新型JNK抑制剂分子的制作方法
技术领域
本发明涉及酶抑制领域,尤其涉及c-Jun氨基末端激酶(JNK)的(多-)肽抑制剂。本发明还涉及培养抗这种(多-)肽抑制剂的抗体的方法,以及对应的抗体和生产所述抗体的细胞。
背景技术:
c-Jun氨基末端激酶(JNK)是丝裂原活化蛋白(MAP)激酶的应激活化群组的成员。这些激酶已经涉及对细胞生长和分化的控制,更广泛地说,涉及细胞对环境刺激的反应。JNK信号传导路径通过对环境应激的反应而被激活,并通过几种类型的细胞表面受体的结合而被激活。这些受体可以包括细胞因子受体、螺旋受体和受体酪氨酸激酶。在哺乳动物细胞中,JNK已经被涉及在生物过程如致癌性转化中,并调节对环境应激的适应性反应。已经将JNK与调节免疫反应相关联,包括免疫细胞的成熟和分化,以及影响识别免疫系统破坏的细胞中的程序性细胞死亡。丝裂原活化蛋白质激酶(MAPK)p38α显示出通过对抗JNK-c-Jun-路径而负面调节细胞增殖。丝裂原活化蛋白质激酶(MAPK)p38α因此看上去在抑制正常和癌症细胞增殖中是起作用的(例如见Hui等人,Nature Genetics,Vol 39,No.6,2007年6月)。还示出的是,c-Jun N-末端激酶(JNK)涉及由脊神经结扎(SNL)造成的神经性疼痛,其中SNL诱导JNK尤其是JNK1的缓慢和持续性的活化,而在SNL之后在脊髓小胶质细胞中发现p38丝裂原活化蛋白质激酶活化,其在21天降至接近基础水平(Zhuang等人,The Journal of Neuroscience,2006年3月29日,26(13):3551-3560))。
作为现有技术中已知的JNK信号路径的抑制剂,尤其包括例如上游激酶抑制剂(例如CEP-1347);JNK的小的化学抑制剂(SP600125和AS601245),其直接影响激酶活性例如通过与蛋白质激酶的ATP-结合位点竞争;以及JNK和其底物之间相互作用的肽抑制剂(例如见Kuan等人,Current Drug Targets–CNS&Neurological Disorders,2005年2月,第4卷,第1号,第63-67页;WO 2007/031280;全部通过引用并入本文)。WO 2007/031280公开了小的细胞渗透性融合肽,包括来源于HIV-TAT蛋白质的基础运输序列的所谓的TAT转运序列、和IB1的氨基酸抑制性序列。
WO 2007/031280公开了尤其是两种特殊的序列:L-TAT-IB1(GRKKRRQRRRPPRPKRPTTLNLFPQVPRSQD,本文的SEQ ID NO:196)和D-TAT-IB1(dqsrpvqpflnlttprkprpprrrqrrkkrg,本文的SEQ ID NO:197),后一种是L-TAT-IB1的逆反序列。由于HIV TAT衍生的转运序列,所以这些融合肽更有效地转运至靶细胞中,在那里保持有效直至蛋白质降解。
由于JNK的ATP独立的肽抑制剂通常是更特殊的抑制剂,如果要抑制JNK它们经常是第一选择。然而,甚至WO 2007/031280中公开的肽抑制剂也不是最理想的。例如,只由L-氨基酸构成的复合物L-TAT-IB1(本文的SEQ ID NO:196)快速地蛋白质降解。为了克服这个问题,WO 2007/031280的发明人还提出了D-TAT-IB1(本文的SEQ ID NO:197),其包括D-氨基酸。更准确地,D-TAT-IB1显示出L-TAT-IB1的逆反(retro-inverso)序列。D-氨基酸的引入由于立体化学的改变可能导致功能丧失的事实而变得困难。逆反方法可以用于降低所述风险,这是因为使用i)只是D-氨基酸ii)但是在反向肽序列中,与将一个或更多个D-氨基酸引入原始序列中相比,可以更有可能产生原始肽的可接受的构象类似物。在WO 2007/031280的情况下,该方法带来了与L-TAT-IB1相比抑制能力上的显著降低(见图4)。另外,逆反肽对蛋白质消化极其稳定,结果是例如在时间敏感的试验中控制的消化几乎是不可能的。
因此,本领域仍然需要比例如L-TAT-IB1(本文为SEQ ID NO:196)更稳定的JNK的肽抑制剂。另一方面,需要比例如D-TAT-IB1(本文为SEQ ID NO:197)更有效同时比其更不稳定的JNK的肽抑制剂。
技术实现要素:
因此,本发明要解决的问题是提供另外的JNK的(肽)抑制剂,它对蛋白水解降解优选不如WO 2007/031280中公开的L-TAT-IB1敏感,但同时优选比WO 2007/031280中公开的D-TAT-IB1对蛋白质水解降解更敏感和/或更有活性。
本发明的目的由发明人借助于所附的权利要求中列出的主题得以解决。
附图说明
下面给出对附图的简要说明。附图旨在更详细地说明本发明。但附图不旨在以任何方式限制本发明的主题。
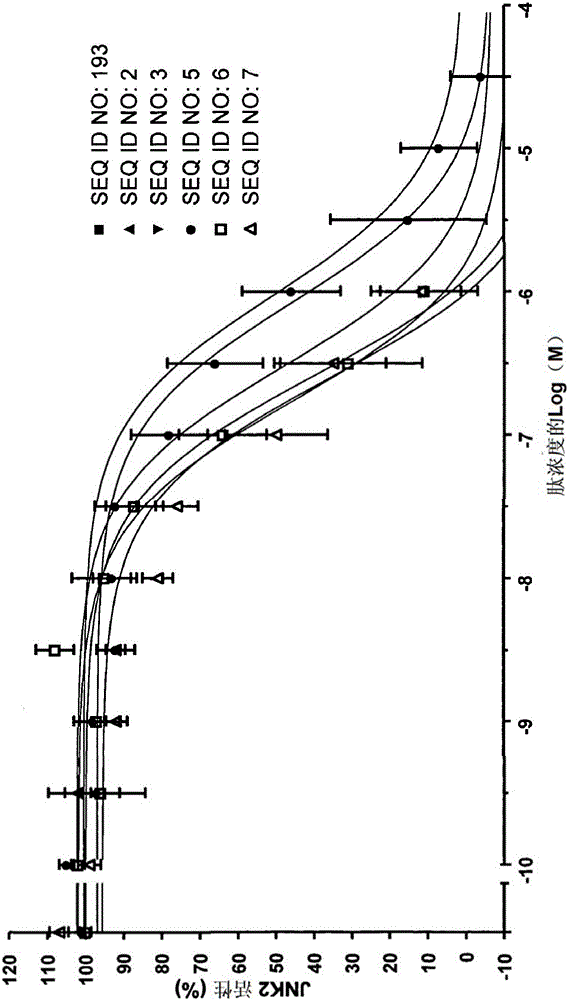
图1:几种根据本发明的JNK抑制剂的抑制效率的图解说明,通过体外AlphaScreen试验(增强的发光临近均相筛选试验(Amplified Luminescence Proximity Homogeneous-Screen Assay))进行调查研究。
图1A:通过SEQ ID NO:193、2、3、5、6和7对JNK1的抑制。
图1B:通过SEQ ID NO:193、2、3、5、6和7对JNK2的抑制。
图1C:通过SEQ ID NO:193、2、3、5、6和7对JNK3的抑制。
图2:表格说明了几种根据本发明的JNK抑制剂(SEQ ID NO:193、2、3、5、6和7)的抑制效率。给出的是nM范围内的IC50值,平均值的各个标准误差和所实施的试验数目(n)。
图3:几种根据本发明的JNK抑制剂的抑制效率的图解说明,它们是JNK抑制性(多-)肽序列和转运序列的融合蛋白。借助体外AlphaScreen试验(Amplified Luminescence Proximity Homogeneous-Screen Assay,增强的发光临近均相筛选试验)确定抑制效率。
图3A:通过SEQ ID NO:194、195、172、200、46、173、174、175、176、177、178、179、180、181和197对JNK1的抑制。
图3B:通过SEQ ID NO:194、195、172、200、46、173、174、175、176、177、178、179、180、181和197对JNK2的抑制。
图3C:通过SEQ ID NO:194、195、172、200、46、173、174、175、176、177、178、179、180、181和197对JNK3的抑制。
图3D:通过SEQ ID NO:194、195、172、200、46、182、183、184、185、186、187、188、189、190和197对JNK1的抑制。
图3E:通过SEQ ID NO:194、195、172、200、46、182、183、184、185、186、187、188、189、190和197对JNK2的抑制。
图3F:通过SEQ ID NO:194、195、172、200、46、182、183、184、185、186、187、188、189、190和197对JNK3的抑制。
图4:表格说明了几种根据本发明的JNK抑制剂的抑制效率,它们是JNK抑制性(多-)肽序列和转运序列的融合蛋白。给出的是nM范围内的IC50值,平均值的各个标准误差和所实施的试验数目(n)。
图5:具有SEQ ID NO:172、196和197的JNK抑制剂在50%人血清中的稳定性。具有SEQ ID NO:196的JNK抑制剂在6个小时内完全降解为氨基酸残基(A)。具有SEQ ID NO:172的JNK抑制剂只有在14天之后才完全降解(B)。具有SEQ ID NO:197的JNK抑制剂稳定至少长达30天(B)。
图6:示出内化实验,其使用TAT衍生的转运体构建如SEQ ID NO:30中表示的D-氨基酸/L-氨基酸模型。经分析的转运序列对应于SEQ IDNO:52-94加上SEQ ID NO:45、47、46、43和99(图6a)以及SEQ ID NO:100-147(图6b)。如可以看出的,具有共有序列rXXXrXXXr(SEQ ID NO:31)的所有转运体表现出比L-TAT转运体(SEQ ID NO:43)更高的内化能力。在包含10mM各种转运体的96孔板中将Hela细胞孵育24小时。随后用酸性缓冲液(0.2M甘氨酸,0.15M NaCl,pH 3.0)清洗细胞两次和用PBS清洗细胞两次。通过添加RIPA裂解缓冲液使细胞破裂。然后通过读取每种提取物的荧光强度(Fusion Alpha读板仪;PerkinElmer)随后进行背景扣除来确定内化的肽的相对量。
图7具有SEQ ID NO:172序列的JNK抑制剂阻断LPS-诱导的细胞因子和趋化因子在THP1-PMA-分化的巨噬细胞中释放。图7A:TNF释放(THP1pma 6h 3ng/ml LPS);图7B:TNFa释放(THP1pma 6h 10ng/ml LPS);图7C:IL 6释放(THP1pma 6h 10ng/ml LPS);图7D:MCP1释放(THP1pma 6h 3ng/ml LPS)。
图8 SEQ ID NO:172的JNK抑制剂阻断LPS-诱导的IL6在THP1分化的巨噬细胞中释放,且比D-TAT-IB1(SEQ ID NO:197)、dTAT(SEQ ID NO:45)和SP 600125的潜能更高。添加LPS 6小时(10ng/ml)。
图9 SEQ ID NO:172的JNK抑制剂阻断LPS-诱导的TNFα在THP1分化的巨噬细胞中释放,且比D-TAT-IB1(SEQ ID NO:197)、dTAT(SEQ ID NO:45)和SP 600125的潜能更高。添加LPS 6小时(10ng/ml)。
图10 SEQ ID NO:172的JNK抑制剂阻断LPS-诱导的IL-6在PMA分化的巨噬细胞中释放,且比D-TAT-IB1(SEQ ID NO:197)和L-TAT-IB1(SEQ ID NO:196)的潜能更高。添加LPS 6小时。
图11 SEQ ID NO:172的JNK抑制剂阻断LPS-诱导的TNFα在PMA分化的巨噬细胞中释放,且比D-TAT-IB1(SEQ ID NO:197)和L-TAT-IB1(SEQ ID NO:196)的潜能更高。
图12 SEQ ID NO:172的JNK抑制剂阻断3ng/ml的LPS-诱导的TNFα在原代大鼠全血细胞中释放。给出的是在不同的LPS水平(ng/ml)下对照、1μM SEQ ID NO:172、3μM SEQ ID NO:172和10μM SEQ ID NO:172的结果。
图13 SEQ ID NO:172的JNK抑制剂阻断通过人类原代T-细胞对PMA/离子霉素的反应的IL2分泌。
图14 SEQ ID NO:172的JNK抑制剂阻断通过人类原代T-细胞对CD3/CD28刺激的反应的IL2分泌。通过它们的SEQ ID NO:172和197指出所用的JNK抑制剂。
图15具有SEQ ID NO:172的JNK抑制剂对大鼠原代淋巴结纯化的T细胞中CD3/CD28-诱导的IL-2释放的剂量-依赖性抑制。将对照大鼠牺牲并收集淋巴结。进一步纯化T-细胞(使用磁性阴性选择)并以200.000细胞/孔放入96孔板。用抗-大鼠CD3和抗-大鼠CD28抗体(2μg/mL)处理细胞。在CD3/CD28处理之前1小时将具有SEQ ID NO:172的JNK抑制剂添加到培养物中,并在处理之后24小时评价上清液中的IL-2释放。
图16大鼠原代淋巴结纯化的T细胞中CD3/CD28-诱导的IL-2释放的剂量-依赖性抑制:几种JNK抑制剂的比较,也就是SEQ ID NO:172、197和SP600125。
图17用PMA+离子霉素刺激的大鼠全血中IL-2释放的剂量依赖性抑制。在用PMA+离子霉素刺激之前1小时,以三种不同的浓度(即1、3和10μM)添加具有SEQ ID NO:172的JNK抑制剂。添加三种剂量的活化剂(25/500ng/mL、50/750ng/mL和50/1000ng/mL)4小时。在上清液中评价IL-2释放。具有SEQ ID NO:172的JNK抑制剂在10μM时确实有效地降低了三种被测试的活化剂浓度下PMA-离子霉素诱导的IL-2释放。
图18人类全血中的JNK抑制和IL-6释放。在用LPS(0.02ng/mL)进行4小时全血刺激之前1小时,以三种不同浓度(即1、3和10μM)添加具有SEQ ID NO:172的JNK抑制剂。具有SEQ ID NO:172的JNK抑制剂确实以剂量-依赖方式降低了LPS-诱导的IL-6释放。
图19人类全血中的JNK抑制和IL-2释放。在用PMA+离子霉素(25/700ng/mL、50/800ng/ml和50/1000ng/mL)进行4小时全血刺激之前1小时,以三种不同浓度(即1、3和10μM)添加具有SEQ ID NO:172的JNK抑制剂。具有SEQ ID NO:172的JNK抑制剂确实以剂量-依赖方式降低了PMA+离子霉素诱导的IL-2释放。
图20人类全血中的JNK抑制和IFN-γ释放。在用PMA+离子霉素(25/700ng/mL、50/800ng/ml和50/1000ng/mL)进行4小时全血刺激之前1小时,以三种不同浓度(即1、3和10μM)添加具有SEQ ID NO:172的JNK抑制剂。具有SEQ ID NO:172的JNK抑制剂确实以剂量-依赖方式降低了PMA+离子霉素诱导的IFN-γ释放。
图21人类全血中的JNK抑制和TNF-α释放。在用PMA+离子霉素(25/700ng/mL、50/800ng/ml和50/1000ng/mL)进行4小时全血刺激之前1小时,以三种不同浓度(即1、3和10μM)添加具有SEQ ID NO:172的JNK抑制剂。具有SEQ ID NO:172的JNK抑制剂确实以剂量-依赖方式降低了PMA+离子霉素诱导的TNF-α释放。
图22人类全血中的JNK抑制和TNF-α释放。在用PHA-L(5μg/mL)进行3天的全血刺激之前1小时,以三种不同浓度(即1、3和10μM)添加具有SEQ ID NO:172的JNK抑制剂。具有SEQ ID NO:172的JNK抑制剂确实以剂量-依赖方式降低了PHA-L诱导的TNF-α释放。
图23人类全血中的JNK抑制和IL-2释放。在用PHA-L(5μg/mL)进行3天的全血刺激之前1小时,以三种不同浓度(即1、3和10μM)添加具有SEQ ID NO:172的JNK抑制剂。具有SEQ ID NO:172的JNK抑制剂确实以剂量-依赖方式降低了PHA-L诱导的IL-2释放。
图24人类全血中的JNK抑制和TNF-α释放。在用CD3+/-CD28抗体(2μg/mL)进行3天的全血刺激之前1小时,以三种不同浓度(即1、3和10μM)添加具有SEQ ID NO:172的JNK抑制剂。具有SEQ ID NO:172的JNK抑制剂确实以剂量-依赖方式降低了CD3/CD28诱导的TNF-α释放。
具体实施方式
JNK抑制剂
本发明的第一方面涉及JNK抑制剂,其包括根据下列通式的抑制性(多-)肽序列:
X1-X2-X3-R-X4-X5-X6-L-X7-L-X8(SEQ ID NO:1),
其中X1是选自氨基酸R、P、Q和r的氨基酸,
其中X2是选自氨基酸R、P、G和r的氨基酸,
其中X3是选自氨基酸K、R、k和r的氨基酸,
其中X4是选自氨基酸P和K的氨基酸,
其中X5是选自氨基酸T、a、s、q、k的氨基酸,或者是不存在的,
其中X6是选自氨基酸T、D和A的氨基酸,
其中X7是选自氨基酸N、n、r和K的氨基酸;和
其中X8是选自F、f和w的氨基酸,
条件是:至少一种、至少两种、至少三种、至少四种、至少五种或六种选自X1、X2、X3、X5、X7和X8的氨基酸是D-氨基酸,优选地条件是,至少一种、至少两种、至少三种或四种选自X3、X5、X7和X8的氨基酸是D-氨基酸。
根据本发明的JNK抑制剂的抑制性(多-)肽序列包括L-氨基酸和在大多数实施方案中包括D-氨基酸。除非特殊指出,在本文中用大写字母表示L-氨基酸残基,而用小写字母表示D-氨基酸残基。甘氨酸可以用大写或小写字母表示(由于没有D-或L-甘氨酸)。除非特殊指出,本文公开的氨基酸序列通常从N-端至C-端(从左到右)给出。所给出的氨基酸序列在C-端和/或N-端可以是经修饰或未经修饰的,例如在C-端乙酰化和/或在N-端酰胺化或用巯基乙胺化(cysteamide)修饰。为了清楚起见,本文公开的氨基酸序列的C-端和/或N-端的这种可能的但完全是任选的修饰为清楚起见没有具体指出。
本发明的JNK抑制剂是c-Jun N-末端激酶(JNK)的(多-)肽抑制剂。所述抑制剂抑制c-Jun N-末端激酶(JNK)的激酶活性,也就是说,阻止或减少JNK底物例如c-Jun、ATF2和/或Elk-1的磷酸化的程度。本领域的技术人员应理解,本文所用的术语“抑制剂”不包括不可逆地破坏c-Jun N-末端激酶(JNK)分子和/或激酶活性的化合物。此外,本文所用的术语“抑制JNK活性”指的是c-Jun N-末端激酶(JNK)的激酶活性的抑制。
此外,本文所用的JNK抑制剂包括至少一个氨基酸的聚合物的功能单元,也就是(多-)肽序列。而且,该至少一个氨基酸的功能聚合物提供对JNK活性的抑制。所述抑制性(多-)肽序列的氨基酸单体通常经由肽键互相连接,但是可以容忍所述肽键或所述侧链残基的(化学)修饰,只要抑制活性(JNK活性的抑制)没有完全丧失,也就是说得到的化学实体仍然在本文定义的功能上适合作为JNK抑制剂。术语“(多-)肽”不应解释为对(多-)肽单元的长度的限制。优选地,本发明的JNK抑制剂的抑制性(多-)肽序列小于500、490、480、470、460、450、440、430、420、410、400、390、380、370、360、350、340、330、320、310、300、290、280、270、260、250、240、230、220、210、200、190、180、170、160、150、140、130、120、110、100、95、90、85、80、75、70、65、60、55、50、49、48、47、46、45、44、43、42、41、40、39、38、37、36、35、34、33、32、31、30、29、28、27、26、25、24、23、22、21、20、19、18、17、16、15、14、13,或者小于12个氨基酸长。优选地,该抑制性(多-)肽序列不小于10个氨基酸残基,更优选地不少于11个氨基酸残基。
此外,本发明的“JNK抑制剂”抑制JNK活性,例如显示出对人类JNK调节的c-Jun底物(SEQ ID NO:198)的磷酸化以下列的IC 50值的抑制:
a)关于人类JNK1的抑制,小于3000nM,更优选小于2000nM,甚至更优选小于1000nM,甚至更优选小于500nM,甚至更优选小于250nM,甚至更优选小于200nM,甚至更优选小于150nM,最优选小于100nM,
b)关于人类JNK2的抑制,小于3000nM,更优选小于2000nM,甚至更优选小于1000nM,甚至更优选小于500nM,甚至更优选小于250nM,甚至更优选小于200nM,甚至更优选小于150nM,最优选小于100nM,和/或
c)关于人类JNK3的抑制,小于3000nM,更优选小于2000nM,甚至更优选小于1000nM,甚至更优选小于500nM,甚至更优选小于250nM,甚至更优选小于200nM,甚至更优选小于150nM,最优选小于100nM。
对于一些应用,优选该抑制剂根据上面的定义抑制人类JNK2和/或JNK3,但是不根据上面的定义抑制JNK1。
本领域技术人员可以容易地评估JNK活性是否受到抑制。本领域公知有几种方法。一个实例是放射性激酶试验或非放射性激酶试验(例如Alpha筛选测试;例如见Guenat等人,J Biomol Screen,2006;11:第1015-1026页)。
根据本发明的JNK抑制剂可以因此例如包括根据SEQ ID NO:2至27中任意项的抑制性(多-)肽序列(见表1)。
根据本发明的JNK抑制剂还可以是JNK抑制剂(变体),其包括与选自SEQ ID NO:1-27的序列,尤其是SEQ ID NO:8分享序列一致性的抑制性(多-)肽序列,所述序列一致性为至少50%,更优选至少55%,更优选至少60%,更优选至少65%,更优选至少70%,更优选至少75%,更优选至少80%,更优选至少85%,最优选至少90%,
条件是,对于选自SEQ ID NO:1-27的各个序列,与其分享序列一致性的这种抑制性(多-)肽序列
a)将L-精氨酸(R)残基保持在位置4上,
b)将两个L-亮氨酸(L)残基保持在位置8和10处(关于SEQ ID NO:25-27为位置7和9),
c)在对应于选自SEQ ID NO:1的X1、X2、X3、X5、X7和X8的氨基酸的各个位置和SEQ ID NO:2-27的各个位置处显示出一个、两个、三个、四个、五个或六个D-氨基酸,更优选地在对应于选自SEQ ID NO:1的X3、X5、X7和X8的氨基酸的位置和SEQ ID NO:2-27的各个位置处显示出一个、两个、三个或四个D-氨基酸,和
d)仍然抑制JNK活性(即,为如本文定义的JNK抑制剂)。
当然,本文公开的变体(尤其是包括在上述定义范围内与选自SEQ ID NO:1-27的序列分享一定程度的序列一致性的抑制性(多-)肽序列的JNK抑制剂变体),它优选与各个参考序列分享小于100%的序列一致性。
考虑到所述定义且为了清楚起见,表1中在包括SEQ ID NO:1-27(见上面定义中的a)和b))的JNK抑制剂的变体中不可以改变的残基被加上了下划线。
不一致的氨基酸优选是保守性氨基酸取代的结果。
如本文所用的,保守性氨基酸取代基可以包括在具有足够相似的物理化学性质的组内的氨基酸残基,从而该组的成员之间的取代会维持分子的生物活性(例如见Grantham,R.(1974),Science 185,862-864)。特别地,保守性氨基酸取代优选是其中的氨基酸源于相同类氨基酸(例如碱性氨基酸、酸性氨基酸、极性氨基酸、具有脂肪族侧链的氨基酸、具有带正电或带负电的侧链的氨基酸、在侧链中具有芳香族基团的氨基酸、其侧链可以成为氢桥的氨基酸例如具有羟基官能的侧链等)的取代。保守性取代在本发明情况下例如用碱性氨基酸残基(Lys、Arg、His)取代另一种碱性氨基酸残基(Lys、Arg、His),用脂肪族氨基酸残基(Gly、Ala、Val、Leu、Ile)取代另一种脂肪族氨基酸残基,用芳香族氨基酸残基(Phe、Tyr、Trp)取代另一种芳香族氨基酸残基,通过丝氨酸取代苏氨酸或者通过异亮氨酸取代亮氨酸。本领域技术人员应已知其他的保守性氨基酸交换。应优选保持异构体形式,例如K优选取代R或H,而k优选取代r和h。
在用于JNK抑制剂变体的以上定义内的其他可能的取代例如是,如果:
a)SEQ ID NO:1的X1、X2、X3、X4、X5、X6、X7和/或X8中的一个、两个或更多个,或者在选自SEQ ID NO:2-27的各个序列中的相应位置取代为A或a,
b)删除SEQ ID NO:1的X1或X8,或者删除选自SEQ ID NO:2-27的各个序列中的相应位置;
c)SEQ ID NO:1的X5或选自SEQ ID NO:2-27的各个序列中的相应位置是E、Y、L、V、F或K;
d)SEQ ID NO:1的X5或选自SEQ ID NO:2-27的各个序列中的相应位置是E、L、V、F或K;或者
e)SEQ ID NO:1的X1、X2、X3的一个、两个或三个,或者选自SEQID NO:2-27的各个序列中的相应位置是中性氨基酸。
如本文所用的术语“序列一致性%”应如下理解:将两个待比对的序列对齐,以给出序列之间的最大关联性。这可以包括在一个或两个序列中插入“缺口”,以增加对齐程度。一致性%可以随后在进行比对的每个序列的全长上(所谓的总体对齐)确定,这尤其适合用于相同的或类似的长度的序列;或者在较短的、限定的长度上(所谓局部比对)确定,这更适合用于不等的长度的序列。在上述情况下,与问询氨基酸序列具有至少例如95%“序列一致性”的氨基酸序列意在表明,除了主题氨基酸序列可以每问询氨基酸序列的每100个氨基酸包括最多5个氨基酸交替之外,主题氨基酸序列的序列与问询序列是相同的。换句话说,为了获得与问询氨基酸序列具有至少95%的一致性的序列的氨基酸序列,在主题序列中可以插入或用其他氨基酸取代或删除最多5%(100个中的5个)的氨基酸残基。为了确定序列一致性,用L-氨基酸取代D-氨基酸(反之亦然)被认为是产生了非一致性的残基,虽然仅仅是同一氨基酸的D-(或L-异构体)。
比较两个或更多个序列的一致性和同源性的方法在本领域是众所周知的。可以例如通过使用数学算法确定两个序列一致的百分比。优选但是不限定的可使用的数学算法的实例是Karlin等人(1993)的算法,PNAS USA,90:5873-5877。这种算法被整合至程序的BLAST家族中,例如BLAST或NBLAST程序(另外见Altschul等人,1990,J.Mol.Biol.215,403-410或Altschul等人(1997),Nucleic Acids Res,25:3389-3402),通过万维网的NCBI主页ncbi.nlm.nih.gov可以访问,以及FASTA(Pearson(1990),Methods Enzymol.183,63-98;Pearson和Lipman(1988),Proc.Natl.Acad.Sci.U.S.A 85,2444-2448.)。通过这些程序可以识别在一定程度上与其他序列一致的序列。此外,在Wisconsin序列分析软件包,9.1版本(Devereux等人,1984,Nucleic Acids Res.,387-395)中可用的程序,例如程序BESTFIT和GAP可以用于确定两个多肽序列之间的一致性%。BESTFIT使用“局部同源性”算法(Smith和Waterman(1981),J.Mol.Biol.147,195-197.),并在两个序列之间发现相似度的最佳单区。
当然,根据本发明的JNK抑制剂除了包括上面提到的抑制性(多-)肽序列之外,还可以包括附加的序列、结构域、标记物(例如荧光或辐射标记物)、表位等,只要不丧失如本文定义的抑制JNK活性的能力即可。例如,根据本发明的JNK抑制剂还可以包括转运序列。本文所用的“转运序列”是为其所附着的分子提供跨生物膜的易位的(多-)肽序列。因此,根据本发明的包括转运序列的JNK抑制剂优选能够跨生物膜易位。因此,本发明的这种JNK抑制剂可以更容易进入细胞、细胞亚室和/或进入细胞的核。
所述转运序列可以例如(例如直接地)N-端或(例如直接地)C-端连接至JNK抑制剂的抑制性(多-)肽序列。转运序列和抑制性(多-)肽序列还可以间隔开,例如可以通过中间序列隔开。还设计可以将转运序列放置在JNK抑制剂分子中完全不同于抑制性(多-)肽序列的别的位置,尤其是如果JNK抑制剂是更复杂的分子(例如包括几个结构域、是多聚缀合物等)。还可以设计,只要保持JNK抑制剂活性,转运序列和抑制性(多-)肽序列可以重叠。下面进一步给出这种重叠的实例。
与本发明的JNK抑制剂一起使用的转运序列可以选自,但不限于来源于,HIV TAT(HIV)的转运序列,例如天然蛋白质如TAT蛋白质(例如美国专利号5,804,604和5,674,980中描述的,这些参考文献的每一个通过引用并入本文)、HSV VP22(单纯性疱疹(Herpes simplex))(例如WO 97/05265;Elliott和O'Hare,Cell 88:223-233(1997)中描述的)、非病毒蛋白质(Jackson等人,Proc.Natl.Acad.Sci.USA 89:10691-10695(1992));来源于的转运序列,尤其是来源于果蝇的控制触角的基因(Drosophila antennapedia)(例如其控制触角的基因的载体序列)、FGF、乳铁蛋白等;或者来源于碱性肽,例如长度为5至15个氨基酸、优选10至12个氨基酸且包括至少80%、更优选85%或甚至90%碱性氨基酸的肽,例如精氨酸、赖氨酸和/或组氨酸;或者可以选自例如富精氨酸的肽序列,例如RRRRRRRRR(R9;SEQ ID NO:152)、RRRRRRRR(R8;SEQ ID NO:153)、RRRRRRR(R7;SEQ ID NO:154)、RRRRRR(R6,SEQ ID NO:155)、RRRRR(R5,SEQ ID NO:156)等;来自VP22、来自PTD-4蛋白质或肽、来自RGD-K16、来自PEPT1/2或PEPT1/2蛋白质或肽、来自SynB3或SynB3蛋白质或肽、来自PC抑制剂、来自P21衍生的蛋白质或肽、或来自JNKI蛋白质或肽。
用在本发明的JNK抑制剂中的转运序列的实例尤其是,但不限于,来源于HIV-1TAT蛋白质的碱性转运序列。优选地,HIV-1TAT蛋白质的碱性转运序列可以包括来自人类免疫缺陷病毒HIV-1TAT蛋白质的序列,例如美国专利号5,804,604和5,674,980中描述的,每个都通过引用并入本文。在本文范围内,全长HIV-1TAT蛋白质具有由HIV TAT基因的两个外显子编码的86个氨基酸残基。TAT氨基酸1-72由外显子1编码,而氨基酸73-86由外显子2编码。全长TAT蛋白质的特征在于,包含两个赖氨酸和六个精氨酸(氨基酸49-57)的碱性区域与包含七个半胱氨酸残基(氨基酸22-37)的富半胱氨酸区域。碱性区域(即氨基酸49-57)被认为对核定位是重要的。Ruben,S.等人,J.Virol.63:1-8(1989);Hauber,J.等人,J.Virol.63 1181-1187(1989)。富半胱氨酸区域调节金属连接的二聚体的体外形成(Frankel,A.D.等人,Science 240:70-73(1988);Frankel,A.D.等人,Proc.Natl.Acad.SciUSA 85:6297-6300(1988)),且对其作为反式作用因子的活性是重要的(Garcia,J.A.等人,EMBO J.7:3143(1988);Sadaie,M.R.等人,J.Virol.63:1(1989))。如在其他调节蛋白中,N-端区域可以涉及对细胞内蛋白酶的保护(Bachmair,A.等人,Cell 56:1019-1032(1989))。用在本发明的JNK抑制剂中的优选TAT转运序列的特征优选在于,存在TAT碱性区域氨基酸序列(天然存在的TAT蛋白质的氨基酸49-57);不存在TAT富半胱氨酸区域氨基酸序列(天然存在的TAT蛋白质的氨基酸22-36)和不存在TAT外显子2-编码的羧基-末端结构域(天然存在的TAT蛋白质的氨基酸73-86)。更优选地,本发明的JNK抑制剂中的转运序列可以选自包含TAT残基48-57或49至57的氨基酸序列或其变体。
优选地,在本发明的给定的JNK抑制剂中转运序列还显示出D-氨基酸,例如为了改善对蛋白酶的稳定性。尤其优选的是,显示出交替的D-氨基酸和L-氨基酸的特殊顺序的转运序列。这种交替的D-氨基酸和L-氨基酸的顺序(基序)可以遵循,但不限于,SEQ ID NO:28-30中任一个的模式:
dlLLLxdmLLLydn(SEQ ID NO:28);
dLLLd(LLLd)a(SEQ ID NO:29);和/或
dLLLdLLLd(SEQ ID NO:30);
其中:d是D-氨基酸;
L是L-氨基酸;
a是0至3,优选是0至2,更优选是0、1、2或3,甚至更优选是0、1或2,和最优选是1;
l、m和n互相独立地是1或2,优选是1;
x和y互相独立地是0、1或2,优选是1。
在转运序列是合成的时,D-氨基酸和L-氨基酸的所述顺序(基序)变得相关,也就是说,当氨基酸序列(即侧链残基的类型)保持不变时,各个异构体相互替代。例如,来源于HIV TAT的已知的转运序列是RKKRRQRRR(SEQ ID NO:43)。对其施加SEQ ID NO:30的D-/L-氨基酸顺序产生rKKRrQRRr(SEQ ID NO:46)。
在特定实施方案中,本发明的JNK抑制剂的转运序列可以包括至少一个根据rXXXrXXXr的序列(SEQ ID NO:31),其中:
r代表D-对映体精氨酸;
X是任意的L-氨基酸(包括甘氨酸);
其中每个X可以单独地且独立于SEQ ID NO:31内的任何其他X来选择。SEQ ID NO:31内所述6个X L-氨基酸中的至少4个是K或R。在另一实施方案中,根据本发明的JNK抑制剂包括转运序列rX1X2X3rX4X5X6r(SEQ ID NO:32),其中X1是K,X2是K,X3是R和X4、X5和X6是互相独立地选择的L-氨基酸(包括甘氨酸)。类似地,根据本发明的JNK抑制剂的转运序列可以包括序列rX1X2X3rX4X5X6r(SEQ ID NO:33),其中X4是Q,X5是R,X6是R且X1、X2和X3是互相独立地选择的L-氨基酸(包括甘氨酸)。本发明的JNK抑制剂还可以包括序列rX1X2X3rX4X5X6r(SEQ ID NO:34),其中1个、2个、3个、4个、5个或6个X氨基酸残基选自下列组:X1是K,X2是K,X3是R,X4是Q,X5是R,X6是R,同时上面的组未选自的剩余的X氨基酸残基可以是任意的L-氨基酸(包括甘氨酸),且互相独立地选择。于是,X1优选是Y和/或X4优选是K或R。
用在本发明的JNK抑制剂分子中的转运序列的实例可以选自,但不限于,下面表2中给出的序列(SEQ ID NO:31-170)或选自其任意片段或变体或化学修饰的衍生物(优选保留跨生物膜易位的功能)。
如上面提及的,转运序列还可以选自表2的上述序列的片段或变体(条件是,该片段或变体优选保留提供跨生物膜易位的功能)。在该特定上下文中,那些转运序列的变体和/或片段优选包括在如表2中定义的这些转运序列的整个序列长度上分享至少10%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%、至少80%或至少85%、优选至少90%、更优选至少95%和最优选至少99%的序列一致性的变体和/或片段。在该特定上下文中,如表2中定义的转运序列的“片段”优选应理解为其截短序列,即与原始序列的氨基酸序列相比较,N-末端、C-末端和/或序列内部被截断的氨基酸序列。
此外,如上定义的转运序列的“变体”或其片段优选理解为,其中在一个或更多个变异体中变体的氨基酸序列不同于如本文定义的原始的转运序列或其片段的序列,例如一个或更多个氨基酸被取代(或者如果需要,插入和/或缺失)。优选地,如上定义的这种转运序列的变体与各自的原始序列相比较,具有相同的生物功能或特定的活性,也就是提供转运例如进入细胞或核。在这种上下文中,如上定义的这种转运序列的变体可以例如包括约1至50、1至20、更优选1至10和最优选1至5、4、3、2或1个氨基酸变化。如上定义的这种转运序列的变体优选可以包括保守性氨基酸取代。保守性氨基酸取代的概念在本领域是公知的,并且在上面已经对于JNK抑制性(多-)肽序列进行了描述,并因此在此应用。
导入本发明的JNK抑制剂中的转运序列的长度可以改变。在一些实施方案中已经设计,在根据本发明的JNK抑制剂的转运序列的长度小于150个氨基酸、小于140个氨基酸、小于130个氨基酸、小于120个氨基酸、小于110个氨基酸、小于100个氨基酸、小于90个氨基酸、小于80个氨基酸、小于70个氨基酸、小于60个氨基酸、小于50个氨基酸、小于40个氨基酸、小于30个氨基酸、小于20个氨基酸和/或小于10个氨基酸。
本领域的技术人员可以轻易地确定,特殊的转运序列在根据本发明的JNK抑制剂的情况下是否仍然具有功能。例如,可以将包括转运结构域的JNK抑制剂融合到标记物,例如荧光蛋白质如GFP、辐射性标记物、酶、荧光体、表位等,其在细胞中可以容易地检测到。随后,将包括转运序列和标记物的JNK抑制剂转染入细胞,或添加至培养物上清夜,细胞膜的渗透可以通过使用生物物理和生物化学标准方法来监控(例如流式细胞术、(免疫)荧光显微镜等)。
根据本发明的JNK抑制剂的特殊实例包括表3中给出的转运序列:
如上所述,在本发明的特定实施方案中,转运序列和抑制性(多-)肽序列可以重叠。换句话说,转运序列的N-端可以与抑制性(多-)肽序列的C-端重叠,或者转运序列的C-端可以与抑制性(多-)肽序列的N-端重叠。尤其优选后一实施方案。优选地,转运序列通过一个、两个或三个氨基酸残基与抑制性(多-)肽序列重叠。在这种情况下,给定的转运序列可以与SEQ ID NO:1或其各个变体在位置1(X1)、位置1和2(X1,X2)、位置1、2和3(X1,X2,X3)处重叠。
SEQ ID NO:174、175、178、179、180、181、182、183、184、188、189和190是根据本发明的JNK抑制剂的良好实例,其中转运序列和抑制性(多-)肽序列重叠,例如rKKRrQRRrRPTTLNLf(SEQ ID NO:174)是SEQ ID NO:46(下划线部分)和SEQ ID NO:11(斜体字部分)的重叠。
当然根据本发明的JNK抑制剂还可以选自是根据SEQ ID NO:171-190的JNK抑制剂中任一个的变体的JNK抑制剂。优选地,该变体与SEQ ID NO:171-190的序列,尤其与SEQ ID NO:172分享至少50%、更优选至少55%、更优选至少60%、更优选至少65%、更优选至少70%、更优选至少75%、更优选至少80%、更优选至少85%、更优选至少90%、更优选至少95%的序列一致性,
条件是,关于所述序列SEQ ID NO:171-190内的抑制性(多-)肽序列(例如参见SEQ ID NO:1的抑制性(多-)肽序列和SEQ ID NO:2-27的特殊实例),与其分享序列一致性的这些序列
a)将L-精氨酸(R)残基保持在抑制性(多-)肽序列内的位置4上,
b)将两个L-亮氨酸(L)残基保持在抑制性(多-)肽序列内的位置8和10(关于SEQ ID NO:25-27在位置7和9)处,
c)在对应于选自SEQ ID NO:1的X1、X2、X3、X5、X7和/或X8的氨基酸的各个位置和SEQ ID NO:2-27的各个位置处显示出至少一个、至少两个、至少三个、至少四个、至少五个或六个D-氨基酸,更优选地在对应于选自SEQ ID NO:1的X3、X5、X7和X8的氨基酸的位置和SEQ ID NO:2-27的各个位置处显示出至少一个、至少两个、至少三个或四个D-氨基酸,和
d)仍然抑制JNK活性(即,如本文定义的JNK抑制剂)。
考虑到所述定义和为清楚起见,在表3中用下划线标出包括SEQ ID NO:171-190的JNK抑制剂的变体中不可改变的残基(见上面定义中的a)和b))。
包括SEQ ID NO:171-190的JNK抑制剂的变体中非一致性的氨基酸优选是保守性氨基酸取代的结果(见上面)。当然,上面提及的其他可能的取代也设计用于包括SEQ ID NO:171-190的JNK抑制剂的变体。同样的,本发明当然还设计,偏离原始序列的根据SEQ ID NO:171-190的JNK抑制剂中任一项的变体不在抑制性(多-)肽序列中或不是排他地在抑制性(多-)肽序列中,但在转运序列中显示出变体残基。转运序列的变体和片段具体见上面各自的公开。
如之前提及的,根据本发明的JNK抑制剂的转运序列和JNK抑制性(多-)肽序列不需要必须直接互相连接。它们还可以间隔开,例如通过中间(多-)肽序列。分隔抑制性(多-)肽序列和其他(功能性)序列例如转运序列的优选的中间序列由长度小于10个氨基酸的短的肽序列构成,例如六聚体、五聚体、四聚体、三肽或者甚至只有二肽或单个氨基酸残基。尤其优选的中间序列是二-脯氨酸、二-甘氨酸、二-精氨酸和/或二-赖氨酸的一个、两个或更多个复制物,所有的都仅仅是L-氨基酸形式,或者所有的都仅仅是D-氨基酸形式,或者具有混合的D-氨基酸和L-氨基酸。当然,还可以使用其他公知的肽间隔序列。
根据本发明特别优选的JNK抑制剂包括SEQ ID NO:8(或在前面进一步定义的范围和限制内与SEQ ID NO:8分享序列一致性的序列)和转运序列。转运序列优选选自SEQ ID No:31-170中的任一个或者其如本文定义的变体,甚至更优选选自SEQ ID NO:31-34和46-151中的任一个。根据本发明的JNK抑制剂的尤其优选的实施方案是包括SEQ ID NO:8和SEQ ID NO:46(或在前面进一步定义的范围和限制内与其分享各个序列一致性的序列)的JNK抑制剂。优选的实例是包括SEQ ID NO:172的序列或如本文定义的其在转运序列和/或抑制性(多-)肽序列中变化的变体的JNK抑制剂。
本发明的另一方面涉及JNK抑制剂,包括:
a)抑制性(多-)肽,其包括选自RPTTLNLF(SEQ ID NO:191)、KRPTTLNLF(SEQ ID NO:192)、RRPTTLNLF和/或RPKRPTTLNLF(SEQ ID NO:193)的序列,和
b)转运序列,优选选自表2中公开的转运序列或其变体/片段的转运序列,甚至更优选选自SEQ ID NO:31-34和46-151或它们各自的变体或片段的转运序列。
转运序列和抑制性(多-)肽序列可以重叠。用于本发明所述实施方案的优选的转运序列尤其是SEQ ID NO:46的转运序列,优选连接(例如直接地)到抑制性(多-)肽序列的N-末端。
本发明的JNK抑制剂还可以是包括或由序列GRKKRRQRRRPPKRPTTLNLFPQVPRSQD(SEQ ID NO:194)或序列GRKKRRQRRRPTTLNLFPQVPRSQD(SEQ ID NO:195)构成的JNK抑制剂。
本发明的另一方面涉及包括选自序列rKKRrQRr(SEQ ID NO:148)、rKKRrQRrK(SEQ ID NO:149)和/或rKKRrQRrR(SEQ ID NO:150)的转运序列的(多-)肽。
如本文所用,包括特定序列或特定SEQ ID NO:通常表明存在所述序列的(至少)一个复制品,例如在JNK抑制剂分子中。例如,一个抑制性(多-)肽序列通常足以达到足够的JNK活性的抑制。然而,本发明人当然设计,还可以使用各个序列的两个或更多个复制物(例如不同或相同类型的抑制性(多-)肽序列的两个或更多个复制物,和/或不同或相同类型的转运序列的两个或更多个复制物),只要得到的分子对抑制JNK活性的整体能力不废止(即各个分子仍然是如本文定义的JNK抑制剂)即可。
本发明的JNK抑制剂可以通过本领域众所周知的方法获得或生产,例如通过化学合成,其经由使用Fmoc(9-芴基甲氧基羰基)策略的固相肽合成,也就是通过Fmoc去保护和Fmoc-氨基酸偶联循环的连续执行。很多公司提供这种肽合成的商业服务,例如PolyPeptide公司(斯特拉斯堡,法国)。
抗体
本发明的另一方面涉及培养用于抗本发明的JNK抑制剂的抗体的生产,即生产识别本发明的JNK抑制剂的抗体的方法。生产抗体的方法在本领域是完全众所周知的。
因此,本发明还涉及用根据本发明的JNK抑制剂免疫非人类动物的方法,所述方法包括下列步骤:
-使适用于抗体生产的非人类动物与本发明的JNK抑制剂接触(免疫),更优选与包括或由具有选自SEQ ID NO:1-27中任一项的序列的(多-)肽构成的JNK抑制剂接触(免疫),所述非人类动物尤其是非人类哺乳动物,更优选是选自山羊和例如小鼠、大鼠和兔子的啮齿动物的动物。
本文所用的“免疫”理解为非治疗性质,因为根据本发明的JNK抑制剂不是病原体(即不需要治疗)。
本发明还涉及生产识别根据本发明的JNK抑制剂的(多克隆)抗体的方法,所述方法包括下列步骤:
-从适用于抗体生产的非人类动物分离识别所述JNK抑制剂的(多克隆)抗体,所述非人类动物尤其是非人类哺乳动物,更优选是选自山羊和例如小鼠、大鼠和兔子的啮齿动物的动物,
所述非人类动物事先已经与本发明的JNK抑制剂接触(免疫),更优选与包括或由具有选自SEQ ID NO:1-27中任一项的序列的(多-)肽构成的JNK抑制剂接触(免疫)。
本发明还涉及分离产生识别根据本发明的JNK抑制剂的抗体的细胞的方法,所述方法包括下列步骤:
-从适用于抗体生产的非人类动物分离产生识别所述JNK抑制剂的抗体的细胞,以及任选地使所述细胞永生化,所述非人类动物尤其是非人类哺乳动物,更优选是选自山羊和例如小鼠、大鼠和兔子的啮齿动物的动物,
所述非人类动物事先已经与本发明的JNK抑制剂接触(免疫),更优选与包括或由具有选自SEQ ID NO:1-27中任一项的序列的(多-)肽构成的JNK抑制剂接触(免疫)。
本发明还涉及生产识别根据本发明的JNK抑制剂的(单克隆)抗体的方法,所述方法包括下列步骤:
从生产所述抗体的细胞的细胞培养上清液分离抗体,所述细胞任选地是永生化的,所述抗体识别本发明的JNK抑制剂,更优选地识别包括或由具有选自SEQ ID NO:1-27中任一项的序列的(多-)肽构成的JNK抑制剂。
本领域技术人员应理解,本文公开的使非人类动物免疫的方法和生产(多克隆)抗体的方法可以连续地实施。类似地,可以将使非人类动物免疫的方法、分离生产抗体的细胞的方法以及生产(单克隆)抗体的方法结合。
本发明的另一方面涉及用根据本发明的用于生产多克隆或单克隆抗体的方法可以生产(和/或生产)的抗体,其中该抗体识别包括或由选自SEQ ID NO:1-27中任一项的序列构成的至少一个(多-)肽,但的确优选不识别(或至少在较小范围内,例如至少在一个数量级内)在SEQ ID NO:1-27的各个序列伸展中用L-氨基酸代替D-氨基酸的基本相同的(多-)肽。优选地,该抗体确实识别本发明的JNK抑制剂,但确实不识别(或至少在较小范围内,例如至少在一个数量级内)包括序列RPKRPTTLNLF(SEQ ID NO:193)的(多-)肽。尤其优选的抗体(单克隆或多克隆)确实识别包括序列SEQ ID NO:8的JNK抑制剂(例如包括序列SEQ ID NO:172的JNK抑制剂),但不识别(或至少在较小范围内,例如至少在一个数量级内)包括用L-氨基酸代替D-氨基酸的同样序列的(多-)肽。尤其优选的是这样的多克隆或单克隆抗体,其识别包括SEQ ID NO:172的(多-)肽,但不识别(或至少识别较小范围,例如至少在一个数量级内)包括序列RKKRRQRRRRPKRPATLNLF(SEQ ID NO:199)的(多-)肽。
本发明还涉及根据上面分离生产识别根据本发明的JNK抑制剂的抗体的细胞的特定方法分离的细胞,其中所述细胞产生抗体,该抗体优选识别选自SEQ ID NO:1-27中任一项的至少一个(多-)肽,但不识别在对应于SEQ ID NO:1的序列中用L-氨基酸代替D-氨基酸的基本相同的(多-)肽,(例如,确实识别包括序列RPkRPaTLNLf(SEQ ID NO:8)的(多-)肽,但不识别(或至少在较小范围内,例如至少在一个数量级内)包括序列RPKRPTTLNLF(SEQ ID NO:193)的(多-)肽。
本发明还设计生产抗特定的转运序列的抗体,从而允许识别例如表3中公开的JNK抑制剂。因此,以上讨论的关于识别本发明的JNK抑制剂的抗体(尤其是包括或由选自SEQ ID NO:1-27中任一项的序列构成的至少一个(多-)肽)的所有方面(单克隆或多克隆抗体;生产单克隆或多克隆抗体的方法;产生单克隆或多克隆抗体的的细胞等),在上下文中都可以应用在包括或由选自SEQ ID NO:31-34和46-151中任一项的序列构成的(多-)肽上。当然,必须不被识别的参考序列(或者至少在较小的范围内,例如至少一个数量级内)在这种情况下还是同样的序列,但是在各个转运序列延伸中用L-氨基酸代替D-氨基酸。
测试(单克隆和/或多克隆)抗体的结合亲合力的方法在本领域是众所周知的。其中一种可能性是借助于夹心ELISA表征抗体的结合亲合力,所述夹心ELISA通过使用靶肽以及阴性对照(例如只具有L-氨基酸的相同的肽)。不限于此,可以如下在空白复制品上计算ELISA极限:
ELISA极限=平均(阴性对照)+(3×阴性对照的标准偏差)。
如果样品值小于或等于ELISA极限,则可以认为被测试的抗体对靶肽没有亲合力。如果样品值超过ELISA极限,则可以认为被测试的抗体对靶肽显示出亲合力。而且,样品值越高,被测试的抗体对靶的亲合力越强。
提供单克隆抗体或多克隆抗体的生产的商业服务机构例如是Eurogentec(比利时,瑟兰)。
本文引用的所有参考文献通过引用并入本文。
实施例
下面给出了说明本发明的各种实施方案和方面的具体的实施例。然而,本发明不应限于本文所描述的特定实施方案的范围。事实上,除本文描述的那些之外,本领域的技术人员从前面的描述、附图和下面的实施例中很容易得出本发明的各种修改方案。所有的这些修改方案均落入所附的权利要求的范围内。
实施例1:JNK抑制剂SEQ ID NO:172的合成
下面给出具有SEQ ID NO:172的JNK抑制剂的合成作为说明性实施例。本领域技术人员应了解,所述合成还可以用于并容易地适用于根据本发明的任何其他JNK抑制剂的合成。
具有SEQ ID NO:172的JNK抑制剂是通过使用Fmoc(9-芴基甲氧基羰基)策略的固相肽合成制造的。肽和树脂之间的连接物是Rink酰胺连接物(p-[Fmoc-2,3-二甲氧基苄基]-苯氧基乙酸)。通过连续的Fmoc去保护和Fmoc氨基酸偶合循环合成肽。在合成结束时,直接利用三氟乙酸(TFA)切割完整的肽,获得粗制的C-端酰胺,随后通过制备反相HPLC将其纯化。将经过纯化的级分混合成均匀的批次,通过离子交换色谱处理获得其醋酸盐。然后将肽冻干。
1.1肽的固相合成
除非指出,该制造在室温(22℃±7℃)下在空气过滤的环境中进行。合成的规模为0.7毫摩尔起始氨基酸在树脂上,期望的产量为约1g纯化的肽。合成通常在装备有烧结盘并且以机械搅拌和/或氮气鼓泡的30–50mL反应器中手动进行。
1.2树脂的制备
首先用二氯甲烷/二甲基甲酰胺/二异丙基乙胺在氮气下清洗对甲基二苯甲基酰胺树脂(MBHA-树脂)。随后将经过清洗的树脂在PyBOB(六氟磷酸苯并三唑-1-基-氧-三-吡咯烷)/二异丙基-乙胺/1-羟基苯并三唑中偶联至Rink酰胺连接物(p-[Fmox-2,4-甲氧基苄基]-苯氧基乙酸),以产生Fmoc-Rink酰胺-MBHA树脂。
1.3氨基酸的偶联
使用下列循环将氨基酸偶联至树脂:
通过在35%(v/v)的哌啶/二甲基甲酰胺中清洗,随后用二甲基甲酰胺清洗使Fmoc-Rink酰胺-MBHA树脂去保护。去保护反应进行约16分钟。将二甲基甲酰胺/二氯甲烷(50/50)中的Fmoc-保护的氨基酸(例如2当量的氨基酸和HOBt(1-羟基苯并三唑)加入树脂,随后添加2当量偶联剂二异丙基碳二亚胺(DIC)。根据所添加的各种氨基酸不同,偶联反应进行1个小时至过夜。在0.5mL/100mg肽-树脂的基础上计算体积,并在每个循环后调节。偶联之后,用DMF清洗树脂3次。通过在伯胺上的茚三酮测试(或Kaiser测试1)和在仲胺上的chloranyl测试2来测试偶联的完成。在一些情况下,chloranyl测试可以与茚三酮测试联合,作为安全控制。在偶联测试显示反应不完全的情况下,用在二甲基甲酰胺/二氯甲烷和二异丙基乙胺中稍微过量(0.5-1当量)的氨基酸、PYBOP、HOBT重复偶联。测量树脂的官能性,根据树脂的初始载荷通常为0.6–0.2meq/g。在最后一种氨基酸已经偶联之后,如常见的对肽-树脂去保护,并然后在30℃的真空炉中干燥之前用DCM清洗5次。在肽-树脂干燥之后,以肽树脂的重量增加相对于由树脂的初始载量计算的理论重量增加的比率来计算固相合成的产率。产率可以接近100%。
1.4切割和去保护
在三氟乙酸/1,2-乙二硫醇(ethanedthiol)/茴香硫醚/水/苯酚(88/2.2/4.4/4.4/7v/v)混合物中,也称为TFA/K试剂中,在室温下4小时从树脂切割肽。反应体积为1mL/100mg肽树脂。向试剂中加入树脂期间,将混合物温度调节至保持低于30℃。
1.5从树脂提取肽:
由通过烧结盘的过滤从树脂提取肽。在旋转蒸发器上浓缩至其体积的1/3之后,通过冷的叔丁基甲基醚沉淀肽并且过滤。随后在30℃真空下干燥粗制的肽。
1.6制备HPLC纯化:
然后通过反向HPLC纯化粗制的肽至纯度≥95%。在旋转蒸发器上浓缩经纯化的级分并冻干。
1.7离子交换色谱
将序列SEQ ID NO:172的具有纯化的肽的经浓缩并且冻干的混合物溶解在水中,并在Dowex乙酸盐、50–100目树脂上通过离子交换色谱纯化。
合成所需的起始试剂是:
本发明的其它JNK抑制剂可以以类似方式制备。
实施例2:选择的根据本发明的JNK抑制剂的抑制效率
以下列出如何测量根据本发明的JNK抑制剂的抑制效率的标准操作步骤。该方法允许在非辐射性标准化试验中体外测量候选化合物降低c-Jun特定底物通过JNK磷酸化的能力。此外,应说明如何确定JNK的选定化合物的抑制效率(IC50)和Ki。该方法适用于证实,候选化合物是否抑制或不抑制JNK活性,本领域技术人员当然应理解如何使得下面的方法适应其特殊的目的和需求。
2.1材料
AlphaScreen试剂和板:
-His-JNK1(编号14-327,Upstate,10μg在100μl中:浓度:2.2μM)最终5nM
-His-JNK2(编号14-329,Upstate,10μg在100μl中:浓度:2μM)最终5nM
-His-JNK3(编号14-501,Upstate,10μg在100μl中:浓度:1.88μM)最终5nM
-Anti-Phospho-cJun(编号06-828,Upstate,批号DAM1503356,浓度:44.5μM)最终10nM
-Biotin-cJun(29-67):
序列:Biotin–SNPKILKQSMTLNLADPVGSLKPHLRAKNSDLLTSPDVG(SEQ ID NO:198),批号100509(mw 4382.11,P 99.28%)溶解在H2O中,浓度:10mM)最终30nM
-ATP(编号AS001A,Invitrogen,批号50860B,浓度:100mM))最终5μM
-SAD珠(编号6760617M,PerkinElmer,批号540-460-A,浓度:5mg/ml)最终20μg/ml
-AprotA珠(编号6760617M,PerkinElmer,批号540-460-A,浓度:5mg/ml)最终20μg/ml
-Optiplate 384孔白板(编号6007299,PerkinElmer,批号654280/2008)
-用于肽稀释的96孔板(编号82.1581,Sarstedt)
-TopSeals-A(编号6005185,Perkin Elmer,批号65673)
-生物发光能量转移读取
-在Fusion Alpha读板仪(Perkin Elmer)上读取生物发光能量转移。
移液管:
-使用电子的EDP3移液管20-300(编号17007243;Rainin),用酶-抗体混合物、底物-ATP混合物和珠填充板。
-使用Ultra多通道8X20(编号21040;Gilson)填充具有抑制性化合物的板。
缓冲液和溶液
-激酶缓冲液:20mM Tris-base pH 7.4,10mM MgCl2,1mM DTT,100μM Na3VO4,0.01%Tween,(1%DMSO)
-终止缓冲液:20mM Tris-base pH 7.4,200mM NaCl,80mM EDTA-K(pH 8,用KOH代替NaOH),0.3%BSA
-JNK稀释激酶缓冲液:50mM Tris-base pH 7.4,150mM NaCl,0.1mM EGTA,0.03%Brij-35,270mM蔗糖,0.1%β-巯基乙醇。
2.2方法
为了评价肽的抑制作用,实施标准AlphaScreen试验(例如见Guenat等人,J Biomol Screen,2006;11:第1015-1026页)。按照指示制备不同的组分并随后混合。将板密封并如下面所示的进行孵育:
5μl JNK+抗体
5μl TP激酶+/-抑制剂预孵育30分钟
5μl Biotin-cJun+ATP在24℃孵育60分钟
10μl珠SAD+A protA在黑暗中在24℃孵育60分钟
为了避免污染,用移液管将混合物添加到孔的不同角落。在用每种混合物填充板之后,轻敲板(保持一侧固定,使相反侧轻敲桌子)使得混合物下落到孔的壁。
在Fusion Alpha读板仪(Perkin Elmer)上读取生物发光能量转移。
所有化合物应在3个独立的实验中对JNK的每种异构体至少测试3次。待测试的化合物的可能浓度为0、0.03nM、0.1nM、0.3nM、1nM、3nM、10nM、30nM、100nM、300nM、1μM、3μM、10μM、30μM和100μM。对照是没有JNK的样品或没有底物(c-Jun)的样品。
混合物制备
JNK1、JNK2和JNK35nM
Biotin-cJun 30nM
ATP 5μM;抗phospho-cJun(S63)10nM
Bille SAD/AprotA 20μg/ml
抗体[最终]=10nM(抗Phospho cJun(S63))
检测部分:[混合物]X5(5μl,最终体积为25μl)
[母料]=44.5μM(编号06-828,Upstate,批号DAM1503356)
10nM→50nM在激酶缓冲液中
JNK1、JNK2和JNK3[最终]=5nM
反应部分:[混合物]X3(5μl,最终体积为15μl)
[母料]=2.2μM JNK1(编号14-327,Upstate,批号D7KN022CU)
2.0μM JNK2(编号14-329,Upstate,批号33221CU)
1.88μM JNK3(编号14-501,Upstate,批号D7CN041CU)
5nM→15nM在抗体缓冲液中
抑制剂:
反应部分:[混合物]X3(5μl,最终体积为15μl)
[母料]=10mM
100μM→300μM在激酶缓冲液中
30μM→90μM在激酶缓冲液中
10μM→30μM在激酶缓冲液中
…
0.03nM→0.09nM在激酶缓冲液中
和0nM→激酶缓冲液
在96孔板中实施两个系列的10次系列稀释,一次从300μM开始至0nM,第二次从90μM开始至0.03nM。用8通道的重复吸液管(编号F14401,Gilson,8X20)将肽加入384个板中。
ATP[最终]=5μM
反应部分:[混合物]X3(5μl,最终体积为15μl)
[母料]=100mM(编号AS001A,Invitrogen,批号50860B)
5μM→15μM在激酶缓冲液中
Biotin c-Jun[最终]=30nM
反应部分:[混合物]X3(5μl,最终体积为15μl)
[母料]=10mM
30nM→30nM在ATP缓冲液中
珠SAD/A ProtA[最终]=20μg/ml(光敏的)
检测部分:[混合物]X 2.5(10μl,最终体积为25μl)
[母料]=5mg/ml→20μg/ml 50μg/ml在终止缓冲液中
在暗室(绿色光)或黑暗中混合。
IC50曲线的分析:
通过GraphPad Prism4软件利用下面的方程进行分析:S形剂量-反应(无约束)。
Y=底部+(顶部-底部)/(1+10^((LogEC50-X)))
使用Grugg测试避免离群数据。
IC50的比对:
通过GraphPad Prism4软件利用下面的测试进行分析:单向ANOVA测试,之后是Tukey多重比对测试。P<0.05被认定为是有效的。
在报道AlphaScreen标准化试验中确定用于JNK的ATP的Km和biotin-cJun特定肽的Km。
Ki和IC50之间的数学关系(Ki=IC50/(1+([底物]/底物的Km))可以用来计算Ki值。
实施例3:内化实验和分析
3.1用于摄入实验的材料和方法
a)细胞系:
用于本实验的细胞系是HL-60(编号CCL-240,ATCC,批号116523)
b)培养介质和培养板
RPMI(编号21875-091,Invitrogen,批号8296)或DMEM(编号41965,Invitrogen,批号13481)在05.05.2008补充以下物质:
10%FBS(编号A64906-0098,PAA,批号A15-151):在04.04.2008在56℃下去补体30分钟。
1mM丙酮酸钠(编号S8636,Sigma,批号56K2386)
青霉素(100单位/ml)/链霉素(100 g/ml)(编号P4333,Sigma,批号106K2321)
PBS 10X(编号70011,Invitrogen,批号8277):用无菌H2O稀释至1X
Trypsine-0.05% EDTA(编号L-11660,PAA,批号L66007-1194)
6孔培养板(编号140675,Nunc,批号102613)
24孔培养板(编号142475,Nunc,批号095849)
96孔培养板(编号167008,Nunc,批号083310)
用于蛋白质剂量添加的96孔板(编号82.1581,Sarstedt)
用于荧光度测量的96孔板(编号6005279,Perkin Elmer)
c)溶液
聚-D-赖氨酸涂布溶液(Sigma P9011批号095K5104):25μg/ml最终稀释在PBS中1X
酸性清洗缓冲液:0.2M甘氨酸,0.15M NaCl,pH 3.0
Ripa裂解缓冲液:10mM NaH2PO4pH 7.2,150mM NaCl,1%TritonX-100,1mM EDTA pH 8.0,200μM Na3VO2,0.1%SDS,1X蛋白酶抑制剂混合物(编号11873580001,Roche,批号13732700)
d)显微镜和荧光读板仪
用倒置显微镜观察细胞并计数(Axiovert 40CFL;Zeiss;20X).
利用Fusion Alpha读板仪(Perkin Elmer)读取荧光。
e)方法
在悬浮细胞上研究FITC标记的肽内化。将细胞以1x 106细胞/ml的浓度平板接种在聚-DL-赖氨酸涂布的培养皿内。随后在添加已知浓度的肽之前,在37℃、5%CO2和100%相对湿度下孵育板24小时。在添加肽之后,在37℃、5%CO2和100%相对湿度下孵育细胞30分钟、1小时、6小时或24小时。随后用酸性缓冲液(甘氨酸0.2M,NaCl 0.15M,pH 3.0)清洗细胞两次,以除去吸附于细胞表面的肽(见Kameyama等人,(2007),Biopolymers,88,98-107)。由于富碱性氨基酸的肽强烈地吸附在细胞表面上(这经常导致对内化肽的过高估测),所以使用酸性缓冲液。使用酸性缓冲液清洗的细胞因此被用于除去细胞表面吸附的肽。进行酸清洗用于确定Fab/细胞-渗透肽缀合物的细胞摄入,随后进行两次PBS清洗。通过添加RIPA裂解缓冲液使细胞破裂。然后在背景扣除和蛋白质含量归一化之后通过荧光确定内化肽的相对量。
步骤因此为:1.细胞培养
2.酸性清洗和细胞提取
3.用荧光读板仪分析肽内化
f)细胞培养和肽处理
用3ml聚-D-赖氨酸(Sigma P9011;25μg/ml在PBS中)涂布6孔板,用600μl聚-D-赖氨酸涂布24孔培养板和用125μl聚-D-赖氨酸涂布96孔板,并在37℃、CO2 5%和100%相对湿度下孵育4小时。
4小时之后用3.5ml PBS、700μl PBS或150μl PBS分别清洗6孔板、24孔板或96孔板两次。
将细胞以用于悬浮细胞的1’000’000细胞/ml的平板接种密度平板接种在2.4ml培养基(RPMI)的培养皿中。接种之后,在添加肽之前将板在37℃、CO2 5%和100%相对湿度下孵育24小时。在处理之日贴壁细胞应该为90-95%的密度,并平板接种在DMEM中:
用期望浓度的FITC标记的肽(浓度为在H2O中10mM的母液)处理细胞。
肽添加之后,在37℃、CO2 5%和100%相对湿度下孵育细胞0至24小时(例如,30分钟,1、6或24小时)。
酸性清洗和细胞提取物:
在冰上冷却细胞提取物。
悬浮细胞(或确实很好附着于培养皿的细胞):
将细胞转移至《Falcon 15ml》。为了回收最多的细胞,用1ml PBS清洗培养皿。
以最大2400rpm(转每分钟)下收集细胞2分钟。
将细胞悬浮在1ml冷的PBS中。
将细胞转移至涂布的“Eppendorf管”(用1ml聚D-赖氨酸涂布4小时并用1ml PBS清洗两次)。
用1ml冷的酸性清洗缓冲液清洗3次,并在最大2400rpm下离心2分钟。小心细胞在“eppendorf”中的铺展。
用1ml冷的PBS清洗两次至中性。
添加50μl裂解RIPA缓冲液。
在冰上孵育30分钟至1小时,同时搅拌。
贴壁细胞:
用3ml、1ml或200μl冷的酸性清洗缓冲液分别清洗6孔板、24孔板或96孔板3次。小心与培养皿分离的细胞。
用1ml冷的PBS分别清洗6孔板、24孔板或96孔板两次至中性。
添加50μl裂解RIPA缓冲液。
在冰上孵育30分钟至1小时,同时搅拌。
用冷的刮板刮下细胞。在4℃以4000rpm直接离心24孔板和96孔板15分钟,以除去细胞碎片。随后将上清液(100ml或50ml分别用于24孔板或96孔板)直接转移至深色的96孔板中。通过荧光读板仪(FusionAlpha,Perkin Elmer)读取板。
将裂解物转移至涂布的“eppendorf”(用1ml聚D-赖氨酸涂布4小时并用1ml PBS清洗两次)。
随后在4℃以10000g离心已裂解的细胞30分钟,以除去细胞碎片。
除去上清液并在-80℃储存在“Eppendorf管”中(用1ml聚D-赖氨酸涂布4小时并用1ml PBS清洗两次)。
用荧光读板仪分析肽内化:
按照制造商的用法说明,通过标准BCA试验(试剂盒N°23225,Pierce)确定每种蛋白质提取物的含量。
在荧光读板仪(Fusion Alpha,Perkin Elmer)中读取10μl的每种样品,背景扣除和通过蛋白质浓度进行归一化之后,确定每种样品的相对荧光。
3.2摄入实验
如上所述利用SEQ ID NO:52-96、43和45-47的序列转运肽进行将FITC-标记的TAT衍生的转运体构建在HL-60细胞系的细胞内的时间依赖型内化(摄入)。下面的表4中列出这些序列。
在上表中,D-氨基酸由各个氨基酸残基之前的小写的“d”表示(例如dR=D-Arg)。
由于技术原因,一些序列合成在第一步不幸失败。这些序列在图6中的缩写为1、2、3、4、5、6、7、8、43、52、53、54、55、56、57、85、86、87、88、89和90。但在内化实验中使用其余的序列。
图6示出了结果。
如图6中可见的,孵育24小时之后,具有共有序列rXXXrXXXr(SEQ ID NO:31)的所有转运体示出了比L-TAT转运体(SEQ ID NO:43)更高的内化能力。在96孔板中用10mM r3-L-TAT-衍生的转运体孵育Hela细胞24小时。细胞随后用酸性缓冲液(0.2M甘氨酸,0.15M NaCl,pH 3.0)清洗两次和用PBS清洗两次。通过添加RIPA裂解缓冲液使细胞破裂。通过读取每种提取物的荧光强度(Fusion Alpha读板仪;PerkinElmer),随后进行背景扣除来确定内化的肽的相对量。
如图6中可见的,一个位置看上去对于最高的转运活性和改善的转运活性动力学是关键的:位置2中的Y(对应SEQ ID NO:142的肽N°91)。
下面是这次实验的总结:
·在孵育24小时之后,具有共有序列rXXXrXXXr(SEQ ID NO:31)的所有转运体(见表2中可能序列的选择)示出了比L-TAT转运体(SEQ ID NO:43)更高的内化能力(图6)。那些结果充分验证了共有序列rXXXrXXXr(SEQ ID NO:31)。
·一个位置对于最高的转运活性是关键的(图6):位置2中的Y(对应SEQ ID NO:142的肽N°91)。
因此,优选如表4中所示的TAT衍生的序列,其表现出位置2中的Y,尤其是在序列显示出9aa和具有共有序列rXXXrXXXr(SEQ ID NO:31)时。
实施例4:细胞因子和趋化因子释放的测量
下面列出的步骤描述了,如何在配体诱导的人类细胞(血、WBC、PBMC、纯化的原代淋巴细胞、细胞系等)分泌之后测量几种人类细胞因子的释放量。
使用的技术是夹心ELISA,它允许测量抗体的两个层(即捕捉抗体和检测抗体)之间的抗原量。待测量的抗原必须包含至少两个能够结合至抗体的抗原位点,这是因为至少两个抗体作用在夹心体中。单克隆抗体或多克隆抗体都可以用作夹心ELISA体系中的捕捉抗体和检测抗体。单克隆抗体识别单个表位,允许精细检测和抗原中小的差异的量化。多克隆抗体通常用作捕捉抗体,用于拉下尽可能多的抗原。夹心ELISA的优势在于,分析之前不需要纯化样品,且试验可是非常灵敏(比直接或间接灵敏达到2至5倍)。
该方法可以用于确定本发明的JNK抑制剂在体外/在细胞培养物中的作用。在非毒性剂量下,通过与未处理的样品相比较的细胞因子水平的降低(光密度的改变(在450nm的吸光度))来表示化合物的效能,并通过ELISA监测。结果以ng/ml表示。
4.1材料
●96孔板:
用于收集上清液(编号82.1581,Sarstedt)
用于ELISA(F96maxisorp,编号442404,Nunc)
●顶部密封-A:96孔微板密封(编号600585,PerkinElmer).
●ELISA试剂
涂布缓冲液ELISA:0.1M碳酸钠pH 9.5(=7.13g NaHCO3(编号71627,Fluka)+1.59g Na2CO3(编号71345,Fluka)在1升H2O中,用NaOH浓缩pH至9.5)
清洗缓冲液ELISA:PBS 1X+0.01%Tween20。制备1升PBS 1X(PBS10X:编号70011,GIBCO)并缓慢添加100ul Tween20(编号P1379,Sigma),同时用磁性搅拌器混合)
试剂稀释液:PBS 1X+10%FBS(编号A15-151,PAA,在56℃去补体,30分钟).
DAKO TMB(编号S1599,DAKO):商用底物溶液
终止溶液:1M H3PO4(→200ml=177ml H2O+23ml H3PO485%(编号345245,Aldrich).
●ELISA试剂盒(用于20个板的试剂)
IFN-γ:人IFN-γELISA组,BD OptEIATM(编号555142,DB).
IL-1β:人IL-1βELISA组II,BD OptEIATM(编号557953,BD)
IL-10:人IL-10ELISA组II,BD OptEIATM(编号555157,DB).
IL-12:人IL-12(p70)ELISA组,BD OptEIATM(编号555183,DB).
IL-15:人IL-15ELISA组,BD OptEIATM(编号559268,DB).
IL-2:人IL-2ELISA组,BD OptEIATM(编号555190,DB).
IL-4:人IL-4ELISA组,BD OptEIATM(编号555194,DB).
IL-5:人IL-5ELISA组,BD OptEIATM(编号555202,DB).
IL-6:人IL-6ELISA组I,BD OptEIATM(编号555220,DB).
IL-8:人IL-8ELISA组,BD OptEIATM(编号555244,DB).
MCP-1:人MCP-1ELISA组,BD OptEIATM(编号555179,BD)
TNF-α:试剂盒人TNF ELISA组,BD OptEIATM(编号555212,DB).
●读取吸光度:在Fusion Alpha读板仪(Perkin Elmer)上读取吸光度。
●重复移液管、数字移液管或多通道移液管。
4.2方法
样品的制备
样品是来自经培养的人类细胞(典型地是全血、WBC、PBMC、WBC的经纯化的亚型、癌细胞系)的培养基上清。通过离心(4℃,400g下5分钟)除去任何颗粒状材料,立即分析或在≤-20℃储存样品。避免重复冻融循环。
使用之前一小时,将样品在冰上解冻并离心。在步骤11,在试验稀释液中将样品直接稀释至板中(首先添加试验稀释液,之后是样品并用移液管上下抽吸):
标准物的制备
在将冻干的标准物加温至室温之后,小心地打开小瓶以避免材料损失。重构的冻干标准物具有预计体积的去离子水,由此得到母料标准物。允许在进行稀释之前使标准物平衡至少15分钟。轻轻地进行漩涡振荡来混合。在重构之后,立即将标准母料等分至聚丙烯小瓶中,每瓶50μl并在-20℃冷冻达6个月。如果需要,在等分/冷冻之前在2-8℃下储存最多8小时。不要将重构的标准物保留在室温下。
在马上要使用之前,用在试剂稀释液中2倍的系列稀释得到10个点标准曲线。推荐4000pg/ml的高标准。
检测器混合物的制备
Biotin/SAv试剂的一步孵育。向试剂稀释液中添加所需体积的检测抗体。在使用前15分钟内,添加所需量的酶制剂,漩涡振荡或很好地混合。对于推荐的稀释,见批号指定的(lot-specific)用法说明/分析证书。使用之后丢弃所有剩余的工作检测器。
用捕捉抗体涂布
1.用每孔100μl的在涂布缓冲液中稀释的捕捉抗体涂布PVC微量滴定板的孔。对于推荐的抗体涂布稀释,见批号指定(lot-specific)的用法说明/分析证书。
2.用粘合塑料覆盖所述板并在4℃孵育过夜。
3.除去涂布溶液,通过用150μl的清洗缓冲液填充孔来清洗所述板。
4.在水槽上轻敲所述板来除去溶液或清洗物。
5.重复所述过程两次,共计清洗三次。
6.最后一次清洗之后,在纸巾上轻拍所述板来除去任何剩余的清洗缓冲液。
阻断
7.通过每孔添加100μl试剂稀释液来阻断经涂布的孔中剩余的蛋白质结合位点。
8.用粘合塑料覆盖所述板并在室温下孵育1小时。
9.在孵育期间,开始制备标准物。
添加样品
10.如步骤3用150μl清洗缓冲液清洗一次。所述板现在已经能够用于样品添加。
11.向每个孔添加50μl在试验稀释液中适当稀释的样品。为了得到准确的量化结果,总是将未知样品的信号与标准曲线的信号比较。标准(三次)和空白必须每种细胞因子都运行,以确保准确。
12.用粘合塑料覆盖板并在室温下孵育2小时。
用检测抗体和第二抗体孵育
13.如步骤3用150μl清洗缓冲液清洗板4次.
14.以推荐的稀释(见批号指定的用法说明/分析证书)向每个孔中添加50μl的检测器混合物(在试验稀释液中的检测抗体+第二链霉亲和素-HRP抗体)。
15.用粘合塑料覆盖板并在室温下光保护地孵育1小时。
16.如步骤3用150μl清洗缓冲液清洗板6次。
17.向每个孔中添加50μl DAKO TMB溶液,在室温黑暗中不密封地孵育15至20分钟。
18.向每个孔中添加50μl终止溶液。轻敲板以确保彻底混合。
19.在板混合器上以500rpm将板混合5分钟。
20.在450nm读取光密度。(程序:Fusion Alpha读板仪上的Cytokine-ELISA)。
数据分析
对每个标准对照和每个样品的三次读数取平均值。减去平均空白标准光密度(O.D)。制成绘制细胞因子浓度的log对O.D的log的标准曲线,最佳拟合线可以通过回归分析来确定。如果样品已经稀释,则来自标准曲线的浓度读数必须乘以稀释系数。标准曲线应对于每组所分析的样品来产生。使用Grugg测试避免离群数据。放弃不在SD两倍间隔中的数据。如果阳性对照显示之前观察到的数据,则考虑独立实验。将独立实验进行合并(N>3)。
与没有抑制剂处理的诱导条件相比,数据以pg/ml细胞因子释放或以%来给出。
实施例5:THP1分化–刺激细胞因子释放
下面列出的步骤将描述如何诱导经过LPS挑战6小时的人PMA分化的THP1细胞的细胞因子产生,以测试本发明的JNK抑制剂、尤其是具有SEQ ID NO:172的JNK抑制剂减少刺激诱导的细胞因子释放的能力。通过不同的配体对THP1细胞进行离体刺激来读出细胞释放。在非毒性剂量下,通过与未经处理的样品比较的细胞因子水平的降低来指示JNK抑制剂效率,并通过ELISA监测。通过产生紫色的tretazolium盐(MTS)到甲臜的还原来评估化合物的毒性。
步骤:
a.材料
●细胞系:THP-1(编号TIB-202,ATCC,批号57731475)
●培养基、试剂和板
RPMI(编号21875-091,Invitrogen)补充有:
10%FBS(编号A15-151,PAA):在56℃下去补体30分钟。
10mM Hepes(编号H0887,Sigma)
50 M-巯基乙醇(编号63690,Fluka:母料为14.3M):添加560l储备在-20℃的在PBS中的50mM的等份试样)
1mM丙酮酸钠(编号S8636,Sigma)
青霉素(100单位/ml)/链霉素(100g/ml)(编号P4333,Sigma)
之后用0.22M过滤器(编号SCGPU05RE,Millipore)过滤RPMI培养基。
PBS 10X(编号70011,Invitrogen):用无菌H2O稀释至1X
DMSO:编号41444,Fluka
PMA(佛波醇12-豆蔻酸酯13-乙酸酯,编号P1585,Sigma,浓度1mM=616.8ug/ml在DMSO中,在-20℃)。以RPMI中100nM的最终浓度(1μl在10ml介质中)直接使用。
LPS超纯(脂多糖,编号tlrl-eklps,Invivogen,浓度为5mg/ml):LPS的母液:3g/ml在PBS中,4℃。直接使用来制备40ng/ml在RPMI介质中的4X浓缩溶液(最少1800l/板:对5个板:125l的LPS 3g/ml+9250l RPMI)。
96孔板:
用于贴壁细胞培养(编号167008,Nunc)
用于收集上清液(编号82.1581,Sarstedt)
用于ELISA(F96maxisorp,编号442404,Nunc)
涂布溶液:聚-D-赖氨酸(编号P9011,Sigma):25g/ml最终在PBS 1x中稀释
●ELISA试剂和试剂盒
涂布缓冲ELISA:0.1M碳酸钠pH 9.5(=7.13g NaHCO3(编号71627,Fluka)+1.59g Na2CO3(编号71345,Fluka)在1升H2O中,用NaOH浓缩pH至9.5)
清洗缓冲ELISA:PBS 1X+0.01%Tween20(编号P1379,Sigma,批号094K0052)(=制备1升PBS 1X并缓慢添加100μl Tween20,同时用磁性搅拌器混合)
试剂稀释液:PBS 1X+10%FBS(编号A15-151,PAA,在56℃去补体,30分钟)
DAKO TMB(编号S1599,DAKO):商用底物溶液
终止溶液:1M H3PO4(→200ml=177ml H2O+23ml H3PO485%(编号345245,Aldrich)
TNF-:试剂盒。人TNF ELISA组,BD OptEIA(编号555212,DB)
●细胞毒性测量:CellTiter 96试剂(编号G3581,Promega)
●对照化合物:SP600125(编号ALX-270-339-M025,Alexis,浓度:20mM DMSO)
●读取吸光度:在Fusion Alpha读板仪(Perkin Elmer)上读取吸光度。
●重复移液管、数字移液管或多通道移液管。
●顶部密封-A:96孔微板密封(编号600585,PerkinElmer)。
b.方法
孔涂布
板已经用200l聚-D-赖氨酸(1x)涂布,并在37℃,CO2 5%和100%相对湿度下孵育两小时。
细胞平板接种
2小时之后用200l PBS 1X清洗孔两次(立即使用或留在37℃的200l的PBS 1X中直至使用,但是不要超过3天)。
对细胞计数。取期望数目的细胞并重悬在获得1’000’000细胞/ml稀释度所需量的培养基中。添加100nM的PMA以诱导THP1从悬浮的单细胞向贴壁巨噬细胞的分化。将细胞以100’000细胞/孔的平板接种密度平板接种在100l培养基的孔中。接种之后,在添加实验药物之前将板在37℃,CO2 5%和100%相对湿度下孵育3天,使其分化。
细胞处理
3天后,用显微镜观察到贴壁细胞。抽出包含PMA的培养基并用100l没有PMA的新鲜RPMI培养基(没有使用PBS 1X的清洗步骤)置换。
实验药物以在H2O或DMSO中10mM的浓度来制备并在-80℃储存。每次日常使用之前,将一等份的JNK抑制剂解冻,并稀释至达到在RPMI培养基中4X的浓缩溶液(120M),随后稀释至RPMI中期望的浓度。稀释SP600125达到在RPMI培养基中4X的浓缩溶液(40M),随后稀释至包含0.8%DMSO的RPMI中期望的浓度。
用50l培养基或4X的最终期望的药物浓度(对于JNK化合物最终为0、100nM、1、3、10或30M,或者对于SP600125阳性对照最终为0、10、100nM、1、3或10M)来处理所述板。随后添加药物,在37℃、5%CO2和100%相对湿度下孵育所述板。
1小时之后,通过添加50l 4X倍浓缩的LPS超纯稀释液(3ng/ml最终)来诱导TNF的分泌。
试验
6小时后,将100l上清液转移至新的96孔板。将那些板密封并储存在-20°直至通过细胞因子分泌的ELISA(例如见实施例4)进行分析。
通过MTS吸光度估测化合物的细胞毒性作用(例如见实施例4),并使用倒置显微镜(Axiovert 40CFL;Zeiss;10X)观察细胞。
数据分析
如ELISA中指示的(见实施例4)进行数据的分析。简要地,对于ELISA:对每个标准对照和每个样品的三次读数取平均值。减去平均空白标准光密度(O.D)。制成绘制细胞因子浓度的log对O.D的log的标准曲线,最佳拟合线可以通过回归分析来确定。如果样品已经经过稀释,则来自标准曲线的浓度读数必须乘以稀释系数。标准曲线应由每组所分析的样品来产生。使用Grugg测试避免离群数据。放弃不在SD的两倍的间隔中的数据。如果阳性对照显示之前观察到的数据,则考虑独立实验。将独立实验进行合并(N>3)。
对于细胞毒作用评估:在考虑到细胞因子释放实验分析的每个独立实验的每个板上,只是将介质的吸光度平均值看作背景,并减去得到每个吸光度值。将每种化合物的未经处理细胞的三次平均值作为100%生存力。每种化合物的三次平均值通过其100%来归一化。使用Grugg测试避免离群数据。放弃不在SD两倍间隔中的数据。将独立实验进行合并(N>3)。
对条件的所有统计学比对都通过GraphPad Prism4软件利用下面的测试进行:单向ANOVA测试,之后是Tukey多重比对测试。P<0.05被认定为是有效的。
实施例6:SEQ ID NO:172的JNK抑制剂和TNFα在原代大鼠或人全血细胞中的释放
从麻醉大鼠或人类健康志愿者收集全血,使用连接了包含柠檬酸钠的预先标记的真空管的静脉穿刺。通过倒转7-8次轻柔地使管混合;随后保持在室温下直至进行刺激。制备在PBS中6倍浓缩的SEQ ID NO:172的JNK抑制剂,将混合物以30μl/孔加入96孔板中。通过在PBS中以1:2稀释全血,并将120μl经过稀释的血液加入到每个孔中,包括只有PBS的或已经预先加入SEQ ID NO:172的JNK抑制剂的孔。在37℃、85rpm(Stuart Orbital恒温箱SI500)下孵育全血60分钟。制备活化剂(LPS),30μl/孔的LPS,6倍浓缩。孵育60分钟之后,将LPS加入到血液中,通过上下抽取混合血液,随后在37℃搅拌(85rpm)下保持4小时。孵育4小时之后,在约770g、4℃下在预先冷却的离心机中将板离心15分钟。最后收集上清液并保持在-20℃直至细胞因子测量。之后使用标准Elisa试剂盒(例如来自R&D Systems的DuoSet Elisas;或来自BD Biosciences的BD Opteia Set Elisa)测量细胞因子(IL-6、IL-2、IFNγ和TNFα)。结果以所测量的细胞因子的上清液的pg/ml来表示。
用PMA+离子霉素代替LPS作为活化剂/刺激物实施类似的实验。
实施例7:本文公开的具体JNK抑制剂的半衰期
具有序列SEQ ID NO:196、197和172的JNK抑制剂(最终浓度0.1mM)在人血清(10和50%在PBS 1x中)中被消化。如Tugyi等人(Proc Natl Acad Sci U S A,2005,413-418)描述的进行实验。通过UPLC-MS量化剩余的完整的肽。同样地但在两个分开的试验中评估SEQ ID NO:196、197和172的稳定性。具有SEQ ID NO:196的JNK抑制剂在6小时内完全降解为氨基酸残基,具有SEQ ID NO:172的JNK抑制剂在14天之后才彻底降解。具有SEQ ID NO:197的JNK抑制剂在30天之后仍然是稳定的。
实施例8:具有序列SEQ ID NO:172的JNK抑制剂对CD3/CD28-诱导的在原代大鼠T-细胞中IL-2释放的剂量依赖性抑制
将对照动物牺牲,收集淋巴结(LN)并保持在完全RPMI培养基中。在使用5ml活塞的70μm过滤器上用完全RPMI捣碎LN。加入几滴培养基保持过滤网(strainer)湿润。在450g和4℃下离心细胞7分钟。将丸重悬在5ml新鲜培养基中。使细胞再次通过细胞过滤网。对细胞的等份试样进行计数,同时在1400rpm和4℃下再次离心细胞10分钟。将细胞重悬在MACS缓冲液(每107个细胞80μl MACS缓冲液)中。每1千万个细胞加入10μl抗-大鼠MHC微珠,在4℃-8℃孵育细胞15分钟。用15ml MACS缓冲液清洗细胞并在700g和4℃下离心7分钟。将丸重悬在每108个细胞500μl MACS缓冲液中。在每个动物的MACS分离器的磁场中放置一个LS柱。首先用3ml MACS缓冲液冲洗柱。将一个管放置在柱下方的冰中来收集细胞(T细胞)(阴性选择,因此收集洗出物)。加入细胞悬浮液并在冰上收集洗出物。用3mL MACS缓冲液清洗柱3次。在700g和4℃下离心洗出的T细胞7分钟。对重悬的细胞计数,并以200000细胞/孔的密度在100μl完全培养基中平板接种。在实验前一天用2μg/mL的CD3抗体预先涂布板,在实验当天用PBS清洗板3次。用100μl的(多-)肽JNK抑制剂(SEQ ID NO:172)处理细胞,在配体活化之前两次浓缩1小时。用(多-)肽JNK抑制剂(SEQ ID NO:172)预处理1小时之后,用2μg/mL的抗CD28抗体刺激细胞24小时。刺激24小时之后,收集上清液并在-20℃下储存直至分析。随后使用标准Elisa试剂盒测量细胞因子。结果以所测量的细胞因子的上清液的pg/ml来表示。
在其他实验中,主要使用如上面列出的相同的方案,除了具有SEQ ID NO:172的(多-)肽JNK抑制剂之外,还测试了具有序列SEQ ID NO:197的JNK抑制剂和药物分子SP600125,因此允许比较这些抑制剂对CD3/CD28-诱导的IL-2释放的抑制。
实施例9:JNK抑制剂和人类全血中TNFα/IL-2的释放:
从人类健康志愿者收集全血,使用连接了包含柠檬酸钠的预先标记的真空管的静脉穿刺。通过倒转7-8次轻轻地使管混合;随后保持在室温下直至进行刺激。将350μl的RPMI+P/S加入1.2ml-96孔板中。在RPMI+P/S(每孔50μl)中制备10倍浓缩的SEQ ID NO:172。将50μl加入1.2ml-96孔板中。随后在每个孔中加入50μl全血,包括只有培养基的或已经预先加入JNK抑制剂的孔。在37℃、5%CO2下孵育全血60分钟。制备在RPMI+P/S中稀释的50μl/孔的配体,对应10倍浓缩的最终稀释。孵育60分钟之后,加入配体;随后通过上下抽取血液使孔混合。在37℃下孵育全血3天(每天一次通过对每个孔上下抽取使孔混合)。在孵育结束时,混合板并在预先冷却的离心机中在2500rpm、4℃下离心15分钟。之后使用标准Elisa试剂盒测量细胞因子。结果以所测量的细胞因子的上清液的pg/ml来表示。
稍微改动来实施类似的实验。在CD3/CD8刺激的情况下,以在PBS中2μg/mL在4℃涂布CD3抗体过夜。实验当天,用PBS清洗孔3次,并在37℃下保留在PBS中直至使用。在SEQ ID NO:172之后1小时以2μg/mL的最终浓度添加CD28抗体;刺激3天后收集上清液。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种靶向锚定剂及其制备方法和应用与流程
- 一种超低分子量插层抑制剂及其制备方法与流程
- 具有胶原特异结合能力的神经再生抑制分子的拮抗蛋白CBD-PlexinB1LBD及应用的制作方法与工艺
- 具有胶原特异结合能力的神经再生抑制分子的拮抗蛋白CBD-EphA4LBD及应用的制作方法与工艺
- 一种KDM1A小分子抑制剂在抑制肿瘤细胞生长和转移中的应用的制作方法与工艺
- 一种抑制多药耐受基因表达的靶向抗肿瘤纳米药物的制作方法与工艺
- MALT1靶向抑制物在制备MALT1依赖性肿瘤治疗药物中的应用的制作方法与工艺
- 作为ASGPR靶向剂的被取代的‑6,8‑二氧杂双环[3.2.1]辛烷‑2,3‑二醇化合物的制作方法与工艺
- 小分子C‑Myc抑制剂的制作方法与工艺
- 一种RNA抑制剂及其在肿瘤中的应用的制作方法与工艺