一种基因热点突变的检测方法与流程
本发明涉及生物技术领域,具体涉及分子生物学和临床检验医学领域,涉及基因工程和医学诊断,尤其涉及一种基因热点突变的检测方法。
背景技术:
肿瘤的发生、发展与基因突变相关,包括遗传性基因突变和后天的体细胞基因突变。目前表明有多种基因的热点突变,其发生频率较高,不但与多种肿瘤的发生发展相关,还与肿瘤的治疗选择密切相关。目前对基因突变的常用分析方法,包括直接测序分析,位点特异性引物扩增,PCR产物熔点曲线分析等。这些技术各有优缺点,但是对肿瘤早期筛查和治疗后效果监测领域,还需要研发没有假阳性而同时又具有高效率的新技术。
PCR扩增技术、限制性内切酶技术和基因克隆技术,广泛单独或与基因测序技术相结合,用于基因分析和基因突变分析。
PCR的引物与待扩增核酸通过碱基互补的杂交原理,扩增产物对模板和引物具有相互配对的要求。此外,通过位点特异性引物以及结合针对扩增产物的标记探针等手段,利用PCR技术能通过多种方式用于基因分析。
限制性内切酶因其对识别位点的高度特异性辨认,广泛用于限制性酶切谱分析,限制性内切酶片段长度多态性分析等。
连接酶在突变分析中,利用一种连接酶链式反应(Ligation chain reaction,LCR),用于突变分析。并且,连接酶还广泛用于亚克隆以及第一代测序及高通量测序分析。
基因亚克隆在基因突变分析中具有多种方式。一方面,可以对克隆形成的菌落直接测序。另外,也可利用蓝白筛选技术,选择性的挑选特定颜色的菌落进行测序。
蓝白斑筛选(β-半乳糖苷酶系统)是分子生物学实验中较常用的筛选标记,具有高效和简单的优势,主要用于基础研究,包括提高克隆效率。近来,蓝白斑筛选用于体细胞突变的体外基础研究,后者如big-blue小鼠和大鼠(ref);以及用于人类遗传性突变的基因分析。但蓝白斑筛选技术用于人类KRAS基因突变,还是一种有待研发的新技术。
在离体研究中,此类载体携带lacZ基因,它编码β-半乳糖苷酶的N-末端(α-肽),并且处于诱导物IPTG(异丙基-β-D-硫代半乳糖苷,与乳糖结构类似但不能被β-半乳糖苷酶降解)诱导的启动子调控之下。质粒转化的宿主菌为LacZ△M15基因型(含有编码N-末端缺陷型的β-半乳糖苷酶多肽的基因)。未重组的质粒转化宿主菌后,在IPTG的诱导下,质粒与宿主菌分别表达缺陷但可以相互补偿的两个肽(即α-互补),形成了有功能的β-半乳糖苷酶,该β-半乳糖苷酶能把培养基中无色的X-gal(5-溴-4-氯-3-吲哚-β-D-半乳糖苷)底物分解成半乳糖和深蓝色的5-溴-4-氯-靛蓝,菌落呈现蓝色。但当外源DNA插入质粒位于α-肽编码序列中的多克隆位点时,由于破坏了α-肽的阅读框架,使其编码的α-肽失去活性,则重组子所在的细菌不能完成α互补,不能形成有活性的β-半乳糖苷酶,致使重组菌形成白色菌落。因此,可以借助蓝白斑筛选出重组子。
在多种肿瘤相关突变中,如缺失突变,插入突变,和多种点突变,或者可以直接导致TAA,TAG,TGA等终止密码子的出现与丧失,或者通过人工移码技术获得野生基因与突变基因具有含或不含终止密码子的情形。这样,利用位于突变序列上下游的扩增引物对对其进行扩增,并将扩增片段插入原核表达载体,便可以得到蓝白颜色不同的菌落。
本发明针对涉及终止密码子的热点突变,采用结果PCR扩增,限制性内切酶消化,对消化产物进行去磷酸化,亚克隆技术相结合,可使形成的克隆中含有突变的克隆得到大幅提升,在突变涉及终止密码子时,这种提升效果由于菌落蓝白颜色的不同,而得到进一步的提高。对克隆菌落的测序,可以起到对突变的确认的作用。
技术实现要素:
本发明要解决的技术问题是提供一种可以在可能混合有野生与突变基因片段的生物标本中进行热点突变的检测方法,且具有较高的检测效率。
为了解决上述的技术问题,本发明提供了一种对含有野生型基因和突变基因的生物标本中进行突变基因分析的方法,其特征在于,其包括如下步骤:
(1)在生物标本中加入限制性内切酶进行消化,
(2)对消化切口进行阻碍连接酶反应的处理,
(3)用连接酶介导连接反应,最后对亚克隆所形成的菌落进行突变分析。
在本发明的一个优选实施中,所述步骤(1)中的生物标本中含有野生型基因和突变基因。
本发明的一个优选实施例中,所述步骤(1)中,与所述突变基因相对应的所述野生型基因被限制性内切酶消化,而所述突变基因的片段因不含有酶切位点,而不能被消化。
本发明的一个优选实施例中,所述步骤(1)中,所述突变基因的片段含有酶切位点被限制性内切酶消化,而与所述突变基因相对应的所述野生型基因不含有酶切位点不能被限制性内切酶消化。
本发明的一个优选实施例中,所述的步骤(2)对消化切口进行阻碍连接酶反应的处理为对消化切口进行五末端去磷酸化处理。
本发明的一个优选实施例中,所述的步骤(3)的突变分析为菌落PCR初筛和第一代测序分析。
本发明的一个优选实施例中,所述的野生型基因为KARS基因。
本发明提供了一种可利用限制性内切酶消化与突变基因相对应的野生基因片段,并对酶切缺口进行阻碍连接酶反应的核酸片段5末端去磷酸化处理,通过DNA连接酶将带分析的基因片段亚克隆至质粒体载体,最后对形成的菌落进行基因分析。菌落的基因分析采用菌落PCR初筛和第一代测序分析。
本发明可以用于基因突变涉及限制性酶切位点消失,和基因突变与野生序列存在通过人工引入突变得到突变基因片段产物不含有酶切位点而其相对应的野生基因片段在人工引入突变后形成酶切位点两种情形。
对于基因突变涉及到限制性内切酶酶切位点消失的情形,本发明在对待分析生物标本对其进行扩增。在得到扩增产物后,对其进行限制性内切酶消化,使野生产物消化成两个较小片段,而突变基因片段引物不含有限制性内切酶位点,不能被消化而保持完整。
本发明的应用例针对KRAS基因热点突变,通过设计一对序列特异性引物,在KRAS基因热点突变的侧翼开始进行PCR扩增反应;与突变相对应的野生型序列通过扩增反应引入酶切位点,并对酶切缺口进行阻碍连接酶反应的核酸片段5末端去磷酸化处理;再将去磷酸化后的产物亚克隆至具有蓝白筛选功能的原核载体,对待测标本中的野生序列片段与突变序列片段进行蓝白菌落斑筛选。
本发明提供的引物对具有为突变相对应的野生型模板引入酶切位点的能力;PCR产物中野生型基因片段经限制性内切酶酶切,再经虾碱性磷酸酶去磷酸化,可使形成的克隆中含有突变的克隆得到大幅提升,从而具有从数以千计和数以万计的野生基因片段中选择性对突变序列形成白色菌落的能力,蓝白斑筛选得到的白色菌落通过基因测序分析,可以确定该待测标本中有无基因突变。
本发明的技术关键为将直接含有终止密码子或通过人工引入终止密码子的基因片段与相对应的无终止密码子的不同基因型基因片段通过体外扩增,限制性内切酶消化,消化末端的去磷酸化处理,亚克隆后将不同基因型的基因片段转换成为颜色不同的大肠杆菌菌落。该技术实施的措施为设计具有引入酶切位点,还同时能通过亚克隆调整以适应贝塔半乳糖苷酶基因阅读框架能力的PCR引物对,将突变相对应的野生型基因片段引入的酶切位点酶切,并对酶切缺口进行阻碍连接酶反应的核酸片段5末端经虾碱性磷酸酶去磷酸化处理。
用于亚克隆的蓝白斑菌落筛选实验,主要用于检测亚克隆后插入片段是否成功插入。其原理是插入片段尤其是长的插入片段的成功插入,一般都会由于插入而引入新的终止密码子导致贝塔半乳糖苷酶基因产物表达的终止,这样大肠杆菌菌落因为缺乏贝塔半乳糖苷酶而不能将没有颜色的化学物质5-溴-4-氯-3-吲哚-β-D-半乳糖苷通过酶化学反应转换为具有蓝色的5-溴-4-氯-靛蓝。我们最近的研究表明,在插入较短片段时,通过引入终止密码子,同样可以进行蓝白斑筛选。
亚克隆常用方法为T/A克隆和利用限制性内切酶位点定点克隆。前者的优点是PCR产物不需要后处理,直接进行连接反应亚克隆至相应T载体之上。该方法的原理是PCR低保真DNA聚合酶的反应产物中有大约一半的产物的三末端带有一个非模板依赖性的A碱基,而克隆载体的五末端以预先设置了一个突出的T碱基,通过TA碱基互补配对,达到对PCR产物直接克隆的效果。利用限制性酶切位点进行的亚克隆,缺点是需要对PCR产物继续后处理,先利用限制性内切酶对PCR产物酶切,然后进行亚克隆。限制性酶切位点进行的亚克隆方案的优点是能够保证PCR产物在亚克隆后按照设计要求,适应克隆载体的阅读框架。
本发明实施例提供的引物对e、f,如Seq No.6,Seq No.7所示;本发明实施例提供的引物对g、h,如Seq No.10,Seq No.11所示所示。引物对e、f,和g、h的5末端,采用上游引物5末端为限制性内切酶HindIII和下游引物为5末端为限制性内切酶BamHI。限制性内切酶可以产生两个不同粘性末端的技术方案。不同粘性末端的使用,避免了对载体进行去磷酸化处理,保证高效率的同时,还能减低非特异性克隆产物。
本发明所包含的引物对e和f,具有对KRAS基因12号热点突变所在区域进行序列特异性扩增能力,同时因为KRAS基因热点突变具有通过蓝白载体进行大肠杆菌菌落颜色转换的能力。本发明的PCR引物对扩增的产物,在亚克隆至蓝白筛选载体上后,只增加34个氨基酸,不形成终止密码子,也因此不影响贝塔半乳糖苷酶相关基因产物的表达,转化后相应的大肠杆菌菌落为蓝色。然而,该引物对扩增含有KRAS基因12位密码子的热点突变时,对于GG/GA突变,将产生TGA终止密码子,这些PCR扩增产物在亚克隆至原核克隆载体,理论上转化后得到的相应大肠杆菌菌落为白色。
本发明所包含的引物对g和h具有对KRAS基因13号热点突变所在区域进行序列特异性扩增能力,同时因为KRAS基因热点突变具有通过蓝白载体进行大肠杆菌菌落颜色转换的能力。本发明的PCR引物对扩增的产物,在亚克隆至蓝白筛选载体上后,只增加34个氨基酸,不形成终止密码子,也因此不影响贝塔半乳糖苷酶相关基因产物的表达,转化后相应的大肠杆菌菌落为蓝色。然而,该引物对扩增含有KRAS基因13位密码子的热点突变时,对于GG/GA突变,将产生TGA终止密码子,这些PCR扩增产物在亚克隆至原核克隆载体,理论上转化后得到的相应大肠杆菌菌落为白色。
在肿瘤诊断的临床应用方面,待测标本主要为肿瘤组织和血中游离核酸两类。对肿瘤组织而言,当活检标本中大于百分之十以上均为肿瘤细胞时,基因突变检测的几乎所有相关技术均可以用于该标本的基因突变检测,其中最为临床接受的金标准为部分链终止法测序技术。然而,若活检标本中的肿瘤细胞含量低于百分之十,但仍高于百分之一时,新一代高通量测序技术可用于该类标本的基因突变检测。
对活检标本中肿瘤细胞含量低于百分之一的标本,以及血中游离核酸标本,由于其中肿瘤突变基因部分与正常基因的比例低于百分之一,很多用于分析遗传性基因异常的基因诊断技术均难以有效检测这些微量的突变基因。蓝白筛选技术具有从大量野生菌落中挑选可能含有突变基因片段的菌落的能力。对常用的10厘米细菌生长平板,可生长的单克隆菌落可以达到2000-3000个菌落。从这些菌落中挑选一个至数个可能的白色菌落,通过进一步的测序分析,可以对稀有突变进行有效的分析。本发明方法借助于一个引入酶切位点的引物对,借助于PCR扩增的高效率,借助于内切酶酶切的高效率和去磷酸化的高效率,借助蓝白筛选的高效率,和测序技术的排除假阳性的高特异性,为含量稀有的肿瘤突变基因标本的基因诊断,提供了一种高效而又可靠的技术。
本发明的方法分别在KRAS基因第12位和第13位密码子热点突变的检测中得到了证实,可以检测到三万比一的低含量突变。引入酶切位点的引物对e和f引物对KRAS基因第12位密码子热点突变中,可通过采用本发明的限制性内切酶BSTNI酶切,去磷酸化后使PCR产物转化为蓝白菌落的差异。引入酶切位点的引物对g和h引物对KRAS基因第13位密码子热点突变中,可通过采用本发明的限制性内切酶haeIII酶切,去磷酸化后使PCR产物转化为蓝白菌落的差异。
在与肿瘤突变相关的基因突变中,存在多种热点突变可以通过体外调整贝塔半乳糖苷酶基因的阅读框架而达到采用蓝白区分相应基因的野生与突变基因片段的目的。因此,降低野生序列连接反应的基因热点突变检测技术可以作为一种技术方案,通过针对不同基因突变设计不同的引物对引入酶切位点,经PCR扩增后,限制性内切酶消化与突变相对应的野生基因片段,并对酶切缺口进行阻碍连接酶反应的核酸片段5末端去磷酸化处理,通过DNA连接酶将带分析的基因片段亚克隆至质粒体载体,最后对形成的菌落进行基因分析。可以根据不同的基因,设计出相应的引物对,经相应的内切酶酶切,并对酶切缺口进行阻碍连接酶反应的核酸片段5末端去磷酸化处理研发相应的基因检测技术。
本发明通过采用体外调整基因阅读框架的原理,将利用限制性内切酶消化与突变相对应的野生基因片段,并对酶切缺口进行阻碍连接酶反应的核酸片段5末端去磷酸化处理,通过DNA连接酶将带分析的基因片段亚克隆至质粒体载体检测肿瘤突变KRAS基因第12位和13位氨基酸编码子的热点突变,使野生产物消化成两个较小片段,而突变基因片段引物不含有限制性内切酶位点,不能被消化而保持完整。与亚克隆技术相结合,可使形成的克隆中含有突变的克隆得到大幅提升,在突变涉及终止密码子时,这种提升效果由于菌落蓝白颜色的不同,而得到进一步的提高。本发明采用不直接含有酶切位点的KRAS热点突变作为实施例。对于直接含有酶切位点的热点突变,本发明应用时不需人工引入酶切位点。在应用本发明是,依据终止密码子是定位于野生序列还是突变序列,决定测序挑取的克隆总是对应于突变序列相对应的克隆。
附图说明
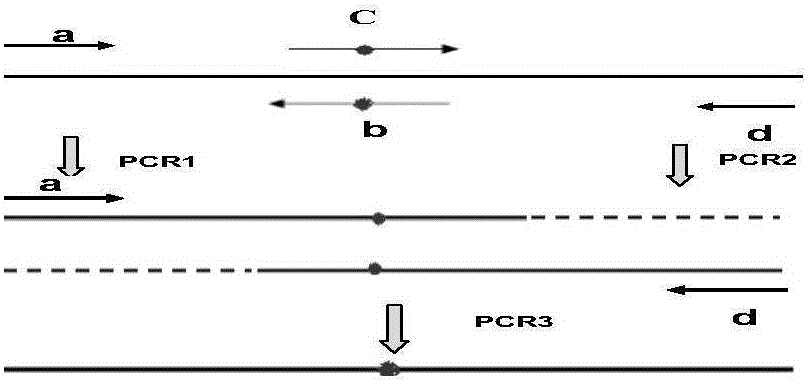
图1为重叠延伸PCR工作原理示意图。
图2为a,d引物扩增的野生型PCR产品电泳图谱。
图3为第一轮PCR电泳图。1,2泳道为产物ab;3,4泳道为产物cd;6泳道为100bp DNA marker。
图4为KRAS野生模板质粒测序图。
图5为KRAS第12位密码子模板质粒测序图。
图6为第13位突变型质粒的测序图。
图7A为KRAS纯野生型质粒酶切后去磷酸化的蓝白斑检测菌落形成结果示意图。
图7B为KRAS野生型与12位密码子突变基因片段质粒为3000:1的蓝白斑检测菌落形成结果示意图
图7C为KRAS野生型与12位密码子突变基因片段质粒为10000:1的蓝白斑检测菌落形成结果示意图。
图7D为KRAS野生型与12位密码子突变基因片段质粒为30000:1的蓝白斑检测菌落形成结果示意图。
图8A为KRAS纯野生型质粒酶切后去磷酸化的蓝白斑检测菌落形成结果示意图。
图8B为KRAS野生型与13位密码子突变基因片段质粒为3000:1的蓝白斑检测菌落形成结果示意图。
图8C为KRAS野生型与13位密码子突变基因片段质粒为10000:1的蓝白斑检测菌落形成结果示意图
图8D为KRAS野生型与13位密码子突变基因片段质粒为30000:1的蓝白斑检测菌落形成结果示意图。
图9为采用蓝白斑检测30000:1野生与12位密码子突变基因的白色菌落的菌液PCR结果示意图。
图10为采用蓝白斑检测30000:1野生与12位密码子突变基因的白色菌落菌液PCR经内切酶BstnI酶切的结果示意图,显示第12位有5个阳性克隆。
图11为采用蓝白斑检测30000:1野生与13位密码子突变基因的白色菌落的菌液PCR结果示意图。
图12为采用蓝白斑检测30000:1野生与13位密码子突变基因的白色菌落菌液PCR经内切酶HaeIII酶切的结果示意图,显示第13位有7个阳性克隆。
图13a和图13b是采用蓝白斑检测30000:1野生与12位密码子突变基因的白色菌落测序结果示意图,显示第12位密码子TGG突变为TGA。
图14a至图14e是采用蓝白斑检测30000:1野生与13位密码子突变基因的白色菌落测序结果示意图,显示第13位密码子TGG突变为TGA。
具体实施方式
以下结合具体实施例对上述方案做进一步说明。应理解,这些实施例是用于说明本发明而不限于限制本发明的范围。实施例中采用的实施条件可以根据具体应用要求的条件做进一步调整,未注明的实施条件通常为常规实验中的条件。
一、材料和仪器:HindIII和BamHI内切酶及BstnI内切酶购于New England Biolabs公司、PMD-19T质粒购于TAKARA;PFU酶、Tag酶、HaeIII、rSAP,T4DNA连接酶购于thermoscientific公司。X-gal,IPTG,氨苄青霉素、Tris-bas、EDTA、DMSO、电泳级琼脂糖、酵母提取物、胰蛋白胨、琼脂粉等为国产分析纯。
E Coli.DH5α菌株购自北京天根公司。
PCR仪-BioRad公司(美国),凝胶成像系统-BioRad公司(美国),DYY-6C型电泳仪-上海六一仪器厂。
二、具体方法
实施例一
1.KRAS基因12位密码子热点突变和对应的野生序列质粒构建
1.1包含有KRAS基因热点突变的基因序列如Seq No1所示。
引物a TAGCCAAGCTTTGACTGCATACAAACTTGT,如Seq No 2所示;
引物d CGTCAGGATCCTTGGACCATATTCGTCCACA,Seq No3所示。
以人血基因组为模板,引物a,d进行PCR,构建野生模板。
以上体系在PCR仪上按如下程序反应:95℃预变性5min,进入三步循环(95℃-15s、58℃-15s、72℃-15s共30个循环),最后72℃-1min。取5μL反应液在琼脂糖凝胶电泳,如图2所示:1泳道为100bp DNA marker;2,3泳道为野生型PCR产物。
PCR产物纯化,后连接PMD-19T质粒,最后转化DH5α大肠杆菌,挑取克隆测序结果见图见图4。
1.2KRAS基因第12位密码子突变的质粒构建(重叠延伸PCR)
构建模板引物序列如下:
引物a:TAGCCAAGCTTTGACTGCATACAAACTTGT,Seq No.2所示;
引物b:ACGCCATCAGCTCCAACTA,如Seq No.4所示;
引物c:TTGGAGCTGATGGCGTAGG,如Seq No.5所示;
引物d:CGTCAGGATCCTTGGACCATATTCGTCCACA,如Seq No.3所示。
1.2.1第一轮PCR:
PCR扩增体系如下:人血基因组为模板,引物a、b,进行PCR。产物为ab。
以上体系在PCR仪上按如下程序反应:95℃预变性5min,进入三步循环(95℃-15s、58℃-15s、72℃-15s共30个循环),最后72℃-1min。取5μL反应液在琼脂糖凝胶电泳,如图1所示:1泳道为100bp DNA marker;2,3泳道为野生型PCR产物。
PCR扩增体系如下:基因组为模板,引物c、d,进行PCR。产物为cd。
以上体系在PCR仪上按如下程序反应:95℃预变性5min,进入三步循环(95℃-15s、58℃-15s、72℃-15s共30个循环),最后72℃-1min。
1.2.2第二轮PCR
以第一轮PCR的产物ab和cd互为模板相互延伸形成ad。将产物ab和产物cd分别100倍稀释,再各取1.0μL为模板。
PCR扩增体系如下:
以上体系在PCR仪上按如下程序反应:95℃预变性5min,进入三步循环(95℃-15s、58℃-15s、72℃-15s共30个循环),最后72℃-1min。
1.2.3PCR产物纯化、连接
PCR产物用天根的DNA纯化试剂盒纯化产物,用HindIII和BamHI酶切PCR产物;同时用HindIII和BamHI酶切PMD-19T质粒,切胶纯化。将经DNA纯化试剂盒纯化后的PCR产物,与酶切纯化后的载体连接。其连接体系如下:
其反应条件:22℃反应30分钟
1.2.4转化DH5α大肠杆菌
将上一步连接产物转化入DH5α大肠杆菌,37℃过夜培养。克隆后产物挑菌落,送测序,测序结果见图5。
2.1PCR扩增野生与含第12位密码子突变的基因片段
引物e:AAGCTTTGACTGCATACAAACTTGTGGTAGTTGGAC,如Seq No.6所示;
引物f:GGATCCTTAGACCATATTCGTCCACA,如Seq No.7所示。
PCR扩增体系如下:
以上体系在PCR仪按如下程序反应:95℃预变性5min,进入三步循环(95℃-15s、58℃-15s、72℃-15s共30个循环),最后72℃-1min。
模板为实施例一构建的野生型质粒和第12位密码子突变的质粒。先测定模板质粒OD,调整浓度到每微升20ng。将野生型质粒与突变型质粒按分子拷贝数混合为野生型:突变型分别为:3000:1,10000:1,30000:1。其中野生模板质粒在25微升反应体系中的总量为20ng,相对应的突变基因片段依次为6.67pg,2pg,0.67pg.
2.2PCR扩增片段的酶切
2.2.1PCR产物纯化酶切:将前述步骤PCR产物用天根的DNA纯化试剂盒纯化产物。用限制性内切酶BstnI酶切PCR产物,酶切体系如下:
其反应条件:37℃反应60分钟。
2.2.2酶切后的PCR产物纯化
将前述步骤PCR产物用天根的DNA纯化试剂盒纯化产物。
2.3酶切后的PCR产物去磷酸化
将前述步骤PCR产物用thermoscientific公司的rSAP去磷酸化。
去磷酸化体系如下:
其反应条件:37℃反应4小时,80度30分钟热失活
2.4PCR扩增片段的亚克隆蓝白筛选实验
2.4.1PCR产物与蓝白载体的亚克隆
2.4.1.1纯化、酶切:将前述步骤PCR产物用天根的DNA纯化试剂盒纯化产物,用HindIII和BamHI酶切PCR产物;同时用HindIII和BamHI酶切PMD-19T质粒,切胶纯化。
2.4.1.2连接,将经DNA纯化试剂盒纯化后的PCR产物,与前述纯化后的载体连接。其连接体系如下:
其反应条件:22℃反应30分钟
2.4.1.3转化、蓝白斑筛选
取50μl感受态细胞并于冰上融化10min。将10μl的连接体系全部加入上述感受态细胞,轻弹混匀。并置于冰上30min。42℃水浴热击45sec,迅速置于冰上冷却15min。加入800L 37℃预温的LB培养基,180rpm振荡培养40min。低速离心,弃除上清液500μl。用剩余300μlLB吹打沉淀,并将其全部涂布于含有X-Gal、IPTG、Amp的L-琼脂平板培养基上,倒置于37℃培养箱过夜培养。
实施例二
1.KRAS基因第13位密码子热点突变和对应的野生序列质粒构建同上,选用的中间引物引物b'、c'序列如下:
b':ACGTCACCAGCTCCAACTA,如Seq No.8所示;
c':TTGGAGCTGGTGACGTAGG,如Seq No.9所示。
2.PCR扩增野生与含第13位密码子突变的基因片段
2.1引物g:AGACATCGTAGGTAGTGACATAGCCAAGCTTTGACTGCATAC,如Seq No.10所示;
引物h:AAGGGATCCTTGGACCATATTCGTCCACAAAATGATTCTGAATTAGCTGTATCGTCAAGGCACTCTTGCCTAGG,如Seq No.11所示。
PCR扩增体系如下:
-
以上体系在PCR仪按如下程序反应:95℃预变性5min,进入三步循环(95℃-15s、58℃-15s、72℃-15s共30个循环),最后72℃-1min。
模板为实施例一构建的野生型质粒和实施例二构建的第13位密码子突变的质粒。先测定模板质粒OD,调整浓度到每微升20ng。将野生型质粒与突变型质粒按分子拷贝数混合为野生型:突变型分别为:3000:1,10000:1,30000:1。其中野生模板质粒在25微升反应体系中的总量为20ng,相对应的突变基因片段依次为6.67pg,2pg,0.67pg.
2.2PCR扩增片段的酶切
2.2.1PCR产物纯化酶切:将前述步骤PCR产物用天根的DNA纯化试剂盒纯化产物。用限制性内切酶HaeIII酶切PCR产物;
采用60μL的酶切反应体系,酶切体系如下:
2.4PCR扩增片段的亚克隆蓝白筛选实验
其反应条件:37℃反应60分钟
2.2.2酶切后的PCR产物纯化
2.3酶切后的PCR产物去磷酸化
2.4PCR扩增片段的亚克隆蓝白筛选实验
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。
本行业的技术人员应该了解,本发明不受上述实例的限制,上述实例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等同物界定。
序列表
<110> 南华大学
<120> 一种基因热点突变的检测方法
<160> 11
<210> 1
<211> 102
<212> DNA
<213> 人工合成
<400> 1
atgactgaat acaaacttgt ggtagttgga gctggtggcg taggcaagag tgccttgacg 60
atacagctaa ttcagaatca ttttgtggac gaatatggtc ca 102
<210> 2
<211> 30
<212> DNA
<213> 人工合成
<400> 2
tagccaagct ttgactgcat acaaacttgt 30
<210> 3
<211> 19
<212> DNA
<213> 人工合成
<400> 3
acgtcaccag ctccaacta 19
<210> 4
<211> 19
<212> DNA
<213> 人工合成
<400> 4
ttggagctgg tgacgtagg 19
<210> 5
<211> 31
<212> DNA
<213> 人工合成
<400> 5
cgtcaggatc cttggaccat attcgtccac a 31
<210> 6
<211> 36
<212> DNA
<213> 人工合成
<400> 6
aagctttgac tgcatacaaa cttgtggtag ttggac 36
<210> 7
<211> 26
<212> DNA
<213> 人工合成
<400> 7
ggatccttag accatattcg tccaca 26
<210> 8
<211> 19
<212> DNA
<213> 人工合成
<400> 8
acgtcaccag ctccaacta 19
<210> 9
<211> 19
<212> DNA
<213> 人工合成
<400> 9
ttggagctgg tgacgtagg 19
<210> 10
<211> 42
<212> DNA
<213> 人工合成
<400> 10
agacatcgta ggtagtgaca tagccaagct ttgactgcat ac 42
<210> 11
<211> 74
<212> DNA
<213> 人工合成
<400> 11
aagggatcct tggaccatat tcgtccacaa aatgattctg aattagctgt atcgtcaagg 60
cactcttgcc tagg 74
- 还没有人留言评论。精彩留言会获得点赞!