单细胞全基因组线性扩增法的制作方法
技术领域
本公开的实施方案包括核酸扩增、核酸操作、遗传学、医学等领域。本公开的实施方案的领域涉及包括例如来自一个或多个细胞的基因组或转录组的扩增。
发明背景
出于对单细胞异质性的巨大兴趣,人们最近尝试利用全基因组扩增和稳健性(robustness)进行单细胞基因组测序(Navin等人,2011;Fan等人,2011;Lao等人,2008;Hou等人,2012;Cheng等人,2011;Telenius等人,1992;Zhang等人,2006;Zhang等人,1992)。然而,迄今为止,所使用的方法通常都受限于相对较低的覆盖率。聚合酶链反应(PCR)是对特定区域的DNA扩增的金标准。依赖于使用随机引物的指数扩增,基于PCR的全基因组扩增方法引入了强的序列依赖性偏差,因此对整个基因组均一代表是不理想的。已经开发了多重置换扩增(MDA)以克服PCR的这些缺点(Dean等人,2002;Dean等人,2001),但是MDA仍然显示有不可忽略的偏差。出于这些原因,对允许精确检测单个核苷酸变体(SNV)的单个体类细胞全基因组测序还没有可靠的报道。
为实现用于单细胞的全基因组SNV,并且具有可媲美大量测序的精确度,主要的技术障碍在于现有技术中在非线性扩增中产生并繁衍的扩增错误。在非线性扩增中,聚合酶产生的错误会在随后的循环中以新合成产物用作模板时被拷贝。对于常规PCR扩增有成千上万的模板可用于起始,这些错误不会引起任何问题,因为具体拷贝中的每个随机错误都由大量的其他独立拷贝所稀释。然而,对于单细胞扩增,情况却并不相同,因为一个细胞只有每个独特染色体的一份拷贝作为模板。在非线性扩增中,第一个循环中产生的错误将被半数的DNA产物所有,而这些错误将持续被以类似的百分比拷贝。最终在测序数据中,不能将这些错误与该单细胞中真正的杂合子变体相区分。更重要地,这种假阳性率不能通过简单地增加测序深度而减少。为了克服该技术问题,需要进行线性扩增。当扩增是线性时,所有DNA产物都直接由原始模板拷贝而来。因此,扩增错误是独立产生的,并可在线性扩增产物中稀释。
发明简述
本发明针对的是用于扩增大量核酸的系统、方法和组合物。在具体实施方案中,所述多种核酸是一个或多个细胞或无细胞材料的完全或部分基因组或者一个或多个细胞或无细胞材料的转录组,例如或表观基因组,即在二硫化物转化后的基因组中的稀有核酸材料,又例如选定的基因组,即染色质沉淀后基因组中的稀有核酸材料。所述基因组或转录组可以来自单细胞或多个细胞,例如两个细胞、三个细胞、四个细胞、五个细胞等。在其中基因组或转录组或表观基因组或基因组的靶向区来自多个细胞的实施方案中,所述多个细胞可以是相同的类型、基因型或表型。在其中核酸源自多个细胞的情况下,例如,所述细胞可来自相同来源或来自相同组织。在某些实施方案中,细胞是胎儿细胞、癌细胞、疑似癌性细胞等。无细胞核酸材料包括存在于血液或其他体液中的核酸。
本公开的实施方案一般涉及用于扩增基因组序列,如单细胞或任选多细胞的全基因组的方法和组合物。本公开的实施方案一般还涉及用于扩增部分或全部转录组,如单细胞或任选多细胞的整个转录组的方法和组合物。本领域技术人员会认识到本公开的方法允许从基因组或转录组中对具体核酸进行线性扩增,其中本发明的所得产物是大量扩增子(代表部分或全部的各自基因组或转录组),且至少在一些情况下线性产生的扩增子会进一步以线性或非线性方式(如通过PCR)得到扩增。
本公开的实施方案包括用于在整个基因组上以高均一性和高保真度对单细胞(或任选多细胞)进行全基因组扩增的方法。此类方法允许例如拷贝数变异(CNV)和单核苷酸变异(SNV)的精确检测,并且任选地例如通过标准高通量测序平台或微阵列或基于PCR基因分型按照本发明的方法来检测CNV或SNV的存在。
本公开的方法的实施方案提供两队本领域中当前使用的方法的显著改进。例如,通过从覆盖全基因组的原始单细胞DNA片段的线性扩增(即,从单细胞DNA模板产生扩增子的独立拷贝)进行SNV检测的精确度相对于本领域中的方法得到大大提高。线性扩增允许对扩增错误的有效过滤,因此实现例如与用于SNP检测的大量测序相当的精度。本文提供的方法的具体实施方案为每个独立拷贝的扩增子引入“条形码”。该条形码允许确定在基因组上的每个基因座的假阳性率。
本文提供了可用于对提取自一个单细胞的DNA片段进行扩增的方法,尽管在一些情况下DNA可提取自多于一个细胞。因此,至少某些方法允许对一个单细胞进行均一的全基因组扩增,其允许通过标准高通量测序平台精确检测拷贝数变异(CNV)和单核苷酸变异(SNV)。
本公开的实施方案允许使用精确的线性扩增方法,其将为一个且仅一个细胞提供精确的SNV检测。在特定情况下,不需要亲缘细胞来过滤假阳性。本公开的实施方案对本领域的一个改进在于允许使用组织学临床样品作为待扩增核酸的细胞来源。
本文提供了方法用于通过将不同的DNA片段分离成数百万个小体积反应区室并在无不同扩增子之间的干扰和竞争下进行扩增而消除扩增中的偏差。因此,在具体实施方案中,单个扩增子存在于反应孔中用于例如通过PCR而扩增。在某些方面,反应区室中的反应体积为飞升至纳升的体积。
本文提供的方法的实施方案通过在微流体装置中分开的反应区室中进行每个DNA片段的扩增或通过例如乳化剂产生的分离反应液滴而显著改善均一性。在一些情况下,在具有小至飞升体积的数千或更多个反应区室中进行各自的扩增。在具体方面,扩增在每个单独的反应中饱和,非线性扩增(测序依赖性PCR偏差或MDA的扩增偏差)得到最小化。
在具体实施方案中,所述方法使用的聚合酶具有强的置换强度以及高的与引物-DNA复合物的结合常数。
在一个实施方案中,有从一个或多个细胞线性产生扩增子的方法,包括以下步骤:将来自所述一个或多个细胞的核酸接触第一大量引物和包含链置换活性的聚合酶,所述接触发生于0℃至约35℃的温度范围条件下,其中所述引物与所述核酸退火,且所述引物通过聚合酶得到延伸,其中所述第一大量引物具有以下特征:a)富含40%-60%的G或富含40%-60%的C;及b)包含限制性内切核酸酶位点,从而产生包含引物退火的核酸模板的混合物;对引物退火的核酸模板进行两轮或多轮延伸、解链和退火步骤,由此产生核酸模板、线性产生的半扩增子和非线性产生的全扩增子的混合物;将所述混合物置于能使全扩增子的两个末端能够彼此退火的条件下,从而产生成环的全扩增子;将所述成环的全扩增子接触限制性内切核酸酶,从而使得全扩增子不能与第一大量引物或第二大量引物退火,其中第二大量引物富含40%-60%的G或C;以及使第一大量引物或第二大量引物与混合物中剩余的线性产生的半扩增子退火并延伸,其中所述退火和延伸在核酸不会进一步解链的情况下发生,由此产生线性产生的全扩增子。在一个具体的实施方案中,该方法进一步包含使线性产生的全扩增子进行扩增的步骤。在一个实施方案中,本公开的方法中所用的聚合酶缺乏外切核酸酶活性。在具体实施方案中,混合步骤中的接触发生于低于60℃的温度下。在一些情况下,所述方法另外提供了包括从一个或多个细胞获得核酸,例如通过裂解细胞和从细胞提取合适。在具体实施方案中,所述核酸包括基因组DNA。在一些情况下,所述核酸包括RNA且所述方法进一步包括从RNA产生cDNA的步骤。
在本公开的实施方式中,可使用特定的引物,至少包括第一或第二大量引物。在具体实施方案中,第一、第二或二者的引物包含下式:
XnYmZp,
其中n大于2且X为富含40%-60%的G或富含40%-60%的C,其中Y是任意核苷酸并且m为3-8个核苷酸,其中当Xn是富含G时Z是G或者当Xn是富含C时Z是C,p是2-4个核苷酸。在具体情况下,m为5个核苷酸;p是3个核苷酸;n是20-40个核苷酸;n是25-35个核苷酸;或n是24-28个核苷酸。在一个具体的实施方案中,聚合酶是Bst大片段或pyrophage 3173聚合酶。在特定情况下,引物退火的核酸模板的延伸步骤在30℃至65℃的温度范围内发生。在具体实施方案中,在引物退火的核酸模板的延伸之后,核酸在至少90℃的温度下解链。在某些情况下,在核酸解链后,将核酸冷却至低于引物解链温度的温度,并加入可热灭活的聚合酶。在一些情况下,在加入可热灭活的聚合酶之后,在低于PCR引物的Tm的温度和高于PCR引物的Tm的温度之间,例如在58℃-67℃的温度进行热循环。在一些情况下,热循环可以包括10-30个循环。在具体实施方案中,对聚合酶进行热灭活,随后加入限制性内切核酸酶。
在一些实施方案中,对有引物退火的核酸模板进行三至十个连续的延伸、解链和退火步骤。在某些方面,限制性内切核酸酶能够在超过50℃的温度下消化核酸,例如BtsCI/BseGI。在某些实施方案中,通过聚合酶链反应(PCR)或环介导的等温扩增(LAMP)对线性产生的全扩增子进行扩增。在具体实施方案中,至少大多数线性产生的全扩增子是彼此分开的。在特定方面,至少大多数线性产生的全扩增子各自被置于分开的容器中,例如微孔基底的孔中。在某些实施方案中,这些孔包含一种或多种扩增反应试剂。
在具体实施方案中,对分开包含的扩增子进行扩增,例如进行PCR或LAMP。在一些实施方案中,对线性产生的全扩增子经受尿嘧啶-DNA-糖基化酶和DNA糖基化酶-裂解酶内切核酸酶VIII混合物的作用,然后经受S1核酸酶或T4聚合酶的作用。在具体实施方案中,对线性产生的全扩增子进行测序或进行文库构建。在具体实施方案中,测定一个或多个线性产生的全扩增子的某个核苷酸或核苷酸序列,例如代表核酸突变的扩增子中突变。在具体实施方案中,所述突变是疾病相关突变。在具体实施方案中,所述一个或多个细胞来自胎儿、婴儿、儿童或成人。所述一个或多个细胞可以固定于组织学制备物中。然而,在一些情况下,所述一个或多个细胞是新鲜的。所述一个或多个细胞可以从具有医学病况或怀疑患医学病况(例如遗传性疾病)的个体获得。在特定情况下,所述医学病况包括癌症。
在一个实施方案中,一种方法测定来自个体的核酸从而鉴定个体的医学病况或鉴定个体具有所述医学病况的风险,包括将通过本公开的方法从个体样品所得的线性产生的全扩增子的部分或全部序列与标准品进行比对的步骤。在具体实施方案中,所述核酸包括基因组DNA。在一些情况下,所述核酸包括从来自样品的RNA产生的cDNA。标准品可以包含来自个体正常细胞的核酸,例如来自一个或多个其他个体的正常细胞的核酸。在具体实施方案中,来自个体样品中的细胞中核酸的表达水平以线性产生的全扩增子的数目表示。在一些情况下,比较步骤包括将来自个体样品中的细胞的至少一些线性产生的全扩增子的数目与标准品进行比较。在具体实施方案中,所述至少一些线性产生的全扩增子包含一个或多个具体基因。在某些实施方案中,比较步骤包括测定线性产生的全扩增子与标准品相比其中一种或多种特定核苷酸是否存在
前文已经相当广泛地概述了本发明的特征和技术优势,以便可以更好地理解下文对本发明的详细描述。下文将描述形成本发明权利要求的主题的本发明的附加特征和优势。本领域技术人员应当理解,所公开的概念和具体实施方式可以容易地用作修改或设计用于实现本发明的相同目的的其他结构的基础。本领域技术人员还应该认识到,这样的等价的构建不脱离如所附权利要求中阐述的本发明的精神和范围。被认为是本发明特征的新颖特征当结合附图考虑时,将从下面的描述中更好地得到理解,包括其组织和操作方法,以及进一步的目的和优点。然而,应当清楚地理解,每个附图仅仅是为了说明和描述的目的,而不旨在作为限制本发明的定义。
附图简述
为了更完全地理解本发明,参考以下结合附图的描述,其中:
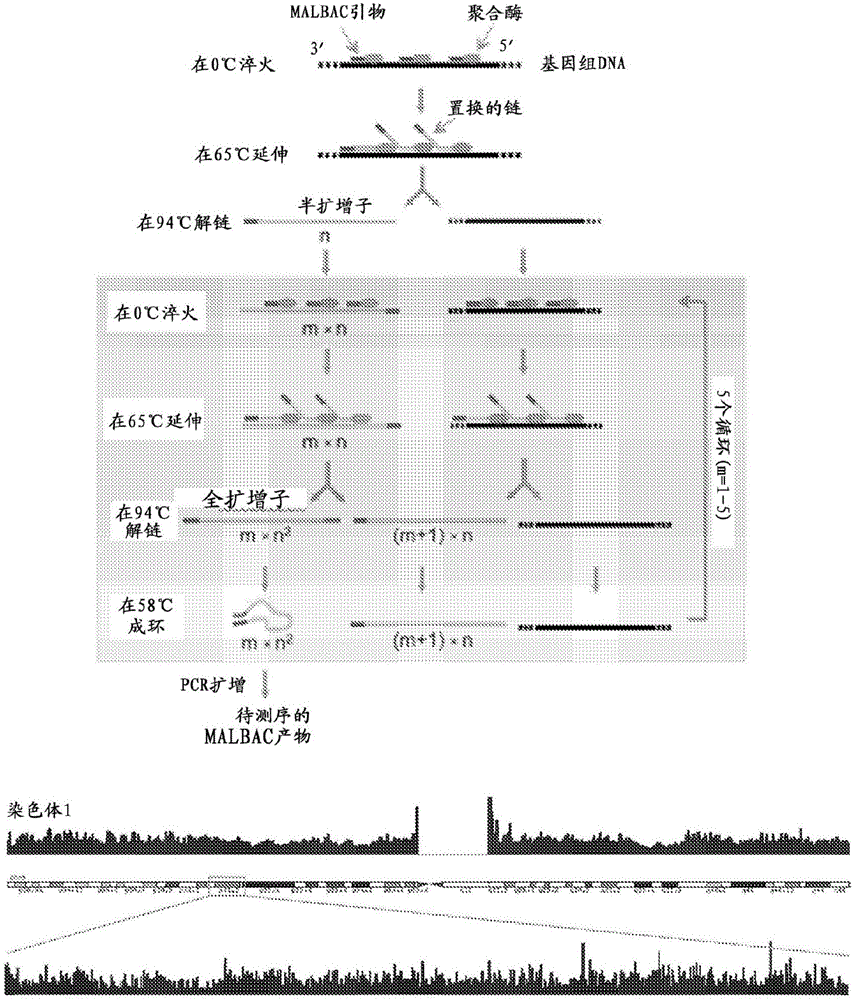
图1示例显示了一种已知的称为基于多重退火和成环的扩增循环(MALBAC)的扩增形式。低偏差单细胞全基因组扩增(WGA)。(左)MALBAC工作流程。将基因组DNA解链为单链DNA分子后,裂解单细胞。在附加的PCR扩增前进行MALBAC预扩增。首先,MALBAC引物随机退火于单链DNA分子上,并由具有置换活性的聚合酶延伸,产生半扩增子。在下一个循环中,生成两端都具有互补序列的单链扩增子。3'末端在中等温度下通过形成环而得到保护,这防止嵌合体的形成和进一步的扩增。将上述循环重复5次以产生具有重叠基因组覆盖的扩增子,其在两端都含有通用互补序列用于随后的PCR扩增;
图2提供了本发明扩增方案的一个实施方案的概况。在该方案中,不使用在MALBAC中产生的全扩增子用于PCR扩增。相反,例如通过消化移除成环的全扩增子的引物序列。之后,可以对于从原始基因组模板线性产生的半扩增子线性地再产生全扩增子。这种完全线性扩增的全扩增子用于下列PCR扩增,例如;
图3示出条形码测序的概况。测序的读数是对齐的。有两种可能的杂合子突变,由红色和蓝色点(在黑白图中,它们分别是各栏中左边和右边的点)表示。在左侧,测序读数不具有条形码,因此得到两种突变。相比较,右侧的测序读出具有条形码α、β、γ和ζ,其代表在线性扩增中生成的独立DNA拷贝。显然,相比于由蓝色点标记的突变(由具有两个不同条形码的读数表示),由红色点标记的突变是明显的假阳性(由仅有一个条形码的读数表示);
图4示出了线性产生半扩增子的实例。
图5示出了线性产生全扩增子的实例。
图6提供了微滴或孔中全基因组扩增的实例。
图7展示了使用具体酶在引物中移除引物序列。
图8:早期肿瘤发展概况。在前体中,可以预期突变数目有限。在发育不良中,可以预期,与瘤形成相比,细胞积累了显著数量的突变和高程度的基因组异质性。
图9:产率和扩增循环数之间的线性关系。该数据表明线性扩增的成功。x轴表示预扩增循环数。y轴表示qPCR测量的扩增子产率。线性关系确认达到了线性扩增。实验步骤按照实施例1中的描述进行。
图10:该图表示例显示了线性扩增后的步骤的实例。线性产生的DNA半扩增子或全扩增子被分到多个管中。因此在不同管中扩增独立的线性拷贝。使用来自各个管的扩增DNA构建测序文库,并用下一代测序仪进行测序。下图示出测序数据的快照结果。可以看到各个染色体间都有类似的平均覆盖。该数据包括六个文库,其由线性扩增的扩增子分入的16管中的6个而构建。
图11:下一代测序结果表明线性扩增允许检测到生殖系/体细胞突变并鉴定出扩增错误。对其作图示出了来自测序实验的读数,如图10中所描述。真突变和假阳性读数的典型形式在作图中表示。右图中的突变显示在与已有数据库相比而测序的细胞中检测到从头开始的突变。
发明详述
I.定义
如本文所使用的,术语“半扩增子”指代仅在一个末端由引物序列延伸而生成的多核苷酸。其是全扩增子的半产物。
如本文所使用的,术语“扩增子”指代用作PCR反应中模板的多核苷酸。
如本文所使用的,术语“全扩增子”指代在两个末端都有(彼此不同或互补)的引物序列,容易进行PCR扩增。
如本文所使用的,术语“线性扩增”表示直接从原始模板拷贝的扩增产物,所以DNA产物的增加是线性的。相反,对于非线性扩增,产物拷贝自原始模板和拷贝而来的产物二者。例如PCR反应就是一种典型的非线性扩增,其中产物以指数形式增加。在特定方面,特定模板的线性扩增被限定为大量的模板拷贝中的每份(或大部分)特定模板的拷贝都是由该特定模板直接产生。在特定模板的非线性扩增中,模板的拷贝可由特定模板的另一份拷贝产生。
II.通用实施方案
本公开的实施方案提供了用于从一个或多个细胞中扩增部分或全部核酸的扩增方法。在具体方面,所述核酸是细胞的部分或全部基因组或者细胞的部分或全部mRNA,由逆转录自mRNA的cDNA表示。这些扩增方法生成线性产生的半扩增子和非线性产生的全扩增子,随后非线性产生的全扩增子几乎都用生物素化的PCR引物转化为双链DNA,然后通过链霉亲和素磁珠反应提取,或在方法的随后步骤中有效地从沉淀物中移除(例如通过使其不能由具体一组引物退火),其后线性产生的半扩增子经过退火和延伸步骤,而不经过解链步骤,从而产生线性产生的全扩增子。然后任选地解链所述线性产生的全扩增子并进行其他方法,如线性或非线性扩增(例如通过PCR)。
根据本公开的某些方面,反应混合物中来自单细胞或多个细胞的DNA由至少一种DNA聚合酶进行扩增。
根据一个方面,单细胞核酸扩增或多个细胞核酸扩增之前将来自样品的双链DNA变性至允许引物退火于DNA的单链状态。随后,反应温度降低至允许第一种引物3’末端随机核苷酸退火至DNA形成杂交双螺旋的温度。杂交双螺旋形成后,在孵育时期内反应混合物中存在的或此时加入的一种或多种DNA聚合酶从第一种引物的3’末端延伸互补DNA链。DNA聚合酶可包含或不含5’-3’外切核酸酶活性或链置换活性。
图1示出一种称为MALBAC的在先技术方法的概况,本发明是对其的改进。裂解单细胞,接着将基因组DNA解链成单链DNA分子。在附加的PCR扩增前进行MALBAC预扩增。首先,MALBAC引物随机退火至单链DNA分子上并由具有置换活性的聚合酶进行延伸,形成半扩增子。在下一个循环,生成两端都有互补序列的单链扩增子。3'末端在中等温度下通过形成环而得到保护,这防止嵌合体的形成和进一步的扩增。将上述循环重复约(例如)5次以产生具有重叠基因组覆盖的扩增子,其在两端都含有通用互补序列用于随后的PCR扩增。
本领域的MALBAC预扩增不是完全线性的。在MALBAC预扩增中,半扩增子直接拷贝自原始DNA模板,因此其是线性产生的。然而,半扩增子在接下来的扩增循环中被用作模板。例如,在第一个循环中产生的半扩增子被用于接下来的五个循环中;在第二个循环中产生的半扩增子被用于接下来的四个循环中,等等。在扩增旗舰,如果聚合酶在第一个循环中在半扩增子中产生错误,错误便在接下来的扩增循环中拷贝五次。由于这个扩增错误占据最终读数中的30%,因此其可能被认为是突变。因此,需要测序例如至少三个亲缘细胞得到精确的单个核苷酸变体(SNV)结果。
为了克服这种技术上产生的变化性,本公开的实施方案提供了实现真正的线性预扩增的新过程。因为半扩增子是线性产生的,所以使这些半扩增子线性地无失真地拷贝为全扩增子是有用的。其实现后将提供完全线性的扩增方案。为了实现该目的,本公开的实施方案包括图2所示的内容;预扩增后,不采取(例如)下游PCR扩增,可以使用合适的限制酶切割成环的全扩增子中的引物序列区域。也可以用生物素化的PCR引物将全扩增子转化为双链DNA,然后通过链霉亲和素磁珠从反应中提取。同时,单链的半扩增子保持完整。在随后的步骤中,可以灭活限制酶,并立即由半扩增子重新产生新的全扩增子以保证线性代表(图2)。可以通过一轮或多轮退火和延伸步骤再产生全扩增子。或者,半扩增子可以有尾巴,并且尾部区域可以与新的引物杂交以产生全扩增子。
因此,本发明的实施方案包括从一个或多个细胞线性扩增核酸的方法。所述方法在一些方面由提供的核酸样品开始,尽管在一些情况下所述核酸必须(例如通过常规方法)获自所述细胞。在扩增DNA时,将从细胞提取的全部核酸经由RNA酶处理。当扩增(以来自mRNA的cDNA形式的)转录组时,将所述全部核酸在mRNA的逆转录之前经由DNA酶处理。来自一个或多个细胞的核酸与第一大量引物接触且所述细胞还与包含链置换活性的聚合酶接触;这一步骤例如在温度范围0℃至约30℃的条件下发生。在这一步骤中,引物与核酸退火,且引物通过聚合酶得到延伸。在特定实施方案中,第一大量引物富含40%-60%的G或富含40%-60%的C并且还包含限制性内切核酸酶位点。当引物接触核酸时,生成包含引物退火的核酸模板的混合物。然后使引物退火的核酸模板进行两轮或多轮的延伸、解链和退火步骤,且这些步骤产生核酸模板、线性产生的半扩增子和非线性产生的全扩增子的混合物。将所述混合物暴露于使得全扩增子的两个末端能够彼此退火的条件下,从而产生成环的全扩增子。将所述成环的全扩增子随后接触能够消化成环的全扩增子的退火末端的限制性内切核酸酶。这样做使得全扩增子不再能够由至少某些引物退火,包括第一大量引物或第二批大量引物,其中第二大量引物富含40%-60%的G或C。留在混合物中的线性产生的半扩增子可由第一大量引物或第二批大量引物退火并延伸。所述退火和延伸在核酸不会进一步解链的情况下发生,由此产生线性产生的全扩增子。在一些情况下,所述线性产生的全扩增子被进一步扩增例如通过线性或者非线性方法,包括例如标准PCR方法。
在本发明的实施方案中,可以实现用于单细胞全基因组(或转录组)扩增的第一个精确线性扩增方法。对于单细胞SNV(例如)检测通过本创新所达到的灵敏度和精确度,本公开的方法和组合物可以在生物学和/或临床研究和用途中产生广泛的影响。
在本公开的实施方案中,提供了能够有效地移除预先存在的引物以允许半扩增子的有效加尾的方法。如果引物没有得到有效消化,残留引物的加尾与半扩增子的加尾相竞争导致在随后步骤中的扩增失败。因此,在特定实施方案中,提供了预先存在的引物的有效消化,因此实现了全扩增子的成功产生和扩增。
可以使用T4聚合酶或在低于(30℃或更低)的低温下具有外切核酸酶活性的其它聚合酶和ExoI外切核酸酶或仅消化单链DNA的其它外切核酸酶。酶可以被加热灭活。加尾研究可以用高浓度的C碱基进行。在特定实施方案中,dC加尾的区域可与GAT5N3G引物杂交而仅产生全扩增子。
III.条形码
尽管图2的方案示意线性的,在一些实施方案中,也可能有其中并非所有半扩增子都有效拷贝成全扩增子的情况,例如在最后几个循环中。作为该方面的结果,至少在一些情况下,可能有有限数目的独立全扩增子。如果该数目小于(例如)四个拷贝,则仍然难以区分真实突变和扩增错误。在具体方面,需要具有原始模板的足够数量独立拷贝以稀释最终读数呈现中的扩增错误。为了解决这个问题,还可以向引物中引入随机的“条形码”(具有可变长度的随机DNA序列(例如NNNNN,其中N表示四个核苷酸的混合物)),其将索引每个线性扩增的半扩增子并将每个全扩增子登记到相应的半扩增子上(图3)。通过用条形码对每个读数进行索引,可以评估读数是否是线性分布的。在有残留非线性的情况下,可以使用条形码鉴定扩增错误并改进SNV结果的准确度,如图3所示。
IV.本公开方法的示例性应用
本公开方法可在研究、临床和/或其它应用中使用。在具体的实施方案中,本公开的方法被用于例如对个体的一种或多种疗法的诊断和/或预后和/或监测、微生物的宏观基因组分析和法医DNA测试。
在本公开的一个或多个方法的应用的一个实例中,所述方法被用于测定来自个体的核酸的含量或表达水平中一种或多种变化;所述变化可以是相对于已知的标准品,例如,如具体核酸的对应野生型序列。含量的变化可以包含一个或多个相比野生型的核苷酸差异,如置换、删除、倒置,等等。表达的变化可包含具体相比于某个具体的已知或确定的标准的正常表达水平的上调或下调。所述标准品可包含已知基因型和/或表型正常细胞中的正常核酸含量或表达水平。
在特定的情况下,要测定的核酸获自来自个体的样品,该个体患医学病况或怀疑患医学病况或有患医学病况的风险或正在接受针对医学病况的治疗。所述样品可以为任意种类,只要能直接或间接从来自样品的一个或多个细胞中获得核酸即可。在具体的实施方案中,核酸获自来自个体的样品中的一个或多个细胞。所述样品可以是血液、组织、毛发、活检标本、尿液、乳头抽吸物、羊水、面颊刮屑、粪便物、或胚胎。
从所述个体获得适当的样品,并可由获得样品的个体直接或间接实行本公开的方法,或者可由另一方或多方实行所述方法。
A.遗传检测
在具体应用中,期望的是获知来自个体的样品中的一个或多个具体核酸的序列。所述个体可以是任何年龄。所述个体可以是进行常规检测,或处于某一具体期望或医学原因而进行测试。所述个体可能怀疑患某具体医学病况,例如由于具有一种或多种与所述医学病况相关的症状和/或具有与所述医学病况相关的个体或家族史。所述个体可能有患某医学病况的风险,如具有所述医学病况的家族病史或具有所述医学病况的一种或多种已知的风险因素,如高胆固醇之于心脏疾病,吸烟之于多种医学病况,高血压之于心脏疾病或中风,具有与所述医学病况相关的遗传标志物,等等。
在特定的情况下,所述个体是胎儿,并且该胎儿可被或不被怀疑具有某一具体核酸序列或核酸表达相比野生型有变化,这种序列内容或表达变化与医学病况相关。在一些情况下,尽管所述胎儿可能出于常规目的需要进行测试,但该胎儿由于例如家族史或环境风险(即辐射)或高龄妊娠,而具有某一具体医学病况的风险。在那些其中期望从胎儿得知具体序列内容或表达水平的情况下,采集包含一个或多个胎儿细胞的样品。所述样品可以来自胎儿的活检标本,尽管在具体情况下,样品是羊水或母体血液或发育早期的胚胎。
在本公开的一方面,从怀孕母亲获得羊水并从中分离一个或多个胎儿细胞。所述胎儿细胞分离可通过本领域常规方法进行,如通过使用胎儿细胞表面的标志物区分胎儿细胞和母体细胞。三种不同类型的胎儿细胞可存在于母体循环中:滋养层细胞、白细胞和胎儿红细胞(有核红细胞)。最可能富集的细胞是胎儿红细胞,在某些实施方案中其可通过尺寸色谱柱鉴定,随后进行CD71-抗体染色或ε球蛋白链免疫分型,然后基于荧光强度进行扫描或分选而鉴定。
一旦分离出胎儿细胞,即从中提取核酸,例如通过本领域常规方法。对来自胎儿细胞核酸使用本公开的方法以产生线性生成的扩增子,其覆盖至少部分、大部分或全部的所述胎儿细胞的基因组。在线性扩增后,可进一步扩增扩增子的一个或多个序列,也可对其测序,至少部分测序,或可实施微阵列技术。在特定的实施方案中,例如测定了SNV或CNV,且测定结果被用于确定相应的胎儿是否患具体医学病况或是否怀疑患具体医学病况。在特定情况下,例如可对胎儿进行所述医学病况的治疗或可对其使用防止或延迟所述医学病况发病的方法,且这可在子宫内和/或分娩后进行。
虽然可对胎儿样品测定SNV或CNV的存在情况(其中任一可为疾病相关的或导致疾病的),在具体实施方案中对胎儿样品测定与任意具体医学病况相关的遗传突变。可测定的与产前医学病况相关的基因实例包括以下中的一个或多个:ACAD8、ACADSB、ACSF3、C7orf10、IFITM5、MTR、CYP11B1、CYP17A1、GNMT、HPD、TAT、AHCY、AGA、PLOD2、ATP5A1、C12orf65、MARS2、MRPL40、MTFMT、SERPINF1、FARS2、ALPL、TYROBP、GFM1、ACAT1、TFB1M、MRRF、MRPS2、MRPS22、MRPL44、MRPS18A、NARS2、HARS2、SARS2、AARS2、KARS、PLOD3、FBN1、FKBP10、RPGRIP1、RPGR、DFNB31、GPR98、PCDH15、USH1C、CERKL、CDHR1、LCA5、PROM1、TTC8、MFRP、ABHD12CEP290、C8orf37、LEMD3、AIPL1、GUCY2D、CTSK、RP2、IMPG2、PDE6B、RBP3、PRCD、RLBP1、RGR、SAG、FLVCR1、ZNF513、MAK、NDUFB6、TMLHE、ALDOA、PGM1、ENO3、LARS2、ATP7A、ATP7B、TNFRSF11B、LMBRD1、MTRR、FAM123B、FAM20C、ANKH、TGFB1、SOST、TNFRSF11A、CA2、OSTM1、CLCN7、PPIB、TCIRG1、SLC39A13.COL1A2、TNFSF11、SLC34A1、NDUFAF5、FOXRED1、NDUFA2、NDUFA8、NDUFA10、NDUFA11、NDUFA13、NDUFAF3、SP7、NDUFS1、NDUFV3、NUBPL、TTC19、UQCRB、UQCRQ、COX4I1、COX4I2、COX7A1、TACO1、COL3A1、SLC9A3R1、CA4、FSCN2、BCKDHA、GUCA1B、KLHL7、IMPDH1、PRPF6、PRPF31、PRPF8、PRPF3、ROM1、SNRNP200、RP9、APRT、RD3、LRAT、TULP1、CRB1、SPATA7、USH1G、ACACB、BCKDHB、ACACA、TOPORS、PRKCG、NRL、NR2E3、RP1、RHO、BEST1、SEMA4A、RPE65、PRPH2、CNGB1、CNGA1、CRX、RDH12、C2orf71、DHDDS、EYS、IDH3B、MERTK、PDE6A、FAM161A、PDE6G、TYMP(ECGF1)、POLG(POLG1、POLGA)、TK2、DGUOK(dGK)、SURF1、SCO2(SCO1L)、SCO1、COX10、BCS1L、ACADM、HADHA、ALDOB、G6PC(GSD1a)、PAH(PH)、OTC、GAMT、SLC6A8、SLC25A13、CPT2、PDHA1、SLC25A4(ANT1)、C10orf2(TWINKLE)、SDHA、SLC25A15、LRPPRC、GALT、PMM2、ATPAF2(ATP12)、GALE、LPIN1、ATP5E、B4GALT7、ATP8B1(ATPIC、PFIC)、ABCB11(ABC16、PFIC-2、PGY4)、ABCB4(GBD1、MDR2、PFIC-3)、MPV17(SYM1)、TIMM8A(DDP、MTS)、CPS1、NAGS、ACADVL、SLC22A5(OCTN2)、CPT1A(CPT1-L、L-CPT1)、CPT1B、SUCLA2、POLG2(HP55、MTPOLB)、ACADL、SUCLG1、MCEE、GAA、PDSS1(COQ1、TPT)、PDSS2(bA59I9.3)、COQ2(CL640、FLJ26072)、RRM2B(p53R2)、ARG1、SLC25A20(CACT)、MMACHC(cblC)、FAH、MPI、GATM、OPA1、TFAM、TOMM20(MAS20P、TOM20)、NDUFAF4(HRPAP20、C6orf66)、NDUFA1(CI-MWFE、MWFE)、SLC25A3(PHC)、BTD、OPA3(FLJ22187、MGA3)、GYS2、NDUFAF2(B17.2L、MMTN)、HLCS(HCS)、COX15、FASTKD2、NDUFS4、NDUFS6、NDUFS3、MMAA(cblA)、MUT、NDUFV1、MOCS1、NDUFS7(PSST)、TAZ(BTHS、G4.5、XAP-2)、MOCS2、COX6B1(COXG)、HADHB、MCCC1(MCCA)、MCCC2(MCCB)、TSFM(EF-TS、EF-Tsmt)、PUS1、ISCU、AGL、SDHAF1、IVD、GCDH、ADSL、DARS2、RARS2、TMEM70、ETHE1、PC、JAG1、MRPS16、PCCA、PCCB、COQ9、LDHA、PYGL、GALK1、PYGM、PGAM2、TUFM、TRMU、PFKM、GBE1、SLC37A4、GYS1、ETFDH、NDUFS8、CABC1(ADCK3)、ETFA、ETFB、DBT、SLC25A19、MMADHC、PDP1、PDHB、ACAD9、AUH、DLAT、PDHX、ACADS、NDUFS2、FBP1、NDUFAF1(CIA30、CGI65)、YARS2、SUCLG2、TCN2、CBS、PHKB、PHKG2、PHKA1、PHKA2、LIPA、ASL、HPRT1、OCRL、PNP、TSHR、ADA、ARSB、ALDH5A1、PNP、AMT、DECR1、HSD17B10、IYD、IL2RG、MGME1、HMGCL、IQCB1、OTX2、KCNJ13、CABP4、NMNAT1、ALG2、DOLK、ABCD4、ALDH4A1、ALG1、GPR143、UBE3A、ARX、GJB2(CX26、NSRD1)、APC、HTT、IKBKG(NEMO)、DMPK、PTPN11、MECP2、MECP2、RECQL4、ATXN1、ATXN10、RMRP、CDKL5、PLP1、GLA、DMD、RUNX2、PLP1、CHD7、ASS1、AIRE、EIF2B、LDLR、HPRT1、RPS19、LMX1B、COL10A1、CRTAP、LEPRE1、PORCN、ASL、CFTR、ARSA、IDUA、IDS、MYO7A、GLANS、GALC、KRAS、SOS1、RAF1、AR、PTEN、BLM、SLC9A6、HRAS、GJC2(GJA12)、NPC1、NPC2、FMR1、FMR1、PLOD1、COL2A1、COL5A1、COL5A2、ABCA4、FOXG1、TINF2、USH2A、CDH23、CLRN1、CREBBP、ABCA4、POU3F4、NRAS、CHRNA7、FOXF1、MEF2C、DHCR7、RAI1、VHL、TYR(OCAIA)、OCA2(BEY、BEY1、BEY2、EYCL)、TYRP1(b-PROTEIN、CATB、GP75、SLC45A2(AIM-1)、PCDH19、SHOC2、BRAF、MAP2K1、MAP2K2、HEXA、STXBP1、ALDH7A1、SLC2A1、WDR62、MAGEL2、SDHB和FH。
B.癌症测试
在本公开的一些实施方案中,对来自患癌症或怀疑患癌症或癌症治疗结果正被监测的个体的样品实施本发明的方法。除了本公开的方法之外,还可以在样品或类似样品中运行其他诊断或预后测试。所述样品可以通过常规方法获得,并且可以包括活检标本,其包含似乎是、怀疑是或已知是癌变的细胞或组织。用于癌症检测的示例性的样品包括血液、尿液、活检标本、粪便物、乳头抽吸物、脸颊刮屑等。在一些情况下,从具有患癌症风险的个体获得样品;该个体可以有家族和/或个体史,可能已经接触已知或怀疑会导致癌症的环境条件,可以已知具有与至少一种癌症类型相关的遗传标志物,等等。具体类型的活检标本包括皮肤、肺、乳房、结肠、子宫颈、肝、肾、前列腺的标本,等等。
在具体实施方案中,对来自个体的待测样品实施本公开的方法以测定其中序列内容相比于已知样品的变化,或测定某一序列表达水平相比于正常水平的变化(如一个或多个基因的上调或下调)。在一些情况下,由本公开的方法产生的扩增子的数量所代表的一个或多个特定基因的表达水平指示存在癌症或者患癌风险情况或对癌症治疗的成功与否。
可测定的与具体癌症相关基因的实例包括APC、MLH1、MSH2、MSH6、PMS2、MUTYH(MYH)、RECQL4、TP53(LFS1、P53)、PTEN、RUNX1、TPMT、VHL、EPCAM(TACSTD1)、ERBB2(HER2/neu)、ALK、RET、EGFR、MET、IGH、ROS1、BRAF、NPM1、JAK2、MPL、AKT1(AKT)、PIK3CA、FLT3、IgVH、CEBPA、MAX、KIT、KRAS、NF1、SDHAF2、SDHB、SDHC、SDHD、TMEM127、BMPR1A、SMAD4、STK11、BRCA1、BRCA2、CDH1、PALB2、CDKN1C、FH、FLCN、GPC3、PALB2、WTL、CDC73、MEN1、PRKAR1A、ATM、NBN、NF2、PHOX2B、PTCH1、SUFU和/或UGT1A1。
V.样品处理和来自本发明细胞的核酸
可以通过任何适当的手段来从用本公开方法进行测试的个体获得一种或多种样品。在某些实施方式中,可在提取核酸前对样品进行处理。在提取核酸时样品可以是新鲜的,或在提取核酸时样品可能经过固定或其他处理技术的处理。
样品可以是任何种类的。在其中一个或多个目的细胞与其他细胞混在一起的实施方案中,可基于所期望的一个或多个细胞的独特特征,如细胞表面表达的蛋白质,分离一个或多个目的细胞。在其中基于细胞标志物而分离胎儿细胞的实施方案中,细胞标志物可以为CD71或ε球蛋白链,等等,在其中基于癌症标志物而分离癌细胞的实施方案中,细胞标志物可以为ER/PR、Her-2/neu、EGFR、KRAS、BRAF、PDFGR、UGT1A1等(Bigbee W,Herberman RB.Tumor markers and immunodiagnosis.In:Bast RC Jr.,Kufe DW,Pollock RE,等人,editors.Cancer Medicine.6th ed.Hamilton,Ontario,Canada:BC Decker Inc.,2003.)。
可将细胞在有表面活性剂(即Trion-X100,tweet-20,NP-40等)和蛋白酶(即蛋白激酶K)的裂解缓冲液中孵育而裂解所分离的细胞。还可通过碱性溶液(即去垢剂十二烷基硫酸钠(C12H25SO4Na)和强碱如氢氧化钠)裂解细胞,这将导致双链DNA的变性。碱性溶液通过乙酸钾或乙酸钠中和。
VI.本发明的试剂盒
本文描述的任意组合物或类似物可以包含在试剂盒中。在一个非限制性实例中,一种或多种用于扩增核酸的方法的反应剂可包含于试剂盒中。这些反应剂可包括酶、缓冲液、核苷酸、盐、引物等等。试剂盒组分在合适的容器形式中提供。
这些试剂盒的一些组分可以在水媒介物中或以冻干形式进行包装。试剂盒的容器形式通常包括至少一个小瓶、试管、烧瓶、瓶、注射器或其他容器形式,其中置有组分,并优选被合适地分装。试剂盒中有超过一种组分时,该试剂盒通常含有第二、第三或其他附加的容器,其中附加组分可分开置于这些容器中。然而,组分的多种组合可包含于小瓶中。本发明的试剂盒还典型地包括用于将这些组分包含在密闭容器中用于商业销售的方式。所述容器可包括注塑或吹塑成型的塑料容器,其中留存有所期望的小瓶。
当所述试剂盒的组分是以一种和/或多种液体溶液提供时,所述液体溶液是水溶液,其中灭菌水溶液尤其有用。在一些情况下,所述容器形式自身可以是注射器、吸头和/或其它类似装置,或可以是具有用于进行所期望的反应的多个区室的基底。
所述试剂盒的一些组分可以以干粉提供。当反应剂和/或组分以干粉提供时,可以通过加入合适的溶剂来重构所述粉末。可以预见的是,所述溶剂也可在其他容器形式中提供。这些试剂盒还可包含第二种容器形式,用于包含灭菌的可接受缓冲液和/或其他稀释剂。
在特定实施方案中,反应剂和材料包括用于扩增期望序列的引物、核苷酸、合适的缓冲液或缓冲反应剂、盐等等,且在一些情况下,所述反应剂包括用于分离具体的期望细胞的装置或反应剂。
在具体实施方案中,试剂盒中有一种或多种装置适用于从个体提取一种或多种样品。该装置可以是针筒、细针头、手术刀等等。
实施例
包括以下实施例以说明本发明的优选实施方案。本领域技术人员应当清楚的是,在那些根据本发明者发现的代表性技术的实施例中公开的那些技术在本发明的实践中能很好的起作用,并且因此可被认为组成优选的实践形式。然而,本领域技术人员根据本公开应当清楚的是,在公开的特定实施方案中可进行多种变化且仍然获得相似或类似的结果而不偏离本发明的精神和范围。
实施例1
从组织样品中分离完整的单细胞
使用商业化冷冻切片机制备组织学组织切片。制备100-200μm厚的多聚甲醛固定的固体组织的切片。用激光微切显微镜(Leica,MMI等)从组织切片中切割出目的的细胞。使用标准细胞解离操作流程将个体细胞从微切组织切片中解离出来。将完整的单细胞收集到各自的管子中。在3至5μl的裂解缓冲液(30mM Tris-Cl PH 7.8,2mM EDTA,20mM KCl,0.2%Triton X-100,12μg/ml Qiagen蛋白酶)中裂解所述个体细胞并将其加入PCR管的边上,离心下来。然后热裂解所捕获的细胞,在PCR仪上使用如下温度程序:50℃2小时,75℃20分钟,80℃5分钟。
线性产生半扩增子的多重循环(图4)
在第一轮扩增中,一对准简并引物用于在整个基因组DNA起始重叠的扩增子。所述引物被记为NG、NT引物,如下:
NG引物5'-GTGAGTGATGGTTGAGGATGAGTGGTNNNNNGGG-3'(SEQ ID NO:1)
NT引物5'-GTGAGTGATGGTTGAGGATGAGTGGTNNNNNTTT-3'(SEQ ID NO:2)
将以下缓冲液加到PCR管中并用于第一次扩增:6.0μl ThermoPol缓冲液(NEB),1.0μl的dNTP(10mM),26μl的H2O(经UV处理)和0.3μl的NG和NT引物(100μΜ)。
将PCR缓冲液加入到含有裂解的单细胞的PCR管后,将样品在94℃下加热1-2分钟,以使DNA变性成单链DNA。立即将样品加入冰中使其淬灭并至约0℃的温度,在此期间发生引物退火。然后将0.6μl的聚合酶Bst大片段的混合物加入到PCR管中。在PCR仪上运行下列温度循环。在第二次和随后的循环中,将PCR管接着转移到冰上以淬灭反应,并起始新的引物引发。将新鲜聚合酶混合物加入到PCR管中并在PCR仪上运行以下循环以产生扩增子。
步骤1:10℃-45秒
步骤2:20℃-45秒
步骤3:30℃-45秒
步骤4:40℃-30秒
步骤5:55℃-30秒
重复步骤1-5,4次
步骤6:65℃-60秒
步骤7:95℃-20秒
步骤8:冰上淬灭并重新添加聚合酶
重复步骤1-8,N次
4℃–∞
重复次数N可根据条件进行调整。在某些实施方案中,N为3。
全扩增子的双链转换
来自上述程序的产物是全扩增子和半扩增子的混合物。将0.3μl的100μΜ PCR引物加入反应中并进行以下热循环过程(例如:58℃20秒-70℃-10秒,重复30次),以将全扩增子转化成双链的DNA,而单链半扩增子保持不变。在上述双链转换后。
PCR引物:GTGAGTGATGGTTGAGGATGAGTGGT(SEQ ID NO:4)
全扩增子的限制性消化
来自上述热循环的产物包括线性扩增的半扩增子和非线性扩增的全扩增子。将温度加热至94℃,持续30秒以解链双链DNA产物,然后保持在50℃。在50℃,全扩增子将由于5’末端序列与3’末端序列的互补形成成环的,而半扩增子以单链DNA存在。引物序列整合有GGATG序列基序,可被限制性内切核酸酶BtsCI识别并切割DNA。其结果是,全扩增子一半以上的引物序列被删除,因此,经消化的DNA产物在下游PCR反应中不再被扩增。消化后将温度升高至72℃至80℃的范围以灭活限制酶。
线性产生全扩增子(图5)
在上述消化后,将样品在94℃加热20秒以使DNA变性为单链DNA。立即将样品加入冰中淬灭并达到约0℃的温度,此期间发生引物退火。此后重新加入聚合酶并实行下面的热循环,从扩增的半扩增子线性再现全扩增子。也可将所述半扩增子分到多个管中并继续进行以下的多个退火步骤,以形成扩增子。扩增子的线性产率示于图9。
步骤1:10℃-45秒
步骤2:20℃-45秒
步骤3:30℃-45秒
步骤4:40℃-30秒
步骤5:55℃-30秒
重复步骤1-5,4次
步骤6:65℃-60秒
备选地,也可将半扩增子分到多个管中,并继续进行以下实施例的包括消化、加尾和延伸步骤,以形成扩增子:
用T4聚合酶和ExoI核酸酶的组合消化预扩增产物,如下:加入0.4μl ExoI,在25℃消化30分钟,然后添加0.4μl T4聚合酶,在25℃消化150分钟。在80℃对酶进行进行20分钟的热灭活,随后进入加尾程序。
10X TdT反应缓冲液由0.35μl的TdT缓冲液、0.4μl 100mM的dCTP、0.1μl TdT末端转移酶及2.75μl H2O构成。将TdT混合物加入样品,混匀并使用以下温度进行加尾反应:37℃15分钟,72℃15分钟。
加尾后,添加下述延伸缓冲液(1.5μl 10X Thermopol,1.25μl的dNTP(各10μM),1.25ul 10μM GAT21 5n3G和12μl H2O)。充分混合反应后,把样品置于模块上95℃加热1min,将温度降至50℃,保持至少20s;添加0.4μl的deepvent DNA聚合酶或其他聚合酶,混匀并进行以下循环:
步骤1:50℃45秒
步骤2:72℃45秒
重复步骤1至2,10次
实施例2
使用标准方法进行扩增子的单管或多管扩增
制备以下反应缓冲液,并加入维持于冰上的PCR管中。
3.0μl ThermoPol缓冲液(NEB)
1.0μl dNTP(10mM)
26μl H2O(经UV处理)
0.1μl引物(100μM)(5’-GTGAGTGATGGTTGAGGATGAGTG-3’;SEQ ID NO:5)
可将扩增缓冲液分到多个管中。线性扩增产物将被分到各管中(图10)。用如下标准PCR程序实行扩增,以生成1-2μg的DNA材料。
94℃-20秒
58℃-20秒
65℃-1分钟
72℃-1分钟
重复上述循环20次
72℃-5分钟
4℃–∞
在第二轮扩增后,可以用Qiagen柱纯化DNA,并储存以用于下一程序以移除DNA扩增子的引物末端。
扩增子产物可以用于全基因组或定向(例如,外显子组和基因面板的任何列表)测序/重测序和基因分型方法,包括Sanger测序,下一代测序和微阵列等。下一代测序的结果示于图10和图11。
实施例3
微滴/微孔中全基因组单一片段扩增
从实施例1中所得线性扩增的扩增子覆盖了单细胞DNA的全基因组,并可用作以下程序的起始材料(见图6)。
可如下使用本领域技术人员已知的其他扩增方法。
将产物分成千万个皮升级微滴/微孔或飞升级微滴/微孔。可使用商业可用的基于微流体的液滴乳化剂或者有皮升或飞升孔的微流体设备。通过在各反应微滴/微孔中达到扩增饱和(或通过可用的引物或dNTP限制扩增)将单个DNA片段扩增到类似的水平。
可如下使用本领域技术人员已知的其他形成大规模个体反应的方法。
可以在1千万个微滴/微孔中的每个中实行PCR反应以扩增单个扩增子。
94℃-20秒
58℃-20秒
65℃-1分钟
72℃-1分钟
重复上述循环20次
72℃-5分钟
4℃–∞
从每个微滴/微孔中收集DNA产物并使用商业纯化柱或乙醇沉淀进行纯化。
实施例4
移除扩增子中引物序列
可以用以下尿嘧啶引物对从实施例II收集的完全扩增的DNA再扩增6个循环(见例如图7):
5'-GTGAGTGATGGTTGAGGATGAGTGGU-3'(SEQ ID NO:3)
扩增后,使用DNA纯化柱纯化DNA产物。尿嘧啶-DNA糖基化酶(UDG)和DNA糖基化酶裂解酶内切核酸酶VIII的混合物用于移除引物序列中的U碱基。UDG催化尿嘧啶基团的切除,而内切核酸酶VIII移除非嘧啶碱基。
用DNA纯化柱纯化所述有缺口的DNA。用Zn++离子作为催化离子的S1核酸酶在切口位点切开单链DNA。由此,在DNA的两个末端的原始引物序列都被移除。
T4聚合酶也可用于移除引物序列。酶会找到缺口并在缺口位点从3’至5’进行消化。移除顶链后,酶将移除3’突出,即底链。
用DNA纯化柱纯化DNA产物。该DNA将可以直接用于文库构建进行下一代测序实验。
实施例5
大量微滴/微孔中分开进行单细胞DNA片段的PCR扩增
可以覆盖单细胞DNA的全基因组的PCR扩增子作为起始材料。
可如下使用本领域技术人员已知的其他扩增方法。
将产物分成千万个皮升级微滴/微孔或飞升级微滴/微孔。可使用商业可用的基于微流体的液滴乳化剂或者有皮升或飞升的孔的微流体设备。通过在各反应微滴/微孔中达到扩增饱和(或通过可用的引物或dNTP限制扩增)将单个DNA片段扩增到类似的水平。
可以在1千万个微滴/微孔中的每个中实行PCR反应以扩增单个扩增子。
94℃-20秒
58℃-20秒
65℃-1分钟
72℃-1分钟
重复上述循环20次
72℃-5分钟
4℃–∞
从每个微滴/微孔中收集DNA产物并使用商业纯化柱或乙醇沉淀进行纯化。
实施例6
大量微滴/微孔中分开进行单细胞DNA片段的SDA
链置换扩增(SDA)用于形成单个片段。然而,与正常SDA反应相比,扩增时间最小化为10至30分钟。这将避免超分支(hyperbranch)的形成和超支化单链DNA引起的扩增偏差。
加入新置换酶(Phi29),并将反应分成1千万个皮升级微滴/微孔或者飞升级微滴/微孔。可使用商业可用的基于微流体的液滴乳化剂或有皮升或飞升的孔的微流体设备。
可在30度下继续进行链置换扩增(SDA)以在各个微滴/微孔中扩增单个DNA片段。大多数个体微滴/微孔中没有DNA片段,或DNA片段数量有限。延长SDA的实行时间(12小时)以达到饱和。
通过在分开的孔中扩增单个片段,该反应避免了片段的干扰和竞争。通过在每个反应微滴/微孔中达到扩增的饱和(或通过可用的引物或dNTP限制扩增)将单个DNA片段扩增到类似的水平。
从每个微滴/微孔中收集DNA产物并使用商业纯化柱或乙醇沉淀进行纯化。
可如下使用本领域技术人员已知的其他扩增方法。可以如本文所述使用本领域技术人员已知的其他形成大规模单个反应的方法。
实施例7
大量微滴/微孔中分开进行单细胞RNA片段的PCR扩增
可用通过单细胞RNA转录物的逆转录产生的cDNA作为起始材料。
可如下使用本领域技术人员已知的其他扩增方法。
将产物分成千万个皮升级微滴/微孔或飞升级微滴/微孔。可使用商业可用的基于微流体的液滴乳化剂或有皮升或飞升的孔的微流体设备。通过在各反应微滴/微孔中达到扩增饱和(或通过可用的引物或dNTP限制扩增)将单个DNA片段扩增到类似的水平。
可以在1千万个微滴/微孔的每个当中实行单个扩增子的PCR反应。
94℃-20秒
58℃-20秒
65℃-1分钟
72℃-1分钟
重复上述循环20次
72℃-5分钟
4℃–∞
从每个微滴/微孔中收集DNA产物并使用商业纯化柱或乙醇沉淀进行纯化。
实施例8
用于人类癌症样品的方法
可使用本实施方案的方法和组合物用于表征肿瘤发生的复杂的演变过程。与实体癌的大规模测序的努力相比,可将测序研究的前沿推向能够在例如临床环境中得到恢复的肿瘤最早期。这仅在有单细胞全基因组扩增测定时变得合理,该测定允许得到上文描述的准确的SNV结果和使用临床样品进行有效的单细胞分离测定法。
从临床组织样品分离单细胞
为了对临床样品应用所述新颖的单细胞分析,可以利用有效方法对目的的细胞进行分离。例如可通过酶解获得组织样本的单细胞悬浮液。然而,这样会失去解离细胞附近的信息。另一种可获得形态学信息的方法是显微激光解剖。然而,就激光显微解剖的可用性,其不能保证得到一个完整的单细胞。为了从目的组织结构中获得完整的细胞,可以在研究中结合所述两种测定。可制备100微米厚的组织切片并从解剖的组织中切割单个组织单元(即单个腺体、管道等)。本节的。通过这样做,可以保证有完整的单细胞在解剖组织中。下一步可应用酶解离。可以通过明场显微镜成像记录整个消化过程。
早期肿瘤发展中的基因组异质性
考虑到组织中有成千上万个细胞,因此器官中的某个细胞随机地进行驱动突变很可能并不奇怪。考虑到基因组是三十亿个碱基的集合,在第一个驱动突变后,此细胞极少会获得第二个驱动突变。事实上,在获得第一个驱动突变后(PanIN中的Kras),细胞可逃避正常的分化并开始增殖和重编程。在扩大后,这群异常的前体细胞可遇到内部和外部的危机,即端粒危机、免疫攻击和缺氧。其结果是,人们可以预料到增加的细胞死亡和减缓的增殖。该阶段的存在可以表现为发育不良,这可能是癌症发展的关键阶段。发育不良可存活多年,积累高度的基因组异质性。
随着对肿瘤的全基因组测序,在不同类型的成人癌症(Stratton,2011)中肿瘤细胞通常积累1000-10000个体细胞突变。该数字设定了前体细胞发展成癌症所必须的突变数量范围。因此,预期成千上万的突变在发育不良细胞的潜伏期积累。争议在于仅当大量前体细胞已经获得广泛突变时,其中某个细胞可以经过下一轮重要的驱动突变。一旦发生,可预期该细胞会得到相比其他巨大细胞的增殖优势并形成大得多的克隆性扩张;该增殖期对应肿瘤形成的时期(图8)。面对各种类型的危机,发育不良和肿瘤形成可能有多重时期且其要在一个肿瘤中共存。然而,由于细胞群体越来越大,克隆性演化加速并最终导致恶性转化。
根据上述早期癌症发展的景象,我们希望在发育不良时期见到高度基因组异质性。在肿瘤性细胞之间异质性可能较少。通过对比发育不良和肿瘤形成之间的突变,我们能找到导致此转化的潜在突变。
在胰腺癌的情况下,Kras、p16、p53和SMAD4基本上是最显著的驱动突变。人们普遍认为,Kras基因突变是第一驱动突变,因为它发生于90%以上的胰腺癌中。即使在低级别PanIN-1A的病变中亦包括Kras基因突变。当肿瘤性本质还没有明确建立时,被记为PanIN/L-1A。该级别对应可以临床样本中采集的最早期的PanIN病变。由于细胞稀少,所以可使用单细胞全基因组测序揭示低级别PanIN/L-1A中的基因组抑制性。从PanIN-1A至PanIN-2期,可获得其他关键突变如p16、p53或SMAD4,此时预期的是肿瘤的发展。
基因组异质性的早期阶段还将包括由于端粒危机或其他压力造成的非整倍体。这种染色体异常已在结直肠癌和乳腺癌腺瘤中被广泛观察到。通过复制或删除大的染色体片段,非整倍体可能会导致细胞死亡,或者备选地对细胞施压朝向重编程方向进行。显然非整倍体会影响进化动力学,如果这些删除发生在有关键肿瘤抑制子基因的区域,其还可起到驱动作用导致恶性转化。非整倍体是否起到导致肿瘤形成的驱动作用或只是起到支持作用以增加发育不良细胞的存活,这一点将在本研究中得到解决。
参考文献
Cheng J,Vanneste E,Konings P,Voet T,Vermeesch JR,Moreau Y.Single-cell copy number variation detection.Genome biology.2011;12(8):R80.doi:10.1186/gb-2011-12-8-r80.PubMed PMID:21854607;PubMed Central PMCID:PMC3245619.
Dean FB,Hosono S,Fang L,Wu X,Faruqi AF,Bray-Ward P,et al.Comprehensive human genome amplification using multiple displacement amplification.Proceedings of the National Academy of Sciences.2002;99(8):5261-6.doi:10.1073/pnas.082089499.
Dean FB,Nelson JR,Giesler TL,Lasken RS.Rapid Amplification of Plasmid and Phage DNA Using Phi29DNA Polymerase and Multiply-Primed Rolling Circle Amplification.Genome Res.2001;11(6):1095-9.doi:10.1101/gr.180501.
Fan HC,Wang J,Potanina A,Quake SR.Whole-genome molecular haplotyping of single cells.Nature biotechnology.2011;29(1):51-7.doi:10.1038/nbt.1739.PubMed PMID:21170043.
Hou Y,Song L,Zhu P,Zhang B,Tao Y,Xu X,et al.Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm.Cell.2012;148(5):873-85.doi:10.1016/j.cell.2012.02.028.PubMed PMID:22385957.
Lao K,Xu N,Straus N.Whole genome amplification using single-primer PCR.Biotechnol Journal.2008;3(3):378.
Navin N,Kendall J,Troge J,Andrews P,Rodgers L,Mclndoo J,et al.Tumour evolution inferred by single cell sequencing.Nature.2011;472(7341):90-4.doi:10.1038/nature09807.PubMed PMID:21399628.
Stratton MR.Exploring the genomes of cancer cells:progress and promise.Science.2011;331(6024):1553-8.doi:10.1126/science.1204040.PubMed PMID:21436442.
Telenius H,Carter NP,Bebb CE,Nordenskjold M,Ponder BA,Tunnacliffe A.Degenerate oligonucleotide-primed PCR:general amplification of target DNA by a single degenerate primer.Genomics.1992;13(3):718-25.PubMed PMID:1639399.
Zhang K,Martiny AC,Reppas NB,Barry KW,Malek J,Chisholm SW,et al.Sequencing genomes from single cells by polymerase cloning.Nature biotechnology.2006;24(6):680-6.doi:10.1038/nbt 1214.PubMed PMID:16732271.
Zhang L,Cui X,Schmitt K,Hubert R,Navidi W,Arnheim N.Whole genome amplification from a single celkimplications for genetic analysis.Proceedings of the National Academy of Sciences of the United States of America.1992;89(13):5847-51.PubMed PMID:1631067;PubMed Central PMCID:PMC49394.
虽然已经详细描述了本发明及其优势,但是应当理解,在不脱离由所附权利要求限定的本发明的精神和范围的情况下,可以对此进行各种变化、替换和改变。此外,本申请的范围不旨在受限于说明书中描述的过程、机器、制作、物质组成、方式、方法和步骤的具体实施方案。本领域普通技术人员从本发明的公开内容中将容易地理解,根据本发明可使用目前存在或以后会开发的过程、机器、制作、物质组成、方式、方法或步骤实行基本上与本文描述的对应实施方案相同的功能或实现基本上相同的结果。因此,所附权利要求旨在将这些过程、机器、制作、物质组成、方式、方法或步骤包括在其范围内。
序列表
<110> Zong, Chenghang
<120> 单细胞全基因组线性扩增法
<130> BAYM.P0124WO
<140> UNKNOWN
<141> 2015-05-05
<150> 61/989,002
<151> 2014-05-06
<160> 5
<170> PatentIn version 3.5
<210> 1
<211> 34
<212> DNA
<213> 人工序列
<220>
<223> 合成的引物
<220>
<221> misc_feature
<222> (27)..(31)
<223> n是a, c, g,或t
<400> 1
gtgagtgatg gttgaggatg agtggtnnnn nggg 34
<210> 2
<211> 34
<212> DNA
<213> 人工序列
<220>
<223> 合成的引物
<220>
<221> misc_feature
<222> (27)..(31)
<223> n是a, c, g,或t
<400> 2
gtgagtgatg gttgaggatg agtggtnnnn nttt 34
<210> 3
<211> 26
<212> DNA
<213> 人工序列
<220>
<223> 合成的引物
<400> 3
gtgagtgatg gttgaggatg agtggu 26
<210> 4
<211> 26
<212> DNA
<213> 人工序列
<220>
<223> 合成的引物
<400> 4
gtgagtgatg gttgaggatg agtggt 26
<210> 5
<211> 24
<212> DNA
<213> 人工序列
<220>
<223> 合成的引物
<400> 5
gtgagtgatg gttgaggatg agtg 24
- 还没有人留言评论。精彩留言会获得点赞!