化合物、例如、正戊烷、异戊烷、正己烷、异己烷、正庚烷、异庚烷、正辛烷、异辛烷、正壬烷、异壬烷、正癸烷、异癸烷等,(c)脂环式烃化合物、例如、乙基环己烷、甲基环己烷、环己烷、环己烯、对薄荷烷(p-menthane)、十氢萘、双戊烯、柠檬烯(limonene)等,(d)醚类,例如二丙基醚、二丁醚、乙醚、甲基叔丁基醚、苯甲醚等。
作为前述二卤代硅烷化合物,使用由通式:Si(R1)(R2)X2(式中,R1、R2各自独立地表示氢原子、烃基、含烃基的甲硅烷基、含烃基的氨基、或者烃氧基,X表示F、Cl、Br、或者I)表示的二卤代硅烷化合物,X为氯原子的二氯硅烷化合物从原料价格这一点等等考虑是优选的。另外,作为前述R1、R2中的烃基,例如列举出甲基、乙基、丙基、3-氯丙基、3,3,3-三氟丙基、3-氰基丙基等、也可被卤素基团、氰基、或者芳基等取代的直链、支链、或者环状烷基,乙烯基、烯丙基等烯基,苄基等芳烷基,苯基等芳基。另外作为烃氧基,例如列举出甲氧基、乙氧基等烷氧基、环烷氧基、芳氧基(aryloxy group)等。
具体性地例示可以优选使用的二氯硅烷化合物时,则例如列举出二氯硅烷、甲基(氢)二氯硅烷、乙基(氢)二氯硅烷、乙烯基(氢)二氯硅烷、烯丙基(氢)二氯硅烷、苯基(氢)二氯硅烷、苄基(氢)二氯硅烷、二甲基二氯硅烷、甲基苯基二氯硅烷、二苯基二氯硅烷、甲基乙烯基二氯硅烷、二乙烯基二氯硅烷、二乙基二氯硅烷、乙基甲基二氯硅烷、乙基苯基二氯硅烷、乙基乙烯基二氯硅烷、乙氧基甲基二氯硅烷、乙氧基乙基二氯硅烷、乙氧基苯基二氯硅烷、乙氧基乙烯基二氯硅烷、甲基3,3,3-三氟丙基二氯硅烷、烯丙基甲基二氯硅烷、3-氯丙基甲基二氯硅烷、甲基丙基二氯硅烷、二乙氧基二氯硅烷、3-氰基丙基甲基二氯硅烷等,但是从反应性以及原料价格等观点考虑,特别优选为二氯硅烷。予以说明,二氯硅烷化合物不受限于上述具体记载的化合物,另外,二卤代硅烷化合物也不受限于二氯硅烷化合物。
由上述通式(IV)表示的伯胺类化合物的R3的烷基如果是烷基即可,没有特别限定,例如,列举出甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、戊基、己基等基团,其中优选为丙基、丁基。具体性地表示可以优选使用的伯胺类化合物时,则例如列举出丙基胺、异丙基胺、仲丁胺、异丁胺等烷基胺类,环己胺等环状烷基胺类,烯丙基胺等烯基胺类,苯胺等芳香族胺类。然而,本发明中使用的伯胺类化合物不受限于这些具体例示出的伯胺类化合物。
关于胺解,作为优选方法而列举如下的方法,即,使由前述通式(III)表示的二卤代硅烷化合物与碱进行反应,使所形成的二卤代硅烷化合物的加合物与伯胺类化合物进行反应的方法(例如参照专利文献1)。一般而言二卤代硅烷为酸性,可通过与碱进行反应而形成加合物。关于该加合物的形成速度以及作为加合物而言的稳定性,依赖于二卤代硅烷的酸性的强度与碱性物质的碱性的强度、立体因素等。关于此情况下的加合物的稳定性,未必表示的是可作为加合物而单独分离的程度的稳定性,而是不但包含稳定地存在于溶剂中的情况,而且包含实质上作为反应中间体而发挥功能的全部的情况。关于使用的碱,根据二卤代硅烷化合物的种类,选择可以容易地形成稳定的加合物、可以经过胺解以及氨解而容易地获得规定的共聚聚硅氮烷的碱,即可。
作为碱,例如列举出路易斯碱、叔胺类、吡啶、甲基吡啶以及它们的衍生物、具有立体阻碍的基团的仲胺类、膦、以及它们的衍生物等,其中从低沸点考虑,优选为伯胺类化合物、碱性比氨小的碱,特别是从处理上以及经济上考虑优选为吡啶以及甲基吡啶。另外,其量相对于二卤代硅烷而言相比于化学计量的量而言过量地存在即可。关于加合物的形成,例如,通过将二卤代硅烷化合物投入于调温为-40~20℃的范围的前述溶剂与碱的混合液中,接着在该温度下搅拌例如30~120分钟从而进行。
予以说明,由于被用作碱的吡啶也优选用作溶剂,因而也可通过向过量的吡啶中加入二卤代硅烷化合物而形成加合物,接着将伯胺类化合物投入于该液中进行胺解,其后向其中注入氨从而进行氨解。
关于胺解,例如,可通过在温度-5℃~15℃反应0.5~2小时来进行。另外,关于氨解,例如,可通过在温度-5℃~15℃将氨注入于反应溶液,将该液在该温度搅拌,反应0.5~2小时从而进行。
关于伯胺类化合物的用量,按相对于二卤代硅烷化合物1摩尔的摩尔比计为0.1~3.0的范围,优选为0.5~2.0的范围,氨的用量按相对于二卤代硅烷化合物1摩尔的摩尔比计为0.2~3.1的范围,优选为1.2~2.7的范围。伯胺类化合物的用量按相对于二氯硅烷化合物1摩尔的摩尔比计不足0.1时,或者氨的用量按相对于二氯硅烷化合物1摩尔的摩尔比计超过3.1时,通过低温焙烧而制造的二氧化硅质膜不具有充分的耐受电压特性和对溶剂的耐受性,相反地伯胺类化合物的用量按相对于二氯硅烷化合物1摩尔的摩尔比计超过3.0或者氨的用量按相对于二氯硅烷化合物1摩尔的摩尔比计不足0.2时,则没有推进分子间聚合并且无法获得适于向基板表面涂布的共聚聚硅氮烷。
予以说明,本发明不受其任何限定,但在本发明的胺解中,可认为生成了-((Si(R1)(R2)-NR3)n-,但是n的值小,根据比较例1的结果,可认为基本上是NR3-Si(R1)(R2)-NR3单体。可认为,通过将通过胺解而获得的化合物进行氨解,从而可促进聚合,使得伯胺类化合物充分地反应,形成具有规定的NR3/SiH1,2比、分子量的共聚物。
这般地,获得NR3/SiH1,2比为0.005~0.3的范围、优选为0.0095~0.26的范围、更优选为0.05~0.1的范围的共聚聚硅氮烷。共聚聚硅氮烷中的NR3/SiH1,2比不足0.005时,通过低温焙烧而制造的二氧化硅质膜不具有充分的耐受电压特性和对溶剂的耐受性,相反地在超过0.3时,与基板的密接性降低因而不适合涂布于基板表面。关于本发明的共聚聚硅氮烷的分子量,聚苯乙烯换算重均分子量(Mw)优选为700~4,000,更优选为1,300~2,500。由此,为了获得具有这样的分子量的共聚聚硅氮烷,除了上述伯胺和氨的用量之外,也可根据需要进行反应温度、反应时间等的调整。本发明的共聚聚硅氮烷的SiH3/SiH1,2比为0.05~0.4的范围,优选为0.06~0.35的范围,更优选为0.14~0.31的范围。另外,本发明的共聚聚硅氮烷的NH/SiH比为0.08~0.16,优选为0.10~0.15的范围,更优选为0.12~0.15的范围。
对于所获得的含有共聚聚硅氮烷的溶液,通过过滤将不溶于溶液的副产物的盐等去除,然后根据需要利用真空蒸馏等将溶剂、碱等去除,从而获得具有规定的浓度的共聚聚硅氮烷溶液、或者共聚聚硅氮烷单一物体。在反应溶剂中包含吡啶与其它的有机溶剂的情况下,通常通过真空蒸馏而去除吡啶,进一步如有必要则也进行有机溶剂的去除。通过利用真空蒸馏将有机溶剂溶液的聚硅氮烷树脂浓度调整为例如5~30重量%的范围,从而可将所获得的含有聚硅氮烷的液体直接用作涂布组合物或者用作涂布组合物的基液。也可向该涂布组合物的基液中,根据需要而加入其它的添加剂或者溶剂而制成涂布组合物。
在本发明的涂布组合物中,除了包含本发明的共聚聚硅氮烷之外,还如前述那样包含溶剂。关于溶剂,如果是将本发明的共聚聚硅氮烷溶解,在进一步使用添加剂的情况下也溶解这些添加剂,且不与共聚聚硅氮烷以及添加剂进行反应的溶剂,则可以是任一种溶剂并且没有特别限定,但是作为优选者而列举出作为在前述反应之时所使用的反应溶剂而列举的溶剂等。当然,如前所述,在反应中应用的溶剂也可直接使用作为涂布组合物的溶剂,也可进一步向其中添加其它的溶剂。
关于这些溶剂,为了调整溶剂的蒸发速度,或者为了降低对人体的有害性,或者为了调整涂布组合物中所含的成分的溶解性,因而可使用适宜混合了两种以上而得到的溶剂。另外,关于涂布组合物中使用的共聚聚硅氮烷溶液,也可通过将本发明的共聚聚硅氮烷溶解于有机溶剂而制造。
在涂布组合物中,也可根据需要添加添加剂。作为添加剂,例如列举出日本特开平6-299118号公报中记载的那样的二氧化硅转化反应促进化合物、例如、金属羧酸、N-杂环状化合物、胺类化合物等。作为其它的添加剂成分,例如列举出粘度调整剂、交联促进剂等。另外,在应用于半导体装置时,也可以以对钠的吸杂效果等为目的,含有磷化合物例如三(三甲基甲硅烷基)磷酸酯等。对于所获得的含有共聚聚硅氮烷的液体,优选使用过滤精度0.1μm以下的过滤器进行循环过滤,将粒径为0.2μm以上的粗大颗粒减低至50个/cc以下。
关于前述涂布组合物的各成分的含量,根据涂布条件和焙烧(固化)条件等而变化。但是,关于共聚聚硅氮烷的含有率,以涂布组合物的总重量为基准而优选为0.1~40%重量,更优选为0.2~30%重量,进一步优选为0.3~25%重量。另外,关于除了共聚聚硅氮烷以外的各种添加剂的含量,虽然根据添加剂的种类等而变化,但是相对于聚硅氮烷化合物的添加量优选为0.001~40%重量,更优选为0.005~30%重量,进一步优选为0.01~20%重量。
关于前述的含共聚聚硅氮烷的涂布组合物,为了在基板上形成二氧化硅质膜,因而涂布于基板表面。作为涂布组合物的涂布方法,适当使用历来公知的方法例如旋涂法、浸渍法、喷雾法、转印法等。它们之中,特别优选的是旋涂法。关于涂膜,可根据需要而反复涂布一次或两次以上而制成所希望的膜厚。作为成为涂布对象的基材,列举出硅基板、玻璃基板、树脂薄膜等,如有必要则也可涂布于在形成半导体元件的过程中的半导体膜、设置了电路等的基板上等等。关于涂膜的厚度,根据膜的使用目的等而不同,但一般而言,按照干燥膜厚,制成为10~2,000nm,优选制成为20~1,000nm。
通过涂布涂布组合物而形成了共聚聚硅氮烷涂膜,然后为了将该涂膜进行干燥,优选将涂膜进行预烘烤(加热处理)。由于预烘烤的目的不是在于将共聚聚硅氮烷进行固化,因而一般通过在低温进行短时间加热而进行。通过具体在70~150℃、优选在100~150℃,加热1~10分钟、优选加热3~5分钟从而进行。
涂布工序,然后将涂膜交付于固化工序。在本发明的聚硅氮烷化合物向二氧化硅质膜的转化方法中,固化工序在300℃以下进行。在本发明中,共聚聚硅氮烷膜向二氧化硅质膜的转化、即、固化是在极其低的温度下进行。固化优选在包含水蒸气、氧气、或者其混合气体的气氛中、即、氧化气氛中进行。在水蒸气氧化中的水蒸气浓度是用于将聚硅氮烷树脂转化为二氧化硅质膜(二氧化硅)的重要的因素,优选为1%以上,更优选为10%以上,最优选为20%以上。特别是在水蒸气浓度为20%以上时,则聚硅氮烷树脂向二氧化硅质膜的转化变得容易进行,孔隙等缺陷的产生变少,二氧化硅质膜的特性得到改良,因而优选。关于固化,通过使用固化炉、热板而进行。在使用非活性气体作为气氛气体的情况下,使用氮气、氩气、或者氦气等。
直到目标温度为止的升温时间一般设为0.1~100℃/分钟,到达目标温度之后的固化时间一般设为1分钟~10小时,优选设为30分钟~3小时。
在本发明中,根据需要,可在该被覆薄膜上,通过进一步反复进行这些工序而形成多层的被覆薄膜。通过这样地堆积两层以上的被覆薄膜,从而可将各被覆薄膜的每一层的堆积膜厚减薄,由此在将各被覆薄膜进行固化时,在该被覆薄膜的厚度方向中的任一个部分都从表面获得充分的氧原子的扩散以及供给,可形成特性更优异的二氧化硅质膜。
这般获得的二氧化硅质膜优选用作半导体装置等电子设备中的层间绝缘膜。另外,所获得的二氧化硅质膜也可优选用作半导体装置等电子设备中的平整化膜、保护膜、钝化膜、硬质掩膜、应力调整膜、牺牲膜等。
实施例
以下列举出实施例、比较例来更具体说明本发明,但本发明不受这些实施例、比较例的任何限定。予以说明,关于通过合成而获得的共聚聚硅氮烷的聚苯乙烯换算重均分子量、FT-IR谱图以及1H NMR谱图的测定,通过使用下述的装置而进行。进一步,关于NR3/SiH1,2比,SiH3/SiH1,2比、NH/SiH比,利用下述的方法而获得。
<重均分子量的测定>
使用(株)岛津制作所制GPC作为测定装置,使用THF作为洗脱液而进行测定。
<FT-IR谱图的测定>
使用日本分光(株)制FT/IR-6100作为测定装置而进行测定。
<1H NMR谱图的测定>
使用日本电子株式会社制NMR(400MHz)作为测定装置而进行测定。
<NR3/SiH1,2比的算出>
在1H NMR谱图中,算出源自NR3基的峰的质子一部分(プロトン一つ分)的峰面积相对于源自SiH1基与SiH2基的4.5~5.3ppm的峰面积之比,对NR3基的导入量进行了评价。
<SiH3/SiH1,2比的算出>
在1H NMR谱图中,将源自SiH3基的4.0~4.5ppm的峰面积相对于源自SiH1基与SiH2基的4.5~5.3ppm的峰面积之比作为SiH3/SiH1,2比而算出。
<NH/SiH比的算出>
将含共聚聚硅氮烷的涂布组合物旋转涂布于硅晶圆,为了去除溶剂因而在150℃保持3分钟,获得了具有膜厚为480~740nm的涂膜的硅晶圆。对于所获得的试样,测定出FT-IR谱图。将源自N-H键的3,200~3,500cm-1的峰面积相对于源自Si-H键的2,000~2,500cm-1的峰面积之比作为NH/SiH比而算出。
实施例1
用干燥氮气将反应容器进行置换,然后加入脱水吡啶40g与脱水二甲苯460g而冷却至0℃以下,投入了二氯硅烷(DCS)25g。将该混合溶液搅拌1.5小时后,投入异丙基胺44g,在反应温度-5~15℃进行了100分钟胺解。接着一边保持-5~15℃一边将NH3以250ml/分钟注入4.4分钟,搅拌100分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)1,700的树脂(树脂A)的20重量%浓度的溶液(溶液A)。根据FT-IR谱图测定,确认了包含了异丙基胺骨架的聚硅氮烷化合物的生成并且NH/SiH比为0.10。另外,根据1H NMR谱图,NPr/SiH1,2比为0.21,SiH3/SiH1,2比为0.11。
实施例2
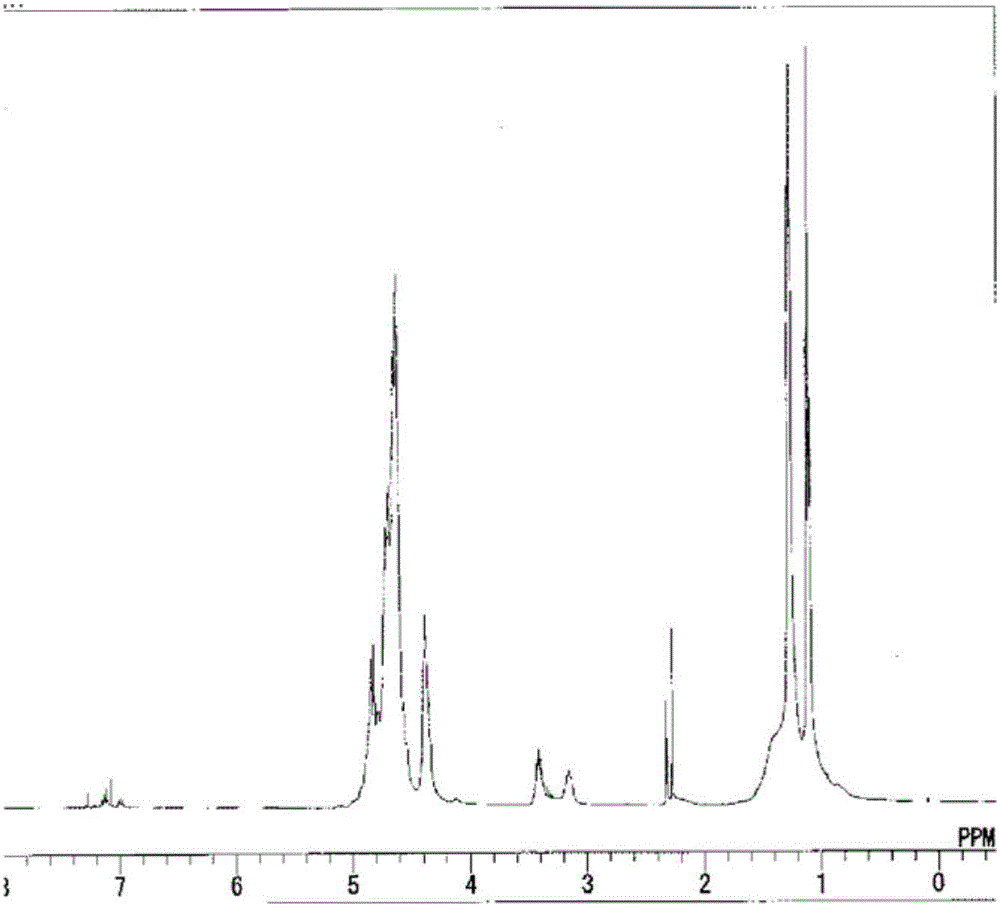
用干燥氮气将反应容器进行置换,然后加入脱水吡啶40g与脱水二甲苯456g而冷却至0℃以下,投入了二氯硅烷(DCS)25g。将该混合溶液搅拌1.5小时后,投入异丙基胺29g,在反应温度-5~15℃进行了100分钟胺解。接着一边保持为-5~15℃一边将NH3以250ml/分钟注入26.8分钟,搅拌100分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)1,350的树脂(树脂B)的20重量%浓度的溶液(溶液B)。进行了树脂B的FT-IR谱图测定、1H NMR测定。将所获得的FT-IR谱图示于图1,另外将1H NMR谱图示于图2。根据FT-IR谱图,确认了包含了异丙基胺骨架的聚硅氮烷化合物的生成,NH/SiH比为0.14。另外,根据1H NMR谱图,NPr/SiH1,2比为0.10,SiH3/SiH1,2比为0.15。
实施例3
用干燥氮气将反应容器进行置换,然后加入脱水吡啶41g与脱水二甲苯456g而冷却至0℃以下,投入了二氯硅烷(DCS)26g。将该混合溶液搅拌1.5小时后,投入异丙基胺7.8g,在反应温度-5~15℃进行了100分钟胺解。接着一边保持为-5~15℃一边将NH3以250ml/分钟注入62.4分钟,搅拌100分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)1,950的树脂(树脂C)的20重量%浓度的溶液(溶液C)。对树脂C进行了FT-IR谱图测定、1H NMR测定。将所获得的FT-IR谱图示于图3,另外将1H NMR谱图示于图4。根据FT-IR谱图,确认了包含了异丙基胺骨架的聚硅氮烷化合物的生成,NH/SiH比为0.13。另外,根据1H NMR谱图,NPr/SiH1,2比为0.05,SiH3/SiH1,2比为0.31。
实施例4
用干燥氮气将反应容器进行置换,然后加入脱水吡啶38g与脱水二甲苯456g而冷却至0℃以下,投入了二氯硅烷(DCS)25g。将该混合溶液搅拌1.5小时后,投入异丙基胺1.5g,在反应温度-5~15℃进行了100分钟胺解。接着一边保持为-5~15℃一边将NH3以250ml/分钟注入68.8分钟,搅拌100分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)2,100的树脂(树脂D)的20重量%浓度的溶液(溶液D)。根据FT-IR谱图测定,确认了包含了异丙基胺骨架的聚硅氮烷化合物的生成,NH/SiH比为0.13。另外,根据1H NMR谱图,NPr/SiH1,2比为0.0095,SiH3/SiH1,2比为0.17。
实施例5
用干燥氮气将反应容器进行置换,然后加入脱水吡啶35g与脱水二甲苯403g而冷却至0℃以下,投入了二氯硅烷(DCS)21g。将该混合溶液搅拌1.5小时后,投入仲丁胺38g,在反应温度-5~15℃进行了60分钟胺解。接着一边保持为-5~15℃一边将NH3以250ml/分钟注入13.2分钟,搅拌60分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)700的树脂(树脂E)的20重量%浓度的溶液(溶液E)。根据FT-IR谱图测定,确认了包含了仲丁胺骨架的聚硅氮烷化合物的生成,NH/SiH比为0.12。另外,根据1H NMR谱图,NsBu/SiH1,2比为0.11,SiH3/SiH1,2比为0.25。
实施例6
用干燥氮气将反应容器进行置换,然后加入脱水吡啶38g与脱水二甲苯456g而冷却至0℃以下,投入了二氯硅烷(DCS)30g。将该混合溶液搅拌1.5小时后,投入丙基胺8.8g,在反应温度-5~15℃进行了100分钟胺解。接着一边保持为-5~15℃一边将NH3以250ml/分钟注入72分钟,搅拌100分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)1,600的树脂(树脂F)的20重量%浓度的溶液(溶液F)。根据FT-IR谱图测定,确认了包含了丙基胺骨架的聚硅氮烷化合物的生成,NH/SiH比为0.091。另外,根据1H NMR谱图,NPr/SiH1,2比为0.26,SiH3/SiH1,2比为0.22。
实施例7
用干燥氮气将反应容器进行置换,然后加入脱水吡啶41g与脱水二甲苯456g而冷却至0℃以下,投入了二氯硅烷(DCS)28g。将该混合溶液搅拌1.5小时后,投入环己胺5.6g,在反应温度-5~15℃进行了100分钟胺解。接着一边保持为-5~15℃一边将NH3以250ml/分钟注入74.4分钟,搅拌100分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)2,500的树脂(树脂G)的20重量%浓度的溶液(溶液G)。根据FT-IR谱图测定,确认了包含了环己胺骨架的聚硅氮烷化合物的生成,NH/SiH比为0.15。另外,根据1H NMR谱图,NHex/SiH1,2比为0.035,SiH3/SiH1,2比为0.20。
实施例8
用干燥氮气将反应容器进行置换,然后加入脱水吡啶41g与脱水二甲苯456g而冷却至0℃以下,投入了二氯硅烷(DCS)21g。将该混合溶液搅拌1.5小时后,投入苯胺3.9g,在反应温度-5~15℃进行了100分钟胺解。接着一边保持为-5~15℃一边将NH3以250ml/分钟注入56分钟,搅拌100分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)1,700的树脂(树脂H)的20重量%浓度的溶液(溶液H)。根据FT-IR谱图测定,确认了包含了苯胺骨架的聚硅氮烷化合物的生成,NH/SiH比为0.14。另外,根据1H NMR谱图,NPh/SiH1,2比为0.027,SiH3/SiH1,2比为0.35。
实施例9
用干燥氮气将反应容器进行置换,然后加入脱水吡啶391g与脱水二甲苯44558g而冷却至0℃以下,花费2.5小时将二氯硅烷(DCS)248g按照反应温度不超过2.5℃的方式投入。将该混合溶液搅拌1.5小时后,投入异丙基胺293g,在反应温度-5~15℃进行了60分钟胺解。接着一边保持为-5~15℃一边将NH3以2.5L/分钟注入27.0分钟,搅拌100分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)3,600的树脂(树脂I)的20重量%浓度的溶液(溶液I)。根据FT-IR谱图测定,确认了包含了异丙基胺骨架的聚硅氮烷化合物的生成,NH/SiH比为0.14。另外,根据1H NMR谱图,NPr/SiH1,2比为0.051,SiH3/SiH1,2比为0.066。
比较例1
用干燥氮气将反应容器进行置换,然后加入脱水吡啶16g与脱水二甲苯185g而冷却至0℃以下,投入了二氯硅烷(DCS)10g。将该混合溶液搅拌1.5小时后,投入异丙基胺20g,在反应温度-5~15℃进行了100分钟胺解。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)不足100的树脂(树脂J)的10%重量浓度的溶液(溶液J)。尝试了将溶液J旋转涂布向硅晶圆,但是分子量过低因而无法在基板上形成膜。
比较例2
准备了通过日本特许第2613787号中记载的方法而获得了的重均分子量(Mw)为5,450的聚(全氢硅氮烷)溶液(设为溶液K)作为比较例2的涂布组合物。
比较例3
用干燥氮气将反应容器进行置换,然后加入脱水吡啶39g与脱水二甲苯455g而冷却至0℃以下,投入了二氯硅烷(DCS)25g。将该混合溶液搅拌2小时后,投入丙基胺29.6g,在反应温度-5~15℃进行了90分钟胺解。接着一边保持为-5~15℃一边将NH3以250ml/分钟注入26.8分钟,搅拌80分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)510的树脂(树脂L)的20重量%浓度的溶液(溶液L)。根据FT-IR谱图测定,确认了包含了丙基胺骨架的聚硅氮烷化合物的生成,NH/SiH比为0.10。另外,根据1H NMR谱图,NPr/SiH1,2比为0.43,SiH3/SiH1,2比为0.49。尝试了将溶液L旋转涂布向硅晶圆,但是无法在基板上形成膜。
比较例4
用干燥氮气将反应容器进行置换,然后加入脱水吡啶39g与脱水二甲苯456g而冷却至0℃以下,投入了二氯硅烷(DCS)25g。将该混合溶液搅拌1.5小时后,将异丙基胺0.3g混合溶解于20g的二甲苯并且投入,在反应温度-5~15℃进行了100分钟胺解。接着一边保持为-5~15℃一边将NH3以250ml/分钟注入70.4分钟,搅拌100分钟。进行过滤,将二甲苯混合于所获得的滤液而加热为40℃,在5mmHg的减压下蒸馏而去除吡啶,获得了包含重均分子量(Mw)2,000的树脂(树脂M)的20重量%浓度的溶液(溶液M)。根据FT-IR谱图测定,确认了包含了异丙基胺骨架的聚硅氮烷化合物的生成,NPr/SiH1,2比为0.002。
<焙烧膜的耐受电压以及湿法蚀刻率的测定>
将由实施例1~9制作出的溶液A~I、以及比较例2的溶液K、比较例4的溶液M分别旋转涂布于硅晶圆,为了去除溶剂因而加热为150℃,保持3分钟的(旋转涂布机=Mikasa制1H-DX2、热板=AS ONE Corporation制Shamal HHP-412)。利用水蒸气焙烧炉(Koyo Thermo Systems Co.,Ltd.制272A-M100)将所获得的试样在300℃、80%水蒸气气氛下,进行了1小时的退火。其后,通过下述方法而实施了各自的膜的耐受电压特性以及湿法蚀刻率的测定。结果示于表1。
(耐受电压特性)
使用日本SMS制SSM495 272A-M100进行测定。将电流密度超过了1E-6(A/cm2)时的电场(Field)设为Fbd(MV/cm)。
(湿法蚀刻率)
将所获得的二氧化硅质膜浸没于0.5%氟化氢水溶液,测定出膜厚减少速度。
表1
根据表1可知,与使用由比较例2表示的历来公知的无机聚硅氮烷树脂而在300℃的焙烧温度获得二氧化硅质膜的情况相比,通过使用在聚合物中含有在氮原子上被氢原子以外的基团取代了的骨架的本发明的共聚聚硅氮烷树脂,可在300℃以下的低温下的焙烧条件下较大地改善耐受电压特性和对溶剂的耐受性。另外可知,相比较于由比较例3表示的NR/SiH1,2比大的、在聚合物中含有在氮原子上被氢原子以外的基团取代的骨架的共聚聚硅氮烷树脂、由比较例4表示的NR/SiH1,2比低的、在聚合物中含有在氮原子上被氢原子以外的基团取代的骨架的共聚聚硅氮烷树脂而言,本发明的共聚聚硅氮烷树脂具有有利于在300℃以下的低温下的焙烧条件下进行氧化的结构。
- 还没有人留言评论。精彩留言会获得点赞!