可用作S100‑抑制剂的新型化合物的制作方法
本发明涉及新型的N-(2-氧代-3-(三氟甲基)-2,3-二氢-1H-苯并[d]咪唑并[1,2-a]咪唑-3-基)酰胺衍生物、这些衍生物的药物组合物及其作为药物的用途。更具体地,本发明涉及用于治疗癌症、自身免疫性病症、炎性病症和神经退行性病症的此类衍生物。
发明背景
S100A9属于钙结合蛋白的S100-家族且已经被公认为是用于治疗例如自身免疫、炎性疾病、神经退行性疾病和癌症的有吸引力的新治疗靶点。其它S100蛋白在许多不同的生物过程中具有不同的作用且与许多疾病包括癌症、心肌病、动脉粥样硬化、阿茨海默氏疾病和炎性疾病相关。二十一种人类基因,包括S100A9,位于染色体区域1q21中,其在肿瘤中经常改变(Marenholz等人,2004)。有趣的是,尽管基本序列(primary sequence)在家族成员之间趋异,但不同蛋白的3D结构非常类似。
S100A9经常与S100A8(S100蛋白家族的另一成员)共同表达,且它们在骨髓细胞诸如嗜中性粒细胞和单核细胞中高度表达,但也可在其它细胞或组织中诱导(Srikrishna, 2012)。它们形成可响应细胞活化而特异性释放的非共价同源复合物(homocomplex)和异源复合物(heterocomplex) (Foell等人,2007,Ryckman等人,2003)。S100A9可在功能上描述为损伤相关分子模式(DAMP)分子,其在组织中释放并通过与受体诸如RAGE和TLR4相互作用而诱导信号传导(Foell等人,2007,下文)。至于许多其它DAMP分子,除了胞外功能以外,S100A9还具有细胞内作用,例如通过结合细胞骨架并影响细胞骨架重排且由此影响细胞迁移(Srikrishna, 2012)。
S100A9的促炎作用得到炎性疾病中升高的S100A9血清水平和局部炎症位点(例如在类风湿性关节炎患者的滑液中)S100A9的高浓度的支持(Foell等人,2004)。在各种形式的癌症中也已经发现高水平的S100A9,且已经显示高表达水平与在这些癌症形式中的一些中的不良肿瘤分化相关(Arai等人,2008)。在慢性炎症的病理学病况中以及在癌症中的升高的S100A9水平支持在炎症相关致癌作用中的可能作用。
S100A9在免疫系统和癌症之间的关联中的作用也得到显示S100A8和S100A9在源自骨髓的抑制细胞(MDSC)中高度表达并对其功能重要的研究的支持(Cheng等人,2008;Sinha等人,2008;Wang等人,2013),源自骨髓的抑制细胞是抑制T-细胞和NK-细胞激活且促进血管生成和肿瘤生长的未成熟骨髓细胞的混合物。通过干扰肿瘤浸润性MDSC的S100A9调节的积累,这些过程之间的平衡可有利于肿瘤进展受抑制的抗血管生成和较低免疫抑制环境而变化。此外,有数据表明S100A9在将炎性细胞和肿瘤细胞募集至转移性位点中的作用(Hiratsuka等人,2006;Acharyya等人,2012;Hibino等人,2013)。因此,阻断S100A9的功能可提供预防转移的新方法。
尽管已经提议了S100A9的许多可能的生物学功能,但S100A9在炎症、癌症和其它疾病中的确切作用仍然未知。已经报道S100蛋白家族的成员与促炎分子RAGE相互作用且研究显示在生理水平的Ca2+和Zn2+存在的情况下S100A9是S100家族内最强的RAGE结合剂(Björk等人,2009)。这些研究进一步证明S100A9与toll样受体4 (TLR4)相互作用。关于S100A9-RAGE相互作用,S100A9-TLR4相互作用看起来严格地取决于生理水平的Ca2+和Zn2+两者的存在。在癌症中可能重要的S100A9的另一受体为EMMPRIN (CD147),该蛋白在不同细胞类型中表达且已经显示S100A9-EMMPRIN相互作用参与黑色素瘤转移(Hibino等人,2013)。
S100A8和S100A9蛋白已经主要被描述为在激活时从骨髓细胞分泌的胞质蛋白。通常相信与炎症有关的主要生物学功能需要将S100蛋白释放至胞外空间。在该模型中,胞外S100A9将结合例如促炎受体RAGE和TLR4且导致炎症反应。这得到显示S100A8/A9在人外周血液单核细胞中经由TLR4诱导TNFα产生的研究的支持(Vogl等人,2017)。并且,与S100A8复合的S100A9已经显示经由RAGE信号传导直接对肿瘤细胞的生长促进活性(Ghavami等人,2008)。S100A9还以膜相关形式存在于单核细胞上(Bhardwaj等人,1992)。膜相关的S100A9打开了涉及S100A9的细胞-细胞或细胞-ECM信号传导的可能性。
收集的数据表明S100A9在炎症、癌症生长、癌症转移及它们的关联中具有重要作用。抑制S100A9在这些过程中的活性且由此扰乱肿瘤微环境的新型化合物将在不同类型的癌症的治疗中有吸引力。
除了癌症、炎症和自身免疫以外,S100A9还与神经退行性疾病具有强烈关联。S100A9在阿茨海默氏疾病(AD)患者中和小鼠疾病模型中的大脑中上调(Shepherd等人,2006;Ha等人,2010)。此外,在小鼠AD模型中S100A9的敲除或缺失抑制认知下降和大脑中的斑块负荷(Ha等人,2010;Chang等人,2012)。RAGE的作用在AD中也是显而易见的,其中RAGE的抑制降低在小鼠AD模型中的疾病(Deane等人,2013)。S100A9的抑制及其相互作用代表治疗性干预AD及其它神经退行性疾病的新的有前景的方法。
发明概述
在第一个方面,提供了式(I)化合物
或其药学上可接受的盐,其中
RA、RB和RC独立地选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基,或
RA和RC中的一个与RB一起形成双基(biradical)-(CH2)m-,其中m是3至5的整数,且RA和RC中的另一个选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基;
每个R16独立地选自卤素、氰基、硝基、R17O、任选地被R17O取代的C1-C6烷基、任选地被R17O取代的C3-C6环烷基、R18C(O)、R19S、R20S(O)2、R21OC(O)、(R22ON)C(R23)、R24R25NC(O)、R26R27N、R28S(O)2NR29和R30S(O)2NR31C(O);
R1-R31中的每一个独立地选自H、C1-C6 烷基和C3-C6 环烷基;
W是直接键或X1-X2-X3;
X1是任选地被C1-C4 烷基或R32O取代的C1-C2亚烷基;
X2是O或不存在;
X3是直接键或任选地被C1-C4烷基或R32O取代的C1-C2亚烷基;
R32选自H和C1-C4烷基;
RD是C1-C6烷基或选自苯基、4-至6-元杂环基、C4-C6 环烷基或C5-C6 环烯基的环状基团,其中所述环状基团任选地被一个或多个R33取代;
每个R33独立地选自卤素、氰基、任选地被R34O取代的C1-C6烷基、C3-C6 环烷基、苯基、5-或6-元杂环基、R34O、R35OC(O)、R36S(O)2、R37C(O)、R38R39N、R40R41N(CO);且连接至一个且相同碳原子的两个R33可以与它们两者均连接的碳原子一起形成在环中任选地含有一个或多个杂原子的4-至6-元环;
R34独立地选自H、C1-C6烷基和C3-C6 环烷基;其中所述烷基或环烷基任选地被R42O取代;
R35-R36中的每一个独立地选自H、C1-C6 烷基和C3-C6 环烷基;
R37独立地选自C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R43O取代;
R38和R39独立地选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R44R45N或R46O取代,或
R38和R39与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基取代的4-至6-元杂环;
R40和R41独立地选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R47R48N或R49O取代,或
R40和R41与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基取代的4-至6-元杂环;
R44和R45独立地选自H、C1-C6 烷基和C3-C6 环烷基,或
R44和R45与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基取代的4-至6-元饱和杂环;
R47和R48独立地选自H、C1-C6 烷基和C3-C6 环烷基,或
R47和R48与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基取代的4-至6-元饱和杂环;
R42、R43、R46和R49独立地选自H、C1-C6 烷基和C3-C6 环烷基;
任何烷基任选地被一个或多个F取代。
如本文上面定义的式(I)化合物用作S100A9和相互作用搭档诸如RAGE、TLR4和EMMPRIN之间的相互作用的抑制剂。因此,根据又一个方面,提供如本文上面定义的式(I)化合物,其用作S100A9与其相互作用搭档的相互作用的抑制剂并用于治疗与S100A9功能相关的病症,例如炎性疾病、神经退行性疾病、自身免疫性疾病和癌症。
根据一个方面,提供式(I)化合物,其用于治疗中,例如用于治疗炎性疾病、神经退行性疾病、自身免疫性疾病和癌症。
根据又一个方面,提供药物组合物,其包含式(I)化合物或其药学上可接受的盐和至少一种药学上可接受的赋形剂。本发明的药物组合物可用于治疗选自炎性疾病、自身免疫性疾病、神经退行性疾病和癌症的疾病。
附图简述
图1是使用小分子S100A9结合剂抑制生物素化人类S100A9和人类RAGE-Fc之间的相互作用的测定的示意图。
图2是显示在(A) RAGE和(B) TLR4存在的情况下实施例13的化合物(ABR 238219)与S100A9的竞争性结合的图。在该测定中,将S100A9以~ 1.3 µg/mL注入(2 min; 30 µL/min)胺偶联的人类RAGE/Fc (密度~ 4.2 kRU))或TLR4 (密度~ 2900共振单位) + 0.391-200 µM ABR-238219上。将表示为结合至(A) RAGE或(B) TLR4的% S100A9的响应(Y-轴)相对于竞争剂浓度作图,并拟合至S形剂量-响应模型。测定缓冲液 - 10 mM HEPES、0.15 M NaCl, pH 7.4 (HBS缓冲液),其含有0.005% v/v表面活性剂P20、1 mM Ca2+和20 µM Zn2+。每个循环之后,通过HBS缓冲液中的3 mM EDTA的30 µL脉冲进行再生。
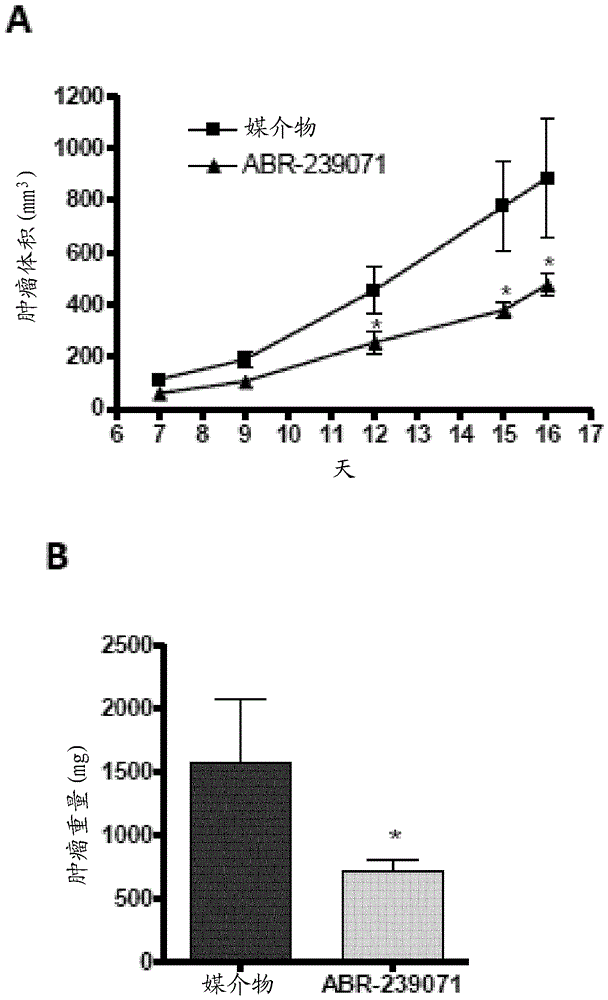
图3是(A)显示肿瘤生长、随后连续卡尺测量的图。将小鼠用肿瘤细胞皮下接种,并且在第二天开始用实施例118(a)的化合物(ABR 239071)治疗。每日口服给予治疗(30 mg/kg)。(A)通过连续卡尺测量肿瘤生长;和(B)显示终点(第16天)的肿瘤重量的条形图。治疗组之间肿瘤生长和重量的差异通过非参数Mann-Whitney U检验统计学评估,*p < 0.05。误差条表示SEM。
发明详述
本文下面提供本文使用的一些术语的定义。列举并不是穷尽的,并且注意,本文使用的任何术语和表述应当给予其通常含义,除非另有指明或从上下文清楚地显而易见。因此,例如,术语烷基,单独或作为基团的一部分,包括通式CnH2n+1的直链或支链烷基。
术语烯基是指式-(CnH2n)-的双基。例如,C2烯基是式-CH2CH2-的基团,即:
。
术语C1-C6烷基包括具有1、2、3、4、5或6个碳原子的任何烷基。
术语C1-C4烷基包括甲基、乙基、正丙基、异丙基、正丁基、异丁基和叔丁基。
术语C1-C3烷基包括甲基、乙基、正丙基和异丙基。
术语环烷基是指通式CnH2n-1的环状烷基。
术语环烯基是指通式CnH2n-3的环状烯基。
术语C3-C6环烷基是指环丙基、环丁基、环戊基和环己基。
术语苯基是指下式的C6H5基团
。
术语杂环基是指在环中含有至少一个杂原子的饱和或不饱和且芳族或非芳族环状基团。
术语杂芳基是指芳族杂环基,例如吡啶基(pyridyl)(也称为吡啶基(pyridinyl))、四唑基、呋喃基或嘧啶基(pyridiminyl)。
术语吡啶基是指下式的C5NH5基团
包括2-吡啶基、3-吡啶基和4-吡啶基。
术语嘧啶基是指下式的C4N2H4基团
包括2-嘧啶基、4-嘧啶基和5-嘧啶基。
术语噻唑基是指1,2-噻唑基和1,3-噻唑基。
术语1,2-噻唑基是指下式的基团
包括1,2-噻唑-3-基、1,2-噻唑-4-基和1,2-噻唑-5-基。
术语1,3-噻唑基是指下式的基团
包括1,3-噻唑-2-基、1,3-噻唑-4-基和1,3-噻唑-5-基。
术语卤素是指F、Cl、Br和I,优选F、Cl和Br。
术语羟基是指式-OH的基团。
术语烷氧基是指式RO的基团,其中R为烷基。
术语RO是指下式的基团
术语氰基是指式(即-CN)的基团。
术语RC(O)是指下式的基团
。
基团RS是下式的基团
。
术语RS(O)2是指下式的基团
。
术语ROC(O)是指下式的基团
。
术语(RON)C(R’)是指下式的基团
。
术语RR´N是指下式的基团
。
术语RR´NC(O)
。
术语RS(O)2NR´是指下式的基团
。
术语RS(O)2NR´C(O)是指下式的基团
。
“任选的”或“任选地”意指随后描述的事件或情况可能发生,但未必发生,且该描述包括其中该事件或情况发生的情况及其中该事件或情况不发生的情况。
“药学上可接受的”意指可用于制备药物组合物,其通常为安全、无毒且既不是生物学上不期望也不是其它方式不期望并且包括为兽医以及人药物用途所接受的那些。
术语化合物的药学上可接受的盐是指如本文定义在药学上可接受的且具有期望的母体化合物的药理学活性的盐。药学上可接受的盐包括与无机酸例如盐酸、氢溴酸、硫酸、硝酸、磷酸形成的酸加成盐;或与有机酸例如乙酸、苯磺酸、苯甲酸、樟脑磺酸、柠檬酸、乙磺酸、富马酸、葡庚糖酸、葡糖酸、谷氨酸、乙醇酸、羟基萘甲酸、2-羟基乙磺酸、乳酸、马来酸、苹果酸、丙二酸、扁桃酸、甲磺酸、粘康酸、2-萘磺酸、丙酸、水杨酸、丁二酸、酒石酸、对甲苯磺酸、三甲基乙酸形成的酸加成盐;或当母体化合物中存在的酸性质子被金属离子例如碱金属离子、碱土离子或铝离子替代;或与有机或无机碱配位时形成的盐。可接受的有机碱包括例如二乙醇胺、乙醇胺、N-甲基葡糖胺、三乙醇胺和氨基丁三醇。可接受的无机碱包括例如氢氧化铝、氢氧化钙、氢氧化钾、碳酸钠和氢氧化钠。
每当手性碳存在于化学结构中时,除非另有指明,该结构意欲涵盖与该手性碳相关的所有立体异构体。使用Cahn-Ingold-Prelog RS命名系统,任何不对称碳原子都可以(R)-或(S)-构型存在,且化合物可作为其立体异构体的混合物例如外消旋混合物或者仅一种立体异构体存在。
本发明化合物中的一些可以互变异构形式存在。任何此类互变异构体都涵盖在本发明的范围内。
并且,在如本文定义的式(I)化合物中,任何氢原子都可被氘(2H)替代且包含代替相应数目的氢原子的一个或多个氘的任何此类氘化的式(I)化合物都被视为在本发明的范围内。
“治疗有效量”意指在给药至对象用于治疗疾病状态时足以实现对该疾病状态的此类治疗的化合物的量。“治疗有效量”将根据化合物、所治疗的疾病状态、所治疗的疾病的严重程度、对象的年龄和相对健康、给药途径和形式、主治医生或执业兽医的判断等而变化。
本文使用的术语“治疗(treatment)”或“治疗(treating)”是获得有益或期望结果、包括临床结果的方法。有益或期望临床结果可包括但不限于一种或多种症状或病况的缓解或改善、疾病程度的减轻、疾病的稳定化(即,不恶化)状态、防止疾病传播、疾病进展的延迟或减慢、疾病状态的改善或减缓和缓解(无论是局部缓解,还是整体缓解),无论是可检测的还是不可检测的。该术语还可意指与在没有治疗的情况下的预期存活相比的延长存活。
术语哺乳动物是指人或任何哺乳动物,例如灵长类动物、家畜、玩赏动物或实验室动物。此类动物的实例为猴、奶牛、绵羊、马、猪、狗、猫、兔、小鼠、大鼠等。优选所述哺乳动物为人。
术语癌症是指由异常和不受控制的细胞分裂引起的任何恶性生长或肿瘤;其可通过淋巴系统或血流扩展到身体的其它部位且包括实体肿瘤和血液肿瘤两者。示例性癌症包括肾上腺皮质癌、AIDS相关癌症、AIDS相关淋巴瘤、肛门癌、肛门直肠癌、阑尾癌、儿童小脑星形细胞瘤、儿童大脑星形细胞瘤、基底细胞癌、胆癌、肝外胆管癌、肝内胆管癌、泌尿膀胱癌、骨骼和关节癌、骨肉瘤和恶性纤维组织细胞瘤、脑肿瘤、脑干胶质瘤、小脑星形细胞瘤、大脑星形细胞瘤/恶性胶质瘤、室管膜瘤、成神经管细胞瘤、视路和下丘脑胶质瘤、乳腺癌、支气管腺瘤/类癌瘤、神经系统癌、神经系统淋巴瘤、中枢神经系统癌、中枢神经系统淋巴瘤、宫颈癌、儿童癌症、慢性淋巴细胞性白血病、慢性髓细胞性白血病、慢性骨髓增生病、结肠癌、结肠直肠癌、皮肤T-细胞淋巴瘤、淋巴瘤、阿利贝尔氏病(mycosis fungoides)、塞扎里综合征(Sezary syndrome)、子宫内膜癌、食道癌、颅外生殖细胞肿瘤、性腺外生殖细胞肿瘤、眼癌、成视网膜细胞瘤、胆囊癌、胃(gastric)(胃(stomach))癌、胃肠类癌瘤、胃肠间质肿瘤(GIST)、生殖细胞肿瘤、卵巢生殖细胞肿瘤、妊娠期滋养叶瘤胶质瘤、头颈部癌、肝细胞性(肝)癌、霍奇金氏淋巴瘤(Hodgkin's lymphoma)、咽下部癌症、眼睛癌症(ocular cancer)、卡波西肉瘤(Kaposi's sarcoma)、肾癌、喉癌、急性淋巴细胞性白血病、急性骨髓性白血病、毛细胞白血病、唇和口腔癌、肺癌、非小细胞肺癌、小细胞肺癌、非霍奇金氏淋巴瘤、原发性中枢神经系统淋巴瘤、瓦尔登斯特伦氏巨球蛋白血症(Waldenstrom's macroglobulinemia)、眼内(眼睛)黑色素瘤、梅克尔细胞癌(Merkel cell carcinoma)、恶性间皮瘤、转移性鳞状颈癌、舌癌、多发性内分泌瘤综合征、骨髓增生异常综合征、骨髓增生异常/骨髓及外骨髓增生性疾病、鼻咽癌、成神经细胞瘤、口癌(oral cancer)、口腔癌(oral cavity cancer)、口咽癌、卵巢癌、卵巢上皮癌、卵巢低恶性潜在肿瘤、胰腺癌、岛细胞胰腺癌、鼻窦和鼻腔癌、甲状旁腺癌症、阴茎癌、嗜铬细胞瘤、成松果体细胞瘤和幕上原发性神经外胚叶肿瘤、垂体瘤、浆细胞瘤/多发性骨髓瘤、胸膜肺母细胞瘤、前列腺癌、横纹肌肉瘤、唾腺癌、肿瘤的尤文肉瘤家族、软组织肉瘤、子宫癌、子宫肉瘤、皮肤癌(非黑色素瘤)、皮肤癌(黑色素瘤)、小肠癌、鳞状细胞癌、睾丸癌、咽喉癌、胸腺瘤、胸腺瘤和胸腺癌、甲状腺癌、肾盂和输尿管及其它泌尿器官的移行细胞癌、妊娠期滋养叶瘤、尿道癌、阴道癌、外阴癌和威姆氏肿瘤(Wilm's tumor)。
术语自身免疫性病症(或自身免疫性疾病)是指由身体对通常存在于身体中的物质和组织的不当免疫反应(自身免疫)产生的任何病症。此类反应可局限于某些器官或涉及在不同位置的特定组织。示例性自身免疫性病症为急性播散性脑脊髓炎(ADEM)、阿狄森氏病(Addison's disease)、丙种球蛋白缺乏症、斑秃、肌萎缩性侧索硬化症、强硬性脊柱炎、抗磷脂综合征、抗合成酶综合征、特异性过敏、特异性皮炎、自身免疫性再生障碍性贫血、自身免疫性心肌病、自身免疫性肠病、自身免疫性溶血性贫血、自身免疫性肝炎、自身免疫性内耳疾病、自身免疫性淋巴组织增生综合征、自身免疫性周围神经病、自身免疫性胰腺炎、自身免疫性多内分泌腺综合征、自身免疫性黄体酮皮炎、自身免疫血小板减少性紫癜、自身免疫性荨麻疹、自身免疫性葡萄膜炎、巴洛病(Balo disease)/巴洛同心圆性硬化(Balo concentric sclerosis)、白塞氏病(Behçet's disease)、贝格尔氏病(Berger's disease)、比克斯塔夫氏脑炎(Bickerstaff's encephalitis)、Blau综合征、大疱性类天疱疮、卡斯尔曼氏病(Castleman's disease)、乳糜泻、恰加斯式病(Chagas disease)、慢性炎性脱髓鞘多神经病、慢性复发性多灶骨髓炎、慢性阻塞性肺病、谢格-司托司综合征(Churg-Strauss syndrome)、瘢痕性类天疱疮、科干综合征、冷凝集素病、补充组分2缺乏、接触性皮炎、颅动脉炎、CREST综合征、克罗恩氏病(Crohn's disease)(两种类型的特发性炎性肠病“IBD”中的一种)、库兴氏综合征(Cushing's Syndrome)、皮肤白细胞分裂性脉管炎、德戈氏病(Dego's disease)、德尔肯氏病(Dercum's disease)、疱疹样皮炎、皮肌炎、1型糖尿病、扩散性皮肤全身性硬化、德雷斯勒氏综合征(Dressler's syndrome)、药物诱发性狼疮、盘状红斑狼疮、湿疹、子宫内膜异位、附着点炎症相关关节炎、嗜酸性筋膜炎、嗜酸性肠胃炎、后天性大疱性表皮松解、结节性红斑、胎儿成红细胞增多症、原发性混合型冷球蛋白血症(essential mixed cryoglobulinemia)、Evan综合征、进行性肌肉骨化症(fibrodysplasia ossificans progressive)、纤维性肺泡炎(或特发性肺纤维变性)、胃炎、胃肠类天疱疮、肾小球性肾炎、古德帕斯彻氏综合征(Goodpasture's syndrome)、格雷夫斯氏病(Graves's disease)、吉兰-巴雷综合征(Guillain-Barré syndrome,GBS)、桥本氏脑病、桥本氏甲状腺炎(Hashimoto's thyroiditis)、亨-舍二氏紫癜(Henoch-Schonlein purpura)、妊娠疱疹(又名妊娠期类天疱疮)、化脓性汗腺炎、Hughes-Stovin综合征、低丙种球蛋白血症、特发性炎性脱髓鞘病、特发性肺纤维变性、特发性血小板减少性紫癜、IgA肾病、包含体肌炎、慢性炎性脱髓鞘多发性神经病、间质性膀胱炎、幼年特发性类风湿性关节炎(又名幼年型类风湿性关节炎)、川崎病(Kawasaki's disease)、朗伯-伊顿类重症肌无力综合征(Lambert-Eaton myasthenic syndrome)、白细胞分裂性脉管炎、扁平苔癣、硬化性苔藓、线性IgA疾病(LAD)、狼疮样肝炎(又名自身免疫性肝炎)、红斑狼疮、Majeed综合征、梅尼埃病(Ménière's disease)、显微镜下多血管炎、混合型结缔组织病、硬斑病、穆-哈二氏病(Mucha-Habermann disease)(又名急性苔癣痘疹样糠疹)、多发性硬化、重症肌无力、肌炎、嗜眠病、视神经脊髓炎(亦称德维克氏病(Devic's disease))、神经性肌强直、眼瘢痕性类天疱疮、眼肌阵挛综合征、Ord's甲状腺炎、汉-罗二氏综合征(palindromic rheumatism)、PANDAS (与链球菌相关的儿科自身免疫性神经精神病学病症)、副肿瘤小脑变性、阵发性睡眠性血红蛋白尿症(PNH)、Parry Romberg综合征、Parsonage-Turner综合征、睫状体平坦部炎、寻常型天疱疮、恶性贫血、静脉周脑脊髓炎、POEMS综合征、多发性结节性动脉炎、多肌痛风湿病、多肌炎、原发性胆汁性肝硬化、原发性硬化性胆管炎、渐进性炎性神经病、牛皮癣、牛皮癣关节炎、坏疽性脓皮病、纯红细胞再生障碍性贫血、Rasmussen's脑炎、雷诺现象、复发性多软骨炎、莱特尔氏综合征(Reiter's syndrome)、下肢不宁综合征、腹膜后纤维化、类风湿性关节炎、风湿热、结节病、精神分裂症、施密特综合征、另一形式的APS、施尼茨勒氏综合征(Schnitzler syndrome)、巩膜炎、硬皮病、血清病、斯耶格伦氏综合征(Sjögren's syndrome)、脊柱关节病、僵人综合征、亚急性细菌性心内膜炎(SBE)、Susac综合征、Sweet综合征、交感性眼炎、全身性红斑狼疮、高安氏动脉炎(Takayasu's arteritis)、颞动脉炎(也称作“巨细胞动脉炎”)、血小板减少症、Tolosa-Hunt综合征、横贯性脊髓炎、溃疡性结肠炎(两种类型的特发性炎性肠病“IBD”中的一种)、与混合型结缔组织病不同的未分化型结缔组织病、未分化型脊柱关节病、荨麻疹脉管炎、脉管炎、白斑病和韦格纳氏坏死性肉芽肿(Wegener's granulomatosis)。
术语炎性病症(或炎性疾病)是指与炎症相关、通常由白细胞浸润引起的病理状态。所述炎性病症可为急性的或慢性的。示例性炎性病症包括炎性皮肤病,包括而不限于牛皮癣和特异性皮炎;全身性硬皮病和硬化;与炎性肠病(IBD)(例如克罗恩氏病和溃疡性结肠炎)相关的反应;缺血性再灌注病症,包括手术组织再灌注损伤;心肌缺血性病况,例如心肌梗塞、心跳骤停、在心脏手术之后的再灌注和在经皮冠状动脉成形术之后的收缩;中风;和腹主动脉瘤;中风继发性脑水肿;颅脑外伤;低血容量性休克;窒息;成人呼吸窘迫综合征;急性肺损伤;贝切特氏病;皮肌炎;多肌炎;多发性硬化(MS);皮炎;脑膜炎;脑炎;葡萄膜炎;骨关节炎;狼疮肾炎;自身免疫性疾病,例如类风湿性关节炎(RA)、Sjorgen综合征、脉管炎、包括白细胞渗出的疾病;中枢神经系统(CNS)炎性病症;败血病或创伤继发性多器官损伤综合征;酒精性肝炎;细菌性肺炎;抗原-抗体复合物介导的疾病,包括肾小球性肾炎、脓毒症、结节病、组织或器官移植的免疫病理学反应;肺部的炎症,包括胸膜炎、肺泡炎、脉管炎、肺炎、慢性支气管炎、支气管扩张、弥漫性泛细支气管炎、过敏性肺炎、特发性肺纤维变性(IPF)和囊性纤维变性;等。
术语神经组织退行病症(或神经组织退行疾病)是指与影响脑、脊髓或周围神经系统的结构或功能的神经元的结构或功能的渐进性损失相关的病症。示例性神经退行性病症包括线粒体脑肌病和肠道运动功能障碍综合征、共济失调综合征包括弗里德棘希氏共济失调(Friedreich's ataxia)和脊髓小脑共济失调(SCA)、脊髓损伤、家族性和间歇性肌萎缩性侧索硬化(分别为FALS和ALS)、家族性和间歇性帕金森病(Parkinson's disease)、家族性和间歇性阿尔茨海默氏病(Alzheimer's disease);亨廷顿氏病(Huntington's disease)、橄榄体脑桥小脑萎缩、多系统萎缩症、渐进性核上麻痹、弥漫性路易体病和共核蛋白病、唐氏综合征(Down Syndrome)、皮质齿状核变性(corticodentatonigral degeneration)、渐进性家族性肌阵挛型癫痫、纹状体黑质变性、变形性肌张力障碍、家族性颤振、日勒德拉图雷特综合征(Gilles de la Tourette syndrome)和哈勒沃登-施帕茨病(Hallervorden-Spatz disease)。
术语赋形剂是指药学上可接受的化学品,诸如制药领域的普通技术人员已知有助于药物试剂的给药的药学上可接受的化学品。其为可用于制备药物组合物的化合物,其通常为安全、无毒且既不是生物学上、也不是不期望的并且包括为兽医使用以及人类药物使用所接受的赋形剂。示例性赋形剂包括粘合剂、表面活性剂、稀释剂、崩解剂、抗粘附剂和润滑剂。
根据第一个方面,提供了如本文上面定义的式(I)化合物,
或其药学上可接受的盐。
在式(I)化合物中,RA、RB和RC基团独立地选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基;或
RA和RC中的一个与RB一起形成双基-(CH2)m-,其中m是3至5的整数,且RA和RC中的另一个选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,RA、RB和RC独立地选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些其它实施方案中,RA和RC中的一个与RB一起形成双基-(CH2)m-,其中m是3至5的整数,且RA和RC中的另一个选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。
因此,基团RA选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基;或与RB一起形成双基-(CH2)m-,其中m是3至5的整数。
在一些实施方案中,RA选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,RA选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13和R14S(O)2NR15C(O)。
在一些实施方案中,RA选自H、卤素、氰基、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、R12S(O)2NR13和R14S(O)2NR15C(O)。
在一些实施方案中,RA选自H、卤素、C1-C6烷基和R2C(O),例如H、卤素和C1-C6烷基,或H和卤素。
在一些实施方案中,RA是H。
在一些其它实施方案中,RA选自卤素、C1-C6烷基和R2C(O),例如C1-C6 烷基和R2C(O),例如RA是R2C(O),或RA是C1-C6 烷基。
在一些实施方案中,RA选自H、卤素、氰基、C1-C6烷基、C3-C6 环烷基、R2C(O)、R4S(O)2、R5OC(O)、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基;例如选自H、卤素、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基,例如选自H、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基,或选自任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,当RA是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基时,其更具体地是任选地被一个或多个基团R16取代的苯基。在一些实施方案中,当RA是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基时,其更具体地是任选地被一个或多个基团R16取代的5-或6-元杂环基。
基团RB选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6 烷基、任选地被R1O取代的C3-C6 环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基;或与RA或RC一起形成双基-(CH2)m-,其中m是3至5的整数。
在一些实施方案中,RB选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6 环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,RB选自H、卤素、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6 环烷基、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,RB是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,RB选自H、卤素、R1O、任选地被R1O取代的C1-C6烷基和任选地被R1O取代的C3-C6 环烷基,例如选自H、卤素和任选地被R1O取代的C1-C6烷基。
在一些实施方案中、RB选自H、卤素、任选地被R1O取代的C1-C6烷基和任选地被R1O取代的C3-C6 环烷基,例如选自H和卤素。
在一些实施方案中、RB选自H、卤素、C1-C6烷基和C3-C6 环烷基,例如选自H、卤素和C1-C6烷基,例如选自H和C1-C6烷基。
在一些实施方案中,RB选自卤素、任选地被R1O取代的C1-C6烷基和任选地被R1O取代的C3-C6 环烷基,例如选自卤素和任选地被R1O取代的C1-C6烷基;或选自卤素和C1-C6烷基,例如RB是卤素。
在一些实施方案中,RB是H。
基团RC选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基,或与RB一起形成双基-(CH2)m-,其中m是3至5的整数。
在一些实施方案中,RC选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6 环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,RC选自H、卤素、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6 环烷基、R2C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,RC是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,RC选自H、卤素、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6 环烷基和R2C(O)。
在一些实施方案中,RC选自H、卤素、R1O、任选地被R1O取代的C1-C6烷基和任选地被R1O取代的C3-C6 环烷基,例如选自H、卤素、任选地被R1O取代的C1-C6烷基和任选地被R1O取代的C3-C6 环烷基,或选自H、卤素、C1-C6烷基和C3-C6 环烷基,特别是选自H、卤素和C1-C6烷基,或选自H和C1-C6烷基。
在一些实施方案中,RC是H。
在一些实施方案中,RA和RC中的一个与RB一起形成双基-(CH2)m-,其中m是3至5的整数,例如,m是3或4,或m是3。在此类实施方案中,RA和RC中的另一个选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13、R14S(O)2NR15C(O)、任选地被一个或多个基团R16取代的苯基和任选地被一个或多个基团R16取代的5-或6-元杂环基。在这些实施方案中,RA和RC中的另一个优选地选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、任选地被R1O取代的C3-C6 环烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13和R14S(O)2NR15C(O);例如选自H、卤素、氰基、R1O、任选地被R1O取代的C1-C6烷基、R2C(O)、R3S、R4S(O)2、R5OC(O)、(R6ON)C(R7)、R8R9NC(O)、R10R11N、R12S(O)2NR13和R14S(O)2NR15C(O);例如选自H、卤素和C1-C3烷基;诸如H、F和甲基;或H和F,特别是H。
在一些实施方案中,RA、RB和RC中的至少一个是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,RA、RB和RC中的一个是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基。
在一些实施方案中,当RA、RB和RC中的一个是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基时,RA、RB和RC中的另外两个如本文上面所定义,但不是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基。
例如,在一些实施方案中,当RA、RB和RC中的一个是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基时,RA、RB和RC中的另外两个选自H、卤素、C1-C6烷基、C3-C6 环烷基和R2C(O),或选自H、卤素 C1-C6烷基和R2C(O),例如选自H、F、Cl和C1-C3烷基;或选自H、F和甲基;例如两者均为H 或F或两者均为H。
在一些实施方案中,RA、RB和RC独立地选自H、卤素、氰基、C1-C4烷基、R1O、R2C(O)、R3S和R4S(O)2;或RA和RC中的一个与RB一起形成双基-(CH2)m-,其中m是3至5的整数,且RA和RC中的另一个选自H、卤素、氰基、C1-C4烷基、R1O、R2C(O)、R3S和R4S(O)2。
当RA、RB和RC中的任一个选自卤素时,其更具体地可以选自F、Cl 或Br,或选自F和Cl,特别是Cl。
当RA、RB和RC中的任一个选自任选地被R1O取代的C1-C6烷基和任选地被R1O取代的C3-C6 环烷基时,其更具体地可以选自任选地被R1O取代的C1-C6烷基,例如选自任选地被R1O取代的C1-C4烷基或任选地被R1O取代的C1-C3烷基,特别是任选地被R1O取代的甲基。
在一些实施方案中,当RA、RB和RC中的任一个选自任选地被R1O取代的C1-C6烷基和任选地被R1O取代的C3-C6 环烷基时,其更具体地选自C1-C6烷基,或选自C1-C4烷基或C1-C3烷基,特别是甲基。
在一些实施方案中,当RA、RB和RC中的任一个选自任选地被R1O取代的C3-C6 环烷基时,其更具体地选自C3-C6 环烷基,或选自C3-C5 环烷基,或选自C3-C4 环烷基,例如环丙基。
在一些实施方案中,RA、RB和RC都是H,即式(I)化合物是式(Ia)化合物
其中 W和RD如本文所定义。
在一些其它实施方案中,RA、RB和RC中的至少一个不同于H。
在一些实施方案中,RA如本文上面所定义,但不同于H;或RB如本文上面所定义,但不同于H。
在一些实施方案中,RA、RB和RC中的两个不同于H,即RA和RB 不同于H,或 RA和RC 不同于H,或 RB和RC 不同于H。
在一些实施方案中,RA是H,且所述化合物则可以由式(Ib)代表
其中RB、RC、W和RD如本文所定义,例如RB和RC 如本文上面所定义,但RB和RC都不是H。
在一些其它实施方案中,RA不同于H。在一些实施方案中,RA和RB不同于H,且RC是H。在一些其它实施方案中,RA和RC不同于H,且RB是H。
在一些实施方案中,RA不同于H,且RB和RC均为H,即所述化合物可以由式(Ic)代表
其中 RA、W和RD如本文所定义。
当RA、RB和RC中的任一个是R1O、任选地被R1O取代的C1-C6烷基或任选地被R1O取代的C3-C6 环烷基时,基团R1选自H、C1-C6烷基和C3-C6 环烷基,例如选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,基团R1选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是H。
在一些实施方案中,基团R1如本文上面所定义,但不是H;例如R1是CH3。
在一些实施方案中,当RA、RB和RC中的任一个选自R1O、任选地被R1O取代的C1-C6烷基和任选地被R1O取代的C3-C6 环烷基时,其更具体地可以选自R1O和任选地被R1O取代的C1-C6烷基,例如选自R1O和任选地被R1O取代的C1-C3烷基,或选自R1O和任选地被R1O取代的C1-C2烷基,例如其可以选自R1O、R1OCH2和R1OCH(CH3),特别是R1O。
当RA、RB和RC中的任一个是R2C(O)时,R2是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R2选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R2选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H、CH3和(CH3)2CH,特别是CH3。在一些实施方案中,基团R2如本文上面所定义,但不是H。
当RA、RB和RC中的任一个是R3S时,R3是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R3选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R3选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是CH3。在一些实施方案中,基团R3如本文上面所定义,但不是H。
当RA、RB和RC中的任一个是R4SO2时,R4是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R4选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R4选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是CH3。在一些实施方案中,基团R4如本文上面所定义,但不是H。
当RA、RB和RC中的任一个是R5OC(O)时,R5是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R5选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R5选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是H。
当RA、RB和RC中的任一个是(R6ON)C(R7)时,R6是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R6选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R6选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是H。R7是H、C1-C6烷基和C3-C6 环烷基。在一些实施方案中,R7选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R7选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是CH3。在一些实施方案中,基团R7如本文上面所定义,但不是H。在一些实施方案中,R6是H且R7如本文上面所定义,但不同于H,例如R7是CH3。
当RA、RB和RC中的任一个是R8R9NC(O)或R10R11N时,R8、R9、R10和R11中的每一个独立地选自H、C1-C6烷基和C3-C6 环烷基,例如选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,当RA、RB和RC中的任一个是R8R9NC(O)或R10R11N时,R8、R9、R10和R11中的每一个独立地选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3。
当RA、RB和RC中的任一个是R12S(O)2NR13或R14S(O)2NR15C(O)时,R12、R13、R14和R15中的每一个独立地选自H、C1-C6烷基和C3-C6 环烷基,例如选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,当RA、RB和RC中的任一个是R12S(O)2NR13或R14S(O)2NR15C(O)时,R12、R13、R14和R15中的每一个独立地选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3。在一些实施方案中,R12如本文上面所定义,但不是H和R13是H。在一些实施方案中,R14如本文上面所定义,但不是H和R15是H。在一些实施方案中,R12和R14 如本文上面所定义,但不是H且R13和R15均为H。
在一些实施方案中,当RA、RB和RC中的任一个是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基时,所述苯基或杂环基被0、1、2或3个基团R16,例如0、1或2个基团R16或 0或1个基团R16,例如1个基团R16取代。
在一些实施方案中,当RA、RB和RC中的任一个是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基时,其更具体地是被0、1、2或3个基团R16或 0、1或2个基团R16取代的苯基。
在一些实施方案中,当RA、RB和RC中的任一个是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基时,其更具体地是被0、1、2或3个基团R16或例如0、1或2个基团R16取代的5-或6-元杂环基。
在一些实施方案中,当RA、RB和RC中的任一个是被一个基团R16取代的苯基时,所述基团在所述苯基环上的对位。
在一些实施方案中,RA是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基。在这些实施方案中的一些中,式(I)化合物可以由式(Id)代表
其中环B是苯基或5-或6-元杂环基,k是0至3的整数且 R16、RB、RC、W 和 RD如本文所定义。
在一些实施方案中,环B是苯基,且式(Ic)化合物可以由式(Ie)代表
其中k、R16、RB、RC、W和RD如本文所定义。
在式(Ie)化合物的一些实施方案中,k是1且R16在对位,即所述化合物可以由式(If)代表
其中R16、RB、RC、W和RD如本文所定义。
在式(Ic)、(Id)、(Ie)和(If)化合物的一些实施方案中,RB和RC均为H。
在一些实施方案中,当RA、RB和RC中的任一个是任选地被一个或多个基团R16取代的5-或6-元杂环基时,所述杂环基是5-元的。在一些实施方案中,当RA、RB和RC中的任一个是任选地被一个或多个基团R16取代的5-或6-元杂环基时,所述杂环基是6-元的。任何5-或6-元杂环基在环中包含1个或多个杂原子,例如1、2、3或4个杂原子。在一些实施方案中,所述杂环基是芳族的。在一些其它实施方案中,所述杂环基是非芳族的,饱和或不饱和的,例如所述杂环基是非芳族且单不饱和的。例如,所述杂环基可以选自
和。
当RA、RB和RC中的任一个是任选地被一个或多个基团R16取代的苯基或任选地被一个或多个基团R16取代的5-或6-元杂环基时,每个基团R16独立地选自卤素、氰基、硝基、R17O、任选地被R17O取代的C1-C6烷基、任选地被R17O取代的C3-C6 环烷基、R18C(O)、R19S、R20S(O)2、R21OC(O)、(R22ON)C(R23)、R24R25NC(O)、R26R27N、R28S(O)2NR29和R30S(O)2NR31C(O)。
在一些实施方案中,每个R16独立地选自卤素、氰基、硝基、R17O、任选地被R17O取代的C1-C6烷基、任选地被R17O取代的C3-C6 环烷基、R19S、R20S(O)2、R21OC(O)和R26R27N。
在一些实施方案中,每个R16独立地选自卤素、氰基、硝基、R17O、任选地被R17O取代的C1-C6烷基、R19S、R20S(O)2、R21OC(O)和R26R27N。
在一些实施方案中,每个R16独立地选自卤素、氰基、硝基、R17O、任选地被R17O取代的C1-C6烷基、R19S、R20S(O)2、R21OC(O)和R26R27N。
在一些实施方案中,每个R16独立地选自R17O, 任选地被R17O取代的C1-C6烷基和任选地被R17O取代的C3-C6 环烷基,例如选自R17O, 任选地被R17O取代的C1-C6烷基,特别是选自R17O。
当R16是任选地被R17O取代的C1-C6烷基时,其例如可以是任选地被R17O取代的C1-C4烷基,例如任选地被R17O取代的C1-C3烷基。
在一些实施方案中,每个R16独立地选自F、Cl、氰基、硝基、CH3、CF3、(CH3)3C、CH3O、CH3S、CH3S(O)2、COOH、NH2和(CH3)2N。
当任何R16是R17O、任选地被R17O取代的C1-C6烷基或任选地被R17O取代的C3-C6 环烷基时,基团R17是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R17选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R17选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是H。在一些其它实施方案中,基团R17如本文上面所定义,但不是H;例如R17是CH3。
当任何R16是R18C(O)时,基团R18是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R18选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R18选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是H。在一些其它实施方案中,基团R18如本文上面所定义,但不是H;例如R18是CH3。
当任何R16是R19S时,基团R19是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R19选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R19选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是H。在一些其它实施方案中,基团R19如本文上面所定义,但不是H;例如R19是CH3。
当任何R16是R20S(O)2时,基团R20是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R20选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R20选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是H。在一些其它实施方案中,基团R20如本文上面所定义,但不是H;例如R20是CH3。
当任何R16是R21OC(O)时,基团R21是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R21选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R21选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是H。在一些其它实施方案中,基团R21如本文上面所定义,但不是H;例如R21是CH3。
当任何R16是(R22ON)C(R23)时,R22是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R22选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R22选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是H。R23是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R23选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R23选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是CH3。在一些实施方案中,基团R23如本文上面所定义,但不是H。在一些实施方案中,R22是H且R23如本文上面所定义,但不同于H,例如R23是CH3。
当任何R16是R24R25NC(O)或R26R27N时,R24、R25、R26和R27中的每一个独立地选自H、C1-C6烷基和C3-C6 环烷基,例如选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,当任何R16是R24R25NC(O)或R26R27N时,R24、R25、R26和R27中的每一个独立地选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3。
当任何R16是R28S(O)2NR29或R30S(O)2NR31C(O)时,R28、R29、R30和R31中的每一个独立地选自H、C1-C6烷基和C3-C6 环烷基,例如选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,当任何R16是R28S(O)2NR29或R30S(O)2NR31C(O)时,R28、R29、R30和R31中的每一个独立地选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3。在一些实施方案中,R28和R30 如本文上面所定义,但不是H且R29和R31均为H。
当RA和RC中的一个与RB一起形成双基-(CH2)m-(其中m是3至5的整数)时,m例如是3或4,特别是3,例如RB和RC一起形成双基-(CH2)3-。
例如,在式(I)化合物的一些实施方案中,RA、RB和RC中的一个是H且另外两个独立地选自卤素、氰基、C1-C4 烷基、R1O、R2C(O)、R3S和 R4S(O)2;或者,如果相邻,在一些实施方案中,另外两个形成双基-(CH2)m-,其中 m是3至5的整数。
在一些实施方案中,RA、RB和RC中的一个是H且另外两个选自卤素、氰基、C1-C4 烷基和 R1O,例如选自卤素、C1-C3 烷基和 C1-C3 烷氧基,例如选自F、Cl、甲基 和 甲氧基;或选自卤素和 C1-C3 烷基,诸如F、Cl和甲基。
在一些实施方案中,RA、RB和RC中的一个是H 且另外两个均选自卤素,例如两者均为F或Cl,或两者均为Cl。
在其它实施方案中,RA、RB和RC中不同于H的两个均选自C1-C3 烷基和 R1O,例如两者均选自甲基和甲氧基。在一些实施方案中,RA、RB和RC中不同于H的两个与彼此是相同的,例如两者均为Cl,或两者均为甲基,或两者均为R1O,例如甲氧基。
例如,在式(I)化合物、例如式(Ib)化合物的一些实施方案中,RB和RC均选自卤素、氰基、C1-C4烷基和 R1O。例如,RB和 RC可以均选自卤素,例如RB和 RC两者均为F或Cl,或RB和 RC两者均为Cl。在其它实施方案中,RB和RC两者均选自C1-C4烷基和R1O,例如RB和RC两者均选自甲基和甲氧基,例如两者均为甲基。在式(I)化合物、例如式(Ib)化合物中,RB和RC可以与彼此不同,或可以相同。
在式(I)化合物中,连接基团W是直接键或X1-X2-X3。在一些实施方案中,W 是直接键,即所述化合物可以由式(Ig)代表
其中RA、RB、RC和RD如本文所定义。
在一些其它实施方案中,W是X1-X2-X3,且所述化合物可以由式(Ih)代表
其中RA、RB、RC、RD、X1、X2和X3如本文所定义。
在式(Ih)化合物中
X1是任选地被C1-C4 烷基或R32O取代的C1-C2亚烷基;
X2是O或不存在;且
X3是直接键或任选地被C1-C4烷基或R32O取代的C1-C2亚烷基。
在一些实施方案中,X1是任选地被C1-C4烷基取代的C1-C2亚烷基,例如X1是任选地被C1-C4烷基取代的C1亚烷基。
在一些实施方案中,X1是任选地被C1-C4烷基或R32O取代的C1亚烷基。在一些实施方案中,X1是C1-C2亚烷基,例如X1是C1亚烷基(即CH2)。
基团X2不存在或是O。在一些实施方案中,X2不存在,即所述化合物可以由式(Ij)代表
其中RA、RB、RC、RD、X1和X3如本文所定义。
在式(Ij)化合物中,X1是任选地被C1-C4烷基或R32O取代的C1-C2亚烷基且X3是直接键或任选地被C1-C4烷基或R32O取代的C1-C2亚烷基;例如X1是任选地被C1-C4烷基或R32O取代的C1-C2亚烷基且X3是直接键或C1-C2亚烷基,例如X3是C1-C2亚烷基或X3是C1亚烷基。
在式(Ij)化合物的一些其它实施方案中,X1是任选地被C1-C4烷基或R32O取代的C1-C2亚烷基且X3是直接键或C1亚烷基。
在式(Ij)化合物的一些其它实施方案中,X1是任选地被C1-C4烷基或R32O取代的C1-C2亚烷基且X3 是直接键。
在式(Ij)化合物的一些其它实施方案中,X1是任选地被C1-C4烷基或R32O取代的C1亚烷基且X3是直接键或任选地被C1-C4烷基或R32O取代的C1-C2亚烷基;例如X1是任选地被C1-C4烷基或R32O取代的C1亚烷基且X3是直接键或C1-C2亚烷基。
在一些其它实施方案中,X2是O,即所述化合物可以由式(Ik)代表
其中RA、RB、RC、RD、X1和X3如本文所定义。
基团X3是直接键或任选地被C1-C4烷基或R32O取代的C1-C2亚烷基。在一些实施方案中,X3是直接键或任选地被C1-C4烷基或R32O取代的C1亚烷基。在一些实施方案中,X3是直接键或C1亚烷基。在一些实施方案中,X3是直接键,即式(Ih)化合物可以由式(Im)代表
其中RA、RB、RC、RD、X1和X2如本文所定义。
在式(Im)化合物的一些实施方案中,X2是O。在式(Im)化合物的一些其它实施方案中,X2不存在,且所述化合物则可以由式(In)代表
其中RA、RB、RC、RD和X1如本文所定义。
当X1或X3或X1和X3两者被选自C1-C4烷基或R32O的基团取代时,所述基团更具体地可以选自C1-C3烷基和R32O,或选自C1-C2烷基和R32O,特别是选自CH3和R32O。在一些实施方案中,所述基团选自C1-C4烷基或C1-C3烷基,或选自C1-C2烷基或CH3。在一些实施方案中,X1和X3均未被取代。在一些实施方案中,所述基团是R32O。
基团R32选自H和C1-C4烷基。在一些实施方案中,R32选自H和C1-C3烷基,或选自H和C1-C2烷基,特别是选自H和CH3,例如H。在一些其它实施方案中,R32选自C1-C4烷基,例如C1-C3烷基或C1-C2烷基,特别是CH3。
在一些实施方案中,W 是直接键、CH2、CH2CH2、CH2CH2CH2、CH(CH3)、CH(CH3)CH2、CH(OH)、CH(OCH3)、CH(CH3)O或CH(CH3)OCH2;例如W 是直接键、CH2、CH2CH2、CH2CH2CH2、CH(CH3)、CH(CH3)CH2、CH(OH)或CH(OCH3)。
在一些实施方案中,W 是直接键、CH2、CH2CH2或CH2CH2CH2,例如W是CH2CH2或CH2CH2CH2,例如W是CH2CH2。
在一些实施方案中,W 是直接键、CH2或CH2CH2,例如W是直接键或CH2,例如W是CH2。
在一些其它实施方案中,W是CH2CH2、CH2CH2CH2、CH(CH3)、CH(CH3)CH2、CH(OH)、CH(OCH3)、CH(CH3)O或CH(CH3)OCH2。
在式(I)化合物中,基团RD是C1-C6烷基或选自苯基、4-至6-元杂环基、C4-C6 环烷基和C5-C6 环烯基的环状基团,其中所述环状基团任选地被一个或多个R33取代;例如基团RD是C1-C6烷基或选自苯基、4-至6-元杂环基和C4-C6 环烷基的环状基团,其中所述环状基团任选地被一个或多个R33取代。
当RD是C1-C6烷基时,其例如可以选自C3-C6烷基,或选自C3-C5烷基。在一些实施方案中,当RD是C1-C6烷基时,W 是直接键。
在一些实施方案中,基团RD是选自苯基、4-至6-元杂环基、C4-C6 环烷基和C5-C6 环烯基、例如选自苯基、4-至6-元杂环基和C4-C6 环烷基的环状基团,其中所述环状基团任选地被一个或多个R33取代。在一些实施方案中,式(I)化合物可以由式(Io)代表
其中 RA、RB、RC、W和R33如本文所定义,环A是苯基、4-至6-元杂环基、C4-C6 环烷基或C5-C6 环烯基,且n是0至3的整数。
在式(Io)化合物中,环A被n个基团R33取代,其中n是0至3的整数。在一些实施方案中,n是0至2的整数,例如n是0、1或2,或n是1或2。在一些实施方案中,n是0。在其它实施方案中,n是1。在还有其它实施方案中,n是2或3,例如n是2。
在式(Io)化合物中,每个基团R33独立地选自卤素、氰基、任选地被R34O取代的C1-C6烷基、C3-C6 环烷基、苯基、5-或6-元杂环基、R34O、R35OC(O)、R36S(O)2、R37C(O)、R38R39N、R40R41N(CO);且当n至少为2时,连接至一个且相同碳原子的两个R33可以与它们两者均连接的碳原子一起形成在环中任选地含有一个或多个杂原子的4-至6-元环,例如,连接至一个且相同碳原子的两个R33形成双基-CH2-L-CH2-,其中 L是CH2、NH或O,例如L是NH。
在一些实施方案中,每个基团R33独立地选自卤素、氰基、任选地被R34O取代的C1-C6烷基、C3-C6 环烷基、苯基、5-或6-元杂环基、R34O、R35OC(O)、R36S(O)2、R37C(O)、R38R39N、R40R41N(CO);例如每个基团R33独立地选自卤素、氰基、任选地被R34O取代的C1-C6烷基、C3-C6 环烷基、苯基、5-或6-元杂环基、R34O、R36S(O)2、R37C(O)、R38R39N和R40R41N(CO)。
在一些其它实施方案中,当n是2或3时,例如当n是2时,连接至一个且相同碳原子的两个R33可以与它们两者均连接的碳原子一起形成在环中任选地含有一个或多个杂原子的4-至6-元环,例如,连接至一个且相同碳原子的两个R33形成双基-CH2-L-CH2-,其中 L是CH2、NH或O,例如L是NH。
当基团R33是卤素时,所述卤素可以是F、Cl或Br,特别是F或Cl。
当基团R33是任选地被R34O取代的C1-C6烷基时,所述C1-C6烷基具体而言可以是C1-C4烷基,更具体地C1-C3烷基,例如甲基或乙基。
在一些实施方案中,当基团R33是任选地被R34O取代的C1-C6烷基时,其更具体地未被任何R34O取代,即,所述R33是C1-C6烷基或C1-C4烷基或C1-C3烷基,例如甲基或乙基。
在式(Io)化合物的一些实施方案中,当基团R33是任选地被R34O取代的C1-C6烷基时,其例如可以是式R34O(CH2)p的基团,其中 p是1至3的整数,例如p是1或2,或p是1。
在基团R34O中,R34是H、C1-C6烷基或C3-C6 环烷基;其中所述烷基或环烷基任选地被R42O取代。在一些实施方案中,R34是H、C1-C3烷基或C3-C4 环烷基,其中所述烷基或环烷基任选地被R42O取代。在一些实施方案中,R34是H或C1-C6烷基,其中所述烷基任选地被R42O取代,例如R34是H或C1-C3烷基,其中所述烷基任选地被R42O取代。在一些实施方案中,R34是H、CH3、CH3CH2、CF3CH2或CH3OCH2CH2。
在基团R35OC(O)中,R35是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R35选自H、C1-C4烷基和C3-C4 环烷基,或选自H、C1-C3烷基和环丙基。在一些实施方案中,R35选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H和CH3,特别是CH3。在一些实施方案中,基团R35如本文上面所定义,但不是H。
在基团R36S(O)2中,R36是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R36选自H、C1-C4烷基和C4-C6 环烷基,或选自H、C1-C3烷基和环戊基。在一些实施方案中,R36选自H和C1-C6烷基,例如选自H和C1-C4烷基,或选自H和C1-C3烷基,诸如选自H、CH3和CH3CH2。在一些实施方案中,基团R36如本文上面所定义,但不是H,例如R36是甲基、乙基或环戊基。
在基团R37C(O)中,R37选自C1-C6烷基或C3-C6 环烷基,其中所述烷基或环烷基任选地被R43O取代。在一些实施方案中,R37选自C1-C5烷基和C4-C6 环烷基,或选自C1-C4烷基和C4-C5 环烷基,例如选自CH3、CF3CH2、(CH3)2CHCH3或环戊基。在一些实施方案中,R37选自任选地被R43O取代的C1-C6烷基,和C3-C6 环烷基。当C1-C6烷基被R43O取代时,所述烷基例如可以是C1-C4烷基或C1-C3烷基,诸如甲基。
在基团R38R39N中,R38和R39独立地选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R44R45N或R46O取代,或R38和R39与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基、例如C1-C3烷基或C1-C2烷基、例如CH3取代的4-至6-元杂环。
在一些实施方案中,R38和R39独立地选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R44R45N或R46O取代;例如R38选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,且R39选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R44R45N或R46O取代。
在一些实施方案中,R38选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R39选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R44R45N或R46O取代;或R38和R39与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基、例如C1-C3烷基或C1-C2烷基、例如甲基取代的4-至6-元杂环;例如,R38和R39与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子的4-至6-元杂环,或R38和R39与它们两者均连接的氮原子一起形成不含其它杂原子的4-至6-元杂环,诸如氮杂环丁烷基。
在一些实施方案中,R38选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R39选自H、C1-C6烷基和C3-C6 环烷基,或R38和R39与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基、例如C1-C3烷基或C1-C2烷基或CH3取代的4-至6-元杂环;例如,R38和R39与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子的4-至6-元杂环,或R38和R39与它们两者均连接的氮原子一起形成不含其它杂原子的4-至6-元杂环。例如,R38R39N可以选自
和。
在一些实施方案中,R38选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R39选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基被R44R45N或R46O取代;例如NR38R39选自
和
其中R38如本文上面所定义,例如R38是H或C1-C3烷基,特别是R38是H或CH3。
在一些实施方案中,当R39是任选地被R44R45N或R46O取代的C1-C6烷基或C3-C6 环烷基时,R39更具体地是任选地被R44R45N或R46O取代的C1-C6烷基,例如任选地被R44R45N取代的C1-C6烷基,例如任选地被R44R45N取代的C1-C4或任选地被R44R45N取代的C1-C3。
在一些其它实施方案中,当R39是任选地被R44R45N或R46O取代的C1-C6烷基或C3-C6 环烷基时,R39更具体地是任选地被R46O取代的C1-C6烷基,例如任选地被R46O取代的C1-C4,或任选地被R46O取代的C1-C3。
在一些实施方案中,R38选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R39选自H、C1-C6烷基和C3-C6 环烷基,例如选自H和C1-C6烷基,其中所述烷基或环烷基被R44R45N取代。
在一些实施方案中,R38选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R39选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基被R46O取代。
在基团R40R41NC(O)中,R40和R41独立地选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R47R48N或R49O取代,或R40和R41与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基取代的4-至6-元杂环。
在一些实施方案中,R40和R41独立地选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R47R48N或R49O取代;例如R40选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,且R41选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R47R48N或R49O取代。
在一些实施方案中,R40选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R41选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基任选地被R47R48N或R49O取代;或R40和R41与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基、例如C1-C3烷基或C1-C2烷基或CH3取代的4-至6-元杂环;例如,R40和R41与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子的4-至6-元杂环,或R40和R41与它们两者均连接的氮原子一起形成不含其它杂原子的4-至6-元杂环,诸如氮杂环丁烷基。
在一些实施方案中,R40选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R41选自H、C1-C6烷基和C3-C6 环烷基,或R40和R41与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子且任选地被C1-C6烷基、例如C1-C3烷基或C1-C2烷基或CH3取代的4-至6-元杂环;例如,R40和R41与它们两者均连接的氮原子一起形成任选地含有一个或多个其它杂原子的4-至6-元杂环,或R40和R41与它们两者均连接的氮原子一起形成不含其它杂原子的4-至6-元杂环。
在一些实施方案中,R40选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R41选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基被R47R48N或R49O取代;例如R40R41NC(O)选自
和
其中R40如本文上面所定义,例如R40是H或C1-C3烷基,特别是R40是H或CH3。
在一些实施方案中,当R41是任选地被R47R48N或R49O取代的C1-C6烷基或C3-C6 环烷基时,R41更具体地是任选地被R47R48N或R49O取代的C1-C6烷基,例如任选地被R47R48N取代的C1-C6烷基,例如任选地被R47R48N取代的C1-C4或任选地被R47R48N取代的C1-C3。
在一些其它实施方案中,当R41是任选地被R47R48N或R49O取代的C1-C6烷基或C3-C6 环烷基时,R41更具体地是任选地被R49O取代的C1-C6烷基,例如任选地被R49O取代的C1-C4,或任选地被R49O取代的C1-C3。
在一些实施方案中,R40选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R41选自H、C1-C6烷基和C3-C6 环烷基,例如选自H和C1-C6烷基,其中所述烷基或环烷基被R47R48N取代。
在一些实施方案中,R40选自H、C1-C4烷基和C3-C4 环烷基;例如H和C1-C3烷基,诸如H和CH3,例如H;且R41选自H、C1-C6烷基和C3-C6 环烷基,其中所述烷基或环烷基被R49O取代。
在基团R42O、R43O、R46O和R49O中,R42、R43、R46和R49独立地选自H、C1-C6烷基和C3-C6 环烷基。在一些实施方案中,R42、R43、R46和R49中的每一个选自H和C1-C6烷基,例如选自H和C1-C3烷基,或选自H和C1-C2烷基,诸如选自H和CH3。
在基团R42O中,R42是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R42是H、C1-C3烷基或C3-C4 环烷基,例如H、C1-C3烷基或环丙基。在一些实施方案中,R42是H或C1-C6烷基,例如R42是H或C1-C3烷基。在一些实施方案中,R42是H或CH3。
在基团R43O中,R43是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R43是H、C1-C3烷基或C3-C4 环烷基,例如H、C1-C3烷基或环丙基。在一些实施方案中,R43是H或C1-C6烷基,例如R43是H或C1-C3烷基。在一些实施方案中,R43是H或CH3。
在基团R46O中,R46是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R46是H、C1-C3烷基或C3-C4 环烷基,例如H、C1-C3烷基或环丙基。在一些实施方案中,R46是H或C1-C6烷基,例如R46是H或C1-C3烷基。在一些实施方案中,R46是H或CH3。
在基团R49O中,R49是H、C1-C6烷基或C3-C6 环烷基。在一些实施方案中,R49是H、C1-C3烷基或C3-C4 环烷基,例如H、C1-C3烷基或环丙基。在一些实施方案中,R49是H或C1-C6烷基,例如R49是H或C1-C3烷基。在一些实施方案中,R49是H或CH3。
在式(Io)化合物中,环A是C4-C6 环烷基、C5-C6 环烯基、苯基或4-至6-元杂环基。在一些实施方案中,环A是C4-C6 环烷基,例如环A是C5-C6 环烷基。
在其中环A是苯基的实施方案中,式(Io)化合物可以由式(Ip)代表
其中RA、RB、RC、R33、n和W如本文所定义。
在式(Ip)化合物中,n是0至3的整数。在式(Ip)化合物的一些实施方案中,n是0至2的整数,例如n是0、1 或2。
在式(Ip)化合物中,每个R33独立地选自卤素、氰基、任选地被R34O取代的C1-C6烷基、C3-C6 环烷基、苯基、5-或6-元杂环基、R34O、R35OC(O)、R36S(O)2、R37C(O)、R38R39N和R40R41N(CO)。
在式(Ip)化合物的一些实施方案中,每个R33独立地选自卤素、氰基、任选地被R34O取代的C1-C6烷基、苯基、5-或6-元杂环基、R34O、R35OC(O)、R36S(O)2、R37C(O)、R38R39N和R40R41N(CO)。
在式(Ip)化合物的一些实施方案中,每个R33独立地选自卤素、氰基、苯基、任选地被R34O取代的C1-C6烷基、R36S(O)2和R38R39N。在一些实施方案中,每个R33独立地选自卤素、氰基、任选地被R34O取代的C1-C6烷基、R34O、R36S(O)2和R38R39N。在一些实施方案中,每个R33独立地选自卤素、氰基和任选地被R34O取代的C1-C6烷基。在一些实施方案中,每个R33是卤素,例如R33独立地选自F、Cl和Br,特别是,F和Cl。
在式(Ip)化合物的一些实施方案中,当R33是被R34O取代的C1-C6烷基时,R33可以代表为R34O(CH2)p,其中 p是0至3的整数且R34是H或C1-C4烷基,更具体地是C1-C3烷基,特别是甲基。在此类实施方案中,整数p更具体地可以选自0至2,例如p是0。在式(Ip)化合物的一些具体实施方案中,任何R34O(CH2)p是C1-C4 烷氧基,例如C1-C3 烷氧基,特别是甲氧基。
在式(Ip)化合物的一些实施方案中,当R33是R36S(O)2时,R36是H或C1-C4烷基,且更具体地可以选自C1-C4烷基,例如R36是C1-C3烷基,特别是R36是甲基。因此,在式(Ip)化合物的一些具体实施方案中,任何R36S(O)2是CH3(SO)2。
在式(Ip)化合物的一些实施方案中,至少一个R33是R38R39N,其中 R38和R39 如本文上面所定义。在这些实施方案中的一些中,R38是H或C1-C4烷基;且R39是H、任选地被选自R46O和R44R45N的基团取代的C3-C6 环烷基或C1-C4烷基;且R44、R45和R46独立地选自H和C1-C4烷基;或R38和R39一起形成双基-(CH2)s-,其中 s是3至5的整数。在这些实施方案中的一些中,R38是H且R39是任选地被选自R44R45N和R46O的基团取代的C1-C4烷基;具体而言R38是H且R39是任选地被R46O取代的C1-C4烷基。在式(Ip)化合物的一些实施方案中,任何R38R39N选自NHR39,其中 R39如本文所定义,且具体而言是被R46O取代的C1-C4烷基,例如R38R39N是NH(CH2CH2OCH3)。
在式(Ip)化合物中,W如本文上面所定义。在一些实施方案中,W是直接键,且式(Ip)化合物则可以由式(Iq)代表
其中RA、RB、RC、n和R33如本文所定义。
在式(Ip)化合物的一些实施方案中,W是任选地被C1-C4烷基、例如C1-C3烷基、特别是甲基或R32O取代的C1-C3亚烷基,其中 R32是H或C1-C4烷基,H或C1-C3烷基,特别是H或甲基。
在式(Ip)化合物的一些实施方案中,W 是直接键、CH2、CH2CH2、CH(CH3)、CH(OH)或CH(OCH3)。
在式(Ip)化合物的一些实施方案中
RA、RB和RC独立地选自H、卤素、C1-C4烷基和R1O;
R1是C1-C4烷基;
W是直接键或任选地被C1-C4烷基或R32O取代的C1-C3亚烷基;
R32是H或C1-C4烷基;
n是0至3的整数;
每个R33独立地选自卤素、氰基、苯基、R34O(CH2)p、R36S(O)2和R38R39N;
R34是C1-C4烷基;
p是0;
R36 是C1-C4烷基;
R38是H;
R39是被R46O取代的C1-C4烷基;且
R46是C1-C4烷基。
在式(Io)化合物的一些实施方案中,环A是5-或6-元杂环基,其中所述杂环基可以是饱和或不饱和的,且芳族或非芳族的。在一些实施方案中,环A是5-或6-元杂芳基。在这些实施方案中的一些中,环A上的基团R33的数目n是0至2,特别是0或1,更具体地n是1。
在一些实施方案中,当环A是5-至6-元杂芳基时,任何R33独立地选自卤素、任选地被R34O取代的C1-C4烷基、C3-C6 环烷基、R37C(O)、R38R39N和R40R41N(CO)。
在还有其它实施方案中,当环A是5-至6-元杂芳基时,任何R33独立地选自卤素、C1-C4烷基、C3-C6环烷基、任选地被R34O取代的C1-C4烷基、R37C(O)、R38R39N和R40R41N(CO)。
在一些实施方案中,当环A是5-至6-元杂芳基时,任何R33独立地选自卤素、任选地被R34O取代的C1-C4烷基和R38R39N。
在一些具体实施方案中,当环A是5-至6-元杂芳基时,任何R33是R38R39N。
例如,在一些实施方案中,环A是5-至6-元杂芳基,n是1且R33是卤素、任选地被R34O取代的C1-C4烷基或R38R39N;具体而言是R38R39N。
在一些实施方案中,环A是5-或6-元杂芳基,n是1,R33是R38R39N,R38是H,R39是被R46O取代的C1-C4烷基;且R46是C1-C4烷基。
在一些实施方案中,当环A是5-或6-元杂芳基时,
RA、RB和RC独立地选自H、卤素、氰基、C1-C4烷基和R2C(O);例如选自H、卤素、C1-C4烷基和R2C(O);
W是直接键或C1-C3亚烷基;
n是0 或1;
每个R33独立地选自卤素、C1-C4烷基; C3-C6 环烷基、任选地被R34O取代的C1-C4烷基、R37C(O)、R38R39N和R40R41N(CO);
R34是H或C1-C4烷基;
R38是H或C1-C4烷基;且R39是任选地被选自R44R45N和R46O的基团取代的C3-C6 环烷基或C1-C4烷基;或R38和R39一起形成双基-(CH2)s-,其中 s是3至5的整数;
R40是H或C1-C4烷基;
R41是被R49O取代的C1-C4烷基;
R44、R45和R46独立地选自H和C1-C4烷基;且
R49是C1-C4烷基。
在一些具体实施方案中,当环A是5-至6-元杂芳基时,其更具体地是5-元杂芳基。在一些实施方案中,当环A是杂芳基时,其更具体地是噻唑基,特别是1,3-噻唑基。
在一些实施方案中,所述化合物可以由式(Ir)代表
其中RA、RB、RC、R33、W和n如本文所定义。
在式(Ir)化合物的一些实施方案中,W是直接键。在式(Ir)化合物的一些实施方案中,n是1。在式(Ir)化合物的一些实施方案中,环A是1,3-噻唑-4-基。
在式(Ir)化合物的一些实施方案中,环A是1,3-噻唑-4-基且n是1。在一些实施方案中,当环A是1,3-噻唑-4-基且n是1时,R33在2-位,即式(I)化合物可以由式(Id-1)代表
其中RA、RB、RC、R33和W如本文所定义。
在式(Is)化合物的一些具体实施方案中,R33选自卤素,诸如Br, R34O和R38R39N,例如选自卤素和R38R39N,特别是R38R39N。更具体地,R33是R38R39N,其中 R38是H或C1-C3烷基,例如H或甲基,特别是H,R39是任选地被R46O取代的C3-C6 环烷基或C1-C4烷基,且R46是C1-C4烷基。在一些实施方案中,所述化合物如式(It)所代表
其中RA、RB、RC、W、R38和R46如本文所定义。
在式(Io)化合物的一些实施方案中,当环A是5-元杂芳基,例如噻唑基时,
RA、RB和RC独立地选自H、卤素、氰基、C1-C6烷基、R2C(O)或任选地被一个或多个基团R16取代的苯基;例如选自H、卤素、氰基、C1-C6烷基和R2C(O);或选自H、卤素、C1-C4烷基和R2C(O);
W 是直接键;
n是或1;
R33是卤素、R34O或R38R39N,例如卤素或R38R39N;
R38是H;
R39是任选地被R46O取代的C1-C4烷基,或C3-C6环烷基,例如被R46O取代的C1-C4烷基;且
R46是C1-C4烷基。
在一些其它具体实施方案中,当环A是5-或6-元杂芳基时,其更具体地是6-元杂芳基,例如在环中含有一个或2个杂原子(例如,1个或2个N)的6-元杂芳基,例如环A是嘧啶基或吡啶基,特别是吡啶基。在其中环A是吡啶基的实施方案中,式(I)化合物可以由式(Iu)代表
其中RA、RB、RC、W、n和R33如本文所定义。
在这些实施方案中的一些中,W是直接键或C1-C3亚烷基,例如W是-CH2CH2-,特别是W 是直接键。
当环A是吡啶基时,其例如可以是 2-吡啶基或3-吡啶基。在一些实施方案中,环A是2-吡啶基,即式(Iu)化合物可以由式(Iv)代表
其中RA、RB、RC、W、n和R33如本文所定义。
在这些实施方案中的一些中,2-吡啶基仅被一个基团R33取代。例如,在一些实施方案中,2-吡啶基仅被一个连接在吡啶基环上的6-位的基团R33取代,且所述化合物则可以由式(Iw)代表
其中RA、RB、RC、W和R33如本文所定义。
在其中环A是3-吡啶基的实施方案中,式(Iu)化合物可以由式(Ix)代表
其中RA、RB、RC、W、n和R33如本文所定义。
在其中环A是3-吡啶基的实施方案中的一些中,3-吡啶基仅被一个基团R33取代。例如,在一些实施方案中,当3-吡啶基仅被一个基团R33取代时,式(Ix)化合物由式(Iy)代表
其中RA、RB、RC、W和R33如本文所定义。
在式(Iu)、(Iv)、(Iw)、(Ix)或(Iy)化合物中,R33如本文所定义。在一些实施方案中,R33选自卤素、C1-C4烷基、C3-C6 环烷基、R34O(CH2)p、R43O(CH2)qC(O)、R38R39N和R40R41N(CO)。
在式(Iu)化合物的一些实施方案中,RA、RB和RC独立地选自H、卤素、氰基和C1-C4烷基;W是直接键或C1-C3亚烷基;n是0或1;且
R33是卤素、任选地被R34O取代的C1-C4烷基;C3-C6 环烷基、R34O、R37C(O)、R38R39N或R40R41N(CO)。
在式(Io)化合物的一些实施方案中,当环A是4-至6-元杂环基时,环A更具体地是4-至6-元非芳族的,例如饱和杂环基。在一些实施方案中,所述杂环基是4-或5-元的,例如4-元的。在一些其它实施方案中,所述杂环基是5-或6-元的,例如5-元的。在还有其它实施方案中,所述杂环基是6-元的。
当环A是4-至6-元饱和杂环基时,所述环优选在环中含有1或2个杂原子,其中每个环杂原子优选选自N和O。
在一些实施方案中,环A是在环中含有一个N和任选地还有一个O的4-至6-元饱和杂环基。
在一些实施方案中,环A是仅含有一个选自N和O的环杂原子的4-至6-元饱和杂环基。
在一些实施方案中,环A是仅含有一个作为N的环杂原子的4-至6-元饱和杂环基。
当环A是在环中含有N的4-至6-元饱和杂环基时,所述N可以是连接基团W的连接点,或者当其不是连接基团W的连接点时,所述N可以被一个基团R33取代,或未被取代(即携带氢原子)。
在一些实施方案中,当A是在环中含有一个N的4-至6-元杂环基时,式(I)化合物可以由式(Iz)代表
其中RA、RB、RC和W如本文所定义,Z1是(CH2)u且Z2是(CH2)v,其中u和v均为0至4的整数,且u+v是2至4的整数;且R33’是H或R33如上文所定义。
在式(Iz)化合物的一些实施方案中,u和v均为1。在式(Iz)化合物的一些实施方案中,u+v是3或4。在这些实施方案中的一些中,u是0且v是3或4。在这些实施方案中的一些其它中,u是1且v是2或3。在这些实施方案中的还有其它中,u和v均为2。
在式(Iz)化合物中,R33’是H或如上文所定义的R33。在式(Iz)化合物的一些实施方案中,R33’是C1-C4烷基、吡啶基、R36S(O)2或R37C(O)。在一些具体实施方案中,R33’是R37C(O)或R36S(O)2;例如R33’是R37C(O)。
在式(Io)化合物的一些实施方案中,环A是通过作为N的环杂原子连接至连接基团W的4-至6-元杂环基。例如,在此类实施方案中,环A可以是吗啉-4-基、吡咯烷-1-基或氮杂环丁烷-1-基。
在一些实施方案中,环A是吗啉-4-基。在一些实施方案中,环A是吡咯烷-1-基。
在一些实施方案中,当环A是吗啉-4-基或吡咯烷-1-基时,n是0。在一些实施方案中,当环A是吗啉-4-基或吡咯烷-1-基时,W是C1-C3亚烷基。
在一些实施方案中,环A是氮杂环丁烷-1-基。在一些实施方案中,当环A是氮杂环丁烷-1-基时,n是2且两个R33连接至相同碳原子并且是F。
在一些实施方案中,当环A是氮杂环丁烷-1-基时,W是C1-C3亚烷基。
在一些具体实施方案中,当环A是4-至6-元杂环基时,
RA、RB和RC独立地选自H、卤素和C1-C4烷基,
W是直接键或C1-C3亚烷基;
n是0至2的整数;且
每个R33独立地选自卤素、C1-C4烷基、吡啶基和R37C(O)。
当环A是C4-C6环烷基或C5-C6环烯基时,其更优选是C4-C6环烷基。在式(Io)化合物的一些实施方案中,环A是C4至C6环烷基,即环丁基、环戊基或环己基。在这些实施方案中,所述化合物可以由式(Iaa)代表
其中RA、RB、RC、W、R33和n如本文所定义,且r是1至3的整数。
在式(Iaa)化合物中,基团W如本文上面所定义。在一些实施方案中,基团W 是直接键、C1-C3亚烷基或被C1-C4烷基取代的C1-C3亚烷基,例如W是直接键或C1-C3亚烷基,特别是W是直接键、CH2、CH2CH2或CH2CH2CH2。
在式(Iaa)化合物的一些实施方案中,W是直接键。在一些其它实施方案中,W是任选地被C1-C4烷基或R32O取代的C1-C3亚烷基;具体而言W是任选地被C1-C4烷基取代的C1-C3亚烷基,例如,W是任选地被C1-C4烷基取代的CH2、CH2CH2或CH2CH2CH2,特别是W是任选地被C1-C4烷基取代的CH2CH2。在一些实施方案中,W是CH2、CH2CH2或CH2CH2CH2。
在式(Iaa)化合物中,r是1至3的整数。在一些实施方案中,r是2或3,即环A是环戊基或环己基,例如环己基。在一些其它实施方案中,r是1或2,即环A是环丁基或环戊基。在一些具体实施方案中,r是2,即环A为环戊基。
在式(Iaa)化合物的一些具体实施方案中,W是CH2CH2,r是2,且n是如本文上面所定义的整数,优选地n是0、1或2,例如n是0或1。在式(Iaa)化合物的一些实施方案中,n是0。在式(Iaa)化合物的一些其它实施方案中,n是1,例如n是1且R33是R34O,特别是OH。
在式(Iaa)化合物的一些实施方案中,当n是2时,两个R33连接至环A的一个且相同的碳原子且形成双基-CH2-L-CH2-,其中L是CH2、NH或O,例如L是NH。
例如,在式(Iaa)化合物的一些实施方案中,r是1,n是2,且所述化合物中存在的两个R33连接至一个且相同的碳原子且形成双基CH2-L-CH2。在这些实施方案中的一些中,式(Iaa)化合物可以由式(Iab)代表
其中RA、RA、RB、RC、R33、W、r 和L如本文所定义。
在式(Iab)化合物的一些实施方案中,r是1。在式(Iab)化合物的一些实施方案中,L是NH。在式(Iab)化合物的一些具体实施方案中,r是1且L是NH。
在式(Iaa)化合物的一些实施方案中,
RA、RB和RC独立地选自H、卤素、氰基、C1-C4烷基、R1O、R2C(O)、R3S和R4S(O)2;或RA和RC中的一个与RB一起形成双基-(CH2)m-,其中m是3至5的整数,且RA和RC中的另一个选自H、卤素、氰基、C1-C4烷基、R1O、R2C(O)、R3S和R4S(O)2;
R1、R2、R3和R4独立地选自C1-C4烷基;
W是直接键或任选地被C1-C4烷基取代的C1-C3亚烷基;
n是0至2的整数,
R33是OH或,当n是2时,两个R33连接至一个且相同的碳原子且形成双基-CH2-NH-CH2-。
在本发明的化合物中,任何烷基,无论是否是官能团的一部分,都可以任选地被一个或多个F取代。
研究已经显示本发明化合物在小鼠中在体外和体内的功效,并且尽管已经针对S100A9抑制开发了化合物,但它们还可显示对其它S100蛋白的活性。本发明因此涉及作为S100蛋白抑制剂、主要作为S100A9抑制剂的如本文定义的化合物及其用于治疗或预防S100-蛋白相关疾病、特别是与S100A9蛋白的活性相关的疾病的用途。
具体而言,本发明涉及如本文定义的式(I)化合物;包含所述化合物的药物组合物;此类组合物在选自特别是癌症、还有自身免疫疾病、炎性疾病和神经退行性疾病的病况的治疗性治疗中的用途;治疗此类病况的方法;和所述化合物用于治疗选自特别是癌症、还有自身免疫疾病、炎性疾病和神经退行性疾病的病况;以及所述化合物用于制备用于治疗此类病况的药物组合物的用途。
本发明包括药物组合物,其包含至少一种根据式(I)的化合物或其各个异构体、异构体的外消旋或非外消旋混合物或其药学上可接受的盐连同至少一种药学上可接受的赋形剂例如载体和任选其它治疗和/或预防成分。
根据本发明的药物组合物可用于向哺乳动物(特别是人)外用(topical)(局部(local))或全身性给药,例如肠道给药,例如直肠或口服给药,或肠胃外给药,且包含与药学上可接受的赋形剂、例如药学上可接受的载体组合的治疗有效量的作为活性成分的根据本发明的化合物或其药学上可接受的盐。活性成分的治疗有效量如本文上面定义且例如取决于哺乳动物的物种、体重、年龄、个体状况、个体药代动力学数据、待治疗的疾病和给药模式。
对于肠道给药,例如口服给药,本发明的化合物可以配制成各种各样剂型。所述药物组合物和剂型可包含一种或多种本发明的化合物或一种或多种其药学上可接受的盐作为活性组分。所述药学上可接受的载体可以是固体或液体。固体形式制剂包括散剂、片剂、丸剂、锭剂、胶囊剂、扁囊剂、栓剂和分散颗粒剂。固体载体可为也可充当稀释剂、调味剂、增溶剂、润滑剂、悬浮剂、粘合剂、防腐剂、片剂崩解剂或包封材料的一种或多种物质。在散剂中,载体通常为细碎的固体,其为与细碎的活性组分的混合物。在片剂中,活性组分通常与具有必要结合能力的载体以合适比例混合并以期望形状和尺寸压缩。合适的载体包括但不限于碳酸镁、硬脂酸镁、滑石粉、糖、乳糖、果胶、糊精、淀粉、明胶、黄蓍胶、甲基纤维素、羧甲基纤维素钠、低熔点蜡、可可脂等。活性化合物的制剂可包含封装材料作为载体,其提供胶囊剂,其中有或没有载体的活性组分由与其组合的载体包围。
适合于口服给药的其它形式包括液体形式制剂,包括乳剂、糖浆剂、酏剂、水溶剂、水性悬浮剂,或意欲在临使用之前转化成液体形式制剂的固体形式制剂。乳剂可在溶液、例如丙二醇水溶液中制备,或者可含有乳化剂,例如卵磷脂、失水山梨糖醇单油酸酯或阿拉伯胶。水溶液可通过将活性组分溶解于水中并添加合适的着色剂、香料、稳定剂和增稠剂来制备。水性悬浮剂可通过将细碎的活性组分分散于具有粘性材料、诸如天然或合成树胶、树脂、甲基纤维素、羧甲基纤维素钠及其它公知的悬浮剂的水中来制备。固体形式制剂包括溶液剂、悬浮剂和乳剂,且除了活性组分之外,还可含有着色剂、香料、稳定剂、缓冲剂、人造和天然甜味剂、分散剂、增稠剂、增溶剂等。
用于直肠给药的示例性组合物包括栓剂,其可含有例如合适的无刺激性赋形剂,诸如可可脂、合成甘油酯或聚乙二醇,其在常温下为固体,但在直肠腔中液化和/或溶解以释放药物。
本发明的化合物也可肠胃外给药,例如通过吸入、注射或输注给药,例如通过静脉内、动脉内、骨内、肌肉内、脑内、脑室内、滑膜内、胸骨内、鞘内、病灶内、颅内、肿瘤内、皮内和皮下注射或输注给药。
因此,对于肠胃外给药,本发明的药物组合物可以为无菌可注射或可输注制剂的形式,例如作为无菌水性或油性悬浮剂。该悬浮剂可根据本领域已知的技术使用合适的分散剂或湿润剂(例如,吐温80)和悬浮剂来配制。无菌可注射或可输注制剂也可为在无毒的肠胃外可接受的稀释剂或溶剂中的无菌可注射或可输注溶液或悬浮剂。例如,药物组合物可为在1,3-丁二醇中的溶液。可用于本发明的组合物中的可接受的媒介物(vehicle)和溶剂的其它实例包括但不限于甘露醇、水、林格氏溶液和等渗氯化钠溶液。此外,通常将无菌的固定油用作溶剂或悬浮介质。为此目的,可以使用包括合成单甘油酯或二甘油酯的任何温和的固定油。脂肪酸,诸如油酸及其甘油酯衍生物可用于可注射剂的制备中,作为天然的药学上可接受的油,诸如橄榄油或蓖麻油,特别是以其聚氧乙烯化形式。这些油溶液或悬浮剂还可含有长链醇稀释剂或分散剂。
用于肠胃外使用的溶液剂还可含有合适的稳定剂和(如果需要的话)缓冲物质。合适的稳定剂包括单独或组合的抗氧化剂,诸如硫酸氢钠、亚硫酸钠或抗坏血酸;柠檬酸及其盐;和EDTA钠。胃肠外溶液剂还可含有防腐剂,诸如苯扎氯铵、对羟基苯甲酸甲酯或对羟基苯甲酸丙酯和氯丁醇。
对于吸入或经鼻给药,合适的药物制剂为颗粒剂、气溶胶、散剂、烟雾剂(mist)或滴剂,例如具有直径为约10μm或更小的平均尺寸。例如,用于吸入的组合物可使用苯甲醇或其它合适的防腐剂、增强生物利用度的吸收助剂、氟烃和/或本领域已知的其它增溶剂或分散剂制备成盐水中的溶液剂。
本发明的药物组合物也可局部给药至皮肤或粘膜。对于局部给药,所述药物组合物可例如为洗剂、凝胶剂、糊剂、酊剂、透皮贴片、用于经粘膜递送的(transmucosal)凝胶剂。所述组合物可配制成含有悬浮或溶解于载体中的活性组分的合适软膏剂。用于本发明的化合物的局部给药的载体包括但不限于矿物油、液体石油、白凡士林、丙二醇、聚氧乙烯聚氧丙烯化合物、乳化蜡和水。或者,所述药物组合物可配制为含有悬浮或溶解于载体中的活性化合物的合适洗剂或霜剂。合适的载体包括但不限于矿物油、失水山梨糖醇单硬脂酸酯、聚山梨酸酯60、鲸蜡基酯蜡、鲸蜡硬脂醇(cetaryl alcohol)、2-辛基十二烷醇、苯甲醇和水。本发明的药物组合物也可通过直肠栓剂制剂或以合适的灌肠制剂局部应用至低位肠道。
制备药物剂型的合适药物赋形剂例如载体和方法描述于药物配制领域中的标准参考教科书Remington's Pharmaceutical Sciences, Mack Publishing Company中。
药物组合物可包含约1%-约95%、优选约20%-约90%的式(I)化合物连同至少一种药学上可接受的赋形剂。通常,本发明的化合物将通过提供类似效用的试剂的任何可接受的给药模式以治疗有效量给药。合适的日剂量通常范围为1-1000mg,例如每日1-500mg,或每日1-50mg,这取决于许多因素,诸如待治疗的疾病的严重程度、患者的年龄和相对健康、使用的化合物的效力、给药的途径和形式及给药涉及的指标等。治疗此类疾病的本领域普通技术人员将能够在没有过度实验的情况下且依赖个人的知识和本申请的公开内容来针对给定疾病确定本发明化合物的治疗有效量。本发明的化合物可作为药物制剂、包括适合于肠道或肠胃外给药的那些制剂给药。优选的给药方式通常为使用方便的日剂量方案来口服,所述方案可根据痛苦程度来调节。
根据一个方面,本发明涉及治疗对S100蛋白家族成员、例如S100A9的抑制响应的疾病例如癌症、自身免疫性疾病、炎性疾病或神经退行性疾病的方法,所述方法包括将治疗有效量的式(I)化合物或其药学上可接受的盐给药至需要此类治疗的温血动物,例如人。
在一些实施方案中,根据本发明治疗的病症为癌症,诸如如本文上面定义的癌症。
在一些其它实施方案中,根据本发明治疗的病症为自身免疫性病症,例如如本文上面定义的自身免疫性病症。
在一些其它实施方案中,根据本发明治疗的病症为炎性病症,例如如本文上面定义的炎性病症。
在一些其它实施方案中,根据本发明治疗的病症为神经退行性病症,例如如本文上面定义的神经退行性病症。
根据本发明的化合物的制备完全在本领域普通技术人员的能力之内。例如,式(I)化合物可以以如反应方案1中一般举例说明的反应顺序来制备。因此,在用于制备如本文定义的式(I)化合物的一般方法中,首先在有机碱例如吡啶或三乙胺存在的情况下在合适的溶剂介质例如DMF、DMSO或N-甲基吡咯烷中使伯酰胺1与3,3,3-三氟-2-氧代丙酸烷基酯2反应,其中R’为烷基,例如C1-C3烷基,诸如甲基,随后添加试剂诸如亚硫酰氯或草酰氯,以提供通式3的“酰基亚胺中间体”。
然后使酰基亚胺3与氨基苯并咪唑4在合适的溶剂介质例如DMF、DMSO或N-甲基吡咯烷中反应,以提供式(I)化合物,其中RA、RA、RB、RC、R33、n、W和环A如本文所定义。
反应方案1。
以下实施例将使得本领域技术人员能够更清楚地理解并实践本发明。然而,这些实施例不应被视为限制本发明的范围,而其仅为说明性和代表性的。
实施例
使用的所有吡啶都是无水的(在氮气下经活化的4 Ä mol筛储存)。
使用的所有DMF都是无水的(在氮气下经活化的4 Ä mol筛储存)。
分子的所有命名都使用MarvinSketch 14.10.27.0进行。
HPLC方法如下:
“低pH方法A”是指使用由水中的0-100% MeCN的梯度中的0.2%甲酸组成的流动相的HPLC纯化。固定相由Waters Sunfire C18柱、10µm粒径、30 x 100mm组成。
“低pH方法B”是指使用由水中的0-100% MeCN的梯度中的0.1%甲酸组成的流动相的HPLC纯化。固定相由Waters Sunfire C18柱、5µm粒径、19 x 100mm组成。
“高pH”是指使用由水中的5-100% MeCN的梯度中的0.2%氨水组成的流动相的HPLC纯化。固定相由Waters X-bridge C18柱、10µm粒径、30 x 100mm组成。
“中性”是指使用由水中的10-100% MeCN的梯度组成的流动相(无改性剂)的HPLC纯化。固定相由Waters Sunfire C18柱、10µm粒径、30 x 100mm组成。
SFC色谱使用具有含有0.1%甲酸的超临界CO2和MeOH的流动相的Chiralpak AD-H柱实施。
微波反应使用CEM Discover、Biotage Initiator+或Activent 微波装置实施。
术语“酰基亚胺中间体”是指伯酰胺(RCONH2)和3,3,3-三氟-2-氧代丙酸烷基酯之间的反应产物。通式:
例如,苯甲酰胺和3,3,3-三氟-2-氧代丙酸甲酯之间的反应产物,即((2Z)-2-{[(Z)-苯甲酰基]亚氨基}-3,3,3-三氟丙酸甲酯)被认为是“酰基亚胺中间体”。
中间体1
(Z)-2-(苯甲酰基亚氨基)-3,3,3-三氟丙酸甲酯
在氮气下向苯甲酰胺(3.00 g, 24.8 mmol)于DMF (60 mL)中的搅拌溶液中逐滴添加吡啶(2.00 mL, 24.8 mmol),随后逐滴添加3,3,3-三氟-2-氧代丙酸甲酯(3.86 g, 24.8 mmol),并将溶液搅拌1小时。添加额外的3,3,3-三氟-2-氧代丙酸甲酯(1.53 g),并将反应物再搅拌2小时。将所得溶液在氮气下冷却至0℃,然后添加亚硫酰氯(1.80 mL, 24.8 mmol),并将溶液在0℃下再搅拌2小时。将反应混合物浓缩。将残余物过滤通过硅胶短垫,使用EtOAc作为洗脱剂,以提供标题化合物(5.47 g, 85%);在MS中观察到的MeOH加合物:m/z = 291.8 (MH)+。
在MS中观察到的MeOH加合物:
。
中间体2
N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙-2-烯酰胺
在氮气下向丙烯酰胺(1.00 g, 14.1 mmol)于无水DMF (25 mL)中的搅拌溶液中添加吡啶(1.13 mL, 14.1 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(1.45 mL, 14.1 mmol)。将反应混合物在室温下搅拌1小时,然后逐滴添加亚硫酰氯(1.02 mL, 14.1 mmol)。将溶液在室温下搅拌3小时,然后浓缩。将残余物过滤通过硅胶短垫,用EtOAc (100 mL)洗脱,并将滤液浓缩。将剩余的酰基亚胺中间体溶解于无水DMF (20 mL)中,并在氮气下添加2-氨基苯并咪唑(1.87g, 14.1 mmol)和三乙胺(1.87 mL, 14.1 mmol)。将溶液在室温下搅拌16小时,然后浓缩。将残余物溶解于EtOAc (25 mL)中,并用水(25 mL)和盐水(25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱(silica chromatography)纯化,使用DCM中的0-5% MeOH作为洗脱剂,以提供作为白色固体的标题化合物(1.05 g, 24%); m/z = 311.0 (MH)+。
中间体3
3-环戊基丙酰胺
在0℃在氮气下将3-环戊基丙酰氯(5.00 g, 31.1 mmol)于无水THF (40 mL)中的溶液经30分钟逐滴添加至氨水(9.35 mL, 93.4 mmol)和THF (10 mL)中。将溶液温热至室温并搅拌1小时。将反应混合物浓缩并在水(50 mL)和EtOAc (3 x 25 mL)之间分配。将合并的有机相干燥(MgSO4),过滤并浓缩,以提供作为白色固体的3-环戊基丙酰胺(3.95 g, 90%);1H NMR (500 MHz, 氯仿-d) δ 0.97 – 1.11 (m, 2H). 1.39 –1.50 (m, 2H), 1.50 – 1.62 (m, 4H), 1.63 – 1.76 (m, 3H), 2.11 – 2.20 (m, 2H), 5.47 (s, 1H), 5.76 (s, 1H)。
中间体4
(Z)-2-((3-环戊基丙酰基)亚氨基)-3,3,3-三氟丙酸甲酯
使用用于制备(Z)-2-(苯甲酰基亚氨基)-3,3,3-三氟丙酸甲酯的程序,除了使用3-环戊基丙酰胺代替苯甲酰胺。不必要再添加3,3,3-三氟-2-氧代丙酸甲酯。洗脱剂为庚烷(66%)中的50-100% EtOAc;MS中观察到的H2O加合物:m/z = 296.1 (M-1)-。
中间体5
(Z)-3,3,3-三氟-2-((3-苯基丙酰基)亚氨基)丙酸甲酯
使用用于制备(Z)-2-(苯甲酰基亚氨基)-3,3,3-三氟丙酸甲酯的程序,除了使用3-苯基丙酰胺代替苯甲酰胺。不必要再添加3,3,3-三氟-2-氧代丙酸甲酯;(99%);MS中观察到的H2O加合物:m/z = 306.1 (MH)+。
MS中观察到的H2O加合物:
。
中间体6
3-(2-氯苯基)丙酰胺
在氮气下向3-(2-氯苯基)丙酸(1.0 g, 5.42 mmol)于DCM (100 mL)中的溶液中逐滴添加亚硫酰氯(0.39 mL, 5.42 mmol)。将溶液在室温下搅拌3小时,然后浓缩。将残余物溶解于无水THF (10 mL)中,并冷却至0℃。在氮气下逐滴添加甲醇中的7M氨(1.55 mL)。将反应物在室温下搅拌4小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用饱和NaHCO3(水溶液)(2 x 25 mL)和盐水(25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩,以提供作为黄色固体的标题化合物(530 mg, 53%);m/z = 183.9, 185.9 (MH)+。
中间体7
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙-2-烯酰胺
在氮气下向丙烯酰胺(1.00 g, 14.1 mmol)于DMF (25 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(1.45 mL, 14.1 mmol),随后添加吡啶(1.13 mL, 14.1 mmol)。将反应混合物在室温下搅拌1小时,然后冷却至0℃。将亚硫酰氯(1.02 mL, 14.1 mmol)逐滴添加至溶液。将反应混合物温热至室温,再搅拌24小时,然后浓缩。将酰基亚胺中间体溶解于DMF (25 mL)中,并在氮气下添加三乙胺(1.87 mL, 14.1 mmol)和5,6-二甲基-1H-1,3-苯并二唑-2-胺(2.27 g, 14.1 mol)。将反应混合物在室温下搅拌5小时,然后浓缩。将残余物溶解于DCM (100 mL)中,并用水(2 x 100 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-5% MeOH作为洗脱剂,以提供作为白色固体的标题化合物(1.30 g, 27%); m/z = 339.0 (MH)+。
中间体8
6-(2,2,2-三氟乙氧基)吡啶-3-甲酰胺
在氮气下向6-(2,2,2-三氟乙氧基)吡啶-3-甲酸(1.00 g, 4.52 mmol)于DCM (50 mL)和DMF (0.1 mL)中的搅拌溶液中逐滴添加亚硫酰氯(328 µL, 4.52 mmol)。将反应混合物在室温下搅拌3小时,然后浓缩。将残余物溶解于无水THF (25 mL)中,冷却至5℃,并添加过量氨水。将反应混合物温热至室温,并再搅拌3小时。将混合物用EtOAc (3 x 50 mL)萃取,并将合并的有机相用饱和NaHCO3(水溶液)(50 mL)和盐水(50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩,以提供作为白色固体的标题化合物(798 mg, 80%);m/z = 221.0 (MH)+。
中间体9
5,6-二氯-1H-1,3-苯并二唑-2-胺
标题化合物根据国际申请号PCT/US2007/020982 (公开号WO2008042282) (定量收率)制备;m/z = 201.8, 203.9 (MH)+。
中间体10
6-氨基甲酰基-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯
在氮气下向2-[(叔丁氧基)羰基]-2-氮杂螺[3.3]庚烷-6-甲酸 (700 mg, 2.90 mmol)于DCM (100 mL)中的搅拌溶液中添加三乙胺(386 µL, 2.90 mmol)。将反应物冷却至0℃,并逐滴添加氯(乙氧基)甲酮(304 µL, 3.19 mmol)。将反应物温热至室温并搅拌2小时。然后将反应物冷却至0℃,并添加氨水(290 µL, 2.90 mmol)。将反应物温热至室温并搅拌3小时。将反应物用DCM (100 mL)稀释,并用水(3 x 50 mL)和10% NaHCO3(水溶液)(50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩,以提供作为白色固体的标题化合物(601 mg, 86%);m/z = 184.9 (MH-tBu)+。
中间体11
6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯
在氮气下向6-氨基甲酰基-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯(515 mg, 2.14 mmol)于DMF (20 mL)中的搅拌溶液中添加吡啶(173 µL, 2.14 mmol)和3,3,3-三氟-2-氧代丙酸甲酯(669 mg, 4.29 mmol)。将反应混合物在室温下搅拌16小时,然后冷却至0℃。逐滴添加亚硫酰氯(156 mL, 2.14 mmol),并将溶液在0℃下搅拌1小时,然后浓缩。将残余物过滤通过硅胶短垫,用DCM/DMF (10:1)洗脱,并将滤液浓缩。在氮气下将剩余的酰基亚胺中间体溶解于DMF(20mL)中。将5,6-二氯-1H-1,3-苯并二唑-2-胺(325 mg, 1.61 mmol)和三乙胺(285 µL, 2.14 mmol)添加至溶液,并将反应物在室温下搅拌3小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(3 x 50 mL)和盐水(2 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,用DCM中的0-7% MeOH洗脱,以提供作为灰白色固体的标题化合物(411 mg, 35%);m/z 491.8, 493.8 (MH-tBu)+。
中间体12
3-(2,6-二氯苯基)丙酰胺
在氩气下向3-(2,6-二氯苯基)丙酸(1.00 g, 4.59 mmol)于DCM (20 mL)中的搅拌溶液中逐滴添加亚硫酰氯(1.67 mL, 23.0 mmol)。将反应混合物回流3小时,然后浓缩。将残余物溶解于DCM (10 mL)中并冷却至0℃。将氨气鼓泡通过反应物10分钟。将反应物浓缩并用水(10 mL)稀释。将所得白色固体通过过滤收集并干燥,以提供作为白色固体的标题化合物(700 mg, 70%);m/z= 218.3, 220.3 (MH)+。
中间体13
6-(环己基氨基)吡啶-3-甲酰胺
向微波管中的6-氯吡啶-3-甲酰胺(1.00 g, 6.40 mmol)于NMP (5 mL)中的溶液中添加N,N-二异丙基乙胺(2.19 mL, 12.8 mmol)和环己胺(2.93 mL, 25.6 mmol)。将反应混合物在微波中在200℃下加热1小时并浓缩。将残余物通过硅胶色谱纯化,使用DCM中的2% MeOH作为洗脱剂,以提供作为灰白色固体的标题化合物(350 mg, 25%); m/z = 220.2 (MH)+。
中间体14
3-(吡啶-3-基)丙酰胺
在0℃下向3-(吡啶-3-基)丙酸(500 mg, 3.27 mmol)于DCM (10 mL)中的溶液中逐滴添加草酰氯(0.84 ml, 9.81 mmol)。将反应物温热至室温,搅拌3小时,然后浓缩。将残余物溶解于DCM (15 mL)中,冷却至0℃,并将氨气鼓泡通过溶液15分钟。然后将反应物浓缩,并将所得白色固体在水(8 mL)中研磨。将固体过滤并在真空下干燥,以提供作为白色固体的标题化合物(200 mg, 40%);m/z = 151.2 (MH)+。
中间体15
4-(吡咯烷-1-基)丁酰胺
在密封管中将氨水(303 µL, 16.2 mmol)添加至根据国际申请号PCT/US2009/050797 (公开号WO2010009290)制备的4-(吡咯烷-1-基)丁酸乙酯(1.00 g, 5.40 mmol)中。将反应物在室温下搅拌16小时,然后浓缩。将残余物通过硅胶色谱纯化,使用DCM中的5% MeOH作为洗脱剂,以提供作为无色油的标题化合物(750 mg, 89%);1H NMR (400 MHz, DMSO-d6) δ 1.57 - 1.72 (m, 8H), 2.01 – 2.09 (m, 2H), 2.28 – 2.36 (m, 2H), 2.50 - 2.58 (m, 2H), 6.68 (s, 1H), 7.20 (s, 1H)。
中间体16
4-(吗啉-4-基)丁酸乙酯
向4-溴丁酸乙酯(2.93 mL, 20.0 mmol)于甲苯 (30 mL)中的搅拌溶液中添加吗啉(7.14 mL, 80.0 mmol)。将溶液回流10小时。将所得白色固体通过过滤去除,并用二乙醚(100 mL)洗涤。将滤液浓缩,以提供作为黄色油的标题化合物(3.20 g, 78%);1H NMR (400 MHz, DMSO-d6) δ 1.17 (t, 3H), 1.62 – 1.70 (m, 2H), 2.21 – 2.32 (m, 8H), 3.51 – 3.57 (m, 4H), 4.03 (q, 2H)。
中间体17
4-(吗啉-4-基)丁酰胺
将氨水(4mL)添加至4-(吗啉-4-基)丁酸乙酯(700 mg, 3.30 mol),并将反应混合物在室温下搅拌3小时。将混合物浓缩并与甲苯共沸,以提供作为黄色半固体的标题化合物(410 mg, 65%);1H NMR (400 MHz, DMSO-d6) δ 1.62 – 1.70 (m, 2H), 2.21 – 2.32 (m, 8H), 3.51 – 3.57 (m, 4H), 7.22 (s, 2H)。
中间体18
2-[(5,6-二氯-1H-1,3-苯并二唑-2-基)氨基]-3,3,3-三氟-2-[3-(吗啉-4-基)丙酰胺基]丙酸乙酯
向根据文献(You等人,2008)制备的3-(吗啉-4-基)丙酰胺(500 mg, 3.16 mmol)于DMF (5 mL)中的溶液中添加3,3,3-三氟-2-氧代丙酸乙酯(419 µL, 3.16 mmol)和吡啶(269 µL, 3.16 mmol)。将反应混合物在室温在氩气下搅拌2小时,然后将亚硫酰氯(230 µL, 3.16 mmol)逐滴添加至溶液。再继续搅拌24小时,然后将反应物浓缩。将所得酰基亚胺溶解于DMF (5 ml)中,然后添加至5,6-二氯-1H-1,3-苯并二唑-2-胺(615 mg, 3.05 mmol)于DMF (5 mL)中的溶液。添加三乙胺(422 µL, 3.05 mmol),并将反应混合物在室温下搅拌16小时。将反应物用盐水(25 mL)稀释,并用EtOAc (2 x 35 mL)萃取。将合并的有机相用水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的2% MeOH作为洗脱剂,以提供作为棕色油的标题化合物(427 mg, 1%);m/z = 512.5 (MH)+。
中间体19
2-(2-氨基甲酰基乙基)哌啶-1-甲酸叔丁酯
在0℃在氩气下向3-{1-[(叔丁氧基)羰基]哌啶-2-基}丙酸(200 mg, 0.78 mmol)于氯仿 (20 mL)中的搅拌溶液中添加三乙胺(108 µL, 0.78 mmol)和氯甲酸异丁酯(93 µL, 0.78 mmol)。将反应混合物在室温下搅拌2小时,然后将氨气鼓泡通过反应混合物15分钟。将反应混合物用DCM稀释并用5% NaHCO3(水溶液)和1M HCl(水溶液)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的2% MeOH作为洗脱剂,以提供作为无色油的标题化合物(110 mg, 55%);1H NMR (400 MHz, DMSO-d6) δ 1.16 – 1.26 (m, 1H), 1.37 (s, 9H), 1.43 – 1.64 (m, 6H), 1.80 -2.04 (m, 3H), 2.73 (d, 1H), 3.80 (d, 1H), 4.07 (s, 1H), 6.70 (s, 1H), 7.24 (s, 1H)。
中间体20
4-(氨基甲酰基甲基)哌啶-1-甲酸叔丁酯
使用用于制备2-(2-氨基甲酰基乙基)哌啶-1-甲酸叔丁酯的程序,除了使用2-{1-[(叔丁氧基)羰基]哌啶-4-基}乙酸代替3-{1-[(叔丁氧基)羰基]哌啶-2-基}丙酸(72%); 1H NMR (400 MHz, DMSO-d6) δ 0.90 - 1.04 (m, 2H), 1.37 (s, 9H), 1.52 - 1.64 (m, 2H), 1.72 - 1.84 (m, 1H), 1.90 - 2.00 (m, 2H), 2.66 (s, 2H), 3.87 (d, 2H), 6.75 (s, 1H), 7.24 (s, 1H)。
中间体21
1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用2,3-二氢-1H-茚-5,6-二胺代替4,5-二氯苯-1,2-二胺(定量收率); m/z = 173.9 (MH)+。
中间体22
4,5-二甲基-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3,4-二甲基苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(83%); m/z = 162.0 (MH)+。
中间体23
4-甲基-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3-甲基苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(定量收率); m/z = 148.0 (MH)+。
中间体24
4,5-二氟-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3,4-二氟苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(60%); m/z = 170.0 (MH)+。
中间体25
3,4-二氯苯-1,2-二胺
向经由文献方法(Carta等人, 2007)可得的2,3-二氯-6-硝基苯胺(1.08 g, 5.21 mmol)于MeOH (4.40 mL)中的溶液中添加氯化锡(II)二水合物(4.71 g, 20.87 mmol)于浓HCl (6.60 mL)中的溶液。将反应物在70℃下加热4小时。将反应混合物浓缩至一半体积,用5M NaOH(水溶液)碱化,用EtOAc (20 mL)稀释并搅拌30分钟。通过过滤去除所得沉淀。将滤液用盐水(20 mL)洗涤,干燥(Na2SO4),过滤并浓缩,以得到作为浅棕色固体的期望产物(0.87 g, 94%);m/z = 176.9, 178.9 (MH)+。
中间体26
4,5-二氯-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3,4-二氯苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(48%); m/z = 201.9, 203.9 (MH)+。
中间体27
4-氯-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3-氯苯-1,2-二胺(根据国际申请号PCT/US2008/003935 (公开号WO2008118454)中描述的方法可得)代替4,5-二氯苯-1,2-二胺(57%); m/z = 167.9, 169.9 (MH)+。
中间体28
4-氯-1H-咪唑并[4,5-c]吡啶-2-胺
在氮气下向2-氯吡啶-3,4-二胺(0.92 g, 6.42 mmol)于DCE (36 mL)中的溶液中逐滴添加N-硫代羰基(carbothioyl)氨基甲酸乙酯(0.83 mL, 7.1 mmol)。10分钟之后,添加EDC.HCl (1.35 g, 7.1 mmol)和N,N-二异丙基乙胺(5.60 mL, 32.1 mmol)。回流2小时之后,将反应物浓缩。将残余物溶解于1M NaOH(水溶液)(50mL)中,用DCM (2 x 40 mL)洗涤并用1M HCl中和。将所得沉淀通过过滤收集并在DCM中研磨。将氨基甲酸酯溶解(take up)于EtOH(10个体积)中并添加2M NaOH(水溶液)(3当量)。将反应物在回流下加热8小时。将反应物浓缩,溶解于水中,用3M HCl(水溶液)中和,用丙-2-醇/氯仿(3 x 50 mL)萃取,干燥(Na2SO4),过滤并浓缩,以得到作为白色固体的标题化合物(0.43 g, 40%)。m/z = 168.9, 170.9 (MH)+。
中间体29
4,6-二氟-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3,5-二氟苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(48%); m/z = 169.9 (MH)+。
中间体30
氧杂环己烷-4-甲酰胺
在0℃在氩气下氧杂环己烷-4-甲酸(800 mg, 6.15 mmol)于氯仿(5 mL)中的搅拌溶液中逐滴添加三乙胺(1.28 mL, 9.22 mmol)和氯甲酸异丁酯(884 µl, 7.37 mmol)。将反应混合物在0℃下搅拌2小时,然后使氨气通过溶液15分钟。将反应混合物浓缩。将残余物溶解于丙-2-醇/氯仿(1:4, 25 mL)中,并用水、5% NaHCO3(水溶液)、然后1M HCl(水溶液)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为白色固体的标题化合物(1.20 g, 91%);1H NMR (400 MHz, DMSO-d6) δ 1.44 – 1.63 (m, 4H), 2.23 – 2.34 (m, 1H), 3.27 (td, 2H), 3.82 (ddd, 2H), 6.75 (s, 1H), 7.22 (s, 1H)。
中间体31
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙-2-烯酰胺
在室温在氮气下向丙-2-烯酰胺(170 mg, 2.38 mmol)于DMF (8 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(400 µL, 3.96 mmol)和吡啶(190 µL, 2.38 mmol)。将反应混合物搅拌2小时。在0℃下逐滴添加亚硫酰氯(180 µL, 2.38 mmol),并将反应混合物搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (6 mL)中。将酰基中间体的溶液添加至5,6-二氯-1H-1,3-苯并二唑-2-胺(400 mg, 1.98 mmol)于DMF (8 mL)中的溶液,随后添加三乙胺(320 µL, 2.38 mmol)。将反应混合物搅拌17小时,然后浓缩。将残余物溶解于EtOAc (25 mL)中,并用水(30 mL)洗涤。将水相用EtOAc (20 mL)萃取,并将合并的有机萃取物用10%柠檬酸(水溶液)(2 x 15 mL)、盐水(25mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物在DCM中研磨,并通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂,以提供作为浅棕色固体的标题化合物(41 mg, 5%);m/z = 378.8, 380.8 (MH)+。
中间体32
6-[(2-甲氧基乙基)(甲基)氨基]吡啶-2-甲酰胺
向6-氟吡啶-2-甲酰胺(500 mg, 3.57 mmol)于DMF (10 mL)中的溶液中添加(2-甲氧基乙基)(甲基)胺(636 mg, 7.14 mmol)和K2CO3 (1.23 g, 8.92 mmol)。将反应混合物在100℃下搅拌12小时。添加额外的(2-甲氧基乙基)(甲基)胺(636 mg, 7.14 mmol),并将反应物在100℃下再加热7小时。然后将反应混合物浓缩。将残余物溶解于EtOAc (100 mL)中,并用水(3 x 50 mL)和盐水(3 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩,以提供作为白色固体的标题化合物(580 mg, 78%);m/z 210.0 (MH)+。
中间体33
3-氨基甲酰基-3-氟氮杂环丁烷-1-甲酸叔丁酯
在0℃在氮气下向1-[(叔丁氧基)羰基]-3-氟氮杂环丁烷-3-甲酸(经由文献方法: Faming Zhuanli Shenqing, 102731362, 2012年10月17日可得)(1.00 g, 4.56 mmol)于DCM (70 mL) 中的搅拌溶液中添加DMF (200 µl, 2.60 mmol)和草酰氯(490 µl, 5.70 mmol)。将所得溶液在0℃下搅拌2小时,然后将氨水(2.28 ml, 22.81 mmol)逐滴添加至溶液。在0℃下再搅拌30分钟之后,将反应物温热至室温。搅拌1小时之后,将反应混合物用水(30mL)稀释。将有机相用水(3 x 30 mL)洗涤,干燥(Na2SO4),过滤并浓缩,以提供作为黄色固体的标题化合物(990 mg, 99%);m/z = 162.9 (MH-tBu)+。
中间体34
3-环丁基丙酰胺
在0℃在氮气下向3-环丁基丙酸(984 mg, 7.67 mmol)于DCM (50 mL)中的搅拌溶液中逐滴添加三乙胺(1.00 mL, 7.67 mmol)和氯甲酸乙酯(731 µL, 7.67 mmol)。将反应混合物在室温下搅拌2小时。在0℃下添加氨水(769 µL, 7.67 mmol),并将反应物经1小时温热至室温。将反应混合物用DCM(50mL)稀释,用水(3 x 100 mL)和10% NaHCO3(水溶液) (100 mL)萃取。将合并的水相用10%柠檬酸(水溶液)(10mL)酸化,然后用DCM (3 x 75 mL)萃取。将合并的有机萃取物干燥(MgSO4),过滤并浓缩,以提供作为白色固体的标题化合物(548 mg, 56%);m/z = 128.0 (MH)+。
中间体35
3-(3,4,5-三甲氧基苯基)丙酰胺
在0℃在氩气下向3-(3,4,5-三甲氧基苯基)丙酸(1.00 g, 4.16 mmol)于DCM (20 mL)中的搅拌溶液中逐滴添加DMF (80 µl, 1.04 mmol)和草酰氯(719 µl, 8.32 mmol)。将反应物经3小时温热至室温,然后浓缩。将残余物溶解于DCM中,冷却至0℃,并将所得溶液用氨气饱和。将反应混合物搅拌2小时,然后浓缩。将残余物用水稀释,并将所得沉淀通过过滤收集,提供作为白色固体的标题化合物(690 mg, 66%);1H NMR (400 MHz, DMSO-d6) δ 2.35 (t, 2H), 2.74 (t, 2H), 3.62 (s, 3H), 3.75 (s, 6H), 6.53 (s, 2H), 6.78 (s, 1H), 7.30 (s, 1H)。
中间体36
3-甲磺酰基苯甲酰胺
在0℃在氩气下向3-甲磺酰基苯甲酸(1.50 g, 7.49 mmol)于DCM (7 mL)中的搅拌溶液中逐滴添加DMF (144 µl, 1.87 mmol)和草酰氯(1.29 ml, 14.98 mmol)。将反应物经3小时温热至室温,然后浓缩。将残余物溶解于THF中,冷却至0℃,并将所得溶液用氨气饱和。将反应混合物搅拌2小时,然后浓缩。将残余物用MeOH/DCM稀释,并通过过滤去除所得沉淀。将滤液浓缩,以提供作为白色固体的标题化合物(750 mg, 42%);m/z = 200.3 (MH)+。
中间体37
4-溴-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3-溴苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(62%); m/z = 211.8, 213.8 (MH)+。
中间体38
2-氨基-5-氯-1H-1,3-苯并二唑-4-甲腈
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用2,3-二氨基-6-氯苯甲腈 (经由文献方法: PCT国际申请2003051277, 2003年6月26日可得)代替4,5-二氯苯-1,2-二胺(60%); m/z = 192.9, 194.9 (MH)+。
中间体39
4-氟-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3-氟苯-1,2-二胺(经由文献方法: PCT国际申请2002008224, 2002年1月31日可得)代替4,5-二氯苯-1,2-二胺(43%); m/z = 151.9 (MH)+。
中间体40
4,6-二甲基-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3,5-二甲基苯-1,2-二胺(经由文献方法: PCT国际申请2003008413, 2003年1月30日可得)代替4,5-二氯苯-1,2-二胺(定量收率); m/z = 162.0 (MH)+。
中间体41
4-氯-5-氟-3H-1,3-苯并二唑-2-胺
在0℃下将BrCN (198 mg, 1.87 mmol)于MeCN (2 mL)中的溶液逐滴添加至3-氯-4-氟苯-1,2-二胺(经由文献方法: Oriental Journal of Chemistry, 2007, 23, 571-576可得)(300 mg, 1.87 mmol)于MeCN/H2O (11:1, 8 mL)中的搅拌溶液。搅拌18小时之后,将反应混合物用饱和NaHCO3(水溶液)(30mL)稀释。通过过滤去除沉淀,并将滤液浓缩。将残余物用水(30mL)稀释,超声处理,过滤并用水洗涤,以得到作为橙棕色固体的标题化合物(110 mg, 32%);m/z = 185.9, 187.9 (MH)+。
中间体42
5-氯-4-甲基-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用4-氯-3-甲基苯-1,2-二胺(经由文献方法: 美国专利申请公开20060111416, 2006年5月25日可得)代替4,5-二氯苯-1,2-二胺(99%); m/z = 181.9, 183.9 (MH)+。
中间体43
4-氯-3-氟苯-1,2-二胺
将微波管装入1,3-二氯-2-氟-4-硝基苯 (2.00 g, 9.52 mmol)、苯基甲胺(4.17 mL, 38.1 mmol)和THF (20 mL)。将反应混合物在微波中在80℃下加热3小时,然后浓缩。将残余物溶解于EtOAc(40mL)中,并用0.5M HCl(水溶液)(20mL)洗涤,然后用盐水(20mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。
将残余物溶解于MeOH(15mL)中,并将烧瓶抽空并用氮气(x 3)吹扫。将10% Pd/C (60 mg, 0.06 mmol)添加至搅拌溶液。将烧瓶抽空并用氮气(x 3)吹扫,然后抽空并用氢气(x 3)吹扫。搅拌18小时之后,将反应物过滤通过硅藻土,用EtOH冲洗并浓缩,以得到作为棕色固体的粗产物。将粗产物通过硅胶色谱纯化,使用庚烷中的20-60% EtOAc作为洗脱剂,以得到作为红棕色固体的标题化合物(655 mg, 72%);m/z = 160.9, 162.9 (MH)+。
中间体44
5-氯-4-氟-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用4-氯-3-氟苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(99%); m/z = 185.9, 187.9 (MH)+。
中间体45
2-氟-3-甲氧基-6-硝基苯胺
将微波管装入2,3-二氟-1-甲氧基-4-硝基苯(1.00 g, 5.29 mmol)和MeOH中的7M氨(15 mL)。将反应混合物在微波中在80℃下加热40分钟,然后浓缩。将残余物用EtOAc (30 mL)稀释,并用水(30 mL)和盐水(30 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩,以得到作为黄色固体的标题产物(0.97 g, 99%);m/z = 186.9 (MH)+。
中间体46
3-氟-4-甲氧基苯-1,2-二胺
将装有2-氟-3-甲氧基-6-硝基苯胺(0.97 g, 5.26 mmol)和MeOH (25 mL)的烧瓶抽空并用氮气(x 3)吹扫。然后将10% Pd/C (112 mg, 0.11 mmol)添加至搅拌溶液。将烧瓶抽空并用氮气(x 3)吹扫,然后抽空并用氢气(x 3)吹扫。搅拌18小时之后,将反应物过滤通过硅藻土,用MeOH冲洗并浓缩,以得到作为棕色固体的标题物。(822 mg, 97%)。m/z = 157.0 (MH)+。
中间体47
4-氟-5-甲氧基-1H-1,3-苯并二唑-2-胺
在0℃下将BrCN (640 mg, 6.04 mmol)于MeCN (2 mL)中的溶液逐滴添加至3-氟-4-甲氧基苯-1,2-二胺(820 mg, 5.25 mmol)的搅拌溶液。搅拌18小时之后,将反应混合物浓缩。将残余物用饱和NaHCO3(水溶液)(20 mL)稀释并超声处理。将沉淀通过过滤收集并用水洗涤,以提供作为红色固体的标题化合物(970 mg, 定量收率);m/z = 182.0 (MH)+。
中间体48
7-(三氟甲基)-1H-1,3-苯并二唑-2-胺
在0℃下将BrCN于MeCN中的5M溶液 (0.87 mL, 4.35 mmol)逐滴添加至3-(三氟甲基)苯-1,2-二胺(700 mg, 3.97 mmol)于MeCN/水(5:1, 14 mL)中的搅拌溶液。搅拌14小时之后,将反应混合物用饱和NaHCO3(水溶液)(10mL)稀释。将沉淀通过过滤收集并用水(20 mL)和二乙醚(40 mL)洗涤,以提供作为黄色固体的标题化合物(540 mg, 66%);m/z = 202.1 (MH)+。
中间体49
4-(甲基硫烷基)-1H-1,3-苯并二唑-2-胺
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3-(甲基硫烷基)苯-1,2-二胺(经由文献方法: J. Med. Chem. 2005, 48, 8253-8260可得)代替4,5-二氯苯-1,2-二胺(99%); m/z = 178.0 (MH)+。
中间体50
(4S)-4-苄基-3-(3-环戊基丙酰基)-1,3-噁唑烷-2-酮
在-78℃在氮气下向(4S)-4-苄基-1,3-噁唑烷-2-酮 (1.00 g, 5.64 mmol)于无水THF (50 mL)中的搅拌溶液中逐滴添加己烷中的2.5M正丁基锂(2.26 mL, 5.65 mmol)。将反应物在-78℃下搅拌1小时,然后逐滴添加3-环戊基丙酰氯(864 µl, 5.64 mmol)。将反应物温热至室温,并再搅拌1小时。在0℃下将反应物用饱和NH4Cl(水溶液) (25 mL)淬灭,用水(30 mL)稀释并用EtOAc (3 x 30 mL)萃取。将合并的有机萃取物用盐水洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用庚烷中的33% EtOAc作为洗脱剂,以提供作为白色固体的标题化合物(1.55 g, 90%);m/z = 302.0 (MH)+。
中间体51
(4S)-4-苄基-3-[(2S)-2-(环戊基甲基)丙酰基]-1,3-噁唑烷-2-酮
在-78℃在氮气下向(4S)-4-苄基-3-(3-环戊基丙酰基)-1,3-噁唑烷-2-酮(1.50 g, 4.98 mmol)于无水THF (60 mL)中的搅拌溶液中逐滴添加THF中的1M NaHMDS (7.47 mL, 7.47 mmol)。将反应物在-78℃下搅拌30分钟,然后逐滴添加碘甲烷 (930 µl, 14.93 mmol)。在-78℃下3小时之后,将反应物温热至室温。再搅拌2小时之后,在0℃下将反应物用饱和NH4Cl(水溶液)淬灭。温热至室温后,将反应混合物用EtOAc (3 x 50 mL)萃取。将合并的有机萃取物用盐水(50 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用庚烷中的0-40% EtOAc作为洗脱剂,以提供作为白色固体的标题化合物(1.03 g, 65%);m/z = 316.2 (MH)+。
中间体52
(2S)-3-环戊基-2-甲基丙酸
向(4S)-4-苄基-3-[(2S)-2-(环戊基甲基)丙酰基]-1,3-噁唑烷-2-酮(1.00 g, 3.17 mmol)于THF (20 mL)中的搅拌溶液中添加氢氧化锂一水合物(532 mg, 12.68 mmol),随后添加过氧化氢(570 µl, 19.02 mmol)。将反应混合物在室温下搅拌5小时,然后浓缩至一半体积。添加EtOAc (25 mL)和1M HCl(水溶液)(25 mL),并将有机相用盐水洗涤,然后浓缩。将残余物溶解于EtOAc (25 ml)中并用1M NaOH(水溶液)萃取。将水相用1M HCl(水溶液)酸化至pH 3,并用EtOAc (3 x 50 mL)萃取。将合并的有机萃取物干燥(MgSO4),过滤并浓缩,以提供作为无色油的标题化合物(395 mg);m/z = 178.0 (M+Na)+。
中间体53
(2S)-3-环戊基-2-甲基丙酰胺
在0℃在氮气下向(2S)-3-环戊基-2-甲基丙酸(395 mg, 2.53 mmol)于DCM (40 mL)中的搅拌溶液中添加DMF (100 µl, 1.30 mmol)和草酰氯(271 µl, 3.16 mmol)。将反应混合物在0℃下搅拌3小时,然后添加氨水(1.27 ml, 12.64 mmol)。将反应物温热至室温,并再搅拌2小时。将反应混合物用DCM(50mL)稀释,并用水(2 x 50 mL)和10%柠檬酸(水溶液)(2 x 50 mL)洗涤。将合并的水相用DCM (50 mL)反萃取,然后将有机萃取物合并,并干燥(MgSO4),过滤并浓缩,以提供作为灰白色固体的标题化合物(294 mg, 75%);m/z = 156.0 (MH)+。
中间体54
(2S)-1-甲磺酰基吡咯烷-2-甲酰胺
在0℃在氮气下向(2S)-1-甲磺酰基吡咯烷-2-甲酸(1.00 g, 5.18 mmol)于DCM (75 mL)中的溶液中添加氯甲酸乙酯(510 µL, 5.18 mmol)和三乙胺(690 µL, 5.18 mmol)。将反应混合物在室温下搅拌3小时。在0℃下添加氨水(1.55 mL, 15.53 mmol)。将反应混合物搅拌1小时,然后用DCM (25 mL)稀释,并用水(3 x 50 mL)萃取。将合并的有机萃取物浓缩。将残余物用DCM (50 mL)稀释,超声处理并过滤。将滤液干燥(MgSO4),过滤并浓缩,以提供作为米色固体的标题化合物(415 mg, 38%);m/z = 192.9 (MH)+。
中间体55
非对映异构体混合物。(S)-{[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]氨基甲酰基}(苯基)乙酸甲酯
在氮气下向(S)-氨基甲酰基(苯基)乙酸甲酯(经由文献方法: Org. Biomol. Chem.2011, 9, 3011-3019可得) (0.63 g, 3.26 mmol)于DMF (20 mL)中的溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(1.22 g, 7.82 mmol),随后添加吡啶(315 µl, 3.91 mmol)。将溶液在室温下搅拌2小时。在0℃下逐滴添加亚硫酰氯(284 µl, 3.91 mmol),并将反应混合物在0℃下再搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。将酰基中间体的溶液添加至5,6-二甲基-1H-1,3-苯并二唑-2-胺(525 mg, 3.26 mmol)于DMF (20 mL)中的搅拌溶液,随后添加三乙胺(546 µl, 3.91 mmol)。将反应混合物搅拌16小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(4 x 50 mL)、盐水(3 x 50 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过反相C18色谱纯化,使用酸性洗脱剂,以提供作为灰白色固体且作为非对映异构体的混合物的标题化合物(536 mg, 36%); m/z = 461.0 (MH)+。
中间体56
3-环戊基-N-[4-氧代-3-(三氟甲基)-9-{2-[三(丙-2-基)甲硅烷基]乙炔基}-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(239986)
将微波管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、碘化铜(I) (2.5 mg, 0.01 mmol)、Pd(PPh3)2Cl2 (9.2 mg, 0.01 mmol)、PPh3 (11 mg, 0.04 mmol)、脱气DMF (1 mL)、乙炔基三(丙-2-基)硅烷 (121 µL, 0.54 mmol)和二乙胺(346 µL, 3.27 mmol)。将反应物在微波中在120℃下搅拌35分钟。将反应物浓缩。将残余物溶解于DCM (15 mL)中,用水(10 mL)和盐水(10 mL)洗涤,然后干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-2% MeOH作为洗脱剂,以提供作为棕色油的标题化合物(100 mg, 49%); m/z = 561.2 (MH)+。
中间体57
4-(三氟甲氧基)-1H-1,3-苯并二唑-2-胺(239991)
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3-(三氟甲氧基)苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(91%); m/z = 218.0 (MH)+。
中间体58
4-碘-1H-1,3-苯并二唑-2-胺(239995)
在氮气下向4-溴-1H-1,3-苯并二唑-2-胺(1.00 g, 4.72 mmol)于二氧杂环己烷(25 mL)中的搅拌溶液中添加碘化钾 (1.57 g, 9.43 mmol)、碘化铜(I) (90 mg, 0.47 mmol)和N,N'-二甲基乙烷-1,2-二胺(102 µL, 0.94 mmol)。最初,将反应物在100℃下加热2小时。然后将反应物在125℃下再加热38小时。在此时段期间,分批添加额外的碘化钾(2.35 g, 14.15 mmol)、碘化铜(I) (180 mg, 0.94 mmol)和N,N'-二甲基乙烷-1,2-二胺(204 µL, 1.88 mmol)。然后将反应物浓缩,并将残余物用EtOAc (40 mL)和水(40 mL)稀释。通过过滤去除所得沉淀,并将水相用IPA/CHCl3 (1:1, 2 x 40 mL)萃取。将合并的有机萃取物用盐水(25 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为紫色固体的标题化合物(775 mg, 64%);m/z = 259.9 (MH)+。
中间体59
3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸苯酯(239996)
将密封管装入3-环戊基-N-[9-碘-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(200 mg, 0.40 mmol)、无水MeCN (2 mL)、乙酸钯(II)(9 mg, 0.04 mmol)和四氟硼酸三叔丁基鏻(46 mg, 0.16 mmol)。将反应物脱气,并添加甲酸苯酯(108 µL, 0.99 mmol),随后添加三乙胺(137 µL, 0.99 mmol)。将反应物用氮气吹扫,密封并在80℃下搅拌16小时。将反应物冷却至室温并浓缩。将残余物溶解于EtOAc (8 mL)中,并用饱和柠檬酸(水溶液)(2 x 7 mL)、水(7 mL)和盐水(7 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-2% MeOH作为洗脱剂,以提供作为黄色固体的标题化合物(65 mg, 64%); m/z = 501.2 (MH)+。
中间体60
4-溴-6-氯-1H-1,3-苯并二唑-2-胺(239916)
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3-溴-5-氯苯-1,2-二胺(经由文献方法: PCT国际申请2013067260可得)代替4,5-二氯苯-1,2-二胺(84%); m/z = 245.8, 247.8 (MH)+。
中间体61
6-溴-4-氟-1H-1,3-苯并二唑-2-胺(239926)
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用5-溴-3-氟苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(定量收率); m/z = 229.8, 231.8 (MH)+。
中间体62
6-氯-5-甲氧基-1H-1,3-苯并二唑-2-胺(239941)
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用4-氯-5-甲氧基苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(61%); m/z = 198.0, 199.9 (MH)+。
中间体63
2-溴-3-甲氧基-6-硝基苯胺(239933)
在氮气下向2-溴-3-氟-6-硝基苯胺(1.00 g, 4.26 mmol)于MeOH (43 mL)中的搅拌溶液中添加MeOH中的5.4M NaOMe (1.7 mL, 9.4 mmol)。将反应物在90℃下加热1小时,然后浓缩。将残余物用水稀释。将所得沉淀通过过滤收集,用水洗涤,以提供作为黄色固体的标题化合物(1.05 g, 定量收率);m/z = 246.9, 248.8 (MH)+。
中间体64
3-溴-4-甲氧基苯-1,2-二胺(239933)
将2-溴-3-甲氧基-6-硝基苯胺(1.15 g, 4.66 mmol)和氯化锡(II)二水合物(5.25 g, 23.28 mmol)于EtOAc (47 mL)中的搅拌悬浮液在80℃下加热1小时。将反应物冷却至室温,并使用5M NaOH(水溶液)将pH调节至pH14。将所得悬浮液搅拌15分钟,然后过滤通过Celite™,用EtOAc冲洗。将来自滤液的有机相用盐水洗涤,干燥(MgSO4),过滤并浓缩,以提供作为黄色固体的标题化合物(970 mg, 96%);m/z = 216.9, 218.9 (MH)+。
中间体65
4-溴-5-甲氧基-1H-1,3-苯并二唑-2-胺(239933)
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用3-溴-4-甲氧基苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(58%); m/z = 241.9, 243.9 (MH)+。
中间体66
5-溴-4-氟-1H-1,3-苯并二唑-2-胺(239937)
使用用于制备5,6-二氯-1H-1,3-苯并二唑-2-胺的程序,除了使用4-溴-3-氟苯-1,2-二胺代替4,5-二氯苯-1,2-二胺(73%); m/z = 229.8, 231.8 (MH)+。
中间体67
(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸乙酯(239949/239952)
在0℃在氮气下,向氢化钠 (60%, 0.61 g, 15.24 mmol)于THF (20 mL)中的搅拌悬浮液中添加(2S)-2-羟基丙酸乙酯(1.50 g, 12.7 mmol)于THF (5 mL)中的溶液。将反应物在0℃下搅拌30分钟。然后逐滴添加4-溴-2-甲基-2-丁烯(2.27 g, 15.2 mmol)于THF (5 mL)中的溶液,并将反应混合物温热至室温。18小时之后,将反应物用水(20 mL)和EtOAc (20 mL)稀释。将水层用EtOAc (2 x 20 mL)萃取。将合并的有机萃取物用盐水(20 mL)洗涤,然后干燥(MgSO4),过滤并浓缩,以提供作为棕色油的标题化合物(2.00 g, 81%);1H NMR (500 MHz, 氯仿-d) δ 1.24 (t, 3H), 1.35 (d, 3H), 1.63 (s, 3H), 1.70 (s, 3H), 3.88 (dd, 1H), 3.93 (q, 1H), 4.06 (dd, 1H),4.12 – 4.20 (m, 2H), 5.28 -5.35 (m, 1H)。
中间体68
(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸(239949/239952)
在0℃下向(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸乙酯(600 mg, 2.90 mmol)于MeOH (10 mL)中的溶液中添加LiOH.H2O (609 mg, 14.50 mmol)于水(5 mL)中的溶液。将反应物在室温下搅拌18小时。将反应物用EtOAc (20 mL)稀释,并用水(20 mL)萃取。将水层用3M HCl(水溶液)酸化至pH1,然后用EtOAc (4 x 20 mL)萃取。将合并的有机萃取物用盐水(30 mL)洗涤,干燥(MgSO4),过滤并浓缩,以提供作为黄色液体的标题化合物(440 mg, 86%);1H NMR (250 MHz, 氯仿-d) δ 1.43 (d, 3H), 1.66 (s, 3H), 1.74 (s, 3H), 3.95 – 4.17 (m, 3H), 5.34 (ddt, 1H), 11.09 (s, 1H)。
中间体69
(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺(239949/239952)
向(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸(400 mg, 2.28 mmol)于DCM (5 mL)中的搅拌溶液中逐滴添加草酰氯(391 µL, 4.51 mmol)。添加DMF(2滴),并将反应物在室温下搅拌2小时。将反应混合物浓缩,并将残余物溶解于DCM(5 mL)中。将所得溶液逐滴添加至氨水(1.30 mL, 11.4 mmol)于DCM (2 mL)中的溶液。将反应物在室温下搅拌18小时,然后浓缩。将残余物溶解于EtOAc (20 mL)中,并用水(20 mL)洗涤。将水层用EtOAc (3 x 20 mL)萃取。将合并的有机萃取物用盐水(30 mL)洗涤,干燥(MgSO4),过滤并浓缩,以提供作为黄色固体的标题化合物(200 mg, 54%);1H NMR (250 MHz, 氯仿-d) δ 1.40 (d, 3H), 1.68 (s, 3H), 1.76 (s, 3H), 3.86 (q, 1H), 4.04 (d, 2H), 5.27 – 5.40 (m, 1H), 5.60 (s, 1H), 6.54 (s, 1H)。
中间体70
(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺(239949/239952)
在氮气下向酰胺(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺(180 mg, 1.145 mmol)于无水DCM (5 mL)中的溶液中添加吡啶(93 µL, 1.15 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(358 mg, 2.29 mmol)。将反应物在室温下搅拌1.5小时。在0℃下添加亚硫酰氯(84 µL, 1.2 mmol)。将反应物在0℃下搅拌1小时。在氮气下将反应混合物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (3 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(154 mg, 3.57 mmol)于DMF (2 mL)中的溶液,随后添加三乙胺(160 µL, 1.15 mmol)。将反应物搅拌2天,然后浓缩。将残余物溶解于EtOAc中,并用盐水洗涤。将水层用EtOAc (3 x 25 mL)萃取。将合并的有机萃取物干燥(MgSO4),过滤并浓缩。将粗产物溶解于DCM中。通过过滤去除所得沉淀。将滤液浓缩,以提供作为橙色固体的标题化合物(270 mg, 67%);m/z = 425.1 (MH)+。
中间体71
(2R)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸乙酯(239958/239959)
使用用于制备(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸乙酯的程序,除了使用(2R)-2-羟基丙酸乙酯代替(2S)-2-羟基丙酸乙酯(84%); 1H NMR (500 MHz, 氯仿-d) δ 1.25 (t, 3H), 1.36 (d, 3H), 1.63 (s, 3H), 1.69 – 1.72 (m, 3H), 3.90 (dd, 1H), 3.95 (q, 1H), 4.04 – 4.09 ( m, 1H), 4.12 – 4.23 (m, 2H), 5.30 – 5.35 (m, 1H)。
中间体72
(2R)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸(239958/239959)
使用用于制备(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸的程序,除了使用(2R)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸乙酯代替(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸乙酯(91%); 1H NMR (250 MHz, 氯仿-d) δ 1.45 (d, 3H), 1.68 (s, 3H), 1.76 (s, 3H), 3.97 – 4.18 (m, 3H), 5.30 – 5.40 (m, 1H), 8.36 (s, 1H)。
中间体73
(2R)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺(239958/239959)
使用用于制备(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺的程序,除了使用(2R)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸代替乙基 (2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酸(29%); 1H NMR (250 MHz, 氯仿-d) δ 1.34 (d, 3H), 1.62 (s, 3H), 1.70 (s, 3H), 3.80 (q, 1H), 3.98 (d, 2H), 5.16 – 5.38 (m, 1H), 6.44 (s, 1H), 6.56 (s, 1H)。
中间体74
(2R)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺(239958/239959)
使用用于制备(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺的程序,除了使用(2R)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺代替(2S)-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺(68%); m/z = 425.0 (MH)+。
中间体75
(2R)-2-(3-氯苯氧基)丙酰胺(239968/29969)
在0℃在氮气下,向(2S)-2-羟基丙酰胺(500 mg, 5.61 mmol)于无水DCM (50 mL)中的搅拌溶液中添加甲磺酰氯(434 µL, 5.61 mmol),随后添加三乙胺(783 µL, 5.61 mmol)。将反应物温热至室温并搅拌1小时。将反应物用水稀释,并将有机相干燥(MgSO4),过滤并浓缩。将残余物溶解于无水乙腈(50 mL)中,并添加3-氯苯酚(721 mg, 5.61 mmol)和K2CO3(776 mg, 5.61 mmol)。将所得悬浮液在70℃下加热16小时,然后浓缩。将残余物溶解于EtOAc (100 mL)中,并用水(100 mL)和盐水(100 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用庚烷中的0-100% EtOAc作为洗脱剂,以提供作为白色固体的标题化合物(220 mg, 20%);m/z = 200.2, 202.3 (MH)+。
中间体76
(2S)-2-环己基丙酰胺(239985)
在0℃在氮气下向[(叔丁氧基羰基)氨基](环己基)乙酸(500 mg, 3.2 mmol)于DCM (50 mL)中的搅拌溶液中添加DMF(2滴),随后添加亚硫酰氯(0.25 mL, 3.4 mmol)。将所得溶液在室温下搅拌1.5小时,然后浓缩。将残余物溶解于无水THF (20 mL)中。将反应物冷却至0℃,并添加THF中的0.5M氨(32 mL)。将反应物温热至室温并搅拌2.5天。将反应物浓缩。将残余物溶解于DCM (30 mL)中,并用水(30 mL)洗涤。将水层用DCM (30 mL)和IPA/CHCl3(1:1, 30 mL)萃取。将合并的有机萃取物用盐水(30 mL)洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物在EtOAc中研磨,以得到作为白色固体的标题化合物(150 mg, 31%);m/z = 156.0 (MH)+。
中间体77
(2S)-2-(环戊-1-烯-1-基甲氧基)丙酸乙酯(239997/239998)
在0℃在氮气下,向氢化钠(60%, 0.81 g, 20.3 mmol)于THF (35 mL)中的搅拌悬浮液中添加(2S)-2-羟基丙酸乙酯(2.00 g, 16.9 mmol)于THF (5 mL)中的溶液。将混合物在0℃下搅拌30分钟。在0℃下将1-(溴甲基)环戊-1-烯(经由文献方法: J. Am. Chem. Soc., 2013, 135, 10769-10775可得)(3.27 g, 20.3 mmol)于THF (5 mL)中的溶液逐滴添加至反应混合物。然后将反应物温热至室温并搅拌18小时。将水(20 mL)和EtOAc (150 mL)添加至反应混合物。将水层用EtOAc (2 x 20 mL)萃取。将合并的有机萃取物用盐水(20 mL)洗涤,然后干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用庚烷中的5-30% EtOAc作为洗脱剂,以提供作为黄色物的标题化合物(1.75 g, 50%);1H NMR (500 MHz, 氯仿-d) δ 1.31 (t, 3H), 1.42 (d, 3H), 1.86 - 1.98 (m, 2H), 2.29 - 2.39 (m, 4H), 3.97 - 4.04 (m, 2H), 4.16 - 4.20 (m, 1H), 4.20 - 4.26 (m, 2H), 5.64 - 5.70 (m, 1H)。
中间体78
(2S)-2-(环戊-1-烯-1-基甲氧基)丙酸(239997/239998)
向(2S)-2-(环戊-1-烯-1-基甲氧基)丙酸乙酯(1.75 g, 8.39 mmol)于MeOH (12 mL)和水(4 mL)中的溶液中逐滴添加7.5M NaOH(水溶液)(1.34 mL, 10.1 mmol)。将反应物在室温下搅拌3小时。将MeOH蒸发,并用1M HCl(水溶液)将残余物的pH调节至pH1。将酸性水溶液用EtOAc (4 x 20 mL)萃取。将合并的有机萃取物用盐水(10 mL)洗涤,干燥(Na2SO4),过滤并浓缩,以提供作为黄色油的标题化合物(1.09 g, 76%);1H NMR (500 MHz, 氯仿-d) δ 1.48 (d, 3H), 1.89 – 2.00 (m, 2H), 2.28 – 2.42 (m, 4H), 4.07 (q, 1H), 4.11 (d, 1H), 4.21 (d, 1H), 5.66 – 5.74 (m, 1H)。
中间体79
(2S)-2-(环戊-1-烯-1-基甲氧基)丙酰胺(239997/239998)
向(2S)-2-(环戊-1-烯-1-基甲氧基)丙酸(1.10 g, 6.46 mmol)于DCM (15 mL)中的搅拌溶液中逐滴添加草酰氯(832 µL, 9.69 mmol)。添加DMF(2滴),并将反应物在室温下搅拌2小时。将反应混合物浓缩。将残余物溶解于DCM (15 mL)中,并添加氨水(3 mL)。将反应物在室温下搅拌30分钟,然后浓缩。将残余物溶解于DCM (50 mL)中,并用水洗涤。将有机相干燥(Na2SO4),过滤并浓缩,以提供作为棕色固体的标题化合物(1.15 g, 定量收率);1H NMR (500 MHz, 氯仿-d) δ 1.43 (d, 3H), 1.94 (m, 2H), 2.24 – 2.43 (m, 4H), 3.91 (q, 1H), 4.04 – 4.19 (m, 2H), 5.41 (s, 1H), 5.69 (s, 1H), 6.57 (s, 1H)。
中间体80
11-氯-3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸苯酯(240015)
使用用于制备3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸苯酯的程序,除了使用N-[11-氯-9-碘-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.0²,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺代替3-环戊基-N-[9-碘-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺. 将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(39%); m/z = 535.2, 537.2 (MH)+。
中间体81
2-(2-甲氧基乙氧基)吡啶-4-甲腈(240032)
在0℃下向氢化钠(60%, 347 mg, 8.66 mmol)于无水二氧杂环己烷 (10 mL)中的搅拌悬浮液中逐滴添加2-甲氧基乙-1-醇(549 mg, 7.22 mmol)于无水二氧杂环己烷(5 mL)中的溶液。将反应物温热至室温并搅拌2小时。在0℃下将2-氯吡啶-4-甲腈(1.00 g, 7.22 mmol)于无水二氧杂环己烷(5 mL)中的溶液逐滴添加至反应混合物,然后将其在室温下搅拌20小时。将反应物用水(40 mL)和EtOAc (40 mL)稀释。将水相用EtOAc (3 x 50 mL)萃取。将合并的有机萃取物用盐水(30 mL)洗涤,干燥(MgSO4),过滤并浓缩。将EtOAc (10 mL)添加至粗产物。通过过滤去除所得沉淀,并将滤液浓缩。将所得残余物通过硅胶色谱纯化,使用庚烷中的0-100% EtOAc作为洗脱剂,以提供作为绿色油的标题化合物(255 mg, 20%); m/z = 179.0 (MH)+。
中间体82
2-(2-甲氧基乙氧基)吡啶-4-甲酰胺(240032)
向2-(2-甲氧基乙氧基)吡啶-4-甲腈于水/EtOH (1:1, 4 mL)中的搅拌溶液中添加2M NaOH(水溶液) (0.65 mL, 1.3 mmol)和28% H2O2(水溶液)(0.14 mL, 1.3 mmol)。将反应物在室温下搅拌18小时,然后用饱和NaHCO3(水溶液)(20 mL)和EtOAc (20 mL)稀释。将水相用EtOAc (3 x 20 mL)萃取。将合并的有机萃取物用盐水(30 mL)洗涤,干燥(MgSO4),过滤并浓缩,以提供作为黄色固体的标题化合物(180 mg, 73%);m/z = 197.0 (MH)+。
中间体83
3-(3-环戊基丙酰胺基)-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸苯酯(240026)
将密封管装入3-环戊基-N-[9-碘-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(65%, 300 mg, 0.36 mmol)、无水MeCN (2 mL)、乙酸钯(II)(8 mg, 0.04 mmol)和四氟硼酸三叔丁基鏻(42 mg, 0.15 mmol)。将反应物脱气,并添加甲酸苯酯(0.1 mL, 0.9 mmol),随后添加三乙胺(157 µL, 0.91 mmol)。将反应物用氮气吹扫,密封并在80℃下搅拌16小时。添加额外的乙酸钯(II)(8 mg, 0.04 mmol)和四氟硼酸三叔丁基鏻(42 mg, 0.15 mmol)、甲酸苯酯(0.1 mL, 0.9 mmol)和三乙胺(157 µL, 0.91 mmol)。将反应物用氮气吹扫,密封并在80℃下再搅拌6小时。将反应物冷却至室温并浓缩。将残余物溶解于EtOAc (15 mL)中,并用饱和柠檬酸(水溶液)(15 mL)洗涤。将水层用EtOAc (15 mL)萃取。将合并的有机萃取物用水(15 mL)和盐水(25 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过反相C18色谱纯化,使用酸性洗脱剂,以提供作为白色固体的标题化合物(115 mg, 58%);m/z = 531.2 (MH)+。
实施例1
6-氯-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(8),6,9,11-四烯-3-基]吡啶-2-甲酰胺(ABR 239471)
在氩气下向6-氯吡啶-2-甲酰胺(1.00 g, 13.0 mmol)于DMF (8 mL)中的搅拌溶液中逐滴添加吡啶(1.05 mL, 13.0 mmol)和3,3,3-三氟-2-氧代-丙酸乙酯(1.73 mL, 13.0 mmol)。将反应混合物在室温下搅拌1小时,然后逐滴添加亚硫酰氯(0.95 mL, 13.0 mmol)。将反应混合物在室温下再搅拌16小时,然后浓缩以提供酰基亚胺中间体。将酰基亚胺中间体溶解于DMF (5 mL)中并添加至5,6-二甲基-1H-1,3-苯并二唑-2-胺(2.03 g, 13.0 mmol)于DMF (7 mL)中的溶液,随后添加三乙胺(1.80 mL, 13.0 mmol)。将反应混合物在室温下搅拌4小时。将反应物用盐水(25 mL)稀释,并用EtOAc (2 x 50 mL)萃取。将合并的有机相用水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂,以提供作为灰白色固体的标题化合物(0.6 g, 22%);m/z = 423.4, 425.4 (MH)+。
实施例2
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-氟吡啶-2-甲酰胺(ABR 239472)
在氩气下向6-氟吡啶-2-甲酰胺(700 mg, 4.75 mmol)于DMF (7 mL)中的溶液中添加3,3,3-三氟-2-氧代丙酸乙酯(629 µL, 4.75 mmol)和吡啶(383 µL, 4.75 mmol)。将反应混合物在室温下搅拌1小时,然后逐滴添加亚硫酰氯(344 µL, 4.75 mmol)。再继续搅拌2.5小时,然后将反应物浓缩以提供酰基亚胺中间体。将所得酰基亚胺中间体溶解于DMF (5 mL)中,然后添加至5,6-二氯-1H-1,3-苯并二唑-2-胺(519 mg, 2.57 mmol)于DMF (5 mL)中的溶液。添加三乙胺(0.35 ml, 2.56 mmol),并将反应混合物在室温下搅拌2小时。将反应物用盐水(30 mL)稀释,并用EtOAc (2 x 50 mL)萃取。将合并的有机相用水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂,以提供作为灰白色固体的标题化合物(300 mg, 25%);m/z = 448.4, 450.4(MH)+。
实施例3
N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]苯甲酰胺(ABR 238128)
在氮气下向苯甲酰胺(1.00 g, 8.26 mmol)于DMF (40 mL)中的搅拌溶液中逐滴添加吡啶(666 µL, 8.26 mmol),随后逐滴添加3,3,3-三氟-2-氧代丙酸甲酯(1.29 g, 8.26 mmol)。将所得溶液在氮气下搅拌30分钟,然后添加亚硫酰氯(0.60 mL, 8.26 mmol),并将溶液在室温下再搅拌1小时。将1H-1,3-苯并二唑-2-胺(1.10 g, 8.26 mmol)和三乙胺(0.10 mL, 0.72 mmol)添加至反应混合物,并将溶液在室温下搅拌6小时,然后在50℃下搅拌3小时。将反应混合物浓缩,溶解于EtOAc (100 mL)中,并用水(3 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,用庚烷中的20-50% EtOAc洗脱,随后是自动反相HPLC(低pH方法A),以提供作为白色固体的标题化合物(12 mg, 0.4%)。
实施例4
2-环己基-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]乙酰胺(ABR 238883)
在氮气下向2-环己基乙酰胺(425 mg, 3.01 mmol)于DMF (15 mL)中的搅拌溶液中添加吡啶(243 µL, 3.01 mmol)和3,3,3-三氟-2-氧代丙酸甲酯(470 mg, 3.01 mmol)。将所得溶液在室温下搅拌1小时,然后在0℃在氮气下将亚硫酰氯(218 µL, 3.01 mmol)逐滴添加至溶液。将反应混合物温热至室温,然后再搅拌16小时。将反应混合物浓缩,以提供酰基亚胺中间体,其不经进一步纯化即使用。
向DMF (5 mL)中的酰基亚胺中间体的一部分(200 mg, 0.72 mmol)中添加1H-1,3-苯并二唑-2-胺(95 mg, 0.72 mmol),随后添加三乙胺(95 µL, 0.72 mmol)。将反应混合物在室温在氮气下搅拌3小时,然后浓缩。将所得棕色油通过自动反相HPLC(低pH方法A)纯化,以提供作为灰白色固体的标题化合物(58 mg, 22%)。
实施例5
3-(吗啉-4-基)-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238798)
在氮气下向3-(吗啉-4-基)丙酰胺(1.00 g, 6.32 mmol)于DMF (10 mL)中的搅拌溶液中添加吡啶(510 µL, 6.32 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(649 µL, 6.32 mmol)。将反应混合物在室温下搅拌1小时,然后添加亚硫酰氯(459 µL, 6.32 mmol)。将反应混合物搅拌48小时,然后浓缩。将残余物溶解于DCM (10 mL)中,并过滤通过硅藻土短垫。将滤液浓缩,以提供酰基亚胺中间体(1.2 g),其不经进一步纯化即使用。向DMF (3 mL)中的酰基亚胺中间体的一部分(250 mg, 0.84 mmol)中添加2-氨基苯并咪唑(112 mg, 0.84 mmol)和三乙胺(109 µL, 0.84 mmol)。将反应混合物在室温在氮气下搅拌16小时。将反应混合物通过硅胶色谱纯化,使用DCM中的0-5% MeOH作为洗脱剂,以提供作为灰白色固体的标题化合物(98 mg, 30%)。
实施例6
N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(吡咯烷-1-基)丙酰胺(ABR 238799)
在氮气下向根据文献(You等人, 2008)制备的3-(吡咯烷-1-基)丙酰胺(1.00 g, 7.03 mmol)于DMF (10 mL)中的搅拌溶液中添加吡啶(567 µL, 7.03 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(722 µL, 7.03 mmol)。将所得反应混合物在室温下搅拌1小时,然后添加亚硫酰氯(510 µL, 7.03 mmol)。将反应混合物在室温下再搅拌6小时,然后浓缩,以提供酰基亚胺中间体(1.78 g),其不经进一步纯化即使用。在氮气下向DMF (3 mL)中的酰基亚胺中间体的一部分(200 mg, 0.71 mmol)中添加2-氨基苯并咪唑(95 mg, 0.71 mmol),随后添加三乙胺(92 µL, 0.71 mmol)。将反应混合物在室温下搅拌2小时,然后浓缩。将残余物溶解于EtOAc (25 mL)中,并用水(2 x 20 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-5% MeOH作为洗脱剂,以提供作为灰白色固体的标题化合物(49 mg, 18%)。
实施例7
3-(氧杂环己烷-4-基)-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238816)
在氮气下向N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(8),6,9,11-四烯-3-基]丙-2-烯酰胺(25 mg, 0.08 mmol)和4-碘氧杂环己烷 (80 mg, 0.26 mmol)于EtOH (5 mL)和水(1.5 mL)混合物中的搅拌溶液中添加锌粉(101.0 mg, 1.54 mmol)和碘化铜(98.2 mg, 0.51 mmol)。将反应混合物在室温下超声处理5小时。将反应混合物过滤通过硅藻土,并用EtOAc (3 x 25 mL)洗涤。将滤液用盐水(50 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(17 mg, 16%)。
实施例8
2-溴-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]苯甲酰胺(ABR 238803)
向2-溴苯甲酰胺(177 mg, 0.88 mmol)于DMF (6 mL)中的搅拌溶液中逐滴添加吡啶(71 µL, 0.88 mmol)和3,3,3-三氟-2-氧代丙酸乙酯(90 µL, 0.88 mol)。将反应混合物在室温下搅拌1小时,然后逐滴添加亚硫酰氯(64 µµL, 0.88 mmol)。将反应混合物搅拌16小时,然后浓缩以提供酰基亚胺中间体。将酰基亚胺中间体溶解于DMF (4 mL)中并添加至1H-1,3-苯并二唑-2-胺(118 mg, 0.88 mmol)于DMF (2 mL)中的溶液,随后添加三乙胺(118 µL, 0.88 mmol)。将反应混合物在室温下搅拌4小时。将反应物用盐水(25 mL)稀释,并用EtOAc (2 x 50 mL)萃取。将合并的有机相用水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过制备型TLC在DCM中的10%MeOH中纯化,以提供作为棕色固体的标题化合物(10 mg, 3%)。
实施例9
3-环戊基-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙酰胺(ABR 238789)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用3-环戊基丙酰胺代替2-溴苯甲酰胺。将粗产物通过硅胶色谱纯化,使用DCM中的2.5% MeOH作为洗脱剂(12%)。
实施例10
3,5-二甲氧基-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺(ABR 238802)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用3,5-二甲氧基苯甲酰胺代替2-溴苯甲酰胺。将粗产物通过制备型TLC纯化,使用DCM中的5% MeOH作为洗脱剂(2%)。
实施例11
6-甲基-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]吡啶-3-甲酰胺(ABR 238843)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用6-甲基吡啶-3-甲酰胺代替2-溴苯甲酰胺。将粗产物通过制备型TLC纯化,使用DCM中的10% MeOH作为洗脱剂(2%)。
实施例12
3,5-二氯-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]苯甲酰胺(ABR 238895)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用3,5-二氯苯甲酰胺代替2-溴苯甲酰胺。将粗产物通过制备型TLC纯化,使用DCM中的5% MeOH作为洗脱剂(8%)。
实施例13
3-环己基-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]丙酰胺(ABR 238219)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用3-环己基丙酰胺代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌2小时代替16小时。在反应的第二阶段中,使用1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌16小时代替4小时。将粗产物通过制备型TLC纯化,使用DCM中的10% MeOH作为洗脱剂(10%)。
实施例14
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]苯甲酰胺(ABR 238786)
在氮气下向(Z)-苯甲酰基亚氨基-3,3,3-三氟丙酸甲酯(150 mg, 0.58 mmol)和5,6-二甲基-1H-1,3-苯并二唑-2-胺(93 mg, 0.58 mmol)于DMF (2 mL)中的搅拌溶液中添加三乙胺(60 µL, 0.58 mmol)。将反应混合物在室温下搅拌1小时,然后浓缩。将残余物溶解于EtOAc (25 mL)中,用水(2 x 20 mL)和盐水(20 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(47 mg, 21%)。
实施例15
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-3-苯基丙酰胺(ABR 238787)
将3-苯基丙酰胺(500 mg, 3.35 mmol)溶解于DMF (15 mL)中,并在室温在氮气下添加3,3,3-三氟-2-氧代丙酸甲酯(523.03 mg, 3.35 mmol)和吡啶(270 µL, 3.35 mmol)。将反应混合物搅拌1小时,然后添加亚硫酰氯(243 µL, 3.35 mmol)。将反应混合物在室温下再搅拌16小时,然后浓缩。将残余物过滤通过硅胶短柱,用EtOAc洗脱,并将滤液浓缩,以提供酰基亚胺中间体(502 mg),其不经进一步纯化即使用。向5,6-二甲基-1H-1,3-苯并二唑-2-胺(84.18 mg, 0.52 mmol)于DMF (5 mL)中的溶液中添加三乙胺(67.17 µL, 0.52 mmol),随后添加酰基亚胺中间体(150 mg, 0.52 mmol)。将反应混合物在室温下搅拌3小时,然后浓缩。将残余物溶解于EtOAc (20 mL)中,并用10%柠檬酸(水溶液)(20 mL)、水(20 mL)和盐水(20 mL)洗涤。将有机相干燥(MgSO4),浓缩并通过自动反相HPLC(低pH方法A)纯化,以提供作为灰白色固体的标题化合物(76 mg, 35%)。
实施例16
3-(2-氯苯基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙酰胺(ABR 238908)
在氮气下向3-(2-氯苯基)丙酰胺(500 mg, 2.72 mmol)于DMF (10 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(425 mg, 2.72 mmol),随后添加吡啶(220 µL, 2.72 mmol)。将反应混合物在室温下搅拌2小时,然后添加亚硫酰氯(198 µL, 2.72 mmol)。将反应混合物再搅拌16小时,然后浓缩。将残余物过滤通过硅藻土短垫,然后过滤通过硅胶,用EtOAc洗脱,并将滤液浓缩,以提供作为黄色油的酰基亚胺中间体(850 mg),其不经进一步纯化即使用。在氮气下向5,6-二甲基-1H-1,3-苯并二唑-2-胺(50 mg, 0.31 mmol)于DMF (5 mL)中的溶液中添加一部分酰基亚胺中间体(100 mg, 0.31 mmol),随后添加三乙胺(41 µL, 0.31 mmol)。将反应混合物在室温下再搅拌8小时。将反应混合物浓缩并通过自动反相HPLC(低pH方法A)纯化,以提供作为米色固体的标题化合物(30 mg, 21%)。
实施例17
3,5-二氯-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]苯甲酰胺(ABR 239340)
在氮气下向3,5-二氯苯甲酰胺(300 mg, 1.58 mmol)于DMF (20 mL)中的搅拌溶液中添加吡啶(127 µL, 1.58 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(370 mg, 2.37 mmol)。将反应混合物在室温下搅拌2小时,然后冷却至0℃。逐滴添加亚硫酰氯(115 µL, 1.58 mmol),并将溶液在该温度下再搅拌1小时。在氮气下,将反应混合物浓缩,通过硅胶短垫,用EtOAc洗脱。将滤液立即蒸发,并将剩余的酰基亚胺中间体重新溶解于DMF (10 mL)中。向该溶液中添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(191 mg, 1.18 mmol)和三乙胺(210 µL, 1.58 mmol)。将所得反应混合物搅拌3小时,然后浓缩。将残余物溶解于EtOAc (40 mL)中,并用水(2 x 30 mL)和盐水(2 x 30 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(高pH方法)纯化,以提供作为白色固体的期望产物(45 mg, 6%)。
实施例18
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-4-苯基苯甲酰胺(ABR 239078)
在氮气下向4-苯基苯甲酰胺(250 mg, 1.27 mmol)于DMF (15 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(198 mg, 1.27 mmol),随后添加吡啶(102 µL, 1.27 mmol)。将反应混合物在室温下搅拌16小时,然后冷却至0℃。逐滴添加亚硫酰氯(92 µL, 1.27 mmol),并将反应物温热至室温。将反应物再搅拌2小时,然后浓缩。将残余物过滤通过硅胶短垫,用EtOAc洗脱,并将滤液浓缩,以提供酰基亚胺中间体。将酰基亚胺中间体溶解于DMF (10 mL)中,并添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(204 mg, 1.27 mmol),随后添加三乙胺(177 µL, 1.27 mmol)。将反应混合物在室温下搅拌,然后在80℃下搅拌18小时。然后将反应混合物浓缩。将粗产物通过硅胶色谱纯化,用DCM中的0-5% MeOH洗脱,随后是自动反相HPLC(低pH方法A),以提供作为白色固体的标题化合物(38 mg, 6%)。
实施例19
4-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丁酰胺(ABR 238854)
将烘干烧瓶装入N-[10,11-二甲基-3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]丙-2-烯酰胺(80 mg, 0.24 mmol),随后装入EtOH (5 mL)和(碘甲基)环戊烷(99 mg, 0.47 mmol)。然后将锌粉(93 mg, 1.42 mmol)和碘化铜(90 mg, 0.47 mmol)添加至反应混合物,随后添加水(1.5 mL)。将反应混合物用氮气脱气2分钟,然后将悬浮液在室温下置于声波浴中6小时。向反应混合物再装入碘化铜(90 mg, 0.47 mmol)、锌粉(93 mg, 1.42 mmol)和(碘甲基)环戊烷 (99 mg, 0.47 mmol),并将反应物再超声处理3小时。将反应混合物过滤通过硅藻土垫,并进一步用EtOAc (100 mL)洗涤。将有机滤液浓缩并通过硅胶色谱纯化,使用DCM中的5-10% MeOH作为洗脱剂,随后是自动反相HPLC(低pH方法A),以提供作为米色固体的标题化合物(11 mg, 11%)。
实施例20
2-环己基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]乙酰胺(ABR 238884)
在氮气下向烘干的烧瓶中添加2-环己基乙酰胺(425 mg, 3.01 mmol),随后添加DMF (15 mL)和吡啶(243 µL, 3.01 mmol)。将3,3,3-三氟-2-氧代丙酸甲酯(470 mg, 3.01 mmol)添加至溶液并搅拌1小时。在0℃在氮气下将亚硫酰氯(218.4 µL, 3.01 mmol)逐滴添加至溶液。将反应混合物温热至室温,然后再搅拌16小时。将反应混合物浓缩,以提供酰基亚胺中间体,其不经进一步纯化即使用。将酰基亚胺中间体(200 mg, 0.72 mmol)溶解于DMF (5 mL)中,并在氮气下添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(115 mg, 0.72 mmol)和三乙胺(95 µL, 0.72 mmol)。将反应混合物在室温下搅拌3小时。将反应物在高真空下浓缩,然后通过自动反相HPLC(低pH方法A)纯化,以提供作为灰白色固体的标题化合物(150 mg, 51%)。
实施例21
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-(2,2,2-三氟乙氧基)吡啶-3-甲酰胺(ABR 238950)
在氮气下向6-(2,2,2-三氟乙氧基)吡啶-3-甲酰胺(500 mg, 2.27 mmol)于DMF (15 mL)中的溶液中添加吡啶(183 µL, 2.27 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(354 mg, 2.27 mmol)。将溶液在室温下搅拌1小时,然后冷却至0℃。逐滴添加亚硫酰氯(165 µL, 2.27 mmol),并将反应混合物在室温下再搅拌16小时,然后浓缩。将所得酰基亚胺中间体(643 mg, 1.8 mmol)和5,6-二甲基-1H-1,3-苯并二唑-2-胺(289.38 mg, 1.8 mmol)立即溶解于DMF (10 mL)中,并在氮气下添加三乙胺(0.24 mL, 1.8 mmol)。将反应混合物在室温下搅拌16小时,然后浓缩。将所得残余物在DCM/MeOH (20:1)中研磨。通过过滤收集所得灰白色固体。将50 mg粗固体样品通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(25 mg, 3%)。
实施例22
1-环戊烷羰基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡咯烷-3-甲酰胺(ABR 239208)
在0℃在氮气下向N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环- [6.4.0.02,6}]十二碳-1(12),6,8,10-四烯-3-基]吡咯烷-3-甲酰胺盐酸盐(95 mg, 0.23 mmol)于无水DCM (3 mL)中的搅拌溶液中添加环戊烷碳酰氯(29 µL, 0.24 mmol),随后添加三乙胺(63 µL, 0.45 mmol)。将反应混合物温热至室温并搅拌16小时。将水(10 mL)和DCM (10 mL)添加至反应混合物,剧烈搅拌5分钟。分离各层,并用DCM (2 x 20 mL)进一步反萃取水层。将合并的有机相干燥(MgSO4),过滤并浓缩,以提供作为棕色油的粗产物(含有4种立体异构体的混合物)。将其通过使用Chiralpak AD-H柱与CO2和含有0.1%甲酸的MeOH的流动相的SFC纯化,以提供立体化学未知的标题化合物的一种异构体(6 mg, 6%)。
实施例23
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(吗啉-4-基)丙酰胺(ABR 238814)
在氮气下向3-(吗啉-4-基)丙酰胺(1.00 g, 6.32 mmol)于DMF (10 mL)中的搅拌溶液中添加吡啶(510 µL, 6.32 mmol),随后添加3,3,3-三氟-2-氧代-丙酸甲酯(649 µL, 6.32 mmol)。将反应混合物在室温下搅拌1小时,然后添加亚硫酰氯(459 µL, 6.32 mmol)。将反应混合物再搅拌48小时,然后浓缩。将残余物溶解于DCM (10 mL)中,并过滤通过硅藻土短垫。将滤液浓缩,以提供酰基亚胺中间体(1.2 g),其不经进一步纯化即使用。在氮气下向一部分酰基亚胺中间体(250 mg, 0.84 mmol)中添加DMF (3 mL)中的5,6-二甲基-1H-1,3-苯并二唑-2-胺(136 mg, 0.84 mmol),随后添加三乙胺(109 µL, 0.84 mmol)。将反应混合物在室温下搅拌3小时,然后浓缩。将残余物溶解于DCM (25 mL)中,并用水(2 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为浅黄色固体的标题化合物(36 mg, 10%)。
实施例24
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡咯烷-3-甲酰胺盐酸盐(ABR 239077)
在氮气下向3-{[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}吡咯烷-1-甲酸叔丁酯(110 mg, 0.23 mmol)于二乙醚 (10 mL)中的搅拌溶液中逐滴添加乙醚中的2M HCl(114 µL, 0.23 mmol)。将反应混合物在室温下搅拌16小时,然后浓缩,以提供作为白色固体且作为立体异构体的混合物的标题化合物(94 mg, 98%)。
实施例25
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]哌啶-4-甲酰胺盐酸盐(ABR 239205)
在氮气下向4-{[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}哌啶-1-甲酸叔丁酯(715 mg, 1.44 mmol)于DCM (20 mL)和二乙醚 (20 mL)中的搅拌溶液中逐滴添加乙醚中的2M HCl(1.44 µL, 2.88 mmol)。将反应混合物在室温下搅拌16小时。将反应物浓缩,与二乙醚(2 x 20 mL)共沸并在真空下干燥,以提供作为盐酸盐且作为米色固体的标题化合物(609 mg, 98%)。
实施例26
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1-(2-甲氧基乙酰基)哌啶-4-甲酰胺(ABR 239227)
在0℃在氮气下向N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]哌啶-4-甲酰胺盐酸盐(75 mg, 0.17 mmol)于DCM (10 mL)中的搅拌溶液中添加2-甲氧基乙酰氯(23 mg, 0.21 mmol),随后添加三乙胺(46 µL, 0.35 mmol)。将反应混合物温热至室温,然后搅拌4小时。将混合物用DCM (30 mL)稀释并用水(2 x 20 mL)洗涤。将有机相浓缩并通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(17 mg, 21%)。
实施例27
3-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238788)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺并使用3-环戊基丙酰胺代替2-溴苯甲酰胺(10%)。
实施例28
6-氯-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]吡啶-3-甲酰胺(ABR 238911)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺并使用6-氯吡啶-3-甲酰胺代替2-溴苯甲酰胺。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂(13%)。
实施例29
3-(2,6-二氯苯基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙酰胺(ABR 238998)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺并使用3-(2,6-二氯苯基)丙酰胺代替2-溴苯甲酰胺。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂(3%)。
实施例30
2-溴-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]苯甲酰胺(ABR 239004)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂(3%)。
实施例31
6-(环己基氨基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡啶-3-甲酰胺(ABR 239024)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.0²,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺并使用6-(环己基氨基)吡啶-3-甲酰胺代替2-溴苯甲酰胺。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂(6%)。
实施例32
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-甲基吡啶-3-甲酰胺(ABR 239031)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺并使用6-甲基吡啶-3-甲酰胺代替2-溴苯甲酰胺。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂(6%)。
实施例33
3-环己基-N-[10,11-二甲基-3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]丙酰胺(ABR 238804)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用3-环己基丙酰胺代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌2小时代替16小时。在反应的第二阶段中,使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌16小时代替4小时。将粗产物通过制备型TLC纯化,使用DCM中的10% MeOH作为洗脱剂(3%)。
实施例34
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(吡啶-3-基)丙酰胺(ABR 239084)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用3-(吡啶-3-基)丙酰胺代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌2小时代替16小时。在反应的第二阶段中,使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌16小时代替4小时。将粗产物通过硅胶色谱纯化,使用DCM中的4% MeOH作为洗脱剂(1%)。
实施例35
6-(环己基氨基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡啶-2-甲酰胺(ABR 238974)
向6-氯-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]吡啶-2-甲酰胺(200 mg, 0.47 mmol)于NMP (2 mL)中的溶液中添加N,N-二异丙基乙胺(0.16 mL, 1.89 mmol),随后添加环己胺(0.22 mL, 1.89 mmol)。将反应混合物在微波中在200℃下加热1小时,然后浓缩。将残余物通过硅胶色谱纯化,使用DCM中的5% MeOH作为洗脱剂,以提供作为黄色固体的标题化合物(15 mg, 7%)。
实施例36
6-环己基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]吡啶-2-甲酰胺(ABR 239059)
将微波管装入6-氯-N-[10,11-二甲基-4-氧代-3-(三氟-甲基)-2,5,7-三氮杂三环[6.4.0.0²,6]十二碳-1(8),6,9,11-四烯-3-基]吡啶-2-甲酰胺(100 mg, 0.24 mmol)、环己烯硼酸(29 mg, 0.24 mmol)、SPhos (9 mg, 0.02 mmol)、Na2CO3 (50 mg, 0.47 mmol)和DMF/水(9:1, 10 mL)。将反应混合物用氩气脱气15分钟。添加乙酸钯(II)(5 mg, 0.02 mmol),并将反应混合物在微波中在100℃下加热30分钟。将反应物冷却至室温并过滤通过硅藻土垫。将滤液用水(10 mL)稀释并用乙酸乙酯(2 x 15 mL)萃取。将合并的有机相干燥(Na2SO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法B)纯化,以提供作为灰白色固体的标题化合物(10 mg, 9%)。
实施例37
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.0²,6]十二碳-1(8),6,9,11-四烯-3-基]-3-苯基丙酰胺(ABR 239019)
在氮气下向(2E)-3,3,3-三氟-2-[(3-苯基丙酰基)亚氨基]-丙酸甲酯(100 mg, 0.35 mmol)和5,6-二氯-1H-1,3-苯并二唑-2-胺(70 mg, 0.35 mmol)于DMF(5 mL)中的搅拌溶液中添加三乙胺(46 µL, 0.35 mmol)。将反应混合物在室温下搅拌16小时,然后浓缩。将残余物通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂,以提供作为灰白色固体的标题化合物(57 mg, 36%)。
实施例38
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]苯甲酰胺(ABR 238949)
在氮气下向5,6-二氯-1H-1,3-苯并二唑-2-胺(100 mg, 0.49 mmol)和(Z)-苯甲酰基亚氨基-3,3,3-三氟丙酸甲酯(128 mg, 0.49 mmol)于DMF(2 mL)中的搅拌溶液中添加三乙胺(66 µL, 0.49 mmol)。将反应混合物在室温下搅拌16小时,浓缩并通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(24 mg, 11%)。
实施例39
3-环戊基-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238926)
在室温在氮气下向3-环戊基丙酰胺(3.50 g, 24.8 mmol)于DMF(50 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(3.87 g, 24.8 mmol),随后添加吡啶(2.00 mL, 24.8 mmol)。将反应混合物在室温下搅拌4小时,然后冷却至0℃。将亚硫酰氯(1.80 mL, 24.8 mmol)逐滴添加至该溶液,并将反应混合物在室温下搅拌16小时。将反应混合物浓缩并过滤通过硅胶短垫,用DCM (100 mL)洗脱。将滤液浓缩,并在氮气下将剩余的酰基亚胺中间体立即溶解于DMF (30 mL)中。将5,6-二氯-1H-1,3-苯并二唑-2-胺(4.01 g, 19.8 mmol)添加至该溶液,随后添加三乙胺(3.96 mL, 29.7 mmol)。将所得混合物在室温下搅拌2小时,然后浓缩。将残余物溶解于EtOAc (200 mL)中,用水(4 x 100 mL)和盐水(2 x 100 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂,以提供深棕色泡沫。将其溶解于MeOH (100 mL)中并用活性炭脱色。将所得悬浮液过滤通过硅藻土并用MeOH洗涤。将滤液浓缩,以提供作为浅黄色固体的标题化合物(3.2 g, 29%)。
实施例40
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-氮杂螺[3.3]庚烷-6-甲酰胺(ABR 239424)
向6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}-2-氮杂螺[3.3]-庚烷-2-甲酸叔丁酯(100 mg, 0.18 mmol)于DCM(25 mL)中的搅拌溶液中逐滴添加TFA (1.0 mL, 13.1 mmol)。将溶液在室温在氮气下搅拌16小时,然后浓缩。将所得固体与二乙醚共沸并在真空下干燥,以提供作为TFA盐且作为浅粉红色固体的标题化合物(78 mg, 76%)。
实施例41
非对映异构体混合物。(2S)-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡咯烷-2-甲酰胺(ABR 239426)
将(2S)-2-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}吡咯烷-1-甲酸叔丁酯(50 mg, 0.1 mmol)溶解于DCM (10 mL)中,并添加TFA (0.5 mL, 3.3 mmol)。将反应混合物在室温下搅拌16小时,然后浓缩。将所得固体与甲苯(2 x 20 mL)共沸并在真空下干燥,以提供作为TFA盐且作为非对映异构体的混合物的标题化合物(49 mg, 95%)。
实施例42
非对映异构体混合物N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡咯烷-3-甲酰胺(ABR 239136)
在氮气下向3-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}吡咯烷-1-甲酸叔丁酯(466 mg, 0.89 mmol)于二乙醚(10 mL)中的搅拌溶液中逐滴添加乙醚中的2M HCl (0.67 µL, 1.34 mmol)。将反应混合物搅拌24小时,然后浓缩。然后将100 mg样品通过自动反相HPLC(高pH方法)纯化,以提供作为盐酸盐、白色固体和非对映异构体的混合物的标题化合物(34 mg)。
实施例43
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]哌啶-4-甲酰胺(ABR 239206)
将4-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}哌啶-1-甲酸叔丁酯(810 mg, 1.51 mmol)溶解于二乙醚(20 mL)和DCM(20 mL)中。经74小时分批添加乙醚中的2M HCl (6.04 mL, 12.08 mmol)。将反应混合物浓缩,并将所得固体用乙醚研磨。将该材料的样品通过自动反相HPLC(高pH方法)纯化,以提供作为白色固体的标题化合物(7 mg)。
实施例44
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1-(2-甲氧基乙酰基)哌啶-4-甲酰胺(ABR 239228)
在氮气下向N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]哌啶-4-甲酰胺盐酸盐(75 mg, 0.16 mmol)于DCM(10 mL)中的搅拌溶液中添加三乙胺(42 µL, 0.32 mmol),随后添加2-甲氧基乙酰氯(21 mg, 0.19 mmol)。将反应混合物在室温下搅拌2小时,然后用DCM (20 mL)稀释并用水(2 x 20 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的5-10% MeOH作为洗脱剂,以提供纯度约75%的作为黄色固体的标题化合物(16 mg, 20%)。标题化合物未经进一步纯化即在生物测定中测试。
实施例45
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1-(3-甲基丁酰基)哌啶-4-甲酰胺(ABR 239229)
在氮气下向N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]哌啶-4-甲酰胺盐酸盐(75 mg, 0.16 mmol)于DCM(10 mL)中的搅拌溶液中添加3-甲基丁酰氯(23 mg, 0.19 mmol),随后添加三乙胺(42 µL, 0.32 mmol)。将反应混合物在室温下搅拌6小时。将反应混合物用DCM (25 mL)稀释并用水(3 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(高pH方法)纯化,以提供纯度约73%的作为浅黄色固体的标题化合物(29 mg, 35%)。标题化合物未经进一步纯化即在生物测定中测试。
实施例46
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1-(3,3,3-三氟丙酰基)哌啶-4-甲酰胺(ABR 239232)
在氮气下向N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]哌啶-4-甲酰胺盐酸盐(75 mg, 0.16 mmol)于DCM(10 mL)中的搅拌溶液中添加3,3,3-三氟丙酰氯(28 mg, 0.19 mmol),随后添加三乙胺(42 µL, 0.32 mmol)。将溶液在室温下搅拌4小时,然后添加额外的3,3,3-三氟丙酰氯(28 mg, 0.19 mmol)。再搅拌8小时之后,将反应混合物用DCM (20 mL)稀释并用水(3 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗反应产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的期望产物(5 mg, 6%)。
实施例47
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氮杂环丁烷-3-甲酰胺(ABR 239257)
在0℃下向3-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}氮杂环丁烷-1-甲酸叔丁酯(800 mg, 1.57 mmol)于DCM(100 mL)中的搅拌溶液中逐滴添加TFA (3.53 mL, 4.72 mmol)。将溶液在0℃下搅拌1小时,然后温热至室温。3小时之后,将反应混合物蒸发至约15 mL,然后添加二乙醚(50 mL)。将悬浮液浓缩,并将所得固体与二乙醚共沸3次。将固体通过SCX柱游离碱化,用MeOH/DCM (1:1)洗涤,然后用MeOH中的2M氨洗脱。将碱性滤液收集并浓缩,以提供作为白色固体的标题化合物(507 mg, 79%)。
实施例48
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1-(乙磺酰基)氮杂环丁烷-3-甲酰胺(ABR 239402)
向N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氮杂环丁烷-3-甲酰胺(50 mg, 0.12 mmol)于无水THF(5 mL)中的搅拌溶液中添加乙烷磺酰氯(13 µL, 0.13 mmol),随后添加2M NaOH(水溶液)(92 µL, 0.18 mmol)。将反应混合物在室温下搅拌2小时。再添加一部分乙烷磺酰氯(13 µL, 0.13 mmol),随后添加2M NaOH(水溶液)(92 µL, 0.18 mmol)。将反应混合物搅拌2小时,然后浓缩。将残余物溶解于EtOAc (100 mL)中,并用水(2 x 25 mL)和盐水(50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(31 mg, 51%)。
实施例49
1-乙酰基-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]氮杂环丁烷-3-甲酰胺(ABR 239403)
向N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氮杂环丁烷-3-甲酰胺(100 mg, 0.25 mmol)于DCM(10 mL)中的搅拌溶液中添加乙酰氯(19 µL, 0.27 mmol),随后添加三乙胺(49 µL, 0.37 mmol)。将反应混合物在室温下搅拌2小时。将反应混合物用水(2 x 25 mL)洗涤,并将有机层浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,并在水中共沸,以提供作为白色固体的标题化合物(19 mg, 17%)。
实施例50
(外消旋).1-环戊烷羰基-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡咯烷-3-甲酰胺(ABR 239137)
在0℃在氮气下向N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]吡咯烷-3-甲酰胺盐酸盐(95 mg, 0.21 mmol)于DCM(3 mL)中的搅拌溶液中逐滴添加环戊烷碳酰氯(26 µL, 0.22 mmol),随后添加三乙胺(58 µL, 0.41 mmol)。将反应混合物温热至室温,然后再搅拌16小时。将反应混合物用水(20 mL)和DCM(20 mL)稀释,并分离各层。将有机相用水(20 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为非对映异构体的混合物的标题化合物(5 mg, 5%)。未经进一步纯化即测试。
实施例51
(3R)-1-(环戊磺酰基)-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡咯烷-3-甲酰胺(ABR 239139)
向(3R)-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]吡咯烷-3-甲酰胺盐酸盐(817 mg, 1.78 mmol)于THF(20 mL)中的搅拌溶液中添加环戊烷磺酰氯(258 µL, 1.96 mmol),随后添加2M NaOH(水溶液)(1.83 mL, 3.66 mmol)。将反应混合物在室温下搅拌3小时。添加额外的环戊烷-磺酰氯(70 µL, 0.53 mmol),并继续再搅拌16小时。然后将反应混合物浓缩并在EtOAc(50 mL)和水(50 mL)之间分配。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂,以提供作为灰白色固体的标题化合物(304 mg, 31%)。
实施例52
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1-(吡啶-2-基)氮杂环丁烷-3-甲酰胺(ABR 239404)
在室温下将N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氮杂环丁烷-3-甲酰胺(100 mg, 0.25 mmol)和碳酸钾(51 mg, 0.37 mmol)于DMF(2 mL)中的悬浮液搅拌。向悬浮液中添加2-氯吡啶(28 µL, 0.29 mmol),并将反应混合物在120℃下搅拌16小时。将反应混合物浓缩并在EtOAc(25 mL)和水(25 mL)之间分配。将有机相用水(3 x 25 mL)和盐水(2 x 25 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体且作为甲酸盐的期望产物(18 mg, 15%)。
实施例53
3-环己基-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239034)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的程序,除了使用5,6-二氯-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺并使用3-环己基丙酰胺代替2-溴苯甲酰胺。将粗产物通过硅胶色谱纯化,使用DCM中的2.5% MeOH作为洗脱剂(6%)。
实施例54
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]环戊烷甲酰胺(ABR 239114)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用环戊烷甲酰胺代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌2.5小时代替16小时。在反应的第二阶段中,使用5,6-二氯-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌2小时代替4小时。将粗产物通过硅胶色谱纯化,使用DCM中的2% MeOH作为洗脱剂(9%)。
实施例55
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]环己烷甲酰胺(ABR 239115)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用环己烷甲酰胺代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌2.5小时代替16小时。在反应的第二阶段中,使用5,6-二氯-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌16小时代替4小时。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂,随后在DCM/戊烷中研磨(2%)。
实施例56
3-氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]苯甲酰胺(ABR 239414)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用3-氯苯甲酰胺代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌18小时代替16小时。在反应的第二阶段中,使用5,6-二氯-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌18小时代替4小时。将粗产物通过硅胶色谱纯化,使用DCM中的2-4% MeOH作为洗脱剂(3%)。
实施例57
3,5-二氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]苯甲酰胺(ABR 239427)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用3,5-二氯苯甲酰胺代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌18小时代替16小时。在反应的第二阶段中,使用5,6-二氯-1H-1,3-苯并二唑-2-胺代替5,6-二甲基-1H-1,3-苯并二唑-2-胺,并将反应物搅拌18小时代替4小时。将粗产物通过硅胶色谱纯化,使用DCM中的4% MeOH作为洗脱剂(5%)。
实施例58
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-4-(吡咯烷-1-基)丁酰胺(ABR 239155)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用4-(吡咯烷-1-基)丁酰胺代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌72小时代替16小时。在反应的第二阶段中,使用5,6-二氯-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌16小时代替4小时。将粗产物通过自动反相HPLC(低pH方法B)纯化(1%)。
实施例59
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-4-(吗啉-4-基)丁酰胺(ABR 239161)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用4-(吗啉-4-基)丁酰胺代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌2.5小时代替16小时。在反应的第二阶段中,使用5,6-二氯-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌16小时代替4小时。将粗产物通过硅胶色谱纯化,使用DCM中的20% MeOH作为洗脱剂(11%)。
实施例60
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(吗啉-4-基)乙酰胺(ABR 239358)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用2-(吗啉-4-基)乙酰胺(根据文献(Chaudhari等人 2007)制备)代替2-溴苯甲酰胺,并且在添加亚硫酰氯之后,将反应混合物搅拌18小时代替16小时。在反应的第二阶段中,使用5,6-二氯-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌16小时代替4小时。将粗产物通过自动反相HPLC(低pH方法B)纯化(4%)。
实施例61
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(吗啉-4-基)丙酰胺(ABR 239259)
在0℃在氩气下向2-[(5,6-二氯-1H-1,3-苯并二唑-2-基)氨基]-3,3,3-三氟-2-[3-(吗啉-4-基)丙酰胺基]丙酸酯(0.20 g, 0.39 mmol)于甲苯(3 mL)中的溶液中逐滴添加三甲基铝(75 µL, 0.39 mmol)。将反应物温热至室温,在室温下搅拌1小时,然后在100℃下加热16小时。将反应物冷却至室温,并添加冰/水。通过过滤去除所得沉淀,并将滤液用EtOAc (2 x 20 mL)萃取。将合并的有机相分离,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的4% MeOH作为洗脱剂,以提供作为灰色固体的标题化合物(15 mg, 8%)。
实施例62
6-氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]吡啶-2-甲酰胺(ABR 239356)
使用用于制备2-溴-N-[3-氧代-4-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-4-基]苯甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用6-氯吡啶-2-甲酰胺代替2-溴苯甲酰胺。在反应的第二阶段中,使用5,6-二氯-1H-1,3-苯并二唑-2-胺代替1H-1,3-苯并二唑-2-胺,并将反应物搅拌16小时代替4小时。将粗产物通过硅胶色谱纯化,使用DCM中的2% MeOH作为洗脱剂(14%)。
实施例63
6-(氮杂环丁烷-1-基)-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]吡啶-2-甲酰胺(ABR 239354)
向密封管中的6-氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡啶-2-甲酰胺(200 mg, 0.43 mmol)于DMF(3 mL)中的溶液中添加K2CO3(59 mg, 0.43 mmol)和氮杂环丁烷(25 mg, 0.43 mmol)。将反应混合物在100℃下加热16小时。将反应混合物用水(15 mL)稀释并用EtOAc (2 x 25 mL)萃取。将合并的有机相用盐水(15 mL)洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法B)纯化,以提供作为白色固体的标题化合物(70 mg, 33%)。
实施例64
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-(2,2,2-三氟乙氧基)吡啶-2-甲酰胺(ABR 239390)
使用用于制备6-(氮杂环丁烷-1-基)-N-[10,11-二氯-4-氧代-3-(三氟-甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡啶-2-甲酰胺的程序,除了使用2,2,2-三氟乙-1-醇代替氮杂环丁烷 (15%)。
实施例65
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-(二甲基氨基)吡啶-2-甲酰胺(ABR 239391)
使用用于制备N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.0²,6]十二碳-1(12),6,8,10-四烯-3-基]-6-(二甲基氨基)吡啶-2-甲酰胺的程序,除了使用二甲胺(水中的70 wt. %)代替氮杂环丁烷 (52%)。
实施例66
6-(环己基氨基)-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡啶-2-甲酰胺(ABR 239409)
使用用于制备N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-(二甲基氨基)吡啶-2-甲酰胺的程序,除了使用环己胺代替氮杂环丁烷并将反应物加热2小时代替16小时。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂,随后在DCM/MeOH中重结晶,以提供标题化合物(31%)。
实施例67
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-(乙基氨基)吡啶-2-甲酰胺(ABR 239101)
将密封管装入6-氯-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡啶-2-甲酰胺(150 mg, 0.35 mmol)和乙胺(水中的70 wt. %, 56 µL, 1.06 mmol)。将反应物在100℃下加热16小时。将反应混合物用水(10 mL)稀释并用EtOAc (2 x 25 mL)萃取。将合并的有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法B)纯化,以提供作为白色固体的标题化合物(31 mg, 20%)。
实施例68
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(哌啶-4-基)乙酰胺(ABR 239386)
在0℃下将4M HCl于二氧杂环己烷中的溶液(3 mL, 12.0 mmol)添加至4-({[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}甲基)哌啶-1-甲酸叔丁酯(60 mg, 0.11 mmol)。将反应混合物在室温下搅拌3小时并浓缩。将残余物在二乙醚/EtOH中研磨,以提供作为盐酸盐且作为白色固体的标题化合物(41 mg, 77%)。
实施例69
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(哌啶-2-基)丙酰胺(ABR 239393)
在0℃下将4M HCl于二氧杂环己烷中的溶液(5 mL, 20.0 mmol)添加至2-(2-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}乙基)哌啶-1-甲酸叔丁酯(200 mg, 0.35 mmol)。将反应混合物在室温下搅拌3小时并浓缩。将残余物在二乙醚/EtOH中研磨,以提供作为盐酸盐且作为灰白色固体的标题化合物(140 mg, 60%)。
实施例70
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-(乙基氨基)吡啶-2-甲酰胺(ABR 239355)
向密封管中的6-氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡啶-2-甲酰胺(2.00 g, 4.31 mmol)于EtOH(20 mL)中的溶液中添加氢氧化铯一水合物(722 mg, 4.31 mmol)和THF中的2M乙胺(194 mg, 4.31 mmol)。将反应物在100℃下加热16小时。将反应混合物用水(15 mL)稀释并用EtOAc (2 x 25 mL)萃取。将合并的有机相用盐水(15 mL)洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法B)纯化,以提供作为橙色固体的标题化合物(172 mg, 8%)。
实施例71
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-[(2-甲氧基乙基)氨基]吡啶-2-甲酰胺(ABR 239432)
向密封管中的N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-氟吡啶-2-甲酰胺(150 mg, 0.33 mmol)于DMF(2 mL)中的溶液中添加K2CO3(93 mg, 0.67 mmol)和2-甲氧基乙-1-胺(25 mg, 0.34 mmol)。将反应混合物在100℃下加热2小时。将反应混合物用水(15 mL)稀释并用EtOAc (2 x 25 mL)萃取。将合并的有机相用盐水(15 mL)洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法B)纯化,以提供作为灰白色固体的标题化合物(35 mg, 21%)。
实施例72
N-[10,11-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]苯甲酰胺(ABR 238928)
在氮气下向5,6-二氟-1H-1,3-苯并二唑-2-胺(60 mg, 0.35 mmol)于DMF(2 mL)中的搅拌溶液中添加(Z)-苯甲酰基亚氨基-3,3,3-三氟丙酸甲酯(92 mg, 0.35 mmol)和三乙胺(47 µL, 0.35 mmol)。将溶液在室温下搅拌3小时,然后浓缩。将残余物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体的标题化合物(39 mg, 28%)。
实施例73
3-环戊基-N-[10,11-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238925)
在氮气下向5,6-二氟-1H-1,3-苯并二唑-2-胺(100 mg, 0.59 mmol)和(2Z)-2-[(3-环戊基丙酰基)亚氨基]-3,3,3-三氟丙酸甲酯(165 mg, 0.59 mmol)于DMF(2 mL)中的搅拌溶液中添加三乙胺(79 µL, 0.59 mmol)。将反应混合物在室温下再搅拌4小时,浓缩并通过自动反相HPLC(低pH方法A)纯化,以提供作为灰白色固体的标题化合物(44 mg, 18%)。
实施例74
2-环己基-N-[10,11-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(8),6,9,11-四烯-3-基]乙酰胺(ABR 238929)
在氮气下向5,6-二氟-1H-1,3-苯并二唑-2-胺(75 mg, 0.44 mmol)和(2Z)-2-[(2-环己基乙酰基)亚氨基]-3,3,3-三氟丙酸甲酯(124 mg, 0.44 mmol)于DMF(2 mL)中的搅拌溶液中添加三乙胺(59 µL, 0.44 mmol)。将反应混合物在室温下搅拌2小时,然后浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为米色固体的期望产物(85 mg, 46%)。
实施例75
N-[10,11-二甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]苯甲酰胺(ABR 238995)
在氮气下向5,6-二甲氧基-1H-1,3-苯并二唑-2-胺(100 mg, 0.52 mmol)和(Z)-苯甲酰基亚氨基-3,3,3-三氟丙酸甲酯(134.15 mg, 0.52 mmol)于DMF(2 mL)中的搅拌溶液中添加三乙胺(68.91 µL, 0.52 mmol)。将反应混合物在室温下搅拌16小时,然后浓缩。将残余物通过自动反相HPLC(低pH方法A)纯化,随后在MeOH中研磨,以提供作为灰白色固体的标题化合物(15 mg, 7%)。
实施例76
3-环戊基-N-[10,11-二甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238927)
在氮气下向5,6-二甲氧基-1H-1,3-苯并二唑-2-胺(100 mg, 0.52 mmol)和(2Z)-2-[(3-环戊基丙酰基)亚氨基]-3,3,3-三氟丙酸甲酯(145 mg, 0.52 mmol)于DMF(2 mL)中的搅拌溶液中添加三乙胺(69 µL, 0.52 mmol)。将反应混合物搅拌4小时,然后浓缩。将残余物通过自动反相HPLC(低pH方法A)纯化,随后用乙醚研磨,以提供作为黄色固体的标题化合物(31 mg, 14%)。
实施例77
N-[9-溴-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]苯甲酰胺(ABR 239170)
在0℃下向N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]苯甲酰胺(84 mg, 0.22 mmol)于DCM(5 mL)中的搅拌溶液中添加N-溴代琥珀酰亚胺(39 mg, 0.22 mmol)。将反应混合物在室温下搅拌2小时。将反应混合物用DCM (20 mL)稀释并用水(2 x 20 mL)洗涤。将有机相浓缩并通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(41 mg, 41%)。
实施例78
3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]-十五碳-1(9),2,7,14-四烯-11-基]丙酰胺(ABR 239320)
在室温在氮气下向3-环戊基丙酰胺(93 mg, 0.57 mmol)于DMF(1.5 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(72 µL, 0.71 mmol),随后添加吡啶(57 µL, 0.71 mmol)。将反应混合物在室温下搅拌1小时。在0℃下添加亚硫酰氯(51 µL, 0.71 mmol),然后将反应混合物在室温下搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM (50 mL)洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (2 mL)中。添加1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺(98 mg, 0.57 mmol)和三乙胺(113 µL, 0.85 mmol),并将反应混合物在室温下搅拌18小时。将反应混合物用EtOAc(20 mL)稀释,用水(4 x 10 mL)和盐水(10 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用庚烷中的50% EtOAc作为洗脱剂,以提供作为白色固体的标题化合物(66 mg, 28%)。
实施例79
N-[10-氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺和
N-[11-氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239329)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙-酰胺的程序,除了使用5-氯-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。将粗产物通过在最小体积的DCM中研磨而纯化,以提供作为区域异构体的1:1混合物的标题化合物(34%)。
实施例80
3-环戊基-N-[9,10-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239330)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙-酰胺的程序,除了使用4,5-二甲基-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。在初始装填之后1小时,在0℃下添加额外当量的亚硫酰氯,然后将反应物在室温下搅拌1小时,然后浓缩。将粗产物通过在最小体积的DCM中研磨而纯化,以提供标题化合物(38%)。
实施例81
3-环戊基-N-[9-甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239343)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙-酰胺的程序,除了使用4-甲基-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。将粗产物通过在最小体积的DCM中研磨而纯化,以提供标题化合物(17%)。
实施例82
3-环戊基-N-[9,10-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环 [6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239371)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙-酰胺的程序,除了使用4,5-二氟-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。将粗产物通过在最小体积的DCM中研磨而纯化,以提供标题化合物(33%)。
实施例83
3-环戊基-N-[9,10-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239375)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙-酰胺的程序,除了使用4,5-二氯-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。将粗产物通过在最小体积的DCM中研磨而纯化,以提供标题化合物(28%)。
实施例84
3-环戊基-N-[9,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239394)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙-酰胺的程序,除了使用4,6-二氯-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺. 在初始装填之后1小时,在0℃下添加额外当量的亚硫酰氯,然后将反应物在室温下搅拌1小时,然后浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供标题化合物(27%)。
实施例85
N-[9-氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239399)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙-酰胺的程序,除了使用4-氯-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。在初始装填之后1小时,在0℃下添加额外当量的亚硫酰氯,然后将反应物在室温下搅拌1小时,然后浓缩。将粗产物通过在最小体积的DCM中研磨而纯化,以提供标题化合物(19%)。
实施例86
3-环戊基-N-[9,11-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239422)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙-酰胺的程序,除了使用4,6-二氟-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。将粗产物通过硅胶色谱纯化,使用庚烷中的44%EtOAc作为洗脱剂,随后在最小体积的DCM中研磨,以提供标题化合物(11%)。
实施例87
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氧杂环己烷-4-甲酰胺(ABR 239441)
在室温在氩气下向氧杂环己烷-4-甲酰胺(500 mg, 3.87 mmol)于DMF(15 mL)中的搅拌溶液中添加吡啶(312 µl, 3.87 mmol),随后添加3,3,3-三氟-2-氧代丙酸乙酯(658 mg, 3.87 mmol)。将反应混合物在室温下搅拌1小时,然后添加亚硫酰氯(422 µl, 5.81 mmol)。将反应物再搅拌16小时,然后浓缩。在氩气下将剩余的酰基中间体溶解于DMF(5 mL)中。添加5,6-二氯-1H-1,3-苯并二唑-2-胺(587 mg, 2.90 mmol)于DMF(7 mL)中的溶液和三乙胺(861 µl, 6.19 mmol),并将反应混合物在室温下搅拌4小时。将反应混合物用水(10 mL)稀释并用EtOAc (2 x 15 mL)萃取。将合并的有机萃取物用盐水洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化。通过在DCM/MeOH、然后戊烷中研磨而实施进一步纯化,以提供标题化合物(20 mg, 1%)。
实施例88
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-3-(1-羟基环戊基)丙酰胺(ABR 239702)
在0℃在氮气下搅拌环戊酮(234 µl, 2.63 mmol)和DMPU(127 µl, 1.05 mmol)于无水脱气THF(30 mL)和MeOH(3 mL)中的溶液。同时经10分钟逐滴添加0.1M碘化钐于THF中的溶液(10.55 mL, 1.06 mmol)和N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.0²,6]十二碳-1(8),6,9,11-四烯-3-基]丙-2-烯酰胺(100 mg, 0.26 mmol)于无水THF(5 mL)中的溶液。将反应混合物在0℃下搅拌20分钟。将反应物倒入冷水(100 mL)中并用EtOAc (3 x 50 mL)萃取。将合并的有机相用1M HCl(水溶液)和盐水(2 x 50 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(22 mg, 18%)。
实施例89
非对映异构体 1:(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-1-甲磺酰基吡咯烷-2-甲酰胺(ABR 239679)
在室温在氮气下向(2S)-1-甲磺酰基吡咯烷-2-甲酰胺(400 mg, 2.08 mmol)于DMF(6 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(320 µL, 3.13 mmol),随后添加吡啶(170 µL, 2.08 mmol)。将反应混合物在室温下搅拌19小时。在0℃下添加亚硫酰氯(150 µL, 2.08 mmol),并将反应物搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(280 mg, 1.74 mmol)于DMF(5 mL)中的溶液和三乙胺(280 µL, 2.08 mmol)。将反应混合物在室温下搅拌17小时,然后浓缩。将残余物用EtOAc (40 mL)稀释,并用水(2 x 30 mL)、10%柠檬酸(水溶液)(2 x 20 mL)和盐水(20 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以得到非对映异构体的纯净混合物。将非对映异构体的混合物通过自动反相HPLC(低pH方法A)分离,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体1 (82 mg, 10%)。
实施例90
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-[(3-甲氧基丙基)氨基]吡啶-2-甲酰胺(ABR 239523)
向密封管中的N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.0^{2,6}]十二碳-1(12),6,8,10-四烯-3-基]-6-氟吡啶-2-甲酰胺(250 mg, 0.56 mmol)于DMF(3 mL)中的溶液中添加K2CO3(185 mg, 1.34 mmol)和3-甲氧基丙-1-胺(62 mg, 0.70 mmol)。将反应混合物在100℃下加热4小时。添加额外的3-甲氧基丙-1-胺(62 mg, 0.70 mmol)。将反应物在100℃下再加热8小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(3 x 25 mL)、然后盐水(3 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色油的标题化合物(35 mg, 12%)。
实施例91
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-[(2-羟基乙基)氨基]吡啶-2-甲酰胺(ABR 239539)
向密封管中的N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-氟吡啶-2-甲酰胺(100 mg, 0.22 mmol)于DMF(3 mL)中的溶液中添加K2CO3(74 mg, 0.54 mmol)和2-氨基乙-1-醇(27 mg, 0.45 mmol)。将反应混合物在100℃下加热18小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(3 x 25 mL)和盐水(3 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色油的标题化合物(51 mg, 47 %)。
实施例92
非对映异构体混合物。N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-{[(2S)-1-甲氧基丙-2-基]氨基}吡啶-2-甲酰胺(ABR 239540)
向密封管中的N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-氟吡啶-2-甲酰胺(100 mg, 0.22 mmol)于DMF(3 mL)中的溶液中添加K2CO3(74 mg, 0.54 mmol)和(2S)-1-甲氧基丙-2-胺(40 mg, 0.45 mmol)。最初,将反应混合物在100℃下加热16小时。然后将反应物再加热20小时。在此时段期间,在再处理期间在室温下随着反应分批添加额外的(2S)-1-甲氧基丙-2-胺(80 mg, 0.90 mmol)和K2CO3(74 mg, 0.54 mmol)。然后将反应物浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(3 x 25 mL)和盐水(2 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体且作为非对映异构体的混合物的标题化合物(15 mg, 13%)。
实施例93
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-{[2-(二甲基氨基)乙基]氨基}吡啶-2-甲酰胺(ABR 239587)
向密封管中的N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-氟吡啶-2-甲酰胺(100 mg, 0.22 mmol)于DMF(3 mL)中的溶液中添加K2CO3(93 mg, 0.67 mmol)和(2-氨基乙基)二甲胺(59 mg, 0.67 mmol)。将反应混合物在100℃下加热16小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(2 x 25 mL)和盐水(2 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法A)纯化,以提供作为甲酸盐且作为粉红色固体的标题化合物(38 mg, 33%)。
实施例94
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-[(2-甲氧基乙基)(甲基)氨基]吡啶-2-甲酰胺(ABR 239537)
在室温在氮气下向6-[(2-甲氧基乙基)(甲基)氨基]吡啶-2-甲酰胺(571 mg, 2.73 mmol)于DMF(10 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(852 mg, 5.46 mmol),随后添加吡啶(220 µl, 2.73 mmol)。将反应混合物在室温下搅拌16小时。在0℃下添加亚硫酰氯(198 µl, 2.73 mmol),然后将反应混合物在室温下搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加5,6-二氯-1H-1,3-苯并二唑-2-胺(441 mg, 2.18 mmol)和三乙胺(381 µl, 2.72 mmol),并将反应混合物在室温下搅拌2小时。将反应混合物浓缩。将残余物溶解于EtOAc (100 mL)中,用水(3 x 50 mL)、然后盐水(3 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂。使用自动反相HPLC(高pH)实施进一步纯化,以提供作为黄色固体的标题化合物(24 mg, 2%)。
实施例95
6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]氨基甲酰基}吡啶-2-甲酸甲酯(ABR 239572)
在室温在氮气下向6-氨基甲酰基吡啶-2-甲酸甲酯(1.00 g, 5.55 mmol)于DMF(15 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(1.14 mL, 11.11 mmol),随后添加吡啶(450 µL, 5.55 mmol)。将反应混合物在室温下搅拌1.5小时。在0℃下添加亚硫酰氯(405 µL, 5.55 mmol),然后将反应混合物搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (15 mL)中。添加5,6-二氯-1H-1,3-苯并二唑-2-胺(935 mg, 4.63 mmol)和三乙胺(739 µL, 5.55 mmol),并将反应混合物在室温下搅拌20小时。将反应混合物浓缩。将残余物用EtOAc (50 mL)稀释,用水(3 x 50 mL)和盐水(2 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-3% MeOH作为洗脱剂,以得到作为棕色油的标题化合物(35 mg, 2%)。
实施例96
2-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-N-(2-甲氧基乙基)吡啶-2,6-二甲酰胺(ABR 239588)
在氮气下将2-甲氧基乙-1-胺(13 µL, 0.15 mmol)添加至1,4-二氮杂双环[2.2.2]辛烷-三甲基铝(1:2, 39 mg, 0.15 mmol)于THF(2 mL)中的搅拌悬浮液。将反应物在40℃下加热30分钟。添加6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]氨基甲酰基}吡啶-2-甲酸甲酯(50 mg, 0.1 mmol)于THF(0.5 mL)中的溶液,然后将反应物在70℃下加热16小时。添加额外的1,4-二氮杂双环[2.2.2]辛烷-三甲基铝(1:2, 39 mg, 0.15 mmol)和2-甲氧基乙-1-胺(13 µL, 0.15 mmol),并将反应物在70℃下再加热4小时。将反应物通过逐滴添加冷水而淬灭,并用EtOAc (3 x 10 mL)洗涤。将水层使用10%柠檬酸(水溶液)(5 mL)酸化,并用EtOAc (3 x 10 mL)萃取。将合并的有机相用盐水(2 x 25 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(高pH)纯化,以提供作为棕色固体的标题化合物(13 mg, 15%)。
实施例97
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-(羟基甲基)吡啶-2-甲酰胺(ABR 239602)
在0℃下将四氢硼酸钠(110 mg, 2.87 mmol)添加至6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]氨基甲酰基}吡啶-2-甲酸甲酯(200 mg, 0.41 mmol)于THF/MeOH(4:1, 10 mL)中的搅拌溶液中。将反应物搅拌24小时。然后在0℃下经40小时时段分批添加额外的四氢硼酸钠(220 mg, 5.74 mmol)。将反应物通过逐滴添加饱和NH4Cl(水溶液)来淬灭,用水稀释并使用10%柠檬酸(水溶液)酸化。将水相用EtOAc (3 x 25 mL)萃取。将合并的有机萃取物干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(中性pH)纯化,以提供作为白色固体的标题化合物(35 mg, 19%)。
实施例98
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-氟苯甲酰胺(ABR 239440)
在室温在氩气下向3-氟苯甲酰胺(500 mg, 3.59 mmol)于DMF(5 mL)中的搅拌溶液中添加吡啶(306 µl, 3.59 mmol),随后添加3,3,3-三氟-2-氧代丙酸乙酯(476 µl, 3.59 mmol)。将反应混合物在室温下搅拌1小时。在0℃下添加亚硫酰氯(231 µl, 3.18 mmol),然后将反应混合物在室温下搅拌2.5小时。将反应混合物浓缩。在氩气下将剩余的酰基中间体溶解于DMF(2 mL)中。添加5,6-二氯-1H-1,3-苯并二唑-2-胺(545 mg, 2.70 mmol)和三乙胺(503 µl, 3.59 mmol),并将反应混合物在室温下搅拌16小时。将反应混合物用水稀释并用EtOAc萃取。将有机相用盐水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂。通过在DCM/MeOH、然后戊烷中研磨而实施进一步纯化,以提供作为灰白色固体的标题化合物(22 mg, 1%)。
实施例99
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3,5-二氟苯甲酰胺(ABR 239446)
在室温在氩气下向3,5-二氟苯甲酰胺(500 mg, 3.18 mmol)于DMF(5 mL)中的搅拌溶液中添加吡啶(271 µl, 3.18 mmol),随后添加3,3,3-三氟-2-氧代丙酸乙酯(422 µl, 3.18 mmol)。将反应混合物在室温下搅拌1小时。在0℃下添加亚硫酰氯(231 µl, 3.18 mmol)。将反应混合物在室温下搅拌16小时,然后将反应混合物浓缩。在氩气下将剩余的酰基中间体溶解于DMF(2 mL)中。添加5,6-二氯-1H-1,3-苯并二唑-2-胺(482 mg, 2.39 mmol)和三乙胺(668 µl, 4.77 mmol),并将反应混合物在室温下搅拌4小时。将反应混合物用水稀释并用EtOAc萃取。将有机相用盐水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂。通过从DCM/MeOH、然后戊烷研磨而实施进一步纯化,以提供作为灰白色固体的标题化合物(20 mg, 1%)。
实施例100
3-氰基-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]苯甲酰胺(ABR 239453)
在室温在氩气下向3-氰基苯甲酰胺(650 mg, 4.45 mmol)于DMF(7 mL)中的搅拌溶液中添加吡啶(378 µl, 4.45 mmol),随后添加3,3,3-三氟-2-氧代丙酸乙酯(590 µl, 4.45 mmol)。将反应混合物在室温下搅拌1小时。在0℃下添加亚硫酰氯(323 µl, 4.45 mmol)。然后将反应混合物在室温下再搅拌16小时,然后将反应混合物浓缩。在氩气下将剩余的酰基中间体溶解于DMF(5 mL)中。添加5,6-二氯-1H-1,3-苯并二唑-2-胺(674 mg, 3.34 mmol)和三乙胺(934 µl, 6.67 mmol),并将反应混合物在室温下搅拌16小时。将反应混合物用水稀释并用EtOAc萃取。将有机相用盐水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂。通过在DCM/MeOH、然后戊烷中研磨而实施进一步纯化,以得到作为白色固体的标题化合物(25 mg, 1%)。
实施例101
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-[(2-甲氧基乙基)氨基]-1,3-噻唑-4-甲酰胺(ABR 239601)
在室温在氮气下向2-溴-1,3-噻唑-4-甲酰胺(360 mg, 1.74 mmol)于DMF(15 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(543 mg, 3.48 mmol),随后添加吡啶(140 µl, 1.74 mmol)。将反应混合物在室温下搅拌16小时。在0℃下添加亚硫酰氯(126 µl, 1.74 mmol),然后将反应混合物搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加5,6-二氯-1H-1,3-苯并二唑-2-胺(293 mg, 1.45 mmol)于DMF(10 mL)中的溶液和三乙胺(231 µl, 1.74 mmol),并将反应混合物在室温下搅拌2小时。将反应混合物浓缩。将残余物用EtOAc (50 mL)稀释,用10%柠檬酸(水溶液)(2 x 25 mL)、水(2 x 25 mL)和盐水(2 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物在DCM中研磨,以提供2-溴-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-1,3-噻唑-4-甲酰胺和2-氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-1,3-噻唑-4-甲酰胺的混合物(746 mg)。
将微波管装入一部分2-溴-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-1,3-噻唑-4-甲酰胺和2-氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-1,3-噻唑-4-甲酰胺的混合物(80 mg)、K2CO3(64 mg, 0.47 mmol)、2-甲氧基乙-1-胺(40 µl, 0.47 mmol)和二氧杂环己烷(2 mL)。最初,将反应物在微波中在130℃下加热1小时。然后将反应物再加热4小时。在此时段期间,在再处理期间在室温下随着反应分批添加额外的2-甲氧基乙-1-胺(160 µl, 1.88 mmol)和K2CO3(256 mg, 1.88 mmol)。然后将反应混合物浓缩。将残余物溶解于EtOAc (25 mL)中,并用水(25 mL)、10%柠檬酸(水溶液)(25 mL)和盐水(25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(27 mg)。
实施例102
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1-(2,2-二氟乙基)氮杂环丁烷-3-甲酰胺(ABR 239437)
向N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氮杂环丁烷-3-甲酰胺(75 mg, 0.18 mmol)于DMF(2 mL)中的搅拌溶液中添加1,1-二氟-2-碘乙烷(39 mg, 0.20 mmol),随后添加K2CO3(51 mg, 0.37 mmol)。将反应物在80℃下加热16小时,然后浓缩。将残余物溶解于EtOAc (25 mL)中,并用水(2 x 25 mL)和盐水(3 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体且作为甲酸盐的标题化合物(11 mg, 13%)。
实施例103
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-3-氟氮杂环丁烷-3-甲酰胺(ABR 239689)
在0℃在氮气下向3-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]氨基甲酰基}-3-氟氮杂环丁烷-1-甲酸叔丁酯(58 mg, 0.11 mmol)于DCM(20 mL)中的搅拌溶液中添加TFA (1 mL)。将反应物温热至室温并搅拌16小时。然后将反应混合物浓缩,并与甲苯(3 x 20 mL)共沸。将粗产物通过自动反相HPLC(高pH)纯化,以提供作为白色固体的标题化合物(11 mg, 23%)。
实施例104
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-3-(3,3-二氟氮杂环丁烷-1-基)丙酰胺(ABR 239705)
向密封管中的N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(8),6,9,11-四烯-3-基]丙-2-烯酰胺(125 mg, 0.33 mmol)于MeCN(10 mL)中的溶液中添加3,3-二氟氮杂环丁烷盐酸盐(56 mg, 0.43 mmol)、二氧化硅(16 mg)和三乙胺(88 µl, 0.66 mmol)。将反应物在80℃下加热3小时。通过过滤去除二氧化硅,并将滤液浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(22 mg, 14%)。
实施例105
3-(3,3-二氟氮杂环丁烷-1-基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙酰胺(ABR 239647)
向密封管中的N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙-2-烯酰胺(125 mg, 0.37 mmol)于MeCN(10 mL)中的溶液中添加3,3-二氟氮杂环丁烷盐酸盐(62 mg, 0.48 mmol)、二氧化硅(18 mg)和三乙胺(98 µl, 0.74 mmol)。将反应物在70℃下加热3小时。通过过滤去除二氧化硅,并将滤液浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(45 mg, 28%)。
实施例106
3-环丁基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(8),6,9,11-四烯-3-基]丙酰胺(ABR 239536)
在室温在氮气下向3-环丁基丙酰胺(250 mg, 1.97 mmol)于DMF(20 mL)中的搅拌溶液中添加吡啶(159 µL, 1.97 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(614 mg, 3.93 mmol)。将反应混合物在室温下搅拌16小时。在0℃下添加亚硫酰氯(147 µL, 1.97 mmol),并将反应物在0℃下搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(254 mg, 1.57 mmol)和三乙胺(261 µl, 1.97 mmol)。将反应混合物在室温下搅拌2小时,然后浓缩。将残余物用EtOAc (50 mL)稀释,并用水(3 x 50 mL)和盐水(2 x 50 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂,以提供作为白色固体的标题化合物(60 mg, 8%)。
实施例107
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-3-(1-羟基环戊基)丙酰胺(ABR 239578)
在0℃在氮气下搅拌环戊酮(210 µl, 2.36 mmol)和DMPU(285 µl, 2.36 mmol)于无水脱气THF(20 mL)和MeOH(5 mL)中的溶液。同时经10分钟逐滴添加0.1M碘化钐于THF中的溶液(23.6 mL, 2.36 mmol)和N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙-2-烯酰胺(200 mg, 0.59 mmol)于无水THF(5 mL)中的溶液。将反应混合物在0℃下搅拌20分钟,然后倒入冷水(100 mL)中并用EtOAc (3 x 50 mL)萃取。将合并的有机相用盐水(2 x 50 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(70 mg, 31%)。
实施例108
2-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(8),6,9,11-四烯-3-基]乙酰胺(ABR 239558)
在室温在氮气下向2-环戊基乙酰胺(350 mg, 2.75 mmol)于DMF(20 mL)中的搅拌溶液中添加吡啶(102 µL, 1.27 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(395 mg, 2.53 mmol)。将反应混合物在室温下搅拌16小时。经3天时段分批添加额外的吡啶(204 µL, 2.54 mmol)和3,3,3-三氟-2-氧代丙酸甲酯(395 mg, 2.53 mmol)。在0℃下添加亚硫酰氯(92 µl, 1.27 mmol),并将反应物在0℃下搅拌1小时。在0℃下添加额外的亚硫酰氯(92 µl, 1.27 mmol),并将反应物在室温下搅拌18小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(355 mg, 2.20 mmol)和三乙胺(366 µl, 2.75 mmol),并将反应混合物在室温下搅拌18小时。然后将反应混合物用EtOAc (30 mL)稀释,并用水(4 x 50 mL)和盐水(2 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-8% MeOH作为洗脱剂。通过在DCM中研磨而实施进一步纯化,以提供作为白色固体的标题化合物(36 mg, 3%)。
实施例109
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-3,5-二氟苯甲酰胺(ABR 239456)
使用用于制备N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氧杂环己烷-4-甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用2-(3,5-二氟苯基)乙酰胺代替氧杂环己烷-4-甲酰胺。在反应的第二阶段中,使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替5,6-二氯-1H-1,3-苯并二唑-2-胺。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂。通过在DCM/MeOH、然后戊烷中研磨而实施进一步纯化,以提供标题化合物(4%)。
实施例110
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-氟吡啶-2-甲酰胺(ABR 239784)
在氮气下向6-氟吡啶-2-甲酰胺(1.51 g, 10.79 mmol)于DMF(5 mL)中的搅拌溶液中添加吡啶(870 µL, 10.79 mmol)和3,3,3-三氟-2-氧代丙酸甲酯(1.65 mL, 16.19 mmol)。将反应物在室温下搅拌16小时。在0℃下逐滴添加亚硫酰氯(790 µL, 10.79 mmol)。将反应混合物再搅拌2小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。将酰基中间体的溶液添加至5,6-二甲基-1H-1,3-苯并二唑-2-胺(1.45 g, 8.99 mmol)于DMF(10 mL)中的溶液,随后添加三乙胺(1.45 mL, 10.79 mmol)。将反应混合物再搅拌16小时,然后浓缩。将残余物溶解于EtOAc (150 mL)中,用水(2 x 100 mL)、10%柠檬酸(水溶液)(50 mL)和盐水(100 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为白色固体的标题化合物(2.35 g, 61%)。
实施例111
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-[(2-甲氧基乙基)氨基]吡啶-2-甲酰胺(ABR 239496)
向密封管中的N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(8),6,9,11-四烯-3-基]-6-氟吡啶-2-甲酰胺(300 mg, 0.85 mmol)于DMF(7 mL)中的溶液中添加K2CO3 (235 mg, 1.70 mmol)和2-甲氧基乙-1-胺(128 mg, 1.70 mmol)。将反应混合物在90℃下加热4小时。将反应混合物用水稀释并用EtOAc萃取。将合并的有机萃取物用盐水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法B)纯化,以提供作为白色固体的标题化合物(60 mg, 15%)。
实施例112
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-苯基乙酰胺(ABR 239500)
在室温在氩气下向2-苯基乙酰胺(1.20 g, 8.88 mmol)于DMF(15 mL)中的搅拌溶液中添加吡啶(755 µl, 8.88 mmol),随后添加3,3,3-三氟-2-氧代丙酸乙酯(1.51 g, 8.88 mmol)。将反应混合物在室温下搅拌1小时。添加亚硫酰氯(644 µl, 8.88 mmol)。将反应物在室温下再搅拌16小时,然后浓缩。在氩气下将剩余的酰基中间体溶解于DMF(5 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(1.07 g, 6.66 mmol)和三乙胺(1.62 mL, 11.54 mmol),并将反应混合物在室温下搅拌4小时。将反应混合物用水稀释并用EtOAc萃取。将合并的有机萃取物用盐水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂,以提供作为白色固体的标题化合物(225 mg, 3%)。
实施例113
2-(3,5-二氯苯基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]乙酰胺(ABR 239503)
使用用于制备N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氧杂环己烷-4-甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用2-(3,5-二氯苯基)乙酰胺代替氧杂环己烷-4-甲酰胺。在反应的第二阶段中,使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替5,6-二氯-1H-1,3-苯并二唑-2-胺。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂,以提供作为白色固体的标题化合物(4%)。
实施例114
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(3,4,5-三甲氧基苯基)丙酰胺(ABR 239529)
使用用于制备N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氧杂环己烷-4-甲酰胺的一般程序,但具有以下变化。在反应的第一阶段中,为了形成酰基亚胺中间体,使用3-(3,4,5-三甲氧基苯基)丙酰胺代替氧杂环己烷-4-甲酰胺。在反应的第二阶段中,使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替5,6-二氯-1H-1,3-苯并二唑-2-胺。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂,以提供作为白色固体的标题化合物(10%)。
实施例115
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-3-甲磺酰基苯甲酰胺(ABR 239489)
在室温在氩气下向3-甲磺酰基苯甲酰胺(800 mg, 4.02 mmol)于DMF(10 mL)中的搅拌溶液中添加吡啶(342 µl, 4.02 mmol),随后添加3,3,3-三氟-2-氧代丙酸乙酯(532 µl, 4.02 mmol)。将反应混合物在室温下搅拌1小时,然后在0℃下添加亚硫酰氯(292 µl, 4.02 mmol)。将反应物在室温下再搅拌18小时,然后浓缩。在氩气下将剩余的酰基中间体溶解于DMF(10 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(485 mg, 3.01 mmol)和三乙胺(562 µl, 4.02 mmol),并将反应混合物在室温下搅拌16小时。将反应混合物用水稀释并用EtOAc萃取。将合并的有机萃取物用盐水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的3% MeOH作为洗脱剂。通过自动反相HPLC(低pH方法B)实施进一步纯化,以提供作为白色固体的标题化合物(20 mg, 0.5%)。
实施例116
3‐溴‐N‐[10,11‐二甲基‐4‐氧代‐3‐(三氟甲基)‐2,5,7‐三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11‐四烯-3-基]苯甲酰胺(ABR 239785)
在室温在氮气下向3-溴苯甲酰胺(777 mg, 3.88 mmol)于DMF(7 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(395 µl, 3.88 mmol),随后添加吡啶(313 µl, 3.88 mmol)。将反应混合物在室温下搅拌1小时。在0℃下添加亚硫酰氯(282 µl, 3.88 mmol),然后将反应混合物在0℃下搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (7 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(98 mg, 0.57 mmol)于DMF(7 mL)中的溶液和三乙胺(619 µl, 4.65 mmol),并将反应混合物在室温下搅拌18小时。将反应混合物用EtOAc(80 mL)和水(40 mL)稀释。沉淀通过过滤去除并保留。将有机相用水(3 x 40 mL)和盐水(40 mL)洗涤。然后将有机相干燥(Na2SO4),过滤,浓缩并用EtOAc研磨。将所得固体与先前沉淀合并,溶解于EtOAc (10 mL)中,并用10%柠檬酸(水溶液)(10 mL)、水(10 mL)和盐水(10 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩,以提供作为白色固体的标题化合物(120 mg, 8%)。
实施例117
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-3-[(2-甲氧基乙基)氨基]苯甲酰胺(ABR 239638)
将微波管装入3‐溴‐N‐[10,11‐二甲基‐4‐氧代‐3‐(三氟-甲基)‐2,5,7‐三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11‐四烯-3-基]苯甲酰胺(300 mg, 0.64 mmol)、CuI(24 mg, 0.13 mmol)、L-脯氨酸(30 mg, 0.26 mmol)和K3PO4(273 mg, 1.28 mmol)。添加脱气的DMSO (9 mL)和2-甲氧基乙-1-胺(193 mg, 2.57 mmol)。将反应混合物在微波中在80℃下加热2小时,然后用EtOAc(100 mL)和水(80 mL)稀释。将有机层用2M氨水(80 mL)、水(80 mL)和盐水(80 mL)洗涤。然后将有机层干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为灰色固体的标题化合物(60 mg, 20%)。
实施例118
2‐溴‐N‐[10,11‐二甲基‐4‐氧代‐3‐(三氟甲基)‐2,5,7‐三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11‐四烯-3-基]‐1,3‐噻唑‐4‐甲酰胺(ABR 239786)
在室温在氮气下向2-溴-1,3-噻唑-4-甲酰胺(456 mg, 2.20 mmol)于DMF(5 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(431 µl, 4.23 mmol)和吡啶(178 µl, 2.20 mmol)。将反应混合物在室温下搅拌1小时。在0℃下添加亚硫酰溴(171 µl, 2.20 mmol),然后将反应混合物在室温下搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(284 mg, 1.76 mmol)于DMF(10 mL)中的溶液和三乙胺(352 µl, 2.64 mmol),并将反应混合物在室温下搅拌18小时。将反应混合物浓缩。将残余物用EtOAc (50 mL)稀释,并用10% 柠檬酸(水溶液)(15 ml)、水(2 x 20 mL)和盐水(20 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的5-20% MeOH作为洗脱剂,以提供作为白色固体的标题化合物(200 mg, 24%)。
实施例119
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-[(2-甲氧基乙基)氨基]-1,3-噻唑-4-甲酰胺(ABR 239655)
向密封管中的2‐溴‐N‐[10,11‐二甲基‐4‐氧代‐3‐(三氟甲基)‐2,5,7‐三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11‐四烯-3-基]‐1,3‐噻唑‐4‐甲酰胺(263 mg, 0.55 mmol)于DMF(4 mL)中的溶液中添加K2CO3(153 mg, 1.11 mmol)和2-甲氧基乙-1-胺(96 µl, 1.11 mmol)。将反应混合物在90℃下加热24小时,然后浓缩。将残余物用水稀释,并用10% 柠檬酸(水溶液)将pH调节至pH 6。将水相用EtOAc (2 x 20 mL)萃取。将合并的有机萃取物用水(2 x 15 mL)和盐水(15 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(45 mg, 17%)。
实施例120
N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239508)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙酰胺的程序,除了使用4-溴-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。将粗产物通过在DCM中研磨而纯化,以提供标题化合物(17%)。
实施例121
N-[9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239436)
将微波管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(150 mg, 0.33 mmol)、Zn(CN)2(58 mg, 0.49 mmol)、Pd(dppf)Cl2(40 mg, 0.05 mmol)和脱气DMF(3 mL)。将反应混合物在微波中在180℃下加热10分钟。将反应混合物用Zn(CN)2 (58 mg, 0.49 mmol)和Pd(dppf)Cl2 (40 mg, 0.05 mmol)再处理,并在微波中在180℃下再加热10分钟。将反应物过滤。将滤液用EtOAc (15 mL)稀释并用水(5 x 10 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗化合物通过硅胶色谱纯化,使用庚烷中的40% EtOAc作为洗脱剂,以提供作为白色固体的标题化合物(18 mg, 14%)。
实施例122
N-[9-乙酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239448)
在氮气下将微波管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、乙酸钯(II)(1.4 mg, 0.006 mmol)、丙烷-1,3-二基双(二苯基膦(phosphane))(5.7 mg, 0.014 mmol)、K2CO3(36 mg, 0.26 mmol)、1-(乙烯基氧基)丁烷(142 µl, 1.09 mmol)和脱气DMF/水(4:1, 1 mL)。将反应物在微波中在100℃下加热1小时。将反应物用EtOAc (17 ml)稀释,并用水(4 x 5 mL)、盐水(6 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(56 mg, 61%)。
实施例123
N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239521)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙酰胺的程序,除了使用2-氨基-5-氯-1H-1,3-苯并二唑-4-甲腈代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。添加亚硫酰氯之后,将反应物在0℃下搅拌1小时,然后浓缩。从水性后处理沉淀固体,并通过过滤收集,以提供标题化合物(36%)。
实施例124
3-环戊基-N-[9-氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239535)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙酰胺的程序,除了使用4-氟-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。添加亚硫酰氯之后,将反应物在0℃下搅拌1小时,然后浓缩。将粗产物通过在DCM中研磨而纯化,以提供标题化合物(19%)。
实施例125
3-环戊基-N-[9,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239545)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙酰胺的程序,除了使用4,6-二甲基-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。在初始装填之后1小时,在0℃下添加额外当量的亚硫酰氯,然后将反应物在0℃下再搅拌1小时,然后浓缩。将粗产物通过在最小体积的DCM中研磨而纯化,以提供标题化合物(26%)。
实施例126
N-[9-氯-10-氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239559)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙酰胺的程序,除了使用4-氯-5-氟-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。添加亚硫酰氯之后,将反应物在0℃下搅拌1小时,然后浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为白色固体的标题化合物(16%)。
实施例127
N-[10-氯-9-甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239569)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙酰胺的程序,除了使用5-氯-4-甲基-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。在初始装填之后1小时,在0℃下添加额外当量的亚硫酰氯,然后将反应物在0℃下再搅拌1小时,然后浓缩。将粗产物通过在DCM中研磨而纯化,以提供标题化合物(35%)。
实施例128
3-环戊基-N-[4-氧代-3,9-双(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙酰胺(ABR 239544)
在室温在氮气下向3-环戊基丙酰胺(200 mg, 1.42 mmol)于DMF(20 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(442 mg, 2.83 mmol),随后添加吡啶(114 µl, 1.42 mmol)。将反应混合物在室温下搅拌2小时。在0℃下添加亚硫酰氯(103 µl, 1.42 mmol),然后将反应混合物在0℃下搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (15 mL)中。添加4-(三氟甲基)-1H-1,3-苯并二唑-2-胺(228 mg, 1.13 mmol)和三乙胺(188 µl, 1.42 mmol),并将反应混合物在室温下搅拌2小时。将反应混合物浓缩。将残余物用EtOAc (100 mL)稀释,并用水(3 x 100 mL)和盐水(3 x 100 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物在DCM中研磨,以提供作为白色固体的标题化合物(194 mg, 31%)。
实施例129
N-[11-溴-4-氧代-3,9-双(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-3-环戊基丙酰胺(ABR 239577)
向3-环戊基-N-[4-氧代-3,9-双(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]丙酰胺(50 mg, 0.11 mmol)于DCM(5 mL)中的搅拌溶液中添加NBS (30 mg, 0.17 mmol)。将所得悬浮液在室温下搅拌48小时。经随后8天时段添加额外的NBS (60 mg, 0.34 mmol)和DCM (25 mL)。然后将反应混合物用DCM (50 mL)稀释,并用水(3 x 25 mL)和饱和NaHCO3(水溶液)(25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(14 mg, 24%)。
实施例130
N-[10-氯-9-氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239603)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙酰胺的程序,除了使用5-氯-4-氟-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺. 添加亚硫酰氯之后,将反应物在0℃下搅拌1小时,然后浓缩。将粗产物通过在DCM中研磨而纯化,以提供标题化合物(3%)。
实施例131
3-环戊基-N-[9-氟-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239551)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙酰胺的程序,除了使用4-氟-5-甲氧基-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。添加亚硫酰氯之后,将反应物在0℃下搅拌1小时,然后浓缩。在添加4-氟-5-甲氧基-1H-1,3-苯并二唑-2-胺之后2小时进行最后步骤。将粗产物通过在DCM中研磨而纯化,以提供标题化合物(10%)。
实施例132
3-环戊基-N-[9-(甲基硫烷基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239636)
使用用于制备3-环戊基-N-[12-氧代-11-(三氟甲基)-10,13,15-三氮杂四环[7.6.0.03,7.010,14]十五碳-1(9),2,7,14-四烯-11-基]丙酰胺的程序,除了使用4-(甲基硫烷基)-1H-1,3-苯并二唑-2-胺代替1H,5H,6H,7H-茚并[5,6-d]咪唑-2-胺。添加亚硫酰氯之后,将反应物在0℃下搅拌1小时,然后浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的1-8% MeOH作为洗脱剂,以提供作为浅棕色固体的标题化合物(定量收率)。
实施例133
3-环戊基-N-[9-甲磺酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239639)
在0℃下向3-环戊基-N-[9-(甲基硫烷基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(100 mg, 0.23 mmol)于MeOH(2 mL)中的搅拌溶液中逐滴添加过硫酸氢钾制剂(359 mg, 0.59 mmol)于水(2 mL)中的溶液。搅拌4小时之后,将反应物用EtOAc (30 ml)稀释,用水(2 x 10 mL)、然后盐水(10 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩,以得到作为浅棕色固体的标题产物(100 mg, 93%)。
实施例134
N-[9-溴-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239730)
向3-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(300 mg, 0.71 mmol)于DCM(30 mL)中的搅拌溶液中添加NBS (165 mg, 0.93 mmol)。将所得悬浮液在室温下搅拌18小时。添加额外的NBS (165 mg, 0.93 mmol)。将反应物再搅拌4小时,然后浓缩。将残余物溶解于EtOAc (100 mL)中,并用水(2 x 50 mL)、饱和柠檬酸(水溶液)(50 mL)和盐水(50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为白色固体的标题化合物(160 mg, 46%)。
实施例135
N-[9,10-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-氟吡啶-2-甲酰胺(ABR 239538)
在室温在氮气下向6-氟吡啶-2-甲酰胺(140 mg, 1.00 mmol)于DMF(10 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(312 mg, 2.00 mmol),随后添加吡啶(81 µl, 1.00 mmol)。将反应混合物在室温下搅拌16小时。在0℃下添加亚硫酰氯(87 µl, 1.20 mmol),然后将反应混合物在0℃下搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加4,5-二氯-1H-1,3-苯并二唑-2-胺(151 mg, 0.75 mmol)和三乙胺(133 µl, 1.00 mmol)。将反应混合物在室温下再搅拌2小时,然后浓缩。将残余物用EtOAc (100 mL)溶解,并用水(3 x 50 mL)和盐水(3 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(122 mg, 27%)。
实施例136
N-[9,10-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-[(2-甲氧基乙基)氨基]吡啶-2-甲酰胺(ABR 239542)
向密封管中的N-[9,10-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]-十二碳-1(8),6,9,11-四烯-3-基]-6-氟吡啶-2-甲酰胺(80 mg, 0.18 mmol)于DMF(3 mL)中的溶液中添加K2CO3 (62 mg, 0.45 mmol)和2-甲氧基乙-1-胺(27 mg, 0.36 mmol)。将反应混合物在100℃下加热12小时。添加额外的2-甲氧基乙-1-胺(27 mg, 0.36 mmol)和K2CO3(62 mg, 0.45 mmol)。将反应物在100℃下再加热6小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(3 x 25 mL)、然后盐水(2 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗化合物通过自动反相HPLC(高pH)纯化,以提供作为白色固体的标题化合物(20 mg, 22%)。
实施例137
N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-氟吡啶-2-甲酰胺(ABR 239579)
在室温在氮气下向6-氟吡啶-2-甲酰胺(175 mg, 1.25 mmol)于DMF(10 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(390 mg, 2.49 mmol),随后添加吡啶(100 µl, 1.52 mmol)。将反应混合物在室温下搅拌2小时。在0℃下添加亚硫酰氯(91 µl, 1.25 mmol),然后将反应混合物在0℃下搅拌1小时。将反应混合物浓缩,并在惰性气氛下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在惰性气氛下将剩余的酰基中间体溶解于DMF (10 mL)中。添加2-氨基-5-氯-1H-1,3-苯并二唑-4-甲腈(200 mg, 1.04 mmol)于DMF(2 ml)中的溶液和三乙胺(133 µl, 1.00 mmol),并将反应混合物在室温下搅拌4小时。将反应混合物浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(3 x 50 mL)和盐水(3 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂。使用自动反相HPLC(低pH方法A)实施进一步纯化,以提供作为棕色固体的标题化合物(115 mg, 25%)。
实施例138
N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-[(2-甲氧基乙基)氨基]吡啶-2-甲酰胺(ABR 239585)
向密封管中的N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-氟吡啶-2-甲酰胺(75 mg, 0.17 mmol)于DMF(2 mL)中的溶液中添加K2CO3 (71 mg, 0.51 mmol)和2-甲氧基乙-1-胺(39 mg, 0.51 mmol)。最初,将反应混合物在100℃下加热2小时。然后将反应物再加热20小时。在此时段期间,在再处理期间在室温下随着反应分批添加额外的2-甲氧基乙-1-胺(80 mg, 1.02 mmol)。然后将反应混合物浓缩。将残余物溶解于EtOAc (100 mL)中,并用水(3 x 50 mL)、然后盐水(3 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(22 mg, 26%)。
实施例139
N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-[(2-甲氧基乙基)氨基]-1,3-噻唑-4-甲酰胺(ABR 239624)
在氮气下向2-溴-1,3-噻唑-4-甲酰胺(194 mg, 0.93 mmol)于DMF(10 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(291 mg, 1.86 mmol),随后添加吡啶(75 µL, 0.93 mmol)。将反应混合物在室温下搅拌16小时。在0℃下逐滴添加亚硫酰氯(68 µL, 0.93 mmol)。将溶液在0℃下再搅拌1小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加2-氨基-5-氯-1H-1,3-苯并二唑-4-甲腈(150 mg, 0.78 mmol)于DMF(5 mL)中的溶液和三乙胺(124 µl, 0.935 mmol),并将反应混合物在室温下搅拌2小时。将反应物浓缩,并将残余物溶解于EtOAc (50 mL)中,并用10% 柠檬酸(水溶液)(25 mL)、水(2 x 25 mL)、然后盐水(2 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的2-6% MeOH作为洗脱剂,以提供2-溴-N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-1,3-噻唑-4-甲酰胺和2-氯-N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-1,3-噻唑-4-甲酰胺的混合物(84 mg)。
将微波管装入一部分2-溴-N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-1,3-噻唑-4-甲酰胺和2-氯-N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-1,3-噻唑-4-甲酰胺的混合物(70 mg)、K2CO3(57 mg, 0.42 mmol)、2-甲氧基乙-1-胺(36 µl, 0.42 mmol)和二氧杂环己烷(2 mL)。将反应物在微波中在130℃下加热1小时。添加额外的2-甲氧基乙-1-胺(36 µl, 0.42 mmol)和K2CO3(57 mg, 0.42 mmol)。将反应物再加热1小时,然后浓缩。将残余物溶解于EtOAc (25 mL)中,并用水(25 mL)、10%柠檬酸(水溶液)(25 mL)和盐水(25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(3 mg)。
实施例140
非对映异构体混合物。N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂-三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-{[(2S)-1-甲氧基丙-2-基]氨基}-吡啶-2-甲酰胺(ABR 239586)
向密封管中的N-[10-氯-9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-氟吡啶-2-甲酰胺(75 mg, 0.55 mmol)于DMF(2 mL)中的溶液中添加K2CO3(185 mg, 1.34 mmol)和(2S)-1-甲氧基丙-2-胺(49 mg, 0.55 mmol)。最初,将反应混合物在100℃下加热6小时。然后将反应物再加热22小时。在此时段期间,在再处理期间在室温下随着反应分批添加额外的K2CO3(150 mg, 1.10 mmol)和(2S)-1-甲氧基丙-2-胺(98 mg, 1.10 mmol)。然后将反应混合物浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(3 x 25 mL)、然后盐水(3 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体且作为非对映异构体的混合物的标题化合物(14 mg, 15%)。
实施例141
N-[9,10-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-氟吡啶-2-甲酰胺(ABR 239787)
在室温在氮气下向6-氟吡啶-2-甲酰胺(190 mg, 1.36 mmol)于DMF(2 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(138 µl, 1.36 mmol),随后添加吡啶(109 µl, 1.36 mmol)。将反应混合物在室温下搅拌1小时。在0℃下添加亚硫酰氯(99 µl, 1.36 mmol),然后将反应混合物在室温下搅拌1.5小时。将反应混合物浓缩,并在惰性气氛下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。添加4,5-二氟-1H-1,3-苯并二唑-2-胺(153 mg, 0.90 mmol)于DMF(4 mL)中的溶液和三乙胺(181 µl, 1.36 mmol),并将反应混合物在室温下搅拌18小时。将反应混合物用EtOAc (30 mL)稀释,用水(4 x 20 mL)和盐水(20 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为白色固体的标题化合物(123 mg, 33%)。
实施例142
N-[9,10-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-[(2-甲氧基乙基)氨基]吡啶-2-甲酰胺(ABR 239632)
向密封管中的N-[9,10-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-6-氟吡啶-2-甲酰胺(123 mg, 0.30 mmol)于DMF(3 mL)中的溶液中添加K2CO3(102 mg, 0.74 mmol)和2-甲氧基乙-1-胺(45 mg, 0.59 mmol)。将反应混合物在100℃下加热18小时。将反应物用EtOAc (50 mL)稀释,并用水(3 x 25 mL)、然后盐水(3 x 25 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗化合物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(36 mg, 26%)。
实施例143
非对映异构体 1 :(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-苯基丙酰胺(ABR 239609)。
实施例144
非对映异构体 2:(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-苯基丙酰胺(ABR 239621)
在室温在氮气下向(2S)-2-苯基丙酰胺(222 mg, 1.49 mmol)于DMF(10 mL)中的搅拌溶液中添加吡啶(120 µl, 1.49 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(465 mg, 2.98 mmol)。将反应混合物在室温下搅拌2小时。在0℃下添加亚硫酰氯(108 µl, 1.49 mmol),然后将反应混合物在0℃下搅拌1小时。然后将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(200 mg, 1.24 mmol)于DMF(5 mL)中的溶液和三乙胺(198 µl, 1.49 mmol),并将反应混合物在室温下搅拌3小时。然后将反应混合物浓缩。将残余物用EtOAc (50 mL)稀释,用水(2 x 25 mL)、10%柠檬酸(水溶液)(2 x 25 mL)和盐水(2 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供非对映异构体的纯净混合物。EtOAc/庚烷中的重结晶提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体1 (95 mg, 18%)。将母液浓缩并通过自动反相HPLC(低pH方法A)纯化,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体2 (33 mg, 6%)。
实施例145
非对映异构体混合物。(2S)-3-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-甲基丙酰胺(ABR 239666)
(a) 非对映异构体 1:(2S)-3-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-甲基丙酰胺(ABR 239667)
(b) 非对映异构体 2:(2S)-3-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-甲基丙酰胺(ABR 239668)
在室温在氮气下向(2S)-3-环戊基-2-甲基丙酰胺(254 mg, 1.64 mmol)于DMF(15 mL)中的搅拌溶液中添加吡啶(132 µL, 1.64 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(511 mg, 3.28 mmol)。将反应混合物在室温下搅拌2小时。在0℃下添加亚硫酰氯(118 µl, 1.64 mmol),并将反应物在0℃下搅拌1小时。然后将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(220 mg, 1.36 mmol)于DMF(10 mL)中的溶液和三乙胺(229 µl, 1.64 mmol)。将反应混合物在室温下搅拌2小时,然后浓缩。将残余物用EtOAc (50 mL)稀释,并用水(3 x 50 mL)、然后盐水(3 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为黄色固体且作为非对映异构体的纯净混合物的标题化合物。将一部分保留为非对映异构体混合物(61 mg, 11%),并将剩余材料通过使用Chiralpak AD-H柱与CO2和含有0.1%甲酸的MeOH的流动相的SFC纯化,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体1 (26 mg, 5%)和作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体2 (40 mg, 7%)。
实施例146
(a) 非对映异构体 1:(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-羟基-2-苯基乙酰胺(ABR 239664)
(b) 非对映异构体 2:(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-羟基-2-苯基乙酰胺(ABR 239665)
向(S)-{[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]氨基甲酰基}(苯基)乙酸甲酯(500 mg, 1.09 mmol)于MeOH(20 mL)中的搅拌溶液中添加2M NaOH(水溶液)(597 µl, 1.20 mmol)。将反应混合物在40℃下加热3小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用10% 柠檬酸(水溶液)(2 x 30 mL)、然后盐水(2 x 30 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过反相C18色谱纯化,使用酸性洗脱剂,以得到非对映异构体的纯净混合物。将非对映异构体的混合物通过自动反相HPLC(低pH方法A)分离,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体1 (38 mg, 8%)和作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体2 (80 mg, 18%)。
实施例147
(a) 非对映异构体 1:(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-甲氧基-2-苯基乙酰胺(ABR 239622)
(b) 非对映异构体 2:(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]-2-甲氧基-2-苯基乙酰胺(ABR 239623)
在室温在氮气下向(2S)-2-甲氧基-2-苯基乙酰胺(184 mg, 1.12 mmol)于DMF(10 mL)中的搅拌溶液中添加吡啶(90 µl, 1.12 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(349 mg, 2.23 mmol)。将反应混合物在室温下搅拌2小时。在0℃下添加亚硫酰氯(81 µl, 1.12 mmol),并将反应物在0℃下搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(150 mg, 0.93 mmol)于DMF(5 mL)中的溶液和三乙胺(149 µl, 1.12 mmol)。将反应混合物在室温下搅拌2小时,然后浓缩。将残余物用EtOAc (50 mL)稀释,并用水(2 x 25 mL)、10%柠檬酸(水溶液)(2 x 25 mL)和盐水(2 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以得到非对映异构体的纯净混合物。将非对映异构体的混合物通过自动反相HPLC(低pH方法A)分离,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体1 (50 mg, 12%)和作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体2 (27 mg, 7%)。
实施例148
3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸甲酯(ABR 239934)
在氮气下向3-环戊基丙酰胺(2.22 g, 15.7 mmol)于无水DCM(50 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(2.40 mL, 23.5 mmol),随后添加吡啶(1.27 mL, 15.7 mmol)。将反应物在室温下搅拌2小时。在0℃下添加亚硫酰氯(1.14 mL, 15.69 mmol)。将反应物在0℃下搅拌1小时,然后在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (25 mL)中。将酰基中间体的溶液添加至2-氨基-1H-1,3-苯并二唑-4-甲酸甲酯(经由文献方法: PCT国际申请2008157270可得)(2.50 g, 13.1 mmol)于DMF(25 mL)中的溶液,随后添加三乙胺(5.22 mL, 39.2 mmol)。将反应混合物再搅拌18小时,然后浓缩。将残余物溶解于EtOAc (100 mL)中,并用饱和柠檬酸(水溶液)(30 mL)洗涤。将水相用EtOAc (100 mL)萃取。将合并的有机萃取物用盐水(2 x 15 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂,以提供作为绿色固体的标题化合物(990 mg, 17%)。
实施例149
3-环戊基-N-[9-碘-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 240060)
在氮气下向3-环戊基丙酰胺(748 mg, 5.29 mmol)于无水DCM(30 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(0.65 mL, 6.35 mmol),随后添加吡啶(0.43 mL, 5.29 mmol)。将反应混合物在室温下搅拌16小时。在0℃下添加亚硫酰氯(0.38 mL, 5.29 mmol)。将反应物在0℃下搅拌2小时,然后在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。将酰基中间体的溶液添加至4-碘-1H-1,3-苯并二唑-2-胺(1.10 g, 4.23 mmol)于DMF(10 mL)中的溶液,随后添加三乙胺(0.70 mL, 5.29 mmol)。将反应混合物再搅拌4小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用10%柠檬酸(水溶液)(50 mL)洗涤。通过过滤去除所得沉淀。将水层用EtOAc (25 mL)萃取。将合并的有机萃取物用盐水(2 x 50 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为白色固体的标题化合物(798 mg, 27%)。
实施例150
N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-甲氧基吡啶-3-甲酰胺(ABR 240061)
在氮气下向2-甲氧基吡啶-3-甲酰胺(经由文献方法: PCT国际申请2010101949可得)(300 mg, 1.97 mmol)于DMF(5 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(0.24 mL, 2.37 mmol),随后添加吡啶(0.16 mL, 1.97 mmol)。将反应物在室温下搅拌16小时。在0℃下添加亚硫酰氯(0.14 mL, 1.97 mmol)。将反应物在0℃下搅拌1小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (6 mL)中。将酰基中间体的溶液添加至4-溴-1H-1,3-苯并二唑-2-胺(334 mg, 1.58 mmol)于DMF(5 mL)中的溶液,随后添加三乙胺(0.26 mL, 1.97 mmol)。将反应混合物再搅拌2小时,然后浓缩。将残余物溶解于EtOAc (30 mL)中,并用饱和柠檬酸(水溶液)(15 mL)洗涤。将水层用EtOAc (2 x 15 mL)萃取。将合并的有机萃取物用水(2 x 20 mL)和盐水(2 x 20 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为白色固体的标题化合物(334 mg, 44%)。
实施例151
N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-羟基吡啶-3-甲酰胺(ABR 240062)
在0℃在氮气下向N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-甲氧基吡啶-3-甲酰胺(334 mg, 0.7 mmol)于DCM(5 mL)中的搅拌悬浮液中添加DCM中的1M三溴硼烷(2.09 mL, 2.09 mmol)。将反应物温热至室温并搅拌4小时。将反应物冷却至0℃,并添加额外的DCM中的1M三溴硼烷(2.09 mL, 2.09 mmol)。将反应物温热至室温并搅拌4小时。在0℃下用水(15 mL)淬灭反应物。将所得沉淀通过过滤收集,并用水、DCM、EtOAc和水冲洗,以得提供作为白色固体的标题化合物(208 mg, 65%)。
实施例152
(2S)-N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-环己基丙酰胺(ABR 240063)
在氮气下向(2S)-2-环己基丙酰胺(280 mg, 1.64 mmol)于DMF(5 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(251 µL, 2.46 mmol),随后添加吡啶(132 µL, 1.64 mmol)。将反应物在室温下搅拌1小时。在0℃下添加亚硫酰氯(119 µL, 1.64 mmol)。将反应物在0℃下搅拌1小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (4 mL)中。将酰基中间体的溶液添加至4-溴-1H-1,3-苯并二唑-2-胺(290 mg, 1.37 mmol)于DMF(2 mL)中的溶液,随后添加三乙胺(204 µL, 1.53 mmol)。将反应物搅拌16小时,然后浓缩。将残余物溶解于EtOAc (15 mL)中,并用饱和柠檬酸(水溶液)(15 mL)洗涤。将水层用EtOAc (15 mL)萃取。将合并的有机萃取物用盐水(2 x 15 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为白色固体的标题化合物(236 mg, 35%)。
实施例153
(2S)-2-(环戊-1-烯-1-基甲氧基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 240068)
在氮气下向酰胺(2S)-2-(环戊-1-烯-1-基甲氧基)丙酰胺(346 mg, 2.05 mmol)于无水DCM(5 mL)中的溶液中添加吡啶(165 µL, 2.05 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(639 mg, 4.09 mmol)。将反应物在室温下搅拌18小时。在0℃下添加亚硫酰氯(149 µL, 2.05 mmol)。将反应物在0℃下搅拌1小时。在氮气下将反应物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (3 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(275 mg, 1.71 mmol)于DMF (2 mL)中的溶液,随后添加三乙胺(286 µL, 2.05 mmol)。将反应物搅拌18小时,然后浓缩。将残余物溶解于EtOAc (30 mL)中,并用水(4 x 20 mL)和盐水(20 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩,以提供作为橙色固体的标题化合物(750 mg, 定量收率)。
实施例154
N-[11-氯-9-碘-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 240064)
在氮气下向N-[9-溴-11-氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(250 mg, 0.50 mmol)于二氧杂环己烷(4 mL)中的搅拌溶液中添加碘化钾(250 mg, 1.5 mmol)、碘化铜(I)(38 mg, 0.20 mmol)和N,N'-二甲基乙烷-1,2-二胺(43 µL, 0.4 mmol)。最初,将反应物在150℃下加热2小时。然后,将温度降低至120℃,持续2小时,并进一步降低至100℃,最终持续2小时。然后将反应物浓缩,并将残余物用EtOAc (15 mL)和饱和柠檬酸(水溶液)(25 mL)稀释。将水层用EtOAc (15 mL)萃取。将合并的有机萃取物用水(15 mL)和盐水(30 mL)洗涤,然后干燥(MgSO4),过滤并浓缩,以提供作为橙色固体的标题化合物(239 mg, 70%)。
实施例155
N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丁酰胺(ABR 240065)
在氮气下向丁酰胺(205 mg, 2.36 mmol)于无水DCM(6 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(0.24 mL, 2.4 mmol),随后添加吡啶(0.18 mL, 2.3 mmol)。将反应物在室温下搅拌1小时。在0℃下添加亚硫酰氯(0.16 mL, 2.3 mmol)。将反应物在0℃下搅拌1小时,然后在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (3 mL)中。将酰基中间体的溶液添加至4-溴-1H-1,3-苯并二唑-2-胺(400 mg, 1.89 mmol)于DMF(5 mL)中的溶液,随后添加三乙胺(0.30 mL, 2.3 mmol)。将反应混合物再搅拌1小时,然后浓缩。将残余物用EtOAc(60 mL)和1M HCl(水溶液)(40 mL)稀释。将有机相用水(2 x 20 mL)和盐水(20 mL)洗涤,然后干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-15% MeOH作为洗脱剂,以提供作为黄色油的标题化合物(650 mg, 76%)。
实施例156
N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(环戊基氨基)-1,3-噻唑-4-甲酰胺(ABR 240066)
在氮气下向2-溴-1,3-噻唑-4-甲酰胺(2.93 g, 14.2 mmol)于DMF(80 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(4.42 g, 28.3 mmol),随后添加吡啶(1.14 mL, 14.2 mmol)。将反应物在室温下搅拌16小时。添加额外的3,3,3-三氟-2-氧代丙酸甲酯(4.42 g, 28.3 mmol),并将反应物再搅拌4小时。在0℃下添加亚硫酰氯(1.03 mL, 14.2 mmol)。将反应物在0℃下搅拌2小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。将酰基中间体的溶液添加至4-溴-1H-1,3-苯并二唑-2-胺(2.50 g, 11.8 mmol)于DMF(40 mL)中的溶液,随后添加三乙胺(1.88 mL, 14.2 mmol)。将反应混合物再搅拌2小时,然后浓缩。将残余物溶解于EtOAc (200 mL)中,用10%柠檬酸(水溶液)(2 x 100 mL)、水(2 x 100 mL)和盐水(2 x 100 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供2-溴-N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺和N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-氯-1,3-噻唑-4-甲酰胺的混合物(3.70 g)。
将密封管装入一部分2-溴-N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺和N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-氯-1,3-噻唑-4-甲酰胺的混合物(500 mg)、环戊胺(0.39 mL, 4.0 mmol)、K2CO3(546 mg, 3.95 mmol)和二氧杂环己烷(5 mL)。将该管用氮气吹扫,密封并在130℃下搅拌20小时。添加额外的环戊胺(0.16 mL, 1.6 mmol)和K2CO3(219 mg, 1.58 mmol),然后将反应物在130℃下搅拌4小时并在120℃下搅拌16小时。然后将反应混合物浓缩,并将所得残余物用EtOAc(30 mL)和水(30 mL)稀释。将水相用EtOAc (30 mL)萃取。将合并的有机萃取物用水(2 x 25 mL)和盐水(50 mL)洗涤,然后干燥(Na2SO4),过滤并浓缩。将粗产物通过反相C18色谱纯化,使用酸性洗脱剂,以提供作为橙色固体的标题化合物(150 mg, 33 %)。
实施例157
3-环戊基-N-[9-碘-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 240067)
将密封管装入N-[9-溴-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(405 mg, 0.83 mmol)、碘化钾(412 mg, 2.48 mmol)、碘化铜(I)(63 mg, 0.33 mmol)、N,N'-二甲基乙烷-1,2-二胺(71 µl, 0.66 mmol)和无水二氧杂环己烷(4 mL)。将该管用氮气吹扫,密封并在120 ℃下搅拌16小时,然后在100℃下搅拌4小时。然后将反应混合物浓缩。将所得残余物用EtOAc (20 mL)和饱和柠檬酸(水溶液)(20 mL)稀释。将水相用EtOAc (15 mL)萃取。将合并的有机萃取物用水(2 x 15 mL)洗涤,干燥(MgSO4),过滤并浓缩,以提供作为绿色固体的标题化合物(350 mg, 51%)。
实施例158
3-环戊基-N-[9-(羟基甲基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239922)
在0℃在氮气下向3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸甲酯(480 mg, 1.09 mmol)于无水THF(5 mL)中的搅拌溶液中添加THF中的1M四氢铝酸锂(4.38 mL, 4.38 mmol)。将反应物经2小时温热至室温。然后将反应混合物用二乙醚(20 mL)稀释。在0℃下缓慢添加水,随后添加15% NaOH(水溶液)。将反应混合物搅拌1小时。将沉淀通过过滤收集,然后溶解于EtOAc (30 mL)中,并使用2M HCl(水溶液)酸化至pH2。将有机相用盐水(10 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以得到作为黄色固体的标题化合物(180 mg, 40%)。
实施例159
N-[9-丁基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239938)
向密封管中的N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(300 mg, 0.65 mmol)、2-甲基丙-1-烯-1-基乙酸酯(149 mg, 1.31 mmol)和三丁基(甲氧基)锡烷(375 µL, 1.31 mmol)于脱气DMSO(1 mL)中的溶液中添加二氯双(三邻甲苯基膦)钯(II)(16 mg, 33 µmol)。将该管用氮气吹扫并密封。将反应混合物在100℃下搅拌16小时。将反应混合物用EtOAc (5 mL)稀释,并添加4M KF(水溶液)(1 mL)。搅拌1小时之后,将反应混合物过滤通过Celite™,并用EtOAc冲洗。将有机相用水(2 x 5 mL)和盐水(5 mL)洗涤,然后干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(30 mg, 11%)。
实施例160
3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸(ABR 239943)
向3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸甲酯(500 mg, 1.14 mmol)于MeOH/H2O(1:1, 12 mL)中的搅拌溶液中添加LiOH.H2O (144 mg, 3.42 mmol)。将反应混合物在60℃下搅拌16小时,然后浓缩。将残余物溶解于水中,并用1M HCl(水溶液)酸化至pH1。将所得沉淀通过过滤收集,用水和己烷洗涤,以提供作为棕色固体的标题化合物(422 mg, 87%)。
实施例161
3-环戊基-N-{9-[1-(甲氧基亚氨基)乙基]-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基}丙酰胺(ABR 239950)
向N-[9-乙酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(70%, 175 mg, 0.29 mmol)和吡啶(82 µL, 1.0 mmol)于EtOH(2 mL)中的搅拌溶液中添加O-甲基羟胺盐酸盐(92 mg, 1.1 mmol)。将反应物在80℃下加热1小时,然后浓缩。将残余物溶解于EtOAc (15 mL)中,并用水(15 mL)洗涤。将水层用EtOAc (2 x 10 mL)萃取。将合并的有机萃取物用盐水(20 mL)洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(61 mg, 47%)。
实施例162
3-(3-环戊基丙酰胺基)-N-甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酰胺(ABR 239951)
将密封管装入丙基膦酸酐于EtOAc中的50%溶液(90 mg, 0.14 mmol)、MeCN(1.2 mL)、三乙胺(66 µL, 0.47 mmol)和3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸(50 mg, 0.12 mmol)。将反应物在室温下搅拌30分钟,然后添加甲胺盐酸盐(8 mg, 0.1 mmol)。将反应混合物再搅拌60小时。将反应混合物用EtOAc稀释。将有机相用1M HCl(水溶液)和盐水洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,随后通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂,以提供作为棕色固体的标题化合物(15 mg, 28%)。
实施例163
3-环戊基-N-[9-乙炔基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239986)
在0℃在氮气下向3-环戊基-N-[4-氧代-3-(三氟甲基)-9-{2-[三(丙-2-基)甲硅烷基]乙炔基}-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(100 mg, 0.18 mmol)于THF(1 mL)中的搅拌溶液中添加THF中的1M TBAF(232 µL, 0.23 mmol)。将反应物在室温下搅拌10分钟。将反应物冷却至0℃,并用THF中的1M TBAF(232 µL, 0.23 mmol)再处理。将反应物在室温下搅拌1小时,然后浓缩。将残余物溶解于DCM (10 mL)中,用水(5 mL)和盐水(5 mL)洗涤,然后干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-2% MeOH作为洗脱剂,随后通过自动反相HPLC(低pH方法A)纯化,以提供作为棕色固体的标题化合物(22 mg, 31%)。
实施例164
3-环戊基-N-[4-氧代-9-(三氟甲氧基)-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239991)
在室温在氮气下向3-环戊基丙酰胺(98 mg, 0.69 mmol)于无水DCM(10 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(141 µL, 1.38 mmol),随后添加吡啶(56 µL, 0.69 mmol)。将反应混合物在室温下搅拌2小时。在0℃下添加亚硫酰氯(50 µL, 0.69 mmol),然后将反应混合物在0℃下搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。将酰基中间体的溶液添加至4-(三氟甲氧基)-1H-1,3-苯并二唑-2-胺(125 mg, 0.58 mmol)于DMF(10 mL)中的溶液,随后添加三乙胺(230 µL, 1.73 mmol)。将反应混合物在室温下搅拌18小时,然后浓缩。将残余物用EtOAc (50 mL)稀释,并用饱和柠檬酸(水溶液)(30 mL)洗涤。将水相用EtOAc (50 mL)萃取。将合并的有机萃取物用盐水(2 x 15 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物在DCM中研磨。一部分该材料进行随后通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(50 mg)。
实施例165
3-环戊基-N-[9-环丙烷亚磺酰氨基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239995)
向密封管中的3-环戊基-N-[9-碘-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(150 mg, 0.30 mmol)于2-甲基四氢呋喃(3 mL)中的溶液中添加环丙基磺酰胺(43 mg, 0.36 mmol),随后添加K2CO3(82 mg, 0.59 mmol)。将所得悬浮液用氮气脱气,然后添加二-叔丁基[2',4',6'-三(丙-2-基)联苯-2-基]膦(3 mg, 0.01 mmol),随后添加双(氯(丙-2-烯-1-基)钯)(7 mg, 0.02 mmol)。将反应物用氮气脱气,密封并在80℃下搅拌3小时。添加额外的二-叔丁基[2',4',6'-三(丙-2-基)联苯-2-基]膦(3 mg, 0.01 mmol)和双(氯(丙-2-烯-1-基)钯)(7 mg, 0.02 mmol)。将反应混合物脱气,并在80℃下再搅拌16小时。然后将反应物浓缩。将残余物溶解于EtOAc (30 mL)中,并用1M HCl(水溶液)(30 mL)、水(2 x 30 mL)和盐水(2 x 30 mL)洗涤。将合并的水洗液用EtOAc (30 mL)萃取。将合并的有机萃取物用盐水(30 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(24 mg, 16%)。
实施例166
3-(3-环戊基丙酰胺基)-N-甲磺酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酰胺(ABR 239996)
在0℃在氮气下,向甲磺酰胺(114 mg, 1.2 mmol)于DMF(10 mL)中的搅拌溶液中添加氢化钠(60%, 48 mg, 1.2 mmol)。在0℃下搅拌15分钟之后,添加3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸苯酯(150 mg, 0.3 mmol)于DMF(2 mL)中的溶液。将反应混合物温热至室温,然后在50℃下加热。1小时之后,将反应混合物冷却至0℃,并用饱和NH4Cl(水溶液)淬灭。将反应混合物浓缩。将残余物溶解于EtOAc (50 mL)中,用水(3 x 25 mL)和盐水(2 x 25 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(19 mg, 13%)。
实施例167
3-环戊基-N-[9-甲烷亚磺酰氨基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239999)
将N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(200 mg, 0.44 mmol)、甲磺酰胺(50 mg, 0.52 mmol)、二-叔丁基[2',4',6'-三(丙-2-基)联苯-2-基]膦(2 mg, 4 μmol)、双(氯(丙-2-烯-1-基)钯)(5 mg, 13 μmol)和K2CO3(120 mg, 0.87 mmol)于脱气2-甲基四氢呋喃(3 mL)中的搅拌悬浮液在80℃下加热5小时。将反应物用甲磺酰胺(50 mg, 0.52 mmol)、二-叔丁基[2',4',6'-三(丙-2-基)联苯-2-基]膦(2 mg, 4 μmol)和双(氯(丙-2-烯-1-基)钯)(5 mg, 13 μmol)再处理,脱气并在80℃下加热18小时。将反应物用EtOAc (15 mL)稀释,并用1M HCl(水溶液)(15 mL)洗涤。将水相用EtOAc (2 x 15 mL)萃取。将合并的有机萃取物用盐水(30 mL)洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(22 mg, 11%)。
实施例168
N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(2-甲氧基乙基)氨基]-1,3-噻唑-4-甲酰胺(ABR 240004)
在氮气下向2-溴-1,3-噻唑-4-甲酰胺(2.93 g, 14.2 mmol)于DMF(80 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(4.42 g, 28.3 mmol),随后添加吡啶(1.14 mL, 14.2 mmol)。将反应物在室温下搅拌16小时。添加额外的3,3,3-三氟-2-氧代丙酸甲酯(4.42 g, 28.3 mmol),并将反应物再搅拌4小时。在0℃下添加亚硫酰氯(1.03 mL, 14.2 mmol)。将反应物在0℃下搅拌2小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。将酰基中间体的溶液添加至4-溴-1H-1,3-苯并二唑-2-胺(2.50 g, 11.8 mmol)于DMF(40 mL)中的溶液,随后添加三乙胺(1.88 mL, 14.2 mmol)。将反应混合物再搅拌2小时,然后浓缩。将残余物溶解于EtOAc (200 mL)中,用10%柠檬酸(水溶液)(2 x 100 mL)、水(2 x 100 mL)和盐水(2 x 100 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供2-溴-N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺和N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-氯-1,3-噻唑-4-甲酰胺的混合物(3.70 g)。
将密封管装入一部分2-溴-N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺和N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-氯-1,3-噻唑-4-甲酰胺的混合物(0.40 g)、K2CO3(0.53 g, 3.8 mmol)、2-甲氧基乙-1-胺(327 µL, 3.81 mmol)和二氧杂环己烷(5 mL)。最初,将反应物在130℃下加热3小时。然后将反应物在130℃下再加热9小时。在此时段期间,在再处理期间在室温下随着反应分批添加额外的2-甲氧基乙-1-胺(982 µL, 11.4 mmol)和K2CO3(1.58 g, 11.4 mmol)。然后将反应混合物在130℃下加热16小时,未经进一步再处理,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用饱和NH4Cl(水溶液)(50 mL)、水(50 mL)和盐水(50 mL)洗涤。将合并的水洗液用EtOAc (25 mL)萃取,并将这些有机萃取物用盐水(50 mL)洗涤。将合并的有机萃取物干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体的标题化合物(130 mg)。
实施例169
N-[9-乙酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(2-甲氧基乙基)氨基]-1,3-噻唑-4-甲酰胺(ABR 240005)
在氮气下将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(2-甲氧基乙基)氨基]-1,3-噻唑-4-甲酰胺(110 mg, 0.21 mmol), 乙酸钯(II)(2.4 mg, 0.01 mmol)、丙烷-1,3-二基双(二苯基膦)(8.7 mg, 0.02 mmol)、K2CO3(41 mg, 0.30 mmol)、1-(乙烯基氧基)丁烷(138 µL, 1.06 mmol)和脱气DMF/水(4:1, 1.3 mL)。将反应物在100℃下搅拌3小时,然后浓缩。将残余物溶解于THF (20 mL)中,并添加1M HCl(水溶液)(5 mL)。将反应物在室温下搅拌1小时,然后用EtOAc (25 mL)稀释并用水(25 mL)洗涤。将有机相用1M HCl(水溶液)(25 mL)、水(25 mL)和盐水(25 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(47 mg, 46%)。
实施例170
3-(3,3-二氟吡咯烷-1-基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239907)
向密封管中的N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙-2-烯酰胺(125 mg, 0.37 mmol)于MeCN(10 mL)中的溶液中添加3,3-二氟吡咯烷盐酸盐(69 mg, 0.48 mmol)、二氧化硅(18 mg)和三乙胺(98 µL, 0.74 mmol)。将反应物在80℃下加热3小时。通过过滤去除二氧化硅,并将滤液浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(42 mg, 26%)。
实施例171
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(3-氟氮杂环丁烷-1-基)丙酰胺(ABR 239909)
使用用于制备3-(3,3-二氟吡咯烷-1-基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺的程序,除了使用3-氟氮杂环丁烷盐酸盐代替3,3-二氟吡咯烷盐酸盐(61%)。
实施例172
3-(3,3-二氟哌啶-1-基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239911)
使用用于制备3-(3,3-二氟吡咯烷-1-基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺的程序,除了使用3,3-二氟哌啶盐酸盐代替3,3-二氟吡咯烷盐酸盐(13%)。
实施例173
N-[9-乙酰基-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239928)
在氮气下将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(500 mg, 1.03 mmol)、乙酸钯(II)(7 mg, 0.03 mmol)、丙烷-1,3-二基双(二苯基膦)(27 mg, 0.06 mmol)、K2CO3(184 mg, 1.33 mmol)、1-(乙烯基氧基)丁烷(667 µL, 5.13 mmol)和脱气DMF/水(4:1, 4 mL)。将反应物在100℃下搅拌3小时。添加额外的乙酸钯(II)(7 mg, 0.03 mmol)、丙烷-1,3-二基双(二苯基膦)(27 mg, 0.06 mmol)和1-(乙烯基氧基)丁烷(667 µL, 5.13 mmol)。将反应物在100℃下再加热13小时,然后浓缩。将残余物溶解于THF (25 mL)中,并添加1M HCl(水溶液)(10 mL)。将反应物在室温下搅拌3小时,然后用EtOAc (100 mL)稀释。将水相用EtOAc (2 x 50 mL)萃取。将合并的有机萃取物用盐水(50 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-5% MeOH作为洗脱剂,随后在DCM中研磨,以提供作为棕色固体的标题化合物(160 mg, 35%)。
实施例174
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-甲氧基吡啶-3-甲酰胺(ABR 239987)
在氮气下向2-甲氧基吡啶-3-甲酰胺(经由文献方法: PCT国际申请2010101949可得)(300 mg, 1.97 mmol)于DMF(5 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(0.24 mL, 2.4 mmol),随后添加吡啶(0.16 mL, 2.0 mmol)。将反应物在室温下搅拌16小时。在0℃下添加亚硫酰氯(0.14 mL, 2.0 mmol)。将反应物在0℃下搅拌1小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。将酰基中间体的溶液添加至5,6-二甲基-1H-苯并咪唑-2-胺(254 mg, 1.58 mmol)于DMF (5 mL)中的溶液,随后添加三乙胺(0.26 mL, 2.0 mmol)。将反应混合物再搅拌4小时,然后浓缩。将残余物溶解于EtOAc (30 mL)中,用饱和柠檬酸(水溶液)(30 mL)洗涤。将水相用EtOAc (2 x 20 mL)萃取。将合并的有机萃取物用水(2 x 20 mL)和盐水(2 x 20 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过在二乙醚中研磨而纯化,以提供作为粉红色固体的标题化合物(215 mg, 30%)。
实施例175
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-羟基吡啶-3-甲酰胺(ABR 239989)
在0℃在氮气下向N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-甲氧基吡啶-3-甲酰胺(235 mg, 0.52 mmol)于DCM(5 mL)中的搅拌溶液中添加DCM中的1M三溴硼烷(1.54 mL, 1.54 mmol)。将反应混合物温热至室温并搅拌18小时。然后将反应物在室温下再搅拌8小时。在此时段期间,在再处理期间在0℃下随着反应分批添加额外的DCM中的1M 三溴硼烷(3.08 mL, 3.08 mmol)。然后将反应混合物冷却至0℃,并添加水(10 mL)和2M HCl(水溶液)(10 mL)。将所得悬浮液在室温下搅拌16小时。将沉淀通过过滤收集,并用DCM(10 mL)和水(10 mL)洗涤。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(80 mg, 42%)。
实施例176
N-[11-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239914)
向3-环戊基-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(2.5 g, 6.6 mmol)于DCM(250 mL)中的溶液中添加NBS (1.4 g, 7.9 mmol)。将反应混合物在室温下搅拌5小时,然后浓缩。将残余物溶解于EtOAc (250 mL)中,用水(2 x 250 mL)和盐水(2 x 150 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂,随后通过自动反相HPLC(低pH方法A),以提供作为黄色固体的标题化合物(1.2 g, 40%)。
实施例177
N-[10-溴-9-氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239927)
在氮气下向3-环戊基丙酰胺(368 mg, 2.61 mmol)于DMF(10 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(0.40 mL, 3.9 mmol),随后添加吡啶(0.21 mL, 2.6 mmol)。将反应物在室温下搅拌1.5小时。在0℃下添加亚硫酰氯(0.19 mL, 2.6 mmol),然后将反应混合物在0℃下搅拌1.5小时。将反应混合物浓缩并在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。将酰基中间体的溶液添加至5-溴-4-氟-1H-1,3-苯并二唑-2-胺(500 mg, 2.17 mmol)于DMF(10 mL)中的溶液,随后添加三乙胺(0.35 mL, 2.6 mmol)。将反应混合物搅拌2小时,然后浓缩。将残余物溶解于EtOAc (20 mL)中,用10%柠檬酸(水溶液) (20 mL)洗涤。将水层用EtOAc (2 x 20 mL)萃取。将合并的有机萃取物用10% 柠檬酸(水溶液)(2 x 20 mL)和盐水(2 x 20 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物在DCM中研磨,以得到作为棕色固体的标题化合物(401 mg, 39%)。
实施例178
N-[9-溴-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239933)
在氮气下向3-环戊基丙酰胺(0.44 g, 3.1 mmol)于无水DCM(17 mL)中的溶液中添加吡啶(250 µL, 3.10 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(967 mg, 6.20 mmol)。将反应物在室温下搅拌2小时。在0℃下添加亚硫酰氯(245 µL, 3.10 mmol)。将反应混合物在0℃下搅拌1小时,然后在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (14 mL)中。添加4-溴-5-甲氧基-1H-1,3-苯并二唑-2-胺(625 mg, 2.6 mmol)于DMF(3 mL)中的溶液,随后添加三乙胺(432 µL, 3.1 mmol)。将反应物搅拌16小时,然后浓缩。将残余物溶解于EtOAc中,并用盐水(4 x)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为黄色固体的标题化合物(0.76 g, 60%)。
实施例179
3-环戊基-N-[9-氟-10-(1-羟基乙基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239937)
在氮气下将密封管装入N-[10-溴-9-氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(156 mg, 0.30 mmol)、乙酸钯(II)(2 mg, 0.01 mmol)、丙烷-1,3-二基双(二苯基膦)(8 mg, 0.02 mmol)、K2CO3(54 mg, 0.39 mmol)、1-(乙烯基氧基)丁烷(196 µl, 1.51 mmol)和脱气DMF/水(3:1, 4 mL)。将反应物在100℃下搅拌3小时。添加额外的乙酸钯(II)(2 mg, 0.01 mmol)、丙烷-1,3-二基双(二苯基膦)(8 mg, 0.02 mmol)、K2CO3(27 mg, 0.2 mmol)和1-(乙烯基氧基)丁烷(78 µl, 0.6 mmol)。将反应物脱气并在100℃下再搅拌16小时。将反应物浓缩。将所得残余物溶解于EtOAc (10 mL)中,并用盐水(10 mL)洗涤。将水相用EtOAc (2 x 15 mL)萃取。将合并的有机萃取物用盐水(2 x 15 mL)洗涤,干燥(MgSO4),过滤并浓缩,以提供作为棕色固体的N-[10-乙酰基-9-氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(152 mg)。
在0℃在氮气下向N-[10-乙酰基-9-氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(25%, 152 mg, 0.09 mmol)于无水MeOH(3 mL)中的搅拌溶液中添加四氢硼酸钠(33 mg, 0.86 mmol)。将所得溶液经16小时温热至室温。在0℃下添加额外的四氢硼酸钠(49 mg, 1.3 mmol)。将反应物经4小时温热至室温,然后浓缩。将残余物溶解于EtOAc (10 mL)中,并用盐水(10 mL)洗涤。将水相用EtOAc (2 x 10 mL)萃取。将合并的有机萃取物用盐水(2 x 10 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-50% MeOH作为洗脱剂,随后通过自动反相HPLC(高pH)纯化,以提供作为白色固体的标题化合物(8 mg, 19%)。
实施例180
N-[9-乙酰基-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239946)
在氮气下将密封管装入N-[9-溴-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(2.00 g, 0.40 mmol)、Pd(PPh3)2Cl2(29 mg, 0.04 mmol)、三丁基(1-乙氧基乙烯基)锡烷(275 µL, 0.80 mmol)和甲苯(2 mL)。最初,将反应物在110℃下搅拌16小时。然后将反应物再加热30小时。在此时段期间,在再处理期间在室温下随着反应分批添加额外的Pd(PPh3)2Cl2 (44 mg, 0.06 mmol)。将反应混合物用2M HCl(水溶液)(2 mL)稀释,并在室温下搅拌1小时。添加EtOAc,并将所得悬浮液过滤通过Celite™。将滤液的有机相用盐水洗涤,干燥(MgSO4),过滤并浓缩。最初将粗产物通过自动反相HPLC(低pH方法A)纯化。随后通过硅胶色谱纯化,使用DCM中的0-50% DCM/MeOH/AcOH/H2O (90:18:3:2)作为洗脱剂,然后在冷DCM中研磨,以提供作为白色固体的标题化合物(33 mg, 18%)。
实施例181
N-[9-溴-11-氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239916)
在氮气下向3-环戊基丙酰胺(0.61 g, 4.29 mmol)于无水DCM(24 mL)中的搅拌溶液中添加吡啶(346 µL, 4.29 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(1.34 g, 8.58 mmol)。将反应混合物搅拌2小时。在0℃下添加亚硫酰氯(311 µL, 4.29 mmol),然后将反应混合物在0℃下搅拌1小时。在氮气下将反应混合物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (20 mL)中。添加4-溴-6-氯-1H-1,3-苯并二唑-2-胺(881 mg, 3.57 mmol)于DMF(4 mL)中的溶液,随后添加三乙胺(343 µL, 2.46 mmol)。将反应混合物在室温下搅拌16小时,然后浓缩。将残余物用EtOAc稀释并用盐水(4 x)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物用DCM研磨,以提供作为黄色固体的标题化合物(884 mg, 50%)。
实施例182
N-[9-乙酰基-11-氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239936)
使用用于制备N-[9-乙酰基-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺的程序,除了使用N-[9-溴-11-氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺代替N-[9-溴-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺。不必要再处理。将粗产物通过自动反相HPLC(低pH方法A)纯化(23%)。
实施例183
N-[11-溴-9-氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239926)
在氮气下向3-环戊基丙酰胺(1.01 g, 7.17 mmol)于DMF(20 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(878 µL, 8.61 mmol),随后添加吡啶(578 µL, 7.17 mmol)。将反应物在室温下搅拌16小时。在0℃下添加亚硫酰氯(521 µL, 7.17 mmol),然后将反应混合物在0℃下搅拌1小时。将反应混合物浓缩并在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (20 mL)中。添加6-溴-4-氟-1H-1,3-苯并二唑-2-胺(1.32 g, 5.74 mmol)于DMF(20 mL)中的溶液,随后添加三乙胺(1.15 mL, 8.61 mmol)。将反应混合物在室温下搅拌2小时,然后浓缩。将残余物溶解于EtOAc (200 mL)中,用水(3 x 100 mL)和盐水(100 mL)洗涤,然后干燥(Na2SO4),过滤并浓缩。将粗产物在DCM中研磨,以得到作为白色固体的标题化合物(1.20 g, 44%)。
实施例184
N-[11-乙酰基-4-氧代-3,9-双(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239942)
在氮气下将密封管装入N-[11-溴-4-氧代-3,9-双(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(150 mg, 0.28 mmol)、乙酸钯(II)(2 mg, 8.5 µmol), 丙烷-1,3-二基双(二苯基膦)(8 mg, 0.02 mmol)、K2CO3(51 mg, 0.37 mmol)、1-(乙烯基氧基)丁烷(185 µL, 1.42 mmol)和脱气DMF/水(3:1, 4 mL)。最初,将反应物在100℃下搅拌4小时。然后将反应物再加热4小时。在此时段期间,在再处理期间在室温下随着反应分批添加额外的乙酸钯(II)(2 mg, 8.5 µmol)、丙烷-1,3-二基双(二苯基膦)(8 mg, 0.02 mmol)和1-(乙烯基氧基)丁烷(185 µL, 1.42 mmol)。将反应物冷却至室温,然后浓缩。将残余物溶解于THF (15 mL)中,并添加1M HCl(水溶液)(4 mL)。将反应物在室温下搅拌20分钟,然后浓缩。将残余物溶解于EtOAc (10 mL)中,并用盐水(10 mL)洗涤。将水相用EtOAc (15 mL)萃取。将合并的有机萃取物用盐水(15 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过反相C18色谱纯化,使用酸性洗脱剂。通过在DCM中研磨和随后的自动反相HPLC(高pH)进一步纯化,以提供作为白色固体的标题化合物(29 mg, 21%)。
实施例185
N-[11-氯-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺和N-[10-氯-11-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239941)
在氮气下向3-环戊基丙酰胺(128 mg, 0.90 mmol)于DCM(1.5 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(138 µL, 1.35 mmol),随后添加吡啶(73 µL, 0.9 mmol)。将反应物在室温下搅拌1小时。在0℃下添加亚硫酰氯(66 µL, 0.90 mmol)。将反应物在0℃下搅拌1小时,然后在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (2 mL)中。将酰基中间体的溶液添加至6-氯-5-甲氧基-1H-1,3-苯并二唑-2-胺(150 mg, 0.75 mmol)于DMF(2 mL)中的溶液,随后添加三乙胺(120 µL, 0.90 mmol)。将反应混合物再搅拌4小时,然后浓缩。将残余物溶解于EtOAc (15 mL)中,并用饱和柠檬酸(水溶液)(15 mL)洗涤。将水相用EtOAc (15 mL)萃取。将合并的有机萃取物用盐水(2 x 15 mL)洗涤,然后干燥(MgSO4),过滤并浓缩,以提供作为区域异构体的1:1混合物且作为棕色固体的标题化合物(52 mg, 15%)。
实施例186
3-环戊基-N-[9-(4-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239731)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(50 mg, 0.11 mmol)、(4-甲氧基苯基)硼酸(21 mg, 0.14 mmol)、Na2CO3(21 mg, 0.2 mmol)、水(0.4 mL)、DME(1.4 mL)和EtOH(0.6 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (7 mg, 0.01 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌6小时。将反应混合物用EtOAc (10 mL)稀释并用水(5 mL)洗涤。将水相用EtOAc (3 x 5 mL)萃取。将合并的有机萃取物用盐水(2 x 15 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(18 mg, 34%)。
实施例187
3-环戊基-N-[9-(1-甲基-1H-吡唑-4-基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239732)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、(1-甲基-1H-吡唑-4-基)硼酸(39 mg, 0.3 mmol)、Na2CO3(42 mg, 0.39 mmol)、水(0.8 mL)、DME(2.8 mL)和EtOH(1.2 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (25 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌2小时。将反应混合物用EtOAc (10 mL)稀释并用水(10 mL)洗涤。将水相用EtOAc (2 x 10 mL)萃取。将合并的有机萃取物用盐水(2 x 10 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(42 mg, 41%)。
实施例188
3-环戊基-N-[9-(6-甲氧基吡啶-3-基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239944)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(160 mg, 0.15 mmol)、(6-甲氧基吡啶-3-基)硼酸(66 mg, 0.44 mmol)、Na2CO3(66 mg, 0.69 mmol)、水(1.8 mL)、DME(6.0 mL)和EtOH(2.5 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (40 mg, 0.04 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌2小时,然后浓缩。将残余物溶解于EtOAc (100 mL)中,并用水(50 mL)、饱和柠檬酸(水溶液)(50 mL)和盐水(2 x 10 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,随后通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(100 mg, 59%)。
实施例189
3-环戊基-N-[9-(3-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239948)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、(3-甲氧基苯基)硼酸(46 mg, 0.3 mmol)、Na2CO3(46 mg, 0.44 mmol)、水(0.8 mL)、DME(2.8 mL)和EtOH(1.2 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (13 mg, 0.01 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌16小时。将反应混合物浓缩。将残余物溶解于EtOAc (50 mL)中,用水(25 mL)、1M HCl(水溶液)(25 mL)和盐水(25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体的标题化合物(44 mg, 42%)。
实施例190
3-环戊基-N-[9-(1,3-噁唑-2-基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239953)
向N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(200 mg, 0.44 mmol)、叔丁醇钠(84 mg, 0.87 mmol)和Pd(PPh3)4(25 mg, 0.02 mmol)于脱气二氧杂环己烷(3 mL)中的悬浮液中添加1,3-噁唑(43 µL, 0.65 mmol)。将反应物用氮气吹扫,密封并在100℃下加热4小时。将反应物用叔丁醇钠(84 mg, 0.87 mmol)、Pd(PPh3)4(25 mg, 0.02 mmol)和1,3-噁唑(43 µL, 0.65 mmol)再处理,脱气并在100℃下再加热3.5小时。将反应物用EtOAc (30 mL)稀释,并用水(15 mL)和盐水(15 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(83 mg, 43%)。
实施例191
N-[9-(4-叔丁基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239954)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(50 mg, 0.11 mmol)、(4-叔丁基苯基)硼酸(24 mg, 0.14 mmol)、Na2CO3(21 mg, 0.20 mmol)、水(0.90 mL)、DME(3.00 mL)和EtOH(1.25 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (13 mg, 0.01 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌18小时。将反应混合物过滤通过Celite™,用MeOH (30 mL)冲洗,并将滤液浓缩。将残余物溶解于EtOAc (20 mL)中,用水(30 mL)、饱和柠檬酸(水溶液)(30 mL)和盐水(30 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用庚烷中的0-100% EtOAc作为洗脱剂,以提供作为白色固体的标题化合物(39 mg, 70%)。
实施例192
N-[9-(4-氰基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239956)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(50 mg, 0.11 mmol)、(4-氰基苯基)硼酸(21 mg, 0.14 mmol)、Na2CO3(21 mg, 0.20 mmol)、水(0.90 mL)、DME(3.00 mL)和EtOH(1.25 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (13 mg, 0.01 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌18小时。将反应混合物过滤通过Celite™,用MeOH (30 mL)冲洗,并将滤液浓缩。将残余物溶解于EtOAc (20 mL)中,用水(30 mL)、饱和柠檬酸(水溶液)(30 mL)和盐水(30 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用庚烷中的0-100% EtOAc作为洗脱剂,以提供作为棕色固体的标题化合物(21 mg, 40%)。
实施例193
3-环戊基-N-[9-(4-硝基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239957)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、(4-硝基苯基)硼酸(46 mg, 0.27 mmol)、Na2CO3(42 mg, 0.39 mmol)、水(0.90 mL)、DME(3.00 mL)和EtOH(1.25 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (25 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌18小时。将反应混合物过滤通过Celite™,用MeOH (30 mL)冲洗,并将滤液浓缩。将残余物溶解于EtOAc (20 mL)中,用水(30 mL)、饱和柠檬酸(水溶液)(30 mL)和盐水(30 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用庚烷中的0-100% EtOAc作为洗脱剂,以提供作为橙色固体的标题化合物(70 mg, 64%)。
实施例194
N-[9-(4-氨基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239965)
向3-环戊基-N-[9-(4-硝基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(50 mg, 0.10 mmol)于EtOAc(5 mL)中的搅拌溶液中添加氯化锡(II)二水合物(86 mg, 0.38 mmol)。将反应物在70℃下加热18小时。将反应物冷却至室温,用EtOAc (10 mL)稀释,并过滤通过Celite™,用EtOAc (30 mL)冲洗。将滤液浓缩。将粗产物装载至酸性SCX-2柱上,用DCM中的50% MeOH洗涤,然后用MeOH中的3.5M氨洗脱。随后通过自动反相HPLC(低pH方法A)纯化,提供作为棕色固体的标题化合物(8 mg, 17%)。
实施例195
3-环戊基-N-[4-氧代-9-苯基-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239967)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(150 mg, 0.32 mmol)、苯基硼酸(55 mg, 0.45 mmol)、Na2CO3(68 mg, 0.64 mmol)、水(0.7 mL)、DME(1.4 mL)和EtOH(0.6 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (37 mg, 0.03 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌3小时。添加额外的苯基硼酸(27 mg, 0.22 mmol)、Na2CO3(34 mg, 0.32 mmol)和Pd(PPh3)4(18 mg, 0.02 mmol),并将反应物在100℃下再搅拌16小时。将反应混合物浓缩。将残余物溶解于EtOAc (25 mL)中,并用盐水(25 mL)洗涤。将水层用EtOAc (25 mL)萃取。将合并的有机萃取物用盐水(2 x 50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为橙色固体的标题化合物(47 mg, 32%)。
实施例196
3-环戊基-N-[9-(2-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239970)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、(2-甲氧基苯基)硼酸(46 mg, 0.3 mmol)、Na2CO3(46 mg, 0.44 mmol)、水(0.8 mL)、DME(2.8 mL)和EtOH(1.2 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (13 mg, 0.01 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌16小时。将反应混合物浓缩。将残余物溶解于EtOAc (50 mL)中,用水(25 mL)、1M HCl(水溶液)(25 mL)和盐水(25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(80 mg, 75%)。
实施例197
2-羟基-N-[9-(4-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]吡啶-3-甲酰胺(ABR 239988)
将密封管装入N-[9-(4-溴苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-羟基吡啶-3-甲酰胺(100 mg, 0.22 mmol)、(4-甲氧基苯基)硼酸(47 mg, 0.31 mmol)、Na2CO3(46 mg, 0.44 mmol)、水(0.2 mL)、DME(2.8 mL)和EtOH(1.2 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (13 mg, 0.01 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌4小时。添加额外的(4-甲氧基苯基)硼酸(23 mg, 0.15 mmol)、Na2CO3(46 mg, 0.44 mmol)和Pd(PPh3)4(13 mg, 0.01 mmol),并将反应物在100℃下再搅拌16小时。将反应混合物浓缩。将残余物溶解于EtOAc (25 mL)中,并用水(25 mL)洗涤。将水相用EtOAc (2 x 20 mL)和IPA/CHCl3 (1:1, 5 x 20 mL)萃取。将合并的有机萃取物干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(高pH)纯化,以提供作为白色固体的标题化合物(24 mg, 22%)。
实施例198
3-环戊基-N-[9-(2-羟基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239978)
在氮气下向3-环戊基-N-[9-(2-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(55 mg, 0.11 mmol)于DCM(10 mL)中的搅拌溶液中添加DCM中的1M三溴化硼(0.34 mL, 0.34 mmol)。最初,将反应物在室温下搅拌1小时。然后将反应物再搅拌4小时。在此时段期间,分批添加额外的DCM中的1M三溴化硼(1.02 mL, 1.02 mmol)。然后将反应物在0℃下用水淬灭。将有机相用1M HCl(水溶液)(25 mL)和水(25 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(29 mg, 54%)。
实施例199
3-环戊基-N-[4-氧代-9-(1H-1,2,3,4-四唑-5-基)-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239990)
在氮气下向密封管中的N-[9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(500 mg, 1.23 mmol)和二丁基(氧代)锡烷(353 µL, 1.23 mmol)于二甲苯(10 mL)中的搅拌悬浮液中添加叠氮基(三甲基)硅烷(490 µL, 3.70 mmol)。将反应物在130℃下加热1.5小时。将反应物在室温下用MeOH (10 mL)稀释。将反应物搅拌2小时,然后浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-20% MeOH作为洗脱剂,以提供作为米色固体的标题化合物(142 mg, 25%)。
实施例200
3-[3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-基]苯甲酸(ABR 239992)
向3-[3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-基]苯甲酸甲酯(48 mg, 0.09 mmol)于MeOH(3 mL)中的搅拌溶液中添加2M NaOH(水溶液)(0.28 mL, 0.56 mmol)。将反应混合物在50℃下搅拌4小时,然后浓缩。将残余物溶解于水中,并使用1M HCl(水溶液)酸化至pH2。通过过滤收集所得沉淀。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体的标题化合物(14 mg, 30%)。
实施例201
N-[9-(4-氯苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 240000)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、(4-氯苯基)硼酸(43 mg, 0.27 mmol)、Na2CO3(42 mg, 0.39 mmol)、水(0.20 mL)、DME(1.00 mL)和EtOH(0.35 mL),并将溶液用氮气脱气。添加Pd(PPh3)4 (25 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌18小时。将反应混合物过滤通过Celite™,用MeOH (30 mL)冲洗,并将滤液浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体的标题化合物(60 mg, 56%)。
实施例202
3-环戊基-N-[9-(4-甲基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 240001)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、(4-甲基苯基)硼酸(37 mg, 0.27 mmol)、Na2CO3(42 mg, 0.39 mmol)、水(0.20 mL)、DME (1.00 mL)和EtOH (0.35 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (25 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌18小时。将反应混合物过滤通过Celite™,用MeOH (30 mL)冲洗,并将滤液浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体的标题化合物(50 mg, 49%)。
实施例203
3-环戊基-N-[9-(3,4-二氯苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 240002)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、(3,4-二氯苯基)硼酸(52 mg, 0.27 mmol)、Na2CO3(42 mg, 0.39 mmol)、水(0.20 mL)、DME(1.00 mL)和EtOH(0.35 mL),并将溶液用氮气脱气。添加Pd(PPh3)4 (25 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌18小时。将反应混合物过滤通过Celite™,用MeOH (30 mL)冲洗,并将滤液浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(56 mg, 49%)。
实施例204
3-环戊基-N-[9-(3,4-二氢-2H-吡喃-5-基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239733)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(50 mg, 0.11 mmol)、4-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷(borolan)-2-基)-3,6-二氢-2H-吡喃(30 mg, 0.14 mmol)、Na2CO3 (21 mg, 0.20 mmol)、水(0.4 mL)、DME (1.4 mL)和EtOH (0.6 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (13 mg, 0.01 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌18小时。将反应混合物用EtOAc (10 mL)稀释并用水(10 mL)洗涤。将水相用EtOAc (2 x 10 mL)萃取。将合并的有机萃取物用盐水(2 x10 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为棕色固体的标题化合物(10 mg, 20%)。
实施例205
3-环戊基-N-[10,11-二甲基-9-(1-甲基-1H-吡唑-4-基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239729)
将密封管装入N-[9-溴-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(75 mg, 0.15 mmol)、(1-甲基-1H-吡唑-4-基)硼酸(24 mg, 0.19 mmol)、Na2CO3(29 mg, 0.28 mmol)、水(0.8 mL)、DME(2.8 mL)和EtOH(1.2 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (18 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌2小时。将反应混合物浓缩。将残余物溶解于EtOAc (100 mL)中,用水(50 mL)、饱和柠檬酸(水溶液)(50 mL)和盐水(50 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(35 mg, 47%)。
实施例206
3-环戊基-N-[10,11-二甲基-4-氧代-9-(1,2-噻唑-4-基)-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239738)
将密封管装入N-[9-溴-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(75 mg, 0.15 mmol)、4-(四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1,2-噻唑(42 mg, 0.20 mmol)、Na2CO3(29 mg, 0.28 mmol)、水(0.8 mL)、DME(2.8 mL)和EtOH(1.2 mL)。将溶液用氮气脱气。添加Pd(PPh3)4(18 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌2小时。将反应混合物浓缩。将残余物溶解于EtOAc (100 mL)中,用水(50 mL)、饱和柠檬酸(水溶液)(50 mL)和盐水(50 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(29 mg, 38%)。
实施例207
3-环戊基-N-[9-(呋喃-3-基)-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239739)
将密封管装入N-[9-溴-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(75 mg, 0.15 mmol)、(呋喃-3-基)硼酸(22 mg, 0.2 mmol)、Na2CO3(29 mg, 0.28 mmol)、水(0.8 mL)、DME(2.8 mL)和EtOH(1.2 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (18 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌2小时。将反应混合物浓缩。将残余物溶解于EtOAc (100 mL)中,用水(50 mL)、饱和柠檬酸(水溶液)(50 mL)和盐水(50 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(18 mg, 25%)。
实施例208
N-[9-(4-甲氧基苯基)-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]乙酰胺(ABR 239929)
将密封管装入N-[9-溴-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]乙酰胺(100 mg, 0.25 mmol)、(4-甲氧基苯基)硼酸(47 mg, 0.31 mmol)、Na2CO3(47 mg, 0.44 mmol)、水(0.9 mL)、DME(3.0 mL)和EtOH(1.3 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (29 mg, 0.03 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌2小时。将反应混合物浓缩。将残余物溶解于EtOAc (100 mL)中,用水(50 mL)、饱和柠檬酸(水溶液)(50 mL)和盐水(50 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(28 mg, 26%)。
实施例209
3-环戊基-N-[9-氟-10-(4-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239924)
将密封管装入N-[10-溴-9-氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(194 mg, 0.39 mmol)、(4-甲氧基苯基)硼酸(90 mg, 0.59 mmol)、Na2CO3(84 mg, 0.79 mmol)、水(0.40 mL)、DME(1.50 mL)和EtOH(0.50 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (46 mg, 0.04 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌16小时。添加额外的(4-甲氧基苯基)硼酸(45 mg, 0.30 mmol)、Na2CO3(42 mg, 0.38 mmol)和Pd(PPh3)4(23 mg, 0.02 mmol)。将溶液用氮气脱气,并将管密封。将反应物在100℃下搅拌3小时。将反应混合物浓缩。将残余物溶解于EtOAc (10 mL)中,并用水(10 mL)洗涤。将水相用EtOAc (2 x 10 mL)萃取。将合并的有机萃取物用水(15 mL)和盐水(2 x15 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-5% MeOH作为洗脱剂,随后在MeOH中研磨,以提供作为奶油色固体的标题化合物(11 mg, 5%)。
实施例210
3-环戊基-N-[11-(4-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239923)
将密封管装入N-[11-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(200 mg, 0.44 mmol)、(4-甲氧基苯基)硼酸(83 mg, 0.54 mmol)、Na2CO3(83 mg, 0.78 mmol)、水(1.80 mL)、DME(6.0 mL)和EtOH(1.50 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (50 mg, 0.04 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌3小时。将反应混合物浓缩。将残余物溶解于EtOAc (100 mL)中,用水(50 mL)、饱和柠檬酸(水溶液)(50 mL)、水(50 mL)和盐水(50 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为米色固体的标题化合物(74 mg, 35%)。
实施例211
(a) 非对映异构体 1:(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(3-甲基丁氧基)丙酰胺(ABR 239949)
(b) 非对映异构体 2:(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(3-甲基丁氧基)丙酰胺(ABR 239952)
将装入(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺(250 mg, 0.589 mmol)于乙醇(10 mL)中的溶液的烧瓶抽空并用氮气(x 3)吹扫。将10% Pd/C (35 mg, 0.33 mmol)添加至搅拌溶液。将烧瓶抽空并用氮气(x 3)吹扫,然后抽空并用氢气(x 3)吹扫。搅拌20小时之后,将反应物用10% Pd/C (10 mg, 0.09 mmol)再处理,并在氢气气氛下再搅拌2小时。然后将反应物过滤通过Celite™,用EtOH冲洗并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-5% MeOH作为洗脱剂,随后通过自动反相HPLC(低pH方法A)纯化,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体1 (34 mg, 14%)和作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体2 (29 mg, 11%)。
(c) 非对映异构体 3:(2R)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(3-甲基丁氧基)丙酰胺(ABR 239958)
(d) 非对映异构体 4:(2R)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(3-甲基丁氧基)丙酰胺(ABR 239959)
使用用于制备(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(3-甲基丁氧基)丙酰胺的程序,除了使用(2R)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺代替(2S)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(3-甲基丁-2-烯-1-基)氧基]丙酰胺。纯化提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体3 (8%)和作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体4 (6%)。
实施例212
(a) 非对映异构体 1:(2R)-2-(3-氯苯氧基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239968)
(b) 非对映异构体 2:(2R)-2-(3-氯苯氧基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239969)
在氮气下向(2R)-2-(3-氯苯氧基)丙酰胺(0.22 g, 1.1 mmol)于DMF(5 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(0.16 mL, 1.6 mmol),随后添加吡啶(0.09 mL, 1.1 mmol)。将反应物在室温下搅拌2小时。在0℃下添加亚硫酰氯(0.08 mL, 1.1 mmol)。将反应物在0℃下搅拌1小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。将酰基中间体的溶液添加至5,6-二甲基-1H-1,3-苯并二唑-2-胺(0.14 g, 0.87 mmol)于DMF(5 mL)中的溶液,随后添加三乙胺(0.17 mL, 1.3 mmol)。将反应混合物再搅拌3小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,用10%柠檬酸(水溶液)(2 x 25 mL)、水(3 x 25 mL)和盐水(3 x 25 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的1-3% MeOH作为洗脱剂,随后通过自动反相HPLC(低pH方法A)纯化,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体1 (33 mg, 8%)和作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体2 (32 mg, 6%)。
实施例213
(2S)-N-[9-乙酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-环己基丙酰胺(ABR 239985)
在氮气下将密封管装入(2S)-N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-环己基丙酰胺(230 mg, 0.47 mmol)、乙酸钯(II)(11 mg, 47 µmol)、丙烷-1,3-二基双(二苯基膦)(39 mg, 0.09 mmol)、K2CO3(85 mg, 0.61 mmol)、1-(乙烯基氧基)丁烷(307 µL, 2.36 mmol)和脱气DMF/水(3:1, 4 mL)。将反应物在100℃下搅拌16小时,然后浓缩。将残余物溶解于EtOAc (10 mL)中,并用盐水(10 mL)洗涤。将水相用EtOAc (15 mL)萃取。将合并的有机萃取物用盐水(2 x 15 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的标题化合物(34 mg, 16%)。
实施例214
(2S)-3-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-羟基丙酰胺(ABR 239994)
向(2S)-2-(乙酰基氧基)-3-环戊基丙酸(经由文献方法: PCT国际申请2000059502可得)(1.00 g, 5.40 mmol)于DCM(15 mL)中的搅拌溶液中添加草酰氯(732 mg, 5.77 mmol),随后添加DMF(2滴)。将反应物搅拌30分钟,然后浓缩。将所得残余物溶解于DCM (3 mL)中,并添加氨水(0.5 mL)。将反应物浓缩,并将残余物在DCM (10 mL)中搅拌并过滤。将滤液浓缩,以提供(1S)-1-氨基甲酰基-2-环戊基乙酸乙酯(838 mg)。
在氮气下向(1S)-1-氨基甲酰基-2-环戊基乙酸乙酯(408 mg, 2.05 mmol)于DMF (4 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(0.34 mL, 3.4 mmol),随后添加吡啶(0.18 mL, 2.2 mmol)。将反应物在室温下搅拌1小时。在0℃下添加亚硫酰氯(0.16 mL, 2.2 mmol)。将反应物在0℃下搅拌1小时,然后在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (3 mL)中。将酰基中间体的溶液添加至5,6-二甲基-1H-1,3-苯并二唑-2-胺(300 mg, 1.86 mmol)于DMF(5 mL)中的溶液,随后添加三乙胺(0.30 mL, 2.2 mmol)。将反应混合物再搅拌1小时,然后浓缩。将残余物溶解于MeOH(10 mL)和水(5 mL)中。添加2M NaOH(水溶液)(5 mL),并将所得溶液在室温下搅拌1小时。将反应物浓缩,并将残余物溶解于EtOAc (60 mL)中。将有机相用水(3 x 20 mL)和盐水(2 x 20 mL)洗涤,然后干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂,随后通过自动反相HPLC(高pH)纯化,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的标题化合物(28 mg, 4%)。
实施例215
(a) 非对映异构体 1:(2S)-2-(环戊基甲氧基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239997)
(b) 非对映异构体 2:(2S)-2-(环戊基甲氧基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239998)
将装入(2S)-2-(环戊-1-烯-1-基甲氧基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(750 mg, 1.72 mmol)和EtOH(10 mL)的溶液的烧瓶抽空并用氮气(x 3)吹扫。然后将二氧代铂(dioxoplatinum)(39 mg, 0.17 mmol)添加至搅拌溶液。将烧瓶抽空并用氮气(x 3)吹扫,然后抽空并用氢气(x 3)吹扫。搅拌18小时之后,将反应物过滤通过Celite™,用EtOH冲洗并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体1 (260 mg, 35%)和作为四价中心的立体化学未定义的单一非对映异构体且作为白色固体的非对映异构体2 (197 mg, 26%)。
实施例216
(a) 对映异构体1: 3-环戊基-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238851)
(b) 对映异构体2: 3-环戊基-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238856)
通过SFC使用Chiralpak AD-H柱以23% 乙醇/77% CO2作为洗脱剂拆分(resolve)3-环戊基-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(40 mg, 0.11 mmol)。对映异构体1被分离为白色固体(15 mg, 38%)。对映异构体2被分离为白色固体(13 mg, 33%)。
实施例217
(a) 对映异构体1: 3-环己基-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238924)
(b) 对映异构体2: 3-环己基-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 238941)
通过SFC使用Chiralpak AD-H柱以23% EtOH/77% CO2作为流动相拆分3-环己基-N-[4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(20 mg, 0.05 mmol)。对映异构体1被分离为棕色固体(4 mg, 21%)。对映异构体2被分离为白色固体(5 mg, 24%)。
实施例218
(a) 对映异构体1: 3-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239071)
(b) 对映异构体2: 3-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239105)
通过SFC使用Chiralpak AD-H柱以15% EtOH/85% CO2作为流动相拆分3-环戊基-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(200 mg, 0.49 mmol)。对映异构体1被分离为白色固体(77 mg, 39%)。对映异构体2经历通过自动反相HPLC(低pH方法A)进一步纯化,以得到作为白色固体的标题化合物(67 mg, 34%)。
实施例219
(a) 对映异构体1: 3-环戊基-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239396)
(b) 对映异构体2: 3-环戊基-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239397)
通过SFC使用Chiralpak AD-H柱以7% MeOH/93% CO2作为流动相拆分3-环戊基-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(120 mg, 0.27 mmol)。对映异构体1被分离为白色固体(46 mg, 38%)。对映异构体2被分离为白色固体(46 mg, 38%)。
实施例220
(a) 对映异构体1: N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-[(2-甲氧基乙基)氨基]吡啶-2-甲酰胺(ABR 239662)
(b) 对映异构体2: N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-[(2-甲氧基乙基)氨基]吡啶-2-甲酰胺(ABR 239663)
通过SFC使用Chiralpak IC柱以12% MeOH/88% CO2作为流动相拆分N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-6-[(2-甲氧基乙基)氨基]吡啶-2-甲酰胺(100 mg, 0.22 mmol)。对映异构体1被分离为白色固体(40 mg, 40%)。对映异构体2被分离为白色固体(46 mg, 40%)。
实施例221
(a) 对映异构体1: N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(1-羟基环戊基)丙酰胺(ABR 239707)
(b) 对映异构体2: N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(1-羟基环戊基)丙酰胺(ABR 239714)
通过SFC使用ChiralPak AS柱以80% MeOH/20% CO2作为流动相拆分N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-(1-羟基环戊基)丙酰胺(125 mg, 0.29 mmol)。对映异构体1被分离为白色固体(44 mg, 35%)。对映异构体2经历通过自动反相HPLC(低pH方法A)进一步纯化,以得到作为白色固体的标题化合物(6 mg, 4%)。
实施例222
(a) 对映异构体1: 3-环戊基-N-[9-甲磺酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239709)
(b) 对映异构体2: 3-环戊基-N-[9-甲磺酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239710)
通过SFC使用ChiralPak AS柱以80% MeOH/20% CO2作为流动相拆分3-环戊基-N-[9-甲磺酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(110 mg, 0.23 mmol)。对映异构体1被分离为米色固体(26 mg, 24%)。对映异构体2被分离为米色固体(34 mg, 32%)。
实施例223
(a) 对映异构体1: 3-环戊基-N-[9,10-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239712)
(b) 对映异构体2: 3-环戊基-N-[9,10-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239713)
通过SFC使用ChiralPak AD柱以70% MeOH/30% CO2作为流动相拆分3-环戊基-N-[9,10-二氟-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(110 mg, 0.26 mmol)。对映异构体1被分离为白色固体(53 mg, 48%)。对映异构体2被分离为白色固体(53 mg, 48%)。
实施例224a
(a) 对映异构体1: N-[9-乙酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239727)
(b) 对映异构体2: N-[9-乙酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239728)
通过SFC使用ChiralPak IC柱以30% IPA/70% CO2作为流动相拆分N-[9-乙酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(110 mg, 0.26 mmol)。对映异构体1被分离为白色固体(40 mg, 36%)。对映异构体2被分离为白色固体(41 mg, 37%)。
实施例225a
(a)对映异构体1: 3-(3,3-二氟氮杂环丁烷-1-基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239825)
(b) 对映异构体2: 3-(3,3-二氟氮杂环丁烷-1-基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239824)
通过SFC使用ChiralPak IC柱以20% IPA/80% CO2作为流动相拆分3-(3,3-二氟氮杂环丁烷-1-基)-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(116 mg, 0.27 mmol)。对映异构体1被分离为白色固体(35 mg, 30%)。对映异构体2被分离为白色固体(37 mg, 32%)。
实施例226
(a) 对映异构体1: N-[9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239962)
(b) 对映异构体2: N-[9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 239961)
通过SFC使用Amy-C柱以70% IPA/30% CO2作为流动相拆分N-[9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.25 mmol)。对映异构体1被分离为白色固体(32 mg, 32%)。将对映异构体2溶解于EtOAc (2 mL)中,并用0.5M HCl(水溶液)(1 mL)、水(1 mL)和盐水(1 mL)洗涤。将有机相浓缩,以得到作为白色固体的标题产物(19 mg, 19%)。
实施例227
(a) 对映异构体1: 3-环戊基-N-[9-(2-甲基丙酰基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239975)
(b) 对映异构体2: 3-环戊基-N-[9-(2-甲基丙酰基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239972)
在0℃在氮气下向N-[9-氰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(177 mg, 0.44 mmol)于THF(5 mL)中的搅拌溶液中逐滴添加THF中的2M 氯(丙-2-基)镁(655 µL, 1.31 mmol)。添加溴化铜(I) (0.6 mg, 4 µmol),并将反应物在80℃下加热15分钟。将反应物冷却至0℃,并通过添加水(3 mL)来淬灭。添加1M HCl(水溶液)(3 mL),并将反应物在100℃下加热1小时。将反应物用饱和NaHCO3(水溶液)碱化,并用EtOAc (3 x 15 mL)萃取。将合并的有机萃取物用盐水(20 mL)洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为外消旋混合物、黄色固体的3-环戊基-N-[9-(2-甲基丙酰基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(70 mg, 36%)。
通过SFC使用Chiralpak AD-H柱以30% MeOH/70% CO2作为流动相拆分3-环戊基-N-[9-(2-甲基丙酰基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(60 mg, 0.13 mmol)。对映异构体1被分离为白色固体(24 mg, 40%)。对映异构体2被分离为白色固体(26 mg, 43%)。
实施例228
(a) 对映异构体1: N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-苯基丙酰胺(ABR 239976)
(b) 对映异构体2: N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-苯基丙酰胺(ABR 239974)
通过SFC使用Amy-C柱以75% MeOH/25% CO2作为流动相和二乙胺作为改性剂拆分N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-苯基丙酰胺(206 mg, 0.50 mmol)。将对映异构体1溶解于EtOAc中并用1M HCl(水溶液)(2 x)和盐水洗涤。将有机相干燥(MgSO4)并浓缩,以得到作为棕色固体的标题产物(75 mg, 36%)。将对映异构体2溶解于EtOAc中并用1M HCl(水溶液)(2 x)和盐水洗涤。将有机相干燥(MgSO4)并浓缩,以得到作为棕色固体的标题产物(88 mg, 43%)。
实施例229
(a) 对映异构体1: 3,5-二氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]苯甲酰胺(ABR 239981)
(b) 对映异构体2: 3,5-二氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]苯甲酰胺(ABR 239980)
通过SFC使用Amy-C柱以85% MeOH/15% CO2作为流动相拆分3,5-二氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]苯甲酰胺(62 mg, 0.13 mmol)。对映异构体1被分离为白色固体(20 mg, 32%)。对映异构体2被分离为白色固体(22 mg, 35%)。
实施例230
(a) 对映异构体1: 3-环戊基-N-[9-(4-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239984)
(b) 对映异构体2: 3-环戊基-N-[9-(4-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 239979)
通过SFC使用Chiralcel OJ-H柱以15% MeOH/85% CO2作为流动相拆分3-环戊基-N-[9-(4-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(96 mg, 0.20 mmol)。对映异构体1被分离为白色固体(38 mg, 40%)。对映异构体2被分离为白色固体(42 mg, 44%)。
实施例231
(a) 对映异构体1: 3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸(ABR 240007)
(b) 对映异构体2: 3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸(ABR 240006)
通过SFC使用Chiralpak AD-H柱以15% MeOH/85% CO2作为流动相和二乙胺作为改性剂拆分3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸(180 mg, 0.42 mmol)。对映异构体1被分离为白色固体(70 mg, 37%)。对映异构体2被分离为白色固体(60 mg, 32%)。
实施例232
3-环戊基-N-{9-[4-(甲基硫烷基)苯基]-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基}丙酰胺(ABR 240016)
将密封管装入N-[9-溴-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]乙酰胺(100 mg, 0.25 mmol)、[4-(甲基硫烷基)苯基]硼酸(32 mg, 0.20 mmol)、2M Na2CO3(水溶液)(326 µL, 0.65 mmol)和无水二氧杂环己烷(0.82 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (19 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在110℃下搅拌16小时。将反应物冷却至室温。添加2M HCl(水溶液)(2 mL),并将所得混合物搅拌1小时。将反应物用EtOAc稀释。将有机相用盐水洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-5% MeOH作为洗脱剂,随后通过自动反相HPLC(低pH方法A)纯化,以提供作为棕色固体的标题化合物(50 mg, 60%)。
实施例233
11-氯-3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸(ABR 240015)
在氮气下向11-氯-3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸苯酯(74 mg, 0.14 mmol)于THF(5 mL)中的搅拌溶液中添加2M NaOH(水溶液) (343 µL, 0.69 mmol)。将反应物在室温下搅拌1小时,然后浓缩。将残余物用2M HCl(水溶液)(2 mL)稀释,并用EtOAc (2 x 5 mL)萃取。将合并的有机萃取物用水(5 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(3 mg, 5%)。
实施例234
N-[9-(4-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丁酰胺(ABR 240014)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丁酰胺(650 mg, 1.60 mmol)、(4-甲氧基苯基)硼酸(341 mg, 2.25 mmol)、Na2CO3(680 mg, 6.42 mmol)、水(2.0 mL)、DME(7.0 mL)和EtOH(3.5 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (93 mg, 80 µmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌4小时。将反应混合物浓缩。将残余物溶解于EtOAc (25 mL)中,并用水(25 mL)洗涤。将水层用EtOAc (2 x 20 mL)萃取。将合并的有机萃取物用盐水(30 mL)洗涤,然后干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,随后通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(35 mg, 5%)。
实施例235
2-[(2-甲氧基乙基)氨基]-N-[9-(4-甲氧基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺
(ABR 240013)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(2-甲氧基乙基)氨基]-1,3-噻唑-4-甲酰胺(150 mg, 0.29 mmol)、(4-甲氧基苯基)硼酸(66 mg, 0.43 mmol)、Na2CO3(61 mg, 0.58 mmol)、水(1 mL)、DME(3 mL)和EtOH(2 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (17 mg, 0.01 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌16小时。添加额外的(4-甲氧基苯基)硼酸(66 mg, 0.43 mmol)、Na2CO3(61 mg, 0.58 mmol)和Pd(PPh3)4(17 mg, 0.01 mmol)。将溶液用氮气脱气,并将管密封。将反应物在100℃下搅拌5小时。将反应混合物浓缩。将残余物溶解于EtOAc (50 mL)中,用1M HCl(水溶液)(50 mL)、水(50 mL)和盐水(50 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体的标题化合物(39 mg, 25%)。
实施例236
N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-羟基-1,3-噻唑-4-甲酰胺(ABR 240018)
在氮气下向2-溴-1,3-噻唑-4-甲酰胺(2.93 g, 14.2 mmol)于DMF(80 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(4.42 g, 28.3 mmol),随后添加吡啶(1.14 mL, 14.2 mmol)。将反应物在室温下搅拌16小时。添加额外的3,3,3-三氟-2-氧代丙酸甲酯(4.42 g, 28.3 mmol),并将反应物再搅拌4小时。在0℃下添加亚硫酰氯(1.03 mL, 14.2 mmol)。将反应物在0℃下搅拌2小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。将酰基中间体的溶液添加至4-溴-1H-1,3-苯并二唑-2-胺(2.50 g, 11.8 mmol)于DMF(40 mL)中的溶液,随后添加三乙胺(1.88 mL, 14.2 mmol)。将反应混合物再搅拌2小时,然后浓缩。将残余物溶解于EtOAc (200 mL)中,用10%柠檬酸(水溶液)(2 x 100 mL)、水(2 x 100 mL)和盐水(2 x 100 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供2-溴-N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺和N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.0²,6]十二碳-1(12),6,8,10-四烯-3-基]-2-氯-1,3-噻唑-4-甲酰胺的混合物(3.70 g)。
在0℃在氮气下,向NaH(60%, 11 mg, 0.27 mmol)于DMF(2 mL)中的悬浮液中添加甲磺酰胺(24 mg, 0.25 mmol)。在0℃下搅拌10分钟之后,添加一部分2-溴-N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺和N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-氯-1,3-噻唑-4-甲酰胺的混合物(100 mg)。将反应物温热至室温并搅拌2小时。然后将温度增加至80℃,持续4小时。将反应物在0℃下用饱和NH4Cl(水溶液)淬灭并用EtOAc (50 mL)稀释。将有机相用水(2 x 25 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(11 mg, 11%)。
实施例237
N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(2-甲基丙基)氨基]-1,3-噻唑-4-甲酰胺(ABR 240019)
在室温在氮气下向2-溴-1,3-噻唑-4-甲酰胺(360 mg, 1.74 mmol)于DMF(15 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(543 mg, 3.48 mmol),随后添加吡啶(140 µL, 1.74 mmol)。将反应混合物在室温下搅拌16小时。在0℃下添加亚硫酰氯(126 µL, 1.74 mmol),然后将反应混合物搅拌1小时。将反应混合物浓缩,并在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加5,6-二氯-1H-1,3-苯并二唑-2-胺(293 mg, 1.45 mmol)于DMF(10 mL)中的溶液和三乙胺(231 µL, 1.74 mmol),并将反应混合物在室温下搅拌2小时。将反应混合物浓缩。将残余物用EtOAc (50 mL)稀释,用10%柠檬酸(水溶液)(2 x 25 mL)、水(2 x 25 mL)和盐水(2 x 25 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物在DCM中研磨,以提供2-溴-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺和2-氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺的混合物(746 mg)。
将密封管装入一部分2-溴-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺和2-氯-N-[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-1,3-噻唑-4-甲酰胺的混合物(190 mg)、K2CO3(255 mg, 1.84 mmol)、无水二氧杂环己烷(3 mL)和2-甲基丙-1-胺(184 µL, 1.84 mmol)。将管密封并在130℃下搅拌1小时。添加额外的K2CO3 (255 mg, 1.84 mmol)和2-甲基丙-1-胺(184 µL, 1.84 mmol)。将反应物在130℃下再搅拌18小时,然后浓缩。将残余物用EtOAc (30 mL)和盐水(30 mL)稀释。将水层用EtOAc (3 x 30 mL)萃取。将合并的有机萃取物用盐水洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为棕色固体的标题化合物(40 mg, 21%)。
实施例238
3-环戊基-N-{9-[4-(二甲基氨基)苯基]-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基}丙酰胺(ABR 240028)
将密封管装入N-[9-溴-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]乙酰胺(75 mg, 0.16 mmol)、[4-(二甲基氨基)苯基]硼酸(38 mg, 0.23 mmol)、2M Na2CO3(326 µL, 0.65 mmol)、1,4-二氧杂环己烷(0.82 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (38 mg, 0.04 mmol),将溶液用氮气脱气并将管密封。将反应物在110℃下搅拌16小时。将该反应物冷却至室温,并用EtOAc和2M NaHCO3(水溶液)稀释。将有机相用盐水洗涤,干燥(MgSO4),过滤并浓缩。将粗产物首先通过硅胶色谱纯化,使用DCM中的0-5% MeOH作为洗脱剂。随后通过自动反相HPLC(低pH方法A)纯化,然后通过硅胶色谱纯化,使用DCM中的0-20% MeOH作为洗脱剂,提供作为白色固体的标题化合物(20 mg, 25%)。
实施例239
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2,2,2-三氟乙酰胺(ABR 240031)
在氮气下向三氟乙酰胺(210 mg, 1.86 mmol)于无水DCM(8.3 mL)中的搅拌溶液中添加吡啶(120 µL, 1.48 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(380 µL, 3.72 mmol)。将反应物在室温下搅拌2小时。在0℃下添加亚硫酰氯(107 µL, 1.48 mmol)。将反应物在0℃下搅拌1小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (6 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(250 mg, 1.24 mmol)于DMF (2.3 mL)中的溶液和三乙胺(207 µL, 1.48 mmol)。将反应物搅拌60小时,然后浓缩。将残余物溶解于EtOAc中并用盐水(4 x)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的0-10% MeOH作为洗脱剂,以提供作为白色固体的标题化合物(155 mg, 26%)。
实施例240
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(2-甲氧基乙氧基)吡啶-4-甲酰胺(ABR 240032)
在氮气下向2-(2-甲氧基乙氧基)吡啶-4-甲酰胺(190 mg, 0.97 mmol)于无水DCM(5 mL)中的搅拌溶液中添加吡啶(79 µL, 0.97 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(303 mg, 1.94 mmol)。将反应物在室温下搅拌2小时。在0℃下添加亚硫酰氯(70 µL, 0.97 mmol)。将反应物在0℃下搅拌1小时,然后在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (3 mL)中。添加5,6-二甲基-1H-1,3-苯并二唑-2-胺(130 mg, 0.81 mmol)于DMF (2 mL)中的溶液和三乙胺(136 µL, 0.97 mmol)。将反应物搅拌18小时,然后浓缩。将残余物用EtOAc和盐水稀释。将有机相干燥(MgSO4),过滤并浓缩。将粗产物在MeCN中研磨,随后通过自动反相HPLC(低pH方法A)纯化,提供作为白色固体的标题化合物(10 mg, 3%)。
实施例241
3-环戊基-N-[9-(4-甲磺酰基苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 240029)
在0℃下向3-环戊基-N-{9-[4-(甲基硫烷基)苯基]-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基}丙酰胺(40 mg, 80 µmol)于MeOH(5 mL)中的搅拌溶液中添加过硫酸氢钾制剂(122 mg, 0.2 mmol)于水(1 mL)中的溶液。将反应物在室温下搅拌2小时,用饱和Na2S2O3(水溶液)淬灭,然后浓缩。将所得残余物用EtOAc (15 mL)和水(10 mL)稀释。将水相用EtOAc (2 x 15 mL)萃取。将合并的有机萃取物用水(20 mL)、盐水(40 mL)和水(2 x 10 mL)洗涤。然后将有机相干燥(Na2SO4),过滤并浓缩,以提供作为白色固体的标题化合物(4 mg, 11%)。
实施例242
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2,2-二甲基丙酰胺(ABR 240030)
在氮气下向2,2-二甲基丙酰胺(392 mg, 3.88 mmol)于无水DCM(5 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(475 µL, 4.65 mmol),随后添加吡啶(313 µL, 3.88 mmol)。将反应物在室温下搅拌16小时。在0℃下添加亚硫酰氯(281 µL, 3.88 mmol)。将反应物在0℃下搅拌1.5小时,然后在氮气下过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。将酰基中间体的溶液添加至5,6-二甲基-1H-1,3-苯并二唑-2-胺(500 mg, 3.1 mmol)于DMF(5 mL)中的溶液,随后添加三乙胺(516 µL, 3.88 mmol)。将反应混合物再搅拌1小时,然后浓缩。将残余物溶解于EtOAc (20 mL)中,并用饱和柠檬酸(水溶液)(30 mL)洗涤。将水层用EtOAc (20 mL)萃取。将合并的有机萃取物用水(2 x 20 mL)和盐水(25 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为粉红色固体的标题化合物(505 mg, 61%)。
实施例243
N-[9-乙酰基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(环戊基氨基)-1,3-噻唑-4-甲酰胺(ABR 240024)
在氮气下将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-(环戊基氨基)-1,3-噻唑-4-甲酰胺(150 mg, 0.26 mmol)、乙酸钯(II)(6 mg, 26 µmol)、丙烷-1,3-二基双(二苯基膦)(21 mg, 0.05 mmol)、K2CO3(47 mg, 0.34 mmol)、1-(乙烯基氧基)丁烷(170 µL, 1.30 mmol)和脱气DMF/水(3:1, 4 mL)。将反应物在100℃下搅拌20小时,然后浓缩。将所得残余物用EtOAc (10 mL)和盐水(10 mL)稀释。将水相用EtOAc (15 mL)洗涤,并弃去有机洗液。然后将水相用IPA/CHCl3 (2 x 25 mL)萃取。将合并的有机萃取物干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(24 mg, 19%)。
实施例244
N-[9-(3-氯苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(ABR 240025)
将微波管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(75 mg, 0.16 mmol)、(3-氯苯基)硼酸(32 mg, 0.2 mmol)、2M Na2CO3(水溶液)(0.15 mL)、DME(0.80 mL)和EtOH(0.30 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (19 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在微波中在120℃下搅拌1.5小时。将反应混合物过滤通过Celite™,用MeOH (30 mL)冲洗。将滤液浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体的标题化合物(35 mg, 44%)。
实施例245
3-环戊基-N-[4-氧代-3-(三氟甲基)-9-[4-(三氟甲基)苯基]-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 240022)
使用用于制备N-[9-(3-氯苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺的程序,除了使用[4-(三氟甲基)苯基]硼酸代替(3-氯苯基)硼酸(47%)。
实施例246
3-环戊基-N-[9-(4-氟苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 240023)
使用用于制备N-[9-(3-氯苯基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺的程序,除了使用(4-氟苯基)硼酸代替(3-氯苯基)硼酸(29%)。
实施例247
3-(3-环戊基丙酰胺基)-N-甲磺酰基-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酰胺(ABR 240026)
在0℃在氮气下,向甲磺酰胺(50 mg, 0.53 mmol)于DMF(3 mL)中的搅拌溶液中添加NaH (60%, 21 mg, 0.53 mmol)。将反应物在0℃下搅拌30分钟,然后添加3-(3-环戊基丙酰胺基)-10-甲氧基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-甲酸苯酯(70 mg, 0.13 mmol)于DMF(2 mL)中的溶液。将反应物温热至室温,然后在50℃下加热4小时。将反应物在0℃下用饱和NH4Cl(水溶液)淬灭,然后浓缩。将所得残余物用EtOAc (50 mL)和水(25 mL)稀释。将有机层用水(2 x 25 mL)、盐水(2 x 25 mL)洗涤,干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为米色固体的标题化合物(30mg, 43%)。
实施例248
3-环戊基-N-[9-(甲氧基甲基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(ABR 240027)
在0℃下向3-环戊基-N-[9-(羟基甲基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]丙酰胺(60 mg, 0.13 mmol)于MeOH(1.0 mL)中的搅拌溶液中添加浓H2SO4 (0.01 mL, 0.15 mol)。将反应物在50℃下加热24小时。在0℃下添加额外的浓H2SO4 (0.02 mL, 0.30 mol)。将反应物在60℃下加热24小时。将反应物浓缩,将残余物溶解于EtOAc (5 mL)中,并用饱和NaHCO3(水溶液)(3 mL)、水(3 mL)和盐水(3 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗物质通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(13 mg, 23%)。
实施例249
3-{[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}吡咯烷-1-甲酸叔丁酯(ABR 240049)
使用制备6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}-2-氮杂螺- [3.3]庚烷-2-甲酸叔丁酯的程序,除了使用3-氨基甲酰基吡咯烷-1-甲酸叔丁酯代替6-氨基甲酰基-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯和使用5,6-二甲基-1H-1,3-苯并二唑-2-胺代替5,6-二氯-1H-1,3-苯并二唑-2-胺。在初始装填之后2小时,添加额外的1当量的3,3,3-三氟-2-氧代丙酸甲酯。使用自动反相HPLC(高pH方法A)实施最终纯化;(20%);m/z = 426.1 (MH-tBu)+。
实施例250
4-{ [10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}哌啶-1-甲酸叔丁酯(ABR 240050)
使用制备6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯的程序,除了使用4-氨基甲酰基哌啶-1-甲酸叔丁酯代替6-氨基甲酰基-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯并使用5,6-二甲基-1H-苯并[d]咪唑-2-胺代替5,6-二氯-1H-苯并[d]咪唑-2-胺; (40%); m/z = 440.1 (MH-tBu)+。
实施例251
(2S)-2-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}吡咯烷-1-甲酸叔丁酯(ABR 240051)
使用制备6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯的程序,除了使用(2S)-2-氨基甲酰基吡咯烷-1-甲酸叔丁酯代替6-氨基甲酰基-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯; (29%); m/z = 465.8, 467.8 (MH-tBu)+。
实施例252
3-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}吡咯烷-1-甲酸叔丁酯(ABR 240052)
使用制备6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯的程序,除了使用3-氨基甲酰基吡咯烷-1-甲酸叔丁酯代替6-氨基甲酰基-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯. 使用自动反相HPLC(高pH方法A)实施最终纯化,以提供作为非对映异构体的混合物的标题化合物;(32%);m/z = 466.1, 468.1 (MH-tBu)+。
实施例253
4-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}哌啶-1-甲酸叔丁酯(ABR 240053)
使用制备6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯的程序,除了使用4-氨基甲酰基哌啶-1-甲酸叔丁酯代替6-氨基甲酰基-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯; (40%); m/z = 480.1, 482.1 (MH-tBu)+。
实施例254
3-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}氮杂环丁烷-1-甲酸叔丁酯(ABR 240054)
使用制备6-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯的程序,除了使用3-氨基甲酰基氮杂环丁烷-1-甲酸叔丁酯代替6-氨基甲酰基-2-氮杂螺[3.3]庚烷-2-甲酸叔丁酯; (22%); m/z = 451.8, 453.8 (MH-tBu)+。
实施例255
4-({[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}甲基)哌啶-1-甲酸叔丁酯(ABR 240055)
在氩气下向4-(氨基甲酰基甲基)哌啶-1-甲酸叔丁酯(1.00 g, 4.00 mmol)于DMF(8 mL)中的搅拌溶液中添加吡啶(316 µL, 4.00 mmol)和3,3,3-三氟-2-氧代丙酸乙酯(104 µL, 4.00 mmol)。将反应混合物在室温下搅拌1小时,然后逐滴添加亚硫酰氯(300 µL, 4.00 mmol)。将反应混合物在室温下再搅拌16小时,然后浓缩以提供酰基亚胺中间体。将酰基亚胺中间体溶解于DMF (5 mL)中并添加至5,6-二氯-1H-1,3-苯并二唑-2-胺(804 mg, 4.00 mmol)于DMF(7 mL)中的溶液,随后添加三乙胺(0.56 mL, 4.00 mmol)。将反应混合物在室温下搅拌4小时。将反应物用盐水(25 mL)稀释,并用EtOAc (2 x 50 mL)萃取。将合并的有机相用水洗涤,干燥(Na2SO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用DCM中的2% MeOH作为洗脱剂,以提供作为红色固体的标题化合物(65 mg, 6%);m/z = 450.4, 452.4 (MH-CO2tBu)+。
实施例256
2-(2-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环-[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]氨基甲酰基}乙基)哌啶-1-甲酸叔丁酯(ABR 240056)
使用用于制备4-({[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-氨基甲酰基}甲基)哌啶-1-甲酸叔丁酯的程序,除了使用2-(2-氨基甲酰基乙基)哌啶-1-甲酸叔丁酯代替4-(氨基甲酰基-甲基)哌啶-1-甲酸叔丁酯(17%); m/z = 463.4, 465.4 (MH-CO2tBu)+。
实施例257
3-{[10,11-二氯-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(8),6,9,11-四烯-3-基]氨基甲酰基}-3-氟氮杂环丁烷-1-甲酸叔丁酯(ABR 240057)
在氮气下向3-氨基甲酰基-3-氟氮杂环丁烷-1-甲酸叔丁酯(285 mg, 1.30 mmol)于DMF(25 mL)中的搅拌溶液中添加吡啶(105 μL, 1.30 mmol),随后添加3,3,3-三氟-2-氧代丙酸甲酯(407 mg, 2.61 mmol)。将反应物在室温下搅拌2小时。在0℃下添加亚硫酰氯(95 µl, 1.30 mmol)。将反应物在0℃下搅拌1小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (10 mL)中。添加5,6-二氯-1H-1,3-苯并二唑-2-胺(220 mg, 1.08 mmol)于DMF(10 mL)中的溶液和三乙胺(182 µl, 1.30 mmol)。将反应物搅拌2小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(4 x 50 mL)和盐水(3 x 50 mL)洗涤。将有机相干燥(Na2SO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为黄色固体的标题化合物(59 mg, 10%);m/z = 469.8, 471.8 (MH-tBu)+。
实施例258
3-[3-(3-环戊基丙酰胺基)-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-9-基]苯甲酸甲酯(ABR 240058)
将密封管装入N-[9-溴-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-3-环戊基丙酰胺(100 mg, 0.22 mmol)、[3-(甲氧基羰基)苯基]硼酸(49 mg, 0.27 mmol)、Na2CO3(42 mg, 0.39 mmol)、水(0.20 mL)、DME(3.00 mL)和EtOH(1.25 mL)。将溶液用氮气脱气。添加Pd(PPh3)4 (25 mg, 0.02 mmol),将溶液用氮气脱气并将管密封。将反应物在100℃下搅拌18小时。将反应混合物过滤通过Celite™,用MeOH (30 mL)冲洗,并将滤液浓缩。将所得残余物溶解于EtOAc (20 mL)中,用水(30 mL)、饱和柠檬酸(水溶液)(30 mL)和盐水(30 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过硅胶色谱纯化,使用庚烷中的0-100% EtOAc、然后DCM中的0-10% MeOH作为洗脱剂,以提供作为棕色固体的标题化合物(50 mg, 30%);m/z = 515.3 (MH)+。
实施例259
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]乙酰胺(ABR 238786)
在氮气下向乙酰胺(220 mg, 3.72 mmol)于DMF(50 mL)中的溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(678 mg, 4.34 mmol),随后添加吡啶(300 µL, 3.72 mmol)。将反应物在室温下搅拌2小时。在0℃下添加亚硫酰氯(270 µL, 3.72 mmol)。将反应物在0℃下搅拌1小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (25 mL)中。将酰基中间体的溶液添加至5,6-二甲基-1H-1,3-苯并二唑-2-胺(0.50 g, 3.1 mmol)于DMF(25 mL)中的溶液,随后添加三乙胺(519 µL, 3.72 mmol)。将反应混合物再搅拌16小时,然后浓缩。将残余物溶解于EtOAc (100 mL)中,用10%柠檬酸(水溶液)(3 x 50 mL)、水(3 x 50 mL)和盐水(3 x 50 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过在DCM中研磨而纯化,以提供作为白色固体的标题化合物(182 mg, 18%);m/z = 327.0 (MH)+。
实施例260
N-[9-溴-10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]乙酰胺(ABR 240059)
向N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]乙酰胺(150 mg, 0.46 mmol)于DCM(25 mL)中的搅拌溶液中添加NBS (82 mg, 0.46 mmol)。最初,将反应物在室温下搅拌3小时。然后将反应物再搅拌20小时。在此时段期间,分批添加额外的NBS (164 mg, 0.92 mmol)。然后将反应物浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(2 x 50 mL)和盐水(50 mL)洗涤。将有机相干燥(MgSO4),过滤并浓缩。将粗产物在DCM中研磨,以提供作为黄色固体的标题化合物(122 mg, 66%);m/z = 404.8, 406.8 (MH)+。
实施例261
N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.02,6]十二碳-1(12),6,8,10-四烯-3-基]-2-[(2-甲氧基乙基)氨基]嘧啶-4-甲酰胺(ABR 239626)
在氮气下向2-氯嘧啶-4-甲酰胺(经由文献方法: PCT国际申请2001068612可得)(330 mg, 2.08 mmol)于DMF(6 mL)中的搅拌溶液中添加3,3,3-三氟-2-氧代丙酸甲酯(355 µL, 3.47 mmol),随后添加吡啶(170 µL, 2.08 mmol)。将反应物在室温下搅拌2小时。在0℃下添加亚硫酰氯(150 µL, 2.08 mmol)。将反应物在0℃下搅拌1小时,然后浓缩。在氮气下将残余物过滤通过硅胶短垫,用DCM洗脱。将滤液浓缩,并在氮气下将剩余的酰基中间体溶解于DMF (5 mL)中。将酰基中间体的溶液添加至5,6-二甲基-1H-1,3-苯并二唑-2-胺(280 mg, 1.74 mmol)于DMF(8 mL)中的溶液,随后添加三乙胺(280 µL, 2.08 mmol)。将反应混合物再搅拌16小时,然后浓缩。将残余物溶解于EtOAc (50 mL)中,并用水(3 x 50 mL)和盐水(2 x 50 mL)洗涤,然后干燥(MgSO4),过滤并浓缩,以提供2-氯-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.0²,6]十二碳-1(12),6,8,10-四烯-3-基]嘧啶-4-甲酰胺。(260 mg, 15%)。m/z = 424.95 (MH)+。
将可密封管装入一部分2-氯-N-[10,11-二甲基-4-氧代-3-(三氟甲基)-2,5,7-三氮杂三环[6.4.0.0²,6]十二碳-1(12),6,8,10-四烯-3-基]嘧啶-4-甲酰胺(250 mg, 0.35 mmol)、2-甲氧基乙-1-胺(92 µL, 1.06 mmol)、K2CO3(150 mg, 1.06 mmol)和DMF(5 mL)。将该管用氮气吹扫,密封并在100℃下搅拌6小时。然后将反应混合物浓缩,并将所得残余物用EtOAc (20 mL)和水(20 mL)稀释。将水相用EtOAc (2 x 10 mL)萃取。将合并的有机萃取物用10 % 柠檬酸(水溶液)(2 x 20 mL)和盐水(2 x 20 mL)洗涤,然后干燥(MgSO4),过滤并浓缩。将粗产物通过自动反相HPLC(低pH方法A)纯化,以提供作为白色固体的标题化合物(50 mg, 31 %)。
上述实施例的化合物的结构式显示于表1中。
本发明实施例的NMR和质谱数据显示于表2中。
生物学测定
制备并纯化用于S100A9相关测定的生物学试剂
重组人类S100A9野生型
培养:rhS100A9 wt的表达通过在0.25mM IPTG诱导的情况下摇瓶培养工作细胞库BL21 (DE3)/pET1120 (pLR757)进行。将细胞团块冷冻。
包含体的纯化:将大肠杆菌团块在室温下用150mL裂解缓冲液(50mM Tris/HCl,1mM EDTA,25% Saccarose,pH 8.0)解冻并在烧杯中在冰下超声处理3 x 15秒。此后,添加10μL 1M MgCl2 (10mM最终浓度)/mL团块溶液、1μL 1M MnCl2 (1mM最终浓度)/mL团块溶液和1μL 10mg/mL DNase I (10μg/mL最终浓度)/mL团块溶液。在室温下培育30分钟之后,以1:1体积比添加洗涤剂缓冲液(20mM Tris/HCl,pH 7.5,2mM EDTA,1% Nonidet P-40)和蛋白酶抑制剂(Complete Mini Protease Inhibitors, Roche) 1-2片/25mL。将溶液在14,000 x g、5℃下离心20分钟。将团块用90mL 0.5% Triton X-100、1mM EDTA再悬浮,超声处理3 x 15秒并再次离心下来(spin down)。将该洗涤和超声处理程序再重复5次。
再悬浮和折叠:在所有溶液和透析步骤中使用Milli-Q水。将最终团块再悬浮在100mL的500mM NaH2PO4缓冲液(pH 1.8)中的8M尿素、40mM DTT中。当溶液澄清时,将其在20,000 x g、5℃下离心25分钟。将含有再悬浮的包含体的上清液用500mM磷酸盐缓冲液(pH 1.8)设定至pH 2。
上清液的第一次透析针对5L的50mM NaH2PO4缓冲液、1.5mM DTT, pH 2进行6小时。第二次透析针对5L的10mM乙酸钠缓冲液、150mM NaCl、1.5mM DTT, pH 4进行15小时。第三次透析针对5L的10mM乙酸钠缓冲液、150mM NaCl、1.5mM DTT, pH 4进行8小时。第四次透析针对5L的20mM Tris/HCl、150mM NaCl、1.5mM DTT, pH 7.2进行16小时。第五次透析针对5L的20mM Tris/HCl、1mM EDTA、1mM EGTA、1.5mM DTT, pH 8.5进行6小时。离心在22,000 x g、5℃下进行30分钟。
通过色谱纯化:所有色谱柱和树脂都购自GE HealtCare, 瑞典。添加DTT至1.5mM的最终浓度。在HiPrep Q FF 16/10柱上的阴离子交换色谱以1.5mL/min的流速进行,使用20mM Tris、1mM EDTA、1mM EGTA、1.5mM DTT中的0-1M NaCl梯度, pH 8.5用于洗脱蛋白。在洗脱之前使用没有NaCl的相同缓冲液来平衡并洗涤。将含有rhS100A9wt的合并级分使用Centriprep YM-3 (Amicon, USA)浓缩至1.5mL。
Superdex 75 16/790柱上的尺寸排阻色谱使用补充有10mM DTT的HBS-N缓冲液(10mM Hepes、150mM NaCl, pH 7.4)以0.5mL/min的流速进行。运行PD-10用于缓冲液交换至10mM Hepes、150mM NaCl, pH 7.5。
Biacore结合测定
使用表面等离子共振(SPR)技术(Björk等人,2009)研究S100A9与其假定目标受体(例如,RAGE和TLR4)的Ca2+和Zn2+依赖性相互作用。简言之,将S100A9注入RAGE和TLR4上,在生理学浓度的Ca2+和Zn2+存在的情况下经由Biacore传感器芯片上的伯胺固定化,允许无标记且实时分析这些相互作用。显然,可以以将S100A9固定化并注入RAGE和TLR4的方式使测定反转。在图1中,举例说明对S100A9和RAGE之间的相互作用的抑制影响的测定 – 本文下面详细描述该测定。本领域普通技术人员将基本上能够进行涉及S100A9和TLR4的相互作用的相同测定。
测定显示研究的本发明化合物分别对S100A9与RAGE或TLR4之间的蛋白-蛋白相互作用的抑制作用,参考图2。
抑制测定,biot-hS100A9:hRAGE-Fc
原理. AlphaScreen (增强型发光邻近均质化测定)含有两种类型的珠粒:α供体珠粒和受体珠粒(PerkinElmer)。在680nm下激光激发时,供体珠粒中的光敏剂使周围氧转化成更加激发的单线态。单线态氧分子扩散(最大200nm)以便与受体珠粒中的二甲基噻吩衍生物反应并产生化学发光反应。受体珠粒中的荧光团随后发出520-620nm下的光,其可在EnVision® 多标记微孔板检测仪(Multilabel plate reader)(PerkinElmer)中检测。这些珠粒为光敏的且使用珠粒的所有工作都在暗光条件下或对于光源使用绿色滤光器(Roscolux Chroma Green #389, Rosco)进行。
在本文所述的AlphaScreen抑制测定中,将蛋白A(金黄色葡萄球菌(Staphylococcus aureus))缀合的受体珠粒与链霉抗生物素蛋白涂覆的供体珠粒(Perkin Elmer 6760617M)一起使用。将受体珠粒与Fc标记的重组人类RAGE (rhRAGE-Fc)预培育,允许rhRAGE-Fc结合至珠粒上的蛋白A。将生物素化人类S100A9 (biot-hS100A9)与低分子测试化合物预培育。随后将预混合物添加至微板的孔中并培育,允许biot-hS100A9和rhRAGE-Fc之间相互作用。随后添加链霉抗生物素蛋白涂覆的供体珠粒,引起链霉抗生物素蛋白与生物素化hS100A9结合。在额外培育之后,测量信号。
在没有抑制性化合物的情况下,biot-hS100A9与rhRAGE-Fc之间的相互作用将使得受体珠粒和供体珠粒密切邻近,因此产生高信号。在存在抑制剂的情况下,将不会形成复合物,产生减小的信号。
化学品和试剂
AlphaScreen® General IgG (蛋白A)检测试剂盒(PerkinElmer 6760617M)
HBS-P缓冲液(GE Healthcare, BR-1003-68)
HBS-N缓冲液(GE Healthcare, BR-1003-69)
HBS-P中的CaCl2
Milli-Q水中的ZnCl2
DMSO
生物素化hS100A9,(通过EZ-连接IA-PEO2-生物素试剂,Pierce Biotechnology经由半胱氨酸生物素化),在HBS-N中
rhRAGE-Fc (R&D Systems, 1145-RG-50),在HBS-P中
程序. 使用AlphaScreen测定法来筛选固定浓度的不同化合物样品的抑制效果或通过改变化合物浓度测定IC50。测试化合物和参考物的样品由DMSO中的溶液制备。将相关参考抑制剂和DMSO分别用作测定中定义的抑制和非抑制的对照。在测定中测试化合物和参考物的抑制百分数通过比较其获得的测定信号与仅用DMSO(无化合物)的情况下的对照的信号值来计算。
生物素化hS100A9和rhRAGE-Fc的测定浓度是批次依赖性的,且使用该AlphaScreen抑制方法通过分开的交叉滴定实验来测定并定义以证实对于信号强度的最佳设置和用相关参考化合物实现定义的抑制。受体珠粒和供体珠粒的最终测定浓度为20µg/mL。
溶液和珠粒的筛选、制备的实验设置.
通过将CaCl2和ZnCl2添加至HBS-P中制备测定缓冲液并将新鲜制备的缓冲液用于该实验中。
-该实验的Biotin-hS100A9溶液通过在测定缓冲液(具有CaCl2和ZnCl2)中稀释适当量的储备溶液biot-hS100A9并在室温下培育30分钟来制备。
-该实验的rhRAGE-Fc溶液通过在测定缓冲液中稀释适当量的rhRAGE-Fc储备物来制备。
-将蛋白A受体珠粒稀释于测定缓冲液中并将其添加至等体积的制备的稀释的rhRAGE-Fc溶液中。珠粒为光敏的。将小瓶用铝箔覆盖并在室温下在暗处培育,直至biot-hS100A9+化合物培育完成(参见下文)。
-链霉抗生物素蛋白涂覆的供体珠粒稀释于测定缓冲液中。珠粒为非常光敏性的。将小瓶用铝箔覆盖并在室温下在暗处培育直至使用(参见下文)。
稀释样品并与biot-hS100A9培育
-将测试化合物、适当参考物和DMSO对照的样品稀释于测定缓冲液中。
-将稀释的测试化合物、参考物和DMSO对照添加至Greiner微量滴定96孔板(PP,u型底(编号650201))的孔中并将适当量的稀释的biot-hS100A9溶液添加至具有样品的各孔中(最终DMSO浓度≤1.25% (v/v))。将板用板密封物覆盖并在暗处在轨道板震荡器上在室温下培育1小时。
在Optiplate中培育biot-hS100A9+化合物样品和rhRAGE-Fc-受体珠粒
-当biot-hS100A9+化合物培育完成时,将溶液转移至Optiplate (Optiplate 384白色,Perkin Elmer 编号6007299)中并将rhRAGE-Fc-受体珠粒溶液添加至各孔中(使用绿色过滤的光)。将板用板密封物覆盖并在暗处在板培育箱中在标称25℃下培育40分钟。
在Optiplate中培育biot-hS100A9+化合物样品和rhRAGE-Fc-受体珠粒和供体珠粒
-在培育之后,将供体珠粒溶液添加至各孔中(使用绿色过滤的光)。将板用板密封物覆盖并在暗处在板培育箱中在标称25℃下培育。在50分钟之后,将板在紧挨着EnVision®仪的实验台上培育(在暗处)10分钟,用于温度平衡。
在EnVision® 多标记微孔板检测仪中读取Optiplate
-除去板密封物并在读取之前将板置于EnVision®中5分钟。
计算. 每种样品(测试化合物或参考物)的抑制百分比(%)使用下式计算:1-(样品信号/DMSO信号) x 100%。
在S100A9-RAGE抑制测定中本发明的许多化合物的IC50值列于表3中。
细胞毒性的测定
原理. 通过该方法中描述的测定,研究直接毒性(24小时培育)和抗增殖效果(72小时培育)。将细胞与不同浓度的测试化合物培育之后,添加细胞增殖试剂。然后通过分光光度计测量代谢活性细胞产生的甲的形成。
化学品和试剂. Jurkat细胞,DMSO,细胞增殖试剂(Roche参考号11644807001)。
溶液. R10培养基=补充有10%胎牛血清、5%丙酮酸钠(Lonza,比利时)的RPMI 1640培养基。台盼蓝溶液0.4%,Sigma。测试化合物的DMSO储备溶液。抗增殖阳性的DMSO储备溶液。阴性对照的DMSO储备溶液。
程序. 将Jurkat细胞转移至50 mL试管。将小体积稀释于台盼蓝溶液中,并计数细胞。将细胞浓度调节至0.2 x106个细胞/mL的最终浓度。通过将细胞悬浮液添加至所有孔(除了其中仅添加R10且无细胞的一对孔(空白)),将细胞接种于96孔平底无菌板(10,000个细胞/孔)。
将储备溶液在96孔板中连续稀释至0.070- 25 mM区间内的浓度。然后将DMSO稀释板中的溶液在R10培养基中进一步稀释,以得到0.28-200 µM区间内的浓度。
将来自R10稀释板的溶液添加至细胞接种板的孔中,除了仅含有R10的孔。化合物的最终浓度为0.14-100µM,且培养板中的DMSO的最终浓度为0.2%,这被认为不显著影响细胞增殖。将制备两个相同板,一个用于24小时培育,一个用于72小时培育。将板在37℃、5% CO2和90 % Rh下培育。
测量. 24小时培育之后,将10μL细胞增殖试剂/孔添加至两个相同板之一,然后将板培育(37℃、5% CO2和90 % Rh)。2小时培育之后,在分光光度计中在450 nm下测量吸光度。72小时培育之后,对另一板重复该程序。
计算. 计算背景值。将对于每个浓度(包括对照(有细胞且无化合物的孔))测量的吸光度针对背景校正。对于每个浓度,将校正的吸光度值除以对照的校正的吸光度值,并将“对照的%”的值相对于浓度作图。然后计算IC50值。
体内模型MC38/小鼠.
购买约7周龄的雌性C57Bl/6小鼠。在研究开始之前,将小鼠在实验室中驯化至少一周。常规使用8-12周龄的小鼠。在所有实验中,随机选择小鼠的对照组。完全如治疗组一样处理对照组,但不给药任何药物化合物。通过用100μl 人工基质胶(Matrigel)中的约500 000个MC38-C215细胞皮下注射来进行引发肿瘤疾病(第0天)。该细胞系是C215-转染的鼠MC38结肠腺癌细胞,将其在R10培养基(补充有10%胎牛血清、50μM β-巯基乙醇和0.5mg/ml G418硫酸盐的具有Ultraglutamine的RPMI-1640)中培养。从第7天起,用卡尺一周三次测量肿瘤生长并计算肿瘤体积。肿瘤体积计算为V = LxW2x0.4,其中V为体积(mm3),L为长度(mm)且W为宽度(mm),且L >或= W (Attia 1966)。当对照组中的肿瘤已达到合适大小时,实验完成,且将所有小鼠处死(通常在第12-16天)并将肿瘤剖出,且测定肿瘤质量。在图3中显示使用实施例218a的化合物获得的结果。
使用的缩写
aq 水溶液
CHRM 冷藏保存的肝细胞恢复培养基
DCE 1,2-二氯乙烷
DCM 二氯甲烷
DME 二甲氧基乙烷
DMF N,N-二甲基甲酰胺
DMSO 二甲亚砜
DMPU 1,3-二甲基四氢嘧啶-2(1H)-酮
dppf 1,1'-双(二苯基膦基)二茂铁
DTT 二硫苏糖醇
EDC.HCl N-[3-(二甲基氨基)丙基]-N'-乙基碳二亚胺盐酸盐(1:1)
EDTA 乙二胺四乙酸
EGTA 乙二醇四乙酸
EtOAc 乙酸乙酯
EtOH 乙醇
FACS 荧光激活细胞分选
h 小时
HPLC 高效液相色谱
IPA 丙-2-醇
IPTG 异丙基β-D-1-硫代吡喃半乳糖苷
KHB Krebs-Henseleit碳酸氢盐缓冲液
MeCN 乙腈
MeOH 甲醇
min 分钟
NaHMDS 双(三甲基甲硅烷基)氨基化钠
NBS 1-溴-2,5-吡咯烷二酮
NMP 1-甲基吡咯烷-2-酮
Ph 苯基
NMR 核磁共振
PBS 磷酸盐缓冲盐水
PBST 磷酸盐缓冲盐水Tween-20
RT 室温
SCX 强阳离子交换
SFC 超临界流体色谱
SPhos 二环己基(2',6'-二甲氧基联苯-2-基)膦
tBu 叔丁基
TFA 三氟乙酸
THF 四氢呋喃
TLC 薄层色谱
wt 重量。
参考文献
Acharyya S等人, A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150(1), 165-7
Arai K等人, S100A8 and S100A9 overexpression is associated with poor pathological parameters in invasive ductal carcinoma of the breast. Curr Cancer Drug Targets 2008, 8(4): 243-52
Attia M,等人 (1966). Cancer Res., 26: 1787-1800
Bhardwaj RS等人, The calcium-binding proteins MRP8 and MRP14 form a membrane-associated heterodimer in a subset of monocytes/macrophages present in acute but absent in chronic inflammatory lesions. Eur J Immunol 1992, 22:1891-97
Björk P等人, Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009, 7(4):e97
Carta A等人, Design, synthesis, and preliminary in vitro and in silico antiviral activity of [4,7]phenantrolines and 1-oxo-1,4-dihydro-[4,7]phenantrolines against single-stranded positive-sense RNA genome viruses Bioorg. Med. Chem. (2007) 15:1914-1927
Chang KA等人, The role of S100a9 in the pathogenesis in Alzheimer’s disease: the therapeutic effects of S100a9 knockdown or knockout. Neurodegener Dis 2012, 10(1-4):27-9
Chaudhari K等人, Novel and Facile Transformation of N,N-Disubstituted Glycylamides into Corresponding Cyanamides by Using Pentavalent Iodine Reagents in Combination with Tetraethylammonium Bromide. Synlett 2007, 18, 2815-2818
Cheng P等人, Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 2008, 205(10), 2235-49
Deane R等人, A multimodal RAGE-specific inhibitor reduces amyloid b-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 2013. 122(4):1377-92
Foell D等人, S100 proteins in phagocytes: a novel group of damage-associated moleculat pattern molecules. J Leukoc Biol 2007, 81:28-37
Foell D等人, Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum 2004, 50, 3762-3771
Ghavami S等人, S100A8/S100A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J Leukoc Biol 2008, 83(6), 1484-92
Ha T等人, S100a9 knockdown decreases the memory impairment and the neuropathology in Tg2576 mice, AD animal model. PLoS one 2010, 5(1):e8840
Hibino T等人, S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res 2012 Nov 7 Epub ahead of print
Hiratsuka S等人, Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 8(12), 1369-75 (2006)
国际申请号PCT/US2007/020982 (公开号WO2008042282)
国际申请号PCT/US2009/050797 (公开号WO2010009290)
国际申请号PCT/US2008/003935 (公开号WO2008118454)
Marenholz I等人, S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). BBRC 2004, 322:1111-22
Ryckman C等人, Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion J. Immunol. 170, 3233-42 (2003)
Shepherd CE等人, inflammatory S100A9 and S100A12 proteins in Alzheimers disease. Neurobiol Aging 2006, 27:1554-1563
Sinha P等人, Proinflammatory S100 proteins regulate the ackumulation of myeloid-derived suppressor cells. J Immunol 2008, 181:4666-4675
Srikrishna G等人, S100A8 and S100A9: New insights into their roles in malignancy. J Innate Immun 2012, 4:31-40
Vogl T等人, Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 2007, 13(9):1042-9
Wang L等人, Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol 2013, 190:794-804
You L等人, Silica gel accelerated aza-Michael addition of amines to α,β-unsaturated amides. Tetrahedron Lett. 2008, 49, 5147-5149。
- 还没有人留言评论。精彩留言会获得点赞!