一株高耐受酿酒酵母菌株及其构建方法与流程
本发明涉及基因工程领域,具体是涉及一株高耐受酿酒酵母菌株及其构建方法。
背景技术:
:
乙醇是重要的化工原料、有机溶剂、消毒剂及饮料酒勾兑的原料。随着地球上化石能源(石油、煤炭、天燃气等)的日趋枯竭和全球环境保护意识的不断加强乙醇将成为未来主要的可再生能源之一。然而,例如高温、高渗透压、高乙醇浓度等胁迫条件都会影响酵母细胞的生长,进而影响乙醇的产量。在浓醪发酵过程中,温度过高就需要用冷水降温,这也会降低乙醇生产的经济效益。因此,提高工业生产酿酒酵母的高温耐受性,可以从节约冷却水方面提高生物乙醇生产的经济效益。
对于调节酵母细胞的耐受性,Ras-cAMP信号通路起着关键作用,CYR1基因编码的腺苷酸环化酶是该通路的关键酶,催化cAMP的合成。研究发现,cAMP作为酿酒酵母生理活动的第二信使,在调节细胞生长、代谢和压力抗性方面起着重要的作用。有报道将反转录转座子Ty元件插入到CYR1基因的5’端编码区或非编码区可以构建出具有多种压力抗性的突变菌株,这样做会导致腺苷酸环化酶活性减弱(约比野生型菌株低三倍),这些突变株不但对致死热激具有抗性,而且对其它的压力条件如紫外照射,高乙醇浓度等也具有抗性。刘莹等研究发现将酵母细胞的CYR1基因敲除会具有较高的热抵抗能力。
Ras-cAMP信号通路活性的改变会影响酵母细胞的耐受性。通过对CYR1基因的表达调控,实现酵母细胞对胁迫环境的耐受性的改变。改变启动子强度是实现基因表达调控的重要途径。融合PCR可以实现基因片段的无缝拼接,且不受有限的酶切位点选择的限制,从而避免了传统的质粒构建过程中重复的酶切和连接操作。利用融合PCR和整合型载体质粒YIplac211介导的两步整合方法,在不残留标记基因的前提下,实现对CYR1启动子3’端序列的不同程度敲除,进而构建带有CYR1启动子强度弱化的酿酒酵母菌株。构建的高耐受酿酒酵母菌株可安全用于工业生产,同时融合PCR介导的两步整合方法可广泛应用于酵母及其他微生物启动子调控,促进了基因精确修饰的发展。
技术实现要素:
:
本发明解决的技术问题是提供一株高耐受酿酒酵母菌株及其构建方法。
为解决上述技术问题,本发明采用如下技术方案:
本发明提供的高耐受酿酒酵母菌株是对高温和乙醇具有一定耐受性的酵母菌株。获得高耐受酿酒酵母菌株的出发酵母菌株为酿酒酵母(Saccharomyces cerevisiae)CICC32315,保存于中国工业微生物菌种保藏管理中心,公众可购买获得。
所述高耐受酿酒酵母菌株在正常的生长性能不受影响的情况下,对55℃热击以及8%(v/v)乙醇都有一定的耐受性,并且在40℃玉米原料高温浓醪发酵中乙醇产量相对亲本菌株提高了14.6%。
所述高耐受酿酒酵母菌株具体可以通过对出发菌株酿酒酵母(Saccharomyces cerevisiae)CICC32315的腺苷酸环化酶编码基因CYR1启动子的-150bp至-30bp(ATG=+1)这一序列无痕敲除来实现。
上述基因无痕敲除可以用以下方法实现。各步骤所涉及具体操作方法参考现有文献报道,如Joseph Sambrook等,《分子克隆实验指南》第二版,科学出版社,1995。
用PCR方法获得突变ura3片段,将其通过醋酸锂化学转化法导入到酿酒酵母出发菌株,通过同源重组实现出发菌株的URA3基因突变;用PCR方法分别扩增靶序列上游和下游长度约为1.2kb的同源序列片段,然后通过融合PCR,将上、下游序列融合成无缝片段;将该片段克隆至酵母整合质粒YIplac211上,获得能够进行整合敲除的质粒;在整合敲除质粒的上游同源臂或者下游同源臂中,选择合适的单一酶切位点,将质粒酶切线性化;通过醋酸锂化学转化法,将线性化的整合敲除质粒导入酿酒酵母中,用不含尿嘧啶的酵母合成培养基筛选发生第一步整合重组的酵母突变株;将经过鉴定的发生第一步整合重组的酵母突变株,经含有5-氟乳清酸合成培养基平板反向筛选获得发生第二步整合重组的酵母突变株;用PCR方法获得出发菌株正常的URA3基因片段,将其导入到发生了二步整合重组的酵母突变株中,以回复突变的ura3标记基因。
本发明所述酿酒酵母菌株可应用于生物燃料乙醇生产中。
有益效果:
1、本发明提供一种高耐受的酿酒酵母菌株,克服了普通酿酒酵母在胁迫条件下生长不好的问题。
2、本发明提供的高耐受的酿酒酵母菌株是在保持优良发酵性能的前提下,腺苷酸环化酶的编码基因CYR1的表达量有一定程度的降低,进而降低了酵母细胞的Ras-cAMP信号通路的活性,使得酵母细胞的耐受性发生变化,为生物燃料乙醇的生产奠定了理论基础,而且具有重要的市场价值。
3、本发明所使用的靶序列敲除方法,避免了酵母传统敲除过程中筛选标记残留的问题,满足自克隆的条件,可以安全的用于工业生产。并且本方法在基因精确修饰方面具有广阔的应用前景。
附图说明:
图1为AY15aΔU的表型验证;
图2为重组质粒的构建流程示意图;
图3为重组质粒的酶切验证图:
泳道M为DNA maker;1:SalI和BamHI双酶切载体质粒YIplac211;2-6:分别是SalI和BamHI双酶切YIplac211-UD-30、YIplac211-UD-60、YIplac211-UD-90、YIplac211-UD-120、YIplac211-UD-150;
图4为酵母重组菌株的构建流程示意图;
图5为发生第一步整合重组的菌株的电泳验证图:
泳道M为DNA maker;1:AY15aΔU为模板;2-6:模板分别为AY15aΔU-U-30、AY15aΔU-U-60、AY15aΔU-U-90、AY15aΔU-U-120、AY15aΔU-U-150,CYR1-U-F和YIp-D-F为验证引物;7:AY15aΔU为模板;8-13:模板分别为AY15aΔU-U-30、AY15aΔU-U-60、AY15aΔU-U-90、AY15aΔU-U-120、AY15aΔU-U-150,YIp-U-R和CYR1-D-R为验证引物;
图6为发生第二步整合重组的菌株的电泳验证图:
泳道M为DNA maker;1:AY15aΔU为模板;2-6:模板分别为AY15aΔU-30、AY15aΔU-60、AY15aΔU-90、AY15aΔU-120和AY15aΔU-150,CYR1P-U和CYR1P-D为验证引物;
图7为酵母重组菌株的测序验证;
图8为亲本菌株和酵母重组菌株的生长曲线:
其中,A:培养温度为30℃,B:培养温度为40℃;
图9为亲本菌株和酵母重组菌株的耐受性实验结果图。
具体实施方式:
下面通过具体的实施方案叙述本发明。除非特别说明,本发明中所用的技术手段均为本领域技术人员所公知的方法。另外,实施方案应理解为说明性的,而非限制本发明的范围,本发明的实质和范围仅由权利要求书所限定。对于本领域技术人员而言,在不背离本发明实质和范围的前提下,对这些实施方案中的物料成分和用量进行的各种改变或改动也属于本发明的保护范围。
实施例1:CYR1启动子部分缺失酿酒酵母菌株的构建
本实例所用的出发菌株CICC32315。所述大肠杆菌DH5a购自Takara公司。所述YPD培养基为通用的完全培养基,固体培养基含2%进口琼脂粉。
根据Genebank中的酵母基因组数据和整合质粒序列,设计了下述引物。
表1本实施例中所用到的引物
注:小写字母表不酶切位点,下划线表不融合PCR所用引物的重叠序列。
(1)带有缺陷ura3基因的酿酒酵母AY15aΔU的构建
使用Solarbio公司的酵母基因组提取试剂盒,提取实验室菌株W303-1a的基因组。利用引物对URA3-F和URA3-R,以W303-1a的基因组为模板,PCR扩增菌株W303-1a突变ura3片段。经过试剂盒回收以后,将片段通过醋酸锂化学转化法导入到亲本菌株CICC32315中,通过突变ura3与正常URA3之间的同源重组,实现标准菌株的URA3基因突变。转化后的菌悬液,涂布于补加了5-氟乳清酸(5-FOA)和尿嘧啶(uracil)酵母合成培养基(SD)平板上,30℃培养48h,获得带有缺陷ura3基因的酿酒酵母AY15aΔU。获得的突变株经过表型验证确定正确,如图1所示AY15aΔU在酵母合成培养基上不长,在YPD平板上生长良好。
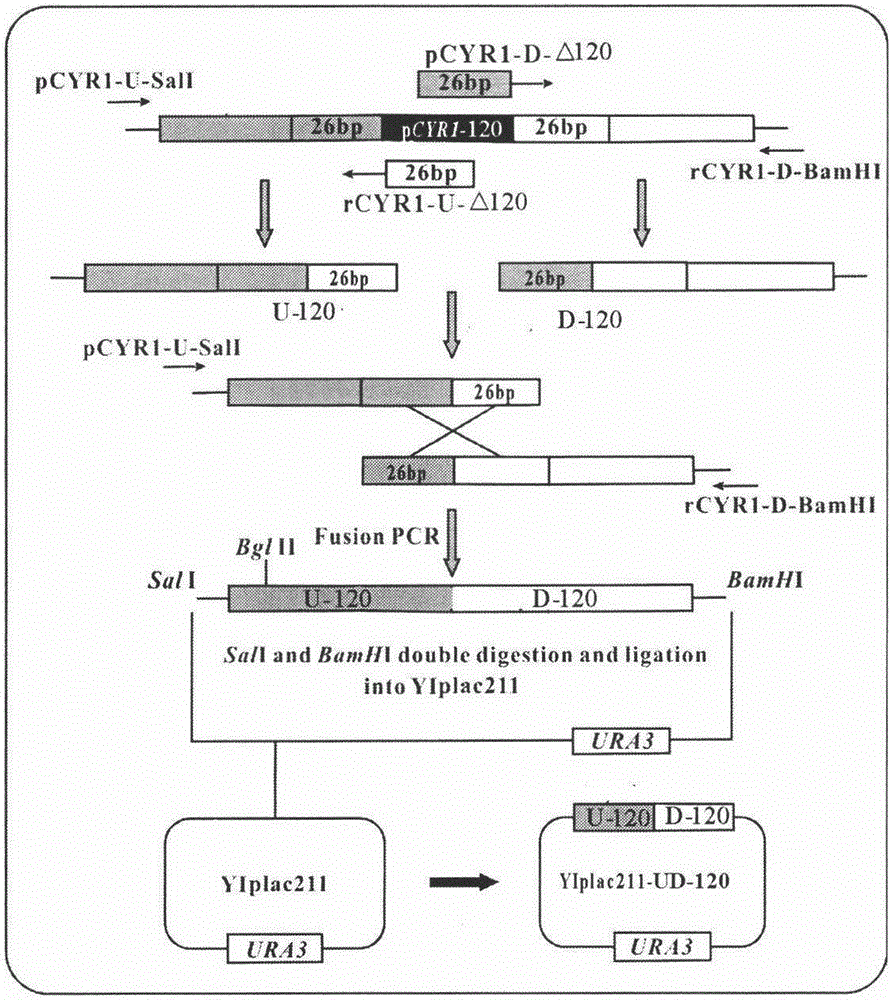
(2)整合敲除质粒YIplac211-UD-120的构建
首先,以亲本菌株CICC32315的基因组为模板,使用引物pCYR1-U-SalI和rCYR1-U-Δ120通过PCR获得靶序列pCYR1-120左侧1151bp的基因序列,PCR反应条件:95℃5min;94℃40s,53℃1min,72℃80s,30个循环;72℃10min;同时使用引物pCYR1-D-Δ120和rCYR1-D-BamI扩增靶序列右侧1201bp的基因序列,PCR反应条件:95℃5min;94℃40s,53℃1min,72℃80s,30个循环,72℃10min,分别命名为U-120和D-120。然后,以U-120和D-120的混合物为模板,加入引物pCYR1-U-SalI和rCYR1-U-Δ120进行融合PCR,获得无缝融合片段。经过切胶回收(试剂盒)后,分别用SalI和BamHI双酶切,最后亚克隆到整合质粒YIplac211的相应酶切位点中,整合敲除质粒YIplac211-UD-120构建成功,缺失序列大小为120bp,构建流程如图2所示。为了确定最优的CYR1启动子强度,用同样的质粒构建方法构建了其他四种重组质粒,,分别为YIplac211-UD-30(缺失序列大小为30bp)、YIplac211-UD-60(缺失序列大小为60bp)、YIplac211-UD-90(缺失序列大小为90bp)、YIplac211-UD-150(缺失序列大小为150bp),将这五种质粒统称为YIplac211-UD*。图3为重组质粒YIplac211-UD*的酶切验证图。
上述融合PCR法为本领域公知的采用具有互补末端的引物,形成具有重叠序列PCR产物,通过PCR产物重叠序列延伸,从而将任意DNA片段连接起来的方法,此技术不需要内切酶的消化和连接酶的处理,就可实现DNA片段的体外连接,这使得整合质粒经过两步整合重组后,酵母基因组不会残留外源酶切位点。
(3)CYR1启动子部分缺失酵母突变株的构建
BglII酶切重组质粒YIplac211-UD*;用醋酸锂化学转化法将线性化的重组质粒YIplac211-UD*导入缺陷型酵母菌株AY15aΔU中,经过两步整合重组后,得到CYR1基因启动子3’端部分缺失的酿酒酵母,两步整合重组过程如图4。
第一步整合重组的发生,是由于导入的线性化质粒与酵母基因组的同源部分发生整合,从而将整个质粒带入基因组。转化后的菌悬液,涂布于不含尿嘧啶的酵母合成培养基平板上,30℃培养48h,获得发生第一步整合重组的酵母菌株,分别命名为AY15aΔU-U-30、AY15aΔU-U-60、AY15aΔU-U-90、AY15aΔU-U-120和AY15aΔU-U-150,这5株菌统称为AY15aΔU-U*。采用CYR1-U-F和YIp-D-F,YIp-U-R和CYR1-D-R为验证引物,进行PCR验证筛选。当使用引物对CYR1-U-F和YIp-D-F进行PCR验证时,因为下游引物YIp-D-F的退火序列在YIplac211载体质粒上,所以酵母菌株AY15aΔU无特异性条带,而一步整合重组菌株AY15aΔU-U*应有一条大小为1800bp左右的条带;当使用引物对YIp-U-R和CYR1-D-R进行PCR验证时,因为上游引物YIp-U-R的退火序列在YIplac211载体质粒上,所以酵母菌株AY15aΔU无特异性条带,而一步整合重组菌株AY15aΔU-U*应有一条大小为3000bp左右条带。PCR产物进行0.8%琼脂糖凝胶电泳结果见图5。转化子与对照比较结果显示,线性化的质粒整合到目的位点。
经过筛选和鉴定的一步整合重组酵母菌株AY15aΔU-U*,取一环接到5ml液体YPD培养基中,30℃下200rpm振荡12h后,稀释10倍涂布于含有5-氟乳清酸和尿嘧啶的酵母合成培养基平板上,30℃培养48h,获得发生第二步整合重组的酵母菌株,第二步整合重组后,出现两种结果:一,如果上游同源序列(U)形成的重复序列之间发生整合,则酵母回复成出发菌株AY15a;二,如果下游同源序列(D)形成的重复序列之间整合,则中间的所有序列弹出,实现启动子的精确敲除,同时靶位置上不引入任何外源基因。挑选所得到的单菌落,采用引物对CYR1P-U和CYR1P-D,进行PCR验证,PCR产物条带大小应该分别为366bp、336bp、306bp、276bp、246bp和216bp。筛选出启动子靶序列部分得到精确敲除的转化子,分别命名为AY15aΔU-30、AY15aΔU-60、AY15aΔU-90、AY15aΔU-120和AY15aΔU-150。PCR产物进行2%琼脂糖凝胶电泳结果见图6。转化子与对照比较结果显示,CYR1的启动子部分得到不同程度的敲除。
(4)二步重组酿酒酵母菌株突变ura3基因的回复
同本实例中(1)所述的方法,利用引物对URA3-F和URA3-R,扩增AY15a正常URA3基因片段,通过醋酸锂化学转化,以回复二步重组酵母菌株突变的ura3基因,最终构建出不残留任何外源基因序列的无痕敲除菌株,分别命名为AY15a-30、AY15a-60、AY15a-90、AY15a-120、AY15a-150。
分别提取二步整合重组酵母菌株的基因组,使用引物对CYR1P-F和CYR1P-R进行PCR反应。PCR反应产物经华大基因公司测序,测序结果与NCBI公布序列比对,验证结果如图7。从图7可以看出了AY15a-30、AY15a-60、AY15a-90、AY15a-120、AY15a-150的CYR1的启动子缺失情况分别为30bp、60bp、90bp、120bp、150bp。
实施例2:CYR1启动子部分缺失酿酒酵母菌株生长性能测定及耐受性实验
(1)CYR1启动子部分缺失酿酒酵母菌株的生长性能测定
测定亲本菌株CICC32315及其转化子AY15a-30、AY15a-60、AY15a-90、AY15a-120、AY15a-150在30℃和40℃条件下的生长性能,结果如图8。结果显示,对CYR1启动子的部分敲除在没有明显影响酵母在30℃的生长性能的前提下,40℃下的酵母细胞的生长性能都得到了一定程度的提高。
(2)CYR1启动子部分缺失酿酒酵母菌株的耐受性实验
利用稀释点板实验测定亲本菌株CICC32315及其转化子AY15a-30、AY15a-60、AY15a-90、AY15a-120、AY15a-150的耐受性变化。
培养条件:
一级:取一环菌泥接于5mLYPD液体中,30℃,180rpm,培养12h。
二级:取200μL一级菌液接于5mLYPD液体中,30℃,180rpm,培养6-8h至酵母细胞的对数期。
调节对数期的酵母细胞的OD600=1.2,用于做耐受性实验。对单倍体亲本及其转化子各取200μL用于热击实验,55℃热击3min,并且稀释至10-4,然后从不同稀释度各取3μL未做热击实验的菌液和做热击实验菌液分别点接于YPD平板上;再从不同稀释度各取3μL未做热击实验的菌液分别点接于含有260g/L葡萄糖或者8%(v/v)乙醇的YPD平板上,30℃培养3天,拍照。结果如图9。结果显示AY15a-90和AY15a-120不但表现出很好的耐热性能,而且对8%(v/v)乙醇的耐受性也有一定程度的提高。
实施案例3:菌株AY15a-90和AY15a-120玉米原料浓醪发酵实验
(1)AY15a-90和AY15a-120与亲本菌株的玉米原料浓醪发酵实验
将亲本菌株CICC32315及转化子AY15a-90和AY15a-120分别同时进行玉米浓醪发酵实验,发酵工艺路线图:玉米粉→浸泡→液化→糖化→冷却→接菌→发酵→蒸酒→测定指标;
工艺条件:
浸泡条件:60~70℃,浸渍20min;液化条件:85~90℃,加入耐高温α-淀粉酶,液化90min;糖化条件:55~60℃,加入糖化酶,糖化20min;
配料:玉米粉60g,水180mL,耐高温α-淀粉酶2×105U/mL,30μL,糖化酶200U/mL,90μL,2.5×103U/mL酸性蛋白酶1.2mL;营养盐1mL(MgSO4 150g/L、KH2PO4 75g/L、尿素81g/L,过滤,4℃保存);接种量:7.5%,30℃放置12h,再放入40℃,发酵3天,发酵结束后测定各菌株的发酵性能(CO2失重、乙醇产量、残糖);结果见表2。
表2菌株发酵结果比较
注:所示数据为三个平行试验的结果。
表2显示,在40℃下的高温浓醪发酵条件下,转化子AY15a-90和AY15a-120的CO2失重和乙醇产量都较亲本菌株有所提高,AY15a-90和AY15a-120的乙醇产量相对亲本菌株分别提高了14.0%和14.6%,说明AY15a-120是优于AY15a-90的高耐受酿酒酵母菌株。
- 还没有人留言评论。精彩留言会获得点赞!