卵巢癌标志物及其用途的制作方法
本发明涉及高级别浆液性卵巢癌(HG-SOC)的生物标志物及其用于诊断高级别浆液性卵巢癌(HG-SOC)和/或确定患有高级别浆液性卵巢癌(HG-SOC)的受试者的预后的方法和用途。
背景技术:
卵巢癌(其中高级别浆液性卵巢癌(high-grade serous ovarian cancer,HG-SOC)是最普遍的)是目前世界上最致命的妇科疾病之一。高级别浆液性卵巢癌(HG-SOC),上皮卵巢癌(EOC)的一种主要的组织学类型,是一种表征较差的、异质的且致命的疾病,其中TP53的体细胞突变是9.5-13%的EOC患者中的有癌症倾向的BRCA1/2中的常见的且遗传性的功能丧失突变(Bolton等人JAMA 2012 25;307(4):832-90)。然而,由于遗传性或散发性突变所致的疾病的总体负荷是未知的。尽管在高通量生物技术和致癌基因组(oncogenomic)研究中取得了显著进展,但是这种复杂疾病的遗传背景知之甚少,并且用于早期检测、差异诊断学、预后和疾病预测的生物标志物尚未在临床实践中实施。诊断为HG-SOC的患者面临严峻的统计值,即,即使利用标准化学疗法和放射疗法他们中的仅30%在初始诊断后会存活超过5年。原因可能是由于高肿瘤异质性、未知的组织来源位点、无症状的肿瘤生长、晚期临床检测和诊断、以及初级化学疗法后对复发的高易感性。
事实上,HG-SOC肿瘤的异质性和可靠的早期检测、预后和预测性生物标志物的缺乏表明,患者的临床状态是变化的,并且肿瘤常常对标准治疗的响应差。因此,用于风险评估和疾病形成/复发的风险的高置信度分子标志物的鉴定在从预防性到患者临床管理的各种领域中变得重要。因此,患者基于他们的存活模式的分层在从患者临床管理到特定肿瘤亚型的科学发现的各种领域中变得重要。
最近的技术进步已经促进了这种复杂疾病的研究,并且高级别浆液性卵巢癌(HG-SOC)是已经由肿瘤基因图谱(The Cancer Genome Atlas,TCGA)研究网络全面研究的癌症疾病之一。这些研究的结果表明,通过mRNA数据的表达谱,可以将患者分为四个具有生物学意义和不同的肿瘤/基因亚组:分化的、免疫反应性的、间充质的或增殖的(TCGA research network Nature 2011Vol 474;609-15)。然而,存活分析没有表明TCGA数据集中这些转录亚型之间的显著差异。基于TCGA和几个其它分组(cohort)的miRNA和mRNA表达谱的元分析(meta-analysis),已经将HG-SOC患者可靠分类为三个预后亚组,其中患者的总体存活与特定的途径和治疗结果相关联(Tang等人Int J Cancer.2014;134(2):306-18)。尽管进行了集中的研究和努力,但是与具有HG-SOC的专利相关的信息在当今不比10年前更好,因为没有可用的临床批准的预后。
最近对TCGA患者分组的HG-SOC的突变研究揭示了诸如TP53,NF1,RB1,FAT3,CSMD3,GABRA6,CDK12,BRCA1,BRCA2,SMARCB1,KRAS,NRAS,CREBBP和ERBB2的突变基因。在另一项研究中通过大量平行测序也鉴定出肿瘤抑制基因的其它突变,如BRIP,CHEK2,MRE11A,MSH6,NBN,PALB2,RAD50和RAD51C。然而,这些和其它突变尚未在其提供HG-SOC临床结果的预后的能力的背景下系统性研究。研究已经显示,在HG-SOC中,在几乎所有HG-SOC患者中报道TP53体细胞突变,并且虽然其在诸如早期诊断或形成疾病的风险预测的领域中将是有用的,但是其在患者存活预测中的应用受到限制。此外,最近报道了BRCA1或BRCA2的常规的“驱动”突变相对于野生型变体反常地与更好的患者存活相关。通常,就疾病病因学、诊断或预后而言的突变数据的研究可能由于缺乏合适的和/或高质量肿瘤样品而面临典型的统计学问题。当特定基因或基因变体的突变为罕见时,该问题可以进一步恶化。
先前研究了CHEK2突变在卵巢癌患者分组中的作用,由此,CHEK2I157T的错义变体与卵巢囊腺瘤,边缘卵巢癌和低度侵入性癌症显著相关,但不与高度卵巢癌相关(Szymanska-Pasternak等人Gynecol Oncol.2006;102(3):429-31)。在另一项研究中,Baysal等人通过焦磷酸测序进行单核苷酸多态性基因型分型并鉴定CHEK2的del1100C和A252G变体(Baysal等人Gynecol Oncol.2004;95(1):62-9)。然而,由于在与对照相比时,变体频率的统计学差异不显著,提示了CHEK2中的变异与卵巢癌的发病机制不相关。在俄罗斯卵巢癌患者中,研究了CHEK2 1100delC对卵巢癌发病机制的影响,但没有观察到关联(Krylova等人Herd Cancer Clin Pract.2007;5(3):153-56)。这些研究主要聚焦于筛选一些完善报道的CHEK2基因变体,例如,del1100C,A252G和I157T。此外,在这些以前的报告中,作者仅研究了关于疾病发病机制的特定变体的关联。然而,由于CHEK2突变的影响,HG-SOC患者的预后目前不清楚或不显著。
相关基因的互连性和相互作用是正常或肿瘤组织中生物学过程的共同特征。在生物学过程中涉及的或与HG-SOC的预后意义相关的许多潜在基因,特别是能够进行患者分层的基因使得对这些基因的鉴定成为令人生畏的任务。卵巢癌是一种高度致死的疾病,比女性生殖系统的任何其它癌症造成更多死亡,并且在女性中的癌症死亡中排名第五。在这方面,迫切需要用于预测和鉴定高级别浆液性卵巢癌(HG-SOC)患者的癌症风险评估,分层,总体存活预后和治疗响应预测的新方法。
技术实现要素:
本发明的第一方面涉及用于确定罹患高级别浆液性卵巢癌(HG-SOC)的患者的预后的方法,所述方法包括从所述患者获得的样品中确定选自CHEK2、ERN2、ADAMTSL3、ATR、ENAH、GLI2、GYPB、KIAA1324L、LRRN2、MAP3K6、MAPK15、MET、MLL4、NIPBL、PCDH15、PPP1CC、PTCH1、PTK2B、RPS6KA2、RSU1和TNC的基因中的突变的存在或不存在,其中所述ERN2基因中的突变的存在指示所述患者的预后良好,并且所述CHEK2、ADAMTSL3、ATR、ENAH、GLI2、GYPB、KIAA1324L、LRRN2、MAP3K6、MAPK15、MET、MLL4、NIPBL、PCDH15、PPP1CC、PTCH1、PTK2B、RPS6KA2、RSU1和TNC基因的任一种中的突变的存在指示所述患者的预后不良。
本发明的另一个方面涉及用于实施本文中所述的方法的试剂盒,所述试剂盒包含与选自下组的突变基因的mRNA互补的至少一种核酸探针:CHEK2、ERN2、ADAMTSL3、ATR、ENAH、GLI2、GYPB、KIAA1324L、LRRN2、MAP3K6、MAPK15、MET、MLL4、NIPBL、PCDH15、PPP1CC、PTCH1、PTK2B、RPS6KA2、RSU1和TNC。
本发明的另一个方面涉及用于预测患者发生高级别浆液性卵巢癌(HG-SOC)的风险的方法,所述方法包括从所述患者获得的样品中确定选自下组的基因中的种系突变:CHEK2、RPS6KA2和MLL4。
本发明的另一个方面涉及用于实施诊断方法的试剂盒,所述试剂盒包含与选自下组的突变基因的mRNA互补的至少一种核酸探针:CHEK2、RPS6KA2和MLL4。
参考以下附图和各个非限制性实施方案的描述,本发明的其它方面对于本领域技术人员将是显而易见的。
附图说明
在下面的描述中,参考以下图描述本发明的各个实施方案。
图1:从TCGA数据门户(data portal)下载的高级别浆液性卵巢癌(HG-SOC)的突变数据。
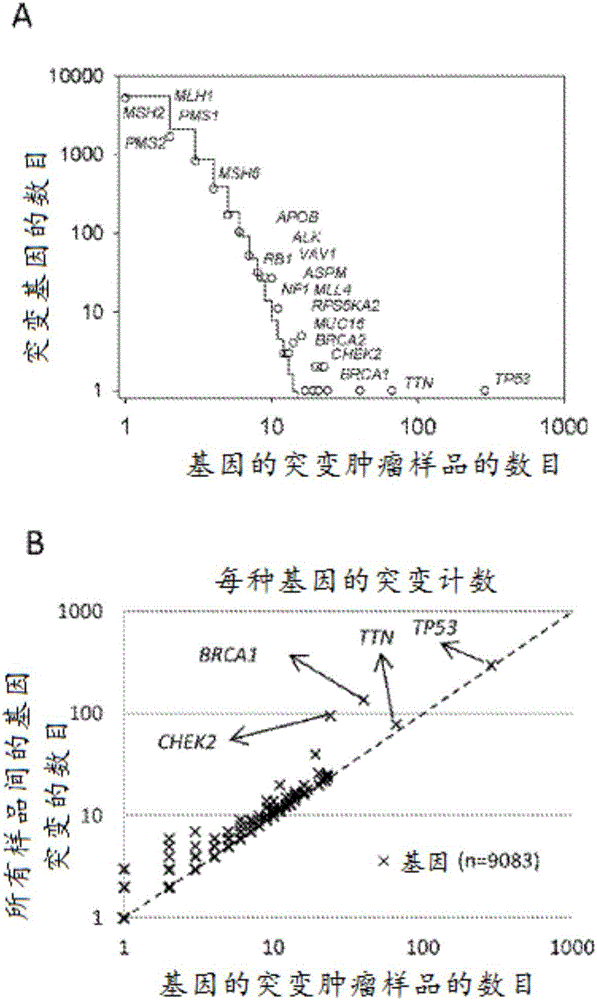
图2:HG-SOC基因中的突变的统计特征。
(A)易感驱动基因中的突变的频率分布。
(B)针对突变样品数目的不同突变的数目。
基因的散点图,其中垂直轴对应于所有样品间的突变数目,并且水平轴对应于对于给定基因具有至少一个突变的样品的数目。对角线表示每个基因的每个样品的突变数为1的假设情况。两个轴均进行log10转化。
图3:具有CHEK2突变的患者与在BRCA1,BRCA2,RPS6KA2或MLL4基因中具有突变的患者的κ相关性。
列联表中的值表示对应于行和列标签的独特样品ID的数目。计算加权κ作为一致性(agreement)的量度,并且通过Mantel-Haenszel(MH)检验估计显著性。使用StatXact-9(计算的权:平方差,得分:相等间隔)实施计算。
图4:对于455种高度突变的基因(在至少5名患者中突变)和334名患者观察到的种系,LOH或体细胞突变的热图。
图的强度对应于对于该基因和患者观察到的突变(包括沉默突变)的数目。
图5:(A)沿着TP53基因基因座的特定位点突变的样品的频率
(B)在各种基因内的特定位点处鉴定的突变。
图6:(A)通过分层聚类(hierarchical clustering)(Kendall-tau距离,完全连锁)排列的属于58种基因和22名患者的突变矩阵的提取子簇。图的强度对应于对于该基因和患者观察到的突变(包括沉默突变)的数目。
(B)从突变子簇鉴定的21种基因的子集的直接相互作用基因网络。
图7:在突变亚群中鉴定的58个基因符号的注释。
图8:通过(A)DAVID生物信息学,(B)MetaCore途径分析,(C)MetaCore过程网络分析和(D)MetaCore疾病生物标志物分析,在突变子簇中58种基因的富集分析。
图9:19种直接相互作用基因与DNA损伤信号传导,修复,凋亡,细胞增殖或免疫过程的关联。
图10:在临床、拷贝数变异、突变和表达数据集之间的合并的CHEK2信息。
图11:基于(A)CHEK2,(B)TP53,(C)BRCA1和(D)MUC16的非沉默突变706状态的TCGA HG-SOC患者的Kaplan-Meier存活曲线。
图12:(A)CHEK2突变和(B)非沉默CHEK2突变与治疗抗性的Κ相关性。列联表中的值表示对应于行和列标签的独特样品ID的数目。
图13:(A)基于CHEK2拷贝数的330名TCGA HG-SOC患者的患者分层。
(B)具有CHEK2缺失,扩增或不显著改变的样品的CHEK2表达。
(C)378个样品的肿瘤类型间的CHEK2mRNA的表达谱。378个样品来自8个输卵管样品和370个具有肿瘤级别和阶段信息的HG-SOC样品。(D)基于CHEK2表达数据的358名HG-SOC患者的预后分层。12名没有存活时间和事件的HG-SOC患者从分析中排除。高CHEK2mRNA表达与较高的风险相关,而低CHEK2mRNA表达与较低的风险相关。
图14:TCGA突变位点与已知或预测的CHEK2区域的共定位。
图15:(A)来自UCSC基因组浏览器的CHEK2的基因组基因座。显示了单个同种型(isoform)的内含子-外显子-UTR结构。
(B)来自TCGA数据库的263名高级别浆液性卵巢癌患者间的CHEK2同种型的RNA-seq表达。
图16:(A)沿着CHEK2基因座的基因组图式的DNA突变的位置。外显子块从5’到3’顺序编号。倒置三角形代表外显子上突变的位置。倒置三角形上方的数字指示具有突变(包括同义突变)的患者的数目。
(B)氨基酸序列上预期突变的位置。倒置三角形中的字母表指示参考氨基酸残基,而具有非同义突变的患者的数目显示在倒置三角形上方。矩形块中的数字指示氨基酸残基跨度。
(C)计算建模和分子动力学模拟后Chk2蛋白松弛状态的代表性晶体结构。所有Chk2突变由有色球体表示,其指示对应于翻译后的DNA突变的残基的位置。使用CHEK2同种型1(NM_007194/NP_009125/O96017)作为参照同种型。叉头相关(forkhead-associated,FHA)域,激酶域和核定位信号(NLS)分别标记为粉红色,蓝色和青色。文氏图(Venn diagram)将患者的数目与在两个不同的核苷酸位置处观察到的突变进行比较。图没有按比例绘制。
图17:基于非沉默突变状态的21种存活显著基因的预后显著性(对数秩统计p值≤0.05,#突变≥5和#非突变≥5)。
图18:(A)鉴定的基因突变簇与突变状态呈预后显著的基因之间的共同基因的文氏图。
(B)基于21-基因标签(21-gene signature)的突变状态的预后分层。
(C)基于CHEK2基因和20-基因标签的突变的预后分层。
图19:由21种基因突变标签分类的患者与治疗抗性的K相关性。列联表中的值表示对应于行和列标签的独特样品ID的数目。
图20:在预后标签中的21个基因符号的注释。
图21:通过(A)DAVID生物信息学,(B)MetaCore途径分析,(C)MetaCore过程网络分析和(D)MetaCore对21种存活显著基因的富集分析。
图22:(A)种系,LOH和体细胞突变,(B)种系突变,(C)LOH和(D)体细胞突变的不良预后亚组中21种预后基因和58名患者的非沉默突变的簇。基因和患者通过分层聚类(kendall-tau距离和完全连锁)进行排序。
图23:CHEK2-MLL4-RPS6KA2确定的EOC肿瘤亚类的遗传和临床特征(G:种系,S:体细胞,L:LOH)。
图24:(A)参与各种卵巢癌亚型的病因学的关键基因。
(B)在肿瘤等级和阶段的HG-SOC样品间CHEK2mRNA的表达(在红色箱线图中表示)。通过Mann-Whitney检验计算正常和肿瘤样品之间的差异表达。
具体实施方式
来自TCGA的HG-SOC患者的全基因组突变和临床数据集的综合生物信息学和统计分析允许鉴定其突变状态可将患者分层到不同存活亚组中的预后基因(生物标志物)。还鉴定了与患者的不良预后相关的基因标签,其中不同的肿瘤亚组得到表征并且潜在地由这些标签基因的种系或体细胞突变驱动。
基于它们的存活模式的用于风险评估和患病患者分层的新型分子标志物的鉴定在从发现特定肿瘤类别和亚型到改进的预防,早期诊断和临床管理的各个领域中变得重要。
本发明的第一个方面涉及用于确定罹患高级别浆液性卵巢癌(HG-SOC)的患者的预后的方法,所述方法包括从所述患者获得的样品中确定选自CHEK2、ERN2、ADAMTSL3、ATR、ENAH、GLI2、GYPB、KIAA1324L、LRRN2、MAP3K6、MAPK15、MET、MLL4、NIPBL、PCDH15、PPP1CC、PTCH1、PTK2B、RPS6KA2、RSU1和TNC的基因中的突变的存在或不存在,其中所述ERN2基因中的突变的存在指示所述患者的预后良好,并且所述CHEK2、ADAMTSL3、ATR、ENAH、GLI2、GYPB、KIAA1324L、LRRN2、MAP3K6、MAPK15、MET、MLL4、NIPBL、PCDH15、PPP1CC、PTCH1、PTK2B、RPS6KA2、RSU1和TNC基因的任一种中的突变的存在指示所述患者的预后不良。
这些突变基因或标志物基因可以在组织和/或体液样品中,例如在血液样品中检测,并且因此提供了用于预后罹患HG-SOC的患者的新方法。由于此类方法不需要昂贵的设备,新方法可以由任何内科医生进行。优选地,直接(即在DNA水平上),或依靠基因产物(包括mRNA或蛋白质)检测基因或标志物基因。优选地,通过测序方法,如通过Illumina或ABI SOLID测序平台测序来检测突变。任何合适的方法,如PCT熔化技术也可以适合于确定突变或本领域已知的用于确定序列变异的任何其它方法。
对于本发明的基因标志物的检测,可以采用特异性结合配偶体。在一些实施方案中,特异性结合配偶体可用于检测样品中标志物的存在,其中标志物是蛋白质或RNA。标志物及其结合配偶体表示分子的结合对,其通过多种分子力(包括例如离子,共价,疏水,范德华力和氢键键合)的任一种彼此相互作用。优选地,此结合是特异性的。“特异性结合”是指结合对的成员优先彼此结合,即通常比对非特异性结合配偶体以显著更高的亲和力结合。因此,对特异性结合配偶体的结合亲和力通常比非特异性结合配偶体的结合亲和力高至少10倍,优选至少100倍。结合配偶体也可以是特异性的,因为它们以比非突变形式更高的亲和力结合基因产物(即RNA或蛋白质)的突变形式,优选地,差异是亲和力增加至少10倍。
确定预后包括风险分层和预测不良结果的可能性。这可以就某个时间段而言做出。在多个实施方案中,时间段是5年。本发明意义上的不良或不利结果包括患者状况的恶化,例如如本文中所述,由于在诊断或预后确定后5年内的转移或死亡。有利或积极的结果包括维持或改善患者的状况,例如由于积极响应化学疗法如顺铂疗法,或5年或更长时间的存活。
总体上,技术可以改善患者风险评估,管理和咨询,以及为临床环境中治疗人卵巢癌的个性化药物策略提供优化解决方案。
在多个实施方案中,检测CHEK2基因中的突变。检查点激酶2(CHEK2)编码参与细胞周期检查点控制,DNA损伤反应信号转导和细胞凋亡调节的核丝氨酸/苏氨酸蛋白激酶。CHEK2。在DNA损伤的存在下,CHEK2磷酸化下游细胞周期调节剂如p53,Cdc25和BRCA1以激活检查点修复或恢复应答,以及同时延迟进入有丝分裂。偏离其正常生理功能可能有助于疾病发病机制。
在多个实施方案中,CHEK2标志物基因包含如SEQ ID NO.1(NM_001005735)、或SEQ ID NO.67(NM_001257387)、或SEQ ID NO.68(NM_007194)、或SEQ ID NO.69(NM_145862)的任一项中所示的序列。这些序列是CHEK2已知的最常见的序列,并且这里已经证明了这些标准序列的突变或变异与罹患高级别浆液性卵巢癌的患者的不良存活相关。CHEK2基因的畸变先前没有与HG-SOC中的总体存活时间或治疗响应的预后相关联。334名HG-SOC患者的相对大和设计良好的分组允许鉴定先前未鉴定的患者亚类,其具有潜在非常差的治疗反应和总体存活(5年总体存活率为0%)。当在CHEK2标志物基因中检测到突变时,可以建议患者依靠姑息护理。这将为患者节省在用化学疗法的进取性治疗的情况下牵涉的不必要的费用和痛苦。
在多个实施方案中,ADAMTSL3标志物基因包含如SEQ ID NO 5(NM_001301110)和SEQ ID NO.71(NM_207517)的任一项中所示的序列。
在多个实施方案中,ATR标志物基因包含如SEQ ID NO.6(NM_001184)中所示的序列。
在多个实施方案中,ENAH标志物基因包含如SEQ ID NO.7(NM_001008493)和SEQ ID NO.72(NM_018212)的任一项中所示的序列。
在多个实施方案中,GLI2标志物基因包含如SEQ ID NO.8(NM_005270)中所示的序列。
在多个实施方案中,GYPB标志物基因包含如SEQ ID NO.9(NM_001304382)和SEQ ID NO.73(NM_002100)的任一项中所示的序列。
在多个实施方案中,KIAA1324L标志物基因包含如SEQ ID NO.10(NM_001142749)、SEQ ID NO.74(NM_001291990)、SEQ ID NO.75(NM_001291991)、和SEQ ID NO.76(NM_152748)的任一项中所示的序列。
在多个实施方案中,LRRN2标志物基因包含如SEQ ID NO.11(NM_006338)和SEQ ID NO.77(NM_201630)的任一项中所示的序列。
在多个实施方案中,MAP3K6标志物基因包含如SEQ ID NO.12(NM_001297609)和SEQ ID NO.78(NM_004672)的任一项中所示的序列。
在多个实施方案中,MAPK15标志物基因包含如SEQ ID NO.13(NM_139021)中所示的序列。
在多个实施方案中,MET标志物基因包含如SEQ ID NO.14(NM_000245)和SEQ ID NO.79(NM_001127500)的任一项中所示的序列。
在多个实施方案中,MLL4标志物基因包含如SEQ ID NO.4(NM_014727)中所示的序列。标志物也可以称作KMT2B。
在多个实施方案中,NIPBL标志物基因包含如SEQ ID NO.15(NM_015384)和SEQ ID NO.80(NM_133433)的任一项中所示的序列。
在多个实施方案中,PCDH15标志物基因包含如SEQ ID NO.16(NM_001142763)、SEQ ID NO.81(NM_001142764)、SEQ ID NO.82(NM_001142765)、SEQ ID NO.83(NM_001142766)、SEQ ID NO.84(NM_001142767)、SEQ ID NO.85(NM_001142768)、SEQ ID NO.86(NM_001142769)、SEQ ID NO.87(NM_001142770)、SEQ ID NO.88(NM_001142771)、SEQ ID NO.89(NM_001142772)、SEQ ID NO.90(NM_001142773)、和SEQ ID NO.91(NM_033056)的任一项中所示的序列。
在多个实施方案中,PPP1CC标志物基因包含如SEQ ID NO.17(NM_001244974)和SEQ ID NO.92(NM_002710)的任一项中所示的序列。
在多个实施方案中,PTCH1标志物基因包含如SEQ ID NO.18(NM_000264)、SEQ ID NO.93(NM_001083602)、SEQ ID NO.94(NM_001083603)、SEQ ID NO.95(NM_001083604)、SEQ ID NO.96(NM_001083605)、SEQ ID NO.97(NM_001083606)、和SEQ ID NO.98(NM_001083607)的任一项中所示的序列。
在多个实施方案中,PTK2B标志物基因包含如SEQ ID NO.19(NM_004103)、SEQ ID NO.99(NM_173174)、SEQ ID NO.100(NM_173175)、和SEQ ID NO.101(NM_173176)的任一项中所示的序列。
在多个实施方案中,RPS6KA2标志物基因包含如SEQ ID NO.3(NM_001006932)和SEQ ID NO.70(NM_021135)的任一项中所示的序列。
在多个实施方案中,RSU1标志物基因包含如SEQ ID NO.20(NM_012425)和SEQ ID NO.102(NM_152724)的任一项中所示的序列。
在多个实施方案中,TNC标志物基因包含如SEQ ID NO.21(NM_002160)中所示的序列。
所有前述序列是相应的野生型序列,其可以用作检测这些基因中的突变的参考。括号中的代码表示其各自的数据库条目编号(databank entry number)。
在多个实施方案中,ERN2标志物基因中的突变指示患者的有利的治疗结果。在多个实施方案中,ERN2标志物包含SEQ ID NO.2(NM_033266)中所示的核酸序列。该序列是对于ERN2已知的最常见的序列,并且此处已经证明来自该标准野生型序列的突变或变异与罹患高级别浆液性卵巢癌的患者的更好的存活相关,总体5年存活率为37%。由于ERN2突变与患者的更好的总体存活期相关,所以被鉴定为在ERN2标志物中具有突变的HG-SOC患者可以用化学疗法和其它治疗例如放射治疗和切除进行治疗。
在多个实施方案中,该方法还包括如下步骤:通过卵巢组织活体组织切片的显微分析或通过超声或确认患者中的卵巢癌特别是HG-SOC的预后的本领域中已知的任何其它方法来确认预后。超声可以在外部进行或优选在阴道内进行,以更好地确定任何肿瘤生长的大小。确认预后的方法还可以包括检测卵巢癌,优选HG-SOC的已知标志物中的突变,例如TP53,BRCA1或BRCA2中的突变。结果指示,CHEK2和BRCA1是相互排斥的突变,其可以能够对会或不会良好地响应化学疗法的患者进行分层。具有CHEK2标志物突变的患者通常不良好响应化学疗法。
在多个实施方案中,CHEK2标志物中的突变位于CHEK2标志物的外显子10、11或15中。在多个实施方案中,CHEK2基因的末端外显子15表达核定位序列。HG-SOC患者中的CHEK2突变是患者存活预后的强不良指标,并且与疗法抗性相关。假设但不限于任何理论,它可能是由于核定位位点的突变,这防止蛋白的核输入,并随后导致单倍不足(haplo-insufficiency)。在多个实施方案中,突变位于对应于编码氨基酸R346,T383,R406,R519,P522,R535和/或P536的密码子的序列位置。这些氨基酸存在于CHEK2的核定位位点中。
在多个实施方案中,本发明的方法可以进一步包括确定上文所示的那些标志物的一种或多种额外的标志物中突变的存在。如果检测到一个或多个额外的突变标志物基因,那么这可以提高该方法的准确性。在本发明方法的某些实施方案中,确定至少2、3、4、5、6、7、8、9、10、20、30、40、45、50或58个或更多个额外标志物中的突变。
在确定CHEK2标志物中的突变的多个实施方案中,所述方法还包括确定选自下组的基因的任一种中的突变:ABCA3、ADAM15、ADAMTSL3、ALK、ANKHD1-EIF4EBP3、ANKMY2、ANXA7、ASPM、CDC27、CHD6、CHL1、DPYSL4、ENAH、EP400、ERBB2IP、FN1、FOXO3、GCLC、GLI2、GLI3、GYPB、GZMB、HLA-G、HNF1A、INPP5D、INSR、ITGB2、KIF3B、KIF4B、KTN1、LRRN2、MAD1L1、MAP3K6、MAPK15、MET、MKL1、MLL4、MYO5C、NUMA1、PDGFRA、PHLPP、PIK3C2B、PKP4、PLAGL2、PPARA、PRKCI、PTK2B、RAB3D、ROR2、RPS6KA2、RSU1、SPTB、TBK1、TNK2、TP53、VAV1和ZC3H11A。
在多个实施方案中,ABCA3标志物基因包含如SEQ ID NO.22(NM_001089)中所示的序列。
在多个实施方案中,ADAM15标志物基因包含如SEQ ID NO.23(NM_001261464)、SEQ ID NO103(NM_001261465)、SEQ ID NO.104(NM_001261466)、SEQ ID NO.105(NM_003815)、SEQ ID NO.106(NM_207191)、SEQ ID NO.107(NM_207194)、SEQ ID NO.108(NM_207195)、SEQ ID NO.109(NM_207196)、SEQ ID NO.110(NM_207197)、SEQ ID NO.111(NR_048577)、SEQ ID NO.112(NR_048578)、和SEQ ID NO.113(NR_048579)中所示的序列。
在多个实施方案中,ADAMTSL3标志物基因包含如SEQ ID NO 5(NM_001301110)和SEQ ID NO.71(NM_207517)的任一项中所示的序列。
在多个实施方案中,ALK标志物基因包含如SEQ ID NO.24(NM_004304)中所示的序列。
在多个实施方案中,ANKHD1-EIF4EBP3标志物基因包含如SEQ ID NO.25(NM_020690)中所示的序列。
在多个实施方案中,ANKMY2标志物基因包含如SEQ ID NO.26(NM_020319)中所示的序列。
在多个实施方案中,ANXA7标志物基因包含如SEQ ID NO.27(NM_001156)和SEQ ID NO.114(NM_004034)中所示的序列。
在多个实施方案中,ASPM标志物基因包含如SEQ ID NO.28(NM_001206846)和SEQ ID NO.115(NM_018136)中所示的序列。
在多个实施方案中,CDC27标志物基因包含如SEQ ID NO.29(NM_001114091)、SEQ ID NO.116(NM_001256)、SEQ ID NO.117(NM_001293089)、和SEQ ID NO.118(NM_001293091)中所示的序列。
在多个实施方案中,CHD6标志物基因包含如SEQ ID NO.30(NM_032221)中所示的序列。
在多个实施方案中,CHL1标志物基因包含如SEQ ID NO.31(NM_001253387)、SEQ ID NO.119(NM_001253388)、SEQ ID NO.120(NM_006614)、和SEQ ID NO.121(NR_045572)中所示的序列。
在多个实施方案中,DPYSL4标志物基因包含如SEQ ID NO.32(NM_006426)中所示的序列。
在多个实施方案中,ENAH标志物基因包含如SEQ ID NO.7(NM_001008493)和SEQ ID NO.72(NM_018212)的任一项中所示的序列。
在多个实施方案中,GLI2标志物基因包含如SEQ ID NO.8(NM_005270)中所示的序列。
在多个实施方案中,EP400标志物基因包含如SEQ ID NO.33(NM_015409)中所示的序列。
在多个实施方案中,ERBB2IP标志物基因包含如SEQ ID NO.34(NM_001006600)、SEQ ID NO.122(NM_001253697)、SEQ ID NO.123(NM_001253698)、SEQ ID NO.124(NM_001253699)、SEQ ID NO.125(NM_001253701)和SEQ ID NO.126(NM_018695)中所示的序列。
在多个实施方案中,FN1标志物基因包含如SEQ ID NO.35(NM_002026)、SEQ ID NO.127(NM_054034)、SEQ ID NO.128(NM_212474)、SEQ ID NO.129(NM_212476)、SEQ ID NO.130(NM_212478)、和SEQ ID NO.131(NM_212482)中所示的序列。
在多个实施方案中,FOXO3标志物基因包含如SEQ ID NO.36(NM_001455)和SEQ ID NO.132(NM_201559)中所示的序列。
在多个实施方案中,GCLC标志物基因包含如SEQ ID NO.37(NM_001197115)和SEQ ID NO.133(NM_001498)中所示的序列。
在多个实施方案中,GLI3标志物基因包含如SEQ ID NO.38(NM_000168)中所示的序列。
在多个实施方案中,GYPB标志物基因包含如SEQ ID NO.9(NM_001304382)和SEQ ID NO.73(NM_002100)的任一项中所示的序列。
在多个实施方案中,GZMB标志物基因包含如SEQ ID NO.39(NM_004131)中所示的序列。
在多个实施方案中,HLA-G标志物基因包含如SEQ ID NO.40(NM_002127)中所示的序列。
在多个实施方案中,HNF1A标志物基因包含如SEQ ID NO.41(NM_000545)中所示的序列。
在多个实施方案中,INPP5D标志物基因包含如SEQ ID NO.42(NM_001017915)和SEQ ID NO.134(NM_005541)中所示的序列。
在多个实施方案中,INSR标志物基因包含如SEQ ID NO.43(NM_000208)和SEQ ID NO.135(NM_001079817)中所示的序列。
在多个实施方案中,ITGB2标志物基因包含如SEQ ID NO.44(NM_000211)、SEQ ID NO.136(NM_001127491)、和SEQ ID NO.137(NM_001303238)中所示的序列。
在多个实施方案中,KIF3B标志物基因包含如SEQ ID NO.45(NM_004798)中所示的序列。
在多个实施方案中,KIF4B标志物基因包含如SEQ ID NO.46(NM_001099293)中所示的序列。
在多个实施方案中,KTN1标志物基因包含如SEQ ID NO.47(NM_001079521)、SEQ ID NO.138(NM_001079522)、SEQ ID NO.139(NM_001271014)、SEQ ID NO.140(NM_004986)、SEQ ID NO.141(NR_073128)、和SEQ ID NO.142(NR_073129)中所示的序列。
在多个实施方案中,LRRN2标志物基因包含如SEQ ID NO.11(NM_006338)和SEQ ID NO.77(NM_201630)的任一项中所示的序列。
在多个实施方案中,MAD1L1标志物基因包含如SEQ ID NO.48(NM_001013836)、SEQ ID NO.143(NM_001013837)、SEQ ID NO.144(NM_001304523)、SEQ ID NO.145(NM_001304524)、SEQ ID NO.146(NM_001304525)、和SEQ ID NO.147(NM_003550)中所示的序列。
在多个实施方案中,MAP3K6标志物基因包含如SEQ ID NO.12(NM_001297609)和SEQ ID NO.78(NM_004672)的任一项中所示的序列。
在多个实施方案中,MAPK15标志物基因包含如SEQ ID NO.13(NM_139021)中所示的序列。
在多个实施方案中,MET标志物基因包含如SEQ ID NO.14(NM_000245)和SEQ ID NO.79(NM_001127500)的任一项中所示的序列。
在多个实施方案中,MKL1标志物基因包含如SEQ ID NO.49(NM_001282660)、SEQ ID NO.148(NM_001282661)、SEQ ID NO.149(NM_001282662)、和SEQ ID NO.150(NM_020831)中所示的序列。
在多个实施方案中,MLL4标志物基因包含如SEQ ID NO.4(NM_014727)中所示的序列。该基因也可以称作KMT2B。
在多个实施方案中,MYO5C标志物基因包含如SEQ ID NO.50(NM_018728)中所示的序列。
在多个实施方案中,NUMA1标志物基因包含如SEQ ID NO.51(NM_001286561)、SEQ ID NO.151(NM_006185)、和SEQ ID NO.152(NR_104476)中所示的序列。
在多个实施方案中,PDGFRA标志物基因包含如SEQ ID NO.52(NM_006206)中所示的序列。
在多个实施方案中,PHLPP标志物基因包含如SEQ ID NO.53(NM_194449)中所示的序列。该基因也可以称作PHLPP1。
在多个实施方案中,PIK3C2B标志物基因包含如SEQ ID NO.54(NM_002646)中所示的序列。
在多个实施方案中,PKP4标志物基因包含如SEQ ID NO.55(NM_001005476)、SEQ ID NO.153(NM_001304969)、SEQ ID NO.154(NM_001304970)、SEQ ID NO.155(NM_001304971)、和SEQ ID NO.156(NM_003628)中所示的序列。
在多个实施方案中,PLAGL2标志物基因包含如SEQ ID NO.56(NM_002657)中所示的序列。
在多个实施方案中,PPARA标志物基因包含如SEQ ID NO.57(NM_001001928),SEQ ID NO.157(NM_005036)中所示的序列。
在多个实施方案中,PRKCI标志物基因包含如SEQ ID NO.58(NM_002740)中所示的序列。
在多个实施方案中,PTK2B标志物基因包含如SEQ ID NO.19(NM_004103)、SEQ ID NO.99(NM_173174)、SEQ ID NO.100(NM_173175)、和SEQ ID NO.101(NM_173176)的任一项中所示的序列。
在多个实施方案中,RAB3D标志物基因包含如SEQ ID NO.59(NM_004283)中所示的序列。
在多个实施方案中,ROR2标志物基因包含如SEQ ID NO.60(NM_004560)中所示的序列。
在多个实施方案中,RPS6KA2标志物基因包含如SEQ ID NO.3(NM_001006932)和SEQ ID NO.70(NM_021135)的任一项中所示的序列。
在多个实施方案中,RSU1标志物基因包含如SEQ ID NO.20(NM_012425)和SEQ ID NO.102(NM_152724)中所示的序列。
在多个实施方案中,SPTB标志物基因包含如SEQ ID NO.61(NM_000347)和SEQ ID NO.158(NM_001024858)中所示的序列。
在多个实施方案中,TBK1标志物基因包含如SEQ ID NO.62(NM_013254)中所示的序列。
在多个实施方案中,TNK2标志物基因包含如SEQ ID NO.63(NM_001010938)和SEQ ID NO.159(NM_005781)中所示的序列。
在多个实施方案中,TP53标志物基因包含如SEQ ID NO.64(NM_000546)、SEQ ID NO.160(NM_001126112)、SEQ ID NO.161(NM_001126113)、SEQ ID NO.162(NM_001126114)、SEQ ID NO.163(NM_001126115)、SEQ ID NO.164(NM_001126116)、SEQ ID NO.165(NM_001126117)、SEQ ID NO.166(NM_001126118)、SEQ ID NO.167(NM_001276695)、SEQ ID NO.168(NM_001276696)、SEQ ID NO.169(NM_001276697)、SEQ ID NO.170(NM_001276698)、SEQ ID NO.171(NM_001276699)、SEQ ID NO.172(NM_001276760)、和SEQ ID NO.173(NM_001276761)中所示的序列。
在多个实施方案中,VAV1标志物基因包含如SEQ ID NO.65(NM_001258206)、SEQ ID NO.174(NM_001258207)和SEQ ID NO.175(NM_005428)中所示的序列。
在多个实施方案中,ZC3H11A标志物基因包含如SEQ ID NO.66(NM_014827)中所示的序列。
所有前述序列是相应的野生型序列,其可以用作检测这些基因中的突变的参考。括号中的代码表示其各自的数据库条目编号。
在多个实施方案中,确定一组标志物基因的任一种中的突变的存在或不存在,包括检测选自下组的核苷酸序列的突变的存在或不存在:SEQ ID NO.3-5、7-9、11-14、19、20、22-73、77-79、99-175中所示的核酸序列。
在确定CHEK2标志物中的突变的多个实施方案中,该方法还包括确定选自下组的基因的任一种中的突变:ADAMTSL3、ATR、ENAH、ERN2、GLI2、GYPB、KIAA1324L、LRRN2、MAP3K6、MAPK15、MET、MLL4、NIPBL、PCDH15、PPP1CC、PTCH1、PTK2B、RPS6KA2、RSU1和TNC。在一些实施方案中,另外确定至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或所有20种基因中的突变。
在多个实施方案中,该方法还包括确定以下标志物的每种中的突变的存在:ADAMTSL3,ATR,CHEK2,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC。任何前述基因中的任何突变的检测指示患者的不利的治疗结果。在多个实施方案中,标志物包含SEQ ID NO.3-21,或70-102中所示的核酸序列。上述标志物的任一种中的突变与患者的较差的总体存活相关联。
在多个实施方案中,该方法还包括确定选自下组的标志物序列的任一种中的突变:SEQ ID NO.1、3-21和67-102中所示的核酸序列。
在多个实施方案中,该方法包括确定基因标志物的组中的突变的存在或不存在,所述基因标志物的组包含CHEK2,ERN2,ADAMTSL3,ATR,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC的任何2种或更多种,3种或更多种,4种或更多种,5种或更多种,6种或更多种,7种或更多种,8种或更多种,9种或更多种,10种或更多种,11种或更多种,12种或更多种,13种或更多种,14种或更多种,15种或更多种,16种或更多种,17种或更多种,18种或更多种,19种或更多种,20种或更多种,或全部21种。在此实施方案中,包含21种基因(DNA和/或mRNA和/或蛋白质)的组合突变组或标签可以用于将患者分组分层为低和高风险亚组。如果在ERN2中观察到突变或在ADAMTSL3,ATR,CHEK2,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC中没有观察到突变,那么将HG-SOC患者分类为低风险,如果在ERN2中没有观察到突变,并且在ADAMTSL3,ATR,CHEK2,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC中观察到突变,那么将HG-SOC患者分类为高风险。在多个实施方案中,该方法可包括确定包含SEQ ID NO.:1-21中所示的非突变序列的标志物组中突变的存在。
在多个实施方案中,所述方法包括确定标志物组中的突变,所述标志物组包含ADAMTSL3,ATR,CHEK2,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC,其中在基因标志物组的任一种中的突变表示患者的不利的治疗结果。在多个实施方案中,该方法包括确定具有SEQ ID NO.1和3-21中所示的野生型序列的标志物组中的突变的存在或不存在,其中突变的存在指示所述受试者具有预后不良。如果在ERN2中没有观察到突变,并且在ADAMTSL3,ATR,CHEK2,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC中观察到突变,那么将HG-SOC患者分类为高风险。
在多个实施方案中,通过CHEK2,RPS6KA2和/或MLL4标志物中的种系突变确定第一肿瘤亚型。在多个实施方案中,通过检测如SEQ ID NO:1、3和/或4的任一种中所示的核酸序列中的种系突变的存在确定第一肿瘤亚型,所述序列对应于相应的野生型序列。
在多个实施方案中,CHEK2标志物基因的野生型序列包含如SEQ ID NO.1(NM_001005735)、或SEQ ID NO.67(NM_001257387)、或SEQ ID NO.68(NM_007194)、或SEQ ID NO.69(NM_145862)的任一项中所示的序列。RPS6KA2标志物基因的野生型序列包含如SEQ ID NO.3(NM_001006932),和SEQ ID NO.70(NM_021135)的任一项中所示的序列。MLL4标志物基因的野生型序列包含如SEQ ID NO.4(NM_014727)中所示的序列。该基因也可以称为KMT2B。
在多个实施方案中,所述方法包括确定基因标志物组中突变的存在或不存在,所述基因标志物组包含CHEK2,ABCA3,ADAM15,ADAMTSL3,ALK,ANKHD1-EIF4EBP3,ANKMY2,ANXA7,ASPM,CDC27,CHD6,CHEK2,CHL1,DPYSL4,ENAH,EP400,ERBB2IP,FN1,FOXO3,GCLC,GLI2,GLI3,GYPB,GZMB,HLA-G,HNF1A,INPP5D,INSR,ITGB2,KIF3B,KIF4B,KTN1,LRRN2,MAD1L1,MAP3K6,MAPK15,MET,MKL1,MLL4,MYO5C,NUMA1,PDGFRA,PHLPP,PIK3C2B,PKP4,PLAGL2,PPARA,PRKCI,PTK2B,RAB3D,ROR2,RPS6KA2,RSU1,SPTB,TBK1,TNK2,TP53,VAV1和ZC3H11A。在此实施方案中,组合突变组或标签包含在7%的HG-SOC患者中相对经常突变的58种基因。该组可以用于鉴定具有不良预后的HG-SOC患者。在多个实施方案中,所述方法包括确定基因标志物组中突变的存在或不存在,包括检测SEQ ID NO 3-5、7-9、11-14、19、20、22-73、77-79、99-175中所示的任何一个或多个核苷酸序列中的突变的存在或不存在。
在多个实施方案中,可以用于预后的鉴定突变在图5中相对于其染色体位点位置列出。本领域技术人员可以使用诸如d-chip软件或其它可用软件等标准软件基于此信息容易地获得特异性突变。
本发明的另一方面涉及用于实施本文所述方法的试剂盒,所述试剂盒包含至少一种能够检测下列任一项中的突变的检测试剂,如与野生型或突变的mRNA互补的核酸探针或允许扩增并且然后测序扩增序列的引物,或允许直接测序的引物:ABCA3,ADAM15,ADAMTSL3,ALK,ANKHD1-EIF4EBP3,ANKMY2,ANXA7,ASPM,CDC27,CHD6,CHL1,DPYSL4,ENAH,EP400,ERBB2IP,FN1,FOXO3,GCLC,GLI2,GLI3,GYPB,GZMB,HLA-G,HNF1A,INPP5D,INSR,ITGB2,KIF3B,KIF4B,KTN1,LRRN2,MAD1L1,MAP3K6,MAPK15,MET,MKL1,MLL4,MYO5C,NUMA1,PDGFRA,PHLPP,PIK3C2B,PKP4,PLAGL2,PPARA,PRKCI,PTK2B,RAB3D,ROR2,RPS6KA2,RSU1,SPTB,TBK1,TNK2,TP53,VAV1和ZC3H11A标志物基因。
所述标志物基因中的突变是上文关于本发明方法所描述的突变,并且它们的检测可以通过标准测序方法进行。
在多个实施方案中,检测试剂是与SEQ ID NO.1-21和67-102中所示的任一序列的野生型mRNA互补的核酸探针。
在多个实施方案中,试剂盒包含与ABCA3,ADAM15,ADAMTSL3,ALK,ANKHD1-EIF4EBP3,ANKMY2,ANXA7,ASPM,CDC27,CHD6,CHL1,DPYSL4,ENAH,EP400,ERBB2IP,FN1,FOXO3,GCLC,GLI2,GLI3,GYPB,GZMB,HLA-G,HNF1A,INPP5D,INSR,ITGB2,KIF3B,KIF4B,KTN1,LRRN2,MAD1L1,MAP3K6,MAPK15,MET,MKL1,MLL4,MYO5C,NUMA1,PDGFRA,PHLPP,PIK3C2B,PKP4,PLAGL2,PPARA,PRKCI,PTK2B,RAB3D,ROR2,RPS6KA2,RSU1,SPTB,TBK1,TNK2,TP53,VAV1和ZC3H11A标志物基因的任一种的mRNA互补的至少一种核酸探针。
在多个实施方案中,试剂盒包含与ADAMTSL3,ATR,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC标志物基因的mRNA互补的核酸探针组。
在多个实施方案中,试剂盒包含与SEQ ID NO 1-175中所示的标志物基因序列的任一种的mRNA互补的至少一个核酸探针,以及任选地书面说明,用于:从患者的样品中提取核酸,并将所述核酸与DNA微阵列杂交;并为患者获得总体存活的预后或治疗结果的预测。
在多个实施方案中,试剂盒包含与如SEQ ID NO 1-21,67-102中所示的标志物基因序列的mRNA互补的核酸探针组。
在多个实施方案中,试剂盒还包含与如SEQ ID NO 3-5,7-9,11-14,19,20,22-73,77-79和99-175中所示的标志物基因序列的任一种的mRNA互补的至少一种核酸探针。
在多个实施方案中,探针能够检测标志物中的突变。这可以使用与标志物互补的探针实现,从而当它们与PCR熔解技术一起使用时,在更高温度下突变体和探针之间的杂交亲和力小于标准核酸和探针之间的杂交亲和力,从而可以鉴定突变。备选探针可以包括突变,否则与标志物序列的任一种的野生型mRNA基本上互补。
在多个实施方案中,试剂盒包含与ABCA3,ADAM15,ADAMTSL3,ALK,ANKHD1-EIF4EBP3,ANKMY2,ANXA7,ASPM,CDC27,CHD6,CHL1,DPYSL4,ENAH,EP400,ERBB2IP,FN1,FOXO3,GCLC,GLI2,GLI3,GYPB,GZMB,HLA-G,HNF1A,INPP5D,INSR,ITGB2,KIF3B,KIF4B,KTN1,LRRN2,MAD1L1,MAP3K6,MAPK15,MET,MKL1,MLL4,MYO5C,NUMA1,PDGFRA,PHLPP,PIK3C2B,PKP4,PLAGL2,PPARA,PRKCI,PTK2B,RAB3D,ROR2,RPS6KA2,RSU1,SPTB,TBK1,TNK2,TP53,VAV1和ZC3H11A标志物基因的mRNA互补的核酸探针组。
在多个实施方案中,所述试剂盒包含与如SEQ ID NO 1、3-5、7-9、11-14、19、20和22-69中所示的标志物序列的mRNA互补的核酸探针组。
本发明的另一方面涉及用于预测患者发生高级别浆液性卵巢癌(HG-SOC)的风险的方法,包括确定从所述患者获得的样品中的选自CHEK2,RPS6KA2和MLL4的基因中的种系突变的存在或不存在,其中在CHEK2,RPS6KA2和/或MLL4基因中的突变的存在指示患者患HG-SOC。
在本发明的多个实施方案中,通过分析从患者获得的样品来检测一种或多种标志物基因中的突变。样品通常含有核酸,并且可以例如是体液,细胞或组织样品。体液包括但不限于血液,血浆,血清,脑脊液,耵聍(耳垢),内淋巴和外淋巴,胃液,粘液(包括鼻引流和痰),腹膜液,胸膜液,唾液,皮脂(皮肤油),精液,汗液,眼泪,阴道分泌物,乳头抽吸液,呕吐物和尿液。在上文详述的方法的某些实施方案中,体液选自下组:血液,血清,血浆,尿液和唾液。组织样品可以是卵巢组织,并且细胞样品可以包含来自卵巢或输卵管组织的细胞。
本技术还包括使用CHEK2和/或RPS6KA2和/或MLL4基因(DNA和/或mRNA和/或蛋白)的种系突变作为预测健康女性的HG-SOC启动和形成风险中的危险因素。
在多个实施方案中,种系突变指示所述患者患HG-SOC的风险增加。
在多个实施方案中,可以用于诊断的鉴定的种系突变列于图23中。
如本文中提及,诊断方法可以改进鉴定有卵巢癌的遗传性和体细胞突变的高风险的女性的努力,所述卵巢癌与那些与p53体细胞突变和种系BRAC1/BRAC2突变相关的卵巢癌截然不同。
本发明的另一个方面涉及用于实施诊断方法的试剂盒,所述试剂盒包含与CHEK2、RPS6KA2和MLL4标志物的任一项的mRNA互补的至少一种核酸探针。
在多个实施方案中,与mRNA互补的核酸探针包含与SEQ ID NO 1、3、和/或4中所示的任一种核酸序列的mRNA互补的标志物序列,和任选书面用法说明,用于:从所述患者的样品提取核酸,并且使所述核酸与DNA微阵列杂交;以及获得所述患者患HG-SOC的风险。
应当理解,上面关于本发明的方法或用途公开的所有实施方案类似地适用于每种方法和用途,反之亦然。
如上文已经描述,生物标志物的重要性和定量方法的技术优势对于理解罹患HG-SOC的受试者的病因学,病理生理学以及更重要地,预后和诊断,特别就患者存活事件和时间而言具有巨大希望。
本技术包括方法,所述方法(i)鉴定CHEK2基因(DNA和/或mRNA和/或蛋白质)的突变作为具有HG-SOC的患者的重要风险和不良预后因子,(ii)鉴定包含在7%的HG-SOC中的58种相对经常突变的基因的组合突变标签,所述基因鉴定与不良的预后显著相关的HG-SOC患者,(iii)鉴定包含21种基因(DNA和/或mRNA和/或蛋白质)的组合突变标签,其将患者分组显著分层为低风险和高风险亚组,并且(iv)使用CHEK2基因(DNA和/或mRNA和/或蛋白)或5-8基因标签(DNA和/或mRNA和/或蛋白)或21-基因标签(DNA和/或mRNA和/或蛋白)作为在临床背景中个体HG-SOC患者的总体存活和治疗结果预测中的强制性预后工具。
包含在58-基因和21-基因突变标签中包含的基因与诸如激酶活性和ATP结合等功能相关,并且它们也是诸如细胞周期调节,凋亡控制和DNA损伤修复等生物过程中富集的。使用这些基因突变标签将诊断的HG-SOC患者显著分层为低和高风险亚组。具体来说,21-基因突变标签提供患者到两种疾病形成风险组上的分层,其5年总体存活率分别为37%和6%。此外,高风险亚组中的肿瘤是可能表现对治疗的抗性的约两倍(高风险亚组的15%和低风险亚组的8.7%)。
HG-SOC患者中的CHEK2突变是患者存活预后的强不良指标,并且与治疗抗性相关。假设但不限于任何理论,它可以是由于核定位位点的突变,其防止蛋白质的核输入,并随后导致单倍不足。还鉴定出21-基因突变标签,其与患者的存活模式高度相关(p=7.311e-08)。在这些基因中,诸如激酶活性或ATP结合的蛋白质功能得到富集,这可能指示这些过程在致癌作用中起关键作用并且靶向这些过程可能是有吸引力的治疗策略,以恢复与更高风险亚组相关的失调的细胞增殖的不平衡。
经由CHEK2,RPS6KA2和MLL4的种系突变或其它标签基因的体细胞突变表征HG-SOC的两个亚类。通过CHEK2的种系突变或/和杂合性丢失(LOH)表征的肿瘤亚组的存在提供了潜在的筛选努力来鉴定具有形成HG-SOC的高风险的女性。
分析来自诊断为HG-SOC的患者的9083种基因符号和334个肿瘤组织样品间的突变计数。预期地,发现已知其突变是HG-SOC的限定特征之一的TP53在所有样品间是高度突变的。然而,每个肿瘤样品中TP53突变的频率较低,每个肿瘤样品平均约1个TP53。相比之下,CHEK2和BRCA1基因(其参与DNA损伤修复)在患者的小亚组中以高频率突变。
进一步的非监督分层聚类揭示了58种基因的高度突变簇和主要通过CHEK2突变表征的22名患者(来自334HG-SOC)。基因本体论和网络分析揭示这些基因与激酶和ATP结合相关,并且可以参与与细胞周期,DNA损伤修复,凋亡或免疫反应相关的生物过程。58种基因的簇是:ABCA3、ADAM15、ADAMTSL3、ALK、ANKHD1-EIF4EBP3、ANKMY2、ANXA7、ASPM、CDC27、CHD6、CHEK2、CHL1、DPYSL4、ENAH、EP400、ERBB2IP、FN1、FOXO3、GCLC、GLI2、GLI3、GYPB、GZMB、HLA-G、HNF1A、INPP5D、INSR、ITGB2、KIF3B、KIF4B、KTN1、LRRN2、MAD1L1、MAP3K6、MAPK15、MET、MKL1、MLL4、MYO5C、NUMA1、PDGFRA、PHLPP、PIK3C2B、PKP4、PLAGL2、PPARA、PRKCI、PTK2B、RAB3D、ROR2、RPS6KA2、RSU1、SPTB、TBK1、TNK2、TP53、VAV1和ZC3H11A。
评估了最高度突变的基因(在至少5名患者中)的突变状态的预后意义。鉴定21种基因,所述基因的突变状态可以独立且显著分层患者为低或高风险(p值≤0.05)。这21种基因是:ADAMTSL3、ATR、CHEK2、ENAH、ERN2、GLI2、GYPB、KIAA1324L、LRRN2、MAP3K6、MAPK15、MET、MLL4、NIPBL、PCDH15、PPP1CC、PTCH1、PTK2B、RPS6KA2、RSU1和TNC。除了突变状态与更好的总体存活相关的ERN2外,其它20种基因具有与较差的总体存活相关的突变。这些基因在激酶活性和ATP结合功能中高度富集。它们也在DNA损伤和修复,凋亡和细胞周期的途径或基因网络中显著富集。
随后,组成组合的21-基因突变标签,其中HSG-SOC患者分类为:
如果在ERN2中观察到突变和/或在ADAMTSL3,ATR,CHEK2,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC中没有观察到突变,那么分类为低风险。
如果在ADAMTSL3,ATR,CHEK2,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC中观察到突变和/或在ERN2中没有观察到突变,那么分类为高风险。
对通过21-基因突变标签定义的58高风险患者亚组中的患者的进一步分析揭示了以两种不同肿瘤亚型为特征的两群不同的患者。第一种肿瘤亚型的特征在于CHEK2,RPS6KA2和MLL4的种系突变,而第二种肿瘤亚型的特征在于其它基因的自发体细胞突变。结果还揭示在存在TP53的情况下通常表征HG-SOC肿瘤的两种可能的疾病病因学途径。遗传变体的CHEK2,RPS6KA2和MLL4的筛选可以用作预测健康女性形成疾病的风险中的风险因素。
基因突变标签可以有助于临床设置中用于卵巢癌的早期诊断以及治疗效率和总体存活的预后的潜在应用。本发明还可以用于鉴定不太可能良好响应化学疗法的肿瘤亚类,并因此为科学家提供开发新的临床策略以靶向该肿瘤亚类的工具。基因突变标签可以形成诊断或预后试剂盒,用于实验室或临床环境中。
观察到CHEK2突变与对化学疗法的不良响应和因此不良总体存在高度相关,其中具有CHEK2突变的高级别浆液性卵巢癌(HG-SOC)患者的0%存活超过5年时。具有CHEK2突变的患者的此亚类占所有HG-SOC患者的约7.2%。这允许鉴定诊断为高级别浆液性卵巢癌(HG-SOC)的患者的先前未鉴定的亚类。迫切需要用于患者的此亚类的疗法或临床管理的新线路,因为他们的肿瘤没有良好地响应疗法。因此,这暗示鉴定HG-SOC患者的此亚类的必要性,为这组患者提供更好的临床护理,并且研究肿瘤的此亚组以在未来驱动未来的个性化临床治疗益处。
除了治疗高级别和高阶段浆液性卵巢癌外,在临床干预方面,在最早阶段检测此类癌症也可以对患者有益。测量CHEK2表达水平以提供卵巢癌的早期诊断可以帮助解决此类需要。
“包括”意味着包括但不限于在词语“包括”之后的任何事物。因此,术语“包括”的使用指示列出的要素是必需的或强制性的,但是其它要素是可选的并且可以或不可以存在。
“由...组成”表示包括并限于短语“由......组成”之后的任何事物。因此,短语“由...组成”指示列出的要素是必需的或强制性的,并且不存在其它要素。
本文中示例性描述的本发明可以在没有本文未具体公开的任何一个或多个要素,限制的情况下适当地实施。因此,例如,术语“包括”,“包括”,“含有”等应当广泛且在没有限制的情况下理解。另外,本文中使用的术语和表达已经用作描述性而不是限制性术语,并且在使用这些术语和表达时不意图排除所示和所描述的特征或其部分的任何等同物,而是认识到在所要求保护的本发明的范围内的各种修改是可能的。因此,应当理解,虽然本发明已经通过优选实施方案和可选特征具体公开,但是本文公开的其中体现的本发明的修改和变化可以由本领域技术人员采取,并且这样的修改和变化被认为在本发明的范围内。
本文已广泛和一般性地描述了本发明。落入属的公开内容内的每个较窄的种类和亚属分组也形成本发明的一部分。这包括本发明的一般性描述,其具有从属除去任何主题的附带条件或否定限制,而不管所排除的材料是否在本文中具体描述。
其它实施方案在所附权利要求书和非限制性实施例内。
实施例
基因突变的全基因组突变谱和统计学分布
在人类基因组测序中心(HGSC)对334个HG-SOC肿瘤样品进行通过Illumina或ABI SOLID测序平台的外显子组测序:贝勒医学院(Baylor College of Medicine,BCM),布洛德研究所(Broad Institute)基因组中心(BI)和华盛顿大学基因组研究所(WUSM)。如先前所述(TCGA research network Nature 2011Vol 474;609-15),通过TCGA研究网络分析数据。从TCGA数据门户下载处理的突变数据用于进一步分析。
TCGA数据门户含有所有研究的基因和患者间的21,978种突变。除去突变状态未知的基因,并且剩余的17,639种突变由334名患者间的种系,LOH或体细胞突变和9083种独特的基因符号构成(图1)。这些突变包括所有变体,包括缺失,插入,错义突变或种系、体细胞或杂合性丢失(LOH)起源的沉默突变。该分析包括沉默突变,假定它们可以是条件致病突变,特别是在由常见内源性诱变物(AID/APOBEC胞苷脱氨酶)诱导的DNA损伤,调节信号传导,RNA-蛋白结合修饰,转录后事件和胞质溶胶-核转运的背景中。
为了提供基因内突变发生的相对频率和患者样品间的基因突变的相对频率的全基因组理解,首先产生二维关联矩阵,其中行和列分别对应于9083种独特基因符号和334种独特的肿瘤样品ID(数据未显示)。矩阵的每个室中的整数值表示每个基因和每个肿瘤样品ID的独特突变位点的数目。该表的突变基因的频率分析证明,如果两个突变被认为是置信阈值,则在先前TCGA研究中报道的所有23个突变基因都包括在突变基因的子集中。
随后,对于每个基因,计算在该基因中具有报道的突变的肿瘤样品的数目,以及所有样品间的突变事件的总数。在研究的个体基因(N=9083)中分布的肿瘤样品数目的频率分布函数显示在图2A中。该图显示了HG-SOC样品中具有低,中等和高频率的突变基因的实例。特别地,图2A显示在TCGA患者分组中具有BRCA1(334个肿瘤样品中的40个)或BRCA2(334个肿瘤样品中的23个)基因中的突变的肿瘤样品的相对高频率。相比之下,DNA错配修复基因MLH1,MSH2,MSH6,PMS1和PMS2中的突变发生在少得多的患者(分别为334患者中的1,1,4,2和1名)中。这些基因通常与Lynch综合征相关,并且造成遗传性卵巢癌的亚组。
频率分布是偏斜的,有长右尾,代表下述观察结果:少数基因是高度突变的,而许多其它基因在HG-SOC肿瘤样品中较少突变。此类概率函数属于偏态分布家族,其在许多演进和交互(互连)系统中经常观察到,其中出生-死亡过程发生并且通过朝向复杂性和自组织(self-organization)的演化来驱动系统(参见方法)。在此类模型中,函数的偏斜形式是强烈的群体/样品大小和尺度依赖性的。在癌症驱动突变的情况下,Kolmogorov-Waring(KW)模型允许我们更好地理解突变事件的巨大变异性和可塑性的性质以及癌症起源及其进展中常见和罕见突变的作用。在实际意义上,K-W模型允许估计突变基因的分数,所述突变基因可以在突变肿瘤样品的数目增加时观察到。在这种情况下,最佳拟合的K-W函数产生以下参数:
a=3.944;b=9.50;θ=0.867和
因此,易感性靶基因的总数Ns可以通过下式估计:
Ns=Nb/a=9083x 9.5/3.944=21887种基因。
该结果表明,用于诱变的潜在靶基因的预期数目将包括人类中的整组蛋白编码基因。由于数据仅揭示9083种突变基因,这些差异可能是假阴性,并且可以通过增加样品大小或改进技术来改善。
此外,产生散点图,其中每个点代表每个基因,并且轴代表相对于所有样品间的基因的总突变位点的数目在该基因中具有至少一种突变的患者肿瘤样品的数目(图2B)。对角线表示假设的情况,其中每个样品精确突变每种基因一次(如果有的话)。我们的结果指示虽然TP53是最高度突变的基因,并且在几乎所有HG-SOC患者中观察到,在每个患者样品中具有基因基因座的突变的数目相对较低,即平均仅对每个患者观察到1个TP53突变(285名HG-SOC患者间的298种突变)。此肿瘤抑制剂的改变的功能或功能丧失似乎对HG-SOC致癌作用是关键的。
其它癌症易感性基因BRCA2不太频繁突变,并且在23种HG-SOC突变中仅观察到25种突变。CHEK2和BRCA1突变似乎在HG-SOC患者中是相互排斥的,因为仅18%(4/22)具有非沉默的CHEK2突变的患者具有BRCA1突变(图3)。类似地,仅18%(4/22)具有非沉默CHEK2突变的患者携带BRCA2突变(图3)。
突变簇由参与各种细胞周期相关过程的基因定义
对于在至少5名HG-SOC患者中具有观察到的突变的455种基因的子集,进行基因-患者突变关联矩阵上的无监督分层聚类。455种基因和334名HG-SOC患者的完整热图如图4所示。如预期的,在大多数HG-SOC患者(85%,334名中的285名)中观察到TP53突变。然而,患者中TP53突变的强度较低:通常在给定患者的每个p53基因中仅观察到1个TP53突变。有趣的是,沿着TP53基因座的突变位点似乎随机位于外显子间,并且对于任何特定基因变体似乎没有强阳性克隆选择(图5)。肿瘤样品中其它基因如BRCA1(12.0%,334名中的40名)和CHEK2(7.2%,334名中的24名)的突变频率相对较小。然而,每个基因的这些突变的强度比对于TP53的高3倍以上(平均而言,对于BRCA1和CHEK2分别为每名患者的3.38和3.96个突变)。热图提供了这些发现的视觉呈现。它还证实了我们先前的发现,BRCA1和CHEK2中的突变通常是相互排斥的(图3和4)。
来自分层聚类的结果还揭示了与CHEK2相关(180)的独特的基因-患者簇(图4)。该亚群包括58种基因符号和22名HG-SOC患者(图6)。在该簇内,CHEK2的突变似乎占主导,因为观察到CHEK2的多个区域在这些患者的每一名中是突变的(图6A)。58种基因符号的注释在图7中列出。通过DAVID生物信息学分析58种基因符号,揭示了这些基因在蛋白激酶活性(TBK1,PIK3C2B,MET,PRKCI,CHEK2,ALK,MAP3K6,PTK2B,RPS6KA2,MAPK15,PDGFRA,ROR2,TNK2和INSR)、腺苷酸和嘌呤核糖核苷酸结合(KIF4B,KIF3B,GCLC,TBK1,PIK3C2B,MET,PRKCI,TP53,CHEK2,ALK,ABCA3,MAP3K6,PTK2B,RPS6KA2,MAPK15,PDGFRA,ROR2,TNK2,CHD6,INSR,EP400和MYO5C)和疾病突变(MAD1L1,HNF1A,GCLC,MET,TP53,ITGB2,CHEK2,GLI2,GLI3,ABCA3,ROR2,INSR,SPTB和FN1)中显著富集(图8A)。通过Metacore的进一步分析揭示了,与免疫应答和DNA损伤途径以及凋亡和细胞周期基因网络的显著关联(图8B,C)。这58种基因的网络分析进一步确定了21种基因的紧密直接相互作用网络,主要参与凋亡,细胞周期控制,DNA损伤反应和免疫反应(图6B)。这些生物类别和网络被强烈分配到充分研究的DNA损伤,修复,细胞周期,检查点调节(图9)。
本技术描述了基于检测CHEK2相关的58-突变基因标签的DNA和/或mRNA和/或蛋白质的种系和/或体细胞突变的高级别浆液性卵巢癌(HG-SOC)的风险评估,预后和治疗结果预测的方法,所述58-突变基因标签由以下构成:
ABCA3,ADAM15,ADAMTSL3,ALK,ANKHD1-EIF4EBP3,ANKMY2,ANXA7,ASPM,CDC27,CHD6,CHEK2,CHL1,DPYSL4,ENAH,EP400,ERBB2IP,FN1,FOXO3,GCLC,GLI2,GLI3,GYPB,GZMB,HLA-G,HNF1A,INPP5D,INSR,ITGB2,KIF3B,KIF4B,KTN1,LRRN2,MAD1L1,MAP3K6,MAPK15,MET,MKL1,MLL4,MYO5C,NUMA1,PDGFRA,PHLPP,PIK3C2B,PKP4,PLAGL2,PPARA,PRKCI,PTK2B,RAB3D,ROR2,RPS6KA2,RSU1,SPTB,TBK1,TNK2,TP53,VAV1和ZC3H11A
所述21-突变基因标签由以下构成:
ADAMTSL3,ATR,CHEK2,ENAH,ERN2,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC。
本发明包括所述方法,所得的标签和随后的临床应用,以预后诊断的HG-SOC患者或者筛选健康女性以进行形成疾病的风险预测。
导致CHEK2、58-基因和21-基因突变标签开发的方法包括:
■使用非监督分层聚类和监督统计分析来鉴定58种基因的高度突变簇和以CHEK2的突变为特征的HG-SOC患者。
■HG-SOC患者(至少5名患者)中高度突变的基因的无偏筛选以鉴定21种预后基因(ADAMTSL3,ATR,CHEK2,ENAH,ERN2,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC),其突变状态显著且独立将患者分为低和高风险亚组。
■使用基因本体论,途径和网络分析来评估和确认CHEK2相关突变簇中58种基因和21种预后基因的生物学有效性。
■使用Kaplan-Meier和对数秩检验来确认基因突变在诊断的HG-SOC患者中的预后意义。
组合的21-基因突变标签的组成,其中:
■如果在ERN2中观察到突变或在ADAMTSL3,ATR,CHEK2,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC中没有观察到突变,那么将HG-SOC患者分类为低风险。
■如果在ERN2中没有观察到突变,并且在ADAMTSL3,ATR,CHEK2,ENAH,GLI2,GYPB,KIAA1324L,LRRN2,MAP3K6,MAPK15,MET,MLL4,NIPBL,PCDH15,PPP1CC,PTCH1,PTK2B,RPS6KA2,RSU1和TNC中观察到突变,那么将HG-SOC患者分类为高风险。
本技术提出:
■使用CHEK2突变状态用于已经诊断为HG-SOC的患者的总体存活预后和疗法响应预测的方法。
■使用组合的58基因突变标签鉴定主要与种系或体细胞突变相关的高风险亚组的方法
■使用组合的21基因突变标签将患者分类为低或高风险亚组的方法,其中低和高风险亚组的5年总体存活率分别为37%和6%。
■使用组合的21-基因突变标签,基于21-基因突变标签的基因的种系和/或LOH和/或体细胞突变的表征将高风险患者亚组分类两种进一步的肿瘤亚型的方法。第一种肿瘤亚型与CHEK2和/或RPS6KA2和/或MLL4基因的种系和/或LOH突变相关,而其它肿瘤亚型与其它基因的体细胞突变相关。
■使用CHEK2和/或RPS6KA2和/或MLL4基因的种系突变来鉴定可能对导致HG-SOC的肿瘤的起始、形成和进展易感的健康女性的方法。
CHEK2突变与诊断的HG-SOC患者的不良预后相关
诊断为HG-SOC的患者的突变谱的初始分析揭示了不同的基因-患者群,其中CHEK2突变似乎在几名患者中高度集中。将随后的分析聚焦在CHEK2上,检查该基因中的突变以确定它们是否与患者总体存活时间相关,并且其是否可以用作已经诊断为HG-SOC的患者的预后存活因素。
基于CHEK2基因的非沉默突变状态进行TCGA HG-SOC患者的分层。在这项分析中,研究具有突变数据和临床信息两者的总共311位患者(图10)。在22名TCGA HG-SOC患者中观察到非沉默的CHEK2突变,而未观察到其余289名患者(具有临床信息)中的CHEK2突变。在与没有CHEK2突变的亚组相比时,具有CHEK2突变的患者亚组的Kaplan-Meier存活曲线展现出显著较差的总存活时间(p值≤0.01,图11A)。有效地,来自TCGA数据的回顾性研究的结果表明,对于已经诊断为HG-SOC的患者,CHEK2基因的非沉默突变(种系,LOH或体细胞)对于患者总体存活时间是非常不利的,因为这些患者在初始病理诊断后没有存活超过5年。
在TCGA HG-SOC数据中,诸如TP53,BRCA1或MUC16的基因以比CHEK2更高的频率突变,但与CHEK2不同,这些基因的突变状态不能将HG-SOC患者独立地分为存活显著亚组(图11B-D)。尽管缺乏统计学显著性,但有一些略微的指标,即MUC16(一种已知的卵巢癌临床生物标志物)中的突变可以与较差的患者存活相关。另一方面,具有BRCA1突变的患者似乎与更好的患者存活相关,这与其它几个公开的数据一致。尽管TP53在HG-SOC中经常突变,并且可用于疾病诊断,但是我们的分析揭示,在诊断的患者中,它不能有效作为患者总体存活时间的预后标志物(图11B)。
CHEK2突变与对治疗的不良的响应相关
研究CHEK2突变与治疗抗性之间的关联,并且发现其在HG-SOC中是显著的。从TCGA数据看,HG-SOC患者分为两个亚组。第一亚组由在初步疗法后展现出进行性疾病的患者组成。第二亚组由初步疗法后具有部分响应,稳定疾病或完全响应的患者组成。随后,生成一个2x 2的列联表,其中列代表先前定义的患者的两个亚组,并且这些行对应于CHEK2的突变状态。通过κ相关性测量的分析揭示了CHEK2基因中的突变与具有边界显著性的进行性疾病相关(κ=0.1278,p值=0.05536,图12A)。当从分析中排除沉默突变时,观察到与疗法抗性的略微更加显著的相关性(κ=0.1422,p值=0.03769,图12B)。基本上,25%的具有CHEK2突变的患者(20名中的5名)显示疾病进展,而仅有8.8%的没有CHEK2突变的患者(237名中的21名)显示疾病进展。因此,结果指示CHEK2突变与对疗法的不良的响应相关。
CHEK2的拷贝数和mRNA表达似乎对HG-SOC患者存活没有显著影响
为了了解CHEK2的其它方面是否可以与患者存活相关,合并了来自拷贝数,突变,表达和临床实验的可用数据集间的CHEK2的患者信息(图10),并随后评估它们的预后显著性。
356名患者的拷贝数变异数据可用。对这些患者的拷贝数变异数据的分析揭示,CHEK2在15名患者中显著扩增,并且在130名患者中缺失。其余患者没有展现出显著的拷贝数变异。随后,分析还显示,CHEK2的拷贝数不能提供HG-SOC患者的显著预后分类(图13A)。还预期,具有CHEK2区域的显著扩增的样品展现出较高的mRNA表达,而具有显著缺失的样品具有较低的表达(图13B)。
399个样品的表达数据可用,其由8个正常输卵管和391个HG-SOC样品构成。另外,描述了391个HG-SOC样品中的370个,它们具有肿瘤信息,如肿瘤等级或肿瘤阶段。因此,研究了属于不同级别或阶段的正常输卵管组织和肿瘤组织间的CHEK2mRNA的表达谱(图13C)。相对于输卵管样品,肿瘤中CHEK2的较高mRNA表达指示可能由于补偿作用所致的早期疾病发作时的可能的上调,并且提示使用CHEK2mRNA表达作为HG-SOC的早期诊断生物标志物的可能性。另一方面,CHEK2表达数据将已经诊断为HG-SOC的患者分类为低和高风险亚组的预后能力有限(图13D)。将发表的计算算法应用于属于391名HG-SOC患者的CHEK2mRNA表达,其根据通过最大化两个Kaplan-Meier存活曲线的分离而优化的表达截留,将患者分配为低或高风险。虽然391个样品中的370个注释了临床信息,但是12个是不完整的,因为它们没有存活时间和事件。因此,对具有临床数据的良好注释的358个HG-SOC样品进行存活分析。结果提示CHEK2的mRNA表达不与HG-SOC患者的预后显著相关(p值=0.2057,图13D)。因此,结果提示CHEK2的其它方面,如表达或拷贝数变异258不能用作HG-SOC患者的预后特征。
观察到的CHEK2突变不可能改变磷酸化事件或蛋白质结构
CHEK2是丝氨酸/苏氨酸-蛋白激酶,其在细胞核中发挥功能以响应DNA双链断裂而调节细胞周期,DNA修复和凋亡。由于Chk2蛋白经由磷酸化事件的翻译后活化是其生理功能所需要的,检查CHEK2突变以确定是否有任何突变位于已知或预测的磷酸化位点。从UniProt30和Phospho.ELM的数据库收集已知的CHEK2磷酸化位点。在对CHEK2报告的所有突变中,仅发现一个突变位点与已知的磷酸化位点共定位(图14)。来自TCGA HG-SOC患者的突变数据揭示了CHEK2在编码残基Thr-383的核苷酸处突变(hg18,外显子11处的chr22:27421808-27421808)。已经报道在Chk2激酶结构域的激活环内的Thr-383/Thr-387和Chk2的C末端区域的Ser-516处的Chk2自磷酸化对于Chk2激活是必需的。然而,在仅6名患者中观察到在编码Thr-383的核苷酸处的突变。此外,由于突变是同义的,相同的氨基酸残基苏氨酸将被编码,因此,目前看来这里的突变不会导致Chk2功能的异常。结果还显示,CHEK2突变不与由NetPhos和PHOSIDA33,34 274计算预测的任何其它磷酸化位点共定位(图14),这提示基于我们目前的结果,磷酸化事件的改变可能不是导致改变的Chk2行为的关键机制。
确定所观察到的沿CHEK2的DNA突变是否可以潜在修饰蛋白质结构。使用从RNA测序实验产生并从Sage Bionetworks的Synapse数据库下载的数据,首先检查各种CHEK2同种型和属于262名患者的原发性实体瘤间的表达数据。当与其它CHEK2同种型相比时,鉴定出同种型uc003adu.1(代表同种型1或A)是主要表达的(图15)。收集沿着氨基酸残基的已知的二级结构(同种型1,UniProt ID:O96017),并将其与观察到的DNA突变进行比较(图14)。在CHEK2DNA的8个独特的位点处的DNA突变可以潜在地改变7个不同氨基酸残基处的蛋白质结构(图16A-B)。在蛋白质的结构化位点仅出现氨基酸残基之一(Thr-383)。然而,次级螺旋结构不可能被破坏,因为该区域的DNA突变是沉默的。为了进一步可视化,生成了生理Chk2的代表性蛋白质晶体结构并叠加7个突变残基,以相对于其周围的三维构象研究突变位点(图16C)。根据从Thr89到Glu501的Chk2的初始晶体学结构(PDB代码3i6u36,在3.0A解析),使用Modeller完成少数缺失的环并将激酶的C-末端区域延伸到Leu543。进行分子动力学(MD)模拟,从而获得50ns的蛋白质结构的松弛状态构象(图16C)。从图中可以观察到,突变残基(由有色球体表示)大部分位于蛋白质的非结构区域。因此,来自蛋白质建模和MD模拟的我们的结果提示了CHEK2的DNA突变不可能破坏蛋白质结构并且影响其生理功能。
观察到的CHEK2突变可影响蛋白质的核输入
在这些TCGA HG-SOC患者中调查NLS信号的修饰是可能的。先前已报道NLS3是参与细胞中Chk2的核定位的关键NLS(Zannini等人J Biol Chem.2003278(43):42346-51)。经由PSORT II计算预测的单组分(monopartite)NLS3占据一段短氨基酸,跨越蛋白质的残基515-522(氨基酸序列:PSTSRKRP,图16B)。由Zannini等人进行的突变研究显示该区域的突变导致Chk2蛋白的细胞质定位,这提示改变的Chk2不能转运至细胞核。有趣的是,结果揭示了沿着此短NLS序列,具有属于TCGA HG-SOC患者的突变的3个不同的核苷酸(对应于2个氨基酸残基-R519和P522)位点(图14和16)。在染色体坐标chr22:27413951处观察到的突变是在21名患者中观察到的沉默突变(P522P,在图16A中标记为灰色)。在连续核苷酸位置chr22:27413961和chr22:27413962处的两个非沉默突变分别存在于14名和21名患者中(R519Q/R519G,在图16A中标记为深灰色)。总的来说,观察到21名患者在这2个位点的任一处展现突变,以及与CHEK2突变对患者存活有害的发现一起,结果提示NLS区域中的突变可以不利地影响Chk2的核输入,降低有效且功能性Chk2的蛋白质水平,影响Chk2相关的修复途径,并最终有助于较差的患者存活。
由于在NLS3下游有两个其它突变位点,使用备选的计算工具cNLS40来预测沿Chk2蛋白质序列(同种型A,NP_009125-543氨基酸残基)的NLS。结果揭示来自氨基酸残基517-538(TSRKRPREGEAEGAETTKRPAV,图16B)的功能性二分NLS的可能性。该区域包括由12个氨基酸残基的接头连接的两个碱性残基簇。有趣的是,在观察到在编码R519的核苷酸处具有突变的21名TCGA HG-SOC患者中,对90%(21名中的19名)患者观察到编码R535的核苷酸的同时突变(图16B)。来自该分析的结果提示,有效NLS区域可以比由Zannini等人鉴定的NLS3更长。此外,编码二分NLS的两个关键组分(碱性残基)处的残基的突变的共发生进一步暗示在这些19名HG-SOC患者的肿瘤组织样品中的阳性克隆选择的可能性。
21-基因预后标签的鉴定
用在至少5名具有临床信息的患者中观察到的突变研究282种基因的突变状态的预后显著性。结果揭示了有21种基因,它们在至少5名患者中非沉默突变,并且可以独立地将HG-SOC患者分层为预后显著亚组(p值≤0.05,图17)。在这21个基因中具有预后显著性的前3种突变基因包括分别在22名,23名和20名患者中具有非沉默突变的CHEK2,RPS6KA2和MLL4(图17)。有趣的是,来自分层聚类的结果也揭示了这些基因聚集在一起(图6A)。定量地,κ相关分析进一步揭示了CHEK2突变与RPS6KA2或MLL4突变的高度共发生(κ≥0.75,p值≤5E-20,图3)。
总体上,观察到这些预后显著基因与CHEK2相关突变亚簇的基因的相当大的重叠(p值=6e-08,图18A)。由于具有CHEK2突变的患者通常还会具有突变簇中的基因突变(图6A),仅审阅21种预后基因以产生组合的突变预后标签。在这21种基因中,20种基因的突变状态展现出促致癌性行为(pro-oncogenic behaviour),其中突变与较差的总体存活相关(图17)。相比之下,只有ERN2展现出肿瘤抑制行为,其中突变与更好的总存活相关联。使用这些基因,如果在ERN2中存在突变或在所有20种促致癌基因中没有突变,则将患者分类为较低风险的亚组。另一方面,在20种促致癌基因中的任一个中具有突变并且没有ERN2突变的患者被分类为更高的风险。来自Kaplan-Meier存活图的结果揭示了,21-基因标签定义的患者亚组显著分层并与总体存活时间相关(p值=7.31e-08,图18B)。具体来说,低风险和高风险亚组的5年总体存活率分别为37%和6%。为了研究21-基因标签的预后显著性(图18B)是否由于单独的CHEK2突变的贡献,具有CHEK2突变的患者从标签限定的高风险亚组中排除。我们的结果指示,诊断为具有CHEK2突变或剩余的20-基因标签(排除CHEK2)的任一种中的突变的患者展现出相似的总体存活模式(图19C)。在20基因标签中的任何基因中展现出突变的患者的不良预后提示这些基因在HG-SOC基因组中的异常功能发挥可以相反地影响患者对疗法的手术后响应,不依赖于CHEK2突变的效应并且不管CHEK2突变的效应如何。
患者的这两个亚组的临床特征揭示了由21-基因标签定义的高风险患者与进行性疾病相关。具体来说,与低风险亚组形成对比,通过21-基因突变标签定义为高风险的患者以两倍可能展现出进行性疾病(高风险:50名患者中的8名=15%;低风险:208名患者中的18名=8.7%,图19)。然而,统计学显著性为边界(κ=0.08984,p值=0.06065)。不过,趋势提示这些基因可以是治疗抗性中的重要因素。
21-基因预后标签中的基因的详细注释列于(图20)。随后,使用DAVID生物信息学进行标签的21种基因的基因本体论分析。结果指示这些基因在与激酶活性,ATP结合和磷酸化相关的功能中强烈富集(图21)。平行地,通过MetaCore的分析还揭示了与DNA损伤诱导的响应相关的途径以及与细胞周期,DNA修复和凋亡相关的基因网络的关联(图21B-C)。
鉴定来自标签定义的高风险亚组的两个肿瘤亚类
为了研究通过CHEK2或20基因标签鉴定的不良预后患者亚组的可能的异质性,产生了21种基因和58名定义为高风险的患者的基因-患者突变矩阵(图22A)。就种系、LOH或体细胞突变而言进一步表征这些突变,并且我们的结果显示了在22名具有CHEK2突变的患者的亚组中的基因突变似乎是种系或LOH起源的,而其他36名高风险患者的基因突变似乎是体细胞的(图22B-D)。具体来说,在22名具有CHEK2突变的患者的亚组中,16名患者(73%)展现出非沉默的种系CHEK2突变(图23)。有趣的是,在这16名患者中,在15名(94%)和12名(75%)患者中分别观察到RPS6KA2和MLL4基因中的种系突变的强烈的共存在。重新分析整个基因-患者突变矩阵(455种高度突变基因和334名患者)也揭示了类似的发现,即来自CHEK2相关突变410亚簇的基因与种系或LOH而不是体细胞突变相关(结果未显示)。
我们的结果揭示了通过我们的21-基因标签鉴定的高风险患者中,可以有两个独特的肿瘤亚类,其最初的发病机制可以由CHEK2,RPS6KA2和MLL4的遗传性种系突变,或其它标签基因的自发体细胞突变驱动。
HG-SOC中CHEK2的突变可影响核定位并导致不良的临床结果
许多发表的突变研究仅聚焦于特定类别的突变,如体细胞或种系变体。当分别对出生时形成特定疾病的遗传风险,或者鉴定在生命的后期阶段形成疾病的驱动突变感兴趣时,对种系或体细胞突变的聚焦将适合于特定研究。为了预后的目的,突变是由于早期遗传还是后期环境因素是不太相关的。因此,在预后分层期间包括所有类别的突变。
有趣的是,携带Chk2突变的HG-SOC患者具有更高的死亡风险。但重要的是,它也可以促使进一步研究这些患者的备选靶向治疗。Chk2突变为何与不利的患者预后相关的可能的解释可以是由于化学抗性的诱导,因为在CHEK2和治疗响应之间发现显著相关性(κ=0.1422,p值=0.03769,图12B)。在TCGA HG-SOC患者中观察到的突变没有发生在注释的二级结构,ATP结合位点,活性位点,FHA结构域或激酶结构域中(图14)。因此,似乎没有足够的证据表明在TCGA HG-SOC患者中观察到的CHEK2突变可以破坏蛋白质结构、二聚化过程或激酶活性并且有助于化学抗性。
在卵巢癌中,顺铂用作主要的化学治疗剂。Zhang等人报道顺铂处理可以降解Chk2蛋白,并且降低的Chk2水平可以阻碍细胞周期控制,防止细胞凋亡,并且有助于肿瘤的化疗抗性。Chk2降解可以是大量临床相关肿瘤形成获得性对DNA损伤剂的抗性的主要机制之一。就展现CHEK2突变的患者而言,通过体细胞或种系突变的一个拷贝的功能丧失可以导致细胞核中CHEK2的拷贝减少,并且随后在顺铂处理时,CHEK2降解的作用可以加重,并且最终对患者存活有害。有趣的是,观察到的CHEK2突变为何可能最初促成蛋白质功能丧失的原因可以归因于在细胞核中缺乏蛋白质定位。Chk2的核定位的缺乏可能有助于偏离生理活性并导致不期望的效应。分析揭示了在TCGA分组的21名HG-SOC患者中,CHEK2突变发生在对于蛋白质的核输入关键的核定位信号内(图16B)。核定位信号的突变抑制Chk2蛋白的核输入,导致Chk2在核中的功能性拷贝减少。似乎可能的是,沿着CHEK2基因的核定位信号的突变可导致细胞核中Chk2蛋白水平降低,并且在顺铂治疗时,蛋白质水平将进一步耗尽,这可能潜在地导致在存在顺铂的情况下增强的肿瘤增殖,这模拟化疗抗性并导致不利的患者存活。结果显示,展现出CHEK2突变的HG-SOC患者与不良的临床结果显著相关,并且在初始诊断后没有存活超过5年(图11A和12)。
观察到的CHEK2突变不可能影响翻译后修饰
检查CHEK2与不良患者预后的关联,以观察该突变是否可以是由于磷酸化位点的修饰所致。然而,CHEK2中观察到的突变没有沿着任何当前已知和注释的磷酸化位点发生。因此,收集来自文献的计算鉴定的磷酸化基序并研究沿着基序的任何关键残基在TCGA HG-SOC患者中是否突变。结果揭示了尽管它们非常接近,但观察到的突变无一发生在磷酸化位点或围绕磷酸化位点的关键基序上。此外,分析揭示了围绕CHEK2突变的区域似乎不含强的蛋白质二级结构,因此目前似乎不太可能的是,Chk2蛋白质的翻译后修饰的异常是导致受累患者的不良存活预后的促成因素。然而,可以进一步研究CHEK2突变对蛋白质二聚化或与其它蛋白质配偶体的物理相互作用的影响。
沉默突变的可能影响
虽然假设沿着CHEK2观察到的突变可以影响翻译的蛋白质的核移位,但是也可以牵涉涉及沉默突变的其它机制。
观察到21名HG-SOC患者在chr22:27413951处展现沉默突变(P522P,图16)。传统上,假定编码相同氨基酸残基的沉默DNA突变对蛋白质功能具有可忽略的影响。然而,最近的研究已经提示了沉默突变可以通过各种机制影响下游蛋白功能性。例如,DNA三联体密码子的改变可以改变miRNA的结合位点,导致翻译抑制效率和下游信号网络的改变。研究chr22:27413951(P522P,图16)处的突变是否可以潜在改变miRNA结合位点,来自序列比对的结果指示特定的区域未被任何目前已知的人成熟miRNA靶向(结果未显示)。因此,通过同义突变的miRNA靶位点的改变不可能对检查的突变中的mRNA稳定性及其随后的翻译具有任何影响。
单一同义DNA突变可以影响mRNA二级结构、折叠、稳定性,并因此影响翻译的蛋白质的调节,如对于人多巴胺受体D2基因报道的。还提示同义突变可影响氨基酸残基的翻译效率,这是由于细胞中tRNA丰度的变化和不对称性。即使在同义突变不影响mRNA或蛋白质水平的情况下,翻译的蛋白质的功能也可以改变。在MDR1基因中,显示导致罕见的三联体密码子的同义多态性可以改变MDR1蛋白的底物特异性,可能是由于在该氨基酸残基处的翻译速率的减慢,其继而影响蛋白质折叠。在最后一个外显子处展现沉默突变和非沉默突变两者的14名常见患者中的强烈重叠(图16A)似乎提示了这些突变的可能的阳性选择,并且未来的研究可以集中于阐明沉默突变对最终蛋白表达和功能性的可能影响。
21-基因突变标签用于预后和治疗设计的潜在的临床应用
尽管CHEK2突变就患者基于其存活模式的分类而言似乎是最重要的,但总共鉴定了21种基因,其可以基于其突变状态独立且显著地将患者鉴定为低和高风险亚组(图17)。将21种组合的基因分类器应用于TCGA患者分组导致患者显著分层为两个存活显著亚组,其中低和高风险亚组的5年总体存活率分别为37%和6%(p值=3.8E-09,图18B)。此外,基于单独的CHEK2或剩余的20-基因标签的突变状态的分层允许排除21-基因标签的预后价值是由于单独的CHEK2的贡献的假设(图18C)。确切地,这显示即使在缺乏CHEK2突变的情况下,基于20-基因标签也可以鉴定出不良预后亚组。为了预后的目的,虽然从TCGA患者分组的回顾性研究的患者风险预测中使用21-基因突变标签是有用的,但是前瞻性研究将验证在临床背景中使用该标签。
有趣的是,在其突变状态最适合预后应用的21种基因中,与蛋白激酶活性、ATP结合、磷酸化、DNA损伤应答、细胞凋亡或细胞周期调节相关的基因功能是富集的(图21)。发现激酶如CHEK2或RPS6KA2中的突变与患者存活和患者分层相关。具有CHEK2或RPS6KA2的表征的突变的患者仅代表HG-SOC患者的子集。大多数HG-SOC患者的特征在于仅在几个基因中的突变(图4和22),这与患者-基因突变谱为异质和稀疏的一般共识一致。不过,已经假设个体患者可以在通过对应于癌症标志的基因网络功能相关的不同基因中展现出突变。因此,任何特定的生物过程可以通过其任何成员基因的畸变来影响。对HG-SOC患者中突变的异质性质的理解可以提供未来更有效和靶向治疗的机会。
21-基因突变标签用于形成HSG-SOC的风险预测的潜在临床应用
对通过21-基因标签鉴定的58名高风险HG-SOC患者的分析还揭示了可以源于两种不同的肿瘤病因学因素的两种截然不同的肿瘤亚型。第一个肿瘤亚类(或患者亚组)通过基因如CHEK2、RPS6KA2和MLL4的种系突变或LOH清楚地表征(图22)。相比之下,对于其它肿瘤亚类(或患者亚组),未观察到这些基因的种系突变。确切地,此肿瘤亚类似乎是在通常表征HG-SOC肿瘤的TP53突变的存在下这些基因的自发体细胞突变的结果。
事实上,卵巢癌是高度异质的,具有参与几种癌症亚型的形成的各种驱动基因(图24A)。结果提示了大约11.6%的HG-SOC肿瘤可以由于TP53突变存在下标签中的基因的自发体细胞突变而可能启动。此外,在以TP53点突变和基因组不稳定性为特征的高级别浆液性卵巢癌中,遗传的种系CHEK2突变可能导致易感性,并参与约7.1%的HG-SOC患者中的肿瘤的起始、形成和进展(图24)。结果还提示CHEK2、RPS6KA2或MLL4的种系突变可以用作预测个体形成HG-SOC的风险的风险因子的可能性。对于CHEK2,已经进行研究以研究基因变体对卵巢易感性的影响,但是报道了关联是不显著的,可能是由于CHEK2突变的罕见发生,小肿瘤样品大小,缺乏合适的HG-SOC患者样品,可用样品的变体检测的低分辨率。然而,来自TCGA的高质量HG-SOC数据集的当前结果已经揭示了由于CHEK2种系变体的疾病易感性的先前未表征的关联。
CHEK2表达在HSG-SOC的早期诊断中的潜在临床应用
结果揭示了相对于输卵管正常组织,CHEK2mRNA在肿瘤样品中上调(图24B),这提示了升高的CHEK2mRNA表达可以作为高级别浆液性卵巢癌的早期诊断标志物使用。可能地,升高的CHEK2表达可以是由于对HG-SOC中与TP53突变相关的DNA损伤或基因组不稳定性的响应。通过抑制Chk2在肿瘤细胞中诱导凋亡可有益于预防不可控的细胞增殖,从而可导致更好的患者存活。然而,最近的研究发现,卵巢癌细胞系中的Chk2耗竭减少顺铂敏感性,并且提出Chk2是否可以是顺铂治疗的HG-SOC患者中的有效治疗靶点的进一步怀疑。
TCGA HG-SOC数据源和预处理
从2010年11月24日从TCGA数据门户下载属于334名TCGA HG-SOC患者的处理过的突变数据。序列由贝勒医学院(BCM)的人类基因组测序中心(HGSC),布洛德研究所基因组中心(BI)和华盛顿大学基因组研究所(WUSM)基于Illumina或ABI SOLID测序技术产生。该版本(release)包括105名水平2和91名水平3(BCM)患者,来自BI的172名水平2和158名水平3患者,和作为水平2和水平3WUSM患者的88名。
总共,报告了跨越334名患者和10489个RefSeq基因符号的21978个突变。除去4339种具有未知的突变状态的突变。在9083种基因中观察到剩余的17639种突变,并且涵盖诸如插入、缺失、SNP和沉默突变的变体。还下载对应于每个HG-SOC患者的临床信息。
此外,获得463个原发性实体卵巢癌组织样品的mRNA表达数据(来自11批,每批21-47个样品)。在每批中进行质量评估以鉴定质量差的芯片。从随后的分析中除去74个质量差的芯片。在每个批次内进行背景校正和标准化。最后,使用非参数ComBat软件在批次间消除批次效应。
拷贝数变异分析
使用从TCGA门户下载的肿瘤-血液配对样品。将血液拷贝数变异用于标准化和估计匹配肿瘤样品的拷贝数变异数据的倍数变化富集/呈现不足(under representation)。TCGA SNP阵列数据(CNV平台6)通过PARTEK 6.5程序以公司推荐的参数进行处理。使用PARTEK软件,鉴定了拷贝变异区段的基因组坐标,其形成统计学上显著的缺失或扩增的基因组区域。对于每个肿瘤样品,将这些显著区域定位在人类基因组坐标上,并且通过USCS基因组浏览器定制轨迹显现此类信号的标准化倍数变化。卵巢肿瘤中的改变的拷贝数展现出高水平的染色体不稳定性。20573种基因与改变的CN区段重叠,代表与显著的改变拷贝数区域重叠的约70%的RefSeq蛋白编码基因。
处理过的RNA测序表达数据
基因和基因同种型的处理过的RNA测序表达数据从Sage Bionetworks的Synapse数据库下载。该数据集含有73598种基因同种型的RNA-seq表达数据和对应于263名患者的266个样品。在266个样品中,从原发性实体瘤收集262个样品(来自262个患者),而从复发性实体瘤收集其余的样品。
次级数据来源
从UniProt获得由重要功能位点、二级结构、天然变体、诱变实验数据和磷酸化位点构成的蛋白质注释数据。此外,已知的磷酸化位点从验证的数据库Phopho.ELM下载。使用在线工具NetPhos和PHOSIDA(它们基于机器学习技术,如人工神经网络或支持向量机)进一步预测磷酸化位点。通过在线计算工具PSORT II和cNLS Mapper预测核定位信号。
患者和基因间的突变矩阵
患者和基因间的突变谱在二维矩阵中表示,M由分别代表基因符号和患者样品ID的9083行和334列构成。矩阵中的每个条目,Mij表示第j个患者样品的第i个基因中的独特突变位点的数目。
对易感基因的突变肿瘤样品的数目的频率分布的分析
Kolmogorov-Waring(K-W)概率函数用于拟合突变的肿瘤组织样品数目的分布。该函数描述为:
其中m=0、1、2、...并且b,a,θ是我们模型的参数。B(x)是如前所述的β函数。在b>a>0的情况下,通过公式估计未观察的事件的概率,可以以下面的递归公式的形式呈现等式1,以便容易地计算估计模型参数:
为了将概率函数(等式1)或(等式2)应用于观察数据,假定随机变量X限于样品大小,并且最罕见的事件是未观察的。因此,随机变量X被双截短的,即范围1、2、...、J(J<∞)。使用(等式1),得到的截短分布函数的概率分布函数如下书写:
该概率分布函数对应于在没有检测到发生数值0和J+1,J+2,...的有限分组中分析诱变数据中的典型情况。曲线拟合计算算法的细节以前已经公开。
分层聚类
生成表示患者和基因间的突变模式的数值矩阵。行和列分别对应于基因和患者。矩阵中的每个数值表示对于该患者和基因具有报道的突变的不同位置的数目。使用Kendall-tau作为相似性度量和完全链接作为聚类方法进行分层聚类分析。数学过程在Gene Cluster 3.0中实现,并通过Java TreeView可视化。图的强度对应于该患者和基因的独特的突变位置的数目。
基因富集和网络分析
通过来自GeneGo Inc.的DAVID Bioinformatics和MetaCore进行基因功能富集分析。使用默认的人类基因组基因作为背景设置。使用默认参数。通过MetaCore通过直接交互网络算法生成基因网络。网络的图例可以从http://ftp.genego.com/files/MC_legend.pdf进行评估。
存活分析
患者亚组的存活分析参考其总存活时间(最后一次随访前的年数)和存活事件(最后一次随访的生命状态(vital status))进行。使用Kaplan-Meier存活曲线显现患者亚组的比较存活时间和事件,所述Kaplan-Meier存活曲线表示在初始诊断后在给定时间时患者存活的概率。使用基于chi-sq分布的对数秩检验评价完全存活时间范围间的患者亚组分层的统计学显著性。使用开源R编程语言和软件包实现程序。
一致性检验(agreement test)的测量
使用加权κ相关测量计算具有临床参数,如治疗响应的有序患者亚组之间的相关性。使用Mantel-Haenszel(MH)检验评估统计学显著性。使用StatXact-9(计算的权重:平方差,得分:相等间隔)实施计算。所有p值是单侧的(右尾的),其指示随机κ相关性测量大于实际观察的概率。
蛋白质结构建模
初始结构取自丝氨酸/苏氨酸蛋白激酶chk2的晶体结构。(PDB码3i6u,在3.0A解析)。结晶学单元含有二聚体蛋白质(链A和B)。晶体构建体包含残基Thr89至Glu501。使用程序Modellerhas进行残基的质子化,所述程序Modellerhas已经用于完成少数的缺少环并且扩展激酶的C末端区域到Leu543,以包括核定位信号基序。使用PDB2PQR进行残基的质子化。使用AMBER 12包中的前室(antechamber)和LEaP模块建立MD模拟。该系统在截短的八面体TIP3P水盒中溶剂化并用钠离子中和。使用Amber ff99SB全原子力场的最小化和MD模拟用Amber12包的Sander模块,使用GPU加速版本的程序进行。如前所述,遵循多步方案。提取50ns的构象,并假定沿着轨迹,激酶和特别是C末端尾已经采取松弛状态。已经使用PyMOL可视化和生成图形。
- 还没有人留言评论。精彩留言会获得点赞!