N末端截短的白介素‑38的制作方法
本发明涉及N-末端截短的白介素(IL)-38蛋白、或其功能变体,还涉及编码该截短的IL-38肽的核酸和载体以及包含这些核酸或载体的重组细胞。本发明显示,IL-38经过N末端加工,并且该细胞因子的截短形式在巨噬细胞中起免疫活化拮抗剂的作用。这表明了该截短的细胞因子在治疗和预防自身免疫病症中的用途。本发明还提供了包含该截短的IL-38蛋白的药物组合物,以及用于筛选截短的IL-38的功能的调节剂的方法。
描述
IL-1家族细胞因子和受体为尤其调节免疫的异质性的蛋白组。最初,4种IL-1家族细胞因子被集中地表征(IL-lα、IL-1β、IL-lRa和IL-18),揭示了免疫调节的基本原理,其中的一些已转化为临床应用。剩下的7种IL-1家族细胞因子是通过基因数据库的软件分析(silico analysis)而鉴定(IL-33、IL-36α、IL-36β、IL-36γ、IL-36Ra、IL-37和IL-38)。近些年,进行了一些重要研究来考察它们诱导或调节免疫应答的相关性。结论是,IL-1家族细胞因子在免疫方面展示了广谱功能,包括诱导Th1和Th2炎症以及调节抗炎或前消散效应。在机制水平上,触发炎症是通过具有受体激动剂(IL-lα、IL-1β、IL-18、IL-33、IL-36)功能的IL-1家族细胞因子所介导,这通过IL-1家族受体拮抗剂(IL-lRa,IL-36Ra)来抗衡。值得注意的是,完全的受体激动剂或拮抗剂功能一般要求IL-1家族细胞因子的N末端加工,通常是从前体生成成熟的细胞因子。这些事件中最突出的地方可能是利用炎性体的IL-1β成熟。
IL-1家族受体以存在细胞外免疫球蛋白结构域和信号转导所需的细胞内TIR结构域为特征。IL-1受体家族包括具有已知配体和功能的4个成员:IL-1R1(IL-1RI)、IL-1R4(ST2)、IL-1R5(IL-18R)、IL-1R6(IL-lRrp2);2个共受体:IL-1R3(IL-lRAcP)、IL-1R7(IL-18AcP);1个诱骗受体:IL-1R2(IL-IRII);以及3个孤儿受体TIR8(SIGIRR)、TIGIRR-1(IL-1RAPL2)、TIGIRR-2(IL-1RAPL1)。孤儿受体的命名仍是不明确的。IL-1RAPL1也称为TIGIRR-2,起初命名为IL-1R8。但在最近,其被称作IL-1R9或IL-1R10。为了避免混淆,在当前手稿中,发明人将使用术语IL-1RAPL1。IL-1RAPL1在脑中高度表达,参与了小脑发育、智力迟钝和认知缺陷。与IL-1受体家族其它成员的主要结构差异为IL-1RAPL1的细胞内结构域中C末端150个氨基酸长的延伸,这也存在于其紧密同源物IL-1RAPL2和调节性受体TIR8中。这种结构在细胞内信号转导中的作用还未有描述。功能性研究提示,IL-1RAPL1的活化和下游信号转导机制与IL-1受体家族的其它成员不同。有证据表明,IL-1RAPL1选择性地激活JNK,而JNK参与了免疫活化等。实际上,功能RNAi筛选揭示,IL-lRAPLl调节与凋亡细胞相互作用后的巨噬细胞表型。
IL-38,也称为IL1F10,为IL-1家族的最新增加。其与IL-1Ra共有41%的同源性,与IL-36Ra有43%的同源性,因此被认为是IL-1受体拮抗剂。实际上,最近显示,IL-38可结合IL-1R6,由此减少人白色念珠菌(C.albicans)刺激后或以低浓度施用添加IL-36后的细胞因子生成。然而,注意到LPS刺激联合IL-38后所生成的细胞因子的增加。通常,IL-38多态性与出现自身炎性病理学(例如脊椎关节炎、类风湿性关节炎或牛皮癣性关节炎)的增加的易感性相关,或者与CRP水平相关,提示了IL-38在调节引起这些状况的机制中的作用。
细胞因子包括大量的哺乳动物免疫调节激素,它们由免疫系统的细胞分泌。它们通过与细胞表面的特异受体相互作用来发挥它们的生物学效应。因此,对细胞因子的生物学应答是由细胞因子的存在和其受体分子的表达二者来调节的。包括自身免疫、炎症和癌症疾病在内的很多哺乳动物疾病是与增加或以其它方式改动的细胞因子或细胞因子受体水平相关的,它们可能促进了免疫系统调节紊乱和疾病进展。因而,就这些疾病状态而言,能够阻断细胞因子的免疫调节或炎症效应的化合物应该具有明显的治疗活性。
产生与不希望的免疫应答相关的免疫抑制的常规策略是基于广谱免疫抑制药物。此外,为了维持免疫抑制,免疫抑制药物治疗通常是终生命题。不幸的是,广谱免疫抑制药的使用与严重副作用(例如肿瘤、感染、肾毒性和代谢病症)的风险相关联。于是,新的免疫抑制疗法可能是有益的。
所以,直至今天,没有令人满意的治疗手段来治疗或预防自身免疫疾病,并且始终亟需另外的免疫抑制剂,以便推进对患有与病理学或者甚至长期活化的免疫系统相关的疾病的患者的医疗护理。
在第一方面,以上问题是通过分离的截短的IL-38蛋白、或其功能变体来解决,其中所述截短的IL-38蛋白与SEQ ID NO:1的氨基酸序列比较为N末端截短的。
在本文中,术语“截短的IL-38蛋白”指这样的IL-38多肽,其中已从全长IL-38多肽的氨基末端(或N-末端)区域去除氨基酸残基。在本发明上下文中,“截短的IL-38蛋白”不包括如SEQ ID NO:1中显示的全长序列。
术语蛋白的“功能变体”在本文是指这样的变体蛋白,其中与本发明相关的定义为亲和性和稳定性的功能基本上是保留的。因此,与所述功能无关的一个或多个氨基酸可以被替换。术语“功能变体”还应理解为指来自其它哺乳动物的同源物。优选地,本发明的功能变体保留了本文描述如SEQ ID NO:2显示的截短的IL-38蛋白的免疫抑制能力。
在优选的实施方案中,所述分离的截短的IL-38蛋白或其功能变体包括与SEQ ID NO:1的氨基酸序列(全长IL-38)有至少50%序列相同性的氨基酸序列,特征在于该截短的IL-38蛋白不包括SEQ ID NO:1中所示的全长氨基酸序列。
术语“序列相同性”(或“序列同源性”)表明了对两等长的氨基酸序列或两等长的核苷酸序列之间的相同性程度的定量衡量。如果两对待比较序列不是等长的,则它们必须排列为最佳可能的匹配,带有空位插入或者选择性地在蛋白序列的末端截短。
更优选的最小序列相同性百分比为至少70%,例如至少80%、至少85%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、以及至少99.5%、最优选100%(与SEQ ID NO:1中显示的序列相比),条件是所述截短的IL-38序列不包括如SEQ ID NO:1中所示的IL-38全长序列。
在优选的实施方案中,与野生型IL-38蛋白(SEQ ID NO:1)比较,本发明所述截短的IL-38蛋白在其N末端截短了2至50个氨基酸。优选地,与SEQ ID NO:1中所示的蛋白比较,所述截短的IL-38蛋白在其N末端截短了9、10、11、12、13、14、15、16、17、18、19或20个氨基酸。最优选为19个。
根据本文描述的发明,N末端截短优选包含SEQ ID NO:1的1至30位之间的至少2个、优选5个、最优选10个、最优选20个相邻的氨基酸。
在其它实施方案中,本发明的截短的IL-38蛋白具有不同于SEQ ID NO:1的前100、50、30、20个氨基酸的N末端。
在其它实施方案中,本发明的截短的IL-38蛋白具有这样的N末端,其中该N末端的前50个氨基酸不同于SEQ ID NO:1的前50个氨基酸。
在其它实施方案中,本发明的截短的IL-38蛋白具有这样的N末端,其中该N末端的前20个氨基酸不同于SEQ ID NO:1的前20个氨基酸。
作为选择,本发明的截短的IL-38蛋白不包含与SEQ ID NO:1的1至20位之间的序列至少80%相同的序列。
进一步最优选的是由与SEQ ID NO:2(20-152_IL-38)有至少80%、85%、90%、95%或100%序列相同性的氨基酸序列组成的截短的IL-38蛋白。如果通过重组表达本发明的截短的IL-38蛋白来生产,则该截短的IL-38蛋白的序列特征在于在N末端存在额外的甲硫氨酸。这些蛋白可以在表达后接受纯化过程,以获得本发明的分离的截短的IL-38蛋白。
“分离的”和“纯化的”指与其天然环境分开,并且摆脱了约60%至约99%、优选80%至99%的与其天然结合的其它组分的任何分子或化合物。
因此,在这方面,尤其优选的本发明的截短的IL-38蛋白在1位包含N末端甲硫氨酸;这些蛋白为纯粹人工的,非天然存在。
在本文中,术语“重组的”指重组或者合成产生的蛋白或核酸构建体,例如,对于蛋白的情况,通过在宿主细胞中翻译特定载体或质粒相关的系列指定的核酸元件或表达盒的RNA转录本。在本文中,术语“重组的”不包括天然发生的事件(例如,自发突变、天然转化/转导/转座)引起的细胞或载体变动,例如没有刻意的人类介入而发生的那些情况。
“宿主细胞”指这样的细胞,其含有载体或表达盒,并支持它们的复制和/或表达。宿主细胞可以为原核细胞,例如大肠杆菌(E.coli),或真核细胞,例如酵母、昆虫、两栖动物、植物细胞和哺乳动物细胞。优选地,宿主细胞为细菌或原核细胞。
在本文中,“载体”包括对用在宿主细胞转染中并且其中可插入多核苷酸的核酸的指代。载体一般是复制子。
在本文中,术语“蛋白(protein)”或“蛋白(proteins)”指多肽或其任何部分。
术语“多肽”指氨基酸的聚合物,并不是特定长度的产物;因此,肽、寡肽、和蛋白都包括在多肽的定义中。该术语也并不是多肽的表达后修饰或者排除多肽的表达后修饰,例如糖基化、乙酰化、磷酸化等等。包括在该定义内的例如有:含有一个或多个氨基酸类似物(包括,例如非天然氨基酸等)的多肽,具有取代键的多肽,以及本领域中已知的修饰,无论是天然发生的还是非天然发生的。
在本文中,术语“重组蛋白”是指(1)半合成的或合成起源的多肽,源于采用重组DNA技术连接在一起的不同来源的重组DNA分子的表达;(2)半合成的或合成起源的多肽,由于其来源或者操作,其不与天然结合的蛋白的全部或一部分结合;(3)半合成的或合成起源的多肽,其连接至一多肽,该多肽不同于其天然连接的多肽;或(4)半合成的或合成起源的多肽,其不天然出现。
本发明还涵盖化学上或以其它方式修饰的截短的IL-38蛋白。本文描述的多肽的修饰形式例如为经翻译后修饰的IL-38蛋白。翻译后修饰可以为所表达蛋白的糖基化。
上文描述的现有技术的问题在其一另外方面通过提供包含编码(coding或encoding)本文描述的截短的IL-38蛋白的序列的核酸来解决。术语“编码”(coding或encoding)是指核苷酸序列编码一个或多个氨基酸的能力。该术语不需要起始或终止密码子。氨基酸序列可编码在多核苷酸序列和其互补物提供的6种不同阅读框的任何一种中。
本发明的核酸可以包含这样的序列,当表达时生成由本发明的截短的IL-38蛋白组成的多肽,但其不为SEQ ID NO:1的全长IL-38蛋白。
本发明的另一方面还涉及包含上文描述的核酸的载体。最优选的是,本发明的载体为表达载体。
“表达载体”为重组或合成产生的核酸构建体或序列,具有特定的核酸元件,使得能在宿主细胞中转录和/或表达另一核酸。表达载体可为质粒、病毒、或核酸片段的一部分。在一个实例中,表达载体为DNA载体,例如质粒,其包含至少一个启动子序列和至少一个终止子序列(例如,多聚腺苷酸化序列),以及任选的复制起点(ori)序列,和任选的筛选或可筛选标记序列。任选地,该表达载体可以还包含至少一个感兴趣的核苷酸编码序列,其编码至少一种多肽,其中所述至少一个启动子序列与该至少一个编码序列可操作地连接。术语“表达”包括参与多肽生成的任何步骤,包括但不限于,转录、转录后修饰、翻译、翻译后修饰、和/或分泌。
本文还提供了包含本文描述的核酸或载体或表达载体的重组细胞。本发明的重组细胞优选不为人胚胎干细胞。本发明的优选重组细胞例如为细菌细胞,例如大肠杆菌或其它表达系统,或也可为动物细胞,例如昆虫细胞、哺乳动物细胞和人细胞。
本文描述的化合物和组合物可以用在各个医疗领域,尤其是作为活性治疗剂用于在需要治疗的受试者中治疗或预防以免疫应答病理性激活为特征的疾病。由本发明的化合物治疗的优选疾病将在下文描述。
因此,在另外方面,本发明还提供了药物组合物,其包含本发明前文描述的实施方案的截短的IL-38蛋白、或核酸、载体或重组细胞,用于用在医药中,优选用在治疗或预防免疫或炎性疾病中。
在本发明上下文中,免疫或炎性疾病优选选自自身免疫疾病,例如感染性休克,失血性休克,关节炎,例如脊椎关节炎,类风湿性关节炎,牛皮癣性关节炎或骨关节炎,炎性肠病,多发性硬化和代谢疾病如动脉硬化和I型糖尿病。其它的适应症包括对IL-1β的抑制剂治疗有应答的疾病,例如Muckle-Wells综合征、CAPS(cryopyrin相关周期性发热综合征)、家族性地中海热、斯蒂尔病、白塞病和糖尿病。
关节炎包括骨关节炎,类风湿性关节炎作为损伤或晶体沉积结果的关节炎性关节等为常见的炎性状况,它们将受益于抗炎蛋白的治疗应用,例如本发明的截短的IL-38蛋白。例如,类风湿性关节炎(RA)为影响整个身体的全身性疾病,为最常见形式的关节炎之一。其特征在于衬在关节下的隔膜的炎症,其引起疼痛、僵直、升温、发红和肿胀。炎性细胞释放可以消化骨骼和软骨的酶。作为类风湿关节炎的结果,发炎的关节衬里(滑膜)可侵入并损伤骨骼和软骨,引起关节退化和严重疼痛以及其它生理效应。受累关节可能丧失其形状和排列,导致疼痛和丧失运动能力。
类风湿性关节炎(RA)为免疫介导疾病,尤其以炎症和随后的组织损伤为特征,导致严重残疾和增加的死亡率。各种细胞因子在类风湿关节内局部生成。众多的研究已证实,两种原型促炎细胞因子IL-1和TNF-α在涉及滑膜炎症和进行性关节破坏的机制中起重要作用。实际上,在患有RA的患者中以TNF-α和IL-1抑制剂施用已导致炎症的临床和生物学迹象的明显改善和骨骼侵蚀和软骨破坏的放射迹象的减弱。但是,尽管有这些令人鼓舞的结果,有相当百分比的患者对这些药物无应答,提示其它的调节物也参与了关节炎的病理生理学(Gabay,Expert.Opin.Biol.Ther.2(2):135-149.2002;Astry,J.Interferon Cytokine Res.31(12):927-40.2011)。
对于药物应用,本发明的截短的IL-38蛋白被配制为用于常规方法的胃肠外递送,特别是静脉内或者皮下递送。静脉内施用将通过团注、控释(例如,采用微型泵或其它适合的技术)、或通过在一个至数个小时的典型时间段内输注来进行。通常,药物制剂将包括与药学上可接受的载体(例如,盐水,缓冲盐水,5%葡萄糖水溶液等)联合的造血蛋白。制剂还可以包括一种或多种赋形剂、防腐剂、增溶剂、缓冲剂、防止蛋白在小瓶表面损失的白蛋白,等等。当采用这种联合治疗时,细胞因子可以组合在单一制剂中,或者可以在不同的制剂中施用。配制方法为本领域所公知的,例如在文献(Remington's Pharmaceutical Sciences.Gennaro,ed.,Mack Publishing Co.,Easton PA,1990)中有公开,通过参考将其并入本文。
治疗剂量通常在每天0.1至100mg/kg患者体重的范围内,优选每天0.5-20mg/kg,精确剂量由临床医师根据公认的标准,并考虑待治疗的状况的性质和严重性、患者特征等来确定。剂量确定处于本领域普通技术人员的能力范围内。所述蛋白一般在不超过28天的时间内施用。更常见地,所述蛋白在一周或更短时间内施用,经常在1至3天的时间内施用。通常,治疗上有效量的本发明的截短的IL-38蛋白为足以产生病理性炎症反应的临床显著降低的量。
通常,施用的截短的IL-38蛋白的剂量将随诸如患者年龄、体重、身高、性别、一般医学状况和之前的病史的因素而变化。典型地,希望向接受者提供约1pg/kg至10mg/kg(药物量/患者体重)范围的截短的IL-38蛋白剂量,不过根据情况不同也可以以更低或更高剂量施用。在下文提供本发明药物组合物的具体实施方案。
在另外的方面,本发明提供了在需要治疗的受试者中治疗或预防病理性炎性疾病的方法。本发明的方法可以包括向所述受试者施用治疗有效量的本文描述的化合物或本发明的组合物的任一种的步骤。
本发明的另一方面涉及调节细胞的免疫应答的方法,该方法包括让所述细胞与本发明的截短的IL-38蛋白接触,或者通过在所述细胞中表达本发明的核酸。
在优选的实施方案中,该方法为离体或体外方法。
“调节免疫应答”可以为对JNK信号转导的抑制,尤其是抑制IL-6释放和TH17生成。可以通过在所述细胞中使用JNK报告基因构建体来观察对JNK信号转导的抑制。一种广泛使用的报告基因为AP-1启动子驱动的报告基因。
用在本发明所描述的方法中的细胞优选为哺乳动物细胞,最优选人细胞。还优选该细胞为免疫细胞,最优选白细胞,进一步更优选巨噬细胞。
本发明的另一方面涉及筛选截短的IL-38的活性的调节剂的方法,包括以下步骤:
a.提供细胞,
b.让所述细胞与微生物相关分子模式(MAMP)、病原体相关分子模式(PAMP)、或凋亡细胞上清液(ACM)接触,
c.让所述细胞再与本发明的截短的IL-38蛋白和候选调节剂接触,
d.确定所述细胞中的JNK激活,
其中,所述细胞中JNK激活与对照细胞或参考值相比的增加,表明该候选调节剂为截短的IL-38的拮抗剂,JNK激活与对照细胞或参考值相比的降低,表明该候选调节剂为截短的IL-38的激动剂。
根据本发明的PAMP或MAMP可以选自细菌脂多糖(LPS)、细菌鞭毛蛋白、来自革兰氏阳性细菌的脂磷壁酸、肽聚糖、以及通常与病毒相关的核酸变体,例如双链RNA(dsRNA),或未甲基化的CpG基序。
用在本发明的筛选方法中的所述细胞优选在细胞表面表达截短的IL-38的受体,例如通过在所述细胞中异位表达IL-1RAPL1。
候选调节剂优选小分子,小核酸,例如小RNA,或蛋白,例如抗体。
所述JNK激活优选通过AP-1报告基因构建体来确定。这种报告基因构建体可以为基于荧光素酶的、基于酶的或基于荧光蛋白的。也可以例如通过定量PCR(qPCR)来确定直接的JNK靶基因表达。
本发明还提供了通过本文描述的方法鉴定的截短的IL-38的活性调节剂。
疾病和状况
本发明提供了截短的IL-38蛋白,其可在治疗或预防各种疾病中用作治疗剂。根据本发明,这些化合物和组合物尤其可用于治疗和/或预防以病理性激活的免疫或炎性应答为特征的状况。具体地,本发明寻求提供对以细胞因子的病理学活性为特征的状况的治疗,这些细胞因子例如为白介素-6和白介素-17,它们已被证实参与了多系列的慢性炎性和自身免疫病症。
在本发明的上下文中,自身免疫疾病为自身免疫应答导致的病症。自身免疫疾病为对自身抗原的不恰当的和过度的应答的结果,或者为例如经由细胞因子的免疫应答信号转导的病理性激活的结果。自身免疫疾病的实例包括但不限于,阿狄森氏病、斑秃、强直性脊椎炎、自身免疫性肝炎、自身免疫性腮腺炎、克罗恩病、糖尿病(I型)、营养不良型大疱性表皮松解症、附睾炎、血管球性肾炎、格式(Graves)病、巴雷(Guillain-Barr)综合征、桥本病、溶血性贫血、系统性红斑狼疮、多发性硬化、重症肌无力、寻常型天疱疮、牛皮癣、风湿热、类风湿性关节炎、结节病、硬皮病、斯耶格伦氏综合征、脊柱关节病、甲状腺炎、脉管炎、白癜风、粘液水肿、恶性贫血、溃疡性结肠炎等等。
治疗或预防自身免疫疾病的组合物和试剂盒
本申请的另一方面涉及用于治疗或预防自身免疫或炎性疾病的组合物和试剂盒。在一个实施方案中,该组合物包含本文描述的诸如蛋白、核酸或重组细胞的化合物,任选地联合药学上可接受的载体。
在本文中,用语“药学上可接受的载体”旨在包括与药物管理相符的任何和所有的溶剂、增溶剂、填充剂、稳定剂、粘结剂、吸附剂、碱、缓冲剂、润滑剂、控释载体、稀释剂、乳化剂、湿润剂、润滑剂、分散介质、涂层、抗细菌或抗真菌剂、等张和吸收延缓剂,等等。将这些介质和试剂用于药学上有活性的物质在本领域内是公知的。除了任何常规媒介物或试剂与该活性化合物不相容之外,考虑将其用在该组合物中。补充剂也可掺入到该组合物中。在某些实施方案中,该药学上可接受的载体包括血清白蛋白。
本发明的药物组合物配制为与其预期的施用途径相容。施用途径的实例包括胃肠外施用,例如鞘内、动脉内、静脉内、皮内、皮下施用,口服,透皮(局部)和跨粘膜施用。
用于胃肠外、皮内或皮下应用的溶液或悬浮液可包括以下组分:无菌稀释剂如注射用水,盐水溶液,不挥发油,聚乙二醇,甘油,丙二醇或其它合成溶剂;抗细菌剂例如苯甲醇或对羟基苯甲酸甲酯;抗氧化剂例如抗坏血酸或硫酸氢钠;螯合剂例如乙二胺四乙酸;缓冲剂例如乙酸盐,柠檬酸盐或磷酸盐以及用于调节张力的试剂如氯化钠或右旋糖。可用酸或碱,例如盐酸或氢氧化钠调节pH。胃肠外配制物可封在安瓿瓶、一次性注射器或多重剂量小瓶中,它们由玻璃或塑料制成。
适合于注射使用的药物组合物包括无菌水溶液(当水溶时)或悬浮液以及用于临时制备无菌可注射溶液或悬浮液的无菌粉末。对于静脉内施用,适合的载体包括生理盐水、抑菌水、商品名为Cremophor ELTM的物质(BASF,Parsippany,N.J.)或磷酸盐缓冲盐水(PBS)。所有情况下,可注射组合物应为无菌的,并且应为流体,其程度为存在易注射性。其在生产和保存条件下必须是稳定的,并且必须能免于微生物如细菌和真菌的污染作用。载体可为溶剂或分散介质,含有例如水、乙醇、多元醇(例如,甘油、丙二醇、以及液体聚乙二醇等)、或它们的适当混合物。例如通过使用诸如卵磷脂的涂层、对于悬浮液的情况通过维持所需的粒径以及通过使用表面活性剂可维持适当的流动性。预防微生物作用可通过各种抗细菌和抗真菌剂来实现,例如,对羟基苯甲酸酯、氯丁醇、酚、抗坏血酸、硫柳汞,等。很多情况下,优选在组合物中包括等张剂,例如,蔗糖、多元醇如甘露醇、山梨醇和氯化钠。可注射组合物的延时吸收可通过在组合物中加入延缓吸收的试剂来实现,例如单硬脂酸铝和明胶。
可通过将活性化合物(例如,神经调节蛋白)以所需量与以上列出的成分的一种或组合掺入适合的溶剂中,然后无菌过滤(需要的话),来制备无菌可注射溶液。通常,通过将活性化合物掺入含有基础分散介质和所需的来自以上列出成分的其它成分的无菌载体中来制备分散体。对于用于制备无菌可注射溶液的无菌粉末的情况,优选的制备方法为真空干燥和冷冻干燥,其产生活性成分加上任何额外所需成分(来自其之前无菌过滤的溶液)的粉末。
口服组合物通常包括惰性稀释剂或可食用载体。它们可包封在明胶胶囊中或压缩为片剂。出于口服治疗施用的目的,活性化合物可与赋形剂一起掺混,并以片剂、含片、或胶囊的形式使用。口服组合物还可采用流体载体来制备,以用作漱口水,其中流体载体中的化合物口服施用,在口中来回,并且吐出或咽下。药学上相容的粘结剂和/或佐剂材料可作为组合物的一部分包括进来。片剂、小丸、胶囊、含片等可含有以下成分或具有类似性质的化合物的任何一种:粘结剂如微晶纤维素、黄蓍胶或明胶;赋形剂如淀粉或乳糖;崩解剂如褐藻酸、商品名为Primogel的物质、或玉米淀粉;润滑剂如硬脂酸镁或商品名为Stertes的物质;助流剂如胶体二氧化硅;甜味剂如蔗糖或糖精;或调味剂如薄荷、水杨酸甲酯、或橙香精。
对于通过吸入施用,化合物是以气溶胶喷雾的形式从含有适合的推进剂(例如气体形式,诸如二氧化碳)的压力容器或分配器或喷雾器递送。
全身施用也可通过跨粘膜或透皮方式进行。对于跨粘膜或透皮施用,将适合于待渗透屏障的渗透剂用在制剂中。这些渗透剂通常在本领域是已知的,包括例如用于跨粘膜施用的洗涤剂、胆汁盐、以及梭链孢酸衍生物。跨粘膜施用可通过使用鼻喷剂或者栓剂来完成。对于透皮施用,药物组合物配制为软膏、药膏、凝胶、或乳膏,如本领域所公知的。
在某些实施方案中,药物组合物配制为活性成分的持续或控制释放。可使用生物可降解的、生物相容的聚合物,例如乙烯醋酸乙烯酯、聚酸酐、聚乙醇酸、胶原、聚原酸酯、以及聚乳酸。用于制备这些制剂的方法对本领域技术人员是显而易见的。这些材料也可在商业上从例如阿尔扎公司(Alza Corporation)和诺华制药公司(Nova Pharmaceuticals,Inc.)获得。脂质体悬浮液(包括带有针对病毒抗原的单克隆抗体的靶向感染细胞的脂质体)也可用作药学上可接受的载体。这些可按照本领域技术人员已知的方法来制备。
特别有利的是配制单位剂量形式的口服或胃肠外组合物,以方便施用和剂量一致性。单位剂量形式在本文中包括物理上分开的单位,适合作为单一剂量用于待治疗的受试者;每个单位含有预定量的活性化合物,经计算与所需的药物载体联合会产生期望的治疗效果。本发明单位剂量形式的规格取决于并直接依赖于活性化合物的独特特征和待获得的具体治疗效果以及本领域中将这种活性化合物配制为用于个体治疗的内在限制。
这些化合物的毒性和治疗功效可通过标准药学程序在细胞培养物或试验动物中确定,例如用于确定LD50(50%群体致死剂量)和ED50(50%群体治疗有效剂量)。毒性和治疗效果的剂量比例为治疗指数,其可表示为比例LD50/ED50。显示出大治疗指数的化合物是优选的。尽管可以使用显示出毒副作用的化合物,但应考虑设计将这些化合物靶向受累组织位点的递送系统,以便对非感染细胞的潜在损伤降至最小,并由此减小副作用。
从细胞培养物试验和动物研究获得的数据可用在制定用于人的剂量范围中。这些化合物的剂量优选处于包括ED50的循环浓度的范围内,具有微小毒性或没有毒性。该剂量可以在该范围内随采用的剂型和使用的施用途径而变化。对于用在本发明方法中的任何化合物,治疗有效剂量可从细胞培养测定初步估计。可在动物模型中制定获得包括在细胞培养中确定的IC50(即,测试化合物实现症状的最大半数抑制的浓度)的循环血浆浓度范围的剂量。这种信息可用于更精确地确定人类可用剂量。药物组合物可包括在容器、包装盒、或分散器中,连同施用说明书。
下面将参照附图和序列,在以下实施例中进一步描述本发明,然而并不限于此。出于本发明的目的,本文提到的所有参考文献都通过引用将它们的全文并入。在附图中:
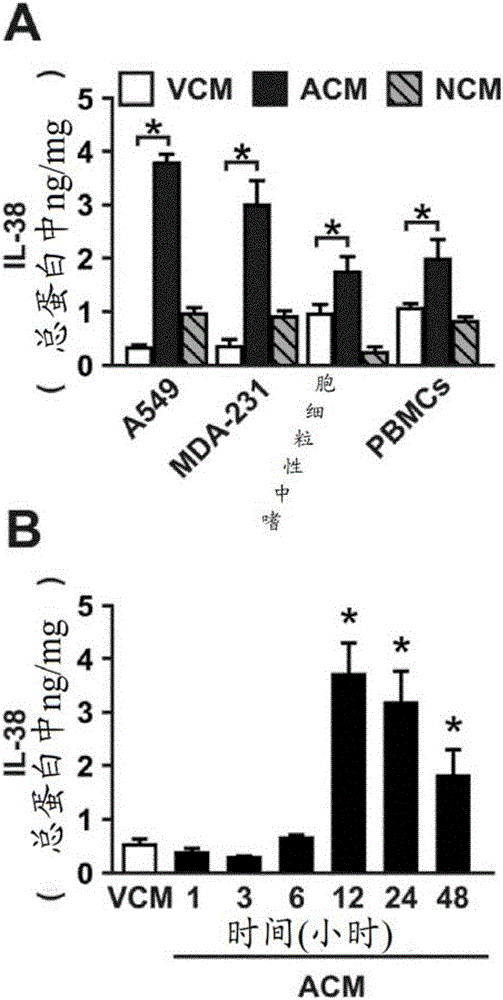
图1:IL-38是从凋亡细胞分泌的。(A)A549人肺癌细胞,MDA-231乳腺癌细胞、人原代PBMC和人原代嗜中性粒细胞保持存活、经TNF-α(20ng/ml)/CHX(10μΜ)处理以诱导凋亡或者在60℃温育30min以诱导坏死。收获存活(VCM)、凋亡(ACM)或坏死(NCM)细胞各自的上清液,并通过ELISA分析IL-38水平。数值为均值±SEM,n=5。(B)在指定的时间通过ELISA分析凋亡的A549细胞的IL-38分泌。数值为均值±SEM,n=5。*p<0.05,ANOVA,有邦弗伦尼(Bonferroni)校正。
图2:凋亡肿瘤细胞释放的IL-38调节巨噬细胞中的细胞因子产生。用(A)单独的LPS(1ng/ml)或联合重组人IL-38(rhIL-38)10ng/ml或者用(B)单独的凋亡A549细胞上清液(ACM)或联合重组人IL-38(rhIL-38)10ng/ml刺激人巨噬细胞。细胞因子的产生采用流式微珠阵列测量,显示了归一化的结果。数值为均值±SEM,n=5。(C)用存活的(VCM)或凋亡的A549细胞(ACM)上清液刺激人巨噬细胞,该A549细胞先经非靶向的siRNA(siCtrl)、针对IL-38的siRNA(silL-38)或IL-38过表达载体(oeIL-38)转染。细胞因子的产生采用流式微珠阵列测量,显示了归一化的结果。数值为均值±SEM,n=5。(D)用AP1报告基因构建体转染人巨噬细胞,并在来自siCtrl和siIL-38A549细胞的ACM刺激24小时后测量荧光素酶活性。从每个实验值中减去从空白对照转染细胞获得的背景测量值。显示了经归一化的结果。数据为均值±SEM,n=5。*p<0.05,ANOVA,有邦弗伦尼校正。
图3:IL-38结合IL-1R6和IL-lRAPLl。(A,B)用LPS或ACM刺激人巨噬细胞6和24h。IL-1R6和IL-lRAPLl的mRNA表达(A)和细胞表面蛋白表达(B)分别通过RT-qPCR和FACS测量。数值为均值±SEM,n=5。(C,D)巨噬细胞为对照(Ctrl)或者与50ng/ml IL-38在冰上温育15min,然后用抗IL-lR6或抗IL-lRAPLl以及它们各自的PE偶联第二抗体染色。显示了代表性的流式细胞直方图(C)和中值PE强度的统计定量(D)。数值为均值±SEM,n=5。(E)IL-38与固定化的IL-1R6和IL-lRAPLl的结合动力学。用0.5μg的人IL-1R6和IL-lRAPLl细胞外结构域-Fc嵌合体包被96孔板,并与所示的递增量的人重组IL-38一起温育。采用生物素化的单克隆IL-38抗体来检测结合至受体的细胞外结构域的IL-38。数据为均值±SEM,n=5。*p<0.05,ANOVA,有邦弗伦尼校正。
图4:IL-1R6和IL-lRAPLl在细胞因子产生中的作用。用非靶向siRNA(siCtrl)或针对(A)IL-lRAPLl(silLlRAPLl)或(B)IL-1R6(siILlR6)的siRNA转染巨噬细胞,并用VCM和ACM刺激24h。细胞因子的产生采用流式微珠阵列测量,显示了归一化的结果。数值为均值±SEM,n=5。*p<0.05,ANOVA,有邦弗伦尼校正。
图5:IL-38调节Th17应答。人T细胞用抗CD3/抗CD28微珠激活,用商品名为eFluor 670的物质进行染色,并用之前经来自对照A549细胞(ACMshCtrl/ΜΦ)或IL-38敲除A549细胞(ACMshIL-38/ΜΦ)的ACM刺激的巨噬细胞的上清液进行刺激。7天后,测量(A)细胞因子的产生和(B,C)细胞增殖。(A)采用流式微珠阵列来定量细胞因子,显示了归一化的结果。数值为均值±SEM,n=10。采用商品名为eFluor 670的稀释液确定T细胞增殖。显示了(B)所有增殖T细胞的统计定量和(C)代表性流式细胞直方图。数值为均值±SEM,n=10。*p<0.05,ANOVA,有邦弗伦尼校正。
图6:IL-38为N末端截短的。(A)显示了IL-38中的IL-36家族特征性一致基序,这确定了该细胞因子在N末端的推测切割位点。(B)C末端带myc标签的IL-38在A549细胞中过表达,然后将其用于ACM生成。在采用抗myc覆盖的微珠免疫沉淀该过表达的IL-38之后,进行2D凝胶电泳(在pH 4-7等电聚焦,接着是聚丙烯酰胺凝胶分离),并将单克隆抗myc抗体用于检测经蛋白转移至硝酸纤维素膜上的免疫沉淀的IL-38。(C)将考马斯染色的2D凝胶用于挑取推测的IL-38斑点,通过质谱仪对其进行分析。显示了对不同的IL-38斑点所鉴定的IL-38N末端肽。
图7:全长和截短的IL-38在细胞因子产生和结合至IL-lRAPLl方面有相反的作用。(A,C)人巨噬细胞(A)未经处理或者(C)先前经非靶向siRNA(siCtrl)或针对IL-lRAPLl(silLlRAPLl)的siRNA转染,并用单独的重组人IL-1β50ng/ml或者联合不同浓度的重组人全长(IL-38aal-152)或剪切的(IL-38aa20-152)IL-38刺激6h。24小时后,上清液中的IL-6浓度采用流式微珠阵列测量,显示了归一化的结果。数值为均值±SEM,n=7。(B)全长和剪切的IL-38与固定化的IL-lRAPLl的结合动力学。用0.5μg的人IL-lRAPLl细胞外结构域-Fc嵌合体包被96孔板,并与所示的递增量的人重组IL-38aal-152或IL-38aa20-152一起温育。采用生物素化的单克隆IL-38抗体对结合至受体的细胞外结构域的IL-38进行检测。数据为均值±SEM,n=5。*p<0.05,ANOVA,有邦弗伦尼校正。
图8:在HEK细胞中由IL-38调节的IL-lRAPLl诱导的信号转导途径。用IL-lRAPLl过表达质粒联合(A,D)AP1、(B,E)NFκB或(C)IL-6报告基因构建体共转染HEK细胞。将转染了报告基因构建体以及空质粒而不是IL-1RAPL1过表达质粒的HEK细胞用作对照(空白对照)。(A,B)用IL-1β(50ng/ml)刺激HEK细胞24h,测量AP1或NFκB活性。显示了经归一化的结果。数值为均值±SEM,n=15。(C)使用在指出的转录结合位点中有点突变的IL-6报告基因构建体。转染后,将细胞再温育24h,并测量IL-6启动子依赖的荧光素酶活性。结果表示为相对于空白对照转染细胞的诱导倍数。数值为均值±SEM,n=10。(D,E)转染后,添加新鲜培养基(Ctrl)或不同浓度的IL-38aal-152或IL-38aa20-152,将细胞再温育24h。结果表示为相对于空白对照转染细胞的诱导倍数。数值为均值±SEM,n=15。(F)ILIRAPL1在HEK细胞中过表达,并且在转染后,向细胞添加新鲜培养基或IL-38aal-152或IL-38aa20-152(25ng/ml),接着再温育24h。进行磷酸化的JNK和p38的细胞内染色并通过FACS测量。结果表示为相对于空白对照转染细胞的诱导倍数。数值为均值±SEM,n=5。*p<0.05,ANOVA,有邦弗伦尼校正。
图9:IL-38调节巨噬细胞中的AP1活性。用空载体、AP1、NFκB或IL-6报告基因构建体转染人巨噬细胞。转染后,用单独的IL-1β(50ng/ml)或者联合IL-38aal-152或IL-38aa20-152(20ng/ml)刺激巨噬细胞24h。测量荧光素酶活性。从每个实验值中减去从空白对照转染细胞获得的背景测量值。显示了经归一化的结果。数值为均值±SEM,n=7。*p<0.05,ANOVA,有邦弗伦尼校正。
SEQ ID NO:1
MCSLPMARYYIIKYADQKALYTRDGQLLVGDPVADNCCAEKICILPNRGLARTKVPIFLGIQGGSRCLACVETEEGPSLQLEDVNIEELYKGGEEATRFTFFQSSSGSAFRLEAAAWPGWFLCGPAEPQQPVQLTKESEPSARTKFYFEQSW
SEQ ID NO:2
LYTRDGQLLVGDPVADNCCAEKICILPNRGLARTKVPIFLGIQGGSRCLACVETEEGPSLQLEDVNIEELYKGGEEATRFTFFQSSSGSAFRLEAAAWPGWFLCGPAEPQQPVQLTKESEPSARTKFYFEQSW
实施例
实施例1:IL-38从凋亡细胞释放
当进行内部ELISA来确定肿瘤细胞系产生的IL-38水平时,发明人注意到对凋亡性细胞死亡的诱导显著增加了IL-38向上清液中分泌。与存活的A549肺癌或MDA.231乳腺癌细胞上清液(VCM)相比,是凋亡细胞上清液(ACM)而不是坏死细胞上清液(NCM)中含有大约高10倍水平的IL-38(图1A)。对于原代人嗜中性粒细胞或PBMCs也是如此,尽管凋亡期间IL-38释放的增加不是一样强烈(图1A)。为了分析IL-38分泌动力学,在凋亡诱导后的不同时间点测量了A549上清液中的IL-38浓度。在凋亡诱导后12h观察到增强的IL-38分泌(图1B),与A549细胞中凋亡标志物的出现吻合(数据未示出)。
实施例2:IL-38调节ACM刺激后的细胞因子的产生
凋亡细胞衍生的调节剂具有调节包括细胞因子产生在内的吞噬细胞应答的能力(26)。发明人分析了IL-38在一组细胞因子产生中的作用,这些细胞因子在巨噬细胞被LPS激活后或者在与凋亡细胞相互作用后产生(27)。其中,IL-6和IL-8产生由IL-38调节。与单独LPS相比,向LPS刺激的巨噬细胞添加重组IL-38,增加了IL-6和IL-8的产生。有趣的是,当用单独的A549细胞ACM或者联合重组人IL-38刺激人巨噬细胞,观察到相反的效果(图2B)。IL-38抑制ACM诱导的IL-6和IL-8从巨噬细胞分泌。由于ACM已经含有IL-38,所以发明人对内源IL-38是否影响巨噬细胞的细胞因子产生感到好奇。为了回答这个问题,在产生ACM前,让IL-38在A549细胞中过表达或者敲除。实际上,用产生自IL-38过表达的A549细胞的ACM刺激人巨噬细胞会导致减少的IL-6和IL-8分泌,而用IL-38敲除的A549细胞的ACM刺激则产生更高的IL-6和IL-8浓度(图2C)。在调节细胞因子产生并由IL-1家族调节的主要转录因子中有NFκB和AP1。由于NFκB在巨噬细胞与凋亡细胞相互作用后被阻断(28),所以发明人想了解内源IL-38是否在应答ACM中调节AP1激活。与对照ACM比较,当将IL-38敲除的A549细胞的ACM应用至经AP1荧光素酶报告基因构建体转染的巨噬细胞时,发明人注意到含有较低水平IL-38的ACM诱导了更明显的AP1激活(图2D)。作为结论,内源IL-38限制了应答凋亡细胞上清液的炎性巨噬细胞激活。
实施例3:IL-38拮抗应答ACM中的ILlRAPLl依赖性细胞因子的产生
发明人假设IL-38通过作为受体拮抗剂来抑制ACM诱导的细胞因子产生。由此,发明人分析了IL-1受体家族的候选者与IL-38的结合。已证实IL-38结合IL-1R6(19),并且发明人观察到孤儿受体IL-lRAPLl调节ACM刺激后巨噬细胞中细胞因子产生(16)。发明人首先采用qPCR在mRNA水平(图3A)和通过FACS在细胞表面可得性水平(图3B)上确定了IL1R6和IL-lRAPLl在ACM或LPS刺激后的巨噬细胞中的表达。IL1R6表达通常较低(图3C),并在ACM或LPS刺激后进一步在mRNA水平上下调,然而在细胞表面表达水平上并不明显。相反地,IL-lRAPLl表达是丰富的(图3C),并且在LPS处理后6h以及ACM处理后6h和24h时被进一步诱导,不论是mRNA水平,还是细胞表面(图3A,B)。此外,细胞表面的IL-lRAPLl表达在LPS刺激后24小时降低。这些实验提示,IL-lRAPLl至少是作为IL-38起作用的另外候选者。接着,发明人通过进行竞争测定和受体结合测定来分析IL-38是否结合IL-lRAPLl。对于竞争测定,在分析IL-1R6或IL-lRAPLl的表面表达之前让人巨噬细胞与重组人IL-38一起温育。基于低水平的IL-1R6表面表达,难以在细胞表面表达中观察到由IL-38预温育导致的差异(图3C,D)。但是,对于IL-lRAPLl,发明人观察到IL-38与用于FACS染色的抗体竞争(图3C,D),表明IL-38可以结合IL-lRAPLl。为了验证这些结果,进行了直接的受体结合测定。用IL-1R6-Fc和IL-lRAPLl-Fc嵌合体包被平板,向孔中添加不同IL-38浓度,并观察结合的IL-38。如最近所证实的(19),IL-38确实结合IL-1R6(图3E)。此外,IL-38还结合IL-lRAPLl(图3E)。这些结果提示,IL-38可能通过结合IL-lRAPLl来调节细胞因子产生,因此发明人想了解IL-lRAPLl在ACM诱导的细胞因子产生中的作用。在人巨噬细胞中进行IL-1R6或IL-lRAPLl的瞬时敲除,并在ACM刺激后测量巨噬细胞培养物上清液中的IL-6和IL-8水平。ACM刺激后的IL-6和IL-8的产生依赖于IL-lRAPLl(图4A),而在发明人的模型中IL-1R6不参与细胞因子的产生(图4B)。
实施例4:IL-38调节Th17应答
接下来,发明人通过分析巨噬细胞上清液对T细胞激活的效果,想了解IL-38依赖的抑制巨噬细胞产生细胞因子的下游后果。发明人分离了原代人T细胞,用抗CD3/抗CD28微珠刺激并将它们与巨噬细胞(先前经ACM和IL-38敲除的A549细胞的ACM刺激)的上清液一起反复温育。在培养7天后,测量T细胞上清液中的IL-10、IL-17和IFN-γ水平。当巨噬细胞经ACM刺激,它们的上清液减少T细胞的IFN-γ和IL-10产生并稍稍升高IL-17水平(图5A)。然而,当用缺乏IL-38的ACM刺激巨噬细胞时,它们的上清液强烈升高T细胞的IL-17的产生,还降低IL-10浓度(图5A)。这些效果与T细胞增殖差异无关。用ACM处理不影响增殖的T细胞的数量(图5B),不过其影响了预分裂T细胞分裂数量,这可能解释了降低的IFN-γ和IL-10水平(图5C)。但是,无论ACM是否含有IL-38,在T细胞增殖方面不存在差异(图5B,C)。这些数据显示来自凋亡细胞的IL-38限制了巨噬细胞依赖的TH17细胞的产生并维持IL-10表达。
实施例5:IL-38在凋亡细胞中经N末端加工
除了IL-IRa,IL-1的全部成员都作为前体生成,它们需要在N末端剪切以达到完全的活性。最近,根据N末端前结构域的大小,将IL-38归类进IL-36亚族中(4,19)。与该亚族的其它成员相同,IL-38具有一致基序,推测这可能决定了N末端切割位点(图6A)。为了确定凋亡是否诱导IL-38加工,在肿瘤细胞中过表达C末端带有myc标记的IL-38,并从这些细胞产生ACM。在免疫沉淀IL38后,进行2D凝胶电泳以显现IL-38亚型。在凝胶中成功地鉴定了两种IL-38亚型,表明IL-38确实在凋亡期间被加工(图6B)。挑取两个代表推测的IL-38亚型的斑点,通过质谱仪(MS)进行分析。在具有较高分子量的斑点中(推测为全长IL-38),在MS分析中发现了两种N末端肽,一种来自氨基酸9至18,另一种来自氨基酸24至41,而在具有较低分子量的样品中仅发现来自氨基酸24至41的肽(图6C)。因此,IL-38在凋亡细胞中是经N末端加工的。
实施例6:全长和截短的IL-38通过IL-1RAPL1对细胞因子产生施加相反的作用
为了确定全长和截短的IL-38是否具有不同的生物学活性,在经单独IL-1β或者联合不同浓度的全长(IL-38aal-152)或截短(IL-38aa20-152)的IL-38刺激的人巨噬细胞上清液中检测IL-6浓度。在IL-β刺激后,较高浓度的IL-38aal-152(20ng/ml,l0ng/ml)显著增加了IL-6的产生,而IL-38aa20-152甚至以较低浓度施用时也降低IL-6产生(图7A)。由于ACM中的IL-38通过与IL-lRAPLl相互作用来调节IL-6产生,所以发明人想了解两种IL-38亚型(isoform)(在细胞因子产生中具有相反作用)是否均结合IL-lRAPLl。发明人进行了受体结合测定,如上文所阐释的。在本测定中,两种IL-38亚型都结合IL-lRAPLl(图7B)。但是,结合动力学似乎稍有区别。当考虑到尽管IL-38aal-152和IL-38aa20-152对细胞因子产生施加相反作用,但它们都能够结合IL-1RAPL1时,另一待分析的关键点为对产生IL-6的影响是否都是IL-lRAPLl依赖的。为此,在巨噬细胞中进行了瞬时IL-lRAPLl敲除,并在单独IL-1β或联合IL-38aal-152或IL-38aa20-152刺激后测量了上清液中的IL-6浓度。巨噬细胞中IL-lRAPLl敲除消除了全长IL-38的IL-6诱导和截短的IL-38的IL-6抑制(图7C)。
实施例7:IL-38调节IL-lRAPLl激活的途径JNK/AP1
发明人得到了IL-38调节与凋亡细胞相互作用后的巨噬细胞中的AP-1的证据(图2D)。为了分析IL-38所影响的与其与IL-1RAPL1相互作用有关的信号转导途径,发明人首先使用了HEK 293T细胞的受体过表达模型。用IL-1RAPL1的过表达构建体和AP1或NFκB报告基因构建体共转染该细胞。将经报告基因构建体转染而没有IL-1RAPL1过表达的HEK细胞用作对照。首先,为了表征该模型,用IL-1β刺激IL-1RAPL1过表达细胞和对照细胞,并测量AP1(图8A)或NFκB(图8B)活性。将IL-1β用作孤儿受体IL-1RAPL1的低亲和配体(14)。IL-1β刺激后,在对照细胞中观察到NFκB而不是AP1活性的显著诱导。然而,当IL-1RAPL1过表达时,观察到AP1激活以及增强的NFκB活性。因此,单独IL-1β以IL-lRAPLl独立的方式诱导NFκB激活,而不是AP1激活,其需要IL-1RAPL1。有趣的是,甚至没有任何刺激物,与对照细胞相比,IL-1RAPL1的存在就足以增加AP1和NFκB活性(图8A,B)。因此,IL-1β刺激后,在HEK细胞中IL-1RAPL1激活AP1而不是NFκB,但是在过表达时诱导AP1和NFκB激活,无需添加外源配体。为了确认这一点,使用了IL-6启动子构建体,在不同的转录因子结合位点(AP1、NFκB、CREB和CEBPβ)中存在或不存在点突变。用IL1RAPL1过表达质粒和IL-6报告基因构建体共转染HEK细胞(29)。在这种设计中,与HEK对照细胞比较,IL-1RAPL1的过表达还激活IL-6启动子。当AP1和NFκB结合位点突变时,消除了这种IL-6启动子诱导(图8C)。这表明存在由HEK细胞产生的IL-1RAPL1的内源配体。接着,发明人想了解IL-38aal-152和IL-38aa20-152是否能够影响IL-1RAPL1下游的AP1和NFκB活性。这种设计中,NFκB启动子活性不受IL-38调节(图8D),但是AP1诱导由两种IL-38亚型负调节(图3E)。重要的是,与全长蛋白比较,IL-38aa20-152能够在更低的浓度调节AP1激活。为了分析导致IL-38依赖的对IL-lRAPL诱导的AP1活性的抑制的信号转导机制,与对照HEK细胞相比较在IL-1RAPL1过表达细胞中进行了磷酸化的JNK和p38的细胞内染色。在IL-1RAPL1过表达后,在磷酸化的JNK中观察到诱导而不是在p38中。这种诱导被IL-38aa20-152显著降低,而不是IL-38aal-152,证实了截短的IL-38的更强调节作用。
实施例8:IL-38调节巨噬细胞中的AP1活性
接下来,发明人将发明人的数据从HEK模型转移进巨噬细胞情况中。用AP1或NFκB报告基因构建体转染人巨噬细胞,并用单独IL-1β或联合IL-38aa20-152或IL-38aal-152刺激。如同在HEK细胞中,IL-38aa20-152在巨噬细胞中降低AP1而不是NFκB活性,而IL-38aal-152是无效的(图9)。因此,只有截短的IL-38抑制巨噬细胞中的AP-1活性,这与发明人发现的经缺乏IL-38的凋亡细胞上清液刺激的巨噬细胞显示出较高水平的AP1活性一致(图2D)。最后,发明人着手这个问题:为什么IL-38aal-152在IL-1β刺激巨噬细胞后增加IL-6产生(图7A),而在HEK细胞模型中,IL-38aal-152既不增强AP1也不增强NFκB激活。为了研究这种差异,用IL-6报告基因构建体转染巨噬细胞,并用单独IL-1β或者联合两种IL-38亚型来刺激。如所预期的,IL-38aa20-152减弱了IL-6启动子活性,而IL-38aal-152完全不影响IL-6启动子诱导(图9),表明IL-38aal-152介导的IL-6的产生的增加不是在转录上调节的。
综上所述,本发明显示了经N末端加工的IL-38,其可用在临床上来限制一般的自身炎症或者例如有缺陷的巨噬细胞和凋亡细胞相互作用导致的自身炎症。
参考文献:
1.Sims,J.E.&Smith,D.E.(2010)Nat Rev Immunol 10,89-102.
2.Dinarello,C.A.(2009)Annu Rev Immunol 27,519-50.
3.Dinarello,C.A.(2013)Seminars in immunology.
4.Garlanda,C,Dinarello,C.A.&Mantovani,A.(2013)Immunity 39,1003-18.
5.Towne,J.E.,Renshaw,B.R.,Douangpanya,J.,Lipsky,B.P.,Shen,M.,Gabel,C.A.&Sims,J.E.(2011)J Biol Chem 286,42594-602.
6.Latz,E.,Xiao,T.S.&Stutz,A.(2013)Nat Rev Immunol 13,397-411.
7.Boraschi,D.&Tagliabue,A.(2013)Seminars in immunology.
8.Pavlowsky,A.,Gianfelice,A.,Pallotto,M.,Zanchi,A.,Vara,H.,Khelfaoui,M.,Val-negri,P.,Rezai,X.,Bassani,S.,Brambilla,D.,Kumpost,J.,Blahos,J.,Roux,M.J.,Humeau,Y.,Chelly,J.,Passafaro,M.,Giustetto,M.,BiUuart,P.&Sala,C.(2010)Current biology:CB 20,103-15.
9.Gambino,F.,Kneib,M.,Pavlowsky,A.,Skala,H.,Heitz,S.,Vitale,N.,Poulain,B.,Khelfaoui,M.,Chelly,J.,BiUuart,P.&Humeau,Y.(2009)The European journal of neuroscience 30,1476-86.
10.Jin,H.,Gardner,R.J.,Viswesvaraiah,R.,Muntoni,F.&Roberts,R.G.(2000)European journal of human genetics:EJHG 8,87-94.
11.Born,T.L.,Smith,D.E.,Garka,K.E.,Renshaw,B.R.,Bertles,J.S.&Sims,J.E.(2000)The Journal of biological chemistry 275,29946-54.
12.Carrie,A.,Jun,L.,Bienvenu,T.,Vinet,M.C,McDonell,N.,Couvert,P.,Zemni,R.,Cardona,A.,Van Buggenhout,G.,Frints,S.,Hamel,B.,Moraine,C,Ropers,H.H.,Strom,T.,Howell,G.R.,Whittaker,A.,Ross,M.T.,Kahn,A.,Fryns,J.P.,Beldjord,C,Marynen,P.&Chelly,J.(1999)Nature genetics 23,25-31.
13.Khan,J.A.,Brint,E.K.,O'Neill,L.A.&Tong,L.(2004)The Journal of biological chemistry 279,31664-70.
14.Pavlowsky,A.,Zanchi,A.,Pallotto,M.,Giustetto,M.,Chelly,J.,Sala,C.&BiUuart,P.(2010)Commun Integr Biol 3,245-7.
15.Arthur,J.S.&Ley,S.C.(2013)Nat Rev Immunol 13,679-92.
16.Ley,S.,Weigert,A.,Heriche,J.K.,Mille-Baker,B.,Janssen,R.A.&Brune,B.(2013)Immunobio logy 218,40-51.
17.Lin,H.,Ho,A.S.,Haley-Vicente,D.,Zhang,J.,Bernal-Fussell,J.,Pace,A.M.,Hansen,D.,Schweighofer,K.,Mize,N.K.&Ford,J.E.(2001)The Journal of biological chemistry 276,20597-602.
18.Bensen,J.T.,Dawson,P.A.,Mychaleckyj,J.C.&Bowden,D.W.(2001)Journal of interferon&cytokine research:the official journal of the International Society for Interferon and Cytokine Research 21,899-904.
19.van de Veerdonk,F.L.,Stoeckman,A.K.,Wu,G.,Boeckermann,A.N.,Azam,T.,Netea,M.G.,Joosten,L.A.,van der Meer,J.W.,Hao,R.,Kalabokis,V.&Dinarello,C.A.(2012)Proc Natl Acad Sci U S A 109,3001-5.
20.Jung,M.Y.,Kang,S.W.,Kim,S.K.,Kim,H.J.,Yun,D.H.,Yim,S.V.,Hong,S.J.&Chung,J.H.(2010)Scandinavian journal of rheumatology 39,190-6.
21.Guo,Z.S.,Li,C,Lin,Z.M.,Huang,J.X.,Wei,Q.J.,Wang,X.W.,Xie,Y.Y.,Liao,Z.T.,Chao,S.Y.&Gu,J.R.(2010)International journal of immunogenetics 37,33-7.
22.Chou,C.T.,Timms,A.E.,Wei,J.C,Tsai,W.C,Wordsworth,B.P.&Brown,M.A.(2006)Annals of the rheumatic diseases 65,1106-9.
23.Monnet,D.,Kadi,A.,Izac,B.,Lebrun,N.,Letourneur,F.,Zinovieva,E.,Said-Nahal,R.,Chiocchia,G.&Breban,M.(2012)Annals of the rheumatic diseases 71,885-90.
24.Rahman,P.,Sun,S.,Peddle,L.,Snelgrove,T.,Melay,W.,Greenwood,C.&Glad-man,D.(2006)Arthritis and rheumatism 54,2321-5.
25.Dehghan,A.,,et al.(2011)Circulation 123,731-8.
26.Zitvogel,L.,Kepp,O.&Kroemer,G.(2010)Cell 140,798-804.
27.Ley,S.,Weigert,A.,Weichand,B.,Henke,N.,Mille-Baker,B.,Janssen,R.A.&Brune,B.(2013)Oncogene 32,631-40.
28.Weigert,A.,Tzieply,N.,von Knethen,A.,Johann,A.M.,Schmidt,H.,Geisslinger,G.&Brune,B.(2007)Mol Biol Cell 18,3810-9.
29.Xiao,W.,Hodge,D.R.,Wang,L.,Yang,X.,Zhang,X.&Farrar,W.L.(2004)Prostate 61,354-70.
- 还没有人留言评论。精彩留言会获得点赞!