新型稠合咪唑并苯并噻唑化合物的制作方法
本发明涉及新型稠合咪唑并苯并噻唑衍生物,它们的药学上可接受的盐,以及它们的异构体,立体异构体,阻转异构体(atropisomer),构象异构体(conformer),互变异构体,多晶型物,水合物,溶剂化物和N-氧化物。本发明也涵盖所述化合物的药学上可接受的组合物和制备新型化合物的方法。本发明进一步涉及以上提及的化合物用于制备用作药品(pharmaceuticals)的药物(medicament)的用途。
背景技术:
尽管治疗取得进展,气道疾病的发病率最近数十年来仍在升高。在所述气道疾病中,哮喘恶化和慢性阻塞性肺部疾病(COPD)是住院的主要原因。哮喘和慢性阻塞性肺病都涉及呼吸道的慢性炎症。尽管表现类似的症状,如呼吸困难、咳嗽、喘息和咳痰(expectoration),但这些气道疾病具有不同的基本病理生理过程。COPD是指特征在于干扰正常呼吸的气流阻塞的一大组肺部疾病的术语。肺气肿和慢性支气管炎是构成COPD(COPD-Chronic Bronchitis&Emphysema;Australian lung foundation,2006)的最重要的病症。COPD涉及外周气道和肺实质的慢性炎症,这导致所述气道逐渐狭窄和气短。另一方面,哮喘的特征在于偶发性气道阻塞和症状,并通常在生命早期开始。所述炎症在哮喘和COPD之间是显著不同的,具有不同的细胞、递质、后果并且,对皮质类固醇激素的响应存在差异(Clinics(Sao Paulo).2012;67(11):1335-43)。然而,最近已经变得很清楚的是,严重哮喘更加类似于COPD,在所述发炎方面具有相似性并且都对皮质类固醇激素响应较差(J Allergy Clin Immunol.2013;131(3):636-45)。有趣的是,分子遗传学的研究目前证明,严重哮喘和COPD共享了多个基因多态性(Comp Funct Genomics.2012;2012:968267)。
慢性阻塞性肺部疾病(COPD)是一种越来越普遍的,特别是发展中国家的主要的全球性健康问题。它是世界上最常见的疾病之一,估计终生风险高达25%,而现在同样影响男性和女性(Nature Reviews 2013;12:543-559)。
COPD的当前治疗形式是相对无效的,因为还没有大大减轻疾病进展或死亡率或对病情加重具有显著影响的药物可用,其是住院最常见的原因之一。
长效支气管扩张剂是目前COPD疗法的支柱。在一天只需要一次给药的β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂的开发方面取得了若干进展。此外,长效β2肾上腺素受体激动剂(LABA)和长效毒蕈碱乙酰胆碱受体拮抗剂(LAMA)对支气管扩张和症状改善具有加效性效应(additive effect),这已导致LABA-LAMA组合吸入剂的开发。然而,尽管这些药物产生了有效的支气管扩张,但它们无法治疗患有COPD的患者中基本的炎性疾病。
可替代地或除了支气管扩张剂之外,口服或吸入的皮质类固醇也能够用作COPD治疗。但是皮质类固醇具有局限性,因为长期口服皮质类固醇治疗是不推荐的并且吸入的皮质类固醇已知与患者肺炎风险增加相关,(www.bcguidelines.ca)。此外,吸入的皮质类固醇据发现在显著大量的COPD患者中作为COPD中抗炎疗法基本无效(Ann Fam Med.2006;4(3):253-62)。磷酸二酯酶抑制剂(PDE-4抑制剂)最近已经证明在COPD中记载临床疗效(document clinical efficacy),但是其效用受阻于种类相关的副作用(International Journal of COPD 2007;2(2):121-129)。
通过更好理解COPD疾病进程的病理生理和认知作为重要特征的发炎,可以预料的是,COPD靶向基本炎症的疾病改善疗法将证明是有效的,所述方式已经在像RA的其它慢性炎性疾病的治疗中取得了成功。
许多激酶参与了促炎性转录因子和炎性基因的调节。所述丝裂原活化的蛋白激酶(MAPK)家族包括所述p38激酶,其由响应于炎性信号活化的高度保守的脯氨酸导向丝氨酸-苏氨酸蛋白激酶构成。所述p38 MAPK途径(其由细胞应激活化),调节涉及COPD的许多炎性基因的表达(Nature Reviews 2013;12:543-559)。促炎性细胞因子/趋化因子和环境应激会通过磷酸化作用而激活p38丝裂原活化的蛋白激酶(MAPK),这进而激活所述p38 MAPK信号通路。p38涉及通过活化和释放促炎性细胞因子/趋化因子、这些基因的翻译后调节和炎性细胞迁移的活化,通过不同刺激诱导的炎性响应。因此,p38抑制剂呈现了对于包括COPD的慢性炎性病症潜在有吸引力的治疗靶。迄今已知的所述四种亚型中,p38α在炎性细胞中是最丰富,并研究最多。
在过去的二十年中,p38 MAPK(丝裂原活化的蛋白激酶(mitogen-activated protein kinase))一直是强烈的多学科研究的课题。p38 MAPK抑制剂已经证明在多个疾病模型,包括类风湿关节炎、牛皮癣、克罗恩氏病和中风中是有效的。最近的研究支持p38 MAPK在许多肺部疾病,如哮喘、囊性纤维化、特发性肺纤维化和慢性阻塞性肺部疾病的发展、维持和/或加重方面的作用。现在有丰富的文献表明p38 MAPK在慢性炎症中会被激活,并且其激活会导致进一步的促炎性细胞因子的精细制作(elaboration)和释放(Expert Opin.Investig.Drugs 2008;17(10):1411-1425)。
虽然靶向p38 MARK的口服给药小分子抑制剂已经证明在最初临床研究中降低获自COPD患者的细胞和组织中发炎的各种参数方面是有效的,但阻碍p38 MAPK抑制剂在治疗人类慢性炎性疾病中的潜在利用的定义和利用的主要障碍是在患者中已经观察到的毒性。这已经足够严重而导致从许多所述化合物已经进展的临床开发中撤出。目前,因为与所选分子相关的一种或其它问题,如毒性或选择性,它们中没有任何一种在世界上任何地方获批(Expert Opin.Investig.Drugs 2008;17(10):1411-1425&Chest 2011;139(6):1470-1479)。
例如,进入临床试验的吡啶基咪唑类的p38 MAPK抑制剂据发现与不可接受的安全曲线相关。对其他类似的p38 MAPK抑制剂临床前和临床两种报告的所述副作用包括肝毒性,心毒性,轻头晕以及其他CNS毒性,皮疹,胃肠道症状和感染。对于这些分子也出现了选择性的一些问题。类似地,BIRB-796,一种ATP的非竞争性p38 MAPK抑制剂,对其活性进行了评价,但它可能因为肝酶升高已经从类风湿性关节炎的II期临床试验中撤回(Expert Opin.Investig.Drugs 2008;17(10):1411-1425&Chest 2011;139(6):1470-1479)。
采用另一p38 MAPK抑制剂VX-745在类风湿性关节炎患者中的II期研究显示出相比于所测试的单一低剂量的安慰剂的显著临床益处。然而,它也因为在6个月的安全性研究期间在犬中未披露的CNS毒性而中断(Expert Opin.Investig.Drugs 2008;17(10):1411-1425)
为了克服与已知的p38 MAPK抑制剂相关的所述靶的毒性和选择性的这些问题,设计了一些替代策略。其中之一是设计p38激酶抑制剂直接剂量给药于所述发炎的器官的治疗方法。
其他策略包括开发具有改进的选择性和较小副作用曲线的新一代p38 MAPK抑制剂。例如,PH-797804和Losmapimod已经证明在临床研究中在治疗最高达12~24周时具有很好的耐受性(Thorax.2013Aug;68(8):738-45&J Clin Pharmacol.2012Mar;52(3):416-24)。
但仍然需要确定和开发提供所需的治疗潜力连同改善的药动学曲线和/或更小的副作用的新型p38 MAPK抑制剂。
WO 200214321公开了作为STAT-6抑制剂用于治疗癌症或对于其他抗癌治疗敏化癌细胞的多环咪唑衍生物。
GB 2232666和GB 2211186公开了作为苯并二氮杂卓反向激动剂用于治疗记忆问题、肥胖或能够用作次要镇静剂(minor tranquillizer)的咪唑并苯并噻唑。
本发明提供了作为p38 MAPK抑制剂的新型稠合咪唑并苯并噻唑衍生物,这已经证实了所需的疗效和安全曲线。
技术实现要素:
在一个实施方式中,本发明提供了式(I)的新型化合物,
它们的药学上可接受的盐以及它们的异构体,立体异构体,阻转异构体,构象异构体,互变异构体,多晶型物,水合物,溶剂化物和N-氧化物;
其中,
X选自O,S(O)n,NH和N(C1-C3)烷基;
R1和R2独立地选自氢,A,CHO,C=N-OH,C=N-O-(C1-C6)烷基,CH2OH,CH2R3,N(R5)CO2R4,CH2-卤素,NR5R6,N(R5)C(O)-A,N(R5)S(O)m-A,N(R5)C(O)N(R5)-A,N(R5)C(S)N(R5)-A,C(O)NR5R6,CO2R7,C(O)-A,CH(OH)-A C(CH3)=N-OH,C(CH3)=N-O-(C1-C6)烷基,C(O)CH2-卤素和C(O)CH2R3;
R独立地选自氢,(C1-C6)烷基,(C3-C10)碳环,CN,CHO,C(O)-A,C(CH3)=N-OH,C(CH3)=N-O-(C1-C6)烷基,C(O)CH2-卤素,C(O)CH2R3,NR5R6,N(R5)C(O)-A,N(R5)S(O)m-A,N(R5)C(O)O-A,N(R5)C(O)N(R5)-A,N(R5)C(S)N(R5)-A,CO2R7,C(O)N(R5)-A,(C1-C6)烷基-OR7,(C1-C6)烷基-卤素,(C1-C6)烷基-N3,(C1-C6)烷基-NR5R6,(C1-C6)烷基-N(R5)C(O)-A,(C1-C6)烷基-N(R5)S(O)m-A,(C1-C6)烷基-N(R5)C(O)O-A,(C1-C6)烷基-N(R5)C(O)N(R5)-A,(C1-C6)烷基-N(R5)C(S)N(R5)-A和(C1-C6)烷基-OC(O)N(R5)-A;
A独立地选自(C1-C6)烷基,(C3-C10)碳环,芳基,杂芳基和杂环,所述(C1-C6)烷基、(C3-C10)碳环、芳基、杂芳基或杂环可以可选地用独立地选自以下各项的1-3个取代基取代:卤素,(C1-C6)烷基,(C3-C10)碳环,芳基,杂芳基,杂环,羟基,CF3,OCF3,O(C1-C6)烷基,O-(C3-C10)碳环,NO2,C(O)-(C1-C6)烷基,C(O)CH2-卤素,C(O)CH2R3,NR5R6,CO2R7,C(O)N(R5)-A,N(R5)S(O)m-A,SH,S(O)n(C1-C6)烷基,S(O)mN(R5)-A,CN,CHO,(C1-C6)烷基-OR7,(C1-C6)烷基-卤素和(C1-C6)烷基-NR5R6,其中每个芳基或杂芳基可以进一步可选地用独立地选自以下各项的1-3个取代基取代:卤素,(C1-C6)烷基,(C3-C10)碳环,芳基,杂芳基,杂环,羟基,CF3,OCF3,O(C1-C6)烷基,O-(C3-C10)碳环,NO2,C(O)-(C1-C6)烷基,C(O)CH2-卤素,C(O)CH2R3,NR5R6,CO2R7,C(O)N(R5)-A,N(R5)S(O)m-A,SH,S(O)n(C1-C6)烷基,S(O)mN(R5)-A,CN,OSO3H,CHO,(C1-C6)烷基-OR7,(C1-C6)烷基-卤素,(C1-C6)烷基-NR5R6和
R3独立地选自O-A,NR5R6,S(O)n-A,S(O)n-(C1-C6)烷基-CO2(C1-C6)烷基,S(O)n-(C1-C6)烷基-OH,S(O)n-(C1-C6)烷基-CO2H,N(R5)C(O)-A,N(R5)C(O)O-A,N(R5)C(O)N(R5)-A,N(R5)S(O)m-A,N(R5)C(O)-杂环和N(R5)C(S)N(R5)-A;
R4是氢或A;
R5和R5′独立地选自氢,(C1-C6)烷基,(C1-C6)烷基-(C3-C10)碳环和(C3-C10)碳环;
R6和R6′独立地选自氢,A,(C1-C6)烷基-OH,(C1-C6)烷基-NR5'R6',CH(CH2OH)-芳基,CH(CH2OH)2,(C1-C6)烷基-芳基,(C1-C6)烷基-杂环和(C1-C6)烷基-杂芳基;
R5和R6或R5′和R6′连同与它们连接的氮原子一起可以形成3~8元单环或8~12元双环杂环,其环可选地含有选自O,S或N的其它杂原子以及所述环可选地被一个或多个R9或R10取代基取代。所述环的氮也可以形成N-氧化物。在双环杂环系统中,所述环能够以螺或稠合方式彼此连接;
R7是氢或A;
每个R8独立地是1-2个取代基并且每一个选自氢,卤素,A,CN,CHO,C(O)-A,C(O)CH2-卤素,C(O)CH2R3,羟基,CF3,OCF3,NR5R6,N(R5)C(O)-A,N(R5)S(O)m-A,C(O)N(R5)-A,O-(C1-C6)烷基,O-(C3-C10)碳环,S(O)n-A和S(O)mN(R5)-A,其中R和R8不同时是氢;
R9独立地选自氢,卤素,A,羟基,CF3,OCF3,O(C1-C6)烷基,O-(C3-C10)碳环,NO2,C(O)-A,C(O)CH2-卤素,C(O)CH2R3,NR5R6,N(R5)C(O)O-A,N(R5)C(O)N(R5)-A,N(R5)C(S)N(R5)-A,CO2R7,C(O)N(R5)-A,CN,CHO,(C1-C6)烷基-OR7,(C1-C6)烷基-卤素,(C1-C6)烷基-芳基,(C1-C6)烷基-NR5R6,(C1-C6)烷基-N(R5)C(O)O-A,(C1-C6)烷基-N(R5)C(O)N(R5)-A,(C1-C6)烷基-N(R5)C(S)N(R5)-A,(C1-C6)烷基-OC(O)N(R5)-A和N(R5)S(O)m-A;
R10选自氢,卤素,A,羟基,(C1-C6)烷基-(C3-C10)碳环,(C1-C6)烷基-芳基,C(O)-A,CO2R7,C(O)N(R5)-A,C(O)(C1-C6)烷基-A,氧代,硫代,=N-OH,=N-O-(C1-C6)烷基,O-(C1-C6)烷基,O-(C3-C10)碳环,O-芳基,O-杂芳基,S(O)n-A,NR5R6,N(R5)C(O)-A,N(R5)C(O)O-A,N(R5)C(O)N(R5)-A,N(R5)S(O)m-A,N(R5)C(O)-杂环和N(R5)C(S)N(R5)-A;
m为1或2;
n为0,1或2;
在另一实施方式中,本发明涉及如上的化合物,然而仅包括其药学上可接受的盐。
在另一实施方式中,本发明涉及如上的化合物,然而仅包括N-氧化物。
在另一实施方式中,本发明包括用于制备式(I)的化合物的合成中间体和制备这些中间体的方法。
本发明的另一个实施方式是制备如本文中方案1~6所述的式(1)的化合物的方法。
本发明的另一个实施方式是包含可选地在与药学上可接受的佐剂或载体混合的式(I)的化合物的药物组合物。
本发明的另一个实施方式是通过向有此需要的哺乳动物,包括人类给予治疗有效量的式(I)的化合物而治疗过敏和非过敏性气道疾病的方法。
本发明的另一个实施方式是通过向有此需要的哺乳动物,包括人类给予治疗有效量的式(I)的化合物而治疗慢性阻塞性肺部疾病和哮喘的方法。
本发明的另一个实施方式是式(I)的化合物用于制备治疗过敏和非过敏性气道疾病的药物的用途。
本发明的另一个实施方式是式(I)的化合物用于制备治疗慢性阻塞性肺部疾病和哮喘的药物的用途。
附图说明
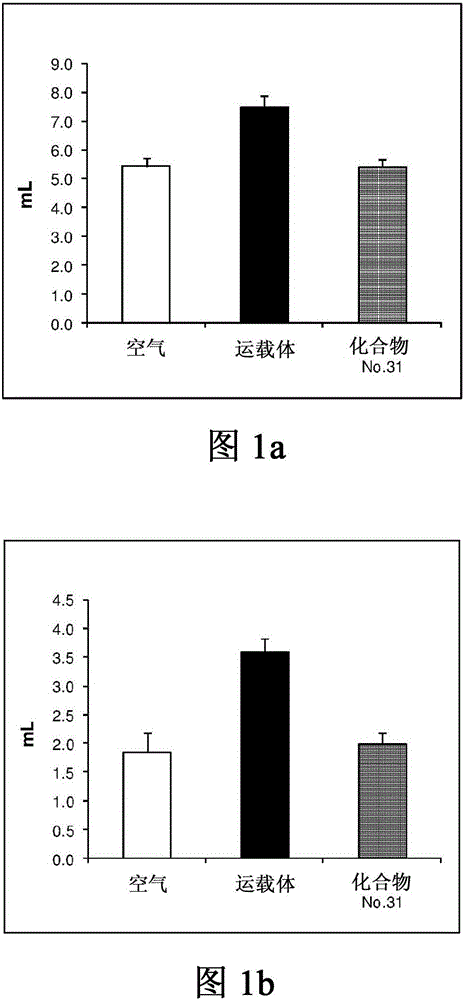
图1:化合物No.31的治疗对肺功能参数的影响;
1.功能性残余容量(图1A),2.肺的残余容量(图1b)。
图2:化合物No.31的治疗对肺功能参数的影响;
1.吸气容量与肺总容量之比(图2a)和2.总肺阻力(图2b)。
具体实施方式
在一个实施方式中,本发明提供了式(I)的新型化合物,
它们的药学上可接受的盐以及它们的异构体,立体异构体,阻转异构体,构象异构体,互变异构体,多晶型物,水合物,溶剂化物和N-氧化物,其中R、R1、R2、R8、R9和X,如以上定义。
在另一实施方式中,本发明提供了式(I)的新型化合物,
它们的药学上可接受的盐以及它们的异构体,立体异构体,阻转异构体,构象异构体,互变异构体,多晶型物,水合物,溶剂化物和N-氧化物;
其中
X是O或S(O)n;
R、R1、R2、R8、R9和n如以上定义。
在优选的实施方式中,本发明提供了式(I)的新型化合物,
它们的药学上可接受的盐以及它们的异构体,立体异构体,阻转异构体,构象异构体,互变异构体,多晶型物,水合物,溶剂化物和N-氧化物;
其中,
X是O,NH或S(O)n;
R1和R2独立地选自氢,A,CHO,CH2OH,CH2R3,CH2-卤素,N(R5)CO2R4,C(O)NR5R6,CO2R7和C(O)-A;
R独立地选自CN,CHO,C(O)-A,NR5R6,N(R5)C(O)-A,N(R5)C(O)O-A,N(R5)C(O)N(R5)-A,C(O)N(R5)-A,(C1-C6)烷基-OR7,(C1-C6)烷基-卤素,(C1-C6)烷基-N3,(C1-C6)烷基-NR5R6,(C1-C6)烷基-N(R5)C(O)-A,(C1-C6)烷基-N(R5)C(O)O-A,(C1-C6)烷基-N(R5)C(O)N(R5)-A和(C1-C6)烷基-OC(O)N(R5)-A;
A独立地选自(C1-C6)烷基,(C3-C10)碳环,芳基,杂芳基和杂环,所述(C1-C6)烷基、芳基或杂芳基可以进一步用独立地选自以下各项的1-3个取代基取代:卤素,(C1-C6)烷基,(C3-C10)碳环,芳基,杂环,羟基,CF3,O(C1-C6)烷基,N(R5)S(O)m-A和(C1-C6)烷基-OR7,每个芳基可以进一步用独立地选自以下各项的1-3个取代基取代:卤素,(C1-C6)烷基,羟基,OSO3H,O(C1-C6)烷基和
R3独立地选自O-A,NR5R6,S(O)n-A,S(O)n-(C1-C6)烷基-CO2(C1-C6)烷基和S(O)n-(C1-C6)烷基-OH;
R4是氢或A;
R5是氢或(C1-C6)烷基;
R6独立地选自氢,A,(C1-C6)烷基-OH,CH(CH2OH)-芳基,CH(CH2OH)2,(C1-C6)烷基-芳基,(C1-C6)烷基-杂环和(C1-C6)烷基-杂芳基;
R5和R6连同它们连接的氮一起可以形成3~8元单环杂环,其环含有选自O、S和N的其它杂原子以及所述环被R9取代;所述环的氮也可以形成N-氧化物;
R7是氢或A,其是(C1-C6)烷基;
R8是氢或A,其是(C1-C6)烷基;
R9是氢,羟基或A,其是(C1-C6)烷基;
n为0;
m为2。
本发明范围内特别感兴趣的特殊化合物家族由以下这些化合物及其药学上可接受的盐构成:
定义
下列定义,除非另外受限于具体情况中,都适用于整个本说明书中使用的所述术语:
本文所使用的术语“化合物”是指由本文中公开的所述通式涵盖的任何化合物。本文中描述的化合物可以包含一个或多个双键,因此,可以作为异构体、立体异构体,如几何异构体,E和Z异构体存在,并且可以具有不对称碳原子(光学中心),因此可以作为对映体、非对映异构体存在。因此,本文中描述的所述化学结构涵盖了所述举例说明的化合物的所有可能的立体异构体,包括立体异构纯的形式(例如,几何纯)和立体异构体混合物(外消旋物)。本文中描述的化合物可以作为构象异构体,如椅式或船式存在。本文中描述的化合物也可以作为阻转异构体存在。所述化合物也可以以多种互变异构体形式,包括烯醇式,酮式及其混合物存在。因此,本文中所述的化学结构涵盖了所举例说明的化合物的所有可能的互变异构体形式。所描述的化合物还包括其中一个或多个原子具有不同于自然界中传统发现的原子质量的原子质量的同位素标记化合物。可以引入本发明的化合物中的同位素实例包括但不限于2H,3H,13C,14C,15N,18O,17O等。化合物可以以非溶剂化形式,以及溶剂化形式,包括水化形式存在。在一般情况下,化合物可以水化或溶剂化。某些化合物可以以多晶或无定形形式存在。在一般情况下,所有物理形式对于本文中设想的用途是等同的并且预想都在本发明的范围之内。
描述本发明的上下文(尤其是在随附权利要求的上下文)中的所述术语“一种”、“一个”和“该”以及类似的指示对象的使用应该解释为包括单数和复数两者,除非本文另外指出或与上下文明显矛盾。
本文中所示的本发明的化合物的命名都是根据具有“logD Suite”的ACD/Lab's ChemDraw(12.0版)进行命名。
“药学上可接受的盐”是指化合物的盐,其具有所述母体化合物的所需药理学活性。这些盐包括:(1)酸加成盐,与无机酸如盐酸、氢溴酸、硫酸、硝酸、碳酸、磷酸等形成;或与有机酸如乙酸、丙酸、异丁酸、己酸、环戊烷丙酸、草酸、乙醇酸、丙酮酸、乳酸、丙二酸、琥珀酸、辛二酸、苹果酸、马来酸、富马酸、酒石酸、柠檬酸、苯甲酸、3-(4-羟基苯甲酰基)苯甲酸、苯二甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、1,2-乙二磺酸、2-羟基乙磺酸、苯磺酸、4-氯苯磺酸、2-萘磺酸、4-甲苯磺酸、樟脑磺酸、4-甲基双环[2.2.2]-辛-2-烯-1-羧酸、葡庚糖酸、3-苯基丙酸、三甲基乙酸、叔丁基乙酸、月桂基硫酸、葡糖酸、葡糖醛酸、半乳糖醛酸、谷氨酸、羟基萘甲酸、水杨酸、硬脂酸、粘康酸等形成;或(2)如下形成的盐,当母体化合物中存在的酸性质子被金属离子,例如碱金属离子、碱土离子或铝离子取代时;或与有机碱,如乙醇胺、二乙醇胺、三乙醇胺、N-甲基葡糖胺等配位。还包括的是氨基酸如精氨酸等的盐(参见,例如,Berge,S.M.,et al.,“Pharmaceutical Salts”,Journal of Pharmaceutical Science,1977,66,1-19)。
正如本文所用,所述术语“多晶型物”涉及具有相同化学式、相同盐类型和具有水合物/溶剂化物相同形式但具有不同结晶学性质的化合物。
如本文所用的,所述术语“水合物”涉及具有多个键合至所述化合物的水分子的化合物。
正如本文中所用,所述术语“溶剂化物”涉及具有多个键合至所述化合物的溶剂分子的化合物。
正如本文中所用,“N-氧化物”是指具有氧化的氮原子的化合物。
本发明还涵盖前药形式的化合物。本文描述的所述化合物的前药是容易经历生理条件下的化学变化(在体内)以提供本发明化合物的那些化合物。此外,前药能够通过在体外环境中的化学或生化方法,例如,具有合适酶或化学品的透皮贴剂储药器而转化成本发明的化合物。前药,在一些情况下,比所述母体药物更容易给药。它们可以,例如,通过口服给药而是生物可利用的,然而所述母体药物却不行。所述前药还可以具有比所述母体药物在药物组合物中改善的溶解性。所述化合物的酯、肽基衍生物等是本发明前药的实例。包含羧基基团的本发明化合物的体内可水解的(或酶切的)酯是,例如,在人或动物体内经过水解而产生所述母体酸的药用酯。
本发明还涵盖S-氧化物形式的化合物。正如本文中所用,“S-氧化物”是指具有氧化的硫原子的化合物。
所述术语“取代的”,正如本文中所用,包括通过所命名的取代基单-和多-取代至这种单和多取代(包括在相同位点上的多个取代)在化学上允许的程度并意味着所指示的原子上任何一个或多个氢用来自所指定的基团的选择进行取代,条件是不超过所指示的原子正常价态,并且所述取代会导致产生稳定的化合物,例如,当取代基是酮基时,则所述原子上的两个氢被取代。所有取代基(R,R1,R2....)以及它们在本文中所描述的进一步的取代基可以在导致形成稳定化合物的任何杂原子或碳原子上连接至所述主结构。
正如本文中所用,“卤素”取代基是选自氯,溴,碘和氟的一价卤素基团。
单独或附连另一基团使用的所述术语“(C1-C6)烷基”是指具有1~6个碳原子并且是未取代的或取代的脂族烃基团。所述“(C1-C6)烷基”可以是直链(例如,甲基,乙基,正丙基,正丁基,正戊基,正己基)或支链(例如,异丙基,异丁基,仲丁基,叔丁基)的,并且它可以包含一个或两个双或三键。所述(C1-C6)烷基也可以包含以螺环方式的(C3-C6)环烷基环。
单独或附连另一基团使用的所述术语“(C3-C10)碳环”是指具有3~10个碳原子并且是未取代的或取代的环状环系统。所述“(C3-C10)碳环”是指在环系统骨架中仅含有碳原子的环状环系统如环丙基,环丁基,环戊基,环己基。碳环可以包括双环稠合环。碳环可以具有任何饱和度,条件是所述环系统中的至少一个环不是芳族的。
所述术语“芳基”是指芳族基团,例如,其是6~10元单环或双环含碳环系统。所述芳基基团包括,但不限于,苯基,萘基,联苯基,四氢萘基和茚满。优选芳基是可以进一步被(C1-C6)烷基、N(R5)S(O)m-A、N(R5)C(O)-A、O(C1-C6)烷基、卤素、羟基、CF3或OCF3取代的苯基。
所述术语“杂芳基”是指芳族基团,例如,其是5~14元单环或二环的环系统,其具有至少一个杂原子。所述术语“杂原子”,正如本文中所用,包括O,N,S。在双环系统中,环能够通过桥杂原子稠合。所述杂芳基基团包括,但不限于吡咯基,呋喃基(furanyl)(呋喃基(furyl)),硫代苯基(噻吩基),吡唑基,咪唑基,噁唑基,异噁唑基,噻唑基,三唑基,噁二唑基,噻二唑基,四唑基,吡啶基(pyridinyl)(吡啶基(pyridyl)),哒嗪基,嘧啶基,吡嗪基,三嗪基,吲哚基,苯并呋喃基,苯并硫代苯基(苯并噻吩基),吲唑基,苯并咪唑基,苯并噁唑基,苯并异噁唑基,苯并噻唑基,喹啉基,异喹啉基,噌啉基,喹唑啉基,喹喔啉基,酞嗪基或萘啶基。优选杂芳基是吡唑基和异噁唑基,最优选杂芳基是吡唑基。
所述术语“杂环状”或“杂环”是指完全或部分饱和的环状基团,例如,其是一个3~14元单环或双环系统,其具有至少一个杂原子。正如本文中所用的所述术语“杂原子”包括O,N,S。在双环杂环系统中,至少一个环不是芳族的,并且所述环也能够以螺环方式彼此连接。杂环或杂环基团包括,但不限于,环氧乙烷基,氮丙啶基,氧杂环丁基,氮杂环丁烷基,吡咯烷基,二氢吡咯基,四氢呋喃基,二氢呋喃基,四氢噻吩基,二氢噻吩基,吡唑烷基,咪唑烷基,噁唑烷基,异噁唑烷基,噻唑烷基,三唑烷基,噁二唑烷基,哌啶基,四氢吡啶基,二氢吡啶基,哌嗪基,四氢吡喃基,二噁烷基,吗啉基,三嗪烷基(triazinanyl),氮杂环庚烷基,二氮杂环庚烷基,二氮杂卓基(diazepinyi),氧杂庚烷基(oxepanyl),二氧杂环庚烷基(dioxepanyl),氧杂氮杂环庚烷基(oxazepanyl),氧杂氮杂卓基(oxazepinyl),吲哚啉基,苯并吗啉基,四氢喹啉基或四氢异喹啉基。
正如本文中所用,“羟基(hydroxyl)”或“羟基(hydroxy)”是指-OH基团。
正如本文中所用,“室温”指的是20~30℃之间的温度。
正如本文中所用,所述术语“哺乳动物”是指人或动物如猴,灵长类,狗,猫,马,牛等。
正如本文中所用的所述术语任何疾病或失调的“治疗”或“治疗处理”是指向有此需要的哺乳动物,包括人类给药化合物。所述化合物可以经过给药而由此提供完全或部分防止或延缓疾病(disease)或失调(disorder)或病征(sign)或其症状(symptom)发作的预防效果;和/或所述化合物可以经过给药而由此提供对疾病或失调和/或可归因于所述失调的有害影响的部分或完全治愈。
所述短语“治疗有效量”是指化合物,当给予患者用于治疗疾病时,足以实现对所述疾病的这种治疗的量。所述“治疗有效量”将根据所述化合物、给药模式、所述疾病及其严重度和所要治疗的患者年龄、体重等而变化。
贯穿本说明书和所附的权利要求书中,要理解的是,所述词语“包含(comprise)”和“包括(include)”及其变体如“包含(comprises)”,“包含了(comprising)”,“包括(includes)”,“包括了(including)”都包含性地进行解释,除非上下文另有要求。即,这些词的使用可以暗示包括没有专门引述的一个或多个要素。
在另一实施方式中,本发明提供了制备式(I)的化合物的方法。
以下的反应方案用于公开根据本发明所述化合物的合成。
因此,本发明式(I)的化合物可以按照以下这些方案所述进行制备。
式(I)的化合物的示例性实施方式包括式Ia、式Ib、式Ic、式Id、式Ie、式If、式Ig、式Ih、式Ii、式Ii-1、式Ij、式laa、式lab、式lac、式lad、式lae、式laf、式lag、式Iba、式Ibb、式Ibc和式Ibd的化合物。其中所述取代基如结合结构通式(I)和方案1~6进行定义。
方案-1
式Ia-Ih的各种化合物的合成,其中R是L-N(R5)C(O)N(R5)-A,如方案1中所示,其中L在所述方案中定义而R5是H。式1a的所述化合物由式Iaa或Iba的化合物,分别与式Ila或Ila′的化合物在合适的碱如N,N-二异丙基乙胺(DIEA)和合适的溶剂如四氢呋喃(THF)或乙酸乙酯(EtOAc)存在下在回流温度下的反应而合成。另外,式Ia的化合物能够使用常规的合成方法通过其他合适的氨基甲酸酯的反应而合成。式Ila的化合物由适当取代的胺(A-NH2)与氯甲酸三氯乙酯在碱性水溶液如在溶剂如处于乙酸乙酯(EtOAc)中的碳酸氢钠/碳酸氢钾的存在下在室温(RT)下的反应而合成(J.Med.Chem.,2011,54,7797或WO 2007/091152)。式ILa'的化合物使用对于合成式ILa的化合物所述的类似条件由合适的氯甲酸苯酯与胺(A-NH2)的反应而合成。式lb的化合物由式Ia的化合物用合适的还原剂如金属硼氢化物如硼氢化锂(LiBH4)或氢化锂铝(LAH)或在适宜溶剂如四氢呋喃(THF)存在下在惰性气氛中回流或更低温度,通常在回流温度下还原而制备。式Ic的化合物由式Ib的化合物用合适的氯化剂如亚硫酰二氯在回流温度或更低温度下在合适的溶剂如二氯甲烷(DCM)存在下进行合成。式Id的化合物由式Ic的化合物与合适的R3H反应而合成。所述反应在合适的溶剂或溶剂混合物如THF、二甲基甲酰胺(DMF)中在室温(RT)至根据R3H性质的回流温度下进行。式Id的化合物的氧化,当R3是含硫(S)基团时,能够提供式Id的相应单-或二氧化物(S(O)m,其中m为1或2)化合物。式Ie和Ig的化合物能够通过式Ia的化合物水解而制得。所述水解反应采用合适的碱如氢氧化锂单水合物(LiOH·H2O)使用合适的溶剂如甲醇(MeOH)在RT至回流之间的温度下,通常在室温下完成。式If和Ih的化合物获自式Ie和Ig的化合物分别与合适的胺(R5R6NH)和合适的偶联剂如1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDCI.HCl)在合适的碱如Ν,Ν-二异丙基乙胺(DIEA)和催化性的4-二甲基氨基吡啶(DMAP)和合适的溶剂如THF存在下于惰性气氛中0℃至室温的偶联反应。当R2为CH2OCH3时,式Ib的一些化合物还能够由式Ia的化合物的去甲基化而合成。去甲基化使用在合适的溶剂如DCM中的三溴化硼(BBr3)在0℃至RT下进行。
方案-2
式Id的化合物也由式Ii的氨基甲酸酯或取代的异氰酸酯(A-N=C=O)如方案2所示进行合成。式Id的化合物能够由式Ii的化合物或式Ii-1的化合物与合适胺(A-NH2)在合适的碱如DIEA和合适的溶剂如甲苯存在下在室温至回流之间的温度,通常在回流下的反应进行制备。式Ii的化合物由式Iaa或Iba的化合物与合适的氯甲酸酯如氯甲酸苯酯或氯甲酸4-硝基苯酯在合适的碱如吡啶和合适的溶剂如THF存在下在0℃至室温下的反应而合成。式Ii-1的化合物能够由式Iaa或Iba的化合物与合适的氯甲酸酯如氯甲酸三氯乙酯在合适的碱如碳酸氢钠和合适的溶剂如EtOAc存在下的反应而合成。另一方面,当式Iaa或Iba的化合物用适当取代的异氰酸酯(A-N=C=O;其中A如以上定义)处理时,它在合适的溶剂如THF或甲苯-DMF存在下于升高的温度下会产生式Id的相应化合物。一些取代的异氰酸酯已经通过将取代羧酸与氯甲酸乙酯在叠氮化钠和三乙基胺(TEA)的存在下使用DMF作为溶剂进行反应而制备出来。在其它方法中,取代异氰酸酯也通过取代胺在三光气和TEA的存在下使用DCM作为溶剂于0℃至RT的温度下的反应而制备。
方案-3
式Ij和Ibc的化合物的合成,其中X是O,如方案3中所述。式Ij的化合物由式Ibc的化合物分两个步骤进行合成。式Ibc的化合物使用氢氧化钠水溶液(NaOH)作为碱和MeOH作为溶剂在RT下水解成相应酸衍生物(Ibd)。所述相应的酸衍生物与合适的取代胺(A-NH2)使用合适的偶联剂如EDCI·HCl和合适的碱如DIEA和催化性的DMAP在溶剂如THF中于0℃至室温下的惰性气氛中进行偶联。式Ibc的化合物由式-N的化合物与4-氟苯甲酸甲酯在碱如碳酸钾(K2CO3)或碳酸钠的存在下利用DMF作为溶剂在升高的温度下的反应而制备。式-N的化合物由式-M的化合物使用在非质子溶剂如DCM中BBr3在0℃至室温下的去甲基化而合成。式-M的化合物由式-C的化合物与氢氧化锂单水合物使用水性MeOH的碱性水解而制备。此外,所述水解产物在0℃至RT下与合适的取代胺(R5R6NH)使用合适的偶联剂如EDCI·HCl和合适的碱如DIEA和催化性的DMAP在非质子溶剂如THF中进行偶联。
方案-4
式Iaa、Iab、Iac、lad、Iae、Iaf和Iba的多种化合物按照方案4中所述进行合成。式Iaa的所述氨基苄基化合物通过不同的合成方法制备。在第一种方法中,其中任一位置-2上R是腈(CN)基团的式Lag的化合物,或其中位置-4上R是腈(CN)基团的式Ibb的化合物,使用硼氢化钠、三氟化硼-醚合物或硼烷-THF或硼氢化钠、三氟乙酸(TFA)在非质子溶剂如THF中在室温至回流进行还原而提供式Laa相应的2-或4-苄氨基化合物。在第二种方法中,其中位置-2上R是醛(CHO)基团的式Iag的化合物,通过在质子溶剂如MeOH中的硼氢化钠在0℃至室温下还原成式Iab的化合物。此外,一旦式Iab的化合物与二苯基磷酰基叠氮化物(DPPA)在1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)存在下在非质子溶剂如THF中于0℃至室温下发生反应,它就会提供式Iaa相应的2-苄氨基化合物。在第三种方法中,式Iab的化合物使用合适的氯化剂如亚硫酰氯在非质子溶剂如二氯甲烷中室温下进行氯化而提供相应的氯代衍生物Iac并且它在用氨在压力下处理之后提供式Iaa相应的2-苄氨基化合物。在第四方法中,氯代衍生物Iac与叠氮化钠在溶剂DMF中反应而提供相应的叠氮化物衍生物Iad,其通过三苯基膦(TPP)在溶剂如含水四氢呋喃中转化为式Iaa的相应2-苄氨化合物。其中位置-2上R是CN基团而R2是CO2Et基团的式Iag的化合物,一旦通过氢氧化锂单水合物在含水MeOH中RT下水解,就会产生式Iae的相应酸化合物。式Iae的化合物使用氯甲酸烷基酯如氯甲酸乙酯在叠氮化钠和TEA存在下在溶剂DMF中接着用合适R4H在甲苯中进行处理而转化成式Iaf的化合物。式Iaf的化合物的腈基团使用硼氢化钠、三氟化硼醚合物在溶剂THF中于回流温度下进行还原而获得式Iaa的相应2-苄氨化合物。另一方面,式Iba的4-氨基或2-氨基化合物由式Ibb和Iag的化合物分别进行制备。当分别地式Iag和Ibb的化合物中位置-2或位置-4任一位置上R是硝基(NO2)基团时,它通过还原剂如氯化亚锡在醇溶剂如MeOH中进行还原而提供式Iba的相应4-氨基或2-氨基化合物。此外,有机合成领域技术人员应该理解的是,各种其它R官能团都能够合成。
方案-5
其中X是O的式Iag和Ibb的各种化合物,按照方案5中所示进行合成。式Iag的一些化合物由式-D的化合物或式-G的化合物中任一化合物,分别与合适的2-氟苯基化合物如取代或无取代的2-氟苯甲醛或2-氟苄腈在室温至100℃下在碱如碳酸钾存在下于溶剂DMF中的反应而制备。用类似的步骤,式Ibb的化合物由式-D或G的化合物分别用合适的4-氟苯基化合物如取代或取代的4-氟硝基苯处理而合成。式-C或F的化合物分别用去甲基化剂如三溴化硼在非质子溶剂如DCM中去甲基化,分别获得式-D或G的化合物。式-C的化合物由取代的或未取代的2-氨基-6-甲氧基苯并噻唑与N,N-二甲基甲酰胺二甲基缩醛(DMF-DMA)在溶剂甲苯中反应,接着用溴代乙酸乙酯在升高的温度下季铵化(quaternisation)并最后使用DBU在溶剂如DMF中室温下环化而合成。然而式-F的化合物是由取代或未取代的2-氨基-6-甲氧基苯并噻唑与溴代丙酮酸乙酯在非质子溶剂如DMF中的反应,随后用氢氧化钠在醇溶剂如甲醇和乙醇中处理而合成。其中R2是CH2R3的式Iag的一些化合物,由式-L的化合物分两步进行制备。在第一步骤中,式-L的化合物用合适的R3H在合适的溶剂如DMF中室温下处理而提供其中R2是CH2R3的相应衍生物(式-Ll的化合物)。在当R3H是醇的情况下,所述试剂(R3H)也可以充当溶剂。所述反应在室温至回流下完成。式-L1的这种中间体化合物与合适的2-氟苯基化合物如取代的或未取代的2-氟苯甲醛或2-氟苄腈在碱如碳酸钾的存在下在溶剂DMF中于室温至100℃下的进一步反应,会提供式Iag的相应化合物。式-L的化合物获自使用氯化剂如亚硫酰氯在室温下非质子溶剂如DCM中式-K的化合物的氯化而获得。式-J的化合物的去甲基化,采用去甲基化剂如三溴化硼在非质子溶剂如DCM中,会产生式-K的化合物。当R1=CH3时式-J的化合物由式-H的化合物使用金属氢化物如硼氢化钠在质子溶剂如MeOH中室温下的还原而合成。一旦式-E的化合物用合适的甲酰化剂如POCI3在DMF中甲酰化,就会产生式-H的化合物。式-E的化合物由取代的或未取代的2-氨基-6-甲氧基苯并噻唑用炔丙基溴在溶剂DMF中处理,接着与醇钠如甲醇钠在醇溶剂如MeOH中反应而获得。式-J的化合物由式-C的化合物,当R1是H而R2是CO2Et时使用合适的还原剂如硼氢化锂和合适的溶剂如THF进行还原而合成。其中R2是CH2R3的式Iag的一些化合物,也由其中R1是H的式-J的化合物制备。式-J的化合物的氯化产生式-C的化合物,将其用R3H处理,使用用于由式-K的化合物分别制备式L和式-L1的化合物的类似条件而提供相应的衍生物C2。式-C2的化合物的去甲基化,使用用于由式-J的化合物制备式-K的化合物的类似条件而提供了相应的酚类衍生物L1。
方案-5a
其中X是O而R2是杂芳基的式Iag一些化合物,按照方案5a中所述进行合成。式Iag的化合物,由式-D-1的化合物的反应使用如方案5中所示对于其中X=O的式Iag化合物讨论的类似条件进行制备。式-C6的化合物的去甲基化使用如方案5中对于式-D的化合物讨论的类似条件而产生式-D-1的化合物。式-C4的化合物采用炔丙基胺使用合适的偶联剂如EDCI和HOBt在合适的碱如三乙胺存在下进行偶联而提供式-C5的化合物,其使用Organic Letters,2012,14,4478-4481中描述的类似条件转化为式-C6的化合物。式-C3的化合物的碱性水解获得式-C4的化合物。
方案6:
其中X是S的式Iag或Ibb各种化合物,如方案6中所述进行合成。当R1是H而R2是CO2Et时式Iag或Ibb的化合物,分别获自取代或未取代的2-氨基-6-硫代苯氧基苯并噻唑B和B1。式-B或B1的化合物分别用N,N-二甲基甲酰胺二甲基缩醛(DMF-DMA)在甲苯溶剂中处理,接着用溴代乙酸乙酯在升高的温度下季铵化和最后在非质子溶剂如DMF中使用DBU室温下环化。当R1是CO2Et而R2是H时式Iag和Ibb的化合物,分别获自取代的或未取代的2-氨基-6-硫代苯氧基苯并噻唑B和B1。本文中,式-B或B1的化合物,分别用溴代丙酮酸乙酯在非质子溶剂如DMF中处理,随后将其用氢氧化钠在醇溶剂如甲醇和乙醇中进行处理。
以上给出的方案1-6提供了本发明的化合物的通用制备方法。普通技术人员将会认识到适当取代起始原料中的各种基团如R、A、R8和R9等而制备根据式(I)的所需化合物。作为所给的方案的替代方案,普通技术人员根据本发明使用传统合成有机技术由可商购或可以易于制备的合适起始原料将很容易地合成这些化合物。
本发明的化合物可以具有手性中心并可以以外消旋体、外消旋混合物和作为单个非对映体或对映体出现(所有异构体形式都包括于本发明中)。因此,在化合物是手性的情况下,所述单独的对映体,基本上无其它对映体,都包括于本发明的范围内;进一步包括所述两个对映体的所有混合物。
本发明的所述新型化合物根据如本文中以上所述的方案的步骤使用合适的原料进行制备,并通过以下具体实施例进行进一步举例说明。这些实施例不应该当作或解释为限制所阐述的本发明的范围。
在本发明说明书中一些通用术语采取其本文中以下定义的已知期望的含义使用:
根据本发明制备的化合物的质量采用使用APCI电离技术(电喷雾化学电离探头)的单四极杆质谱仪(Water ZQ 2000instrument)或使用ESI或APCI的Finnigan LXQ(Thermo Instrument Technique)测定。
实施例:
实施例1:7-(2-甲酰基苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物51)
7-甲氧基咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(US 6191124)使用传统O-去甲基化方法例如BBr3/DCM去甲基化而提供7-羟基咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯化合物。
向在DMF(600mL)中的7-羟基咪唑并[2,1-b][1,3]苯并噻唑-3-羧酸乙酯(85g,320mmol)的搅拌溶液中加入碳酸钾(132g,960mmol)和2-氟苯甲醛(44.2g,350mmol),并将反应混合物(RM)在100~105℃下加热20~24h。反应混合物冷却至室温并滤出悬浮固体,用DMF洗涤。母液(ML)倾入水中并将pH调节至6.0~6.5。将所述分离的固体滤出,用水洗涤并干燥,而最后的固体在甲醇(300ml)中室温(RT)搅拌2~3h。将所述搅拌的固体过滤并真空干燥而得到80.0gm白色固体的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ10.40(1H,s),8.96(1H,d),8.05(1H,s),7.94(1H,s),7.89(1H,d),7.70(1H,t),7.30-7.43(2H,m),7.06(1H,d),4.38(2H,q),1.36(3H,t)
ESMS:367.30
实施例2:7-[2-(羟基甲基)苯氧基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物52)
向在甲醇(600ml)中的化合物7-(2-甲酰基苯氧基)咪唑并[2,1-b][1,3]苯并噻唑-3-羧酸乙酯(80g,218mmol)的搅拌溶液中0℃下分批加入硼氢化钠(24g,655mmol)并室温下搅拌10~12h。在60℃下真空除去甲醇,用水(500mL)稀释并用稀HCl将pH调节至5.0~6.0。所述分离的固体,经过过滤,用水洗涤,而最后用10%EtOAc/己烷在真空60℃下干燥而获得白色固体的标题化合物(80g)。
所述产物也可能由于酯交换而含有甲基酯衍生物。然而,当反应使用乙醇作为溶剂进行时,仅仅获得乙基酯。
1H-NMR(400MHz,DMSO-d6):δ8.89(1H,d),8.04(1H,s),7.69(1H,s),7.58(1H,d),7.21-7.32(3H,m),6.94(1H,d),5.19(1H,s),4.54(2H,s),4.38(2H,q),1.35(3H,t)
ESMS:369.32
实施例3:7-[2-(叠氮基甲基)苯氧基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物54)
向在DCM(300mL)中的化合物7-[2-(羟基甲基)苯氧基]咪唑并[2,1-b][1,3]苯并噻唑-3-羧酸乙酯(80gm,217mmol)的搅拌溶液中0~5℃下逐滴加入亚硫酰氯(51gm,434mmol)。反应混合物在室温下搅拌6~8h。反应混合物60℃下真空浓缩至干,而提供7-[2-(氯甲基)苯氧基]咪唑并[2,1-b][1,3]苯并噻唑-3-羧酸乙酯(化合物53)。将所述固体悬浮于DMF(400mL)中并在0~5℃下搅拌。在0℃下加入叠氮化钠(42gm,650mmol),并将反应混合物室温下搅拌10~12h。将反应混合物倾入冷却的水中并用乙酸乙酯(300×2)萃取。所述分离的有机层用水洗涤,用硫酸钠干燥并在真空60℃下蒸发而获得80gm标题化合物。
实施例4:7-[2-(氨基甲基)苯氧基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯盐酸盐(化合物55)
向在THF(400mL)中的化合物7-[2-(叠氮基甲基)苯氧基]咪唑并[2,1-b][1,3]苯并噻唑-3-羧酸乙酯(80gm,203mmol)的搅拌溶液中0~5℃下连同水(100mL)一起加入三苯基膦(80gm,305mmol)。反应混合物室温下搅拌10~12h。在60℃真空下蒸掉溶剂,进一步用水(~200mL)稀释,并用乙酸乙酯(500mL×2)萃取。有机层经硫酸钠干燥,并在60℃真空下浓缩至干,得到粗化合物。此粗化合物溶于THF(800mL)中并在室温下吹扫HCl气体长达2h。由此得到的所述固体经过过滤,用乙酸乙酯(500mL)和己烷(500mL)洗涤,而获得55gm白色固体的标题化合物。
如果7-[2-(叠氮基甲基)苯氧基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物54)连同甲基酯一起使用,所述产物也可能含有甲基酯衍生物。
1H-NMR(400MHz,DMSO-d6):δ8.96(1H,d),8.51(2H,bs),8.07(1H,s),7.88(1H,d),7.65(1H,d),7.36-7.42(2H,m),7.25(1H,t),6.93(1H,d)4.38(2H,q),4.09-4.10(2H,m),1.35(3H,t)
ESMS:368.04
化合物55也由7-[2-(羟基甲基)苯氧基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物52)使用DPPA、DBU和TPP在THF-H2O中使用文献中已知的标准方案而合成。
实施例5:7-[2-({[(2-羟基-1-苯基乙基)氨甲酰基]氨基}甲基)苯氧基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物42)
向在THF(20mL)和吡啶(0.260g,3.27mmol)中的7-[2-(氨基甲基)苯氧基]咪唑并[2,1-b][1,3]苯并噻唑-3-羧酸乙酯盐酸盐(1gm,2.72mmol)的搅拌溶液中在约0~5℃下加入碳酰氯4-硝基苯酯(0.66g,3.27mmol)。反应混合物在室温下搅拌4~6h。60℃下真空蒸掉溶剂。所得的氨基甲酸酯衍生物(0.3g,0.56mmol)悬浮于甲苯(20mL)中。向其中加入(R)-苯基甘氨醇(glycinol)(0.0078g,0.56mmol)和DIEA(0.150gm,1.12mmol)并回流4~6h。反应混合物冷却至室温并倒入水中,并用乙酸乙酯(10ml×2)萃取。所述有机层经硫酸钠干燥,并真空60℃下蒸发,而获得粗化合物,其通过柱色谱纯化,而得到0.110g固体。
1H-NMR(400MHz,DMSO-d6):δ8.90(1H,d),8.04(1H,s),7.71(1H,bs),7.36(1H,d),716-7.30(7H,m),6.94(1H,d),6.53(1H,d),6.46(1H,t),4.87(1H,t),4.65-4.66(1H,q),4.37(2H,q),4.23(2H,d),3.54(2H,m),1.35(3H,t)
ESMS:531.45
实施例6:7-(2-{[(环己基氨甲酰基)氨基]甲基}苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物46)
向在THF(20mL)中的7-[2-(氨基甲基)苯氧基]咪唑并[2,1-b][1,3]苯并噻唑-3-羧酸乙酯盐酸盐(0.7gm,1.9mmol)的搅拌溶液中加入异氰酸环己酯(0.8g,6.4mmol)。反应混合物回流18~20h。反应混合物冷却至室温。60℃下真空蒸掉溶剂,粘状物料(sticky mass)用冷水骤冷并用乙酸乙酯(10mL×2)萃取,并蒸发有机层而得到粗化合物。所述物质经柱层析纯化而得到0.05gm固体标题化合物。
1H-NMR(400MHz,CDC13)δ9.03(1H,d),8.00(1H,s),7.48(1H,d),7.26(1H,与溶剂信号合并),7.09-7.21(3H,m),6.90(1H,d),4.73(1H,t),4.38-4.43(4H,m),4.26(1H,d),3.42(1H,m),1.85-1.87(2H,m),1.54-1.57(1H,与溶剂信号合并),1.43(3H,t),1.23-1.32(3H,m),0.99-1.14(4H,m)
ESMS:493.33
实施例7:7-{2-[({[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]氨甲酰基}氨基)甲基]苯氧基}咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物25)
步骤1:[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]氨基甲酸2,2,2-三氯乙酯(中间体1)
向在乙酸乙酯(1500mL)和2N碳酸氢钠水溶液(500mL)中的4-(5-氨基-3-叔丁基-lH-吡唑-1-基)-2-氯苯酚(60gm,225.5mmol)的搅拌的溶液中室温下加入氯代甲酸三氯乙酯(119.32gm,563.90mmol)并室温下搅拌直至反应完成。分出乙酸乙酯层,硫酸钠干燥并60℃真空蒸发而获得粗化合物。所述粗化合物悬浮于己烷(100mL)中,0~5℃下搅拌以及滤出由此出现的固体并干燥而获得85gm的固体标题化合物。
1H-NMR(400MHz,CDC13):δ7.44(1H,s),7.22-7.24(1H,m),7.03(1H,d),6.70(1H,bs),6.39(2H,bs),4.81(2H,s),1.34(9H,s)
ESMS:442.24/444.21
使用如对于中间体1,[3-叔丁基-1-(3-氯-4-甲氧基苯基)-lH-吡唑-5-基]氨基甲酸2,2,2-三氯乙基酯(中间体2)所述的类似步骤,由相应的氨基吡唑化合物合成[3-叔丁基-1-(4-甲氧基苯基)-1H-吡唑-5-基]氨基甲酸2,2,2-三氯乙酯(中间体3)和[3-叔丁基-1-(4-甲基苯基)-lH-吡唑-5-基]氨基甲酸2,2,2-三氯乙酯(中间体4)。
步骤2:7-{2-[({[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]氨甲酰基}氨基)甲基]苯氧基}咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物25)
向在THF(600mL)中的7-[2-(氨基甲基)苯氧基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯盐酸盐(化合物55)(55gm,136mmol)的搅拌溶液中加入N,N-二异丙基乙胺(70.33gm,545mmol)和中间体1(60gm,136mmol)。反应混合物回流8~12h。60℃真空蒸掉溶剂,粘状物质用冷水骤冷,并使用HCl将pH调节至4~5。滤出由此获得的所述固体,用己烷(500mL)洗涤,真空干燥并通过使用EtOAc结晶而获得72gm白色固体的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ10.50(1H,s),8.90(1H,d),8.15(1H,s),8.04(1H,s),7.69(1H,d),7.39(1H,d),7.29-7.31(2H,m),7.17-7.24(3H,m),7.04(1H,d),6.94-6.96(1H,m),6.89(1H,t),6.15(1H,s)4.37(2H,q),4.27(2H,d),1.35(3H,t),1.21(9H,s)ESMS:656.74/657.98
如果化合物55连同甲基酯一起使用,所述产物也可能含有甲基酯衍生物。
实施例8:7-{2-[({[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]氨甲酰基}氨基)甲基]苯氧基}咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸(化合物19)
向在MeOH(10mL)中的7-{2-[({[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]氨甲酰基}氨基)甲基]苯氧基}咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(0.2gm,0.3mmol)的搅拌溶液中,通过溶解于水(2mL)中加入氢氧化锂单水合物(0.063gm,1.51mmol)。反应混合物室温下搅拌2~4h。反应混合物骤冷于冷水中,酸化,并搅拌。滤出由此获得的所述固体,并干燥而获得0.080gm白色固体的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ13.21(1H,bs),10.50(1H,s),8.98(1H,d),8.17(1H,s),7.99(1H,s),7.68(1H,d),7.40(1H,d),7.29-7.30(2H,m),7.16-7.24(3H,m),7.04(1H,d),6.89-6.95(2H,m),6.15(1H,s),4.27(2H,d),1.21(9H,s)
ESMS:631.50/633.42
实施例9:7-{2-[({[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]氨甲酰基}氨基)甲基]苯氧基}-N-(2-羟基乙基)咪唑并[2,1-b][l,3]苯并噻唑-3-甲酰胺(化合物20)
向在THF(20mL)中的7-{2-[({[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]氨甲酰基}氨基)甲基]苯氧基}咪唑并[2,1-b][1,3]苯并噻唑-3-羧酸(0.8gm,1.26mmol)的搅拌溶液中,0~5℃下加入N,N-二异丙基乙胺(0.49gm,3.8mmol)、1-羟基苯并三唑(0.342gm,2.56mmol)和l-乙基-3-(3-二甲基氨基)丙基碳二酰亚胺盐酸盐(0.486gm,2.65mmol)并持续搅拌45min。加入乙醇胺(0.152gm,2.53mmol)。反应混合物室温下搅拌20~24h。反应混合物骤冷于冷水中,酸化至pH 4~5并搅拌。通过乙酸乙酯(10mL×2)萃取并蒸掉有机层而获得粗化合物,其进一步通过柱色谱纯化而获得0.1gm标题化合物。
1H-NMR(400MHz,DMSO-d6):δ10.53(1H,s),8.94(1H,d),8.53(1H,t),8.18(1H,s),7.95(1H,s),7.65(1H,m),7.39(1H,m),7.29(2H,bs),7.16-7.23(3H,m),7.04(1H,d),6.91-6.93(2H,m),6.17(1H,s),4.78(1H,t),4.28(2H,d),3.54-3.55(2H,m),3.33(2H,隐藏于水信号下),1.22(9H,s)
ESMS:674.62/676.45
实施例10:1-[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]-3-(2-{[3-(羟基甲基)咪唑并[2,1-b][l,3]苯并噻唑-7-基]氧基}苄基)脲(化合物22)
向在THF(400mL)中的7-{2-[({[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]氨甲酰基}氨基)甲基]苯氧基}咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(40gm,60.69mmol)的搅拌溶液中,0~5℃下分批加入硼氢化锂(4.2gm,182mmol)并将反应混合物回流4~5h。所述反应混合物骤冷于冷水中并通过使用在乙酸(50mL)中33%的HBr酸化,在60~65℃下加热4~5h。反应混合物冷却至室温,用水稀释并在EtOAc(250mL×2)中萃取。有机层用盐水洗涤,硫酸钠干燥并60℃真空蒸发。由此获得的所述固体,悬浮于EtOAc(150mL)中,搅拌并过滤而获得27gm白色固体的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ8.23(1H,s),8.01(1H,d),7.66(1H,d),7.42(1H,d),7.14-7.30(6H,m),7.06(1H,d),6.94(1H,t),6.89(1H,d),6.21(1H,s),5.52(1H,bs)4.81(2H,s),4.30(2H,d),1.23(9H,s)
ESMS:615.08/617.08
实施例11:l-[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]-3-(2-{[3-(甲氧基甲基)咪唑并[2,1-b][l,3]苯并噻唑-7-基]氧基}苄基)脲(化合物31)
向在DCM(100mL)和氯仿(10mL)的混合物中的l-[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]-3-(2-{[3-(羟基甲基)咪唑并[2,1-b][l,3]苯并噻唑-7-基]氧基}苄基)脲(10gm,16mmol)的搅拌溶液中,5~10℃下逐滴加入亚硫酰氯(9.5mL)。反应混合物在冷却下搅拌并随后室温下搅拌2~4h。50℃下真空蒸掉亚硫酰氯而获得1-[3-叔丁基-1-(3-氯-4-羟苯基)-1H-吡唑-5-基]-3-(2-{[3-(氯甲基)咪唑并[2,1-b][l,3]苯并噻唑-7-基]氧基}苄基)(化合物57)。向其中在冷却下加入甲醇(60mL)并随后将反应混合物回流2h。所述悬浮固体趁热过滤,进一步用甲醇冲洗,并在真空中充分干燥而获得8gm白色固体的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ10.49(1H,bs),8.20(1H,s),7.82(1H,d),7.69(1H,d),7.40(1H,d),7.27-7.30(3H,m),7.14-7.23(3H,m),7.05(1H,d),6.90-6.92(2H,m),6.19(1H,s),4.77(2H,s),4.28(2H,d),3.30(3H,与水信号部分合并),1.23(9H,s)
ESMS:629.62/630.45
实施例12:l-[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]-3-(2-{[3-(吗啉-4-基甲基)咪唑并[2,1-b][l,3]苯并噻唑-7-基]氧基}苄基)脲(化合物35)
向在DCM(50mL)和氯仿(5mL)的混合物中的l-[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]-3-(2-{[3-(羟基甲基)咪唑并[2,1-b][l,3]苯并噻唑-7-基]氧基}苄基)脲(5gm,8.1mmol)的搅拌溶液中,5~10℃下逐滴加入亚硫酰氯(1.75mL)。反应混合物在冷却下搅拌并随后室温下搅拌2~4h。50℃下真空蒸掉亚硫酰氯而获得l-[3-叔丁基-1-(3-氯-4-羟苯基)-lH-吡唑-5-基]-3-(2-{[3-(氯甲基)咪唑并[2,1-b][l,3]苯并噻唑-7-基]氧基}苄基)(化合物57)。向其中在冷却(5~10℃)下加入DMF(50mL)和吗啉(7.04gm,81mmol)而反应混合物随后室温下搅拌2~4h。反应混合物骤冷于冷水中,搅拌并滤出固体,干燥而获得粗化合物,其在二氯甲烷中结晶而获得~3gm白色固体的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ10.51(1H,s),8.20(1H,s),8.01(1H,d),7.65(1H,bs),7.40(1H,bs),7.28-7.30(2H,m),7.03-7.24(5H,m),6.92-6.94(2H,m),6.18(1H,s),4.28(2H,d),3.79(2H,s),3.52(4H,s),2.44(4H,与溶剂信号部分合并)1.22(9H,s)
ESMS:686.50/688.45
实施例13:7-[(4-氨基苯基)硫烷基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物59)
向在甲醇(20mL)中的7-[(4-硝基苯基)硫烷基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(1.2gm,3mmol)(化合物58)的搅拌溶液中,室温加入氯化亚锡二水合物(2.7gm,12mmol)并将反应混合物回流4~5h。60℃下真空蒸掉溶剂,进一步用水稀释,碱化,用乙酸乙酯萃取,硫酸钠干燥并真空浓缩至干而获得0.9gm标题化合物。
化合物58由4-[(4-硝基苯基)硫烷基]苯胺使用Journal of Current Pharmaceutical Research,2010,3(1),13中所描述的类似步骤进行合成而获得相应的苯并噻唑化合物,并且将其使用US6191124中所述的类似步骤环化。
实施例14:7-{[4-({[3-叔丁基-1-(4-甲氧基苯基)-lH-吡唑-5-基]氨甲酰基}氨基)苯基]硫烷基}咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物60)
化合物60由7-[(4-氨基苯基)硫烷基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯和中间体3使用如对于实施例7中化合物25的合成所述的类似步骤进行反应而合成。
实施例15:7-{[4-({[3-叔丁基-1-(4-甲氧基苯基)-lH-吡唑-5-基]氨甲酰基}氨基)苯基]硫烷基}咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸(化合物10)
化合物10由7-{[4-({[3-叔丁基-1-(4-甲氧基苯基)-lH-吡唑-5-基]氨甲酰基}氨基)苯基]硫烷基}咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯使用如对于实施例8中化合物19的合成所述的类似步骤进行水解而合成。
1H-NMR(400MHz,DMSO-d6):δ9.40(1H,s),9.00(1H,d),8.55(1H,s),7.93-7.96(2H,m),7.39-7.57(8H,m),7.06-7.09(2H,m),6.35(1H,s),3.81(3H,s),1.28(9H,s)
ESMS:613.16
实施例16:7-(4-氨基苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物62)
步骤1:7-(4-硝基苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙基酯(化合物61)
化合物61由7-羟基咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯和4-硝基氟苯使用如对于实施例1中化合物51的合成所述的类似步骤进行合成。
步骤2:7-(4-氨基苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物62)
化合物62由7-(4-硝基苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯,使用如对于实施例13中化合物59的合成所述的类似步骤进行还原而合成。
实施例17:7-[4-({[3-叔丁基-1-(3-氯-4-甲氧基苯基)-lH-吡唑-5-基]氨甲酰基}氨基)苯氧基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物5)
化合物5由7-(4-氨基苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯和中间体2使用如对于实施例7中化合物25的合成所述的类似步骤进行反应而合成。
1H-NMR(400MHz,DMSO-d6):1H-NMR(400MHz,CDC13)δ9.06(1H,d),7.97(1H,s),7.50(1H,d),7.31-7.34(3H,m),7.22-7.26(2H,m),7.16(1H,dd),7.01(2H,d),6.89(1H,d),6.82(1H,s),6.41(1H,s),4.43(2H,q),3.88(3H,s),1.45(3H t),1.31(9H,s)
ESMS:657.45/659.37
实施例18:7-(4-{[(5-甲基-3-苯基-1,2-噁唑-4-基)氨甲酰基]氨基}苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物43)
向在甲苯(15mL)中的7-(4-氨基苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(0.75gm,2.14mmol)的搅拌溶液中加入4-异氰酰基-5-甲基-3-苯基-1,2-噁唑(0.5gm,2.5mmol))并将反应混合物回流4~6h。所述反应混合物冷却至室温。反应混合物用冷水骤冷,并用乙酸乙酯萃取而获得粗化合物,其通过使用二异丙醚溶剂处理进行纯化并随后干燥而获得0.5gm固体的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ8.94(1H,bs),8.89(1H,d),8.03(1H,s),7.84(1H,s),7.71-7.76(3H,m),7.47-7.52(5H,m),7.23(1H,dd),7.02(2H,d),4.36(2H,q),2.37(3H,s),1.35(3H,t)
ESMS:554.25
实施例19:7-(2-{[(苯氧基羰基)氨基]甲基}苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物80)
向在THF(10mL)中的7-[2-(氨基甲基)苯氧基]咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(1gm,2.72mmol)(化合物55)的搅拌溶液中,加入DIEA(0.7gm,5.44mmol)并将反应混合物在0℃下冷却。向所述冷却的反应混合物中加入氯甲酸苯酯(0.42gm,2.72mmol)并在RT下进一步搅拌反应混合物约1h。反应混合物倾入水中,用乙酸乙酯萃取,硫酸钠干燥并真空浓缩至干而获得1.2gm白色固体的标题化合物。
实施例20:7-(2-{[(丙基氨甲酰基)氨基]甲基}苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(化合物68)
向在甲苯(10mL)中的7-(2-{[(苯氧基羰基)氨基]甲基}苯氧基)咪唑并[2,1-b][l,3]苯并噻唑-3-羧酸乙酯(0.5gm,1.02mmol)(化合物80)的搅拌溶液中,室温下加入DIEA(0.26gm,2.05mmol)和正丙胺(0.12gm,2.05mmol)并将反应混合物回流4~5h。反应混合物倾入水中,用乙酸乙酯萃取,硫酸钠干燥并真空浓缩至干而获得粗化合物,其通过柱色谱纯化而获得0.23gm白色固体的标题化合物。
本发明的以下代表性化合物通过使用如上所述的合成方案按照类似方式进行制备:
表1
药物组合物
在另一实施方式中,本发明提供了包含治疗有效量的式(I)的一种或多种化合物的药物组合物。尽管有可能单独地或组合地、直接无任何配方给予治疗有效量的式(I)的化合物,但是通常的做法是以包含药用赋形剂/佐剂或载体和至少一种活性成分的药物剂型的形式给予所述化合物。这些剂型可以通过各种途径,包括口服,局部,经皮,皮下,肌内,静脉内,腹膜内,鼻内,肺部等进行给药。
口服组合物可以是固体或液体剂型的形式。固体剂型可以包括丸剂,小袋(pouch),香囊(sachet)或离散单元如片剂、多粒单元、胶囊(软&硬明胶)等。液体剂型可以是酏剂(elixir),悬浮液,乳液,溶液,糖浆等的形式。预想口服使用的组合物可以根据本技术领域中对于所述组合物的生产已知的任何方法进行制备而这种药物组合物除了活性成分之外还可以包含赋形剂如稀释剂,崩解剂,粘合剂,增溶剂,润滑剂,助流剂(glidant),表面活性剂,悬浮剂,乳化剂,螯合剂,稳定剂,调味剂,甜味剂,色素等。合适的赋形剂的一些实例包括乳糖,纤维素及其衍生物如微晶纤维素、甲基纤维素、羟丙基甲基纤维素和乙基纤维素,磷酸二钙,甘露醇,淀粉,明胶,聚乙烯吡咯烷酮,各种树胶如阿拉伯胶、黄蓍胶、黄原胶、藻酸盐及其衍生物,山梨醇,右旋糖,木糖醇,硬脂酸镁,滑石,胶体二氧化硅,矿物油,单硬脂酸甘油酯,山嵛酸甘油酯,淀粉羟乙酸钠,交联聚维酮,交联羧甲基纤维素,各种乳化剂如聚乙二醇、山梨糖醇、脂肪酸酯、聚乙二醇烷基醚、糖酯、聚氧乙烯聚氧丙基嵌段共聚物、聚乙氧基化脂肪酸单酯,二酯及其混合物。
根据本发明的鼻内或肺部用的组合物能够是适合鼻内给药的液体或固体或半固体组合物的形式。液体组合物能够是水性的、非水性的组合物,悬浮液或乳液,固体组合物能够是粉末形式等而半固体组合物能够为凝胶等的形式。鼻/肺部的组合物也可以形成原位凝胶。所述鼻或肺部的组合物包含可选地与一种或多种选自原位胶凝剂,粘膜粘附剂,聚合物,保湿剂(humectant),缓冲剂,稳定剂,表面活性剂,防腐剂,增稠剂,溶剂,助溶剂(co-solvent),渗透增强剂,螯合剂,粘度改性剂,增甜剂,味道掩蔽剂,增溶剂,调味剂,乳化剂和等渗剂的合适赋形剂一起的式(I)的化合物。
注射用的无菌组合物能够根据传统制药惯例通过将所述活性物质溶解或悬浮于运载体(vehicle)如注射用的水,N-甲基-2-吡咯烷酮,丙二醇和其它二醇,醇类,天然存在的植物油如芝麻油、椰子油、花生油、棉子油或合成脂肪运载体如油酸乙酯等中而进行配制。缓冲剂,抗氧化剂,防腐剂,络合剂如纤维素衍生物、肽、多肽和环糊精等能够根据需要而引入。
所述剂型除了立即释放剂型之外能够具有慢释、延迟释或控释的活性成分。
需要用于达到治疗效果的活性成分的量,当然,将随着具体化合物、给药途径、所治疗的受试者和所述要治疗的具体失调或疾病而变化。本发明的化合物可以通过口服、吸入或肠胃外途径按照0.0005~100mg/kg/天,优选0.0005~50mg/kg/天,更优选0.001~20mg/kg/天,最优选0.001~10mg/kg/天的剂量给药。对于成年人的剂量范围一般为每天5μg~5g,优选剂量范围为每天10μg~2g。
以离散单位提供的剂型介绍可以方便地含有按照这种剂量或其多倍是有效的一定量的本发明化合物,例如,含有5μg~1000mg的单位。
在另一实施方式中,本发明提供了通过向有此需要的哺乳动物,包括人类给予治疗有效量的式(I)的化合物治疗过敏和非过敏性气道疾病的方法。过敏性和非过敏性气道疾病包括过敏性和非过敏性哮喘,慢性阻塞性肺部疾病(COPD),鼻炎,慢性支气管炎,肺气肿,或哮喘样综合征如咳嗽、气喘或呼吸困难。
本发明的优选实施方式是通过向有此需要的哺乳动物,包括人类给予治疗有效量的式(I)的化合物治疗慢性阻塞性肺部疾病和哮喘的方法。
本发明的最优选实施方式是通过向由此需要的哺乳动物,包括人类给予治疗有效量的式(I)的化合物治疗慢性阻塞性肺部疾病的方法。
本发明的另一实施方式是式(I)的化合物用于制备治疗过敏和非过敏性气道疾病的药物的用途。
本发明的一个优选实施方式是式(I)的化合物用于制备治疗慢性阻塞性肺部疾病和哮喘的药物的用途。
本发明的最优选实施方式是式(I)的化合物用于制备治疗慢性阻塞性肺部疾病的药物的用途。
生物学测试:
生物学实施例1:体外研究
p38αMAPK活性的抑制:时间分辨的荧光共振能量转移激酶标准测试(TR-FRET测试)
各种浓度的本发明化合物与DMSO预混合。该实验通过将0.5%~1.0%的DMSO作为运载体/化合物与纯化的重组人p38αMAPK(Millipore,美国)在孔中混合并室温培养15min而开始。此后,将30nM生物素化的GST-ATF2(激活转录因子2)和100μΜATP加入到含有反应混合物的所述孔中,接着室温下再培养60min。反应通过向所述反应混合物中加入10mM EDTA和含有用铕螯合物标记的抗磷酸苏氨酸ATF2抗体(Perkin Elmer,USA)和APC(Allophycocyanin)标记的链霉素的检测试剂而中止,将其进一步室温培养60min。所述底物(GST-ATF2)的磷酸化程度使用Envision多模式酶标仪(Envision multimode reader)(Perkin Elmer)进行测定。p38激酶活性的百分比抑制通过测定特定铕665nm能量转移信号与对照615nm信号的比值而进行计算。结果总结于表2中。
表2
标准:+++++=抑制≧80%≦100%;++++=抑制≧60%<80%;+++=抑制≧40%<60%;++=抑制≧20%<40%;+=抑制<20%
观察:体外数据表明本发明的化合物有效抑制了p38MAPK活性。
生物学实施例2:体内研究
化合物在气道发炎的动物模型中的体内效能评价:
烟草烟雾诱导的气道发炎模型用于化合物的体内效能。许多研究者已经使用急性烟草烟雾(TS)暴露于啮齿类动物中作为COPD抗炎疗法快速筛选的气道发炎的模型(J Pharmacol Exp Ther.2008;324(3):921-9;J Pharmacol Exp Ther.2010;332(3):764-75;Journal of Inflammation 2013,10(Suppl 1):31和Eur Respir J Suppl 2006;663s:3850)。鉴于其作为COPD的主导原因的位置,使用TS暴露的动物模型将似乎是研究的逻辑选择(Respir Res.2004;2;5:18)。
A:气道发炎的急性小鼠模型中的效能研究
小鼠暴露于丙烯酸小室(chamber)中的烟草烟雾(TS)。动物在第1天,第2天,第3天分别暴露于8、12、16只香烟的TS。从第4天起直至第11天,动物暴露于20只香烟/天的TS。在小鼠暴露于TS的11天,相比于空气暴露的对照小鼠,观察到至肺部的显著炎性细胞招募(recruitment),主要是中性粒细胞(BALF中性粒细胞水平,空气对照组中的零相对于烟雾暴露运载体组中的93.8+11.7×103个细胞/动物)。
测试化合物的肺部递送通过小室内全身气雾剂暴露25min实现。小鼠分成不同剂量组,并在小室内用运载体或化合物No.22(0.3mg/mL)或化合物No.22(3mg/mL))或化合物No.31(0.3mg/mL)或化合物No.31(1.0mg/mL)暴露25min。在小室内对于相应组在25min的时间内雾化了总量3.5mL的运载体或测试化合物制剂(具有D90<5μ的悬浮液制剂,Malvern)。从第6天至第11天在TS暴露之前2h给药测试化合物。在最后TS暴露之后24h实施支气管肺泡灌洗(BAL)。
动物的气管使用导管插管。磷酸盐缓冲盐水(PBS)用作灌洗液。0.5mL的体积轻柔灌输和撤回,并收集于置于冰上的微离心管中。此过程再重复2次。
灌洗液通过离心与细胞分离并分离出上清液。所述细胞颗粒再悬浮于已知体积的PBS中。等分部分的细胞使用特克(Turk)溶液染色而总细胞数通过在显微镜下使用血球计数仪对特克染色的等分部分计数而进行计算。
所述残余的细胞悬液再悬浮并使用细胞离心技术(Cytospin 4,Thermo Shandon)制备载玻片。然后将这些载玻片用甲醇固定,空气干燥并用梅格伦沃尔德姬姆萨(May Grunwald Giemsa)染色进行染色。计数最高达300个细胞并使用标准形态技术在光学显微镜下分辨区分。
所有的结果都对于每个动物以单个数据进行提呈并对每个组计算均值。对于化合物No.22&31治疗组相对于运载体组计算所述中性粒细胞的百分数抑制。结果总结于下文:
治疗化合物No.22&31对BAL液中香烟烟雾诱发中性粒细胞聚集的影响。
表3
这些值是平均值±SEM;NA:不适用
观察:据观察,发现本发明的化合物对中性粒细胞内向通量(influx),(肺部炎症的指标)的抑制是有效的。这些结果表明,本发明的化合物具有肺部抗炎活性。
B.(I)在气道发炎的急性豚鼠模型中的效能研究
豚鼠在丙烯酸小室中暴露于烟草烟雾(TS)。动物在第1天,第2天,第3天分别暴露于5、10、15只香烟的TS。从第4天起直至第11天,动物暴露于15只香烟/天的TS。在豚鼠暴露于TS的第11天,相比于空气暴露的对照豚鼠,观察到至肺部的显著炎性细胞招募,主要是中性粒细胞(BALF中性粒细胞水平,空气对照组中的0.23±0.052*106个细胞/动物相对于烟雾暴露运载体组中的3.5±0.62*106个细胞/动物)。
测试化合物的肺部递送通过小室内全身气雾剂暴露75min实现。豚鼠分成不同剂量组,并在小室内用运载体或化合物No.31(6mg/mL)暴露75min。在小室内对于相应组在75min的时间内雾化了总量7.0mL的运载体或测试化合物制剂(具有D90<5μ的悬浮液制剂,Malvern)。从第6天至第11天在TS暴露之前2h给药测试化合物。在最后TS暴露之后24h实施支气管肺泡灌洗(BAL)。
动物的气管使用导管插管。磷酸盐缓冲盐水(PBS)用作灌洗液。5.0mL的体积轻柔灌输和撤回,并收集于置于冰上的微离心管中。此过程再重复5次。
灌洗液通过离心与细胞分离并分离出上清液。所述细胞颗粒再悬浮于已知体积的PBS中。等分部分的细胞使用特克(Turk)溶液染色而总细胞数通过在显微镜下使用血球计数仪对特克染色的等分部分计数而进行计算。
所述残余的细胞悬液再悬浮并使用细胞离心技术(Cytospin 4,Thermo Shandon)制备载玻片。然后将这些载玻片用甲醇固定,空气干燥并用梅格伦沃尔德姬姆萨(May Grunwald Giemsa)染色进行染色。计数最高达300个细胞并使用标准形态技术在光学显微镜下分辨区分。
所有的结果都对于每个动物以单个数据进行提呈并对每个组计算均值。对化合物No.31治疗组相对于运载体组计算所述中性粒细胞的百分数抑制。结果总结于下文:
治疗化合物No.31对BAL液中香烟烟雾诱发中性粒细胞聚集的影响。
表4
这些值是平均值±SEM;NA:不适用
观察:据观察,发现本发明的化合物在气道发炎的豚鼠模型中对中性粒细胞(肺部炎症指标)的抑制是有效的。这些结果表明,本发明的化合物具有肺部抗炎活性。
(II)豚鼠的COPD慢性模型中的效能研究。
豚鼠在丙烯酸小室内暴露于烟草烟雾(TS)和LPS。在一周内按照以下方式提供TS和LPS暴露总共18周。
测试物质的肺部递送通过小室内全身气雾剂暴露75min实现。豚鼠分成不同剂量组,并暴露于运载体或化合物No.31(2mg/mL)。在小室内对于相应组在75min的时间内雾化了总量7.0mL的运载体或化合物No.31(具有D90<5μ的悬浮液制剂,Malvern)。从第9周至第18周在每天一次TS/LPS暴露之前2h给药化合物No.31。对照动物暴露于代替TS的室内空气和代替LPS的PBS。在最后TS暴露之后24h对每个动物实施肺功能和支气管肺泡灌洗(BAL)。
在麻醉和气管切开的动物中使用PFT操作法(BUXCO,美国)进行各种参数如功能残气量(Functional residual capacity)(FRC)、残气量(residual volume)(RV)、压力量(pressure volume)和流量的关系的测定而实施肺功能评价。
动物的气管使用导管插管。磷酸盐缓冲盐水(PBS)用作灌洗液。5.0mL的体积轻柔地灌输和撤回,并收集于置于冰上的微离心管中。此过程再重复5次。
灌洗液通过离心与细胞分离并分离出上清液。所述细胞颗粒再悬浮于已知体积的PBS中。等分部分的细胞使用特克(Turk)溶液染色而总细胞数通过在显微镜下使用血球计数仪对特克染色的等分部分计数而进行计算。
所述残余的细胞悬液再悬浮并使用细胞离心技术(Cytospin 4,Thermo Shandon)制备载玻片。然后将这些载玻片用甲醇固定,空气干燥并用梅格伦沃尔德姬姆萨(May Grunwald Giemsa)染色进行染色。计数最高达300个细胞并使用标准形态技术在光学显微镜下分辨区分。
BALF收集之后肺在定压下使用中性缓冲福尔马林固定,其要进一步用于组织病理学分析。苏木精和伊红染色的载玻片用于肺部炎性细胞内向通量和肺泡&上皮炎性变化的评价。高碘酸-席夫-淀粉酶染色的载玻片用于评价粘蛋白分泌细胞和马森(Masson)的毛状体染色的载玻片用于实质(parenchyma)中的胶原纤维的评价。
所有的结果都对于每个动物以单个数据进行提呈并对每个组计算均值。对化合物No.31组相对于运载体组计算所述中性粒细胞的百分数抑制。结果总结于下文:
A.化合物No.31的治疗对豚鼠中BAL液炎性细胞内向通量的影响。
表5
这些值是平均值±SEM;NA:不适用
B.化合物No.31的治疗对肺功能参数,功能残气量(FRC),残气量(RV),吸气量(inspiratory capacity)(IC)与总肺容量(total lung capacity)(TLC)的比率和总肺阻力(total lung resistance)(RL)的影响提供于图1和2中。(这些值是平均值±SEM)
C.化合物No.31的治疗对肺重塑的影响,使用肺组织及黏液分泌的病理组织学分析,以粘蛋白分泌细胞数通过综合计分(composite score)进行评价。
表6
NA:不适用
观察:在慢性COPD模型中,本发明的化合物在降低至肺部组织的中性粒细胞内向通量中产生了显著的影响,显著改善了肺功能并防止了与COPD相关的肺重塑方面。
- 还没有人留言评论。精彩留言会获得点赞!