用于癌症治疗的小分子抑制剂的制作方法
本申请要求2014年8月7日提交的美国专利申请号62/034,331的优先权,其被引入本文作为参考。
技术领域
本申请涉及用于癌症治疗的小分子抑制剂。具体地,其涉及通过抑制蛋白质向细胞核中的转运而杀死癌细胞的小分子的应用。
发明背景
核转运是蛋白质穿越核膜孔复合体(NPC)的输入或输出。核转运蛋白参与转运过程,并且其决定给定细胞的核转运能力。转运能力可对基因表达、信号转导以及细胞生长和发育具有直接影响。
核周蛋白β1(Kpnβ1)是参与货物蛋白和RNA穿越NPC从细胞质输入至核中的核转运蛋白。Kpnβ1是核转运蛋白的核周蛋白β超家族的成员。核周蛋白β蛋白质家族的成员超过二十个,其可以充当输入或输出受体,介导蛋白质的核进入或退出。Kpnβ1,亦称为输入蛋白β,是细胞中使包含核定位信号(NLS)的蛋白质通过NPC转运到核中的主要核输入受体。依靠Kpnβ1的核输入的一般特征在于Kpnβ1衔接蛋白——核周蛋白α(Kpnα),亦称为输入蛋白α——对货物蛋白上的NLS的识别。在货物识别之后,Kpnα结合Kpnβ1并且三元复合体移位至核中。
Crm1抑制剂细霉素B(LMB)是唯一被接受且市售的已知抑制核转运的化合物。近来,Kpnα/β介导型转运的小分子肽模拟物抑制剂在体外筛选中被鉴定,但是,这些抑制剂具有低效力并且不是细胞可透的,因此在体内不可观察到Kpnα/β介导型核输入的抑制(Ambrus G,Whitby LR,Singer EL,Trott O,Choi E,Olson AJ等,Small molecule peptidomimetic inhibitors of importin alpha/beta mediated nuclear transport.Bioorg Med Chem,2010年11月1日;18(21):7611-20)。
以强亲和力结合Kpnα的肽抑制剂也已被描述,但这些并不直接抑制Kpnβ1(Kosugi S,Hasebe M,Entani T,Takayama S,Tomita M,Yanagawa H.Design of peptide inhibitors for the importin alpha/beta nuclear import path by activity-based profiling.Chem Biol,2008年9月22日;15(9):940-9)。双氢除虫菌素是广谱抗寄生虫剂,其抑制Kpnα/β复合体,但其不呈现阻断Kpnβ1单独介导的输入(Wagstaff KM,Sivakumaran H,Heaton SM,Harrich D,Jans DA.Ivermectin is a specific inhibitor of importin alpha/beta-mediated nuclear import able to inhibit replication of HIV-1and dengue virus.Biochem J,2012年5月1日;443(3):851-6)。核抑制素1A(Karyostatin 1A)(Hintersteiner M,Ambrus G,Bednenko J,Schmied M,Knox AJ,Meisner NC等,Identification of a small molecule inhibitor of importin beta mediated nuclear import by confocal on-bead screening of tagged one-bead one-compound libraries.ACS Chem Biol,2010年10月15日;5(10):967-79)和Importazole(Soderholm JF,Bird SL,Kalab P,Sampathkumar Y,Hasegawa K,Uehara-Bingen M等,Importazole,a small molecule inhibitor of the transport receptor importin-beta.ACS Chem Biol,2011年7月15日;6(7):700-8)是Kpnβ1的被鉴定的第一批小分子抑制剂,但是,其脱靶效果还没有被考察。而且,还没有核输入抑制剂已经被测试过其抗癌效果。因此,仍需要鉴定新型且有效的Kpnβ1抑制剂和测试抑制剂其抗癌活性。
前文对本发明背景的论述仅旨在便于理解本发明。应当理解,该论述不是认可或承认任何提及材料是在本申请优先权日时本领域公知常识的一部分。
发明概述
根据本发明的第一方面,提供了用于治疗癌症的式I化合物或其盐,
其中R1是分支或无分支的、任选地用选自胺、咪唑、醇或吗啉的取代基官能化的C2-C5烷基;并且其中R2是氢或甲基。
本发明的这方面的进一步特征允许R1选自:
乙基、丙基、丁基、异丁基、异戊基、丙醇基和
本发明的进一步特征允许式I化合物选自:
本发明的进一步特征允许该化合物用于结合细胞中的Kpnβ1以及结合发生在Kpnβ1的结合核周蛋白α(Kpnα)和Ran的结合区域。
根据本发明的第二方面,提供用于治疗癌症的式II化合物或其盐。
根据本发明的第三方面,提供用于治疗癌症的式III化合物、其立体异构体或盐。
再进一步特征允许式I、II和/或III的化合物用于抑制蛋白质和转录因子如Kpnα、激活蛋白-1(AP-1)、NF-κB/P65和活化T细胞的核因子(NFAT)向细胞(优选癌细胞)核中的输入。
根据本发明的第四方面,提供用于治疗癌症的式IV的化合物或其盐。
提供用于治疗癌症的式V化合物或其盐。
提供用于治疗癌症的式VI化合物、其立体异构体或盐。
提供用于治疗癌症的式VII化合物、其立体异构体或盐。
提供用于治疗癌症的式VIII化合物、其立体异构体或盐。
提供用于治疗癌症的式IX化合物、其立体异构体或盐。
提供用于治疗癌症的下式化合物或其盐,
其中R是
提供用于治疗癌症的式XII化合物或其盐。
提供用于治疗癌症的式XIII化合物、其立体异构体或盐。
提供用于治疗癌症的式XIV化合物、其立体异构体或盐。
提供用于治疗癌症的式XV化合物或其盐。
提供用于治疗癌症的式XVI化合物或其盐。
仍进一步特征允许,在给予有效量的式I至XVI化合物或化合物组合之后,以上式I至XVI化合物、其立体异构体或盐用于诱导细胞凋亡——优选在癌细胞中;有效量的给予浓度小于100μM,优选小于50μM。
再进一步特征允许式I至XVI化合物用于诱导癌细胞中的细胞凋亡,同时不实质性影响非癌细胞——通过给予有效量的式I至XVI化合物或化合物组合。
仍进一步特征允许以上限定的式I至XVI化合物用于治疗宫颈、食管、卵巢、乳腺和其它的癌症。
进一步特征允许式I至XVI化合物用于治疗选自以下的肿瘤或癌症:胃的癌症、肺的癌症、肝的癌症、黑素瘤、何杰金氏病、非何杰金氏淋巴瘤、急性或慢性淋巴细胞性白血症、多发性骨髓瘤、成神经母细胞瘤、乳腺癌、卵巢癌、肺癌、维尔姆斯氏瘤、宫颈癌、睾丸癌、软组织肉瘤、原发性巨球蛋白血症、膀胱癌、慢性粒细胞性白血病、原发性脑癌、恶性黑素瘤、小细胞肺癌、胃癌、结肠癌、恶性胰腺胰岛素瘤、恶性类癌癌(malignant carcinoid carcinoma)、恶性黑素瘤、绒毛膜癌、蕈样霉菌病、头或颈癌、骨肉瘤、胰腺癌、急性粒细胞性白血病、毛细胞白血病、成神经母细胞瘤、横纹肌肉瘤、卡波西氏肉瘤、泌尿生殖癌、甲状腺癌、食管癌、恶性高钙血综合症、宫颈增生、肾细胞癌、子宫内膜癌、真性红细胞增多症、特发性血小板增多症、肾上腺皮质癌、皮肤的癌症、前列腺癌、喉的癌症、外阴的癌症和睾丸的癌症。
本发明进一步提供药物,其特征在于,其包括如上所述的式I至XVI中任一种化合物、其立体异构体或药学可接受的盐作为活性成分。
本发明还提供用作药物的式I至XVI中任一种的化合物、其立体异构体或药学可接受的盐。
本发明还进一步提供式I至XVI中任一种的化合物、其立体异构体或盐在制备用于治疗癌症的药物中的应用,其中R1是分支或无分支的、任选地用选自胺、咪唑、醇或吗啉的取代基官能化的C2-C5烷基;并且其中R2是氢或甲基。
本发明提供响应患有肿瘤的患者中的Kpnβ1活性抑制来治疗患者的肿瘤的方法,包括给予需要这种治疗的患者治疗有效量的式I至XVI化合物、其立体异构体或其盐的任一种或组合。
本发明还进一步提供治疗癌症的方法,其包括给予对其有需要的患者治疗有效量的式I至XVI中任一种的化合物、其立体异构体或药学可接受的盐,其中R1是分支或无分支的、任选地用选自胺、咪唑、醇或吗啉的取代基官能化的C2-C5烷基;并且其中R2是氢或甲基。
附图简述
现将参考附图(仅作举例)对本发明进行描述,其中:
图1是显示用20nM对照siRNA(Ctl siRNA)或KpnB1siRNA转染的宫颈癌(HeLa、SiHa、CaSki、Ms751、C33A)、食管癌(WHCO5)、转化(SVWI38)和非癌(WI38、CCD-1068SK、FG0)细胞系的相对细胞增殖的柱形图,其中细胞增殖在转染后5天利用MTT试验监测(**p<0.05);
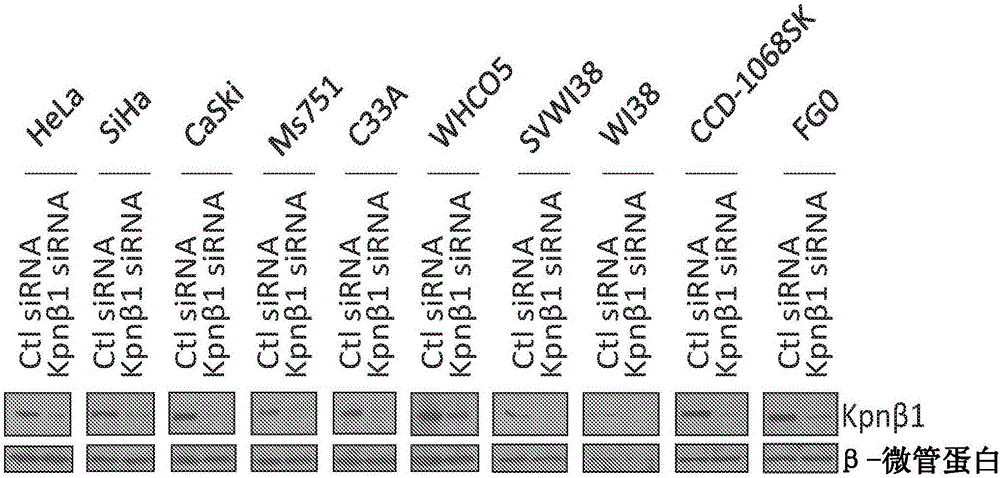
图2是显示在Kpnβ1siRNA转染后Kpnβ1有效敲低(knockdown)的蛋白质印迹分析,其中β-微管蛋白充当蛋白质加载对照;
图3是用递增浓度的喹喔啉处理的HeLa癌细胞的细胞活力的作图(A)和用递增浓度的苯并咪唑处理的HeLa细胞的细胞活力的作图(B);
图4是显示未处理细胞样品和分别用PMA、化合物A、化合物III和化合物II处理的样品中的核和细胞质p65的百分比的柱形图;
图5是显示用化合物A、化合物III和化合物II以IC50和两倍IC50浓度处理导致PARP裂解(作为凋亡指示)的蛋白质印迹分析;
图6是用递增浓度的化合物II处理时利用MTT试验监测的HeLa癌细胞(A)以及FG0非癌细胞(B)的细胞增殖的一对作图;
图7是用递增浓度的化合物III处理时利用MTT试验监测的HeLa癌细胞(A)以及非癌细胞FG0(B)的细胞增殖的一对作图;
图8是显示用pEGFP-Kpnα2稳定转染的CaSki细胞中的GFP-Kpnα2表达的蛋白质印迹分析,其中指示了内源Kpnα2水平并且β-微管蛋白充当蛋白质加载对照;
图9是显示未处理细胞样品和用10μM化合物A处理的细胞样品中的具有细胞质GFP-Kpnα2荧光的细胞的百分比的柱形图(*p<0.05);
图10是未处理细胞样品和用10μM化合物A处理的细胞样品中的核GFP-Kpnα2荧光的柱形图(*p<0.05);
图11是显示未处理细胞和用化合物A处理的细胞的蛋白质裂解物中Kpnα2和NFY的细胞质和核定位的蛋白质印迹,其中β-微管蛋白用作细胞质负载对照,并且兔抗TATA盒结合蛋白(TBP)用作核负载对照;
图12是显示相对于相同提取物中的TBP、未处理细胞和用化合物A处理的细胞中的Kpnα2和NFY的核蛋白质水平的柱形图,其中实验按一式三份进行并且示出平均值和平均值的标准误差(*p<0.05);
图13是显示利用siRNA抑制Kpnβ1表达的蛋白质印迹;
图14是蛋白质印迹分析,显示Kpnβ1表达的siRNA敲低导致Kpnα2和NFY的核定位减少与用化合物A处理所观察到的相似;
图15是显示在以IC50浓度和15μM的化合物A处理后CaSki细胞中的AP1和p65/NFκB转录活性的剂量依赖性降低的柱形图(*p<0.05);
图16是在存在TPA和离子霉素(+TPA/离子霉素)或不存在TPA和离子霉素(-TPA/离子霉素)并且存在不同浓度的化合物A的情况下培育的、用NFAT报告质粒转染的NFAT转染HeLa细胞的NFAT转录活性的柱形图(*p<0.05);
图17是在存在TPA和离子霉素(+TPA/离子霉素)或不存在TPA和离子霉素(-TPA/离子霉素)并且存在不同浓度的化合物II的情况下培育的、用NFAT报告质粒转染的NFAT转染HeLa细胞的NFAT转录活性的柱形图(*p<0.05);
图18是在存在TPA和离子霉素(+TPA/离子霉素)或不存在TPA和离子霉素(-TPA/离子霉素)并且存在不同浓度的化合物III的情况下培育的、用NFAT报告质粒转染的NFAT转染HeLa细胞的NFAT转录活性的柱形图(*p<0.05);
图19是在存在TPA和离子霉素(+TPA/离子霉素)或不存在TPA和离子霉素(-TPA/离子霉素)并且存在不同浓度的双氢除虫菌素(其是Kpnβ1/Kpnα2介导的核转运的一种已知的抑制剂,并且充当阳性对照)的情况下培育的、用NFAT报告质粒转染的NFAT转染HeLa细胞的NFAT转录活性的柱形图;
图20是在存在TPA和离子霉素(+TPA/离子霉素)或不存在TPA和离子霉素(-TPA/离子霉素)并且存在不同浓度的表皮生长因子受体(EGF-R)抑制剂AG1478(其充当阴性对照)的情况下培育的、用NFAT报告质粒转染的NFAT转染HeLa细胞的NFAT转录活性的柱形图;
图21是用递增浓度的化合物A处理时利用MTT试验监测的CaSki(A)、HeLa(B)、KYSE30(C)和WHCO6(D)癌细胞以及DMB(E)和FG0(F)非癌细胞的细胞增殖的作图集合;
图22是在存在10μM化合物A或不存在化合物A的情况下癌细胞(Caski、HeLa、KYSE30和WHCO6)和非癌细胞(FG0和DMB)的细胞增殖的柱形图;
图23是在存在或不存在10μM化合物A的情况下生长的HeLa和KYSE30细胞的锚地不依赖性集落形成的照片;
图24是未处理的CaSki细胞(A)或用10μM化合物A(B)或20μM化合物A(C)处理的CaSki细胞的细胞周期分析;
图25是基于在存在DMSO、IC50浓度的化合物A和20μM化合物A的情况下进行的细胞周期分析、细胞周期的不同阶段中的细胞百分比的柱形图(*p<0.05);
图26是显示化合物A处理导致PARP裂解(作为凋亡指示)的蛋白质印迹分析,其中β-微管蛋白用作蛋白加载对照;
图27是显示细胞色素C向细胞质中的释放增加伴随线粒体细胞色素C水平减少的蛋白质印迹分析,表明内在凋亡途径激活,其中β-微管蛋白和过氧化物氧还蛋白3(Prdx3)分别用作细胞质和线粒体蛋白的加载对照,并且用于确认部分(fractions)的纯度;
图28是未经治疗的或每2-3天用媒介(DMSO)或化合物A治疗3周的WHCO6荷瘤小鼠中的标准化肿瘤体积的作图;和,
图29是在参考图28描述的治疗过程期间每天测量的荷瘤小鼠的体重的作图。
参考附图的详细描述
计算机(in silico)筛选方法揭示了具有结合和抑制Kpnβ1转运蛋白的潜力的小分子的身份。Kpnβ1蛋白结构内的适当区域被选作目标区域。考察Kpnβ1的晶体结构数据,并且Kpnβ1的氨基酸残基331-364被鉴定为其结合至RanGTP、SREBP-2和Kpnα2以转运典型含NLS货物所需的共同区域。进行针对能够结合Kpnβ1的这个区域并且潜在地干扰其核输入功能的小分子的计算机筛选。筛选利用由1400万种化合物组成的文库进行。对筛选的分子基于其预期结合亲和力进行评分。对分子基于其互补形状和对上述结合位点和周围残基的极性进行评分。本文进一步描述的式I至XVI的化合物全部被鉴定为潜在的Kpnβ1抑制剂。这些化合物全部都具有每分子至少两个芳香族区域,并且参与与结合位点和其周围残基上的互补区域的范德华(van der Waals)和静电相互作用。式I至XVI的化合物还包括氢键供体,具体地仲胺和伯胺部分;和氢键受体,如羰基或醚氧原子或叔胺,其在各自分子上适当定位以便参与与构成Kpnβ1上结合位点和其周围残基的部分的互补氢键供体和受体的氢键合相互作用。化合物I至XVI已显示具有抗癌性质,使得其可用于癌症的治疗。
在通过筛选方法鉴定并且稍后证实抑制Kpnβ1的化合物中有喹喔啉衍生物。喹喔啉衍生物是重要的一类杂环化合物,并且喹喔啉结构是装配用于各种应用的多种衍生化合物的前体。大多数喹喔啉衍生物是合成的,而仅有若干天然存在。已知一些喹喔啉衍生物具有生物学活性并且用作抗微生物、抗病毒或抗真菌剂。
通式I的喹喔啉衍生物或其盐现已被发现具有抗癌性质,因此其可用于癌症治疗,
其中R1是分支或无分支的、任选地用选自胺、咪唑、醇或吗啉的取代基官能化的C2-C5烷基;并且其中R2是氢或甲基。
通式I的化合物或其盐抑制蛋白质和转录因子如Kpnα、AP-1、P65、NFAT(已知的Kpnβ1转运蛋白目标)向细胞核中的输入。
式II化合物——(2S,4S)-1-苄基-4-{[(4-甲氧基萘-1-基)甲基]氨基}-N-甲基吡咯烷-2-甲酰胺——或其盐的一种(以下称为化合物II)、和式III化合物——9-[(1-甲基哌啶-3-基)甲氧基]-4-[(6-甲基吡啶-2-基)甲基]-7-(5-甲基噻吩-2-基)-3,5-二氢-2H-1,4-苯并氧杂氮杂——或其立体异构体或盐的一种(以下称为化合物III)类似地抑制蛋白质和转录因子如NFAT和P65/NFKB向至细胞核中的输入。这些化合物同样被发现具有抗癌效果,因此其可被用于癌症治疗。
式IV化合物——({2-[(2-氯苯基)甲氧基]-6H,7H,8H-环戊并[g]喹啉-3-基}甲基)({2H,3H-咪唑并[2,1-b][1,3]噻唑-6-基甲基})胺——或其盐(以下称为化合物IV)具有抗癌效果,从而其可被用于癌症治疗。
以下式V至XVI化合物、其立体异构体(适用时)或盐具有抗癌效果,因此其可用于癌症治疗:
现将进一步描述式I至XVI化合物的抗癌性质及其作用方式。如上所述,由计算机筛选鉴定了所有化合物的Kpnβ1(核转运蛋白)结合亲和力。
Kpnβ1依赖性核输入的特征在于,Kpnβ1衔接蛋白,核周蛋白α(Kpnα),亦称为输入蛋白α,识别货物蛋白上的NLS。在货物识别后,Kpnα结合Kpnβ1并且三元复合体通过Kpnβ1与包括NPC的核孔蛋白(Nups)的相互作用移位至细胞核中。一旦处于NPC的核质侧,复合体因RanGTP与Kpnβ1的结合而解离。RanGTP和Kpnα共享Kpnβ1蛋白上的重叠结合位点,但RanGTP对该位点具有更高的亲和力,因此取代Kpnα,导致货物蛋白被释放(Moroianu J,Blobel G,Radu A.Nuclear protein import:Ran-GTP dissociates the karyopherin alphabeta heterodimer by displacing alpha from an overlapping binding site on beta.Proc Natl Acad Sci U S A,1996年7月9日;93(14):7059-62)。在货物释放后,RanGTP-Kpnβ1移回细胞质中,在此RanGTP被水解成RanGDP并且Kpnβ1被释放用于另一轮核转运。在Kpnβ1与衔接子(adaptor)一起转运其货物蛋白时,其可以靠自己发挥作用——例如在转运固醇调节元件结合蛋白2(SREBP-2)时(Nagoshi E,Imamoto N,Sato R,Yoneda Y.Nuclear import of sterol regulatory element-binding protein-2,a basic helix-loop-helix-leucine zipper(bHLH-Zip)-containing transcription factor,occurs through the direct interaction of importin beta with HLH-Zip.Mol Biol Cell,1999年7月;10(7):2221-33)。
已知Kpnβ1mRNA和蛋白在宫颈肿瘤和细胞系中的表达水平升高(Van der Watt PJ,Maske CP,Hendricks DT,Parker MI,Denny L,Govender D等.The Karyopherin proteins,Crm1 and Karyopherin beta1,are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation.Int J Cancer,2009年4月15日;124(8):1829-4)。启动子在宫颈癌细胞中更为活跃——因为其被细胞周期调节子(E2F)激活(Van der Watt PJ,Ngarande E,Leaner VD.Overexpression of Kpnbeta1and Kpnalpha2Importin Proteins in Cancer Derives from Deregulated E2F Activity.PLoS One 2011;6(11):e27723)。Smith等(2010)发现Kpnβ1 mRNA在卵巢癌细胞系和转化的卵巢细胞中升高,并且Kuusisto等(2012)也描述在几种转化的细胞系中Kpnβ1水平升高(Smith ER,Cai KQ,Smedberg JL,Ribeiro MM,Rula ME,Slater C等.Nuclear entry of activated MAPK is restricted in primary ovarian and mammary epithelial cells.PLoS One 2010;5(2):e9295;Kuusisto HV,Wagstaff KM,Alvisi G,Roth DM,Jans DA.Global enhancement of nuclear localization-dependent nuclear transport in transformed cells.FASEB J,2012年3月;26(3):1181-93)。
Kpnβ1水平升高可表明Kpnβ1蛋白与细胞转化和癌症有关。事实上,癌细胞中Kpnβ1蛋白表达的抑制导致凋亡(Van der Watt等,2009;Angus等,2014),突出了这种蛋白质在癌症发展中的作用。但是,非癌细胞中Kpnβ1蛋白表达的抑制对细胞活力仅具有轻微影响(Van der Watt等,2009;Angus L,van der Watt PJ,Leaner VD.Inhibition of the nuclear transporter,Kpnbeta1,results in prolonged mitotic arrest and activation of the intrinsic apoptotic pathway in cervical cancer cells.Carcinogenesis,2014年1月7日),指出Kpnβ1作为抗癌治疗靶的潜在应用(Van der Watt PJ,Stowell CL,Leaner VD.The nuclear import receptor Kpnbeta1and its potential as an anti-cancer therapeutic target.Crit Rev Eukaryot Gene Expr 2013;23(1):1-10)。
通式I的化合物,化合物II和化合物III,抑制Kpnβ1货物蛋白如p65/NFBK的核输入,通过凋亡诱导癌细胞的细胞死亡,和抑制癌细胞增殖。
在式I至XVI化合物的抗癌研究期间,使用细胞系和组织培养物:
细胞系和组织培养物
通过将XhoI/BamHI切割的人Kpnα2cDNA片段插入pEGFP-C1(Clontech,Mountain View,CA,USA)中来生成GFP-Kpnα2表达质粒(pEGFP-C1-Kpnα2)。将该构建物稳定转染至CaSki宫颈癌细胞(美国典型培养物保藏中心(American Type Culture Collection),ATCC,Rockville,MD,USA)中,通过FACS分析选择表达适度水平的GFP-Kpnα2的细胞库,并将其保持在包含10%胎牛血清(FBS)(Gibco,佩斯利,苏格兰)、100U/ml青霉素、100μg/ml链霉素和200μg/ml G418(Sigma,St Louis,MO,USA)的Dulbecco改良型Eagle培养基(Dulbecco’s Modified Eagle’s Medium,DMEM)中。所有其它细胞系都被保持在包含10%FBS、100U/ml青霉素和100ug/ml链霉素的DMEM中——除了正常hTERT永生化人食管上皮细胞EPC2-hTERT(获自A.K.Rustgi教授(宾夕法尼亚大学(University of Pennysylvania),费城,USA)),其被培养在补充有50μg/ml牛垂体提取物(BPE)、1ng/ml上皮生长因子(EGF)、和100U/ml青霉素和100μg/ml链霉素的角质化细胞无血清培养基(KSFM)中,;和MCF12A良性乳腺癌细胞(获自ATCC),其被保持在补充有5%FBS、100U/ml青霉素、100μg/ml链霉素、20ng/ml EGF(Gibco)、100ng/ml霍乱霉素(Sigma)、10μg/ml胰岛素(Gibco)和500ng/ml氢化可的松(Sigma)的50%HAMS F12和50%DMEM中。KYSE30和KYSE150细胞获自DSMZ(柏林,德国),并且WHCO1、WHCO5和WHCO6食管癌细胞系获自R.Veale教授(Jones GJ,Heiss NS,Veale RB,Thornley AL.Amplification and expression of the TGF-alpha,EGF receptor and c-myc genes in four human oesophageal squamous cell carcinoma lines.Biosci Rep,1993年10月;13(5):303-12)。FG0和DMB正常皮肤成纤维细胞获自医学系(Department of Medicine),UCT。除非如上指定,所有其它细胞系均获自ATCC。
IC50确定如下进行:
将细胞铺平于96孔板,并用不同浓度的测试化合物处理48小时,然后加入MTT染料3-(4,5-二甲基噻唑-2-基)-2,5-二苯基溴化四氮唑(Sigma),并且4小时后利用增溶试剂(10%SLS,在0.01M HCl中)溶解晶体。次日利用BioTek微孔板分光光度计(Winooski,VT,USA)在595nm下测量吸光度,并利用GraphPad Prism生成IC50曲线。
利用siRNA抑制癌细胞中的Kpnβ1表达导致细胞通过凋亡而死亡,而正常细胞中的抑制对细胞生物学仅具有轻微影响。为将此验证,用Kpnβ1siRNA转染一组细胞系,并在转染后5天监测细胞活力。图1显示在宫颈癌、食管癌和转化细胞系中Kpnβ1抑制后细胞数量显著减少,而非癌细胞相对不受影响。通过蛋白质印迹分析确认癌细胞和非癌细胞中的Kpnβ1敲低(图2)。
在利用siRNA的Kpnβ1抑制后癌细胞被选择性杀死表明,Kpnβ1的小分子抑制剂可具有相同的效果。特别是,如果抑制剂结合至对应于Kpnβ1的氨基酸331-363的重叠Ran-和Kpnα2-结合区域。Kpnβ1的这个区域此前经鉴定对其功能至关重要,因为其缺失导致Kpnβ1不能将NLS-HSA货物转运至核中(Moroianu等,1996)。
利用食管癌细胞系(WHCO5和KYSE180)和宫颈癌细胞系(HeLa和CaSki)测试潜在抑制剂化合物对癌细胞活力的影响。细胞活力的MTT试验揭示,化合物A至K显示IC50值小于50μM(表1)。
表1.喹喔啉衍生物在食管癌细胞系(WHCO5和KYSE180)和宫颈癌细胞系(HeLa和CaSki)中的IC50值。
化合物A至K的分子结构包括喹喔啉部分和苯并咪唑部分。测试喹喔啉和苯并咪唑对HeLA细胞的细胞活力的影响,以确定喹喔啉和苯并咪唑分子分别是否可具有抗癌性质(图3)。在上至6.5mM喹喔啉的浓度范围内没有观察到细胞杀伤效果。苯并咪唑在2.078mM的极高IC50浓度下具有细胞杀伤效果。但是,苯并咪唑也可以被认为是无作用的,因为其IC 50远远超过利用化合物A至K和当前化学治疗药物所获得的IC50范围。
利用宫颈癌细胞系CaSki和HeLa,测试化合物II和III其对癌细胞活力的影响。确定基于这些癌细胞系以及非癌细胞系FG0的IC50值。细胞活力的MTT试验揭示,两种化合物均显示癌细胞杀伤效果,并且IC50值小于50μM(表2和3)。
表2.用化合物II处理的CaSki、HeLa和FG0细胞的IC50值。
表3.用化合物III处理的CaSki、HeLa和FG0细胞的IC50值。
与化合物A相似,化合物II和III抑制Kpnβ1货物蛋白的核输入,如图4所示,其中显示在细胞未经处理、用PMA、化合物A、化合物III或化合物II处理时相对于细胞质p65百分比的核p65百分比。
通过图5所示的PARP裂解观察到,化合物A以及化合物II和III诱导癌细胞通过凋亡而死亡。化合物II和II抑制细胞增殖,如图6和7中所示,其中显示在用递增浓度的化合物II和III处理时利用MTT试验监测的HeLa癌细胞(A)以及FG0非癌细胞(B)的细胞增殖。
利用一组不同来源的细胞系测试化合物IV的细胞杀伤效果。化合物IV杀死宫颈(CaSki,HeLa)、食管(WHCO5,KYSE150)和乳腺来源(MCF12A、MCF7、MDA-MB-231)的癌细胞系和转化细胞系(SVW138),并且IC50值在大约8与18μM之间的范围内(表4)。
表4.用化合物IV处理的癌细胞、转化细胞和非癌细胞的IC50值。
显示CasKi宫颈癌细胞的癌细胞杀伤效果的更多化合物列举在表5中。表5中所列的化合物具有50μM以下的IC50值,其在顺铂的IC50范围内。
表5.用不同化合物处理的CaSki细胞的IC50值。
基于包括上皮和成纤维细胞来源的癌细胞系和正常细胞系在内的一组不同细胞系,测试化合物IX–XIII(盐形式)。利用各个化合物的每一种处理时细胞系的IC50值在表6至10中给出。
表6.用化合物IX处理的癌细胞、转化细胞和非癌细胞的IC50值。
表7.用化合物X处理的癌细胞、转化细胞和非癌细胞的IC50值。
表8.用化合物XI处理的癌细胞、转化细胞和非癌细胞的IC50值。
表9.用化合物XII处理的癌细胞、转化细胞和非癌细胞的IC50值。
表10.用化合物XIII处理的癌细胞、转化细胞和非癌细胞的IC50值。
为了证明通式I的化合物——3-(1H-苯并咪唑-2-基)-1-(3-二甲基氨基丙基)吡咯并[5,4-b]喹喔啉-2-胺(以下称为“化合物A”)——的抗癌性质,现将更详细地描述其对输入蛋白的抑制作用。
以下方法被用于研究化合物A的抑制作用:
荧光显微法
使稳定表达GFP-Kpnα2的CaSki细胞在盖玻片上生长,并且用IC50或20μM的化合物A处理24小时。关于对照,使用等体积的DMSO。将细胞在荧光显微前用4%多聚甲醛固定,并将细胞核用0.5μg/ml DAPI染色。由于化合物A在GFP通道中发射荧光,必须通过分离发射光谱而从GFP信号中排除化合物A信号。使用Zeiss LSM 510Meta共聚焦显微镜,并且利用ZEN 2009照相机和关联的AxioVision软件(4.7版)拍摄图像。利用Zeiss图像浏览器编辑照片。关于定量,利用Image J分析来自各条件的100个细胞。
核和细胞质蛋白提取
按照制造商的说明,利用NE-PER核蛋白提取试剂盒(ThermoScientific,Rockford,IL,USA)进行核和细胞质蛋白分离(fractionation)。在蛋白质提取前,将CaSki细胞用IC50的化合物A处理48小时,或用20nM对照siRNA(sc-37007,Santa Cruz Biotechnology)或Kpnβ1siRNA(sc-35736,Santa Cruz Biotechnology)转染96小时。利用山羊抗核周蛋白α2(C-20)抗体(sc-6917,Santa Cruz Biotechnology)和兔抗NFYA(H-209)抗体(sc-10779,Santa Cruz Biotechnology)进行蛋白质印迹分析。抗β-微管蛋白(H-235)抗体(sc-9104,Santa Cruz Biotechnology)被用于细胞质蛋白负载对照,并且兔抗TATA盒结合蛋白(TBP)(N-12)抗体(sc-204,Santa Cruz Biotechnology)被用于核蛋白负载对照。利用Image J进行密度测定。
荧光素酶试验
为分析AP-1和p65荧光素酶活性,将40000个CaSki细胞铺于24孔板,并利用0.16μl TransFectinTM脂质试剂(Bio-Rad,Hercules,CA,USA),用50ng的AP1荧光素酶报道基因(reporter)(含有四拷贝的AP-1结合位点)或50ng的p65荧光素酶报道基因(含有五拷贝的p65结合位点,Clontech,CA,USA)和5ng的pRL-TK(编码Renilla荧光素酶,Promega)转染。次日用10或15μM化合物A处理细胞。按照生厂商说明,利用双荧光素酶R报道基因分析系统(Promega,Madison,WI,USA),在化合物A处理后24小时测量荧光素酶活性。利用VeritasTM微孔板照度计(Promega)测量荧光素酶读数,并将荧光素酶读数相对于相同提取物中的Renilla荧光素酶标准化。
为分析NFAT荧光素酶活性,在24孔板中每孔铺30000个HeLa细胞,并且利用0.4μl GenecellinTM转染试剂(Celtic Molecular Diagnostics,南非),用50ng GFP-NFAT质粒(Addgene质粒#24219,Jerry Crabtree所赠(Beals CR,Clipstone NA,Ho SN,Crabtree GR.Nuclear localization of NF-ATc by a calcineurin-dependent,cyclosporin-sensitive intramolecular interaction.Genes Dev,1997年4月1日;11(7):824-34))、50ng NFAT-荧光素酶(Addgene质粒#10959,Toren Finkel所赠(Ichida M,Finkel T.Ras regulates NFAT3activity in cardiac myocytes.J Biol Chem,2001年2月2日;276(5):3524-30))和5ng pRL-TK进行转染。NFAT-荧光素酶报告质粒包含三个串联重复的、IL-2启动子的30bp片段——已知其结合NFAT。次日将细胞用10或15μM化合物A处理,并且在过夜培育后,用100nM TPA(Sigma)和1.3μM离子霉素(Santa Cruz Biotechnology,Santa Cruz,CA,USA)刺激5小时,然后测量荧光素酶活性。
细胞增殖试验
为分析化合物A处理后的细胞增殖,将5000个细胞/孔铺于96孔板中,并用5、10或15μM化合物A处理,其后利用MTT每24小时监测细胞增殖4天。
细胞周期分析
将CaSki细胞铺于60mm培养皿中,并用适当浓度的化合物A处理。处理后24小时收集细胞,并在100%乙醇中固定,其后将其用碘化丙啶染色,并利用BD流式细胞仪(BD Biosciences,NJ,USA)分析细胞周期谱。利用ModFit 3.3软件进行数据分析。
PARP裂解分析
用不同浓度的化合物A处理CaSki细胞12小时,然后利用RIPA缓冲液(10mM Tris-Cl,pH 7.4,150mM NaCl,1%脱氧胆酸钠,0.1%SDS,1%曲拉通(Triton)X-100,1×完全蛋白酶抑制剂混合物(Roche,曼海姆,德国))收集细胞和细胞漂浮物,并且利用兔抗Parp1/2(H-250)抗体(sc-7150)进行蛋白质印迹分析。
线粒体和细胞质蛋白提取
使细胞在10cm平板中生长,并用不同浓度的化合物A处理24小时。使细胞溶解于亚细胞分离缓冲液(250mM蔗糖、20mM HEPES、10mM KCl、1.5mM MgCl2、1mM EDTA、1mM EGTA、1mM DTT和1×完全蛋白酶抑制剂混合物(Roche))中,通过25G针,并在冰上培育20分钟。在720g下离心5分钟后获得核沉淀物(pellet)。将上清液在10,000g下进一步离心,并将得到的上清液用作细胞质部分。将含有线粒体部分的沉淀物在亚细胞分离缓冲液中洗涤,并使其通过25G针。使线粒体部分在10,000g下形成沉淀物,并重悬浮于RIPA缓冲液中。利用小鼠抗细胞色素C抗体(BD Pharmingen,San Diego,CA)进行蛋白质印迹分析,并且抗β-微管蛋白(H235)(sc-9104,Santa Cruz)和抗过氧化物氧还蛋白-3(PRDX-3)(P1247,Sigma)抗体分别用作细胞质和线粒体部分的蛋白负载对照。
异种移植肿瘤模型
收集WHCO6细胞并将其重悬浮在PBS中。关于肿瘤接种,将癌细胞(每只小鼠5×106)皮下植入至雌性裸鼠的后胁(hind flanks)中。在肿瘤已达到可察觉尺寸后,开始药物治疗,其中荷瘤小鼠被随机分配并且每2-3天腹膜内施以媒介(DMSO)或化合物A(50mg/kg)3周。利用测径器每周两次测量肿瘤体积,并且利用下列公式估测肿瘤体积:体积=(长×宽×宽)/2。在药物治疗开始后3周将小鼠处死。
统计学分析
关于所有数据比较,利用Microsoft Excel进行Student t检验——除了体内研究,其中利用GraphPad Prism 5进行双程ANOVA检验。P值<0.05被认为统计学显著。
化合物A是一种喹喔啉衍生物,其分子式为C22H23N7,分子量为385.465g/mol。该化合物以粉末形式获得,并且可溶于二甲基亚砜(DMSO)。
本领域技术人员将理解,通式I的化合物及其盐——其中R1是分支或无分支的、任选地用选自胺、咪唑、醇或吗啉的取代基官能化的C2-C5烷基;并且其中R2是氢或甲基——将具有与化合物A相同或非常相似的作用方式和癌细胞杀伤效果。
化合物A的作用机制和癌细胞杀伤效果在下文结果章节被进一步描述。本领域技术人员将理解,与化合物A在结构上类似的化合物B至K具有与化合物A相同或非常相似的抑制活性和癌细胞杀伤效果。化合物A至K具有基本上相似的形状和极性——与Kpnβ1上的结合位点和其周围残基互补。本领域技术人员将理解,化合物A至K参与基本上相同的与结合位点和其周围残基的范德华相互作用和氢键合相互作用,从而与其与其缔合或结合并抑制Kpnβ1活性,从而诱导癌细胞的凋亡。
结果
GFP-Kpnα2核移位的阻止
利用基于荧光的筛选试验测试具有细胞杀伤能力的化合物A阻挡核输入的能力。该试验基于如下事实:Kpnα在与Kpnβ1的复合体中进入核。Kpnα2(Impα1或Rch1)带GFP标记(图8),并且其定位在用不同化合物处理后通过荧光显微法监测。预期GFP-Kpnα2定位将主要在核;并且在用有效阻挡Kpnβ1介导的核输入的化合物处理细胞后,GFP-Kpnα2将不能进入核并且将在细胞质中积累。利用这种化合物的处理导致GFP-Kpnα2从核错误定位至细胞质。化合物A抑制核输入。注意,化合物A自身具有荧光,因此必须通过分离发射光谱将化合物荧光信号从GFP-Kpnα2信号中排除。计数含有细胞质GFP-Kpnα2的细胞数(每个条件计数超过100个细胞),并且观察到在化合物A处理后显著增加(图9),以及GFP-Kpnα2核荧光信号显著减小(图10)。
内源性Kpnα2和NFY的核输入减少
为了确认化合物A阻挡核输入,进行独立试验,其中在用化合物A处理细胞后将核和细胞质蛋白部分分离。进行蛋白质印迹分析以确定内源性Kpnα2的定位,因为预期利用核输入有效抑制剂进行的处理将阻止内源性Kpnα2进入核。化合物A处理导致核中的Kpnα2量减少(图11)。但是,细胞质中的Kpnα2量未如预期地增加,这可能因为在不能进入核时Kpnα2的降解增强,或可选地因为其附着于核膜并且因此不被算在核或细胞质蛋白部分内。
独立的Kpnβ1货物NFY-A(Kahle J,Baake M,Doenecke D,Albig W.Subunits of the heterotrimeric transcription factor NF-Y are imported into the nucleus by distinct pathways involving importin beta and importin 13.Mol Cell Biol,2005年7月;25(13):5339-54)的定位也被进行了研究,并且类似地被发现在化合物A处理后在核部分中减少,支持了化合物A可能阻挡核输入的发现(图11)。重复实验中的Kpnα2和NFY水平的密度测定定量确认在化合物A处理后核蛋白水平显著降低(图12)。为了验证化合物A处理后所观察到的带型如同Kpnβ1抑制后的预期,利用siRNA使Kpnβ1沉默,并分离核和细胞质蛋白部分用于蛋白质印迹分析。在利用siRNA进行Kpnβ1抑制后,核中的Kpnα2量较少,支持了关于化合物A的结果。类似地,在Kpnβ1的siRNA转染后,核部分中的NFY量较少(图13和14)。
阻止转录因子进入核
由于Kpnβ1介导的核输入的抑制剂可能影响转录因子向核中的输入,在化合物A处理后进行荧光素酶试验以分析AP-1和p65核活性,因为转录因子已被证实依靠Kpnβ1进行核输入(Forwood JK,Lam MH,Jans DA.Nuclear import of Creb and AP-1transcription factors requires importin-beta 1and Ran but is independent of importin-alpha.Biochemistry,2001年5月1日;40(17):5208-17;Liang P,Zhang H,Wang G,Li S,Cong S,Luo Y等.KPNB1,XPO7and IPO8Mediate the Translocation of NF-kappaB/p65into the Nucleus.Traffic 2013年11月;14(11):1132-43)。在CaSki细胞的化合物A处理后测量AP-1和p65-荧光素酶活性,并且如预期,两者在处理后以剂量依赖性方式降低(图15)。
转录因子NFAT也以Kpnβ1依赖性方式(Ishiguro K,Ando T,Maeda O,Ohmiya N,Niwa Y,Goto H.Acetate inhibits NFAT activation in T cells via importin beta1interference.Eur J Immunol 2007年8月;37(8):2309-16)和Crm1依赖性方式(Kehlenbach RH,Dickmanns A,Gerace L.Nucleocytoplasmic shuttling factors including Ran and CRM1mediate nuclear export of NFAT In vitro.J Cell Biol,1998年5月18日;141(4):863-74)在核和细胞质之间穿梭。但是,其核-细胞质转运是独特的,因为其经由钙暴露来调控(Shibasaki F,Price ER,Milan D,McKeon F.Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4.Nature,1996年7月25日;382(6589):370-3),由此其主要在细胞质。但是,胞内钙水平增加导致其核积累。Soderholm等(2011)通过荧光显微法证实,响应钙离子载体——离子霉素的刺激,通过Kpnβ1抑制核输入会阻止NFAT-GFP的核积累。
在离子霉素和化合物A处理后,利用通过核NFAT活化的NFAT-荧光素酶报道基因进行核NFAT试验。利用佛波醇酯TPA共刺激NFAT,因为此前已证实TPA和离子霉素引起NFAT荧光素酶活性的协同刺激(Northrop JP,Ullman KS,Crabtree GR.Characterization of the nuclear and cytoplasmic components of the lymphoid-specific nuclear factor of activated T cells(NF-AT)complex.J Biol Chem 1993年2月5日;268(4):2917-23)。图15中所示的AP1和p65荧光素酶试验揭示了响应化合物A处理的内源性AP1和p65活性,而NFAT在HeLa细胞中被异位表达。
结果显示,用TPA和离子霉素刺激NFAT转染的HeLa细胞导致NFAT报道基因的激活由于NFAT的核输入而显著增加(图16)。利用化合物A、化合物II和化合物III的处理使NFAT活化以剂量依赖方式显著减少,可能是由于其抑制NFAT的核输入(图16、17和18)。作为阳性对照,用双氢除虫菌素处理细胞,双氢除虫菌素是一种广谱抗寄生虫剂,其近来被发现抑制核周蛋白α/β介导的核转运(Wagstraff等,2012;Wagstaff KM,Rawlinson SM,Hearps AC,Jans DA.An AlphaScreen(R)-based assay for high-throughput screening for specific inhibitors of nuclear import.J Biomol Screen,2011年2月;16(2):192-200)。NFAT试验利用双氢除虫菌素进行,并且与利用化合物A所获得的结果类似,NFAT活性的诱导在双氢除虫菌素处理后显著减少(图19)。最后,在用AG1478(一种EGF-R抑制剂)处理细胞后测量NFAT活化,没有观察到NFAT活化的变化(图20)。
通过细胞周期停滞和凋亡诱导的癌细胞死亡
为了鉴定具有抗癌活性的核输入抑制剂,利用不同来源的一组细胞系测试化合物A的细胞杀伤效果。在测试的细胞系中,化合物A没有显示对具体癌症类型的偏好,并且被发现以大约10μM的IC50杀死宫颈(HeLa、CaSki)、食管(WHCO1、WHCO5、WHCO6)、卵巢(A2780、CP70和OVCAR3)和乳腺来源(MDA-MB-231、MCF7)的癌细胞系(表11)。
表11.用化合物A处理的癌细胞、转化细胞和非癌细胞的IC50值。
重要地,化合物A显示,相对于非癌成纤维细胞系,在癌细胞系或转化细胞系中毒性升高。其显示对癌症或转化的细胞系的选择性相对于非癌成纤维细胞系高约2倍至3倍。
为了确认化合物A相比于非癌细胞更有效杀死癌细胞,使代表性癌细胞系(宫颈和食管)和非癌细胞系在存在化合物A的情况下生长,并且利用MTT试验每天测量细胞增殖五天。图21显示了癌细胞对化合物A高度敏感,因为在24小时内在10和15μM化合物A的浓度下观察到完全细胞死亡(图21A-D)。此效果持续整整5天时间,确认没有细胞群由该处理恢复。
但是,非癌成纤维细胞在存在10μM化合物A的情况下增殖相对正常,但在15μM化合物A下在24-48小时内发生细胞死亡(图21E和F)。结果指出癌和非癌细胞对化合物A的差别敏感性,其中在10μM化合物A处理时,癌细胞发生100%细胞死亡,而非癌细胞相对不受影响(图22)。虽然非癌细胞的IC50此前证实为在20-30μM范围内,但由于在细胞增殖试验中接种了较少细胞,该细胞在15μM时死亡。
响应化合物A处理测量癌细胞的锚地不依赖性增殖,因为以这种方式生长的细胞是更具代表性的体内肿瘤细胞。使细胞在多聚血红素(polyheme)涂覆板上生长,并且利用MTT试验监测处理后9天的细胞增殖。观察到,宫颈和食管癌细胞在存在10μM化合物A的情况下生长时均不能形成集落,不同于其未处理副本(图23),确认了化合物A同时阻挡癌细胞的锚地依赖性和锚地不依赖性增殖的能力。
对化合物A诱导的细胞死亡的机制进行了研究。进行细胞周期分析,并且发现化合物A处理导致细胞周期的G1阶段中细胞百分比增加,和在S和G2/M阶段中的细胞百分比相应减少(图24和25)。细胞周期阻滞的诱导表明凋亡可能被激活;因此通过蛋白质印迹分析测量聚(ADP-核糖)聚合酶(PARP-1)的裂解,作为凋亡的指示。PARP-1在凋亡期间裂解成89和24kDa的片段(Duriez PJ,Shah GM.Cleavage of poly(ADP-ribose)polymerase:a sensitive parameter to study cell death.Biochem Cell Biol 1997;75(4):337-49)。化合物A处理导致89kDa的裂解PARP的水平增加,与未裂解的Parp(113kDa)的存在减少相关(图26)。24kDa带是看不到的。
为了独立地确认凋亡诱导,研究细胞色素C亚细胞定位,因为其向细胞质中的释放是内在线粒体途径介导的凋亡的一个表征特征。用化合物A处理细胞,并且收集线粒体和细胞质蛋白部分用于蛋白质印迹分析。值得注意地,化合物A处理导致细胞色素C以剂量依赖方式从线粒体释放至细胞质中(图27),表明内在线粒体途径在化合物A处理后被激活并且造成癌细胞死亡。
抑制癌症异种移植模型中的肿瘤生长
化合物A显示体外细胞毒性,并且进一步发现其在体内具有抗癌活性——通过小鼠异种移植模型证明。将小鼠接种WHCO6癌细胞,并且在肿瘤达到可察觉尺寸后,每三天用媒介(二甲基亚砜(DMSO))或化合物A治疗小鼠,并且监测肿瘤尺寸三周。化合物A处理显著抑制肿瘤生长(图28)。小鼠的体重在用化合物A处理的过程中没有变化,并且小鼠呈现健康,表明没有经历不良副作用(图29)。
为了确认化合物A阻挡核输入的能力,用化合物A处理细胞,并且监测Kpnα2(在核输入途径中与Kpnβ1形成异源二聚体的辅助蛋白)的定位。因此Kpnα2很大程度上依赖于Kpnβ1以进行其核输入。而且,Kpnα2进入核的快速穿梭使其成为鉴定核输入抑制剂的特别有用的工具。在化合物A处理后检查外源和内源Kpnα2,发现其核定位在存在化合物A的情况下减少,表明化合物A为Kpnβ1核转运的抑制剂。但是,在化合物A处理后分析GFP-Kpnα2时发现,GFP-Kpnα2错误定位至细胞质的程度存在差异,例如,一些细胞在化合物A处理后仅显示细胞质GFP-Kpnα2,而其它细胞则显示核和细胞质GFP-Kpnα2。据报道Kpnα2-Kpnβ1相互作用的本质在细胞周期的不同阶段发生改变(Yasuhara N,Takeda E,Inoue H,Kotera I,Yoneda Y.Importin alpha/beta-mediated nuclear protein import is regulated in a cell cycle-dependent manner.Exp Cell Res,2004年7月1日;297(1):285-93)。因此,在实验中使用异步细胞可导致不同细胞中的不同Kpnα2定位。
此外,已证明Kpnα2可以以Kpnβ1非依赖性方式进入核,可以执行其其它非核转运功能,如其对有丝分裂进展的调控(Miyamoto Y,Hieda M,Harreman MT,Fukumoto M,Saiwaki T,Hodel AE,等人.Importin alpha can migrate into the nucleus in an importin beta-and Ran-independent manner.EMBO J,2002年11月1日;21(21):5833-42)。因此,在不存在Kpnβ1作用的情况下一些Kpnα2仍能够进入核是可能的。
还分析了化合物A处理后其它Kpnβ1依赖性货物蛋白的定位,包括转录因子NFY、AP-1、p65和NFAT,其全部都显示在化合物A处理后核输入减少。这些转录因子在此前研究中都已被证实依赖于Kpnβ1进行核输入,并且在Kpnβ1抑制后其核输入的抑制突出了核输入抑制可影响对癌细胞存活至关重要的细胞过程的程度。
由于发现化合物A抑制核输入,分析其细胞杀伤效果,并且发现癌细胞比非癌细胞对化合物A处理更为敏感。文献中存在大量证据证明靶向Crm1的核输出抑制剂呈现有效的抗癌活性,但对正常细胞仅有轻微影响(Etchin J,Sun Q,Kentsis A,Farmer A,Zhang ZC,Sanda T等.Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells.Leukemia,2013年1月;27(1):66-74;Mutka SC,Yang WQ,Dong SD,Ward SL,Craig DA,Timmermans PB等.Identification of nuclear export inhibitors with potent anti-cancer activity in vivo.Cancer Res,2009年1月15日;69(2):510-7;Pathria G,Wagner C,Wagner SN.Inhibition of CRM1-mediated nucleocytoplasmic transport:triggering human melanoma cell apoptosis by perturbing multiple cellular pathways.J Invest Dermatol,2012年12月;132(12):2780-90)。
此外,利用siRNA抑制Kpnβ1还导致选择性杀死癌细胞,由此在癌细胞中Kpnβ1沉默后诱导凋亡,而非癌细胞中的Kpnβ1抑制对细胞生物学仅具有轻微影响(Van der Watt等,2009)。由于癌症治疗的目标是促进癌细胞死亡,而对正常细胞不造成过多损伤,这种选择性有利于研发具有治疗潜力的抗核输入药物。选择性可源自癌细胞相对于正常细胞的核转运速率增加,因此对核转运机构(machinery)的依赖增加(Kuusisto等,2012)。可选地,对癌细胞的选择性可能是因为癌细胞相对于正常细胞“准备好(primed)”发生凋亡(更接近凋亡阈值)(Ni Chonghaile T,Sarosiek KA,Vo TT,Ryan JA,Tammareddi A,Moore VG,等人.Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy.Science,2011年11月25日;334(6059):1129-33)。
核输入的抑制与癌细胞中多种凋亡标志的增加有关。这些包括G1细胞周期停滞、Parp的裂解和细胞色素C从线粒体向细胞质中的释放。细胞色素C释放是内在线粒体介导凋亡途径的特征,表明化合物A处理导致内在凋亡途径激活。在白血病细胞中的Crm1抑制(Etchin等,2013)以及宫颈癌细胞中利用siRNA的Kpnβ1抑制(Angus等,2014)后,类似地诱导了内在凋亡途径。凋亡可能在核输入抑制并且因此细胞稳态破坏后通过关键蛋白的错误定位被诱导。因此,化合物A至K是新型核输入抑制剂,并且其显示体外抗癌活性。化合物A显示体外和体内抗癌活性。
新型核输入抑制剂具有抗癌效果。具体地,发现上述式I至XVI的化合物显示抗癌效果。已证明化合物A通过抑制核输入和诱导细胞凋亡而显示抗癌活性。本领域技术人员将理解,化合物B至K通过与化合物A相同或相似的作用机制诱导细胞凋亡。已证实化合物II和化合物III具有与核输入抑制相关的抗癌活性。已证明以上表5中所列出的化合物V至XVI具有抗癌活性。
因此提供了通式I化合物(包括化合物A至K)和化合物II至XIV、其立体异构体或盐在治疗癌症中的应用。更具体地,提供了化合物A至K和化合物II至XIV在治疗癌症——具体地,但是不限于,宫颈、食管、卵巢和乳腺的癌症——中的应用。
更具体地,预期通式I化合物——其中R1是分支或无分支的、任选地用选自胺、咪唑、醇或吗啉的取代基官能化的C2-C5烷基;并且其中R2是氢或甲基——和化合物II-XVI、其立体异构体或盐可用于治疗异常生长或肿瘤。
通式I化合物——其中R1是分支或无分支的、任选地用选自胺、咪唑、醇或吗啉的取代基官能化的C2-C5烷基;并且其中R2是氢或甲基——和化合物II-XVI、其立体异构体或盐可用于治疗选自下列的恶性肿瘤或癌症:胃的癌症、肺的癌症、肝的癌症、黑素瘤、何杰金氏病、非何杰金氏淋巴瘤、急性或慢性淋巴细胞性白血症、多发性骨髓瘤、成神经母细胞瘤、乳腺癌、卵巢癌、肺癌、维尔姆斯氏瘤、宫颈癌、睾丸癌、软组织肉瘤、原发性巨球蛋白血症、膀胱癌、慢性粒细胞性白血病、原发性脑癌、恶性黑素瘤、小细胞肺癌、胃癌、结肠癌、恶性胰腺胰岛素瘤、恶性类癌癌、恶性黑素瘤、绒毛膜癌、蕈样霉菌病、头或颈癌、骨肉瘤、胰腺癌、急性粒细胞性白血病、毛细胞白血病、成神经母细胞瘤、横纹肌肉瘤、卡波西氏肉瘤、泌尿生殖癌、甲状腺癌、食管癌、恶性高钙血综合症、宫颈增生、肾细胞癌、子宫内膜癌、真性红细胞增多症、特发性血小板增多症、肾上腺皮质癌、皮肤的癌症、前列腺癌、喉的癌症、外阴的癌症和睾丸的癌症。
这种用于治疗癌症的应用需要化合物A至K通过在Kpnβ1上结合核周蛋白α(Kpnα)和GTP结合核蛋白Ran的的重叠结合位点处结合细胞中的Kpnβ1来抑制核转运蛋白Kpnβ1的活性。在治疗癌症中,化合物A至K、化合物II和化合物III在癌细胞中选择地抑制蛋白质和转录因子Kpnα、AP-1、P65、NFAT向细胞核中的输入并且诱导细胞凋亡。
化合物A至K和化合物II至XIV、其立体异构体或盐中的任一种、或化合物A至K和化合物II至XIV、其立体异构体或盐中的任意两种或更多种的组合的治疗有效浓度根据化合物而变化,但小于100μM,优选小于50μM,最优选约10μM。
式I至XIV化合物、其立体异构体或药学上可接受的盐可以以任何适当方式被给予对其有需要的患者,并且可构成药物的活性成分。这种药物可包括其它成分,包括佐剂和载体。
式I至XIV化合物、其立体异构体或盐中的任一种、或式I至XIV化合物、其立体异构体或盐中的任意两种或更多种的组合可以被用作治疗癌症的药物,并且也可以用于制备用于治疗癌症的药物。这种药物可以具有任何适当形式。
因此,还提供治疗癌症的方法,其包括给予对其有需要的患者治疗有效量的式I至XIV化合物、其立体异构体或盐中的任一种、或式I至XIV化合物、其立体异构体或盐中的任意两种或更多种的组合。
考虑宫颈、食管、卵巢和乳腺的癌症具体地可利用式I至XIV化合物、其立体异构体和/或药学上可接受的盐中的一种或多种来治疗,但是展望上文列举的其它形式的癌症也可用这些化合物治疗。
贯穿说明书和权利要求书,除非内容另有要求,否则用词“包括”或变型如“包含”或“含有”将被理解为隐含包括所述整数或整数组,而非排除任何其它整数或整数组。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种靶向锚定剂及其制备方法和应用与流程
- 一种超低分子量插层抑制剂及其制备方法与流程
- 具有胶原特异结合能力的神经再生抑制分子的拮抗蛋白CBD-PlexinB1LBD及应用的制作方法与工艺
- 具有胶原特异结合能力的神经再生抑制分子的拮抗蛋白CBD-EphA4LBD及应用的制作方法与工艺
- 一种KDM1A小分子抑制剂在抑制肿瘤细胞生长和转移中的应用的制作方法与工艺
- 一种抑制多药耐受基因表达的靶向抗肿瘤纳米药物的制作方法与工艺
- MALT1靶向抑制物在制备MALT1依赖性肿瘤治疗药物中的应用的制作方法与工艺
- 作为ASGPR靶向剂的被取代的‑6,8‑二氧杂双环[3.2.1]辛烷‑2,3‑二醇化合物的制作方法与工艺
- 小分子C‑Myc抑制剂的制作方法与工艺
- 一种RNA抑制剂及其在肿瘤中的应用的制作方法与工艺