一种生物制动器的制作方法
技术领域
本发明涉及一种远程生物制动器。
背景技术:
水凝胶在组织工程学和生物电子学中具有广泛应用[1]。目前,对它们的广泛关注集中于高性能和新功能,诸如用于控制药物释放的刺激-响应性能[2],用于生物电子学或生物传感器的传导性[3],用于心脏组织工程学的传导性和弹性[4,5],压力敏感传导性和用于电子皮肤的自愈[6],用于油/水分离的基质[7],用于生物制动器的磁响应[8]。尽管具有优异传导性或磁响应性能的功能水凝胶被广泛应用,但在一些领域仍有一些由柔性、弹性和响应敏感性不足导致的限制,例如高柔性电子设备、有机压力敏感传感器和响应执行器。它们面临将高度传导性或磁响应性质与高柔性和弹性结合在一起的类似挑战。
最近,报道了一些注射器-可注射大孔柔性网状电子设备,反映了制造的生物电子设备的无手术移植前景[9],但迄今并未报道注射器-可注射预制3D庞大电子设备。尽管报道了诸如海藻酸盐、明胶、壳聚糖和透明质酸晶胶等一些大孔晶胶具有高柔性和注射器-可注射性,它们都不具有传导性[10,11],因为引入诸如碳纳米管(CNT)和聚吡咯(PPY)等具有传导性的刚性成分将增强水凝胶的硬度但同时降低它们的柔性[12]。
在压力敏感传感器领域,已经开发几种方法,诸如中空-球体压缩电阻膜传感器[13]、金纳米线压缩电阻膜传感器[14]、聚二甲基硅氧烷(PDMS)/碳纳米管(CNT)纳米片传感器[15]和纳米线基质膜传感器[16],但这些技术遇到相似问题:复杂的制造工艺、高成本和有限的传导性。此外,它们是用于电皮肤应用的2D膜而不是用于一些环境的3D庞大压力传感器。报道了一些传导性水凝胶,但这些水凝胶并未将良好的压缩弹性/抗性和压力响应传导性结合在一起[17,18]。
此外,报道了可快速响应于外磁场的磁性水凝胶,其通过远程驱动递送药物/细胞,其在约38A/m2的磁场梯度下可达到约70%的变形[19]。还制造了将良好的传导性和磁响应结合在一起的3D石墨烯/氧化铁气凝胶用于软执行器或微开关的潜在应用,其在1.2T磁场强度下具有52.3%径向应变[20]。然而,低至0.2T至3T的中等磁场强度更多可用于常见临床环境,并且的低场MR常应用于MRI[21],因此在实际应用中远程驱动需要更柔性和敏感的磁响应水凝胶。
因此,仍然需要一种能够将传导性或磁响应性与高柔性和弹性良好的结合在一起的新的复合材料。
技术实现要素:
本发明的第一方面提供了一种弹性蛋白复合晶胶(EGC),其包括弹性蛋白、明胶和碳纳米管。其中,“晶胶”是指具有相互连通的大孔的连续凝胶基质。
在本发明的一个实施方案中,弹性蛋白复合晶胶包括30-60重量份的弹性蛋白、30-80重量份的明胶和10-80重量份的碳纳米管。在一个实施方案中,弹性蛋白与明胶的比为30/70至70/30,例如30/70至50/50。该弹性蛋白可选自弹性蛋白肽、原弹性蛋白或消化弹性蛋白。在一个实施方案中,弹性蛋白和明胶通过甲基丙烯酸化改性。在一个实施方案中,甲基丙烯酸化试剂为甲基丙烯酸酐。在一个实施方案中,该弹性蛋白为甲基丙烯酸酯接枝弹性蛋白。在一个实施方案中,该明胶为甲基丙烯酸酯接枝明胶。在一个实施方案中,所述EGC还包括稳定剂,例如30-80重量份的稳定剂。该稳定剂可以是两亲性共聚物,例如聚氧乙烯-聚氧丙烯-聚氧乙烯(PEO-PPO-PEO)三嵌段两亲性共聚物,如F-127-DA。在一个实施方案中,该稳定剂通过丙烯酸化改性。在一个实施方案中,丙烯酸化试剂为丙烯酰氯。在本发明的一个实施方案中,弹性蛋白复合晶胶包括15重量份的弹性蛋白、35重量份的明胶、17.5重量份的CNT和17.5重量份的稳定剂。
EGC水凝胶的SEM形态学显示出与蜂窝状大孔结构相似(图1(e1))并且具有60~130μm的孔径分布以及高于99%的互联性,其可允许变形以及快速排水,并且将CNT(5mg/mL,最终浓度)嵌入在衬底层(如来自图1(e2))的网状网格中。
本发明还提供了一种制备EGC的方法,所述方法包括:
(a)将弹性蛋白、明胶、碳纳米管和稳定剂在水溶液中混合;
(b)在低温下向上述混合物中添加聚合引发剂,使混合物交联;
(c)在低温下使步骤(b)中所得产物固化,以形成EGC。
在本发明的一个实施方案中,在本发明方法的步骤(a)中,将30-60重量份的弹性蛋白、30-80重量份的明胶、10-80重量份的碳纳米管和30-80重量份的稳定剂在水溶液中混合。在一个实施方案中,弹性蛋白和明胶通过甲基丙烯酸化改性。在一个实施方案中,甲基丙烯酸化试剂为甲基丙烯酸酐。在一个实施方案中,该弹性蛋白为甲基丙烯酸酯接枝弹性蛋白。在一个实施方案中,该明胶为甲基丙烯酸酯接枝明胶。在一个实施方案中,该稳定剂是两亲性共聚物,例如PEO-PPO-PEO三嵌段两亲性共聚物,如F-127-DA。在一个实施方案中,所述稳定剂通过丙烯酸化改性。在一个实施方案中,所述聚合引发剂包括过硫酸铵(APS),例如为APS和四甲基乙二胺(TEMED)。在一个实施方案中,步骤(c)的固化持续1-24h,例如16h。
在一个实施方案中,步骤(a)是在低温下进行,例如在0-15℃的温度下进行。在一个实施方案中,步骤(b)在0-15℃下进行。在一个实施方案中,步骤(c)在-30-0℃下进行,优选-20℃。
本发明的第二方面提供了一种晶胶复合材料,其包括EGC和负载于其上的聚吡咯(PPY)。在一个实施方案中,EGC与PPY的重量比约为1:0.15-0.7。
在一个实施方案中,在EGC负载有分散的聚吡咯聚集体(EGC-PPY-FD)。该分散的聚吡咯聚集体可通过快速交联沉积而将聚吡咯聚集体沉积在EGC上。在一个实施方案中,快速交联沉积是通过将溶液中的吡咯单体吸收到EGC上,并通过过硫酸铵(APS)引发吡咯单体聚合并快速原位形成在EGC上,以形成负载在EGC上的聚吡咯聚集体(EGC-PPY-FD)。在一个实施方案中,溶液中的吡咯单体的浓度为3mg/mL-18mg/mL,例如6.7mg/mL-13.4mg/mL,如6.7mg/mL。在一个实施方案中,在EGC-PPY-FD中,EGC与PPY的重量比约为1:0.15-0.30,例如1:0.23-0.29,例如1:0.26。
在另一个实施方案中,在所述EGC的网络结构上形成连续的聚吡咯网络结构。具体而言,刚性PPY成分在软支架上形成第二网络(EGC-PPY-SA)。此时,形成了具有软-硬双连续网络结构的晶胶复合材料。在一个实施方案中,该复合材料是通过快速交联,随后缓慢老化在所述晶胶的网络结构上形成连续的聚吡咯网络结构而形成的。在一个实施方案中,所述复合材料是通过将吡咯单体吸收到EGC上,并加入Fe(NO3)3来引发吡咯交联,随后使剩余未聚合的吡咯单体被Fe(NO3)3缓慢氧化聚合以在所述晶胶的网络结构上形成连续网络结构。在一个实施方案中,溶液中的吡咯单体的浓度为3mg/mL-18mg/mL,例如6.7mg/mL-13.4mg/mL,如13.4mg/mL。在一个实施方案中,在EGC-PPY-SA中,EGC与PPY的重量比约为1:0.32-0.70,例如1:0.32-0.50,例如1:0.38。
在一个实施方案中,所形成的EGC-PPY-FD的孔径分布为50~140μm,其类似于EGC的孔径分布,并且EGC-PPY-SA的孔径分布为20~60μm。
EGC负载的分散的PPY聚集体显示出了高度柔性和注射器-可注射性质以及中等传导性。另一方面,当刚性PPY成分在软支架上形成第二网络时,具有软-硬双连续网络结构的晶胶同时显示可承受高至97.5%压缩应变的出色高弹性和在90%应变下高至50.1±2.9S/cm的突出传导性以及良好的压力敏感传导性。
本发明还提供了制备EGC-PPY-FD的方法,其包括将EGC浸泡于吡咯水溶液中,并加入聚合引发剂(例如APS),以使吡咯单体聚合,并原位沉积于EGC的支架结构上,以形成EGC-PPY-FD。在一个实施方案中,该反应是在低温下进行的,例如在0-15℃下进行的。在一个实施方案中,溶液中的吡咯单体的浓度为3mg/mL-18mg/mL,例如6.7mg/mL-13.4mg/mL,如6.7mg/mL。
本发明还提供了制备EGC-PPY-SA的方法,其包括将EGC浸泡于吡咯水溶液中,并加入Fe(NO3)3来引发吡咯交联,随后使剩余未聚合的吡咯单体被Fe(NO3)3缓慢氧化聚合以在EGC的网络结构上形成连续网络结构。通常,加入Fe(NO3)3来引发吡咯交联是在低温下,例如0-15℃下进行的。老化(如Fe(NO3)3缓慢氧化)可在室温下持续4小时至3天,优选8小时至3天,并且优选8小时、16小时、1天、2天和3天。
本发明的第三方面提供了一种晶胶复合材料(EGC-IONP),其包括EGC和负载于EGC上的氧化铁磁性纳米颗粒(IONP)。在一个实施方案中,EGC-IONP包含30-60重量份的弹性蛋白、30-80重量份的明胶、10-80重量份的碳纳米管、30-120重量份的氧化铁磁性纳米颗粒和30-80重量份的稳定剂。在一个实施方案中,EGC-IONP包含50重量份的弹性蛋白、50重量份的明胶、20重量份的CNT和120重量份的氧化铁磁性纳米颗粒。在一个实施方案中,其还包括聚吡咯,例如约30-80重量份的聚吡咯。在一个实施方案中,该PPY采用与以上类似的快速交联沉积而沉积于EGC-IONP中(EGC-IONP-PPY-FD)。
本发明还提供了一种制备EGC-IONP的方法,所述方法包括:
(a)将弹性蛋白、明胶、碳纳米管、氧化铁磁性纳米颗粒和稳定剂在水溶液中混合;
(b)在低温下向上述混合物中添加聚合引发剂,使混合物交联;
(c)在低温下使步骤(b)中所得产物固化,以形成所述复合材料。
在该方法的步骤(a)中,可将30-60重量份的弹性蛋白、30-80重量份的明胶、10-80重量份的碳纳米管、30-120重量份的氧化铁磁性纳米颗粒和30-80重量份的稳定剂在水溶液中混合。在一个实施方案中,弹性蛋白和明胶通过甲基丙烯酸化改性。在一个实施方案中,甲基丙烯酸化试剂为甲基丙烯酸酐。在一个实施方案中,该弹性蛋白为甲基丙烯酸酯接枝弹性蛋白。在一个实施方案中,该明胶为甲基丙烯酸酯接枝明胶。在一个实施方案中,该稳定剂是两亲性共聚物,例如PEO-PPO-PEO三嵌段两亲性共聚物,如F-127-DA。在一个实施方案中,所述稳定剂通过丙烯酸化改性。在一个实施方案中,所述聚合引发剂包括过硫酸铵(APS),例如APS和TEMED。在一个实施方案中,步骤(c)的固化持续1-24h,例如16h。
在一个实施方案中,步骤(a)和步骤(b)分别在0-15℃下进行。在一个实施方案中,步骤(c)在-30-0℃下进行,优选-20℃。
本发明还提供了一种制备EGC-IONP-PPY-FD的方法,其将PPY通过以上描述的快速交联沉积而沉积于EGC-IONP中,以形成EGC-IONP-PPY-FD。
本发明还涉及EGC、EGC-PPY-FD、EGC-PPY-SA、EGC-IONP和EGC-IONP-PPY-FD用于油/水分离、人工心脏的制造、生物传感器、遥控机器人或生物制动器等的用途。
本发明还涉及一种生物制动器,该生物制动器包括EGC-IONP复合材料。在一个实施方案中,该生物制动器由EGC-IONP复合材料组成。在一个实施方案中,所述EGC-IONP复合材料例如是EGC-IONP-PPY-FD。在本发明中,该生物制动器可以是控制电流回路打开/闭合的装置。
在一个实施方案中,所用EGC-IONP复合材料是通过以下方法制备的:
(a)将弹性蛋白、明胶、碳纳米管、氧化铁磁性纳米颗粒和稳定剂在水溶液中混合;
(b)在低温下向上述混合物中添加聚合引发剂,使混合物交联;
(c)在低温下使步骤(b)中所得产物固化,以形成所述弹性蛋白复合晶胶-氧化铁磁性纳米颗粒复合材料;以及
任选地,将步骤(c)所得的复合材料浸泡于吡咯水溶液中,并加入第二聚合引发剂,以使吡咯单体聚合,并原位沉积于所述复合材料的支架结构上,以形成负载于所述复合材料上的分散的聚吡咯聚集体。
本申请的EGC-IONP复合材料通过采用具有柔性的EGC,并掺入带有磁性的IONP和具有导电性的PPY,形成了同时具有柔性、导电性和磁性的弹性材料。通过施加磁场,可以改变该弹性材料。例如,在常规的弱电场下,例如在约38A/m2的施加的磁场梯度下,本申请的EGC-IONP复合材料能够实现30-180°的弯曲。通过采用这种方式,可使其用于控制远程电路的开关。这种远程控制电路的方法可广泛应用于操作者难以到达的远程电路控制。或者,这种远程控制电路的方法也可以用于操作者难以手动操作或不适宜手动操作的电路,或微电路。此外,由于采用了生物材料,这种生物制动器也是生物相容的。
在一个实施方案中,该生物制动器可制成适合用于电路控制的条形制动器。该制动器的一端可连接于电路中,并且另一端保持打开。当施加常规的弱磁场时,通过控制磁场的变化,可使该条形生物制动器发生变形,进而使其另一端与电路的另一触点接触,使电路闭合并连通。
在一个实施方案中,该生物制动器可作为电路切换开关。当磁场应用于其上时,该生物制动器从打开状态转换到闭合状态,或者从闭合状态转换到打开状态。
在一个实施方案中,本发明公开了一种电路,该电路包括电源、指示器和作为电路切换开关的生物制动器,该生物制动器包括EGC-IONP复合材料。在一个实施方案中,该电源可以是本领域常规的电源,例如电池、电池组、交流电以及其他常用电源。该指示器可以本领域常规的指示器,例如灯泡、发光二极管等等。
本发明还涉及一种超压缩导电性和磁性响应凝胶机器人,其包括主体和连接在其上的三至八条侧臂组成。在一个实施方案中,该遥控机器人为人造章鱼机器人。在一个实施方案中,所述主体材料为EGC-IONP复合材料,如EGC-IONP-PPY-FD。在一个实施方案中,所述主体可为聚氨酯海绵。或者,所述主体可为磁性水凝胶,如EGC-IONP复合材料。在一个实施方案中,所述人造章鱼机器人可采用磁场进行控制和操作。
本发明还涉及一种油/水分离基质,其包括如上所述的EGC-PPY复合材料。在一个实施方案中,所述油/水分离基质为EGC-PPY-FD。本发明还涉及一种用于油/水分离的色谱柱,其中色谱柱中的油/水分离基质包括如上所述的EGC-PPY复合材料。
附图说明
图1是显示了EGC-PPY晶胶的快速弹性、形状记忆性性质和它们的形态的图。(a1)承受压缩前的EGC(30)(弹性蛋白/明胶=30/70)-PPY-SA;(a2)承受压缩的EGC(30)-PPY-SA;(a3)撤销压缩后的EGC(30)-PPY-SA的快速回弹。(b1)排干水的EGC(30)-PPY-SA的无恢复变形;(b2)将排干水的EGC(30)-PPY-SA重新浸入水中;(b3)EGC(30)-PPY-SA恢复形状。(c1)取出变形的EGC(30)-PPY-FD水凝胶;(c2)EGC(30)-PPY-FD水凝胶;(c3)变形的EGC(30)-PPY-FD水凝胶在水中缓慢恢复的形状记忆性行为。(d1)具有“U”或“M”形状的EGC(30)-PPY-SA水凝胶;(d2)变形后的EGC(30)-PPY-SA水凝胶;(d3)在水中恢复到变形前的“U”或“M”形状的EGC(30)-PPY-SA水凝胶。(e1)EGC(30)水凝胶的SEM图像(放大率95倍);(e2)显示出CNT嵌入在衬底层的EGC(30)水凝胶的SEM图像(放大率23000倍)。(f1)通过快速交联的PPY涂覆的EGC水凝胶(EGC(30)-PPY-FD)的SEM图像(放大率190倍);(f2)是显示PPY纳米颗粒聚集体的分散分布在EGC(30)水凝胶上的SEM图像(放大率11000)。(g1)通过缓慢老化的PPY涂覆的EGC水凝胶(EGC(30)-PPY-SA)的SEM图像(放大率800倍),(g2)显示PPY纳米颗粒在支架上沉积形成的发展良好的PPY网络的图像(放大率13000倍)。(h1)对于10次循环,在80%压缩应变下,EGC(30)-PPY-SA的形状记忆性应变固定率随循环次数的变化;(h2)EGC(30)-PPY-SA的形状恢复率随循环次数的变化。
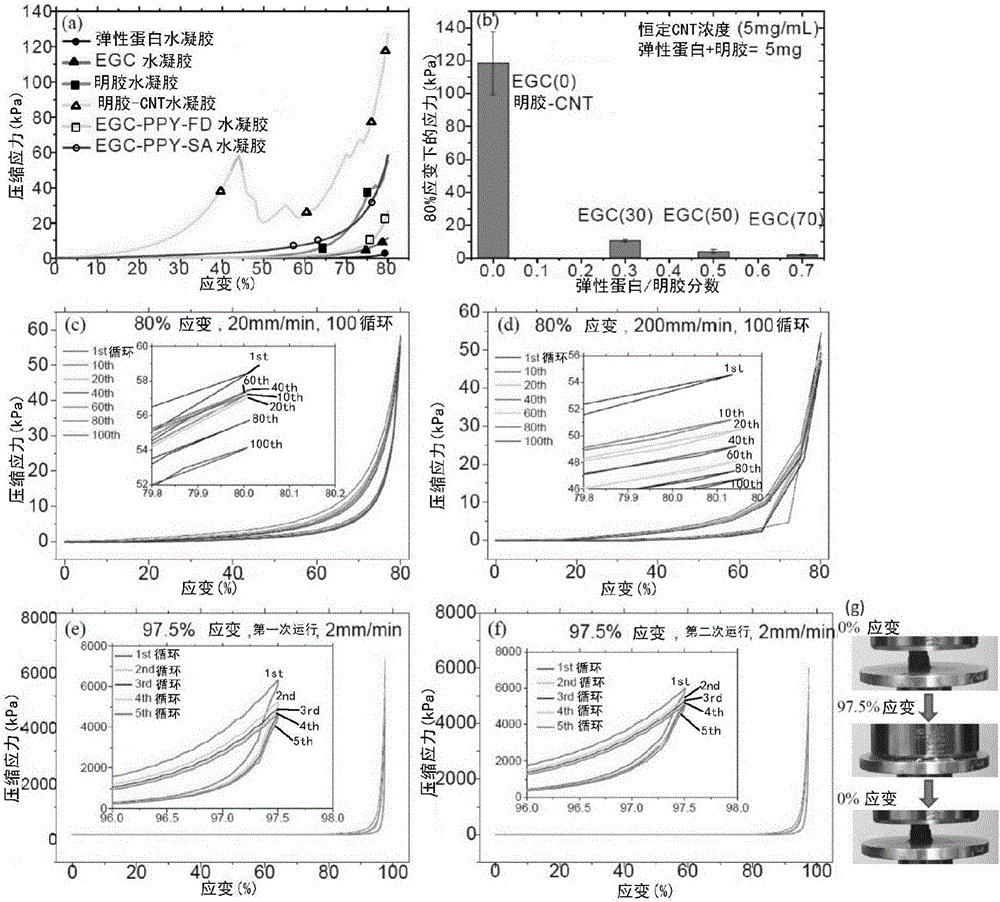
图2是显示了EGC晶胶的机械性质的图。(a)在20mm/min的速率下,具有80%应变的EGC(30)、EGC(30)-PPY-FD、EGC(30)-PPY-SA、弹性蛋白、明胶和明胶-CNT水凝胶的压缩应力应变曲线。(b)在80%应变下,弹性蛋白/明胶质量分数对它们的机械强度的影响(注意:在达到80%应变之前,EGC(0)部分破裂)。(c)在20mm/min和(d)200mm/min的压缩速率下,对于100次循环80%的应变下的EGC(30)-PPY-SA水凝胶的应力-应变曲线。(e)在2mm/min下,对于5次循环,在97.5%高应变下的EGC(30)-PPY-SA水凝胶的应力-应变循环曲线。(f)在24h之后,在2mm/min速率下,对于5次循环,在97.5%的高应变下相同样品的第二压缩-卸荷运行。(g)分别承受0%和97.5%的压缩应变的EGC(30)-PPY-SA的照片。
图3是晶胶的流变学性质的图。(a)EG(30)、(b)EGC(30)、(c)EGC(30)-PPY-FD、(d)EGC(50)、(e)EGC(50)-PPY-FD和(f)EGC(30)-PPY-SA水凝胶。(g1)-(g3)是具有固定形状的EGC(50)-PPY-FD传导性水凝胶的可注射行为。
图4是压力敏感传导性的图.(a1)-(a3)显示了伴随着EGC(30)-PPY-SA逐渐变形,LED的光强度逐渐提高的压力敏感行为。(b):(a1)-(a3)的电路例示。(c1)变形的EGC(30)-PPY-SA水凝胶作为“开/关”电路开关;(c2)显示通过添加DD水恢复原状的EGC(30)-PPY-SA水凝胶开关将电路连同,LED发光;(c3)显示仅使用DD水不能使LED发光。和(d):(c1)-(c3)的电路例示。(e)EGC(30)、EG(50)-PPY-FD、EGC(50)-PPY-FD和EGC(30)-PPY-SA的传导性,通过4探针数字传导性测试仪(n=3(正方向)和n=3(负方向))检测的。(f)EGC(30)-PPY-SA的表观电阻循环试验谱图(50%应变,20mm/min)。(g)EGC(30)-PPY-SA的传导性-压缩应变谱图,由4探针数字传导性测试仪(n=3(正方向)和n=3(负方向))检测的。
图5为水包油乳液的分离的图。(a)植物水包油乳液的分离。(b)分离之前的乳液的光学显微镜图像和(c)分离之后的乳液的光学显微镜图像。(d)油滴尺寸的统计分析(n=4992)。(e)植物油未流入压缩的EGC(30)-PPY-FD水凝胶中,但水立即流入压缩的水凝胶中。
图6为包含IONP的晶胶的磁响应行为。(a)在80%应变下,IONP最终浓度对EGC(50)-IONP压缩强度的影响。(b)在20mm/min速率下,对于10次循环,EGC(50)-IONP的应力-应变循环曲线(20mg/mL)。(c)EGC(50)-IONP的高柔性和压缩性。(d)在弱施加的磁场下,由EGC(50)-IONP制成的人造章鱼机器人。(e)由磁场控制的EGC(50)-IONP-PPY-FD远程传导性生物制动器。
图7为制备多功能弹性蛋白晶胶的示意图。包括高度弹性、可注射、形状记忆性、传导性、压力敏感、油水分离和磁响应性质的EGC-PPY-FD、EGC-PPY-SA和EGC-IONP-PPY晶胶的制造。
图8为在压缩应力下EGC-PPY-SA晶胶的高弹性机制的图示。
图9为0.5mg吡咯作为外标的甲基丙烯酸酯化明胶(明胶-MA,12mg)的1H NMR谱。基于明胶-MA和吡咯的浓度,可通过比较5.4ppm和5.65ppm处的峰强度计算甲基丙烯酸酯的摩尔量,归属于甲基丙烯酸酯的质子,6.2ppm和6.9ppm处的峰强度来自吡咯外标。明胶中甲基丙烯酸酯量的计算值为0.02285mol每100g明胶。
图10为0.5mg吡咯的甲基丙烯酸酯化弹性蛋白(弹性蛋白-MA,18.8mg)的1H NMR谱。基于弹性蛋白-MA和吡咯的浓度,可通过比较5.4ppm和5.65ppm处的峰强度计算甲基丙烯酸酯的摩尔量,归属于甲基丙烯酸酯的质子,6.2ppm和6.9ppm处的峰强度来自吡咯外标。明胶中甲基丙烯酸酯量的计算值为0.02285mol每100g明胶。
图11为F-127二丙烯酸酯的1H NMR谱。
图12是通过CDCl3从Fe(NO3)3引发的缓慢老化反应萃取的残留的吡咯的1H NMR谱(DMSO为外标,0.29mg/mL)。其中,可根据下列方程式计算吡咯的残留浓度:
残留的吡咯的摩尔量
=(强度6.84ppm+强度6.27ppm)/4×(强度2.6ppm/6)×(0.00029/78.13)×67.09,
其中,78.13为DMSO的摩尔分子量。且67.09为吡咯的摩尔分子量。
基于用于萃取的反应溶液的等分体积和用于NMR表征的CDCl3的等分体积,可以计算出反应转化率为96%。
图13是通过CDCl3从APS引发的吡咯快速沉积反应萃取的残留的吡咯的1H NMR谱(DMSO为外标,0.29mg/mL)。
图14是晶胶孔径的统计学分析的图。(a)EGC(30)晶胶,平均孔径,90±14μm,(b)EGC(30)-PPY-FD,平均孔径,98±19μm,(c)EGC(30)-PPY-SA,平均孔径,39±8μm。
图15是GC(30)、EGC(30)-PPY-FD和EGC(30)-PPY-SA的性能图。其中(a)为水含量,(b)为膨胀率(样品制备的体积为360μl)。注意:在计算膨胀率时不考虑PPY重量。
图16是具有不同缓慢老化工艺时间的EGC晶胶的频率扫集流变学谱。(a)EGC(30)-0h,(b)EGC(30)-PPY-SA-8h,(c)EGC(30)-PPY-SA-16h,(d)EGC(30)-PPY-SA-1d,(e)EGC(30)-PPY-SA-2d和(f)EGC(30)-PPY-SA-3d晶胶。
图17是在速率20mm/min下,在承受80%应变之后明胶-CNT和EGC的图像。
图18是EGC(30)-PPY-SA的压缩-卸载循环应力-应变曲线。(a)50%应变,20mm/min,(b)50%应变,200mm/min,(c)70%应变,20mm/min,(d)70%应变,200mm/min,(e)80%应变,200mm/min和(f)80%应变,20mm/min。
图19是对照组PEG-二丙烯酸酯-弹性蛋白-CNT EGC晶胶的机械性质的图。在速率20mm/min下具有80%应变的PEG、PEG-CNT(5mg/mL)、PEG-EL(30)-CNT(5mg/mL)、PEG-EL(30)-CNT-PPY-FD和PEG-EL(30)-CNT-PPY-SA晶胶的压缩应力应变曲线。
图20是对于100次循环,PEG-EL(30)-CNT-PPY-SA的压缩-卸载循环应力-应变曲线,80%应变,20mm/min。
图21是使用不同的水不可溶有机溶剂(密度>1.0)的EGC-PPY的水下疏油性。(a)氯仿,(b)二氯甲烷和(c)二硫化碳。(通过不同颜色染色有机溶剂)。
具体实施方式
为了实现将高柔性和弹性与优异传导性或磁响应行为结合在一起的目标,申请人发现使用具有高负载量的功能纳米成分的高柔性和弹性支架是有效方法。水溶性弹性蛋白(诸如弹性蛋白肽,原弹性蛋白或消化弹性蛋白)是良好的高弹性支架候选物,但它缺乏机械强度[22]。然而,很少使用弹性蛋白晶胶,因为它的机械强度差并且在冷冻凝胶中通过冰晶形成产生的大孔形态甚至进一步降低了机械强度[10,23,24]。
申请人令人惊讶地发现了使用高度水溶性弹性蛋白肽晶胶(EGC晶胶)获得超柔性支架,其通过与明胶、优异和生物相容性凝胶物质和高载入量的CNT复合进一步加强[25]。柔性弹性蛋白肽和明胶链与刚性CNT可形成刚性CNT微区和柔性/软聚合物线圈桥的网络以获得高弹性[26]。此外,水凝胶中高浓度的CNT可形成网络以提高支架中的传导性[4,27]。
然后,将高传导性聚吡咯纳米成分负载在EGC支架上以获得优异的传导性,其中PPY形态学对晶胶的宏观性质发挥关键作用[28]。当EGC负载分散的PPY聚集体时,晶胶显示高度柔性和注射器-可注射性质以及中等传导性。当刚性PPY成分在软支架上形成第二网络时,具有软-硬双连续网络结构的晶胶同时显示可承受至高97.5%压缩应变的非凡高弹性和在90%应变下至高50.1±2.9S/cm的突出传导性以及良好的压力敏感传导性。
申请人还通过相同方法制备EGC-IONP和EGC-IONP-PPY以将高柔性、敏感性、传导性与磁响应性质结合在一起,显示作为远程和软执行器或机器人的潜力。我们还发现EGC-PPY水凝胶是用于“油-水”分离的理想色谱柱[24]。
在本申请中,可以通过甲基丙烯酸化弹性蛋白肽(弹性蛋白-MA,从牛颈韧带水解然后甲基丙烯酸化)、甲基丙烯酸化明胶(明胶-MA)和多壁CNT的晶胶化制备EGC水凝胶。通过F-127-DA聚氧乙烯-聚氧丙烯-聚氧乙烯(PEO-PPO-PEO)三嵌段两亲性共聚物将CNT分散在水中时,疏水PPO嵌段将聚集并且具有与CNT的非共价疏水相互作用并且亲水PEO链在溶液中延伸并且与明胶-MA和弹性蛋白-MA共价键合以形成水凝胶,其将增加CNT与水凝胶基质的亲和力。通过48h在双蒸水(DD水)中的浸泡实验评价CNT与水凝胶基质的亲和力并通过UV-vis测量,其显示>99.9%CNT在水凝胶中稳定[27]。因此,由充当物理交联剂的疏水PPO包围的CNT微区可形成具有软弹性蛋白/明胶/PEO聚合物线桥的弹性网络(CNT制备过程)。支架上的PPY涂层通过(1)快速交联沉积(EGC-PPY-FD)或(2)快速交联,然后缓慢老化(EGC-PPY-SA)。在EGC-PPY-FD的工艺中,将在EGC水凝胶中吸收的吡咯单体引发、聚集、沉淀出并且快速原位吸收在EGC支架上以形成分散的PPY聚集体。快速交联沉积可以在低温下进行,例如在0-15℃下进行。在EGC-PPY-SA工艺中,通过不足量的Fe(NO3)3引发吡咯,其剩余未聚合的单体用于被(NO3)3缓慢氧化聚合以在支架上形成连续网络结构并且缓慢过程允许PPY和明胶/弹性蛋白底物之间的更多分子间氢键相互作用[29,30]。在EGC-PPY-SA工艺中,快速交联可以在低温下进行的,例如在0-15℃下进行,老化可以在室温下进行。
实施例
方法
实验部分
材料:水溶性弹性蛋白肽(Elastin Products Company,CB573,分子量为1000至60000Da。这些肽包含交联氨基酸锁链素和异锁链素,和重复肽Val-Gly-Val-Ala-Pro-Gly)、明胶(类型A,Sigma Aldrich)。所有其他化学品购自Sigma Aldrich,并且直接使用而不进一步纯化。
实施例1:甲基丙烯酸酯接枝明胶(明胶-MA)的合成。
根据前面公开的文献[25],通过与伯胺基团结合将甲基丙烯酸酯基团引入至明胶骨架。简略地说,在50℃下将2g的类型A明胶溶于20mL PBS缓冲液(1×),然后将2mL的甲基丙烯酸酐(MA)添加至明胶溶液并且在50℃下在一起搅拌1hr。在反应之后,将100mL的PBS倒入反应器中以稀释溶液,并且在40℃下,使用10kDa MWCO透析管用DD H2O将明胶溶液透析5天。最后,通过冻干法在-45℃下回收白色多孔泡沫,收率为85%。
实施例2:甲基丙烯酸酯接枝弹性蛋白(弹性蛋白-MA)的合成。
甲基丙烯酸酯接枝弹性蛋白(弹性蛋白-MA)的制备与上述描述的程序相似,然而整个反应在冰浴中发生。简单地说,在冰浴中,将2g的弹性蛋白溶于20mL PBS缓冲液(1×)中,然后将2mL的甲基丙烯酸酐(MA)添加至弹性蛋白溶液并在冰浴中在一起搅拌8hr。在反应后,在4℃下,使用1000Da MWCO透析管用DD H2O将溶液透析48h,并在-45℃下冻干,收率为40%。
实施例3:F-127-二丙烯酸酯的合成。
通过使用丙烯酰氯丙烯酰化F-127(聚(乙二醇)-b-聚(丙二醇)-b-聚(乙二醇))合成F-127-二丙烯酸酯。简略地,在冰浴中,将2.54g(0.2mmol)的F-127和0.061g的三乙胺(0.6mmol)溶于20mL无水二氯甲烷中并通过氮气鼓吹脱气20min。然后,在氮气保护下,将0.05mL丙烯酰氯(0.6mmol)缓慢注射至溶液中。将溶液搅拌24hr,并将反应温度自然升至室温。在反应后,通过旋转蒸发去除溶剂,并通过具有3.5kDa的MWCO的透析管用DD水透析3天将产物纯化,并通过冻干法回收。回收白色固体,收率为2.06g(81%)。
NMR表征。在Bruker Avance 300MHz NMR光谱仪上进行所有1H NMR实验,并将驰豫延迟(relaxation delay,d1)设置为2秒。以10mg/mL的浓度将样品溶于D2O。将大量吡咯添加至明胶-MA的NMR溶液中并且弹性蛋白-MA作为外标用于定量在明胶或弹性蛋白上接枝的甲基丙烯酸酯的量。所有NMR数据参见附图。
实施例4:EGC的制备。
预先制备在DD H2O中的明胶-MA的50mg/mL储备溶液和弹性蛋白-MA的50mg/mL储备溶液,并在使用前在冰箱中储存。将100mg多壁碳纳米管(MWCNT)、100mg F-127-DA和10mL DD H2O添加至50mL塑料管中并在冰浴中通过探针超声波均质器超声处理4h,并在使用前处理另外1h。在典型的制备中,在冰浴中,将30μl的弹性蛋白-MA(50mg/mL)、70μl的明胶-MA(50mg/mL)、75μl的DD H2O和175μl的CNT溶液充分混合。然后,添加5μl的10%TEMED水溶液和5μl的200mg/mL过硫酸铵(APS)水溶液并在冰浴中相继混合,并用移液器将溶液吸移至具体的模具中(颠倒1.5mL EP罐或96-孔板)并密封好,并为了固化在-20℃下储存16hr。然后,使用DD水将水凝胶(表示为EGC(30),弹性蛋白/明胶=30/70)洗涤几次以彻底去除盐。用移液器吸取90μl如上制备的溶液至1.5mL EP管并在-20℃下固化制备可注射样品。制备具有相同浓度和比例的用于97.5%应变的压缩-卸载循环试验的720μl样品溶液。
根据三组冻干样品(3×360μl各个组),EGC晶胶(来自360μl溶液)的实际重量为7.1±0.1mg。通过UV-vis光谱(Ultrospec 4300pro,UV/可见光分光光度计)检测CNT与水凝胶基质的亲和力。使用DD水将选择的样品反复洗涤几次以从反应中去除任何盐和杂质。然后,分别将那些样品(体积:360μl)浸泡在2mL DD水中48h,并且在48h后水保持无色,收集其1mL等分样品用于UV可见光分析,使用CNT的稀释溶液(1mg/mL、0.1mg/mL、0.01mg/mL和0.001mg/mL)作为标准。在48h后,超过99.9%CNT保留在水凝胶中。
采用相同的方法制备了弹性蛋白/明胶=50/50的EGC晶胶(EGC(50))。
实施例5:EGC-PPY-FD的制备。
将实施例4中制备的EGC(30)水凝胶(7.1mg)浸泡在4mL的6.7mg/mL(0.1mol/L)吡咯水溶液中,并在冰浴中将1mL APS(89.6mg/mL、0.4mmol)水溶液倒入吡咯溶液中(实际吡咯浓度为0.08mol/L)。在30min后,将反应停止并使用DD水彻底洗涤水凝胶。根据三组冻干样品(3×360μl各个组),EGC-PPY-FD晶胶的实际重量(来自360μl溶液)为9.0±0.2mg。水凝胶上的PPY负载量为5.2±0.4mg/mL。
实施例6:EGC-PPY-SA的制备。
将实施例4中制备的EGC(30)水凝胶(7.1mg)浸泡在2mL的13.4mg/mL(0.2mol/L)吡咯水溶液中,并在冰浴中将等量的Fe(NO3)3水溶液(0.2mol/L,80.8mg/mL)倒入吡咯溶液中。将溶液充分混合并将水凝胶压缩并释放几次以使吡咯溶液吸收至水凝胶的支架中。将溶液在室温下储存3d,并将EGC-PPY水凝胶移出,并使用DD H2O彻底洗涤。图16中使用的样品具有不同的浸泡时间,包括8h、16h、1d、2d和3d。根据三组冻干样品(3×360μl各个组),EGC-PPY-SA(3d)晶胶的实际重量(来自360μl溶液)为9.8±0.4mg。水凝胶上的PPY负载量为7.4±1.2mg/mL。通过用于CNT的相同方法评价PPY和水凝胶基质的亲和力,并且在48h后>99.9%PPY保留在基质中。
实施例7:IONP纳米颗粒的制备。
在圆底烧瓶中将FeCl3(0.81g,5mmol)和FeCl2·4H2O(0.5g,2.5mmol)溶于10mL DD水中并通过橡胶隔垫帽密封。通过氮气将溶液鼓吹20min以彻底去除空气。在另一个烧瓶中,通过相同方法将20mL 2M氨水水溶液脱气,并在室温下在750rpm高搅拌速率下搅拌。然后,在氮气保护下,通过10mL注射器将铁盐溶液转移至氨水溶液中。在1h后,将反应停止并通过磁铁棒收集黑色固体,使用水将其反复洗涤以彻底去除氨和盐。不干燥,将黑色所得物转移至50mL离心管中并添加DD水以获得总共5mL水性混合物,并添加0.2g F-127-二丙烯酸酯。在冰浴中,通过探针超声波均质器将混合物超声处理2h,然后将0.1mL等分样品转移至EP管中并在真空干燥箱下干燥过夜。基于产物的干重,计算IONP的浓度(收率:0.64g)。将另外的0.44g F-127-二丙烯酸酯添加至管中并添加额外的0.33mL DD水以达到120mg/mL的IONP/F-127浓度。在使用前,在冰浴中将溶液超声处理另外10h。
实施例8:EGC-IONP、EGC-IONP-PPY-FD和生物制动器的制备。
EGC-IONP的制备与EGC晶胶类似。在典型的制备中,在冰浴中,将100μl的弹性蛋白-MA(50mg/mL)、100μl的明胶-MA(50mg/mL)、200μl的CNT溶液(10mg/mL)和200μl的IONP溶液(60mg/mL)充分混合,然后,添加10μl的10%TEMED水溶液和10μl的200mg/mL过硫酸铵(APS)水溶液并在冰浴中相继混合,并用移液器将溶液吸取至1mL注射器筒中并密封好,为了固化在-20℃下储存16hr。可以不同规模但相同比例制备样品。然后,使用DD水将水凝胶洗涤几次以彻底去除盐。将水凝胶浸泡在13.6mg/mL(0.2mol/L)吡咯水溶液中,并在冰浴中将2mL APS(44.8mg/mL)水溶液倒入溶液中,时间为20min,使用DD水彻底洗涤获得的EGC(50)-IONP-PPY-FD晶胶。通过超强力胶水粘附4片EGC(50)-IONP水凝胶(约2cm长度)至一片聚氨酯海绵制备人造章鱼机器人。
另外,将以上制备的EGC(50)-IONP-PPY-FD晶胶制成长条形的生物制动器,并将该生物制动器连接到包括电池,灯泡的简易电路中,其中生物制动器一端与电路相连,并且另一端保持打开。当生物制动器打开时,该灯泡未亮起。而当该生物制动器在外部施加上述磁场的作用下闭合时,该灯泡亮起。具体可参见图6(e)的照片和示意图。
在图6(a)中,通过使用90mg/mL和120mg/mLIONP水溶液制备30mg/mL和40mg/mL最终浓度的IONP晶胶,通过相同浓度的F-127-二丙烯酸酯将其稳定。通过用于CNT的相同方法评价IONP和水凝胶基质的亲和力,并且在48h后>99.9%IONP保留在基质中。
对根据以上制备的本发明的样品采用如下描述的方法进行了分析。并参照附图描述了本发明的有益效果。
PPY快速沉积和缓慢老化沉积的转化率。
通过使用DMSO作为外标的1H NMR检测反应后残留的吡咯计算转化率。由于吡咯在水中具有有限的溶解度,但使用诸如乙醚、氯仿的大量有机溶剂可溶。可通过有机溶剂萃取未反应的吡咯用于NMR表征。对于缓慢老化反应,在两个EP管中分别将6.9μl(6.7mg)的吡咯和40.4mg Fe(NO3)3溶于0.5mL D2O,并在冰浴中将两个溶液混合在一起4h并在室温下总共储存3d。将0.5mL透明上清液转移至另一个EP管,并使用0.3mL氯仿-d(CDCl3)洗涤5次以萃取未反应的吡咯。取出750μL等分样品并与作为外标的10μl DMSO/CDCl3溶液(22mg DMSO/1mL CDCl3)混合以实施1H NMR表征。对于快速沉积反应,在冰浴中将含6.9μl(6.7mg)的吡咯的1mL D2O与250μl APS/D2O溶液(89.6mg/mL)混合30min。黑色PPY聚集体悬浮在溶液中,通过装备有0.45μm PDFE滤片的注射器过滤悬浮溶液。将0.5mL的透明溶液转移至另一个EP管并通过0.3mL CDCl3萃取5次。取出750μL等分样品并与作为外标的10μl DMSO/CDCl3溶液(22mg DMSO/1mL CDCl3)混合以实施1H NMR表征。
SEM表征。
首先将水凝胶样品冻干,然后装载在金属样品容器上以溅射涂覆金的薄层用于表征。在JEOL-5900扫描电子显微镜上实施形态学表征,使用20eV的工作电压。通过图像J软件统计学分析晶胶的孔径信息。
压缩试验和循环试验。
根据上述方案,预先在模具中制备具有圆盘几何结构(4~5mm高度,8mm直径)的水凝胶样品。在20mm/min的速度下,实施压缩试验直至80%压缩应变。在装备有一对压缩板和500N负载元件的万能拉伸试验机(Instron 5965)上实施压缩循环试验。在试验前,在压缩平台上滴下一滴水,并且选择的水凝胶样品分别承受50%、70%或80%的压缩应变,然后在20mm/min或200mm/min的恒定速率下释放至0%,将其重复100次以检测压缩性能和水凝胶的回收。对于具有97.5%的最大应变的循环试验,样品高度分别为9.8mm和9.1mm(相同样品,但有一些永久变形),并且压缩速率为2mm/min。
形状记忆性行为。
具有10mm的初始长度的EGC(30)-PPY-SA水凝胶用于测量。样品的初始标距长度表示为L1。在速率20mm/min下将样品压缩至80%应变,并且压缩的标距长度为L2。将样品保持在该应变下1min,并且挤压的水彻底吸收在纸巾上,然后使样品没有任何负载时间为5min,测量固定的标距长度作为L3。然后,将样品浸泡在水中用于重新水合5min,并且测量恢复的标距长度作为L4。通过两个压缩板的间隙距离测量那些标距长度,将数据取整数至三位小数(mm)(通过仪器自动测量L2,而通过使用精确位置旋钮调节间隙距离手动测量L1、L3和L4,并且实际最小步长为0.003~0.005mm)。根据下列方程式计算形状记忆性固定率和恢复率。
最大压缩应变:εm=(L1–L2)/L1×100%
固定应变:εu=(L1–L3)/L1×100%
残余应变:εp=(L1–L3)/L1×100%
应变固定率:Rf=εu/εm×100%
应变恢复率:Rr=(εm-εp)/εm×100%
流变学特征。
在25℃下,使用8mm直径几何结构的钢平行板在TA发现复合流变仪上进行所有流变学振荡实验。在振荡-频率实验过程中,使样品承受从0.1rad/s慢加速至200rad/s的剪切速率,具有0.5%的恒定应变。
传导性测量。
在0.1μA至100mA的电流范围下,通过具有线性探头(2.0mm间隔)的ST-2258C数字4-探针测试仪(Suzhou Jingge Electrical Co.,Ltd.)检测水凝胶样品的表面电阻。在检测前,必需将所有样品反复洗涤以去除残留的引发剂。可通过方程式σ=1/ρ计算传导性。在70%的压缩应变下检测各个样品,并计算在正电流下检测的三个值的平均结果和在负电流下获得的三个数据的平均结果。在10%、30%、50%、70%和90%的不同压缩应变下检测以4g计的数据。
压力相关的电阻检测。
在室内建造的平台上检测压力响应传导性,包括Instron压缩试验平台,高灵敏度数字万用表及其相关软件、传导铜线和绝缘胶带。将样品压缩并在速率20mm/min下释放,直至50%的最大应变。
植物水包油乳液分离。
使用具有约6.5mm的直径的96孔板作为模具制备样品,并且程序与上述相同。在装载PPY后,将晶胶压缩至1mL注射器筒(出口直径为2mm)中。通过混合0.5mL植物油和9.5mL DD水在一起制备水包油乳液,并且超声处理10mins。通过图像J软件统计学分析晶胶的孔径信息。通过在相同晶胶上5次分离检测流量,每次0.8mL乳液流过晶胶(2mm直径出口),并且在重新使用之前,在包含DD水的器皿中洗涤晶胶。
以下参照附图的描述中涉及的材料EGC、EGC-PPY-FD、EGC-PPY-SA、EGC-IONP、EGC-IONP-PPY-FD均采用以上实施例中描述的具体方法制备,并且按照实验的具体内容制备成相应的形状。其中EGC(30)和EGC(50)分别表示材料中弹性蛋白/明胶=30/70或50/50。
参照图1
为了获得形状记忆性性质,水凝胶应具有(i)大孔海绵样结构以允许水自由地流入/流出,(ii)用于水吸收的高极性和(iii)用于形状恢复的良好弹性[31]。在本发明中,由于EGC-PPY-FD和EGC-PPY-SA的大孔结构和PPY的高极性,两种EGC-PPY水凝胶显示良好的弹性和形状记忆性行为。EGC水凝胶的SEM形态显示出与蜂窝样大孔结构相似(图1(e1))并且具有60~130μm的孔径分布以及高于99%的互联性,其可允许变形以及快速排水,并且将CNT(5mg/mL,最终浓度)嵌入在衬底层,如来自图1(e2)的网状网格。在PPY的涂敷之后,EGC(30)-PPY-FD和EGC(30)-PPY-SA二者都保持与EGC(30)(图1(f1)和(g1))类似的大孔结构,然而EGC(30)-PPY-FD的孔径分布为50~140μm,类似于EGC晶胶,而后者的分布明显降至20~60μm,表明EGC(30)-PPY-SA中累积了更多的PPY。图1(f2)和(g2)分别显示PPY纳米颗粒聚集体的分散分布或支架上沉积形成的发展良好的PPY网络。发展良好的PPY网络的形成由流变学振荡频率扫集实验(rheology oscillation-frequency sweeping experiment)进一步确认。此外,随着PPY缓慢老化时间的增加,储能模量明显增加,并且在缓慢沉积两天之后,在频率范围内储能模量G’常高于损耗模量G”而没有交叉,表明在高剪切速率下没有大规模构象重排并且EGC(30)-PPY的动态机械性质由PPY刚性网络支配,并且形成软-硬双连续结构(图16)。
此外,参照图1中(a1)-(a3),当EGC(30)-PPY-SA圆柱体承受压缩应力,大孔支架变形并且挤出水以释放应力,同时当撤销应力时吸收水并且立即恢复,如图1a所示。从图1b可以看出,当去除水时,变形的EGC(30)-PPY-SA(采用同样方法制备的)失去它的弹性并且保持变形,但再次使用水时,立即恢复它的原始几何结构,显示形状记忆性行为。通过(1)单轴压缩样品达到80%应变,(2)水通过纸巾吸水以保持变形,(3)去除负载5min和(4)将样品重新浸入水中5min用于恢复进一步评价该EGC(30)-PPY-SA的形状记忆性能。计算10个循环的应变固定率和应变恢复率,如在实验部分详细描述的。由于PPY刚性网络,当去除负载时,压缩的水凝胶稍微恢复,并且固定率为约88~90%。当重新浸泡在水中时,形状在约0.3秒内恢复,并且在10次循环之后恢复率保持在约99%,显示良好的形状记忆性(图1(h1)和(h2))[32]。图1c显示该EGC(30)-PPY-FD的类似的形状记忆性行为,其具有较慢的恢复率。可将该EGC(30)-PPY-SA水凝胶模制成“U”和“M”的字母,可使用水将其快速恢复,如图1d所示。
参照图2
为了探查EGC-PPY水凝胶的高压缩弹性,比较EGC(30)、EGC(30)-PPY水凝胶(EGC(30)-PPY-FD和EGC(30)-PPY-SA)和对照样品,包括弹性蛋白、明胶和明胶-CNT水凝胶的压缩性能,其中总弹性蛋白/明胶浓度相同。在图2(a)中,纯明胶水凝胶具有更高的压缩强度,但在75%应变下产生局部机械断裂。通过添加CNT(5mg/mL),明胶-CNT复合水凝胶的机械强度急剧增加,但压缩应力变小。而弹性蛋白肽水凝胶在80%应变下只具有0.9kPa的超低机械强度。因此,引入弹性蛋白急剧降低EGC(30)的机械强度(弹性蛋白/明胶=30/70)至11.4kPa,并且在80%应变下弹性蛋白-基复合水凝胶都不具有任何机械断裂,即便是存在刚性PPY层和CNT成分。具有分散的PPY聚集体涂层或连续PPY网络,EGC(30)-PPY-FD和EGC(30)-PPY-SA在80%应变下分别显示26.7kPa和58.4kPa的压缩强度而没有任何断裂,增加2倍或5倍。为了确认增加的弹性归因于弹性蛋白肽,通过相同方法检验使用PEG-二丙烯酸酯(700)代替明胶的对照组。PEG晶胶显示高度柔性但具有较小的压缩应力并且伴随CNT的掺杂(5mg/mL的最终浓度)产生断裂,但显示与弹性蛋白和PPY相似的高弹性行为(图19)。图2(b)显示弹性蛋白肽成分不可思议地降低晶胶机械强度,并且在存在5mg/mL CNT时EGC(70)只具有2.0±0.4kPa的强度,其中弹性蛋白/明胶的总浓度为5mg/0.35mL。
为了使用快速恢复和稳定的大孔传导性水凝胶进一步评价高弹性,对于100次循环,该EGC(30)-PPY-SA晶胶分别在20mm/min和200mm/min的速率下承受80%变形。200mm/min的高压缩速率要求变形的水凝胶可在1.08s内迅速恢复。如图2(c)和2(d)所示,两个应力-应变谱显示相似的闭合曲线。在100次循环之后,对于20mm/min速率恢复损失8.8%且对于200mm/min速率恢复损失13.8%,并且水凝胶仍保持良好的形状和弹性,表明它的稳定性和在心脏组织工程学中的潜在应用。在较低应变下,该样品显示较好的稳定性和弹性性能,具有较少的恢复损失(图18)。对于5次循环,当同样制备的EGC(30)-PPY-SA晶胶在2mm/min的速率下承受97.5%的超高压缩应变时,在第一次循环时,压缩强度跃至6348.6kPa的极高值,如图2(e)所示。在5次循环后,在97.5%应变下压缩强度的恢复损失为26.8%且样品高度从9.8mm收缩至8.6mm(12.2%变形),但晶胶保持完整形状而没有任何明显断裂,表明PPY层的有限局部断裂。浸泡在水中的样品恢复至9.1mm,具有7.1%永久变形,并且在24h后检查具有高达97.5%的应变的第二压缩循环试验。有趣地,97.5%应变下的压缩强度恢复至6014.5kPa,并且在5次循环之后,压缩强度的恢复损失只为14.6%,如图2(f)所示。在循环试验之后,样品具有另外0.5mm收缩(5.5%),但在水中浸泡后留下小于0.1mm永久变形(<1%永久变形)。表明EGC-PPY-SA晶胶的非凡弹性,其可使用6.35Mpa应力提供97.5%的压缩应变并且晶胶支架保持良好,只具有刚性PPY层的有限局部断裂。
参照图3
通过振荡-频率扫集法检测评价弹性蛋白-明胶(EG(30))、EGC(EGC(30)和EGC(50)、)和EGC-PPY(EGC(30)-PPY-SA、EGC(50)-PPY-FD和EGC(30)-PPY-FD)的动态机械性能。根据图3(a),随着剪切速率的增加,在储能模量和损耗模量痕迹之间有交叉,反映在高剪切速率下的长程分子运动和大规模构象重排并且表明EG(30)水凝胶的高柔性和可注射性质。图3(b)和3(c)显示EGC(30)支架和EGC(30)-PPY-FD的流变学特性,其中G’和G”明显增加,但尽管存在期望的刚性CNT和PPY,仍有高剪切速率下的类似过渡,表明它们的高度柔性和可注射行为。刚性成分仅强化晶胶,但机械性质仍受软支架支配。EGC(50)和EGC(50)-PPY-FD显示相似的流变学行为并且倾向于作为EGC(30)和EGC(30)-PPY-FD,但较少的储能模量。然而,对于EGC(30)-PPY-SA,G’和G”二者几乎增加一个数量级,但G”总是低于G’,表明EGC(30)-PPY-SA由于刚性PPY网络丧失可注射性质。相应地,EGC(50)-PPY-FD显示它的可注射性质,如图3(g)所示。可通过针(ID 1.6mm)将该具有固定形状的传导性水凝胶注入,并且很快恢复它的初始形状。相反,EGC(30)-PPY-SA不能被注入,但具有显著更高的弹性模量和具有快速恢复性质的高弹性。
参照图4
图4(a)显示伴随水凝胶变形,LED改变它的光强度,表明EGC(30)-PPY-SA的压力敏感传导性,其可用作潜在生物传感器。为了进一步展示凝胶的传导性和形状记忆性行为,如图4(c)所示,通过添加DD水,变形的EGC(30)-PPY恢复至它的原始形状,并且当恢复的水凝胶桥接电路时LED灯泡点亮,但纯DD水单独不能照亮灯泡。为了进一步评价压力敏感传导性,在图4(e)中提供EGC(30)、EG(50)-PPY-FD、EGC(50)-PPY-FD和EGC(30)-PPY-SA水凝胶的传导性。在70%的压缩应变下,CNT网状网络向EGC水凝胶提供了0.004±4.4×10-5S/cm的传导性,然而,包含PPY和CNT的EGC(50)-PPY-FD水凝胶的传导性上升两个数量级达到0.375±0.024S/cm的值。没有CNT网状结构,EG(50)-PPY-FD仅具有0.00758±0.00199S/cm的值。伴随发展良好的PPY网络的形成,在70%应变下EGC(30)-PPY-SA水凝胶的传导性显著增加至14.7±1.4S/cm。由于材料具有不均匀横截面,传导性为应变相关的,因此为压力相关的。图4(f)显示反复承受50%压缩应变的EGC(30)-PPY-SA的实时表观电阻谱,每次循环其显示良好的应变相关性和可比较的电阻。将没有应变和实时电阻的最高电阻值分别表示为R0和R,并通过R/R0计算相对电阻,其与水凝胶应变成正比并且在50%应变下接近25%的初始值。根据EGC(30)-PPY-SA的压缩应力-应变曲线,压力敏感性(ΔR/R0每kPa)为约0.086每kPa。
优异的弹性和应力/应变相关性电阻表明PPY负载的EGC水凝胶可为3D庞大压力响应传感器的良好候选[13]。图4g中提供EGC(30)-PPY-SA的传导性vs.应变谱,其并非线性增加,对于最佳样品,获得在90%应变下的50.1±2.9S/cm的最高传导性,据我们掌握的最佳知识,其顶部在目前的传导性压缩水凝胶之间[18,33]。
参照图5
如文献报道的,水下超疏油水凝胶涂覆的网格或滤纸衬底用于油/水分离[34]。本发明的具有固有互连大孔结构和水下疏油性PPY涂覆的晶胶[29,35]也可用于油-水分离而没有多孔支架。当植物水包油乳液(5:95,v:v)通过填充有压缩的EGC(30)-PPY-FD晶胶的注射器时,仅透明的水流过晶胶,如图5a所示。通过光学显微镜的检查,在过滤后具有1~28μm粒度分布的油滴完全消失,如图5b~5d所示。图5e显示晶胶的疏油性,其中植物油不能流入压缩的EGC(30)-PPY-FD水凝胶。分离能力高于99%并且流量可达到404±101L/hr·m2(n=5)。
参照图6
考虑EGC的高柔性和压缩弹性,它可为磁响应材料的良好候选,与前面报道的磁响应海藻酸盐-氧化铁钢化凝胶相比,其在80%应变下具有55~60kPa的模量并且在约38A/m2的施加的磁场梯度下显示约70%的变形[19]。根据实施例8制备EGC(50)-氧化铁(IONP)晶胶,其中将20mg/mL(最终浓度)的Fe3O4纳米颗粒引入至EGC(50),但在80%应变下它的压缩强度仅为5.1±1.2kPa,并且具有40mg/mL IONP的晶胶只具有11.8±2.4kPa的值,显示具有高载量的IONP的高柔性,如图6(a)和(b)所示。由图6(c)可见,在来自永久磁棒的弱磁场下EGC(50)-IONP(20mg/mL)磁性水凝胶圆筒显示超柔性,其容易弯曲至180°,此外在1mL注射器桶的受限环境中,弯曲变形受限制并且当放入磁场中时沿着轴方向整个水凝胶圆筒被轻微压缩至约45%的应变。图6(d)显示高柔性和敏感性能使其成为由施加的磁场控制的模拟人造章鱼机器人。
此外,如图6(e)所示,在涂覆PPY之后,本申请所制备的EGC(50)-IONP-PPY-FD保持高柔性并且在磁场下可弯曲至90°。并且当施加的上述弱磁场控制时,其充当了远程传导性生物制动器[20,36],远程控制了电路的打开与闭合。
因此可以得出结论,水溶性弹性蛋白肽基晶胶大孔支架负载的具有传导性的刚性PPY或IONP可将高弹性、可注射能力和形状记忆性性质与传导性或磁性性质结合在一起。所有PPY涂覆的EGC水凝胶显示优异的弹性、柔性、形状记忆性行为和应力相关的传导性。特别地,当EGC支架负载分散的PPY时,产生的具有固定形状的传导性水凝胶显示优异的柔性和可注射性质,表明它潜在用作注射器-可注射生物传感器或生物电子学;随着EGC-PPY软-硬双连续网络的形成,产品显示高弹性模量和具有快速恢复的非凡弹性,在90%应变下具有50.1±2.9S/cm的优异传导性,并且甚至能承受97.5%的压缩变形,表明用于心脏组织工程学材料或压力敏感生物传感器的良好候选。磁响应水凝胶EGC-IONP还保持高柔性并且显示敏感磁响应行为,并且可用作生物制动器。
本领域技术人员应当知晓,以上提到的各个特征之间可以任意组合,并且这种组合也在本申请的范围之内。本领域技术人员应当意识到在不脱离本发明所附的权利要求所揭示的本发明的范围和精神的情况下所作的更动与润饰,均属本发明的权利要求的保护范围之内。
参考文献:
[1]a)J.Elisseeff,Nat Mater 2008,7,271;b)J.L.Drury,D.J.Mooney,Biomaterials2003,24,4337;c)T.R.Hoare,D.S.Kohane,Polymer 2008,49,1993;d)D.Buenger,F.Topuz,J.Groll,Progress in Polymer Science 2012,37,1678;e)S.Ladet,L.David,A.Domard,Nature2008,452,76.
[2]A.Doring,W.Birnbaum,D.Kuckling,Chemical Society Reviews 2013,42,7391.
[3]a)L.Pan,G.Yu,D.Zhai,H.R.Lee,W.Zhao,N.Liu,H.Wang,B.C.-K.Tee,Y.Shi,Y.Cui,Z.Bao,Proceedings of the National Academy of Sciences 2012,109,9287;b)B.C.-K.Tee,A.Chortos,A.Berndt,A.K.Nguyen,A.Tom,A.McGuire,Z.C.Lin,K.Tien,W.-G.Bae,H.Wang,P.Mei,H.-H.Chou,B.Cui,K.Deisseroth,T.N.Ng,Z.Bao,Science 2015,350,313.
[4]S.R.Shin,S.M.Jung,M.Zalabany,K.Kim,P.Zorlutuna,S.b.Kim,M.Nikkhah,M.Khabiry,M.Azize,J.Kong,K.-t.Wan,T.Palacios,M.R.Dokmeci,H.Bae,X.Tang,A.Khademhosseini,ACS Nano 2013,7,2369.
[5]Z.Li,J.Guan,Polymers 2011,3,740.
[6]B.C.K.Tee,C.Wang,R.Allen,Z.Bao,Nat Nano 2012,7,825.
[7]Y.Si,Q.Fu,X.Wang,J.Zhu,J.Yu,G.Sun,B.Ding,ACS Nano 2015,9,3791.
[8]Y.Li,G.Huang,X.Zhang,B.Li,Y.Chen,T.Lu,T.J.Lu,F.Xu,Advanced Functional Materials 2013,23,660.
[9]a)J.Liu,T.-M.Fu,Z.Cheng,G.Hong,T.Zhou,L.Jin,M.Duvvuri,Z.Jiang,P.Kruskal,C.Xie,Z.Suo,Y.Fang,C.M.Lieber,Nat Nano 2015,10,629;b)G.Hong,T.-M.Fu,T.Zhou,T.G.Schuhmann,J.Huang,C.M.Lieber,Nano Letters 2015,15,6979.
[10]S.A.Bencherif,R.W.Sands,D.Bhatta,P.Arany,C.S.Verbeke,D.A.Edwards,D.J.Mooney,Proceedings of the National Academy of Sciences 2012,109,19590.
[11]a)S.A.Bencherif,R.Warren Sands,O.A.Ali,W.A.Li,S.A.Lewin,T.M.Braschler,T.-Y.Shih,C.S.Verbeke,D.Bhatta,G.Dranoff,D.J.Mooney,Nat Commun 2015,6;b)S.T.Koshy,T.C.Ferrante,S.A.Lewin,D.J.Mooney,Biomaterials 2014,35,2477;c)C.D.Abueva,A.R.Padalhin,Y.-K.Min,B.-T.Lee,Journal of Biomaterials Applications 2015;d)L.Cheng,K.Ji,A.Haddad,T.-Y.Shih,G.Giatsidis,D.J.Mooney,D.P.Orgill,C.S.Nabzdyk,Journal of the American College of Surgeons 2015,221,e119.
[12]a)B.K.Satapathy,R.Weidisch,P.A.Janke,Composites Science and Technology 2007,67,867;b)K.Hu,D.D.Kulkarni,I.Choi,V.V.Tsukruk,Progress in Polymer Science 2014,39,1934.
[13]L.Pan,A.Chortos,G.Yu,Y.Wang,S.Isaacson,R.Allen,Y.Shi,R.Dauskardt,Z.Bao,Nat Commun 2014,5.
[14]S.Gong,W.Schwalb,Y.Wang,Y.Chen,Y.Tang,J.Si,B.Shirinzadeh,W.Cheng,Nature communications 2014,5.
[15]X.Wang,Y.Gu,Z.Xiong,Z.Cui,T.Zhang,Advanced Materials 2014,26,1336.
[16]K.Takei,T.Takahashi,J.C.Ho,H.Ko,A.G.Gillies,P.W.Leu,R.S.Fearing,A.Javey,Nat Mater 2010,9,821.
[17]Y.Tai,M.Mulle,I.Aguilar Ventura,G.Lubineau,Nanoscale 2015,7,14766.
[18]a)Y.Wang,Y.Shi,L.Pan,Y.Ding,Y.Zhao,Y.Li,Y.Shi,G.Yu,Nano Letters 2015,15,7736;X.Zhao,P.Li,B.Guo,P.X.Ma,Acta Biomaterialia 2015,26,236;b)Y.Shi,C.Ma,L.Peng,G.Yu,Advanced Functional Materials 2015,25,1219;c)Y.Shi,M.Wang,C.Ma,Y.Wang,X.Li,G.Yu,Nano Letters 2015,15,6276;d)S.Naficy,J.M.Razal,G.M.Spinks,G.G.Wallace,P.G.Whitten,Chemistry of Materials 2012,24,3425.
[19]X.Zhao,J.Kim,C.A.Cezar,N.Huebsch,K.Lee,K.Bouhadir,D.J.Mooney,Proceedings of the National Academy of Sciences 2011,108,67.
[20]X.Xu,H.Li,Q.Zhang,H.Hu,Z.Zhao,J.Li,J.Li,Y.Qiao,Y.Gogotsi,ACS Nano2015,9,3969.
[21]M.F.Reiser,W.Semmler,H.Hricak,Magnetic resonance tomography,Springer Science&Business Media,2007.
[22]a)S.G.Wise,S.M.Mithieux,A.S.Weiss,in Advances in Protein Chemistry and Structural Biology,Vol.Volume 78(Ed:M.Alexander),Academic Press,2009,1;b)J.-Y.Wang,World Scientific Publishing Co.Pte.Ltd.,Singapore 2014,13;c)Y.-N.Zhang,R.K.Avery,Q.Vallmajo-Martin,A.Assmann,A.Vegh,A.Memic,B.D.Olsen,N.Annabi,A.Khademhosseini,Advanced Functional Materials 2015,25,4814.
[23]G.C.Yeo,B.Aghaei-Ghareh-Bolagh,E.P.Brackenreg,M.A.Hiob,P.Lee,A.S.Weiss,Advanced Healthcare Materials 2015,4,2530.
[24]A.Kumar,A.Srivastava,Nat.Protocols 2010,5,1737.
[25]J.W.Nichol,S.T.Koshy,H.Bae,C.M.Hwang,S.Yamanlar,A.Khademhosseini,Biomaterials 2010,31,5536.
[26]N.Annabi,S.R.Shin,A.Tamayol,M.Miscuglio,M.A.Bakooshli,A.Assmann,P.Mostafalu,J.-Y.Sun,S.Mithieux,L.Cheung,X.Tang,A.S.Weiss,A.Khademhosseini,Advanced Materials 2015,n/a.
[27]Y.Xiao,L.He,J.Che,Journal of Materials Chemistry 2012,22,8076.
[28]D.Ofer,R.M.Crooks,M.S.Wrighton,Journal of the American Chemical Society1990,112,7869.
[29]Y.Lu,W.He,T.Cao,H.Guo,Y.Zhang,Q.Li,Z.Shao,Y.Cui,X.Zhang,Scientific Reports 2014,4,5792.
[30]a)D.Beattie,K.H.Wong,C.Williams,L.A.Poole-Warren,T.P.Davis,C.Barner-Kowollik,M.H.Stenzel,Biomacromolecules 2006,7,1072;b)S.C.Demetgül,M.Timur,N.Carbohydrate Polymers 2010,79,908.
[31]a)R.Luo,J.Wu,N.-D.Dinh,C.-H.Chen,Advanced Functional Materials 2015,25,7272;b)H.Omidian,J.G.Rocca,K.Park,Macromolecular Bioscience 2006,6,703.
[32]A.Lendlein,S.Kelch,Angewandte Chemie International Edition 2002,41,2034.
[33]J.Hur,K.Im,S.W.Kim,J.Kim,D.-Y.Chung,T.-H.Kim,K.H.Jo,J.H.Hahn,Z.Bao,S.Hwang,N.Park,ACS Nano 2014,8,10066.
[34]a)Z.Xue,S.Wang,L.Lin,L.Chen,M.Liu,L.Feng,L.Jiang,Advanced Materials2011,23,4270;b)J.-B.Fan,Y.Song,S.Wang,J.Meng,G.Yang,X.Guo,L.Feng,L.Jiang,Advanced Functional Materials 2015,25,5368.
[35]C.Teng,S.Wang,X.Lu,J.Wang,G.Ren,Y.Zhu,L.Jiang,Soft Matter 2015,11,4290.
[36]P.Ilg,Soft Matter 2013,9,3465.
- 还没有人留言评论。精彩留言会获得点赞!