(2S,3S)‑7‑氟‑2‑(4‑氟苯基)‑3‑(1‑甲基‑1H‑1,2,4‑三唑‑5‑基)‑4‑氧代‑1,2,3,4‑四氢喹啉‑5‑羧酸甲酯的共形成体盐及其制备方法与流程
本申请涉及(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体(coformer)盐,其任选呈溶剂化物形式且额外地任选呈水合物形式,包括结晶形式;以及制备(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯共形成体盐的方法。
背景技术:
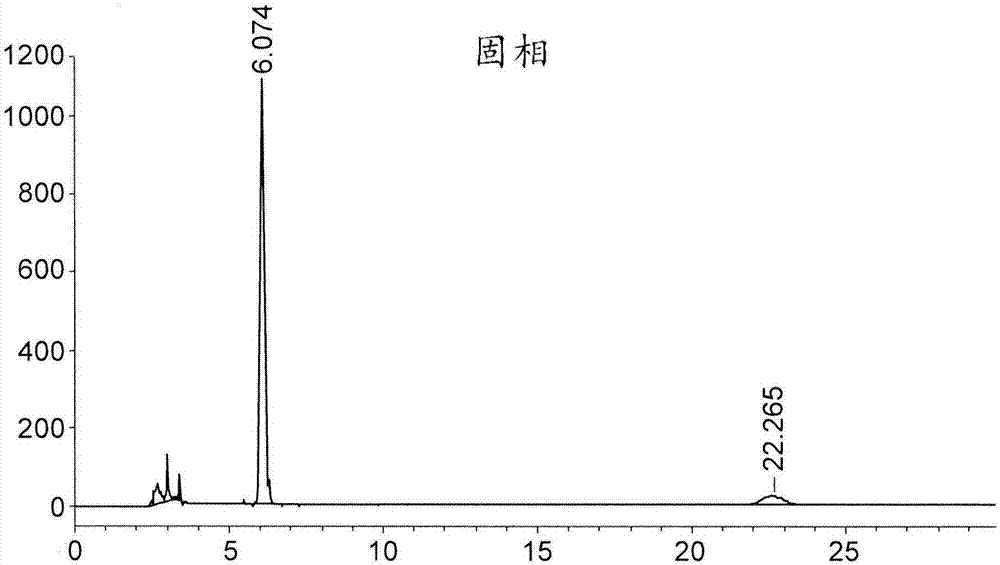
:化合物(8s,9r)-5-氟-8-(4-氟苯基)-9-(1-甲基-1h-1,2,4-三唑-5-基)-8,9-二氢-2h-吡啶并[4,3,2-de]酞嗪-3(7h)-酮甲苯磺酸盐(化合物(a))为聚(adp-核糖)聚合酶(parp)的抑制剂。其制备方法描述于wo2010017055、wo2011097602和wo2012054698。然而,所公开的合成途径需要在制备化合物(a)的途径中的一种合成中间体7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯(中间体(a))进行手性色谱,从而产生手性纯的(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯(化合物(1))使用常规手性色谱通常需要较多溶剂及时间。使用诸如模拟移动床(smb)色谱的更有效的色谱方法仍需要使用昂贵的手性色谱树脂,且对于大规模纯化药物化合物并不可行。此外,色谱过程中长时间保持化合物(1)处于溶液中会导致化合物(1)中9位上的差向异构化及甲酯基团的断裂。用一个或多个结晶步骤替换色谱步骤来纯化化合物(1)是理想的且克服这些问题。因此,希望找到使用手性色谱分离的替代方案以获得对映异构的化合物(1)。本申请披露了(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐及其制备方法,这解决了所述困难。本申请所述的实施方案可显著提高所需化合物的纯度,且可在制备化合物(a)中赋予实现监管批准及销售的额外优点。本申请所述的实施方案允许更一致地生产符合监管机构针对经批准的药品的纯度的标准及准则的化合物。还可在制造时间及费用方面实现明显降低。可实现化合物(1)的“顺/反”异构杂质的显著减少(其中顺式异构体为(2r,3s)及(2s,3r)形式,且反式异构体为(2r,3r)形式)。可实现化合物(1)的较高程度的对映异构选择性。附图说明图1描述了使用xrpd操作2获得的实施例1和3的步骤1a化合物(1a)的xrpd。图2a和2b描述了实施例3中步骤1a化合物(1a)的手性hplc。图3描述了实施例3中步骤1a化合物(1a)的1hnmr。图4描述了实施例3中步骤1a化合物(1a)的tga/dsc。图5描述了使用xrpd操作2获得的实施例3中步骤1b化合物(1a)(上)以及来自实施例1的化合物(1a)的xrpd。图6描述了实施例3中步骤1b化合物(1a)的手性hplc。图7描述了实施例3中步骤2化合物(1)以及中间体(a)的xrpd。图8描述了实施例3中化合物(1)以及中间体(a)的1hnmr。图9描述了使用xrpd操作2获得的实施例5中化合物(1b)、来自实施例1的化合物(1b)以及中间体(a)的xrpd。图10描述了实施例5中化合物(1b)的手性hplc。图11描述了实施例5中化合物(1b)的1hnmr。图12a描述了实施例5中化合物(1b)的tga和dsc。图12b描述了实施例5中化合物(1b)(下)以及实施例1中化合物(1b)的dsc。图13a描述了实施例4中化合物(1a)的1hnmr(在dmso-d6中)。图13b描述了实施例4中化合物(1a)的13cnmr(在dmso-d6中)。图14描述了实施例4中化合物(1a)的ir光谱。图15描述了实施例4中化合物(1a)的dsc。图16描述了实施例4中化合物(1a)的手性hplc。图17a描述了实施例4中化合物(1)的1hnmr(在dmso-d6中)。图17b描述了实施例4中化合物(1)的13cnmr(在dmso-d6中)。图18描述了实施例4中化合物(1)的ir光谱。图19描述了实施例4中化合物(1)的dsc。图20描述了实施例4中化合物(1)的手性hplc。技术实现要素:在一个方面,本申请提供了(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐,其任选呈其溶剂化物且额外地任选呈其水合物的形式。在某些实施方案中,所述共形成体盐呈基本上纯的结晶形式。在某些实施方案中,所述共形成体盐为(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的[(1s)-内]-(+)-3-溴-10-樟脑磺酸盐。在某些实施方案中,所述共形成体酸为[(1s)-内]-(+)-3-溴-10-樟脑磺酸。在某些实施方案中,所述共形成体盐为显示出以下至少一种的结晶形式:在210.3、25.3、21.8、20.8、19.5和18.5ppm±0.2ppm具有峰的固态13cnmr谱;具有在25℃和90℃之间的宽的吸热以及最大值在约135℃和147℃之间的吸热的差示扫描量热分析热分析图;指示为溶剂化物质的热重量分析热分析图;或在6.7、9.7、18.5、19.5和22的2θ角度±0.22θ角度包含峰的x射线粉末衍射图。在一些实施方案中,所述共形成体盐呈显示出以下至少一种的结晶形式:在210.3、25.3、21.8、20.8、19.5和18.5ppm±0.2ppm具有峰的固态13cnmr谱;或在6.7、9.7、18.5、19.5和22的2θ角度±0.22θ角度包含峰的x射线粉末衍射图。在一些实施方案中,所述共形成体盐为(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的(s)-1-苯基乙磺酸盐。在一些实施方案中,所述共形成体酸为(1s)-苯基乙磺酸盐。在另一方面,本申请提供了制备(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐的方法,包括(1)在选自mibk、mek、乙醇和水的一种或多种步骤1a)溶剂中在高温用共形成体处理7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯,形成步骤1a)溶液;(2)使所述步骤1a)溶液在足以使所述共形成体盐以结晶形式析出的条件下静置;和(3)分离呈结晶形式的共形成体盐。在某些实施方案中,所述共形成体盐为化合物(1)的[(1s)-内]-(+)-3-溴-10-樟脑磺酸盐,且所述步骤1a)溶剂选自丙酮、甲基乙基酮、甲基异丁基酮(mibk)、甲醇、乙醇、丙醇、异丙醇和丁醇。在某些实施方案中,所述共形成体盐为化合物(1)的[(1s)-内]-(+)-3-溴-10-樟脑磺酸盐,且所述步骤1a)溶剂为mibk、水和乙醇。在某些实施方案中,所述共形成体盐为化合物(1)的[(1s)-内]-(+)-3-溴-10-樟脑磺酸盐,且所述步骤1a)溶剂为mibk和乙醇。在某些实施方案中,所述方法还包括在一种或多种步骤1b)溶剂中使所述共形成体盐重结晶或重新浆化(reslurry)。在某些实施方案中,所述(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐在步骤1b)溶剂中重结晶或重新浆化后呈结晶形式。在某些实施方案中,所述方法还包括在室温或高温将(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐悬浮于选自水、丙酮、ipa或甲醇的一种或多种步骤2a)溶剂中,形成步骤2a)溶液,并且用选自naoh、nh3(任选25%nh3水溶液)、naco3、naoac或nahco3的碱处理所述步骤2a)溶液;使所述步骤2a)溶液在足以使所述(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的结晶形式析出的条件下静置;和分离(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的结晶形式。在某些实施方案中,所述步骤2a)溶剂选自丙酮、甲基乙基酮、甲基异丁基酮、甲醇、乙醇、丙醇或异丙醇;且所述碱为nh3水溶液。在某些实施方案中,所述步骤2a)溶剂为丙酮、甲醇和2-丙醇;且所述碱为nh3水溶液。在某些实施方案中,所述步骤2a)溶剂为丙酮、甲醇和异丙醇;且所述碱为nh3水溶液。在某些实施方案中,所述方法还包括将(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯在一种或多种步骤2b)溶剂中重结晶或重新浆化。在某些实施方案中,所述(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯在步骤2b)溶剂(s)中重结晶或重新浆化后呈结晶形式。在另一方面,本申请提供了化合物(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯,其任选呈溶剂化物形式且额外地任选呈水合物形式,其通过如下制备:用碱处理(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐,并分离(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯。具体实施方式缩写缩写含义acn乙腈dcm二氯甲烷dmfn,n-二甲基甲酰胺dsc差示扫描量热分析ea乙酸乙酯e.e.对映异构体过量etoh乙醇equiv当量g克ipa异丙醇ir红外mhz兆赫mek甲基乙基酮mibk甲基异丁基酮ml毫升mol摩尔naoh氢氧化钠nmr核磁共振tga热重量分析thf四氢呋喃xrpdx射线粉末衍射定义为便于理解本申请所述的本发明,下文定义多个术语。一般而言,本申请所使用的命名法及本申请所述的有机化学、药物化学及药理学中的实验室操作为本领域中熟知且常用的。除非另外规定,否则本申请所用的所有技术及科学术语一般具有与本发明所属领域的技术人员通常所理解相同的含义。在针对本申请所用的术语存在多个定义的情况下,除非另外说明,否则以此部分中的那些定义为准。如本申请及随附权利要求中通篇所使用,下列术语具有下列含义:除非内容另外明确指出,否则如本申请所用的单数形式“一个”、“一种”及“所述”包括复数个提及物。因此,举例而言,提及“化合物”包括两种或更多种化合物的混合物等。如本申请所用,且除非另有规定,否则术语“约(about)”及“大约(approximately)”当与组合物或剂型的成分的剂量、量或重量百分比结合使用时,意指本领域普通技术人员所认可的提供与由指定剂量、量或重量百分比获得的等效的药理学作用的剂量、量或重量百分比。在某些实施方案中,术语“约”及“大约”在用于此情境时涵盖在指定剂量、量或重量百分比的15%以内、10%以内、5%以内、4%以内、3%以内、2%以内、1%以内或0.5%以内的剂量、量或重量百分比。如本申请所用,且除非另有规定,否则术语“约”及“大约”当与提供用于描述特定固体形式的数值或值范围(例如特定温度或温度范围,诸如描述熔化、脱水、去溶剂化或玻璃化转变;质量变化,诸如作为温度或湿度的函数的质量变化;溶剂或水含量,以例如质量或百分率表示;或峰位置,诸如在13cnmr、dsc、tga及xrpd分析中)结合使用时表明,该值或值范围可偏离至本领域普通技术人员认为合理但仍描述特定固体形式的程度。在某些实施方案中,术语“约”及“大约”在此情境下使用时,指示数值或值范围可变化所述值或值范围的5%、4%、3%、2%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%或0.1%但仍描述特定固体形式。术语“无定形”或“无定形形式”意指例如通过xrpd所测定,所述物质、组分或产物并非实质上结晶的,或当用显微镜检视时,所述物质、组分或产物例如并非为双折射的。在某些实施方案中,包含无定形形式的物质的样品可能实质上不含其他无定形形式和/或结晶形式。术语“结晶形式”或“晶体形式”是指化合物的结晶固体形式,包括但不限于单一组分或多种组分晶体形式(例如化合物的多晶型物);或溶剂化物、水合物、包合物(clathrate)、共晶体、化合物的盐或其多晶型物。本申请中术语“晶体形式”及相关术语是指给定物质的各种结晶变型,包括但不限于多晶型物、溶剂化物、水合物、共晶体及其他分子络合物以及盐、盐的溶剂化物、盐的水合物、盐的其他分子络合物及其多晶型物。可通过如本领域中已知的多种方法获得物质的晶体形式。所述方法包括但不限于熔化重结晶、熔化冷却、溶剂重结晶、受限空间中(诸如在纳米孔或毛细管中)重结晶、表面或模板上(诸如聚合物上)重结晶、在添加剂(诸如共晶抗衡分子)存在下重结晶、去溶剂化、脱水、快速蒸发、快速冷却、缓慢冷却、气相扩散、升华、研磨及溶剂滴落研磨。用于表征晶体形式及无定形形式的技术包括但不限于tga、dsc、xrpd、单晶x射线衍射测定法、振动光谱法(例如ir及拉曼光谱法)、固态nmr、光学显微法、热载台光学显微法、sem、电子结晶及定量分析、psa、表面积分析、溶解性(solubility)研究及溶解(dissolution)研究。如本申请所用且除非另外指明,否则术语“水合物”意指化合物或其盐,其进一步包括化学计量或非化学计量量的通过非共价分子间力结合的水。如本申请所用且除非另外指明,否则术语“溶剂化物”意指由一个或多个溶剂分子与本申请提供的化合物或其盐的缔合而形成的溶剂化物。术语“溶剂化物”包括水合物(例如半水合物、一水合物、二水合物、三水合物、四水合物等)。术语“多晶型物”或“多晶型形式”意指包含相同的一种或多种分子或离子的两种或多种晶体形式中的一种。由于分子或离子在晶格中的排列或构象不同,不同多晶型物可具有不同物理性质,诸如熔化温度、溶解热、溶解度、溶解速率和/或振动光谱。多晶型物所显示的物理性质的差异可影响以下药物参数:诸如储存稳定性、可压缩性、密度(在配制及产品制造中至关重要)及溶出速率(生物利用度中的重要因素)。稳定性的差异可由化学反应性的变化(例如差异氧化,使得包含一种多晶型物的剂型相比包含另一多晶型物的剂型较快褪色)、机械变化(例如片剂在储存时由于动力学上有利的多晶型物转变成热力学上更稳定的多晶型物而破碎)或两者(例如一种多晶型物的片剂更易于在高湿度下破裂)引起。由于溶解性/溶解差异,在极端情况下,一些多晶型转变可导致缺乏效能,或在其他极端情况下产生毒性。另外,结晶形式的物理性质在加工中可能至关重要;举例而言,一种多晶型物可能更有可能形成溶剂化物,或可能难以过滤并洗去杂质(例如,多晶型物之间的粒子形状及尺寸分布可能不同)。如本申请所用的“基本上纯的”是指物质或混合物基本上不含其他化合物、立体异构体、共形成体盐、溶剂化物、水合物或其其他固体形式,包括其他结晶或无定形形式。在某些情况下,“基本上纯的”化合物,诸如基本上纯的(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯或其共形成体盐或溶剂化物可指基本上不含其他化合物,例如可存在于制备所需化合物的方法中的未反应的前体及副产物。在其他情况中,如本申请所用的(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯或其盐或溶剂化物的“基本上纯的”固体形式(例如晶体形式或无定形形式)可指基本上不含(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯或其盐或溶剂化物的其他固体形式。在某些情况下,“立体异构纯的”意指组合物包含化合物的一种立体异构体且基本上不含所述化合物的其他立体异构体。如本申请所用的术语“体积(vol)”是指固体反应物与液体溶剂的重量/体积比。举例而言,10体积的溶剂中250g的固体物质意指该物质溶解于10×250ml或2.5l的溶剂中。应理解的是,(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐包含(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的阳离子(例如,在一个实施方案中,在一个原子位置处质子化,或在其他实施方案中,在多于一个原子位置处质子化)及该共形成体酸的阴离子。实施方案以下段落提出本申请所披露的化合物及方法的多种实施方案且并不意在限制。在一个方面,本发明提供了(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐(此后称为“化合物(1)的共形成体盐”),其任选呈其溶剂化物且额外地任选呈其水合物的形式。在某些实施方案中,所述共形成体盐包含手性酸的阴离子。在某些实施方案中,所述手性酸选自表1。在某些实施方案中,所述手性酸为[(1s)-内]-(+)-3-溴-10-樟脑磺酸或(1s)-苯基乙磺酸。在某些实施方案中,所述共形成体盐为(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的[(1s)-内]-(+)-3-溴-10-樟脑磺酸盐(此后所述共形成体盐称为“化合物(1a)”),其任选呈其溶剂化物且额外地任选呈其水合物的形式。在某些实施方案中,所述共形成体盐为(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的(s)-1-苯基乙磺酸盐(此后所述共形成体盐称为“化合物(1b)”),其任选呈其溶剂化物且额外地任选呈其水合物的形式。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)包含约1:1的阳离子与阴离子的摩尔比。在某些实施方案中,所述阳离子与阴离子的摩尔比为约1:1.1、约1:1.15、约1:1.2或约1:1.3。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)为非溶剂化的。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)为其溶剂化物。在某些实施方案中,所述溶剂化物形式为其水合物。在某些实施方案中,所述溶剂化物形式为其乙醇化物溶剂化物。在某些实施方案中,所述溶剂化物形式为其乙醇化物溶剂化物和水合物。在某些实施方案中,所述化合物(1)的共形成体盐或化合物(1a)或化合物(1b)与乙醇溶剂化物的比为约1:0.4、约1:0.5、约1:0.6或约1:0.7。在某些实施方案中,所述化合物(1)的共形成体盐或化合物(1a)或化合物(1b)与水合物的比为约1:0.4、约1:0.5、约1:0.6或约1:0.7。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物呈固体形式。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物为非结晶的。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物呈晶体形式、无定形形式或其混合物。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)的乙醇化物溶剂化物、水合物或其混合物呈晶体形式、无定形形式或其混合物。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物呈无定形形式。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)的乙醇化物溶剂化物、水合物或其混合物呈无定形形式。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物呈结晶形式。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)的乙醇化物溶剂化物、水合物或其混合物呈结晶形式。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物为基本上纯的。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物的固体形式或晶体形式为基本上纯的。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物的晶体形式为基本上纯的。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)的乙醇化物溶剂化物、水合物或其混合物为基本上纯的。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物为立体化学纯的。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物的固体形式或晶体形式为立体化学纯的。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物的晶体形式为立体化学纯的。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)的乙醇化物溶剂化物、水合物或其混合物其为立体化学纯的。在某些实施方案中,所述基本上纯的共形成体盐包含基本上纯的(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯,其基本上不含其他立体异构体包括例如(2r,3r)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯、(2s,3r)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯和(2r,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)包含化合物(1)的约100重量%的特定立体异构体,其中所述百分比是基于立体化学纯的共形成体盐中组合的立体异构体的总量。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物包含大于约80重量%的化合物(1)且小于约20重量%的化合物(1)的任何立体异构体、大于约90重量%的化合物(1)且小于约10重量%的化合物(1)的任何立体异构体、大于约95重量%的化合物(1)且小于约5重量%的化合物(1)的任何立体异构体、大于约97重量%的化合物(1)且小于约3重量%的化合物(1)的任何立体异构体、大于约99重量%的化合物(1)且小于约1重量%的化合物(1)的任何立体异构体或大于约99.5重量%的化合物(1)且小于约0.5重量%的化合物(1)的任何立体异构体。上述百分比是基于立体化学纯的共形成体盐中组合的立体异构体的总量。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物基本上不含一种或多种其他特定晶体形式、无定形形式和/或其他化合物。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物包含小于约10重量%、小于约5重量%、小于约3重量%、小于约2重量%、小于约1重量%、小于约0.75重量%、小于约0.5重量%、小于约0.25重量%或小于约0.1重量%的(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的一种或多种其他晶体形式或无定形形式和/或可由本申请披露的合成方法所产生的其他化合物。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)的结晶形式基本上不含无定形形式。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物的结晶盐纯度为至少约90重量%、至少约95重量%、至少约97重量%、至少约98重量%、至少约99重量%、至少约99.2重量%、至少约99.5重量%、至少约99.6重量%、至少约99.7重量%或至少约99.8重量%的单一结晶形式,总重量的其余部分可为其他结晶或无定形形式和/或其他化合物。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物的结晶形式基本上为单一组分结晶形式或单一多晶型物。在某些实施方案中,所述化合物(1)的共形成体盐以及化合物(1a)和(1b)及其溶剂化物和水合物的结晶形式为多组分结晶形式,其包含这些共形成体盐的第一结晶形式及这些共形成体盐的至少一种其他结晶和/或无定形形式。在某些实施方案中,所述共形成体盐为具有如下xrpd图的结晶化合物(1a),其包含选自具有根据图1或图5的2θ角度的峰的一个或多个(例如,1个、2个、3个、4个、5个、6个、7个、8个、9个、10个或大于10个;或至少3个、至少4个、至少5个、至少6个或至少7个)特征峰。在某些实施方案中,所述结晶化合物(1a)的xrpd图包含一个或多个(例如1个、2个、3个、4个、5个或至少2个、至少3个或至少4个)选自具有约6.7、9.7、18.5、19.5及22的2θ角度±0.22θ的峰的特征峰。在某些实施方案中,所述结晶化合物(1a)的xrpd图包含选自具有约6.7及9.7的2θ角度±0.22θ的峰的特征峰。在某些实施方案中,所述结晶化合物(1a)的xrpd图基本上如图1或图5中所提供。在某些实施方案中,所述共形成体盐为具有基本上对应于图13b中的谱的13cnmr谱或具有基本上对应于表a中那些峰的峰的谱的结晶化合物(1a),其中具有2个峰的条目表示双重峰:表a在某些实施方案中,所述结晶化合物(1a)的13cnmr谱包含一个或多个峰(例如,至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少11个或至少12个峰),其为选自在约210.3、58.1、56.0、54.7、48.6、47.0、46.3、40.6、25.3、21.8、20.8、19.5及18.5约±0.2ppm处的峰。在某些实施方案中,所述结晶化合物(1a)13cnmr谱包含在约210.3、25.3、21.8、20.8、19.5及18.5约±0.2ppm处的一个或多个峰(例如,至少2个、至少3个、至少4个或至少5个峰)。在某些实施方案中,所述共形成体盐为基于差示扫描量热分析在25℃与约90℃之间具有宽的吸热峰及在约135℃与150℃之间、约140℃与150℃之间或约143℃与147℃之间具有最大值的吸热的结晶化合物(1a)。在某些实施方案中,结晶化合物(1a)具有在约135℃与150℃之间、约140℃与150℃之间或约143℃与147℃之间具有最大值的吸热。在某些实施方案中,所述共形成体盐为具有基本上对应于图4或图15的dsc温度记录图(thermograph)的dsc热分析图(thermogram)的结晶化合物(1a)。在某些实施方案中,所述共形成体盐为具有指示为溶剂化物质的tga热分析图的结晶化合物(1a)。在某些实施方案中,结晶化合物(1a)具有基本上对应于图4的tga温度记录图的tga热分析图。在某些实施方案中,结晶化合物(1a)具有如下tga热分析图,即当由约25℃加热至约90℃的温度时,其呈现逐步重量损失(例如,约2.5%与4.5%之间、约3%与4%之间、约3.5%)。在某些实施方案中,结晶化合物(1a)具有如下tga热分析图,即当由约90℃加热至约160℃的温度时,其呈现逐渐重量损失(例如,约0.5%与2%之间、约0.75%与1.75%之间、约1%与1.5%之间、约1.2%)。在某些实施方案中,所述共形成体盐为具有以下至少一种的结晶化合物(1a):i.在210.3、25.3、21.8、20.8、19.5和18.5ppm±0.2ppm具有峰的固态13cnmr谱;ii.具有在25℃和90℃之间的宽的吸热以及最大值在约135℃和147℃之间的吸热的差示扫描量热分析热分析图;iii.指示为溶剂化物质的热重量分析热分析图;或iv.在6.7、9.7、18.5、19.5和22的2θ角度±0.22θ角度包含峰的x射线粉末衍射图。在某些实施方案中,所述结晶化合物(1a)具有以下的至少一种:i.在210.3、25.3、21.8、20.8、19.5和18.5ppm±0.2ppm具有峰的固态13cnmr谱;或ii.在6.7、9.7、18.5、19.5和22的2θ角度±0.22θ角度包含峰的x射线粉末衍射图。在某些实施方案中,所述共形成体盐为(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的(s)-1-苯基乙磺酸盐(化合物(1b))。在另一方面,本发明提供了基本上纯的(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯(化合物(1)),其通过如下制备:用碱处理化合物(1)的共形成体盐并分离(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯(化合物(1))。在某些实施方案中,所述分离的化合物(1)任选重结晶。制备化合物的方法本申请提供了制备化合物(1)及其共形成体盐的方法。在某些实施方案中,相比于先前报道的(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的合成,所述方法可提供例如改善的产物回收率、产物纯度和/或用于大规模生产的顺应性。在某些实施方案中,任选呈其溶剂化物形式且额外地任选呈水合物形式的(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐以结晶形式制备,产生的化合物(1)的纯度相比于通过手性色谱分离的化合物(1)而言更高。在某些实施方案中,相比于使用手性色谱制备而言,所述使用共形成体的化合物(1)的制备更能顺应大规模生产。方案a提供了用于制备化合物(1)的共形成体盐的方法的示例性阐述。方案a在步骤1a)中,中间体(a)可在室温或在高温(高于室温的温度)在一种或多种步骤1a)溶剂中溶解,其中所述溶剂足以溶解中间体(a)。在某些实施方案中,所述高温为约30℃、约35℃、约40℃、约45℃、约48℃、约50℃、约52℃、约55℃、约60℃、约65℃或约70℃。在某些实施方案中,所述步骤1a)溶剂为c1-6酮、c1-6醇、乙酸乙酯(“ea”)、四氢呋喃(“thf”)、甲苯、乙腈(“acn”)、庚烷、二噁烷或水;或它们的组合。在某些实施方案中,所述c1-6酮为丙酮、甲基乙基酮(“mek”)或甲基异丁基酮(“mibk”)。在某些实施方案中,所述c1-6醇为甲醇、乙醇、丙醇、异丙醇或丁醇。在某些实施方案中,所述c1-6醇为甲醇、乙醇或异丙醇。在某些实施方案中,所述步骤1a)溶剂为乙醇和mibk;或所述溶剂为乙醇、mibk和水。在某些实施方案中,所述mibk/乙醇比为5-20/1;或所述比为5/1或6/1或7/1或8/1或9/1或10/1或11/1或12/1或15/1或20/1。在某些实施方案中,所述mibk/乙醇比为9:1。在某些实施方案中,所述mibk/乙醇/水比为10-15/1-1.5/0.1-0.05;或所述比为12-13/1-1.5/0.1-0.05。在某些实施方案中,所述mibk/乙醇/水比为13/1.5/0.1;或为13/1.5/0.05;或为13/1/0.1;或为13/1/0.05;或为12/1.5/0.1;或为12/1.5/0.05;或为12/1/0.1;或为12/1/0.05。在某些实施方案中,在步骤1a)中,中间体(a)可在高温(例如在约30℃、约35℃、约40℃、约45℃、约48℃、约50℃、约52℃、约55℃、约60℃、约65℃或约70℃)在诸如丙酮、ipa、ea、thf、dmf、甲苯、acn、庚烷、二噁烷、水、mibk、mek或乙醇或它们的组合的一种或多种步骤1a)溶剂中溶解,形成步骤1a)溶液。在某些实施方案中,所述步骤1a)溶剂为mibk、mek、水和/或乙醇。在某些实施方案中,所述mibk:mek:乙醇/水比为20-40:10-20:1-10。在某些实施方案中,所述mibk:mek:乙醇/水比为10-30:20-30:1-5。在某些实施方案中,所述步骤1a)溶剂为mibk、水和/或乙醇。在某些实施方案中,所述步骤1a)溶剂为mibk:乙醇:水,其比为30-50:5-10:1-5或35-45:6-7:1-2或40:6.5:1.6。在某些实施方案中,所述mibk:乙醇:水比为120-130:10-15:0.5-1。在某些实施方案中,所述步骤1a)溶剂为mibk:乙醇,其比为5-20:1或10-20:1或20:1或19:1或18:1或10:1或9:1或8:1。在某些实施方案中,所述步骤1a)溶剂为乙醇和mek。在某些实施方案中,所述乙醇:mek的比为85-99:1-15,或为90-99:1-10,或为95-99:1-5,或为95:5,或为96:4,或为97:3,或为98:2。在某些实施方案中,中间体(a)溶于约5体积的一种或多种步骤1a)溶剂、约7体积的一种或多种步骤1a)溶剂、约10体积的一种或多种步骤1a)溶剂、约12体积的一种或多种步骤1a)溶剂、约14体积的一种或多种步骤1a)溶剂、约16体积的一种或多种步骤1a)溶剂或约20体积的一种或多种步骤1a)溶剂。可添加共形成体酸(约1摩尔当量)并将其溶解于步骤1a)溶液中,以制备步骤1a)共形成体溶液。化合物(1)的共形成体盐的固体形式可通过如下获得:将步骤1a)共形成体溶液用化合物(1)的共形成体盐的结晶种晶,或将步骤1a)共形成体溶液冷却至约室温、约20℃、约15℃、约10℃、约5℃、约0℃、约-5℃、约-10℃或约-15℃。一旦形成固体化合物(1)的共形成体盐,其可经滤过收集,任选用步骤1a)溶剂洗涤,并干燥。在步骤1b)中,化合物(1)的共形成体盐可重新悬浮于步骤1b)溶剂中,以形成步骤1b)溶液。在某些实施方案中,所述步骤1b)溶剂为与一种或多种步骤1a)溶剂相同的溶剂。在某些实施方案中,化合物(1)的共形成体盐在高温(例如在约30℃、约35℃、约40℃、约45℃、约50℃、约55℃、约60℃、约65℃、约70℃)重新悬浮于约5体积的一种或多种步骤1a)溶剂、约7体积的一种或多种步骤1a)溶剂、约10体积的一种或多种步骤1a)溶剂、约12体积的一种或多种步骤1a)溶剂、约14体积的一种或多种步骤1a)溶剂、约16体积的一种或多种步骤1a)溶剂或约20体积的一种或多种步骤1a)溶剂中,以形成步骤1b)溶液。所述步骤1b)溶液可任选冷却至约室温、约20℃、约15℃、约10℃、约5℃、约0℃、约-5℃、约-10℃或约-15℃,以制备化合物(1)的共形成体盐的固体形式。固体共形成体盐可经滤过收集,任选用步骤1b)溶剂洗涤,并干燥。在步骤2a)中,可将碱添加至化合物(1)的共形成体盐溶液中以释放化合物(1)并移去相应的共形成体酸。可采用任何足以释放化合物(1)的碱。在某些实施方案中,所述碱为氨水(如nh4oh)、naoh、naoac、nahco3或na2co3。在某些实施方案中,所述碱为氨水(如nh4oh)。在某些实施方案中,所述碱为naoh。在某些实施方案中,所述步骤2a)溶剂可为足以溶解化合物(1)的共形成体盐或可形成足以允许适合的碱反应以释放化合物(1)的悬浮液的任何溶剂或溶剂的组合。在某些实施方案中,所述步骤2a)溶剂可为任何步骤1a)溶剂。在某些实施方案中,所述步骤2a)溶剂可为c1-6酮、c1-6醇或水;或其组合。在某些实施方案中,所述c1-6酮为丙酮、mibk或mek。在某些实施方案中,所述c1-6酮为丙酮。在某些实施方案中,所述c1-6醇为甲醇、乙醇、2-丙醇或异丙醇。在某些实施方案中,所述c1-6醇为甲醇、2-丙醇或异丙醇。在某些实施方案中,所述步骤2a)溶剂可为丙酮、甲醇、2-丙醇、异丙醇或水;或其组合。在某些实施方案中,所述步骤2a)溶剂可为丙酮和甲醇;或它们可为丙酮、甲醇、2-丙醇和水;或它们可为丙酮、甲醇和异丙醇;或它们可为丙酮、甲醇、异丙醇和水。在步骤2a)中,可通过在选自nh4oh、naoh、naoac、nahco3或na2co3或其组合的碱的存在下将化合物(1)的共形成体盐悬浮于选自c1-6酮、c1-6醇和水或其组合的步骤2a)溶剂中,来释放化合物(1)。在某些实施方案中,所述步骤2a)溶剂为丙酮、甲醇、2-丙醇、异丙醇或水或其组合,且所述碱为nh4oh或naoh水溶液。在某些实施方案中,所述碱为nh4oh。在某些实施方案中,所述步骤2a)溶剂为丙酮、甲醇和异丙醇,且所述碱为nh4oh。在某些实施方案中,所述步骤2a)溶剂为丙酮、甲醇、异丙醇和水,且所述碱为nh4oh。在某些实施方案中,所述步骤2a)溶剂为丙酮、甲醇和2-丙醇,且所述碱为nh4oh。在步骤2a)中,可通过在室温或高温(例如约30℃、约32℃、约35℃、约37℃、约38℃、约40℃、约42℃、约45℃)将化合物(1)的共形成体盐悬浮于约0.5至约10体积或约0.5至约5体积或约0.75至约2.5体积的一种或多种步骤2a)溶剂中以形成步骤2a)溶液,并用约1-1.5当量的适当碱处理所述步骤2a)溶液,来释放化合物(1)。在一些实施方案中,所述共形成体盐在室温或高温(例如约30℃、约32℃、约35℃、约37℃、约38℃、约40℃、约42℃、约45℃)悬浮于约0.75体积或约1体积或约1.5体积或约1.7体积或约2体积或约2.2体积或约2.4体积或约2.5体积的一种或多种步骤2a)溶剂中以形成步骤2a)溶液,并用约1.1当量或约1.2当量或约1.3当量或约1.4当量或约1.5当量的适当碱处理所述步骤2a)溶液。在某些实施方案中,所述共形成体盐在室温或高温(例如约30℃、约32℃、约35℃、约37℃、约38℃、约40℃、约42℃、约45℃)悬浮于约0.5至约10体积或约0.5至约5体积或约0.75至约2.5体积的选自丙酮、甲醇、丙醇、异丙醇和水的一种或多种步骤2a)溶剂中以形成步骤2a)溶液,并用约1-1.5当量的选自naoh、nh3水溶液(任选作为25%nh3水溶液)、naco3、naoac和nahco3的碱处理所述步骤2a)溶液。在某些实施方案中,所述共形成体盐在室温或高温(例如约30℃、约32℃、约35℃、约37℃、约38℃、约40℃、约42℃、约45℃)悬浮于约0.75体积或约1体积或约1.5体积或约1.7体积或约2体积或约2.2体积或约2.4体积或约2.5体积的选自丙酮、甲醇、丙醇、异丙醇和水的一种或多种步骤2a)溶剂中以形成步骤2a)溶液,并用约1当量或约1.1当量或约1.2当量或约1.3当量或约1.4当量或约1.5当量的选自naoh、nh3水溶液(任选作为25%nh3水溶液)、naco3、naoac和nahco3的碱处理所述步骤2a)溶液。在某些实施方案中,在步骤2a)中,可通过在室温或高温(例如约30℃、约35℃、约37℃、约38℃、约40℃、约42℃或约45℃)将化合物(1)的共形成体盐悬浮于约0.75体积、约1体积、约1.5体积、约1.7体积、约2体积、约2.2体积或约2.4体积的诸如水、丙酮、ipa和甲醇的一种或多种步骤2a)溶剂中以形成步骤2a)溶液,并用约1当量、约1.1当量、约1.2当量、约1.3当量或约1.4当量的诸如naoh、nh3(任选25%nh3水溶液)、naco3、naoac或nahco3的碱处理所述步骤2a)溶液,来释放化合物(1)。可任选检查ph,并且若ph≥7,则可添加水(0.55体积)。可将系统冷却至约25℃、约30℃、约35℃或约40℃,并可任选添加化合物(1)的晶种。可在约30分钟内逐滴添加水(3.3体积),在30分钟内冷却悬浮液至约0至5℃的内部温度,历时15分钟搅拌反应混合物。可经过滤收集化合物(1)的固体形式,并用水洗涤3次。在某些实施方案中,所述共形成体盐在室温或高温(例如约30℃、约32℃、约35℃、约37℃、约38℃、约40℃、约42℃、约45℃)悬浮于比为约2-6体积/1-2体积/1-2体积的丙酮/异丙醇/甲醇中以形成步骤2a)溶液,并用约1当量或约1.1当量或约1.2当量或约1.3当量或约1.4当量或约1.5当量的nh3水溶液(任选作为25%nh3水溶液)处理所述步骤2a)溶液。在某些实施方案中,所述丙酮/异丙醇/甲醇比为约2-4体积/1-2体积/1-2体积或为约2-4体积/1体积/1体积或为约2体积/1体积/1体积。在某些实施方案中,所述共形成体盐在室温或高温(例如约30℃、约32℃、约35℃、约37℃、约38℃、约40℃、约42℃、约45℃)悬浮于比为约2体积/1体积/1体积的丙酮/异丙醇/甲醇中以形成步骤2a)溶液,并用约1.3当量的nh3水溶液(任选作为25%nh3水溶液)处理所述步骤2a)溶液。在步骤2b)中,若需要,可在通过使用约4体积、约5体积、约6体积或约7体积的诸如水、丙酮、ipa或甲醇的一种或多种步骤2b)溶剂的任选步骤中来改善化合物(1)的e.e.。例如可将丙酮(4体积)、ipa(1体积)和甲醇(1体积)添加至先前步骤2a)的产物中,并可将反应混合物加热至约38℃至42℃、约35℃、约38℃、约40℃、约42℃或约45℃的内部温度,得到澄清的步骤2b)溶液。可将水(2体积)和化合物(1)的晶种添加至步骤2b)溶液中,并在约35℃的内部温度搅拌系统约15分钟。可在约30分钟内逐滴添加水。然后可将混悬液在30min内冷却至约0至5℃的内部温度,并搅拌额外的15分钟。可将固体经滤过收集,用水洗涤两次,并确定手性纯度。可将固体在约60℃的内部温度减压干燥,得到化合物(1)。在某些实施方案中,所述方法提供了基本上纯的化合物(1)。在某些实施方案中,所述方法提供了具有90-99%e.e.或95%-99%e.e.或97%-99%e.e.或≥96%,e.e.或≥97%e.e.或≥98%e.e.或≥99%e.e或99.5%e.e.的化合物(1)。在另一方面,本申请提供了制备(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯(化合物(1))的共形成体盐的方法,包括(1)在选自mibk、mek、乙醇和水的一种或多种步骤1a)溶剂中在高温用共形成体处理7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯,形成步骤1a)溶液;(2)使步骤1a)溶液在足以使(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯(化合物(1))作为固体析出且在某些实施方案中呈结晶形式析出的条件下静置;和(3)分离呈固体且在某些实施方案中呈结晶形式的化合物(1)。在某些实施方案中,所述共形成体盐为[(1s)-内]-(+)-3-溴-10-樟脑磺酸盐且所述步骤1a)溶剂为mibk、水和乙醇。在某些实施方案中,所述方法还包括使所述共形成体盐在一种或多种步骤1b)溶剂中重结晶或重新浆化。在某些实施方案中,在使所述共形成体盐在一种或多种步骤1b)溶剂中重结晶或重新浆化后,所述(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐呈结晶形式。在某些实施方案中,所述方法还包括在室温或高温将(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐悬浮于选自水、丙酮、ipa或甲醇的一种或多种步骤2a)溶剂中以形成步骤2a)溶液,并用选自naoh、nh3(任选25%nh3水溶液)、naco3、naoac3或nahco3的碱处理所述步骤2a)溶液;使所述步骤2a)溶液在足以使(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯(化合物(1))作为固体析出且在某些实施方案中呈结晶形式析出的条件下静置;和(3)分离呈固体且在某些实施方案中呈结晶形式的化合物(1)。在某些实施方案中,所述方法还包括将化合物(1)在一种或多种步骤2b)溶剂中重结晶或重新浆化。在某些实施方案中,在使所述共形成体盐在一种或多种步骤2b)溶剂中重结晶或重新浆化后,化合物(1)呈结晶形式。化合物的制备以下为示例说明可如何制备和测试本发明的共形成体盐的实施例。尽管实施例仅表示某些实施方案,但应理解以下实施例为示例性的且并不意在限制。在某些实施方案中,所述制备化合物(1)的共形成体盐的方法包括上文及下文所述的各种实施方案中的任一种。本申请所披露的化合物为可商购得到的或可容易由可商购得到的起始物质根据有机合成领域中的已确立的方法来制备。合成本发明化合物的一般方法可见于例如stuartwarrenandpaulwyatt,workbookfororganicsynthesis:thedisconnectionapproach,第二版,wiley,2010。一些化合物的合成详细例示于下文。在某些实施方案中,本发明化合物的各个立体异构体由含有不对称或手性中心的可商购得到的起始物质以合成方式制备,或通过制备外消旋混合物、随后立体选择性地分离成对映异构体而获得。立体选择性分离方法包括,例如(1)使对映异构体混合物结合至手性助剂,通过重结晶或色谱分离所得非对映异构体混合物且由所述助剂释放光学纯的产物;或(2)在手性色谱柱上直接分离光学对映异构体的混合物。x射线粉末衍射(xrpd)除非另外规定,否则当xrpd峰值以2θ角度表示时,应理解使用铜kα1辐射。在某些实施方案中,本申请提供的2θ角度值在约±0.2°θ的范围变化,而仍描述相同xrpd峰。xrpd操作1:在brukeraxsc2gadds衍射器上使用cukα辐射(40kv,40ma)、xyz自动载台、用于自动样品定位的激光视频显微镜及histar二维区检测器来收集x射线粉末衍射图。x射线光学器件由与0.3mm的针孔准直器偶联的单一多层镜组成。使用经鉴定的标准nist1976刚玉(平板)进行每周效能检查。光束发散度,即样品上x射线光束的有效大小为约4mm。采用θ-θ连续扫描模式,其中样品-检测器距离为20cm,产生3.2°至29.7°的有效2θ范围。通常历时120秒样品暴露于x射线光束。xp/20004.1.43软件的gadds用于数据收集,且diffracplusevav13.0.0.2或v15.0.0.0软件用于数据分析及展示。环境条件:使用不进行研磨的原样粉末将在环境条件下运作的样品制备为平板试样;将约1-2mg的样品在载玻片上轻轻按压以获得平坦表面。非环境条件:将在非环境条件下运作的样品与热传导化合物一起安装于硅晶片上。随后以10℃/min将样品加热至适合的温度,并随后等温保持1分钟,之后开始数据收集。xrpd操作2:可选择地,在brukerd8衍射器上使用cukα辐射(40kv,40ma)、θ-2θ测角器及v4及接收狭缝的发散度、ge单色器及lynxeye检测器收集x射线粉末衍射图。使用经鉴定的刚玉标准(nist1976)检查仪器的效能。diffracplusxrdcommanderv.2.6.1软件用于数据收集,且diffracplusevav13.0.0.2或v15.0.0.0软件用于数据分析及呈现。样品利用原样粉末作为平板试样在环境条件下运作。将样品轻轻封装于抛光零背景(510)硅晶片内所切的空腔内。分析期间,使样品在其自身平面中旋转。数据收集详情包括:2至42°2θ的角范围、0.05°2θ的步长及0.5s/步的收集时间。单晶x射线衍射(scxrd)在配备有oxfordcryosystemscobra冷却装置的oxforddiffractionsupernovadualsource(cu为0)的atlasccd衍射器上收集数据。使用mokα辐射收集数据。通常使用shelxs或shelxd程序解析结构,并使用shelxl程序(其为brukeraxsshelxtl程序组(v6.10)的一部分)精修结构。连接至碳的氢原子可以几何学方式放置,且通常允许用riding各向同性位移参数(ridingisotropicdisplacementparameter)精修。连接至杂原子的氢原子位于差值傅里叶(fourier)合成中,且通常允许用各向同性位移参数自由精修。核磁共振对于实施例1-3和5,在配备有自动取样器且由drx400控制台控制的bruker400mhz仪器上收集nmr谱。可使用标准bruker负载实验,使用icon-nmrv4.0.7与topspinv1.3一起运作得到自动实验。对于非常规光谱法,经单独使用topspin获得数据。数据如下以ppm(δ)报道:化学位移(多重性、积分、偶合常数(hz))。在13c固态nmr中,峰位置可取决于诸如信噪比、峰宽、温度、旋转速度、去偶效能、魔角设置、数据处理操作和参数以及软件峰值提取算法的因素而变化。此外,峰位置与化学位移参考操作有关。可使用若干不同化学位移参考标准且将不一定产生相同结果。不同化学位移参考标准的使用可产生相隔若干ppm的峰位置。然而,若使用不同参考标准,或若分析者针对相同标准的参考峰位置使用不同值,则通常所有峰在相同方向的位置将具有系统性变化。在某些实施方案中,本申请所提供的13c固态nmr中的ppm值在约±0.2ppm的程度变化,但仍描述相同峰。差示扫描量热分析(dsc)在配备有34位自动取样器的mettlerdsc823e上收集dsc数据。使用经鉴定的铟校准仪器的能量及温度。在带针孔铝盘中,通常以10℃/min将0.5-2mg的每一样品由25℃加热至300℃。通常在样品上方保持50ml/min的氮气吹扫。starev9.20软件用作仪器控制及数据分析软件。热重量分析(tga)在配备有34位自动取样器的mettlertga/sdta851e上收集tga数据。使用经鉴定的铟对仪器进行温度校准。通常,将3-6mg的每一样品装载至预称重的铝坩埚上,且以10℃/min由环境温度加热至350℃。在样品上方保持50ml/min的氮气吹扫。ir光谱在具有universalatrsamplingaccessory及热电dtgs检测器(氘化硫酸三甘氨酸)的perkinelmerspectrumoneft-ir光谱仪上收集ir数据。经hplc确定手性纯度在配备有二极管阵列检测器的agilenthp1100串联系统上,并使用chemstation软件vb.02.01-sr1或sr2,使用如下详述的方法进行手性hplc分析:用于分析7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的手性hplc方法参数样品制备1.0mg/ml,dcm柱chiralpakic,250x4.6mm柱温(℃)35注射(l)10检测:波长,带宽(nm)235,4流速(ml/min)1.0相a20%/80%etoh/己烷相bn/a合成实施例实施例1:中间体(a)的盐筛选表1中以盐形式供应或制备的共形成体在离子交换树脂上洗脱以分离其游离酸对应物。然而,归因于游离酸的化学不稳定性,含有硫酸的共形成体未直接用作游离酸。取而代之,含有硫酸的共形成体以盐形式溶解于适当溶剂中,并针对每一硫酸基团添加一摩尔当量的hcl(4nhcl/二噁烷)。共形成体ac20、ac125和ac69作为游离酸固体添加。共形成体ac38、ac49、ac111、ac18和ac115作为游离酸在乙醇中的溶液分别以5m、1m、1m、5m和5m的浓度添加。以下共形成体作为游离酸在含水乙醇中的溶液添加:ac70(10%v/v,0.45m)、ac75(10%v/v,0.45m)、ac126(25%v/v,0.8m)、ac4(一水合物,7%v/v,1m)、ac117(20%v/v,0.4m)、ac116(10%v/v,0.45m)和ac127(35%v/v,0.5m)。以下共形成体作为钠盐溶液添加(除了一摩尔当量的4nhcl/二噁烷之外):ac118(0.8m在乙醇中)、ac110(5m在乙醇中)、ac113(3.7m在thf中)、ac114(0.8m在80体积%的含水thf中)和ac119(1.3m在25体积%的含水thf)中。共形成体ac120作为游离酸在水中的0.5m溶液添加。以下共形成体作为铵盐在溶液中添加(除了一摩尔当量的4nhcl/二噁烷之外):ac121(二铵盐,0.7m在38体积%的含水thf中)、ac122(1.4m在水中)、ac112(0.5m在水中)、ac123(1m在50%含水thf中)和ac124(1.3m在水中)。表1.共形成体在50℃制备中间体(a)(30或50mg)在乙醇(20体积)、mek(40体积)和mibk(20体积)中的澄清溶液。在50℃添加如前所述制备的共形成体酸(1.2mol当量)并浆化约1-2小时。将悬浮液冷却至室温并在室温浆化2天。将溶液先后冷却至5℃、20℃并进行缓慢蒸发。将胶状物进行成熟循环(温度循环)。表2.获得结晶化合物(1):(2s,3s)-7-氟-2-(4-氟苯基)-3-(1-甲基-1h-1,2,4-三唑-5-基)-4-氧代-1,2,3,4-四氢喹啉-5-羧酸甲酯的共形成体盐的尝试条件如下方案1描述了使用ac49作为用于制备化合物(1a)且用于手性拆分化合物(1)的共形成体酸。方案1实施例2–使用方案1制备化合物(1)步骤1a在50℃在搅拌下将中间体(a)(5g,12.5mmol)溶于9:1v/vmibk/乙醇(70ml,14体积)并在小于约5分钟内观察到溶解。添加[(1s)-内]-(+)-3-溴-10-樟脑磺酸一水合物(4.1g,12.5mmol)并在约10-20分钟观察到溶解。然后用化合物(1a)(95%e.e.,5mg,0.1%w.)进行种晶并在50℃使体系平衡约1小时,以0.15℃/min冷却至约20℃,然后在20℃平衡2小时。将固相经滤过分离,用乙醇洗涤,并在约50℃和3mbar干燥约2至3小时,得到化合物(1a),其为0.6摩尔当量的etoh溶剂化物和0.6摩尔当量的水合物(93.4%e.e.)。步骤1b然后在50℃在搅拌下将化合物(1a)以95/5体积%悬浮于mibk/乙醇(38ml,10体积)。在50℃保持约2小时后,将悬浮液冷却至约5℃且保持10至15小时。将固相经滤过回收并在约50℃和3mbar干燥约3小时。回收化合物(1a)(97.4%e.e.)。步骤2在不进行步骤1的任选重新浆化的情况下,通过在室温将化合物(1a)(3.9g,5.5mmol)悬浮于20ml水并用5m氢氧化钠/水(1.3ml,1.2mol)处理,来释放化合物(1)。将混合物在室温保持约15小时并将固体经滤过分离且在50℃和3mbar干燥约3小时。回收化合物(1)(94.4%e.e.)。实施例3–使用方案1大规模制备化合物(1)使用3.3kg中间体(a)以及各个溶剂比进行实施例1的操作,得到95.7%e.e.(步骤1a);99.2%e.e.(步骤1b);和99.2%e.e.(步骤2)。实施例4–使用方案1可替换地制备化合物(1)步骤1a在50℃在搅拌下将中间体(a)(751mg,1.86mmol))溶于9:1v/vmibk/乙醇(7.5ml,10体积)。添加[(1s)-内]-(+)-3-溴-10-樟脑磺酸一水合物(620mg,1.88mmol,1当量)。在50℃约1小时观察到形成析出物。然后将体系以0.1℃/min冷却至约5℃,然后在5℃平衡约60小时。将固相经滤过分离并在约50℃和3mbar干燥约2小时,得到化合物(1a)(92%e.e.)。参见图1-4,即xrpd(图1),手性hplc(图2),1hnmr(图3)和tga/dsc分析(图4)。来自实施例3的物质的xrpd图与实施例1的类似,仅在特定衍射峰的位置有略微位移(图1中用黑色箭头突显)。1hnmr与含有0.5摩尔当量的etoh和0.6重量%的残留mibk的化合物(1a)的单盐一致。tga分析显示在25和90℃之间发生3.5%的逐步质量损失(可能表示损失0.5摩尔当量的etoh)且在90℃至160℃之间发生1.2%的逐渐质量损失(可能表示损失所吸附的水)。dsc分析结果具有在25℃至90℃之间表示去溶剂化的宽吸热及在135℃表示熔融/降解的吸热。步骤1b在50℃将化合物(1a)(100.3mg,0.141mmol)重新悬浮于95:5v/vmibk/etoh(1ml,10体积)并搅拌1小时,之后以0.1℃/min冷却至5℃。在5℃保持一晚后,将固体(99.4%e.e.)经滤过回收。不再检测到xrpd衍射峰的位移(图5;相比图1)。图6显示化合物(1a)的手性hplc。步骤2在50℃将步骤1a的化合物(1a)(100.2mg,0.141mmol)悬浮于水(2ml,20体积)并添加5mnaoh/水(34μl,1.2摩尔当量)。将所得的悬浮液在50℃保持一晚,冷却至室温(不受控冷却)并滤过,得到化合物(1)(92%e.e.)。手性纯度不受该步骤的影响且经nmr未检测到[(1s)-内]-(+)-3-溴-10-樟脑磺酸。图7将步骤2的化合物(1)与步骤1的起始物质中间体(a)的xrpd进行比较。图8显示步骤2的化合物(1)与步骤1的起始物质中间体(a)的nmr。实施例5–使用方案1可替换地制备化合物(1)步骤1a在搅拌下将中间体(a)(1当量)添加至mibk(12-13体积)、乙醇(1-1.5体积)和水(0.05-0.10体积)的溶液中并将反应混合物在15分钟内加热至约48℃至约52℃的内部温度。添加[(1s)-内]-(+)-3-溴-10-樟脑磺酸(1当量)并将反应混合物在约48℃至约52℃的内部温度搅拌约5-10分钟直到发生溶解。添加化合物(1a)的晶种并使反应在约48℃至约52℃的内部温度进行1小时。将反应混合物以0.15℃/min的速率冷却至约19-21℃。将悬浮液在约19℃至21℃的内部温度搅拌2小时,然后经滤过收集并用乙醇洗涤两次。将产物通过1hnmr和13cnmr(图13a和13b)、ir光谱(图14)、dsc(图15)和手性hplc(图16)表征。步骤2a向化合物(1a)(1当量)中添加丙酮(1.1体积)、ipa(0.55体积)和甲醇(0.55体积),并将反应混合物加热至约38℃至42℃的内部温度。添加氨水(25%)(1.3当量)并将反应混合物搅拌约10分钟。确认反应混合物的ph且若≥7,则进行下一步。添加水(0.55体积),将反应混合物冷却至约35℃的内部温度,添加化合物(1)的晶种,并将反应混合物搅拌约10分钟。在约30分钟内逐滴添加水(3.3体积),将悬浮液在30分钟内冷却至约0℃至5℃的内部温度,并将反应混合物搅拌15分钟。将固体经滤过收集并用水洗涤三次。步骤2b向步骤2a)的产物中添加丙酮(4体积)、ipa(1体积)和甲醇(1体积),并将反应混合物加热至约38℃至42℃的内部温度,得到澄清溶液。添加水(2体积)和化合物(1)的晶种,并将体系搅拌在约35℃的内部温度约15分钟。在约30分钟内逐滴添加水(342ml)。然后将悬浮液在30min冷却至约0℃至5℃的内部温度,并搅拌额外的15分钟。将固体经滤过收集,用水洗涤两次,并确定手性纯度。如果≥99%e.e.,则将固体在约60℃的内部温度减压干燥,得到化合物(1)。产物通过1hnmr(图19)、13cnmr(图20)、ir(图21)、dsc(图22)、手性hplc(图23)表征。以下方案2描述了使用ac110作为用于制备化合物(1b)且用于手性拆分化合物(1)的共形成体酸。方案2实施例6-使用方案2制备化合物(1)步骤1a在65℃在搅拌下将中间体(a)(102mg,0.256mmol)溶于mibk(1ml,10体积)。添加使用本领域技术人员已知的操作制备的(1s)-苯基乙磺酸在mibk(3.8m,80μl,1摩尔当量)中的溶液并在65℃保持30分钟后观察到悬浮液。将体系在65℃保持另外的30分钟,之后在0.1℃/min冷却至5℃。在5℃保持一晚后,将固体滤过,在50℃、3mbar压力下干燥约2小时,得到化合物(1b)。参见图9-12的xrpd(图9)、手性hplc(图10)、1hnmr(图11)和tga/dsc分析(图12a和12b)。实施例5中所获得的固体的xrpd衍射图与用来自实施例1的盐筛选中的固体所获得的xrpd图不同,而与实施例1和5中不同固体的产生一致。1hnmr与具有0.3重量%的二噁烷残余物的单盐一致。在图12a中,热行为与在201℃显示出熔融/降解的非溶剂化形式一致。图12b将实施例5中化合物(1b)与实施例1中化合物(1b)的熔融图进行比较。可使用与实施例2-5中所使用的类似的操作进行步骤1b和2。实施例7-化合物(1a)的多晶型现象将化合物(1)(92%e.e.,10mg,mmol)置于1.5ml小瓶中并在50℃添加表3的溶剂(1ml或更少)直至达到溶解。在50℃添加固体[(1s)-内]-(+)-3-溴-10-樟脑磺酸。将样品在50℃保持约1小时,之后冷却至室温过夜(不受控的冷却速率)。将澄清溶液先后冷却至4℃、-20℃并在室温蒸发。将蒸发后获得的任何胶状物重新悬浮于乙醚。产生的固相通过xrpd以及(若相关)通过1hnmr和tga/dsc表征。表3.化合物(1a)多晶型现象条件c.s.意指澄清溶液且susp.意指悬浮液。“a”意指xrpd衍射图为新的但与实施例1的ac49的衍射图类似。“b”意指xrpd衍射图与实施例1的ac49的衍射图相同。“m.e.”表示摩尔当量。将观察到溶剂化物(不包括杂溶剂化物)的七种溶剂中的每一种与mibk(90%体积)混合。在50℃在溶剂混合物(10体积)中制备中间体(a)的溶液,并添加[(1s)-内]-(+)-3-溴-10-樟脑磺酸(1摩尔当量)。将所得的澄清溶液以0.2℃/min冷却至5℃。令人惊讶的是,任何样品中均未报道结晶。在约25℃用每一溶剂化物的若干晶体进行种晶。通过xrpd分析固相,并通过手性hplc分析液相。参见表4的结果汇总(其中“dias2”为化合物(1a)的(2r,3r)非对映异构体)。表4.化合物(1a)溶剂化物分析如上表4中所见,乙醇/mibk体系产生93%纯的化合物(1a),其表明化合物(1a)确实作为乙醇化物溶剂化物以极纯的形式结晶。本申请所述的化合物、方法及组合物的其他目标、特征及优点将由以下描述将变得显而易见。然而,应了解,描述及特定实施例尽管指示特定实施方案,但仅为了示例说明而给出,因为由此详细描述,在本发明描述的主旨及范围内的各种变化及修改将变得显而易见。本申请中所引用的所有公开,包括专利、专利申请及公开的专利申请均出于所有目的以引用的方式并入本申请中。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- (R)‑2‑(2,5‑二氟苯基)吡咯烷的合成方法及中间体与流程
- 2‑(2,4‑二氟苯基)‑1,1‑二氟‑1‑(5‑经取代的吡啶‑2‑基)‑3‑(1H‑四唑‑1‑基)丙烷‑2‑醇及其制备方法与流程
- 5-氟-3-苯基-2-[(1S)-1-(9H-嘌呤-6-基氨基)丙基]-4(3H)-喹唑啉酮中间体的制备方法与流程
- 5-氟-3-苯基-2-[(1S)-1-(9H-嘌呤-6-基氨基)丙基]-4(3H)-喹唑啉酮晶型及其制备方法与流程
- 一种新型全氟聚醚基硅烷化合物及其表面组合物的制造方法与工艺
- 1‑(3,4‑二取代)苯基‑2‑(3,4‑二取代)苯基乙烷化合物及其用途的制造方法与工艺
- 一种以全氟代亚苯基为核心的OLED光电材料及其应用的制造方法与工艺
- (2S,3S)‑7‑氟‑2‑(4‑氟苯基)‑3‑(1‑甲基‑1H‑1,2,4‑三唑‑5‑基)‑4‑氧代‑1,2,3,4‑四氢喹啉‑5‑羧酸甲酯的共形成体盐及其制备方法与流程