用于核酸递送的阳离子脂质的制作方法
本发明涉及用于核酸递送的阳离子脂质、包含其的脂质膜结构体及它们的用途。
背景技术:
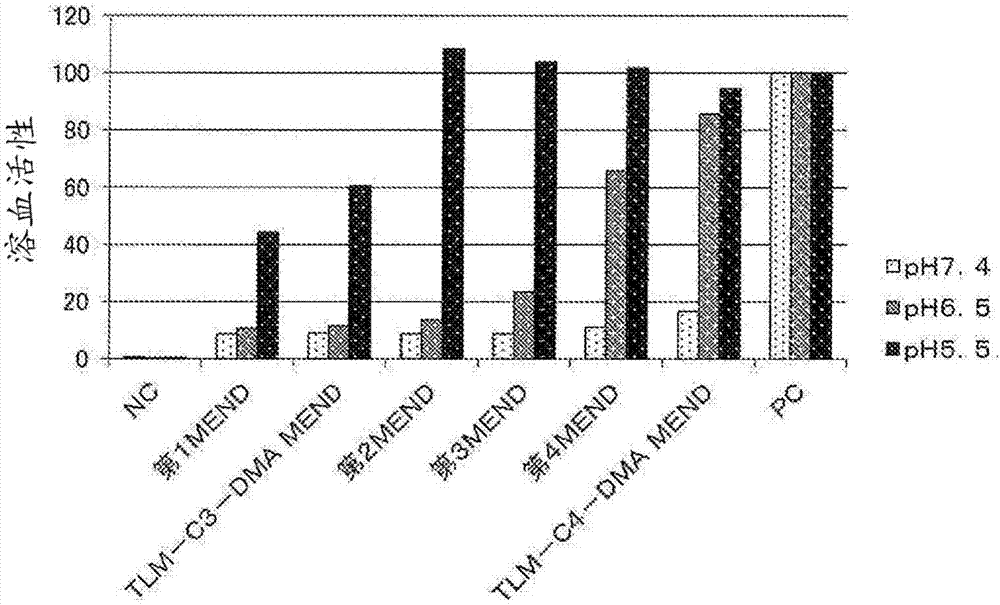
:核酸医药品将功能性核酸递送至细胞质内而抑制致病蛋白质的表达,作为新一代的医药品而受到瞩目,对其已进行了众多研究。由于功能性核酸有在血中的稳定性较低、会迅速分解的缺点,因此,载体是必要的。作为递送核酸的载体,可列举病毒、聚合物胶束、脂质体等脂质膜结构体。虽然病毒载体因为表达效率高而最为常用,但具有致病性、抗原性,并且存在诸如难以赋予功能性的缺点。因此,需要安全性更高的非病毒载体的开发,聚合物胶束、脂质膜结构体等的研究正在开展。聚合物胶束为由聚乙二醇(以下称为“peg”)、聚氨基酸等构成的载体,但临床研究的例子仍较少。脂质膜结构体为由阳离子脂质、磷脂等构成的载体。其优点为易于对构成载体的成分的组成进行改变、利用结构修饰来赋予功能性。迄今为止已进行了多次临床研究,是最常使用的非病毒载体。对将脂质膜结构体用作载体并有效地将功能性核酸递送至细胞内而言,除了改善脂质膜结构体的血中稳定性、肿瘤积累性等药物动力学以外,需要提高细胞的摄取、内质逃逸能力等细胞内动力学。对作为脂质膜结构体的构成成分之一的阳离子脂质而言,以赋予脂质膜结构体以ph响应性的目的使用。通过使用阳离子脂质,一方面使脂质膜结构体在血中等生理环境下稳定地存在。另一方面在细胞内那样的酸性环境下使脂质膜结构体崩溃,从而使药物能够向细胞质内放出。阳离子脂质大致由疏水部分和亲水部分构成,疏水部分为脂肪酸残基、甾醇残基等疏水性基团,亲水部分为氨基或铵基等阳离子性基团。尤其是疏水部分含有2个疏水性基团,且亲水部分含有1个氨基或铵基等阳离子性基团的结构(双链型阳离子脂质)广为人知。作为脂质膜结构体使用的阳离子脂质,可列举作为公知化合物的1,2-二油酰基-3-二甲基氨基丙烷(以下,称为“dodap”)、1,2-二亚油酰基-3-二甲基氨基丙烷(以下,称为“dlindap”)等。对阳离子脂质中包含的氨基而言,通过伴随周围环境的ph的降低而质子化变化为阳离子性,从而赋予脂质膜结构体以ph响应性。给予至活体的脂质膜结构体被内体摄入。已知早期内体随着向高尔基体附近进行移动,而向内部包含有许多内质网的晚期内体成熟,并与溶酶体结合。如果晚期内体与溶酶体结合,则功能性核酸会被溶酶体内的分解酶分解,因此,对于将功能性核酸有效地递送至细胞内,需要在从早期内体起,到晚期内体与溶酶体结合为止的过程中,将功能性核酸向细胞质内放出。但是,非专利文献1主张封装了功能性核酸的脂质膜结构体仅在早期内体的阶段向细胞质内放出功能性核酸,并且放出量为百分之几。像这样,尽管使用了传统的阳离子脂质的脂质膜结构体是该领域中的技术进步,但利用其所实现的向细胞内的核酸递送能力还不足够满意。现有技术文献非专利文献非专利文献1:naturebiotechnology,31,638-646(1july2013)技术实现要素:发明要解决的问题本发明的课题在于提供一种阳离子脂质,其在作为用于递送功能性核酸的载体的脂质膜结构体使用的情况下,可以实现比传统的阳离子脂质更高的细胞内递送效率。解决问题的方法已知具有较小的亲水部分、较大的疏水部分的圆锥型结构的脂质,通常容易从层状相向反向六角相转变。与此相对,认为对传统的双链型阳离子脂质而言,由于氨基在活体内发生质子化而带正电,氨基会彼此发生排斥导致亲水部分扩张,因此脂质膜变得难以发生从层状相向反向六角相的相转变。因而,在包含传统的双链型阳离子脂质的脂质膜结构体中,脂质膜难以发生相转变,脂质膜结构体与内体膜之间的膜融合能力不足,因此包含该脂质膜结构体的核酸导入剂的向细胞质内递送功能性核酸的能力较低。本发明人等对上述课题进行了深入研究,结果发现:开发了为了抑制由静电排斥导致的亲水部分的扩张而具有1个或2个阳离子性基团,并且为了加大疏水部分而导入了1个至4个疏水性基团的阳离子脂质。包含本发明的阳离子脂质的脂质膜结构体,在活体内那样的酸性条件下的膜融合能力较高,相比于使用了包含传统的双链型的阳离子脂质的脂质膜结构体的核酸导入剂,使用该脂质膜结构体的核酸导入剂向细胞质内递送功能性核酸的能力显著升高,从而完成了本发明。即,本发明包含以下内容。[1]由式(1)表示的阳离子脂质。[化学式1](式中,x1~x6中的任意4个各自独立地表示由式(xa)表示的基团、由式(xb)表示的基团或羟基(其中,不包括该4个同时为羟基的情况),其余2个各自独立地表示由式(xc)表示的基团或羟基(其中,不包括这2个同时为羟基的情况))[化学式2]-y1-r1(xa)(式中,r1表示碳原子数8~22个的脂肪族烃基或碳原子数8~22个的酰基;y1表示-o-或-nh-。)[化学式3](式中,r2表示甾醇残基或脂溶性维生素残基;z1表示碳原子数2或3个的亚烷基;y2表示-o-co-或-nh-co-。)[化学式4](式中,r3及r4独立地表示碳原子数1~6个的烷基,r3和r4任选互相结合而形成环;z2表示碳原子数1~6个的亚烷基;y3表示-o-、-o-co-或-nh-co-;n表示0或1。)[2]根据[1]所述的阳离子脂质,其中,x1~x6中的任意4个各自独立地为由式(xa)表示的基团或由式(xb)表示的基团,其余2个各自独立地为由式(xc)表示的基团。[3]根据[1]所述的阳离子脂质,其中,x1~x6中的任意4个各自独立地为由式(xa)表示的基团,其余2个各自独立地为由式(xc)表示的基团。[4]根据[1]~[3]中任一项所述的阳离子脂质,其中,r1为碳原子数10~20个的脂肪族烃基或碳原子数10~20个的酰基。[5]根据[1]~[3]中任一项所述的阳离子脂质,其中,r1为含有不饱和键的碳原子数10~20个的脂肪族烃基、或碳原子数10~20个的酰基。[6]根据[1]~[5]中任一项所述的阳离子脂质,其中,y1为-o-。[7]根据[1]所述的阳离子脂质,其中,x1~x6中的任意4个各自独立地为由式(xb)表示的基团,其余2个各自独立地为由式(xc)表示的基团。[8]包含[1]~[7]中任一项所述的阳离子脂质的脂质膜结构体。[9]包含[8]所述的脂质膜结构体及核酸的核酸导入剂。发明的效果本发明涉及一种包括含有1个至4个疏水性基团的疏水部分,并含有1个或2个阳离子性基团的亲水部分的阳离子脂质,及包含该阳离子脂质的脂质膜结构体、包含该脂质膜结构体及核酸的核酸导入剂。本发明的阳离子脂质可以形成稳定的脂质膜结构体,可以将作为脂质膜结构体的酸解离常数(以下,称为“pka”)调整至中性附近。进一步,在利用使用本发明的阳离子脂质的核酸导入剂进行功能性核酸的导入时,可以仅在内体内那样的弱酸性环境下显示与内体膜的较高的膜融合能力,有效地将功能性核酸向细胞质内放出。即,通过利用使用本发明的阳离子脂质的核酸导入剂进行功能性核酸的导入,可以利用递送至细胞质内的功能性核酸来实现有效的基因敲低(geneknockdown)。附图说明[图1]示出tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend、dlindapmend、dodapmend的溶血活性的图。[图2]示出第1~第4mend、tlm-c3-dmamend、tlm-c4-dmamend的溶血活性的图。[图3]示出由tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、dlindapmend、dodapmend导致的fvii敲低活性的图。[图4]示出由tlm-c3-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend导致的fvii敲低活性的图。[图5]示出由第1~第4mend、tlm-c3-dmamend、tlm-c4-dmamend导致的fvii敲低活性的图。[图6]示出由tlm-c3-dmammend、tdm-c3-dmammend导致的mrna表达活性的图。具体实施方式以下,对本发明的实施方式进行说明。1.本发明的阳离子脂质本发明提供由式(1)表示的阳离子脂质。[化学式5](式中,x1~x6中的任意4个独立地表示由式(xa)表示的基团、由式(xb)表示的基团或羟基(其中,不包括该4个同时为羟基的情况),其余2个独立地表示由式(xc)表示的基团或羟基(其中,不包括这2个同时为羟基的情况)。)[化学式6]-y1-r1(xa)(式中,r1表示碳原子数8~22个的脂肪族烃基或碳原子数8~22个的酰基;y1表示-o-或-nh-。)[化学式7](式中,r2表示甾醇残基或脂溶性维生素残基;z1表示碳原子数2或3个的亚烷基;y2表示-o-co-或-nh-co-。)[化学式8](式中,r3及r4独立地表示碳原子数1~6个的烷基,r3和r4任选互相结合而形成环;z2表示碳原子数1~6个的亚烷基;y3表示-o-、-o-co-或-nh-co-;n表示0或1。)在式(1)中,x1~x6中的任意4个可以是独立地表示由式(xa)表示的基团、由式(xb)表示的基团或羟基(其中,不包括该4个同时为羟基的情况),其余2个可以是独立地表示由式(xc)表示的基团或羟基(其中,不包括这2个同时为羟基的情况),优选x1~x6中的任意4个独立地表示由式(xa)表示的基团或由式(xb)表示的基团,其余2个独立地表示由式(xc)表示的基团或羟基(其中,不包括这2个同时为羟基的情况),更优选x1~x6中的任意4个独立地表示由式(xa)表示的基团或由式(xb)表示的基团,其余2个独立地表示由式(xc)表示的基团。作为式(1)的一个形态,在x1~x6中的任意4个独立地表示由式(xa)表示的基团或由式(xb)表示的基团,其余2个独立地表示由式(xc)表示的基团的情况下,虽然该x1~x6中由式(xa)表示的基团、由式(xb)表示的基团及由式(xc)表示的基团的组合是任意的,但作为该组合的具体例,可列举以下(1)~(8)的组合。(1)x1、x2、x5及x6独立地表示由式(xa)表示的基团,并且x3及x4独立地表示由式(xc)表示的基团。(2)x1、x3、x4及x6独立地表示由式(xa)表示的基团,并且x2及x5独立地表示由式(xc)表示的基团。(3)x2、x3、x4及x5独立地表示由式(xa)表示的基团,并且x1及x6独立地表示由式(xc)表示的基团。(4)x1、x2、x3及x4独立地表示由式(xa)表示的基团,并且x5及x6独立地表示由式(xc)表示的基团。(5)x1、x2、x5及x6独立地表示由式(xb)表示的基团,并且x3及x4独立地表示由式(xc)表示的基团。(6)x1、x3、x4及x6独立地表示由式(xb)表示的基团,并且x2及x5独立地表示由式(xc)表示的基团。(7)x2、x3、x4及x5独立地表示由式(xb)表示的基团,并且x1及x6独立地表示由式(xc)表示的基团。(8)x1、x2、x3及x4独立地表示由式(xb)表示的基团,并且x5及x6独立地表示由式(xc)表示的基团。其中,从原料采购、制造的容易程度的观点考虑,优选(1)的组合,尤其优选其中,x1、x2、x5及x6的由式(xa)表示的基团全部相同,并且x3及x4的由式(xc)表示的基团相同的(1)的组合。另外,从原料采购、制造的容易程度的观点考虑,优选(3)的组合,尤其优选其中,x2、x3、x4及x5的由式(xa)表示的基团全部相同,并且x1及x6的由式(xc)表示的基团相同的(3)的组合。作为式(1)的一个形态,在x1~x6中的任意4个独立地表示由式(xa)表示的基团或由式(xb)表示的基团的情况下,例如,其余2个可以为一方为由式(xc)表示的基团且另一方为羟基,或者也可以为其余2个均为由式(xc)表示的基团。在式(1)中,表示羟基的x1~x6的数量优选为3以下,更优选为1以下,特别优选为0。以下对于式(xa)、式(xb)及式(xc)中的各基团的定义进行详述。[式(xa)]式(xa)呈-y1-r1的结构。r1表示碳原子数8~22个的脂肪族烃基或碳原子数8~22个的酰基。该脂肪族烃基及酰基中包含的碳原子数优选为10~20个。该脂肪族烃基及酰基可以为直链状,也可以具有支链或环,但优选为直链状。该脂肪族烃基及酰基可以含有饱和也可以含有不饱和键,但优选为含有不饱和键。在该脂肪族烃基及酰基含有不饱和键的情况下,其中包含的不饱和键的数量通常为1~6个,优选为1~3个,更优选为1~2个。其中包含的不饱和键优选为碳-碳双键。作为碳原子数8~22个的脂肪族烃基,可列举例如:辛基、壬基、癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基、二十烷基、二十一烷基、二十二烷基、辛烯基、壬烯基、癸烯基、十一碳烯基、十二碳烯基、十三碳烯基、十四碳烯基、十五碳烯基、十六碳烯基、十七碳烯基、十八碳烯基、十九碳烯基、二十碳烯基、二十一碳烯基、二十二碳烯基、辛二烯基、壬二烯基、癸二烯基、十一碳二烯基、十二碳二烯基、十三碳二烯基,十四碳二烯基,十五碳二烯基,十六碳二烯基、十七碳二烯基、十八碳二烯基、十九碳二烯基、二十碳二烯基、二十一碳二烯基、二十二碳二烯基、十八碳三烯基、二十碳三烯基、二十碳四烯基、二十碳五烯基、二十二碳六烯基、异十八烷基、四甲基十六碳烯基(植烷基)等。优选为癸基、十二烷基、十四烷基、十六烷基、十八烷基、二十烷基、癸烯基、十二碳烯基、十四碳烯基、十六碳烯基、十八碳烯基、二十碳烯基、癸二烯基、十二碳二烯基、十四碳二烯基、十六碳二烯基、十八碳二烯基、二十碳二烯基等含有饱和或不饱和键的碳原子数10~20个的脂肪族烃基,更优选为癸烯基、十二烯基、十四碳二烯基、十六碳二烯基、十八碳二烯基等含有不饱和键的碳原子数10~20个的脂肪族烃基。作为碳原子数8~22的酰基,可列举例如:辛酰基、壬酰基、癸酰基、十一碳酰基、十二碳酰基、十三碳酰基、十四碳酰基、十五碳酰基、十六碳酰基、十七碳酰基、十八碳酰基、十九碳酰基、二十碳酰基、二十一碳酰基、二十二碳酰基、辛烯酰基、壬烯酰基、癸烯酰基、十一碳烯酰基、十二碳烯酰基、十三碳烯酰基、十四碳烯酰基、十五碳烯酰基、十六碳烯酰基、十七碳烯酰基、十八碳烯酰基、十九碳烯酰基、二十碳烯酰基、二十一碳烯酰基、二十二碳烯酰基、辛二烯酰基、壬二烯酰基、癸二烯酰基、十一碳二烯酰基、十二碳二烯酰基、十三碳二烯酰基、十四碳二烯酰基、十五碳二烯酰基、十六碳二烯酰基、十七碳二烯酰基、十八碳二烯酰基、十九碳二烯酰基、二十碳二烯酰基、二十一碳二烯酰基、二十二碳二烯酰基、十八碳三烯酰基、二十碳三烯酰基、二十碳四烯酰基、二十碳五烯酰基、二十二碳六烯酰基、异硬脂酰基、四甲基十六碳酰基(植物酰基)、视黄酰基等,优选为癸酰基、十二碳酰基、十四碳酰基、十六碳酰基、十八碳酰基、二十碳酰基、癸烯酰基、十二碳烯酰基、十四碳烯酰基、十六碳烯酰基、十八碳烯酰基、二十碳烯酰基、癸二烯酰基、十二碳二烯酰基、十四碳二烯酰基、十六碳二烯酰基、十八碳二烯酰基、二十碳二烯酰基等含有饱和或不饱和键的碳原子数10~20个的酰基,更优选为癸烯酰基、十二碳烯酰基、十四碳二烯酰基、十六碳二烯酰基、十八碳二烯酰基等含有不饱和键的碳原子数10~20个的酰基。在式(1)包含2个以上由式(xa)表示的基团的情况下,各r1可以相同也可以不同,但优选为全部相同。y1为-o-或-nh-,优选为-o-。在式(1)包含2个以上由式(xa)表示的基团的情况下,各y1可以相同也可以不同,但优选为全部相同。作为优选的由式(xa)表示的基团,为由式(xa)表示的基团中,r1为含有不饱和键的碳原子数10~20的脂肪族烃基或酰基,y1为-o-的基团。在式(1)包含2个以上由式(xa)表示的基团的情况下,各个由式(xa)表示的基团可以相同也可以不同,但优选为全部相同。[式(xb)]式(xb)呈-y2-z1-co-r2的结构。r2表示甾醇残基或脂溶性维生素残基,优选为脂溶性维生素残基。作为甾醇残基,可列举例如:胆甾烯基(cholesteryl)(胆甾醇残基)、胆甾烷基(cholestaryl)(胆甾烷醇残基)、β-谷甾烯基(β-谷甾醇残基)、羊毛甾烯基(羊毛甾醇残基)、麦角甾烯基(麦角甾醇残基)等。甾醇残基优选为胆甾烯基或胆甾烷基。作为脂溶性维生素残基,可列举例如:视黄醇残基、视黄醛残基、麦角甾醇残基、7-羟基胆甾醇残基、7-脱氢胆甾醇残基、钙化醇残基、胆骨化醇残基、二氢麦角钙化醇残基、二氢速固醇残基、生育酚残基、生育三烯酚残基等。脂溶性维生素残基优选为视黄醇残基或生育酚残基。在式(1)包含2个以上由式(xb)表示的基团的情况下,各r2可以相同也可以不同,但优选为全部相同。z1表示碳原子数2或3的亚烷基,该亚烷基可以为直链状也可以具有支链,但优选为直链状。作为碳原子数2或3的亚烷基,例如可列举亚乙基、三亚甲基等,优选为三亚甲基。在式(1)包含2个以上由式(xb)表示的基团的情况下,各z1可以相同也可以不同,但优选为全部相同。y2为-o-co-或-nh-co-,优选为-o-co-。对y2的键的方向没有限制,例如在y2为-o-co-的情况下,式(xb)优选呈-o-co-z1-co-r2的结构。另外,例如在y2为-nh-co-的情况下,式(xb)优选呈-nh-co-z1-co-r2的结构。在式(1)包含2个以上由式(xb)表示的基团的情况下,各y2可以相同也可以不同,但优选为全部相同。作为优选的由式(xb)表示的基团,为r2为甾醇残基(优选为胆甾烯基或胆甾烷基)或脂溶性维生素残基(优选为视黄醇残基或生育酚残基),z1为碳原子数2或3的亚烷基(优选为亚乙基或三亚甲基,更优选为三亚甲基),y2为-o-co-的由式(xb)表示的基团。在式(1)包含2个以上由式(xb)表示的基团的情况下,各由式(xb)表示的基团可以相同也可以不同,但优选为全部相同。[式(xc)]式(xc)呈-(y3-z2)n-nr3r4的结构。r3及r4独立地表示碳原子数1~6的烷基。r3及r4可以为直链状、支链状及环状中的任意,并且也可以r3与r4互相结合而形成环。该烷基的碳原子数优选为1~3。作为直链状或支链状的碳原子数1~6的烷基,可列举例如:甲基、乙基、丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、戊基、异戊基、新戊基、叔戊基、1,2-二甲基丙基、2-甲基丁基、2-甲基戊基、3-甲基戊基、2,2-二甲基丁基、2,3-二甲基丁基、环己基等。作为在r3与r4互相结合并形成环的情况下的-nr3r4的例子,具体可列举:氮丙啶基、氮杂环丁烷基、唑啉基(azolidyl)、哌啶基等。r3及r4优选为甲基、乙基、丙基或异丙基,更优选为甲基。r3和r4可以相同也可以不同,优选r3与r4相同。在式(1)包含2个由式(xc)表示的基团的情况下,各r3可以相同也可以不同,但优选为相同。在式(1)包含2个由式(xc)表示的基团的情况下,各r4可以相同也可以不同,但优选为相同。z2表示碳原子数1~6的亚烷基。z2可以为直链状也可以具有支链,但优选为直链状。作为碳原子数1~6的亚烷基,可列举例如:亚甲基、亚乙基、三亚甲基、异亚丙基、四亚甲基、异亚丁基、五亚甲基、新亚戊基、六亚甲基等。z2优选为亚甲基、亚乙基、三亚甲基、异亚丙基、四亚甲基、六亚甲基,更优选为亚乙基、三亚甲基。在式(1)包含2个由式(xc)表示的基团的情况下,各z2可以相同也可以不同,但优选为相同。y3表示-o-、-o-co-或-nh-co-,优选为-o-co-。对y3的键的方向没有限制,例如在y3为-o-co-的情况下,式(xc)优选呈-(o-co-z2)n-nr3r4的结构。另外,例如在y3为-o-的情况下,式(xc)优选为呈-(o-z2)n-nr3r4的结构,在y3为-nh-co-的情况下,式(xc)优选为呈-(nh-co-z2)n-nr3r4的结构。在式(1)包含2个由式(xc)表示的基团的情况下,各y3可以相同也可以不同,但优选为相同。n为0或1,优选为1。在n为1的情况下,式(xc)呈-y3-z2-nr3r4的结构,在n为0的情况下,式(xc)呈-nr3r4的结构。在式(1)包含2个由式(xc)表示的基团的情况下,各n可以相同也可以不同,但优选为相同。作为优选的由式(xc)表示的基团,为r3及r4独立地表示碳原子数1~3的烷基(优选为甲基、乙基、丙基或异丙基,更优选为甲基),z2为碳原子数1~6的亚烷基(优选为亚甲基、亚乙基、三亚甲基、异亚丙基、四亚甲基、六亚甲基、更优选为亚乙基、三亚甲基),y3为-o-co-,n为1的由式(xc)表示的基团。在式(1)包含2个由式(xc)表示的基团的情况下,各由式(xc)表示的基团可以相同也可以不同,但优选为相同。作为本发明的阳离子脂质的具体例,可列举表1所述的tlm-c2-dma(1,2,5,6-四亚油酰基-3,4-二(二甲基氨基乙酰基)-d-甘露醇)、tlm-c3-dma(1,2,5,6-四亚油酰基-3,4-二(3-二甲基氨基丙酰基)-d-甘露醇)、tlm-c4-dma(1,2,5,6-四亚油酰基-3,4-二(4-二甲基氨基丁酰基)-d-甘露醇),tdm-c3-dma(1,2,5,6-四(癸烯基)-3,4-二(3-二甲基氨基丙酰基)-d-甘露醇)、tlmes-c3-dma(四亚油酰基-二(3-二甲基氨基丙酰基)-d-甘露醇)等。[表1]下面对本发明的阳离子脂质的制造方法进行说明。作为本发明的阳离子脂质的制造方法,可列举向具有6个羟基的由式(1’)表示的化合物中,导入由式(xa)表示的基团和/或由式(xb)表示的基团、以及由式(xc)表示的基团的方法,例如:(i)导入xa和/或xb之后导入xc的方法,(ii)导入xc之后导入xa和/或xb的方法,(iii)同时导入xa和/或xb和xc的方法,或基于这些的方法等。[化学式9]对本发明的阳离子脂质的制造方法没有特别限制,优选为上述(i)方法。虽然以下举出了该(i)方法的具体例,但本发明的阳离子脂质的制造方法并不特别限定于该方法。作为起始化合物,可列举例如,在具有6个羟基的式(1’)表示的化合物中有2个羟基利用保护基保护的化合物,或在式(1’)表示的化合物中有4个羟基利用保护基保护的化合物等。需要说明的是,就这些起始化合物而言,可以容易地取得市售品,或者可以按照本身公知的方法或基于公知的方法的方法进行制造。作为向式(1’)导入的保护基,可列举例如:异亚丙基、亚苄基、苯甲酰基、苄基、三苯甲基、4-甲氧基三苯甲基、4,4’-二甲氧基三苯甲基、三烷基硅烷基(例如,三甲基硅烷基、三乙基硅烷基、叔丁基二甲基硅烷基、叔丁基二苯基硅烷基等)等。对该保护基的导入方法没有特别的限制,可以根据本身公知的方法或基于其的方法进行。例如,在制造作为本发明的阳离子脂质为式(1)中的x1、x2、x5及x6为由式(xa)表示的基团(r1:脂肪族烃基、y1:-o-,此外的符号如式(xc)所定义),x3及x4为由式(xc)表示的基团(y3:-co-o-,n:1)时的式(5)表示的化合物的情况下,对由该式(5)表示的化合物而言,可以使用由式(1a)表示的化合物作为起始化合物,经过以下的工序1(醚化)、工序2(脱保护)、工序3(酯化)、或者工序1(醚化)、工序2(脱保护)、工序4(酯化)、工序5(氨基化)来制造。[化学式10]式中,a表示保护基(例如异亚丙基、亚苄基、苯甲酰基、苄基、三苯甲基、4-甲氧基三苯甲基、4,4’-二甲氧基三苯甲基、三烷基甲硅烷基等),b表示脱离基(例如:碘原子、溴原子、氯原子、甲烷磺酰氧基、对甲苯磺酰氧基、三氟甲烷磺酰氧基等),d表示羟基或卤原子(例如:碘原子、溴原子、氯原子等),e表示由式(e)表示的基团或乙烯基。[化学式11]b-z2-(e)(式中,z2表示碳原子数1~6的亚烷基,b表示脱离基(例如:碘原子、溴原子、氯原子、甲烷磺酰氧基、对甲苯磺酰氧基、三氟甲烷磺酰氧基等)。)工序1(醚化)通过将由式(1a)表示的化合物与由r1-b(式中的r1及b与上述同义)表示的化合物发生反应,得到由式(2)表示的化合物。在反应中可以使用氢氧化钾、氢化钠、叔丁醇钾等碱催化剂,也可以无催化剂进行。优选将氢氧化钾作为催化剂使用。作为使用的催化剂量,相对于由式(1a)表示的化合物通常为6~20摩尔当量,优选为8~12摩尔当量。虽然反应可以使用溶剂,也可以无溶剂进行,但由于由式(1a)表示的化合物为高极性的固体,需要在反应体系中分散,因此优选使用溶剂。作为溶剂,可以使用不阻碍反应,由式(1a)表示的化合物可分散的溶剂,可列举例如:己烷、甲苯、二甲基甲酰胺、二甲亚砜(以下,称为“dmso”)等。这些中,优选甲苯。反应温度通常为20~150℃,优选为40~100℃。反应时间通常为1~50小时,优选为10~30小时。得到的由式(2)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。工序2(脱保护)对由式(2)表示的化合物的保护基脱保护,得到包含2个游离的羟基的由式(3)表示的化合物。在反应中使用酸催化剂。作为酸催化剂,可列举盐酸、乙酸、硫酸、磷酸、对甲苯磺酸一水合物等,优选为盐酸。作为使用的催化剂量,相对于由式(2)表示的化合物通常为1~50摩尔当量,优选为5~20摩尔当量。在反应中使用溶剂。作为溶剂,可列举甲醇、乙醇、异丙醇、水等,优选为甲醇、乙醇。反应温度通常为20~70℃,优选为40~60℃。反应时间通常为1~12小时,优选为4~8小时。得到的由式(3)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。工序3(酯化)可以通过将由式(3)表示的化合物与由式(p)(式中,r3、r4及z2与上述同义)表示的化合物发生反应,得到本发明的式(5)表示的化合物。在反应中,使用二环己基碳二亚胺(以下,称为“dcc”)、二异丙基碳二亚胺(以下,称为“dic”)、1-乙基-3-(二甲基氨基丙基)碳二亚胺盐酸盐(以下,称为“edc”)等缩合剂。在反应中添加碱催化剂。作为碱催化剂,可列举4-二甲基氨基吡啶(以下,称为“dmap”)、吡啶、三乙胺等,优选为dmap。由式(p)表示的化合物的投入量,相对于由式(3)表示的化合物通常为2~10摩尔当量,优选为4~8摩尔当量。虽然反应可以使用溶剂,也可以无溶剂进行,但由于由式(p)表示的化合物为高极性的固体,需要在反应体系中溶解或分散,因此优选使用溶剂。作为可使式(p)表示的化合物溶解或分散的溶剂,可列举例如,氯仿、二氯甲烷、甲苯、乙酸乙酯等,这些中,优选氯仿。反应温度通常为10~60℃,优选为20~40℃。反应时间通常为1~20小时,优选为2~10小时。得到的由式(5)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。工序4(酯化)可以通过将由式(3)表示的化合物与由式(q)(式中的d、e与上述同义)表示的化合物发生反应,得到由式(4)表示的化合物。在由式(q)表示的化合物中的d为羟基的情况下,在反应中,使用dcc、dic、edc等缩合剂。可以使由式(3)表示的化合物与由式(q)表示的化合物直接反应,或也可以形成由式(q)表示的化合物的酸酐后再与式(3)表示的化合物反应。在由式(q)表示的化合物中的d为卤原子的情况下,为了中和副产的卤化氢而添加碱。作为碱,可列举三乙胺、吡啶等。反应使用溶剂。作为溶剂,可列举氯仿、二氯甲烷、甲苯、乙酸乙酯等,优选为甲苯。由式(q)表示的化合物的投入量,相对于由式(3)表示的化合物通常为2~10摩尔当量,优选为2~5摩尔当量。反应温度通常为0~60℃,优选为10~40℃,反应时间通常为1~10小时,优选为1~5小时。得到的由式(4)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。工序5(氨基化)可以通过使含有r3及r4的仲胺与由式(4)表示的化合物发生反应,得到本发明的式(5)表示的化合物。在反应中可以使用碳酸钾、碳酸钠、叔丁醇钾等碱催化剂,也可以无催化剂进行。反应可以使用溶剂,也可以无溶剂进行。对于溶剂,可以使用例如乙酸乙酯、二氯甲烷、氯仿、苯、甲苯、四氢呋喃(以下,称为“thf”)等,这些中,优选甲苯。含有r3及r4的仲胺的投入量,相对于由式(4)表示的化合物通常为1~20摩尔当量,优选为5~10摩尔当量。反应温度通常为10~100℃,优选为60~80℃。反应时间通常为1~10小时,优选为2~6小时。得到的由式(5)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。本领域技术人员可以通过适当选择原料并基于本说明书的实施例的方法进行反应,来制造需要的本发明的阳离子脂质。另外,例如,在制造如下由式(9)表示的化合物的情况下,其中,在由式(9)表示的化合物中,式(1)中的x2、x3、x4及x5为由式(xa)表示的基团(r1:酰基、y1:-o-),x1及x6为由式(xc)表示的基团(n:1,y3:-co-o-,此外的符号如式(xc)所定义),对以该式(9)表示的化合物而言,可以使用由式(1b)表示的化合物作为起始化合物,经过以下的工序6(酯化)、工序7(脱保护)、工序8(酯化)、或者工序6(酯化)、工序7(脱保护)、工序9(酯化)、工序10(氨基化)来制造。[化学式12]式中,a表示保护基(例如异亚丙基、亚苄基、苯甲酰基、苄基、三苯甲基、4-甲氧基三苯甲基、4,4’-二甲氧基三苯甲基、三烷基硅烷基等),d表示羟基或卤原子(例如:碘原子、溴原子、氯原子等),e表示由式(e)表示的基团或乙烯基。[化学式13]b-z2-(e)(式中,z2表示碳原子数1~6的亚烷基,b表示脱离基(例如:碘原子、溴原子、氯原子、甲烷磺酰氧基、对甲苯磺酰氧基、三氟甲烷磺酰氧基等)。)工序6(酯化)通过将由式(1b)表示的化合物与由r1-d(式中的r1及d与上述同义)表示的化合物发生反应,得到由式(6)表示的化合物。在r1-d的d为羟基的情况下,在反应中,使用dcc、dic、edc等缩合剂。对使用量而言,相对于式(1b)表示的化合物,通常为4~10摩尔当量,优选为5~8摩尔当量。在反应中添加碱催化剂。作为碱催化剂,可列举dmap、吡啶、三乙胺等,优选为dmap。在反应中使用溶剂。作为溶剂,只要是不阻碍反应、基质可溶解的溶剂即可,没有特别限定。具体而言,可列举氯仿、二氯甲烷、甲苯、乙酸乙酯等,优选为氯仿。反应温度通常为0~60℃,优选为10~40℃。反应时间通常为1~50小时,优选为10~30小时。得到的由式(6)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。工序7(脱保护)对由式(6)表示的化合物的保护基进行脱保护,得到包含2个游离的羟基的由式(7)表示的化合物。在a为三烷基甲硅烷基的情况下,在反应中使用氟化物。作为氟化物,可列举:四丁基氟化铵、四丙基氟化铵、四乙基氟化铵、四甲基氟化铵、氢氟酸、氟化铯等,优选为四丁基氟化铵。作为氟化物的使用量,相对于式(6)表示的化合物通常为2~10摩尔当量,优选为4~8摩尔当量。在三烷基甲硅烷基的脱保护的过程中,生成为强碱的烷氧化物。由于如果存在烷氧化物,则会发生酯的分解、转移,因此,需要迅速地中和。因而,优选向反应体系中添加作为质子源的酸。由于酸过强则会分解酯,因此优选为弱酸。具体而言,可列举乙酸、乙二酸、柠檬酸、磷酸、苯甲酸、硼酸、三乙胺盐酸盐等,优选为乙酸。作为酸的使用量,相对于式(6)表示的化合物通常为2~10摩尔当量,优选为4~8当量。在反应中使用溶剂。作为溶剂,可列举:四氢呋喃、氯仿、乙酸乙酯、乙醇、甲醇等,优选为四氢呋喃。反应温度通常为0~70℃,优选为10~60℃。反应时间通常为1~12小时,优选为4~8小时。得到的由式(7)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。工序8(酯化)可以通过将由式(7)表示的化合物与由式(p)(式中,r3、r4及z2与上述同义)表示的化合物发生反应,得到本发明的式(9)表示的化合物。在反应中,使用dcc、dic、edc等缩合剂。反应时,可以使由式(7)表示的化合物与由式(p)表示的化合物直接反应,或也可以形成由式(p)表示的化合物的酸酐后再与由式(7)表示的化合物反应。在反应中添加碱催化剂。作为碱催化剂,可列举dmap、吡啶、三乙胺等,优选为dmap。由式(p)表示的化合物的投入量,相对于由式(7)表示的化合物通常为2~10摩尔当量,优选为4~8摩尔当量。虽然反应可以使用溶剂,也可以无溶剂进行,但由于由式(p)表示的化合物为高极性的固体,需要在反应体系中分散,因此优选使用溶剂。作为可使由式(p)表示的化合物分散的溶剂,可列举例如,氯仿、二氯甲烷、甲苯、乙酸乙酯等,这些中,优选氯仿。反应温度通常为10~60℃,优选为20~40℃。反应时间通常为1~20小时,优选为2~10小时。得到的由式(9)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。工序9(酯化)可以通过将由式(7)表示的化合物与由式(q)(式中的d、e与上述同义)表示的化合物发生反应,得到由式(8)表示的化合物。在由式(q)表示的化合物中的d为羟基的情况下,在反应中,使用dcc、dic、edc等缩合剂。可以使由式(7)表示的化合物与由式(q)表示的化合物直接反应,或也可以形成由式(q)表示的化合物的酸酐后再与由式(7)表示的化合物反应。在反应中添加碱催化剂。作为碱催化剂,可列举dmap、吡啶、三乙胺等,优选为dmap。在由式(q)表示的化合物中的d为卤原子的情况下,为了中和副产的卤化氢而添加碱。作为碱,可列举三乙胺、吡啶等。反应使用溶剂。作为溶剂,可列举氯仿、二氯甲烷、甲苯、乙酸乙酯等,优选为甲苯。由式(q)表示的化合物的投入量,相对于由式(7)表示的化合物通常为2~10摩尔当量,优选为2~5摩尔当量。反应温度通常为0~60℃,优选为10~40℃,反应时间通常为1~10小时,优选为1~5小时。得到的由式(8)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。工序10(氨基化)可以通过使含有r3及r4的仲胺与由式(8)表示的化合物发生反应,得到本发明的式(9)表示的化合物。在反应中可以使用碳酸钾、碳酸钠、叔丁醇钾等碱催化剂,也可以无催化剂进行。反应可以使用溶剂,也可以无溶剂进行。对于溶剂,可以使用例如乙酸乙酯、二氯甲烷、氯仿、苯、甲苯、thf等,这些中,优选使用甲苯。含有r3及r4的仲胺的投入量,相对于由式(8)表示的化合物通常为1~20摩尔当量,优选为5~10摩尔当量。反应温度通常为10~100℃,优选为60~80℃。反应时间通常为1~10小时,优选为2~6小时。得到的由式(9)表示的化合物可以通过提取、重结晶、吸附处理、再沉淀、柱色谱等方法进行适当纯化。本领域技术人员可以通过适当选择原料并基于本说明书的实施例的方法进行反应,来制造需要的本发明的阳离子脂质。2.本发明的脂质膜结构体下面对本发明的脂质膜结构体进行说明。本发明的脂质膜结构体,含有由上述式(1)表示的阳离子脂质(即,本发明的阳离子脂质)作为膜的构成脂质。这里,本发明中的“脂质膜结构体”是指,膜的构成脂质的亲水基团朝向界面的水相侧排列的脂质膜结构体。对本发明的脂质膜结构体的形态没有特别限定,例如,作为本发明的阳离子脂质分散在水系溶剂中而成的形态,可列举脂质体(例如:单层脂质体、多层脂质体等)、o/w型乳液、w/o/w型乳液、球状胶束、带状胶束、或不特定的层状结构物(unspecifiedlayerstructure)等。本发明的脂质膜结构体的形态优选为脂质体。本发明的脂质膜结构体除了本发明的阳离子脂质以外,也可以进一步含有这以外的其它构成成分。作为该其它构成成分,可列举例如:脂质(例如:磷脂(例如:磷脂酰乙醇胺、磷脂酰肌醇、磷脂酰丝氨酸、磷脂酸、磷脂酰甘油、磷脂酰胆碱等)、糖脂、肽脂、胆甾醇、除了本发明的阳离子脂质以外的阳离子脂质、peg脂质等)、表面活性剂(例如:chaps、胆酸钠盐、辛基糖苷、n-d-葡萄糖-n-甲基烷酰胺类、poloxamer类、聚氧乙烯山梨糖醇酐脂肪酸酯类等)、peg、蛋白质等。本发明的脂质膜结构体中的该其它构成成分的含量,通常为5~90重量%,优选为10~30重量%。本发明的脂质膜结构体中包含的本发明的阳离子脂质可以仅为1种,也可以组合2种以上使用。通过组合2种以上本发明的阳离子脂质使用,可以将本发明的脂质膜结构体的pka在4~7的范围中任意调整。通过将本发明的脂质膜结构体的pka调整为适于目的的值,能够将功能性核酸高效率地递送至细胞内。对本发明的脂质膜结构体中包含的本发明的阳离子脂质的含量没有特别限定,例如,在将本发明的脂质膜结构体使用于后述的核酸导入剂中的情况下,本发明的脂质膜结构体,优选包含为了导入核酸的充分量的本发明的阳离子脂质。本发明的脂质膜结构体中包含的本发明的阳离子脂质的含量,通常为本发明的脂质膜结构体中包含的总脂质量(totallipidamount)的5~100mol%,优选为30~90mol%,更优选为50~70mol%。本发明的脂质膜结构体,可以通过将本发明的阳离子脂质及其它构成成分(脂质等)溶解或分散于适当的溶剂或分散介质,例如:水性溶剂、醇性溶剂中,并根据需要进行诱导组织化的操作进行制备。作为“诱导组织化的操作”,可列举例如:乙醇稀释法、简单水和法、超声波处理、加热、涡旋震荡(vortex)、醚注入法、法压法(frenchpressmethod)、胆酸法、ca2+融合法、冻融法、反相蒸发法等本身公知的方法。3.本发明的核酸导入剂通过向包含本发明的阳离子脂质的脂质膜结构体导入核酸,并使其与活体内和/或活体外的细胞接触,可以向该细胞内导入核酸。因此,本发明也提供核酸导入剂(以下,称为“本发明的试剂”)。本发明的试剂的主要的特征为含有上述包含本发明的阳离子脂质的脂质膜结构体及核酸。作为一种形态,本发明的试剂可以含有本发明的脂质膜结构体及核酸,在这种情况下,优选该核酸已被导入至本发明的脂质膜结构体。这里,将核酸“导入”至本发明的脂质膜结构体是指将核酸封装到由脂质双重膜形成的空间中。对可导入本发明的脂质膜结构体的核酸没有特别的限制,可以使用任意核酸。作为核酸的种类,可列举例如:dna、rna、dna和rna的嵌合核酸、dna/rna的杂合等,但不限于这些。另外,对核酸而言,可使用1~3条链中的任意,优选为1条链或2条链。核酸可以为例如:作为嘌呤或嘧啶碱的n-糖苷的其它类型的核苷酸,或者具有非核苷酸骨架的其它低聚物(例如:市售的肽核酸(pna)等)或含有特殊的键的其它低聚物(其中,该低聚物含有具有在dna、rna中发现的那样的碱基配对、允许碱基的附着的构造的核苷酸)等。并且该核酸可以为例如:添加了公知的修饰的核酸、具有该领域已知的标记的核酸、带帽的核酸、经甲基化的核酸、将1个以上的天然的核苷酸用类似物进行置换而成的核酸、分子内核苷酸修饰后的核酸、具有非电荷键的核酸(例如,甲基膦酸酯、磷酸三酯、氨基磷酸酯、氨基甲酸酯等)、具有带电荷的键或含硫键(例如:硫代磷酸酯、二硫代磷酸酯等)的核酸、例如蛋白质(例如:核酸酶、核酸酶抑制剂、毒素、抗体、信号肽、聚-l-赖氨酸等)、糖(例如:单糖等)等具有侧链基团的核酸、具有插层化合物(intercalatingcompound)(例如:吖啶、补骨脂素等)的核酸,含有螯合化合物(例如:金属、具有放射活性的金属、硼、氧化性的金属等)的核酸、含有烷基化剂的核酸、具有经修饰的键的核酸(例如:α异头物型核酸等)等。对在本发明中可以使用dna的种类没有特别的限制,可以根据使用的目的适当选择,可列举例如,质粒dna、cdna、反义dna、染色体dna、pac、bac等,优选为质粒dna、cdna、反义dna,更优选为质粒dna。质粒dna等环状dna也可以适当利用限制酶等进行消化,而以线性dna形式使用。在本发明中可以使用的rna的种类没有特别的限制,可以根据使用的目的适当选择,可列举例如sirna、mirna、shrna、反义rna、信使rna(mrna)、单链rna基因组、双链rna基因组、rna复制子、转移rna、核糖体rna等,优选为sirna、mirna、shrna、mrna、反义rna、rna复制子。在本发明中使用的核酸优选通过本领域技术人员通常使用的方法进行纯化。对本发明的试剂而言,例如,可以出于疾病的预防和/或治疗的目的进行活体内(invivo)给予。因此,在本发明中使用的核酸优选相对于某给定的疾病具有预防和/或治疗活性(预防、治疗用核酸)。作为这样的核酸,可列举例如用于所谓基因治疗的核酸等。对向本发明的脂质膜结构体导入核酸的方法没有特别限制,例如,可以通过在形成本发明的脂质膜结构体时,使本发明的脂质膜结构体的构成成分与期望的核酸共存,来向本发明的脂质结构体导入核酸。例如,在利用乙醇稀释法形成本发明的脂质膜结构体时,通过将核酸的水溶液与本发明的脂质膜结构体的构成成分(脂质等)的乙醇溶液利用涡旋震荡等剧烈混合后,将混合物利用适当的缓冲液进行稀释,从而得到导入了核酸的本发明的脂质膜结构体的悬浮液。利用单纯水合法形成本发明的脂质膜结构体时,通过将本发明的脂质膜结构体的构成成分(脂质等)溶解于适当的有机溶剂,并将该溶液放入玻璃容器,通过减压干燥而蒸馏除去溶剂,得到脂质薄膜,然后,这里添加核酸的水溶液,使其水合后,利用超声波发生器进行超声波处理,从而得到导入了核酸的本发明的脂质膜结构体的悬浮液。作为本发明的试剂的一种形态,可列举例如,多功能性包络型纳米结构体(multifunctionalenvelope-typenanodevice,以下,称为“mend”)。该mend例如可以通过将核酸与聚阳离子(例如,鱼精蛋白等)之间的静电复合体导入本发明的脂质膜结构体等来制备(kogureketal.,multifunctionalenvelope-typenanodevice(mend)asanon-viralgenedeliverysystem.adv.drugdeliv.rev.,60,559-571(1march2008)。该结构体(mend)可以用作将核酸等选择地递送至特定的细胞内的药物递送系统,对例如基于向树突细胞导入抗原基因的dna疫苗、肿瘤的基因治疗等有用。导入了核酸的本发明的脂质膜结构体的表面电荷(zeta电位)优选为-10~+10mv,进一步优选为-10~+5mv。在以往的基因导入中,主要使用了表面电荷带正电的粒子。其理由在于促进与具有负电荷的细胞表面的硫酸肝素之间的静电相互作用,虽然作为用于促进细胞的摄取的方法有用,但就正的表面电荷而言,存在产生在细胞内由于与导入基因之间的相互作用而导致转录抑制、由于与mrna之间的相互作用而导致翻译抑制的可能性。通过在上述的范围内调整表面电荷,可解决该问题。表面电荷的测定,可以用metasizernano(malverninstrumentsltd.)进行。通过将本发明的脂质膜结构体的构成成分的组成在不破坏本发明的目的范围内适宜调整,可以将表面电荷调整为期望的值。通过使本发明的试剂与细胞接触,可以将本发明的试剂中包含的核酸导入到细胞内。对该“细胞”的种类没有特别限定,可使用原核生物及真核生物的细胞,优选为真核生物的细胞。对该真核生物的种类也没有特别限定,可列举例如:包括人的哺乳类(例如:人、猴、小鼠、大鼠、仓鼠、牛等)、鸟类(例如:鸡、鸵鸟等)、两栖类(例如:青蛙等)、鱼类(例如:斑马鱼、鳉鱼(稻田鱼)等)等脊椎动物;昆虫(例如:蚕、蛾、果蝇等)等非脊椎动物;植物;微生物(例如:酵母等)等。更优选的是,作为本发明的对象的细胞,优选为动物细胞(例如:脊椎动物细胞等)或者植物细胞,进一步优选为哺乳动物细胞。该细胞可以是包括癌细胞的培养细胞株,也可以是从个体、组织分离而成的细胞,或者组织或者组织片的细胞。另外,该细胞可以为贴壁细胞,也可以为非贴壁细胞。以下对在活体外(invitro)使本发明的试剂与细胞接触的方法进行具体说明。对细胞而言,在与本发明的试剂接触的数天前悬浮于适当的培养基中,以适当的条件进行培养。在与本发明的试剂接触的时候,细胞可以处于增殖期,也可以不是。该接触时的培养液可以是含血清培养基,也可以是不含血清培养基,优选培养基中的血清浓度为30重量%以下,更优选为20重量%以下。如果培养基中含有过量的血清等蛋白质,则存在阻碍本发明的试剂与细胞之间的接触的可能性。对该接触时的细胞密度没有特别限定,可以考虑细胞的种类等进行适当设定,通常为1×104~1×107细胞/ml的范围。向这样制备的细胞,例如,添加上述的导入了核酸的本发明的脂质膜结构体的悬浮液。对该悬浮液的添加量没有特别限定,可以考虑细胞数等进行适当设定。对与细胞接触时该悬浮液中的本发明的脂质膜结构体的浓度而言,只要能够实现目标核酸向细胞内的导入即可,没有特别限定,作为脂质浓度,通常为1~100nmol/ml,优选为10~50nmol/ml,作为核酸的浓度,通常为0.01~100μg/ml,优选为0.1~10μg/ml。将上述的悬浮液添加至细胞后,培养该细胞。对培养时的温度、湿度、co2浓度等而言,可以考虑细胞的种类进行适当设定。在细胞为源自哺乳动物的细胞时,通常温度为约37℃,湿度为约95%,co2浓度为约5%。另外,虽然培养时间也可以考虑使用的细胞的种类等条件进行适当设定,但通常为0.1~24小时的范围,优选为0.25~4小时的范围,更优选为0.5~2小时的范围。如果上述培养时间过短,则核酸不能充分地导入细胞内,如果培养时间过长,则细胞可能变虚弱。通过上述培养将核酸导入细胞内,优选为将培养基与新鲜的培养基交换,或向培养基中添加新鲜的培养基并进一步继续培养。在细胞为源自哺乳动物的细胞时,优选新鲜的培养基含有血清或营养因子。另外,如上所述,通过使用本发明的试剂,不仅在活体外(invitro),在活体内(invivo)中能够也将核酸导入细胞内。即,通过将本发明的试剂给予对象,例如,使导入了核酸的本发明的脂质膜结构体到达并与目标细胞接触,在活体内将已导入该脂质膜结构体中的核酸导入细胞内。对可以给予本发明的试剂的对象没有特别限定,可以举出例如:包括人的哺乳类(例:人、猴、小鼠、大鼠、仓鼠、牛等)、鸟类(例:鸡、鸵鸟等)、两栖类(例:青蛙等)、鱼类(例、斑马鱼、鳉鱼等)等脊椎动物,昆虫(例、蚕、蛾、果蝇等)等无脊椎动物、植物等。本发明的试剂的给予对象优选为人或其它哺乳动物。对目标细胞的种类没有特别限定,可以通过使用本发明的试剂,向各种组织(例如,肝、肾、胰腺、肺、脾、心脏、血液、肌肉、骨、脑,胃、小肠、大肠、皮肤、脂肪组织等)中的细胞导入核酸。另外,本发明的试剂中包含的脂质膜结构体,也可以导入有除了核酸以外的化合物。在本发明的试剂含有导入了除了核酸以外的化合物的脂质膜结构体的情况下,就给予本发明的试剂的对象(例如:脊椎动物、无脊椎动物等)的方法而言,只要是能够使该脂质膜结构体到达并与目标细胞接触,并将已导入至该脂质膜结构体的化合物导入细胞内的方法即可,没有特别限定,可以考虑导入化合物的种类、目标细胞的种类、部位等,适当选择本身公知的给予方法(例如:口服给予、非口服给予(例如:静脉内给予、肌肉内给予、局部给予、经皮给予、皮下给予、腹腔内给予、喷雾等)等)。对本发明的试剂的给予量而言,只要是能够实现化合物向细胞内的导入的范围即可,没有特别限定,可以考虑给予对象的种类、给予方法、导入化合物的种类、目标细胞的种类、部位等进行适当选择。对将化合物导入至本发明的脂质膜结构体的方法没有特别的限制,例如,可以通过与上述的向本发明的脂质膜结构体导入核酸的方法同样的方法,或基于其的方法制备。对本发明的试剂的剂型没有特别限制,可列举例如:注射剂(例如:皮下注射剂、静脉内注射剂、肌肉内注射剂、腹腔内注射剂、点滴注射剂等)等。本发明的试剂可以通过将本发明的脂质膜结构体按照相应于用途(例如研究用试剂、医药等)的常规方法进行制剂化而制造。在将本发明的试剂作为研究用试剂提供的情况下,对该本发明的试剂而言,可以将本发明的脂质膜结构体直接使用,或者使用其与例如水、或者除了水以外的生理上允许的液体(例如,水溶性溶剂(例如,苹果酸缓冲液等)、有机溶剂(例如:甲醇、乙醇、dmso等)或者水溶性溶剂和有机溶剂的混合液等)而成的无菌性溶液或者悬浮液进行提供。本发明的试剂可以适当含有本身公知的生理上允许的添加剂(例如:赋形剂、媒介物、防腐剂、稳定剂、粘合剂等)。另外,在将本发明的试剂作为医药提供的情况下,对该本发明的试剂而言,可以将本发明的脂质膜结构体直接使用,或者与医药上允许的公知的添加剂(例如:载体、香味剂、赋形剂、媒介物、防腐剂、稳定剂、粘合剂等)一起使用,通过以通常认定的制剂实施所要求的单位用量形态进行混合,以口服剂(例如:片剂、胶囊剂等)或者非口服剂(例如注射剂、喷雾剂等)的形式,优选为非口服剂(更优选注射剂)的形式进行制造。本发明的试剂除了成人用以外,也可以作为儿童用的制剂。本发明的试剂也能够以试剂盒的形态进行提供。该试剂盒除了本发明的脂质膜结构体及核酸此外,可以包含向细胞导入核酸时使用的试剂。在一种形态中,本发明的试剂(或试剂盒)可以进一步包含聚阳离子(例如:鱼精蛋白等)。本发明的试剂(或试剂盒),可以通过进一步包含聚阳离子(例如:鱼精蛋白等),容易地将核酸与聚阳离子(例如:鱼精蛋白等)而成的静电复合体导入至本发明的脂质膜结构体而制备mend。该mend可用于核酸的细胞内导入。实施例以下,举出实施例对本发明进行详细说明,但本发明不限于该实施例。在实施例的说明中使用的简称的含义,分别如下所述。lin-ms:甲磺酸亚油酰酯mim:3,4-o-异亚丙基-d-甘露醇tlmim:1,2,5,6-四亚油酰基-3,4-o-异亚丙基-d-甘露醇tlm:1,2,5,6-四亚油酰基-d-甘露醇tlm-c2-br:1,2,5,6-四亚油酰基-3,4-二(溴乙酰基)-d-甘露醇dmap:4-二甲基氨基吡啶chol:胆甾醇peg2000-dmg:1,2-二肉豆蔻酰-sn-丙三醇,甲氧基聚乙二醇(peg分子量:2000)decenyl-ms:甲基磺酸癸烯基酯tdmim:1,2,5,6-四(癸烯基)-3,4-o-异亚丙基-d-甘露醇tdm:1,2,5,6-四(癸烯基)-d-甘露醇tbdps-cl:叔丁基二苯基氯硅烷dipea:二异丙基乙基胺dtbdps-m:1,6-二-(叔丁基二苯基甲硅烷基)-d-甘露醇dtbdps-tlmes:1,6-二-(叔丁基二苯基甲硅烷基)-2,3,4,5-四亚油酰基-d-甘露醇tlmes:四亚油酰基-d-甘露醇tbaf:四丁基氟化铵在表2及表3中,示出了以下的实施例及比较例中制造的阳离子脂质的名称和结构。[表2][表3][制造例1]<甲硅烷基保护>dtbdps-m的合成向d-甘露醇(东京化成工业株式会社制)3.0g(16.5mmol)中添加n,n-二甲基甲酰胺48ml及dipea(关东化学株式会社制)6.4g(49.4mmol),一边搅拌一边冷却至0~10℃。向其中滴加添加将tbdps-cl(东京化成工业株式会社制)13.6g(49.4mmol)溶解于n,n-二甲基甲酰胺16ml而成的溶液并使温度不超过10℃。在滴加结束后,升温至20℃,搅拌2.5小时。通过tlc分析(展开溶剂:氯仿/甲醇=9/1(v/v),以过锰酸钾显色)确认了d-甘露醇及单甲硅烷基体消失,结束反应。向反应溶液加入离子交换水120ml及甲苯60ml,搅拌10分钟,之后静置10分钟使其分层。用离子交换水40ml再次水洗得到的甲苯层之后,浓缩了甲苯层。将得到的浓缩物质溶解于乙腈170ml之后,利用己烷170ml进行5次提取纯化。蒸馏除去得到的乙腈层的溶剂,得到了9.2g的dtbdps-m。通过1h-nmr(600mhz,cdcl3)分析得到的dtbdps-m,确认为目的物质。δ1.03ppm(s,18h,(ch3-)3c-),δ3.79-3.89ppm(m,8h,-o-ch2-ch(-oh)-ch(-oh)-),δ7.25-7.72ppm(m,20h,tbu-si(-ph)2-)[实施例1](tlm-c2-dma的合成)<甲磺酰化>lin-ms的合成向脱水甲苯500g加入亚油酸醇100g(日油株式会社制,纯度≥99%)(0.38mol)、三乙胺(关东化学株式会社制)46g(0.45mol)并溶解,在氮气体氛围中一边搅拌一边冷却至10℃。以温度为30℃以下的方式,经2小时滴加了甲磺酰氯(关东化学株式会社制)47g(0.41mol)。滴加结束后,通过tlc分析(展开溶剂:氯仿,磷酸硫酸铜显色)确认了亚油酸醇的点已消失。添加乙醇5.2g(0.11mol),通过滤纸滤去不溶物。利用离子交换水150g洗涤滤液,并废弃水层。再次进行水洗之后,向得到的有机层添加无水硫酸镁20g进行脱水处理。通过滤纸滤去不溶物,用蒸发仪蒸馏除去滤液的溶剂,得到了120g的lin-ms。通过1h-nmr(600mhz,cdcl3)分析得到的lin-ms,确认为目的物质。δ0.89ppm(t,3h,ch3-ch2-),δ1.41-1.26ppm(m,16h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.75ppm(quint,2h,-ch2-ch2-o-),δ2.05ppm(q,4h,-ch2-ch=ch-ch2-ch=ch-ch2-),δ2.77ppm(t,2h,-ch=ch-ch2-ch=ch-),δ3.00ppm(s,3h,-so2-ch3),δ4.22ppm(t,2h,-ch2-o-),δ5.41-5.31ppm(m,4h,-ch=ch-ch2-ch=ch-)<醚化>tlmim的合成向甲苯40g添加mim(东京化成工业株式会社制)2.0g(9.00mmol),进一步添加氢氧化钾(关东化学株式会社制)4.2g(74.50mmol)、lin-ms18.6g(54.00mmol),在25℃搅拌5分钟。随后升温至80℃,搅拌了14小时。根据1h-nmr分析,确认源自反应产物的峰出现,源自lin-ms峰的积分值的减少停止,并终止反应。向反应溶液添加甲苯60ml和离子交换水100ml,在20℃搅拌10分钟,之后静置10分钟进行分层。除去水层之后,再次进行水洗。接着添加25wt%的食盐水100ml,搅拌10分钟,之后静置10分钟进行分层,除去水层。向得到的有机层添加无水硫酸镁2.0g,进行脱水处理。通过滤纸滤去不溶成分,用蒸发仪蒸馏除去滤液的溶剂,得到了褐色液体11.5g。将得到的褐色液体10g利用硅胶柱色谱进行纯化(洗脱液:己烷/乙酸乙酯=100/0~98/2(v/v)),得到了tlmim3.0g。通过1hnmr(600mhz,cdcl3)分析得到的tlmim,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),δ1.38-1.29ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.38ppm(s,6h,-o-c(ch3)2-o-),δ1.56ppm(m,8h,-ch2-ch2-o-),δ2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),δ2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),δ3.54-3.41ppm(m,10h,-ch2-ch2-o-,-o-ch2-ch-ch-o-c(ch3)2-),δ3.67ppm(m,4h,-o-ch2-ch-ch-o-c(ch3)2-),δ4.06ppm(d,2h,-o-ch2-ch-ch-o-c(ch3)2-),δ5.40-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-)<脱保护>tlm的合成向tlmim6.7g(5.51mmol)添加乙醇67ml、离子交换水4.0g(220.40mmol)、盐酸(4.0m二噁烷溶液)(东京化成工业株式会社制)13.8ml(以盐酸计为55.10mmol),在60℃搅拌6h。通过tlc分析(展开溶剂:氯仿/甲醇=99.5/0.5(v/v),磷酸硫酸铜显色),确认tlmim的点已消失,结束反应。一边将反应液冷却至5℃一边静置13h进行分层,回收了有机层。回收的有机层利用氮气鼓泡而蒸馏除去溶剂,得到了微褐色液体5.1g。将得到的微褐色液体4.6g利用硅胶柱色谱进行纯化(洗脱液:己烷/乙酸乙酯=98/2~9/1(v/v)),得到了tlm3.5g。通过1hnmr(600mhz,cdcl3)分析得到的tlm,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),δ1.38-1.28ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.56ppm(m,8h,-ch2-ch2-o-),2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),3.30ppm(d,2h,-oh),3.51-3.42ppm(m,6h,-o-ch2-ch-ch-oh,-o-ch2-ch-ch-oh),3.68-3.52ppm(m,8h,-ch2-ch2-o-),3.82ppm(d,2h,-o-ch2-ch-ch-oh),5.39-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-)<酯化>tlm-c2-br的合成将tlm1.0g(0.85mmol)和溴乙酸(东京化成工业株式会社制)354.5mg(2.55mmol)溶解于氯仿10ml后,添加dmap(广荣化学工业株式会社制)51.9mg(0.43mmol)、dic(东京化成工业株式会社制)321.8mg(2.55mmol),在25℃搅拌1小时。通过tlc分析(展开溶剂:仅氯仿,磷酸硫酸铜显色),确认tlm的点已消失。随后,利用离子交换水10ml、25wt%食盐水10ml洗涤了反应液,并回收有机层。向回收的有机层添加无水硫酸镁1.0g进行脱水处理,通过滤纸过滤分离不溶成分。用蒸发仪蒸馏除去滤液的溶剂,得到了微褐色液体1.6g。将得到的微褐色液体利用硅胶色谱进行纯化(洗脱液:己烷/乙酸乙酯=100/0~97/3(v/v)),得到了tlm-c2-br980mg。通过1hnmr(600mhz,cdcl3)分析得到的tlm-c2-br,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),1.38-1.27ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),1.52ppm(m,8h,-ch2-ch2-o-),2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),3.51-3.37ppm(m,10h,-ch2-ch2-o-,-o-ch2-ch-ch-o-co-),3.60ppm(m,4h,-o-ch2-ch-ch-o-co-),3.84ppm(s,4h,-o-co-ch2-),5.40-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-),5.51ppm(d,2h,-o-ch2-ch-ch-o-co-)<氨基化>tlm-c2-dma的合成将tlm-c2-br300mg(0.21mmol)溶解于thf2.2ml之后,添加二甲基胺(2.0mthf溶液)(东京化成工业株式会社制)846μl(以二甲基胺计1.68mmol),在25℃搅拌4小时。通过tlc分析(展开溶剂:氯仿/甲醇=96/4(v/v),磷酸硫酸铜显色),确认了反应产物的点出现,以及作为原料的tlm-c2-br的点消失。向反应溶液添加氯仿8ml、离子交换水10ml,搅拌10分钟后,静置10分钟进行分层,除去水层。随后,进行利用离子交换水10ml的水洗3次,利用25wt%食盐水10ml的水洗一次,回收有机层。向回收的有机层添加无水硫酸镁0.5g,进行脱水处理。通过滤纸滤去不溶成分,用蒸发仪蒸馏除去滤液的溶剂,得到了tlm-c2-dma260mg。通过1hnmr(600mhz,cdcl3)分析得到的tlm-c2-dma,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),1.38-1.27ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),1.52ppm(m,8h,-ch2-ch2-o-),2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),2.35ppm(s,12h,-n-(ch3)2),2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),3.16ppm(q,4h,-o-co-ch2-),3.49-3.35ppm(m,10h,-ch2-ch2-o-,-o-ch2-ch-ch-o-co-),3.55ppm(m,4h,-o-ch2-ch-ch-o-co-),5.40-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-),5.47ppm(d,2h,-o-ch2-ch-ch-o-co-)[实施例2](tlm-c3-dma的合成)<酯化>将tlm1.0g(0.85mmol)和二甲基氨基丙酸盐酸盐(东京化成工业株式会社制)783.8mg(5.10mmol)溶解于氯仿10ml之后,添加dmap51.9mg(0.43mmol)、dcc(tama化学工业株式会社制)1.1g(5.10mmol)、在25±5℃搅拌1小时。通过tlc分析(展开溶剂:氯仿/甲醇=85/15(v/v),磷酸硫酸铜显色),确认tlm的点已消失。通过滤纸滤去反应液中的不溶物,向得到的滤液添加氯仿10ml、离子交换水20ml、甲醇30ml洗涤,回收有机层。进一步向有机层添加离子交换水20ml、甲醇40ml洗涤,回收有机层。向回收的有机层添加无水硫酸镁1.0g,进行脱水处理。通过滤纸滤去不溶成分,用蒸发仪蒸馏除去滤液的溶剂,得到了微褐色液体1.6g。将得到的微褐色液体利用硅胶色谱进行纯化(洗脱液:氯仿/甲醇=98/2~96/4(v/v)),得到了tlm-c3-dma102mg。通过1hnmr(600mhz,cdcl3)分析得到的tlm-c3-dma,确认为目的物质。δ0.89(t,12h,ch3-ch2-),δ1.38-1.27(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.52(m,8h,-ch2-ch2-o-),δ2.05(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),δ2.23(s,12h,-n-(ch3)2),δ2.49(t,4h,-o-co-ch2-ch2-),δ2.60(m,4h,-o-co-ch2-ch2-),δ2.77(t,8h,-ch=ch-ch2-ch=ch-),δ3.43(m,8h,-ch2-ch2-o-),δ3.53(m,6h,-o-ch2-ch-ch-o-co-,-o-ch2-ch-ch-o-co-),δ5.39-5.30(m,18h,-ch=ch-ch2-ch=ch-,-o-ch2-ch-ch-o-co-)[实施例3](tlm-c4-dma的合成)<酯化>将tlm0.5g(0.43mmol)和二甲基氨基丁酸盐酸盐(acrosorganics;thermofisherscientific公司制)427.6mg(2.55mmol)溶解于氯仿5ml之后,添加51.9mg的dmap(0.43mmol)、321.8mg的dic(tama2.55mmol),在25℃搅拌4小时。通过tlc分析(展开溶剂:氯仿/甲醇=85/15(v/v),磷酸硫酸铜显色),确认tlm的点已消失。随后,向反应液添加离子交换水5ml、乙醇5ml洗涤,回收有机层。再次进行相同的洗涤,通过向回收的有机层添加硫酸镁0.5g进行脱水处理。通过滤纸滤去不溶成分,用蒸发仪蒸馏除去滤液的溶剂,得到了微褐色液体515.8mg。将得到的微褐色液体中的400mg利用硅胶色谱进行纯化(洗脱液:氯仿/甲醇=96/4~8/2(v/v)),得到了tlm-c4-dma244mg。通过1hnmr(600mhz,cdcl3)分析得到的tlm-c4-dma,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),δ1.38-1.27ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.53ppm(m,8h,-ch2-ch2-o-),δ1.77ppm(quint,4h,-ch2-ch2-ch2-n(ch3)2),δ2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),δ2.21ppm(s,12h,-n-(ch3)2),δ2.28ppm(t,4h,-ch2-ch2-ch2-n(ch3)2),δ2.34ppm(m,4h,-ch2-ch2-ch2-n(ch3)2),δ2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),δ3.54-3.38ppm(m,14h,ch2-ch2-o-,-ch2-ch-ch-o-co-,-ch2-ch-ch-o-co-),δ5.40-5.32ppm(m,18h,-ch=ch-ch2-ch=ch-,-ch2-ch-ch-o-co-)[实施例4](tdm-c3-dma的合成)<甲磺酰化>decenyl-ms的合成向脱水甲苯50g添加癸烯醇(aldrich公司制)10.0g(64.0mol)、三乙胺7.8g(76.8mol)并使其溶解,在氮气体氛围中一边搅拌一边冷却至10℃。以温度为30℃以下的方式,经30分钟滴加了甲磺酰氯8.1g(70.4mol)。滴加结束后,通过tlc分析(展开溶剂:氯仿,磷酸硫酸铜显色)确认了癸烯醇的点已消失。添加乙醇0.9g(19.2mol),通过滤纸滤去不溶物。利用离子交换水20g洗涤滤液,并废弃水层。再次进行水洗之后,向得到的有机层添加无水硫酸镁5g进行脱水处理。通过滤纸滤去不溶物,用蒸发仪蒸馏除去滤液的溶剂,得到了decenyl-ms14.5g。通过1h-nmr(600mhz,cdcl3)分析得到的decenyl-ms,确认为目的物质。δ0.89ppm(t,3h,ch3-ch2-),δ1.26-1.36ppm(m,6h,ch3-ch2-ch2-ch2-),δ1.81ppm(quint,2h,-ch2-ch2-o-),δ2.02ppm(q,2h,ch3-ch2-ch2-ch2-ch=),δ2.16ppm(q,2h,=ch-ch2-ch2-ch2-o-),δ3.00ppm(s,3h,-so2-ch3),δ4.23ppm(t,2h,-ch2-o-),δ5.32ppm,δ5.45ppm(q,2h,-ch=ch-)<醚化>tdmim的合成向mim1.8g(8.1mmol)添加甲苯36g,进一步添加氢氧化钾3.6g(64.8mmol)、decenyl-ms11.4g(48.6mmol),在25℃搅拌5分钟。随后升温至80℃,搅拌14小时。通过tlc分析(展开溶剂:氯仿,磷酸硫酸铜显色),确认decenyl-ms的残存量变得低于10%,结束反应。向反应溶液添加甲苯42ml和离子交换水72ml,在20℃搅拌10分钟,之后静置10分钟进行分层。除去水层之后,再次进行水洗。接着添加20wt%的食盐水72ml,搅拌10分钟,之后静置10分钟进行分层,除去水层。向得到的有机层添加无水硫酸镁3.6g,进行脱水处理。通过滤纸滤去不溶成分,用蒸发仪蒸馏除去滤液的溶剂,得到了褐色液体7.5g。将得到的褐色液体7.5g利用硅胶柱色谱进行纯化(洗脱液:己烷/乙酸乙酯=100/0~98.5/1.5(v/v)),得到了tdmim5.3g。通过1h-nmr(600mhz,cdcl3)分析得到的tdmim,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),δ1.26-1.38ppm(m,24h,ch3-ch2-ch2-ch2-),δ1.38ppm(s,6h,-o-c(ch3)2-o-),δ1.63ppm(m,8h,-ch2-ch2-o-),δ2.02ppm(q,8h,ch3-ch2-ch2-ch2-ch2-),δ2.77ppm(t,8h,-ch2-ch2-ch2-o-),δ3.54-3.41ppm(m,10h,-ch2-ch2-o-,-o-ch2-ch-ch-o-c(ch3)2-),δ3.67ppm(m,4h,-o-ch2-ch-ch-o-c(ch3)2-),δ4.06ppm(d,2h,-o-ch2-ch-ch-o-c(ch3)2-),δ5.40-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-)<脱保护>tdm的合成向tdmim5.0g(6.5mmol)添加乙醇50ml、离子交换水4.6g(258.0mmol)、盐酸(4m二噁烷溶液)16.1ml(64.5mmol),在60℃搅拌3小时。通过tlc分析(展开溶剂:氯仿/甲醇=99.5/0.5(v/v),磷酸硫酸铜显色),确认tdmim和作为中间体的单异亚丙基体消失,结束反应。向反应液添加己烷50ml,在25℃搅拌10分钟,之后静置10分钟进行分层。回收上层(己烷层),向其中添加乙腈50ml。在25℃搅拌10分钟,之后静置10分钟进行分层。除去乙腈层之后,再次进行乙腈洗涤。蒸馏除去得到的己烷层的溶剂,得到了微褐色液体4.1g。将得到的微褐色液体4.0g利用硅胶柱色谱进行纯化(洗脱液:己烷/乙酸乙酯=98/2~95/5(v/v)),得到了tdm2.6g。通过1h-nmr(600mhz,cdcl3)分析得到的tdm,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),δ1.25-1.35ppm(m,24h,ch3-(ch2)3-),δ1.63ppm(m,8h,-ch2-ch2-o-),2.01ppm(q,8h,ch3-(ch2)3-ch2-),δ2.77ppm(t,8h,-ch2-ch2-ch2-o-),3.26-3.84ppm(m,18h,-o-ch2-ch-ch-oh,-ch2-ch2-o-),5.33-5.38ppm(m,8h,-ch=ch-)<酯化>tdm二丙烯酸酯体的合成向脱水甲苯5.0g添加tdm500mg(0.68mmol)、三乙胺275mg(2.72mmol)并搅拌。向其中滴加了溶解于脱水甲苯1.0g的丙烯酰氯246mg(2.72mmol)。在25℃搅拌1h,之后过滤析出物质,得到了tdm二丙烯酸酯体的甲苯溶液。<氨基化>向tdm二丙烯酸酯体的甲苯溶液添加2.0m二甲基胺/四氢呋喃溶液1.7ml(二甲基胺3.40mmol),在70℃搅拌1h。反应溶液冷却至25℃,添加10wt%食盐水5.0g,搅拌10分钟,之后静置10分钟进行分层。除去下层(水层),向上层(甲苯层)添加25wt%食盐水5.0g,搅拌10分钟,之后静置10分钟进行分层。除去下层(水层),利用无水硫酸镁500mg使上层(甲苯层)脱水,过滤之后,浓缩滤液,得到了微黄色液体406mg。将得到的微黄色液体中300mg利用硅胶柱色谱进行纯化(洗脱液:己烷/乙酸乙酯=99/1~95/5(v/v)),得到了241mg的tdm-c3-dma。通过1hnmr(600mhz,cdcl3)分析得到的tdm-c3-dma,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),δ1.27-1.38ppm(m,24h,ch3-(ch2)3-),δ1.63ppm(m,8h,-ch2-ch2-o-),δ2.01ppm(q,8h,ch3-(ch2)3-ch2-),δ2.23ppm(s,12h,-n-(ch3)2),δ2.49ppm(t,4h,-o-co-ch2-ch2-),δ2.60ppm(m,4h,-o-co-ch2-ch2-),δ2.77ppm(t,8h,-ch2-ch2-ch2-o-),δ3.43(m,8h,-ch2-ch2-o-),δ3.53ppm(m,6h,-o-ch2-ch-ch-o-co-),δ5.39-5.30ppm(m,10h,-ch=ch-,-o-ch2-ch-ch-o-co-)[实施例5](tlmes-c3-dma的合成)<酯化>dtbdps-tlmes的合成向氯仿45ml加入dtbdps-m4.0g(6.1mmol)、亚油酸(日油公司制,纯度≥99%)9.4g(33.4mmol)、0.7g的dmap(6.1mmol),使其溶解。向其中添加7.6g的edc(39.5mmol),在30℃搅拌5小时。通过tlc分析(展开溶剂:氯仿,过磷酸硫酸铜显色),确认dtbdps-m及作为中间体的单~三酯体消失,结束反应。从反应溶液蒸馏除去溶剂之后,溶解于己烷60ml中。向己烷溶液添加乙腈30ml,在25℃搅拌10分钟,之后静置10分钟,进行分层。回收己烷层,通过蒸馏除去溶剂,得到微黄色液体11.1g。将得到的微黄色液体10.0g利用硅胶柱色谱进行纯化(洗脱液:己烷/乙酸乙酯=99.5/0.5~99/1(v/v)),得到了6.8g的dtbdps-tlmes。通过1h-nmr(600mhz,cdcl3)分析得到的dtbdps-tlmes,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),δ1.03ppm(s,18h,(ch3-)3c-),δ1.25-1.37ppm(m,64h,ch3-(ch2)3-,=ch-(ch2)4-ch2-ch2-),δ1.47-1.54ppm(m,8h,-ch2-ch2-co-o-),δ2.04ppm(q,16h,-ch2-ch2-ch=ch-),δ2.11-2.32(m,8h,ch2-co-o-),δ2.77ppm(t,8h,=ch-ch2-ch=),δ3.62-3.74ppm(m,4h,-o-ch2-ch-),δ4.99ppm(m,2h,-o-ch2-ch-ch-),δ5.32-5.39ppm(m,16h,-ch=ch-ch2-ch=ch-),δ5.57ppm(m,2h,-o-ch2-ch-ch-),δ7.33-7.63ppm(m,20h,tbu-si(-ph)2-)<脱保护>tlmes的合成将3.00g的dtbdps-tlmes(1.8mmol)溶解于四氢呋喃中,一边搅拌一边冷却至5℃。通过以不超过10℃的方式进行滴加,依次添加了乙酸(关东化学株式会社制)0.7g(12.3mmol)、及tbaf(1m四氢呋喃溶液)(东京化成工业株式会社制)10.5ml(10.5mmol)。滴加后,在25℃搅拌7小时。通过tlc分析(展开溶剂:氯仿,磷酸硫酸铜显色),确认dtbdps-tlmes及作为中间体的单甲硅烷基体消失,结束反应。用氯仿30ml稀释反应溶液之后,添加5wt%碳酸氢钠水溶液30ml,在25℃搅拌10分钟。搅拌后静置10分钟进行分层,回收有机层。得到的有机层进一步利用离子交换水30ml进行水洗,之后蒸馏除去溶剂,得到微黄色液体3.0g。将得到的微黄色液体2.8g利用硅胶柱色谱进行纯化(洗脱液:己烷/乙酸乙酯=97/3~80/20(v/v)),得到了1.9g的tlmes。通过1h-nmr(600mhz,cdcl3)分析得到的tlmes,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),δ1.25-1.37ppm(m,64h,ch3-(ch2)3-,=ch-(ch2)4-ch2-ch2-),δ1.61ppm(m,8h,-ch2-ch2-co-o-),δ2.04ppm(q,16h,-ch2-ch2-ch=ch-),δ2.26-2.37(m,8h,-ch2-co-o-),δ2.77ppm(t,8h,=ch-ch2-ch=),δ2.90-5.23ppm(m,8h,-o-ch2-ch-ch-),δ5.32-5.39ppm(m,16h,-ch=ch-ch2-ch=ch-)<酯化>tlmes二丙烯酸酯体的合成向脱水甲苯17g添加1.7g的tlmes(1.4mmol)、三乙胺0.6g(5.5mmol),在25℃搅拌。向其中滴加了溶解于脱水甲苯3.4g的丙烯酰氯0.5g(5.5mmol)。在25℃搅拌1h之后,过滤析出物质,得到了tlmes二丙烯酸酯体的甲苯溶液。<氨基化>tlmes-c3-dma的合成向tlmes二丙烯酸酯体的甲苯溶液添加2.0m二甲基胺/四氢呋喃溶液6.9ml(13.8mmol),在70℃搅拌1h。将反应溶液冷却至25℃,添加10wt%食盐水17g搅拌10分钟,之后静置10分钟进行分层。除去下层(水层),向上层(甲苯层)添加25wt%食盐水17g,搅拌10分钟,之后静置10分钟进行分层。除去下层(水层),利用无水硫酸镁1.0g使上层(甲苯层)脱水,过滤之后,浓缩滤液,得到了微黄色液体1.4g。将得到的微黄色液体1.4g利用硅胶柱色谱进行纯化(洗脱液:己烷/乙酸乙酯=99/1~97/3(v/v)),得到了0.5g的tlmes-c3-dma。通过1h-nmr(600mhz,cdcl3)分析得到的tlmes-c3-dma,确认为目的物质。δ0.89ppm(t,12h,ch3-ch2-),δ1.25-1.37ppm(m,64h,ch3-(ch2)3-,=ch-(ch2)4-ch2-ch2-),δ1.61ppm(m,8h,-ch2-ch2-co-o-),δ2.06ppm(q,16h,-ch2-ch2-ch=ch-),δ2.22-2.36(m,20h,-(ch2)6-ch2-co-o-,(ch3)2-n-),δ2.47-2.64ppm(m,8h,(ch3)2-n-(ch2)2-co-o-),δ2.78ppm(t,8h,=ch-ch2-ch=),δ4.04ppm,δ4.30ppm,δ5.11ppm,δ5.47ppm(m,8h,-o-ch2-ch-ch-),δ5.32-5.39ppm(m,16h,-ch=ch-ch2-ch=ch-)[实施例6]各种mend的制备(1)由sirna和鱼精蛋白构成的核酸静电复合体的形成将sirna(hokkaidosystemscience株式会社)以2mg/ml、4mg/ml的方式溶解于ultrapurednase/rnase-freedistilledwater(invitrogen;thermofischerscientific公司),制备了sirna溶液。作为载体的核,将该sirna溶液、鱼精蛋白溶液(calbiochem;merckmillipore公司)用10mmhepes缓冲液分别稀释至0.3mg/ml、0.2mg/ml,一边搅拌0.3mg/mlsirna250μl一边每次少量滴加0.2mg/ml鱼精蛋白250μl,制备了sirna与鱼精蛋白的静电复合体(以下,称为“sirna复合体”)(n/p比=1.0)。作为针对factorvii(以下,称为“fvii”)的sirna的序列,使用了akincetal.,moleculartherapy,17(5),872-879(may2009)中记载的序列(未经化学修饰)。(2)封装sirna的mend的制备(作为阳离子脂质,单独使用了tlm-c2-dma、tlm-c3-dma、tlm-c4-dma、tdm-c3-dma、tlmes-c3-dma、dlindap、dodap)将阳离子脂质(tlm-c2-dma(实施例1)、tlm-c3-dma(实施例2)、tlm-c4-dma(实施例3)、tdm-c3-dma(实施例4)、tlmes-c3-dma(实施例5)、dlindap(比较例1)、dodap(比较例2))的90%丁醇溶液和chol溶液,以阳离子脂质:chol=7:3的摩尔比,以总脂质为3000nmol的方式在1.7ml管中混合。进一步,将作为peg脂质的peg2000-dmg溶液以相对于总脂质的3mol%进行添加,分别以使得总量为400μl的方式添加90%丁醇,制成了脂质溶液。在另外的1.7ml管中,将sirna复合体(sirna含量:160μg)与包含130mmnacl的20mm柠檬酸缓冲液(ph4)进行混合,使得总计114μl,制备了sirna溶液。一边通过涡旋震荡混合器搅拌脂质溶液,一边添加sirna溶液进行混合。用1ml进样针(27g)取该混合溶液的总量,缓慢注入正在剧烈搅拌的柠檬酸缓冲液2ml中(5ml管)。用磷酸缓冲生理盐水(以下,称为“pbs”)进行稀释,使用amiconultra-15100kdevice(merckmillipore公司),进行超滤(1000g,15分钟,30℃)而浓缩。随后,用pbs进行稀释,再次进行超滤而浓缩。最后,用pbs调整使得形成为目标脂质浓度,得到了封装sirna的mend。通过该操作制备的mend根据使用的阳离子脂质不同,以下称为“tlm-c2-dmamend”、“tlm-c3-dmamend”、“tlm-c4-dmamend”、“tdm-c3-dmamend”、“tlmes-c3-dmamend”、“dlindapmend”、“dodapmend”。(3)封装sirna的mend的制备(作为阳离子脂质,混合tlm-c2-dma、tlm-c3-dma、tlm-c4-dma中的2种进行使用)将tlm-c2-dma(实施例1)、tlm-c3-dma(实施例2)、tlm-c4-dma(实施例3)的以表4所述的摩尔比混合而成的脂质的90%丁醇溶液(tlm-cx-dmamix),分别以tlm-cx-dmamix:chol=7:3的摩尔比,以总脂质为3000nmol的方式在1.7ml管中混合。进一步,将作为peg脂质的peg2000-dmg以相对于总脂质的3mol%的量进行添加,分别以使得总量为400μl的方式添加90%丁醇,制成了脂质溶液。在另外的1.7ml管中,将sirna复合体(sirna含量:160μg)与10mm的苹果酸缓冲液(ph7.4)进行混合使得总计50μl,制备了sirna溶液。一边用涡旋震荡混合器搅拌脂质溶液,一边添加sirna溶液进行混合。用1ml进样针(27g)取该混合溶液的总量,缓慢注入正在剧烈搅拌的苹果酸缓冲液2ml中(5ml管)。用pbs进行稀释,使用amiconultra-15100kdevice(merckmillipore公司)进行超滤(1000g,10分钟,30℃)而浓缩。随后,用pbs进行稀释,再次进行超滤而浓缩。最后,用pbs调整溶液使得形成为目标脂质浓度,得到了封装sirna的mend。通过该操作制备的mend,如表4记载所述,根据使用的阳离子脂质的比率不同,以下称为“第1mend”、“第2mend”、“第3mend”、“第4mend”。[表4]mend名称脂质混合比第1mendtlm-c2-dma:tlm-c3-dma=0.21:0.79第2mendtlm-c3-dma:tlm-c4-dma=0.75:0.25第3mendtlm-c3-dma:tlm-c4-dma=0.50:0.50第4mendtlm-c3-dma:tlm-c4-dma=0.25:0.75(4)利用乙醇稀释法的mrna封装mend的制备(分别使用tlm-c3-dma、tdm-c3-dma)将阳离子脂质(tlm-c3-dma(实施例2)、tdm-c3-dma(实施例4))的99.5%乙醇溶液以阳离子脂质:dope:chol=3:3:4的比率,以总脂质量为131nmol的方式在5ml管中混合。另外,将作为peg脂质的peg2000-dmg以相对于总脂质的3mol%的量进行添加,使得总量为30ml。准备了这样的4管。另外在另外准备的1.5ml管中,添加编码荧光素酶的mrna(用mmessagemmachinet7ultratranscriptionkit(lifetechnologies公司)制备)3mg,这里添加包含30mmnacl的20mm苹果酸缓冲液(ph3.0)使得总量为45ml,此溶液也与脂质溶液同样准备了4管。将脂质溶液一边涡旋震荡一边与mrna溶液混合,接着添加925ml的100mm2-吗啉代乙磺酸(以下,称为“mes”)缓冲液(ph5.5)进行混合,并滗析进入预先添加了2ml的mes缓冲液的vivaspinturbo15(sartorius公司制。以下,称为“vivaspin”。)中。进一步向该5ml管添加2ml的mes缓冲液,并涡旋震荡进行洗涤,相同地滗析进入vivaspin。该操作重复4次,最后向vivaspin直接添加2ml的mes缓冲液,在25℃,1000g离心进行超滤。进一步添加pbs充分稀释,在相同条件下进行超滤。用pbs调整该溶液使其成为目标浓度,得到了mrna封装mend。通过该操作制备的mend根据使用的阳离子脂质不同,以下称为“tlm-c3-dmammend”、“tdm-c3-dmammend”。作为编码荧光素酶的mrna的序列,使用了miuraetal.,nucleicacidsresearch,43(3),1317-1331(2015)的“supplementarydata”中记载的序列。[实施例7]各种mend的粒径、多分散度、表面电位、sirna封装率、sirna回收率的测定对各种mend的粒径、多分散度以及表面电位用动态光散射法(zetasizernano;malverninstrumentsltd.)进行了测定。对sirna封装率及sirna回收率而言,用ribogreen(invitrogen;thermofischerscientific公司)进行了测定。将实施例6(2)或(3)中制备的各种mend用10mmhepes缓冲液(ph7.4)稀释成1000ng/ml,将其制成样品溶液。另外,将mend的制备中使用的sirna复合体用10mmhepes缓冲液(ph7.4)阶梯性稀释成0~2000ng/ml,将其制成标准曲线溶液。在这些溶液之外,准备了用10mmhepes缓冲液将葡聚糖硫酸、tritonx-100、ribogreen分别稀释成0.08mg/ml、0.4%、5μl/ml而成的测定溶液。另外,也准备了用10mmhepes缓冲液替换tritonx-100而成的溶液。向96孔板添加标准曲线溶液或样品溶液50μl,进一步分别加入含有或者不含tritonx-100的测定溶液50μl并混合,以700rpm搅拌5分钟之后,测定了在激发波长500nm及观测波长525nm的荧光强度。通过用含有tritonx-100的条件下测定时的sirna量除以1000ng/ml,算出了sirna回收率。再从以含有tritonx-100的条件测定时的sirna量减去以不含tritonx-100的条件测定时的sirna量,通过用含有tritonx-100的条件测定时的sirna量除以该值,算出了sirna封装率。结果示于表5及表6。[表5][表6][实施例8]mend的pka评价准备了符合在ph3.0~10.0的范围的各种ph的、包含终浓度150mm的nacl的20mm的柠檬酸缓冲液、磷酸钠缓冲液及trishcl缓冲液。向这些缓冲液中添加了实施例6(2)或(3)中制备的mend使得脂质浓度为30μm,进一步以使得为6μm的方式添加了6-(对甲苯胺基)-2-萘磺酸钠盐,使最终容量为100μl。随后,在37℃测定了在激发波长321nm及观测波长447nm的荧光强度。以各mend中荧光强度的最大值为100%,最小值为0%,以百分率计,算出相对荧光强度。另外,将相对荧光强度为50%的ph作为pka。结果示于表7及表8。[表7]mend名称mend的pkatlm-c2-dmamend4.67tlm-c3-dmamend5.89tlm-c4-dmamend6.83tdm-c3-dmamend6.14tlmes-c3-dmamend5.78dlindapmend5.34dodapmend5.44[表8]mend名称mend的pka第1mend5.81第2mend6.28第3mend6.56第4mend6.74[实施例9]tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend的膜融合能力试验从雄性icr小鼠采血,回收红细胞,悬浮于生理盐水。将包含一定量的红细胞的生理盐水,添加至pbs(ph7.4)或10mm磷酸-10mm苹果酸缓冲生理盐水(ph6.5、5.5)。接下来,添加含有实施例6(2)制备的mend的pbs溶液,使脂质终浓度为300μmol/l。另外,以添加了不含有mend的相同量的pbs的溶液作为阴性对照(nc),将以下液体作为阳性对照(pc):添加了不含有mend的相同量的pbs之后,添加tritonx-100并使得终浓度为0.02%(w/v)、使红细胞溶解的液体。将这些在37℃温育45分钟,之后在4℃,400×g的条件下离心分离5分钟,回收上清,通过测定在545nm的吸光度,定量了血红蛋白从红细胞的渗出量。接下来,以pc的测定值为100%,以百分率表示各样品的测定值(溶血活性)。该百分率越高,显示膜融合能力越高。将结果示于图1。和dodapmend、dlindapmend比较,tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend显示了高膜融合能力。尤其是,tlm-c3-dmamend、tlmes-c3-dmamend仅在ph5.5显示了高膜融合能力。[实施例10]基于第1mend、第2mend、第3mend、第4mend的膜融合能力试验从雄性icr小鼠采血,回收红细胞,悬浮于生理盐水。将pbs调整为ph7.4、6.5、5.5后,添加了包含一定量的红细胞的生理盐水。接下来,分别添加了包含实施例6(2)中制备的tlm-c3-dmamend及tlm-c4-dmamend、实施例6(3)中制备的mend的pbs3.3μl、10μl及30μl。另外,以添加了不含有mend的相同量的pbs的溶液作为阴性对照(nc),将添加了不含有mend的相同量的pbs之后添加tritonx-100至0.5%(w/v)的、使红细胞溶解的液体作为阳性对照(pc)。将这些在37℃温育30分钟,之后在4℃,400×g的条件下离心分离5分钟,回收上清,通过测定在545nm的吸光度,测定了血红蛋白的量。接下来,以pc的测定值为100%,以百分率表示各测定值(溶血活性)。该百分率越高,显示膜融合能力越高。将结果示于图2。在第2mend、第3mend、第4mend中显示了比tlm-c3-dmamend高的膜融合能力。尤其是,与第3mend、第4mend比较,第2mend仅在ph5.5显示了高的膜融合能力。[实施例11]tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、dlindapmend、dodapmend的体内敲低活性试验将包含以实施例6(2)所示的方法制备的mend的溶液,以0.5mg/kg从尾静脉给予4周龄的雄性小鼠。24小时后采血,通过将该血液样品以1000g,10分钟,4℃进行离心,回收上清得到了血浆。对血浆中的factorvii(fvii)的量而言,使用biophenfviichromogenicassay(sysmexbiomed公司)进行定量,将未处理组(nt)的fvii表达量作为1,以相对值(血浆中的相对fvii量)示出了mend给予组的fvii表达量。将结果示于图3。可观察到与dlindapmend、dodapmend比较,tlm-c3-dmamend、tlm-c4-dmamend的fvii表达量降低。尤其是,tlm-c3-dmamend显示了最高的敲低活性。[实施例12]tlm-c3-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend的体内敲低活性试验将包含以实施例6(2)所示的方法制备的mend的溶液,以0.1mg/kg从尾静脉给予4周龄的雄性小鼠。24小时后采血,通过将该血液样品以1000g,10分钟,4℃进行离心,回收上清得到了血浆。对血浆中factorvii(fvii)的量而言,使用biophenfviichromogenicassay(hyphenbiomed公司)进行定量,将未处理组(nt)的fvii表达量作为1,以相对值(血浆中的相对fvii量)示出了mend给予组的fvii表达量。将结果示于图4。可见tdm-c3-dmamend、tlmes-c3-dmamend的fvii表达量也降低,与tlm-c3-dmamend比较,显示出了基本相同的敲低活性。[实施例13]tlm-c3-dmamend、tlm-c4-dmamend、及第1mend、第2mend、第3mend、第4mend的体内敲低活性试验将以实施例6(2)所示的方法制备的tlm-c3-dmamend的溶液及tlm-c4-dmamend溶液、以实施例6(3)所示的方法制备的mend溶液,以0.1mg/kg从尾静脉给予4周龄的雄性小鼠。24小时后采血,通过将该血液样品以1000g,10分钟,4℃进行离心,回收上清得到了血浆。对血浆中factorvii(fvii)的量而言,使用biophenfviichromogenicassay(sysmexbiomed公司)进行定量,将未处理组(nt)的fvii表达量作为1,以相对值(血浆中的相对fvii量)示出了mend给予组的fvii表达量。将结果示于图5。在第1mend、第2mend中显示出了高于tlm-c3-dmamend的敲低活性,第2mend显示了较高较大的敲低活性。[实施例14]体内的mrna表达试验将以实施例6(4)所示的方法制备的tlm-c3-dmammend溶液、tdm-c3-dmammend溶液用pbs稀释至使得mrna为1mg/100ml,将其在颈部进行皮下给予6周龄的雌性icr小鼠。在5.5小时后,将预先制备使得为3mg/200ml/匹的荧光素(vivoglotmluciferin,invivograde,promega公司制)的生理盐水溶液,从腹腔内给予各小鼠,30分钟后,利用ivistmluminaii(caliperlifesciences公司)观察mrna给予部位的发光并进行定量。将结果示于图6。在tlm-c3-dmammend及tdm-c3-dmammend中均确认到发光,均显示了mrna表达活性。其中,在tdm-c3-dmammend中显示最强的发光量,显示mrna表达活性较高。工业实用性本发明的试剂可以高效率地将功能性核酸递送至细胞质内,因此对核酸医药品开发、生化学实验有用。本申请以在日本申请的日本特愿2014-166041(申请日:2014年8月18日)为基础,并将其内容全部包含于本说明书。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1