一类芴乙烯衍生物及其合成方法与流程
本专利属于高分子材料领域,具体涉及一类芴乙烯衍生物以及此类化合物的典型合成路线。
背景技术:
导电高分子是由具有共扼π-键的高分子经化学或电化学“掺杂”使其由绝缘体转变为导体的一类高分子材料。2000年,美国科学家A.G.McDiarmid和A.Heeger以及日本科学家白川英树由于在此领域的开创性工作获得诺贝尔化学奖。
导电高分子是一种性能优良的新型功能材料,并在80-90年代研究进展迅速,成为材料科学的研究中心,促使世界各国科学家致力于导电高分子的实用化。由于导电高分子具有π-共轭的结构,因此,它具有响应速度快(10-13sec)和高的三阶非线性光学系数(i=10-9——10-13esu),可用于信息存贮、调频、光开关和光计算机等技术上。同时,具有高电导的导电高分子可用于电磁屏蔽,防静电、分子导线等技术上的应用。而具有半导体性能的导电高分子,可用于光电子器件(晶体管,整流管),有机发光二极管(organiclight emitting diode,OLED)和有机太阳能电池等方面。
近年来,聚芴类材料成为了导电高分子领域的研究重点之一。这类材料具有良好的热稳定性,在溶液中和薄膜中都有很高的荧光量子产率,发射波长主要集中在蓝光范围。它们的光稳定性及热稳定性要比PPV好。聚芴在每个重复单元中至少包含一个刚性的平面双苯环结构。同PPP相比,芴单元9位的取代基团产生空间阻隔,使聚芴较少有分子间相互作用。聚芴具有独特的液晶性质,这对于制备偏振光器件是有利的。带有辛基的聚芴在170℃进入液晶相,在270~280℃进入均一相,而且可以在这两相之间反复转变。聚芴在室温下制成的膜可以是玻璃态,也可以是半结晶体。聚芴电激发的松弛过程已通过TRFS(Time Resolved Fluorescence Specroscopy)技术测试,为ps数量级。
在九十年代早期,Yoshino等人报告了用FeCl3来氧化单体的方法得到蓝色荧光物质聚芴,相似的方法也可应用于聚噻吩。但是用这种方法得到的聚芴分子量较低(DP~10),而且很难除去痕量的氧化物,不过,还是用这种聚芴制成了器 件。
后来,Yamamoto反应,即镍催化的偶合反应也被用于芴的聚合,并得到了一系列芴的均聚和齐聚物。这种镍催化体系比钯便宜,对于两种共轭基团的偶合反应是比较有效的。
近年来,Suzuki反应也被用于聚芴的合成。非常多的染料都能与芴共聚,包括苯、萘、蒽、苝、噻吩等,由于这些染料的引入,聚芴的发光性能也有了很大的变化,可实现从蓝光到红光的各种颜色光的发射。
用这种方法也合成了一些单分散的齐聚芴,聚合度从3到10不等,主要是考察共轭体系的增大对齐聚芴发光性能的影响。
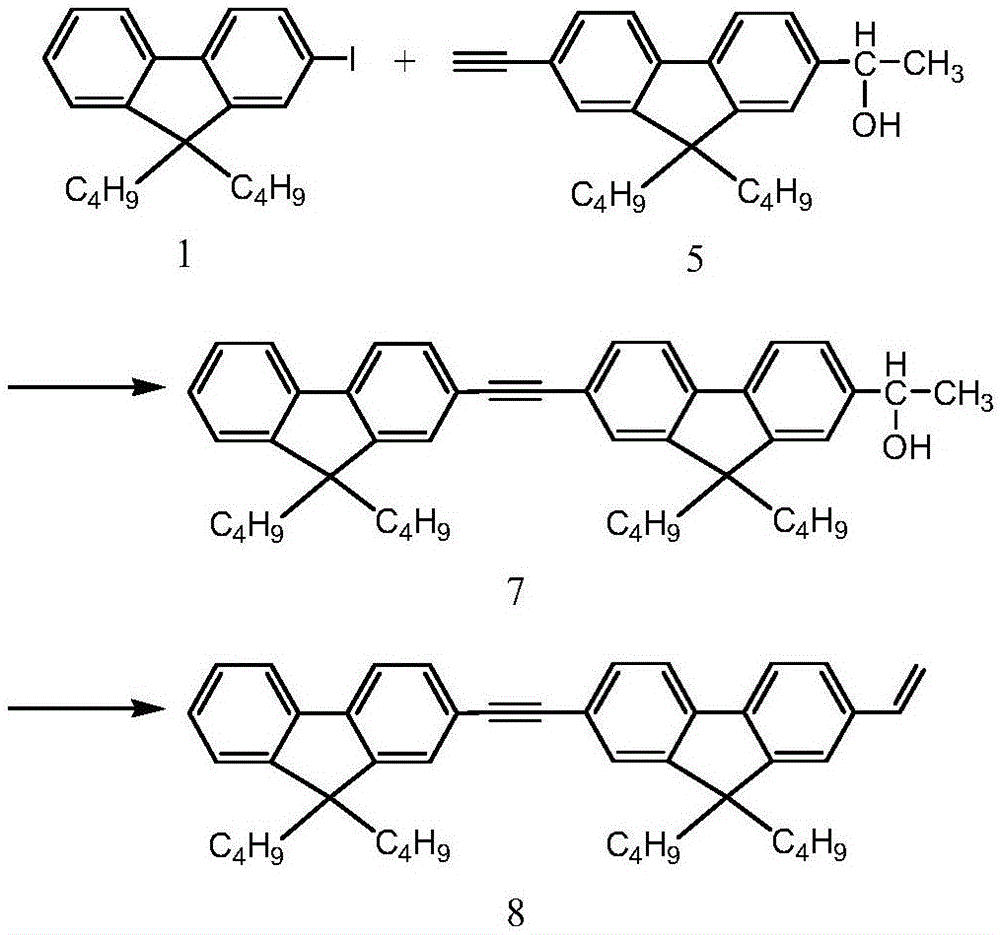
2005年,北京大学胡小丹等人率先报道了一类新型的侧链型聚苯乙烯-芴衍生物(见图1),其特点是,首先合成在分子末端带有卤素的齐聚芴,与对乙炔基苯乙烯偶联后,通过自由基聚合法获得主链为柔性链,共轭基团侧挂于主链上的导电高分子。这种方法具有如下优点:一,与主链型共轭高分子相比合成路径比较简单。二,材料的溶解性由高分子的柔性主链所决定,避免了许多主链型共轭高分子溶解性差的缺点。三,发射光的波长由侧链的低分子荧光团决定,便于在合成中调控。四,当接枝到高分子链上以后,小分子荧光物质易结晶的问题有望得到解决。五,双键的聚合是高分子领域最成熟度技术之一,目前所开发出的活性聚合方法可以方便地获得分子量可控或超高分子量的聚合物,这是Suzuki,Yamamoto等主链型聚合方法所无法做到的,同时也可避免引入金属催化剂残片导致的聚合物光电性能的降低。六,可与多种多样的其他乙烯类单体共聚合实现化学改性。
试验发现,上述结构式所示聚合物中芴基团与苯环间能级匹配差,器件的光电性能较差。为此我们在分子结构上作出改进,去掉苯乙烯结构,直接以芴乙烯衍生物作为聚合单体。本专利中将涉及一系列芴乙烯衍生物及重要的中间产物,以及此类化合物典型的合成路线。
技术实现要素:
针对现有技术存在的问题,本发明提出一系列芴乙烯衍生物及重要的中间产物,以及此类化合物典型的合成路线。
本专利涉及的一类芴乙烯衍生物,其分子结构包括:
其中n为大于1的自然数。
所述的一类芴乙烯衍生物,其合成方法为:
步骤一,在芴环的2位上引入碘原子作为离去基团,得到2-碘芴;
步骤二,用相转移催化法将步骤一中所得2-碘芴9位上的两个氢原子用烷基取代,得到2-碘-9,9-二烷基芴;
步骤三,用傅氏反应在步骤二中所得2-碘-9,9-二烷基芴芴环的7位上引入乙酰基团,得到2-碘-9,9-二烷基芴乙酰;
步骤四,用Sonogashira偶联法将步骤三中所得2-碘-9,9-二烷基芴乙酰芴环2位上的碘原子转变为乙炔基,得到2-乙炔基-9,9-二烷基芴乙酰;
步骤五,用硼氢化钠还原步骤四中所得2-乙炔基-9,9-二烷基芴乙酰芴环7位上的乙酰基,得到中间产物2-乙炔基-9,9-二烷基芴乙醇;
步骤六,在沸腾甲苯中,以对甲苯磺酸为催化剂使烷基取代对乙炔基芴乙醇脱水,获得2-乙炔基-9,9-二烷基芴乙烯;
步骤七,在制备n聚芴乙烯衍生物时,将步骤五所得2-乙炔基-9,9-二烷基芴乙醇与2-碘代n-1聚芴用Sonogashira法偶联,然后重复步骤六。
所述的共轭齐聚芴均采用Sonogashira法偶联,偶联单体之间通过三键桥接,同其他偶联方法,如Suzuki法、Heck法等相比,Sonogashira法条件温和,反应温度低,温度控制在45-50℃,反应在四氢呋喃中进行;且产率高,一般高于80%。
本专利所涉及的共轭齐聚芴,芴9位氢用烷基链取代,优选为正丁基,为调节玻璃化温度及产物溶解性,也可采用正己基、正辛基等。芴9位的氢较为活泼,容易被氧化成酮,导致芴发射光谱红移,为保证光谱的稳定性,通常会将这两个活泼氢用取代基团取代,取代基团包括烷基、芳基等,本合成路线中的烷基取代反应,均采用相转移催化法,这种方法的特点是,工艺流程简单,原料价廉易得,特别适合工业化生产。
在本专利中,均采用碘作为卤素离去基团,碘代芴在乙酰化时,由于碘是一种良好的离去基团,在质子酸的作用下容易形成碘单质析出,最终产物为芴乙酰。故必须严格按照本专利陈述之方法进行,方能获得高转化率。
本专利中双键的制备,均采用甲苯催化脱水法,具体方法为:升温至甲苯沸点以上,通过甲苯的不断蒸出带走反应体系中的水分,使仲醇迅速脱水。仲醇在脱水过程中可能生成烯类或醚类,实验证明,只有迅速、不间断的除去体系中的水分,才能获得高转化率的烯类,否则反应产物将会以醚类为主;使用甲苯而不是苯作为脱水介质,可利用甲苯的高沸点提高水分的脱出速率,从而提高烯类产率。本专利中脱水催化剂采用对甲苯磺酸。
与现有技术相比,本发明具有的有益效果是:本专利所描述的芴乙烯衍生物单体,可通过均聚和共聚等方法,获得大量性能各异的半导体聚合物。这类半导体聚合物具有如下优点:一,与主链型共轭高分子相比合成路径比较简单。二,材料的溶解性由高分子的柔性主链所决定,避免了许多主链型共轭高分子溶解性差的缺点,特别适用于湿法加工。三,发射光的波长由侧链的低分子荧光团决定,便于在合成中调控。四,当接枝到高分子链上以后,小分子荧光物质易结晶的问题有望得到解决。五,双键的聚合是高分子领域最成熟度技术之一,目前所开发出的活性聚合方法可以方便地获得分子量可控或超高分子量的聚合物,这是 Suzuki,Yamamoto等主链型聚合方法所无法做到的,同时也可避免引入金属催化剂残片导致的聚合物光电性能的降低。六,可与多种多样的其他乙烯类单体共聚合实现化学改性。因此此类芴乙烯衍生物单体具有很高的科研价值及应用前景。
附图说明
图1为a单体合成路线图;
图2为b单体合成路线图;
图3为c单体合成路线图;
图4为芴乙烯衍生物溶液荧光光谱;
图5为芴乙烯衍生物溶液紫外光谱;
图6为芴乙烯衍生物膜荧光光谱;
图7为芴乙烯衍生物膜紫外光谱。
具体实施方式
本专利涉及的一类芴乙烯衍生物,其分子结构包括:
其中n为大于1的自然数。
所述的一类芴乙烯衍生物,其合成方法为:
步骤一,在芴环的2位上引入碘原子作为离去基团,得到2-碘芴;
步骤二,用相转移催化法将步骤一中所得2-碘芴9位上的两个氢原子用烷基取代,得到2-碘-9,9-二烷基芴;
步骤三,用傅氏反应在步骤二中所得2-碘-9,9-二烷基芴芴环的7位上引入乙酰基团,得到2-碘-9,9-二烷基芴乙酰;
步骤四,用Sonogashira偶联法将步骤三中所得2-碘-9,9-二烷基芴乙酰芴环2位上的碘原子转变为乙炔基,得到2-乙炔基-9,9-二烷基芴乙酰;
步骤五,用硼氢化钠还原步骤四中所得2-乙炔基-9,9-二烷基芴乙酰芴环7位上的乙酰基,得到中间产物2-乙炔基-9,9-二烷基芴乙醇;
步骤六,在沸腾甲苯中,以对甲苯磺酸为催化剂使烷基取代对乙炔基芴乙醇脱水,获得2-乙炔基-9,9-二烷基芴乙烯;
步骤七,在制备n聚芴乙烯衍生物时,将步骤五所得2-乙炔基-9,9-二烷基芴乙醇与2-碘代n-1聚芴用Sonogashira法偶联,然后重复步骤六。
实施例1:2-碘-9,9-二丁基芴的合成
在25毫升圆底烧瓶中加入1.46g 2-碘芴,2毫升正溴丁烷,0.5毫升DMSO,0.03g四丁基溴化铵,50%NaOH溶液2毫升。80℃反应6小时。反应结束后,加入10毫升水,10毫升石油醚,振荡分液,取上层石油醚相,水相用10毫升石油醚再提取一次。以石油醚为流动相,用色谱柱分离。真空烘箱50℃烘干,产物为浅绿色固体。产率80%。1H NMR(ppm)(400MHz CDCl3):7.65-7.24(m,7H).1.95-1.90(m,4H).1.10-1.04(m,4H).0.68-0.54(m,10H).
实施例2:2-碘-9,9-二丁基芴乙酰的合成
0.73g无水三氯化铝加入20毫升二氯甲烷中,搅拌,冰浴冷却10分钟后,缓慢滴入乙酸酐使混合物变透明。充分冷却后,分批加入1.01g 9,9-二丁基芴,反应12小时。混合物倒入50毫升冰水中,分液,用50毫升二氯甲烷洗涤水相2次,合并油相,旋干溶剂,经柱色谱提纯得到0.95g淡黄色固体。产率85%。洗脱剂为石油醚:二氯甲烷=5:1。1H NMR(ppm)(400MHz CDCl3):7.96-7.94(d,2H).7.74-7.69(m.3H).7.51-7.49(d.1H).2.67(s.3H).2.04-1.92(m.4H).1.10-1.O4(m.4H).0.68-0.65(m.6H).0.57-0.51(m.4H).
实施例3:2-(2-甲基-3-丁炔-2-醇)-9,9-二丁基芴乙酰的合成
50毫升三口瓶中加入2.23g 2-碘-9,9-二丁基芴乙酰,2毫升2-甲基-3-丁炔基-2-醇,25毫升四氢呋喃,5毫升三乙胺。用氩气排氧后,加入0.12g四三苯基膦钯,0.06g碘化亚铜。氩气保护下,45℃反应6小时。反应结束后,混合物倒入烧杯中,加入3g氯化铵,20毫升水和20毫升二氯甲烷,振荡分液,取有机相,用无水硫酸镁干燥。以石油醚:乙酸乙酯2:1为流动相,柱色谱分离提纯。产率80%。 1H NMR(ppm)(400MHz CDCl3):7.97-7.95(m.2H).7.74-7.68(m.2H).7.44-7.43(d.2H).2.67(s.3H).2.07-1.93(m.4H).1.67(s.6H).1.11-1.02(m.4H).0.67-0.63(m.6H).0.53-0.47(m.4H).
实施例4:2-(2-甲基-3-丁炔-2-醇)-9,9-二丁基-芴乙醇的合成
4.02g 2-(2-甲基-3-丁炔-2-醇)-9,9-二丁基-芴乙酰、0.038g硼氢化钠加入20毫升无水乙醇中,室温下搅拌6小时。混合物倒入50毫升去离子水中,用二氯甲烷萃取有机物,无水硫酸镁干燥。以石油醚:乙酸乙酯=2:1为流动相,柱色谱分离提纯。产率95%。1H NMR(ppm)(400MHz CDCl3):7.62-7.59(m.2H).7.40-7.38(d.2H).7.34-7.26(m.2H).4.99-4.98(d.2H).1.97-1.93(t.4H).1.66(s.6H).1.09-1.02(m.4H).0.68-0.62(m.6H).0.60-0.50(m.4H).
实施例5:2-乙炔基-9,9-二丁基-芴乙醇的合成
在装有冷凝管的50毫升三口烧瓶中加入2.02g 2-(2-甲基-3-丁炔-2-醇)-9,9-二丁基-芴乙醇、0.4g粉状氢氧化钾和25毫升异丙醇,80℃下反应15小时。混合物倒入50毫升去离子水中,用二氯甲烷萃取有机物,无水硫酸镁干燥有机相。以石油醚:乙酸乙酯=5:1为流动相,柱色谱分离提纯。产率60%。1H NMR(ppm)(400MHz CDCl3):7.66-7.63(t.2H).7.48-7.46(d.2H).7.35-7.33(t.2H).5.01-4.96(m.1H).3.14(s.1H).1.66(s.6H).1.97-1.93(m.4H).1.11-1.02(m.4H).0.68-0.50(m.10H).
实施例6:2-乙炔基-9,9-二丁基芴乙烯的合成
在装有蒸馏头、冷凝管的250毫升三口烧瓶中加入100毫升甲苯及1.74g 2-乙炔基-9,9-二丁基芴乙醇,油浴加热致甲苯沸腾,待甲苯在蒸镏头中冷凝后开始计时。保持甲苯连续蒸出10分钟,然后加入20毫克对甲苯磺酸。半小时后将反应混合物倒入冰水中,分液,旋干有机相后,以石油醚为流动相,柱色谱分离提纯。产率57%。1H NMR(ppm)(400MHz CDCl3):7.64-7.61(m.2H).7.48-7.46(t.2H).7.41-7.36(m.2H).6.83-6.76(m.1H).5.83-5.79(d.1H).5.29-5.26(d.1H).1.98-1.93(m.4H).1.12-1.02(m.4H).0.68-0.64(t.6H).0.61-0.53(m.4H)
实施例7:2-(2-乙炔基-9,9-二丁基芴)-9,9-二丁基芴乙醇的合成
50毫升三口瓶中加入1.75g 2-乙炔基-9,9-二丁基-芴乙醇,2.02g 2-碘-9,9-二丁基芴,25毫升四氢呋喃,5毫升三乙胺。用氩气排氧后,加入0.12g四三苯基 膦钯,0.06g碘化亚铜。氩气保护下,45℃反应6小时。反应结束后,混合物倒入烧杯中,加入3g氯化铵、20毫升水和20毫升二氯甲烷,振荡分液,取有机相,用无水硫酸镁干燥。以石油醚:乙酸乙酯5:1为流动相,柱色谱分离提纯。产率75%。7.72-7.66(m.4H).7.57-7.55(t.4H).7.36-7.32(m.5H).5.01-4.99(t.1H).2.01-1.97(m.8H).1.87-1.86(d.1H).1.56-1.50(m.3H).1.43(s.1H).1.13-1.04(m.8H).0.70-0.53(m.20H).
实施例8:2-(2-乙炔基-9,9-二丁基芴)-9,9-二丁基芴乙烯的合成
在装有蒸馏头、冷凝管的250毫升三口烧瓶中加入100毫升甲苯及3.11g 2-(2-乙炔基-9,9-二丁基芴)-9,9-二丁基芴乙醇,油浴加热致甲苯沸腾,待甲苯在蒸镏头中冷凝后开始计时。保持甲苯连续蒸出10分钟,然后加入20毫克对甲苯磺酸。半小时后将反应混合物倒入冰水中,分液,旋干有机相后,以石油醚:二氯甲烷=10:1为流动相,柱色谱分离提纯。产率68%。1H NMR(ppm)(400MHz CDCl3):7.71-7.64(m.4H).7.57-7.26(m.4H).7.42-7.33(m.5H).6.85-6.78(m.1H).5.84-5.80(d.1H).5.30-5.27(d.1H).2.01-1.97(t.3H).1.12-1.06(m.8H).0.70-0.57(m.20H).
实施例9:2-(2-甲基-3-丁炔-2-醇)-9,9-二丁基芴的合成
50毫升三口瓶中加入2.02g丁基单碘芴,1.5毫升2-甲基-3-丁炔基-2-醇,25毫升四氢呋喃,5毫升三乙胺。用Ar气排氧后,加入加入0.10g四三苯基膦钯,0.05g碘化亚铜。。Ar气保护下,45℃反应6小时。反应结束后,混合物倒入烧杯中,加入3g氯化铵,20毫升水和20毫升二氯甲烷,振荡分液,取下层二氯甲烷相,水相再用20毫升二氯甲烷提取一次。加入少量无水硫酸镁干燥有机相。除去溶剂,以石油醚:乙酸乙酯=2:1为流动相,用色谱柱分离,真空烘箱60℃烘干。产率76%。
实施例10:2-(乙炔基)-9,9-二丁基芴的合成
在装有冷凝管的100毫升三口烧瓶中加入3.6g 2-(2-甲基-3-丁炔-2-醇)-9,9-二丁基芴、0.84g氢氧化钾粉末和50毫升异丙醇,80℃下反应15小时。混合物 倒入100毫升水中,用100毫升二氯甲烷萃取,分液。用50毫升二氯甲烷洗涤水相,合并有机相。除去溶剂,以石油醚为流动相,柱色谱分离。产物为无色或白色针状晶体。产率70%。1H NMR(ppm)(400MHz CDCl3):7.69-7.63(m.2H).7.49-7.47(m.2H).7.34-7.31(m.3H).3.13(s.1H).1.97-1.94(m.4H).1.08-1.03(m.4H).0.67-0.65(t.6H).0.60-0.55(m.4H).
实施例11:2,7-二碘-9,9-二丁基芴的合成
在25毫升圆底烧瓶中加入2.09g 2,7-二碘芴,2毫升正溴丁烷,0.5毫升DMSO,0.03g四丁基溴化铵,50%NaOH溶液2毫升。80℃反应6小时。反应结束后,加入10毫升水,10毫升石油醚,振荡分液,取上层石油醚相,水相用10毫升石油醚再提取一次。以石油醚为流动相,用色谱柱分离。真空烘箱50℃烘干,产物为浅黄色固体。产率76%。1H NMR(ppm)(400MHz CDCl3):7.66-7.25(m.6H).1.91-1.87(m.4H).1.11-1.05(m.4H).0.70-0.66(t.6H).0.57-0.53(m.4H).
实施例12:2-碘-7-(2-乙炔基-9,9-二丁基芴)-9,9-二丁基芴的合成
50毫升三口瓶中加入1.51g 2-(乙炔基)-9,9-二丁基芴,2.65g 2,7-二碘-9,9-二丁基芴,25毫升四氢呋喃,5毫升三乙胺。用Ar气排氧后,加入0.12g四三苯基膦钯,0.06g碘化亚铜。Ar气保护下,45℃反应6小时。反应结束后,混合物倒入烧杯中,加入3g氯化铵,20毫升水和20毫升二氯甲烷,振荡分液,取下层二氯甲烷相,水相再用20毫升二氯甲烷提取一次。加入少量无水硫酸镁干燥有机相。除去溶剂,以石油醚:二氯甲烷=10:1为流动相,用色谱柱分离,产物为白色固体,真空烘箱60℃烘干。产率45%。1H NMR(ppm)(400MHz CDCl3):7.71-7.65(m.4H).7.57-7.52(m.4H).7.35-7.32(m.5H).2.00-1.95(m.8H).1.11-1.06(m.8H).0.70-0.66(m.12H).0.61-0.56(m.8H).
实施例13:2-(2-乙炔基-7-(2-乙炔基-9,9-二丁基芴)-9,9-二丁基芴)-9,9-二丁基芴乙醇的合成
50毫升三口瓶中加入1.75g 2-乙炔基-9,9-二丁基-芴乙醇,3.52g 2-碘-7-(2-乙炔基-9,9-二丁基芴)-9,9-二丁基芴,25毫升四氢呋喃,5毫升三乙胺。 用氩气排氧后,加入0.12g四三苯基膦钯,0.06g碘化亚铜。氩气保护下,45℃反应6小时。反应结束后,混合物倒入烧杯中,加入3g氯化铵、20毫升水和20毫升二氯甲烷,振荡分液,取有机相,用无水硫酸镁干燥。以石油醚:乙酸乙酯=5:1为流动相,柱色谱分离提纯。产率75%。1H NMR(ppm)(400MHz CDCl3):7.71-7.67(m.6H).7.59-7.56(m.7H).7.38-7.35(m.6H).5.02-5.00(m.1H).2.04-1.98(m.12H).1.86-1.85(d.1H).1.57-1.55(d.3H).1.14-1.05(m.12H).0.71-0.53(m.30H).
实施例14:2-(2-乙炔基-7-(2-乙炔基-9,9-二丁基芴)-9,9-二丁基芴)-9,9-二丁基芴乙烯的合成
在装有蒸馏头、冷凝管的250毫升三口烧瓶中加入100毫升甲苯及4.62g 2-(2-乙炔基-7-(2-乙炔基-9,9-二丁基芴)-9,9-二丁基芴)-9,9-二丁基芴乙醇,油浴加热致甲苯沸腾,待甲苯在蒸镏头中冷凝后开始计时。保持甲苯连续蒸出10分钟,然后加入20毫克对甲苯磺酸。半小时后将反应混合物倒入冰水中,分液,用50毫升甲苯洗涤水相一次。旋干有机相,以石油醚:二氯甲烷=10:1为流动相,柱色谱分离提纯。产率74%。1H NMR(ppm)(400MHz CDCl3):7.71-7.64(m.6H).7.59-7.55(m.7H).7.43-7.32(m.6H).6.85-6.78(m.1H).5.85-5.81(d.1H).5.30-5.27(d.1H).2.04-1.98(m.12H).1.14-1.07(m.12H).0.71-0.61(m.30H)。
- 还没有人留言评论。精彩留言会获得点赞!