灰略红链霉菌果胶裂解酶编码基因及其外源表达和应用的制作方法
本发明涉及基因工程
技术领域:
,具体涉及一种克隆自灰略红链霉菌的果胶裂解酶编码基因序列、含有该基因序列的表达载体、转基因表达菌株和重组果胶裂解酶的特殊酶学活性。
背景技术:
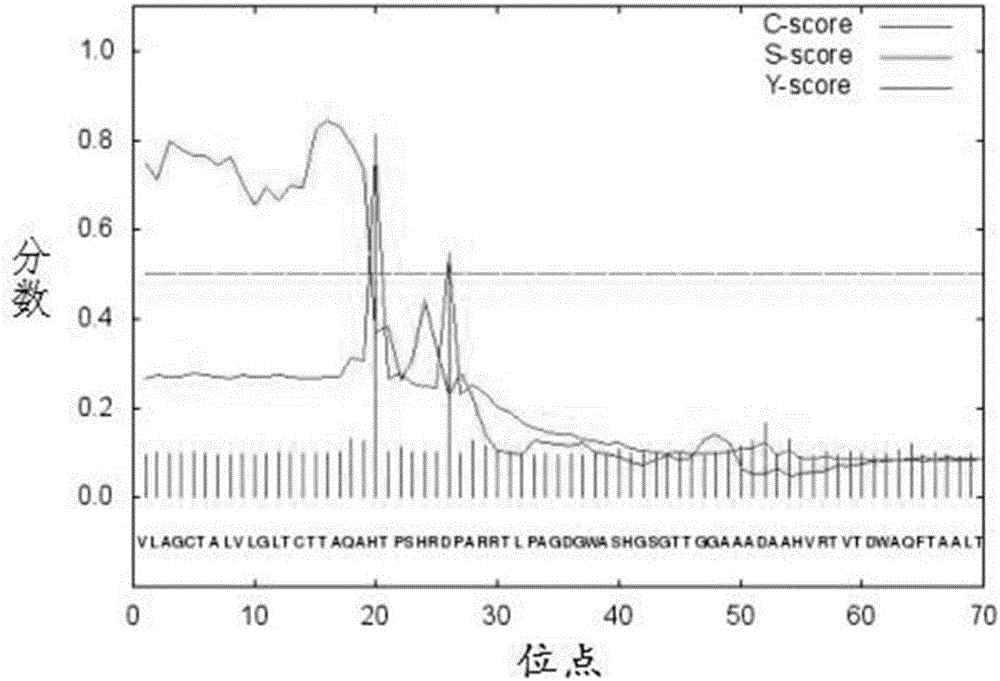
:果胶酶是广泛存在于果实及多种微生物可以作用于果胶质的一类由许多单酶组成的复合酶的总称。果胶酶在苎麻脱胶、木材防腐和酒精发酵等领域具有主要的应用意义,目前已广泛应用于食品行业。果胶裂解酶是果胶酶的重要组成部分,能催化聚半乳糖醛酸α-1,4键的断裂,是一种能降解植物细胞壁,导致植物组织软化甚至死亡的解聚酶。果胶裂解酶作用于半乳糖醛酸的第5位β-碳原子,使β-碳原子上的氢原子转移到糖苷键的氧原子上,糖苷键断裂,在半乳糖醛酸的C-4和C-5之间形成双键。果胶裂解酶在果汁澄清、酶法浸解、酶法提取果汁技术方面的应用广泛,特别是在食品行业中果汁、果酒的澄清及提高水果出汁率等方面具有重要作用。近期研究发现,果胶裂解酶还可以用于植物病毒的纯化、纸浆漂白剂纺织品的生物精炼等,为果胶裂解酶在减少环境污染和降低能源消耗方面的研究指明了新方向。自20世纪90年代以来,人们把研究重点从筛选产碱性果胶酶的菌种逐步转向产果胶裂解酶基因及基因工程上,尤其是随着新型碱性果胶酶产品的快速发展,人们将注意力越来越多的注意力转向了酶的提纯、基因及其克隆技术的研究。果胶裂解酶来源广泛,细菌、真菌和放线菌都已产生果胶裂解酶,但是能产生果胶裂解酶并成功应用于工业生产的实例还很少。研究表明,链霉菌是合成果胶裂解酶的理想菌株。本发明针对一种克隆自灰略红链霉菌的新型果胶裂解酶,对其进行原核表达分析及初步酶学性质分析,为该果胶裂解酶的异源重组表达及生产利用提供了有效的途径。技术实现要素:本发明的目的在于提供一种灰略红链霉菌果胶裂解酶。本发明的另一目的在于提供上述果胶裂解酶的编码基因。本发明的另一目的在于提供含有上述编码基因的重组表达载体或重组大肠杆菌。本发明的另一目的在于提供一种上述的果胶裂解酶编码基因在表达果胶裂解酶中的应用。本发明的另一目的在于提供一种利用上述转基因重组大肠杆菌诱导发酵果胶裂解酶的方法。本发明的目的是通过以上技术方案实现:一种灰略红链霉菌果胶裂解酶,其氨基酸序列为SEQIDNO.2。一种上述灰略红链霉菌果胶裂解酶的编码基因。一种含有所述的果胶裂解酶的编码基因的重组表达载体。一种含有所述的果胶裂解酶的编码基因的大肠杆菌重组菌株。所述的果胶裂解酶及其编码基因克隆自灰略红链霉菌,所述的果胶裂解酶呈现SEQIDNO.2所示的氨基酸序列,该果胶裂解酶由序列表中SEQIDNO.1所示的核苷酸序列编码。所述的SEQIDNO.1克隆自灰略红链霉菌(Streptomycesgriseorubens),命名为JSD-1,现保藏于中国普通微生物菌种保藏管理中心(CGMCC),保藏编号为CGMCCNo.5706,保藏日期为2012年1月9日。本发明所述的果胶裂解酶基因序列是通过全基因组测序和PCR扩增的方式克隆获得。本发明涉及的实验技术包括链霉菌基因组及大肠杆菌质粒DNA的提取、PCR扩增、琼脂糖凝胶回收、连接、质粒酶切、转化、蛋白诱导表达及纯化、聚丙烯酰胺凝胶电泳及重组果胶裂解酶活性的测定等。具体步骤为:以灰略红链霉菌基因组为模板,通过PCR扩增获得该果胶裂解酶编码基因序列,随后将该基因序列连接到表达载体pET-22b(+)上并将重组质粒转化到特定的大肠杆菌表达菌株Transetta(DE3)内,进而获得果胶裂解酶表达菌株,最终通过IPTG诱导实现果胶裂解酶的体外高效表达。收集菌体细胞破碎,离心后获得粗酶液后进行蛋白纯化并通过聚丙烯酰胺凝胶电泳和WesternBlot检测表达情况。最后,测定该果胶裂解酶相关的酶学特征。该果胶裂解酶的编码基因共有1284个核苷酸,序列如SEQIDNO.1,共编码427个氨基酸,序列如SEQIDNO.2所示。所述的重组表达载体为:将所述的果胶裂解酶的编码基因插入大肠杆菌表达载体,进而得到表达果胶裂解酶的重组表达载体;所述的大肠杆菌表达载体为pET-22b(+)载体。所述的大肠杆菌重组菌株为:将含有所述的果胶裂解酶的编码基因的重组表达载体导入大肠杆菌宿主细胞进而得到转基因重组菌株;所述的大肠杆菌宿主细胞为Transetta(DE3)。所述的果胶裂解酶的编码基因在表达果胶裂解酶中的应用。一种表达果胶裂解酶的方法,为培养所述的大肠杆菌重组菌株得到果胶裂解酶。果胶裂解酶在生物体内表达量和酶活性较低,因此严重限制了其使用范围和效果,本发明利用基因工程手段,构建了果胶裂解酶表达工程菌株,大幅度提高了目的蛋白的表达量,为该果胶裂解酶的高效表达与应用奠定了基础。与现有技术相比,本发明的有益效果如下:1.本发明提供了一种新型耐高温碱性果胶裂解酶及其编码基因的核苷酸序列;2.本发明通过构建外源表达载体,实现了上述果胶裂解酶的体外高效表达,并通过添加可溶性标签聚组氨酸标签,保证生物学活性的同时方便后续蛋白的纯化;3.本发明通过载体信号肽实现了目的产物的周质空间表达,保证了果胶裂解酶的活性;4.本发明所述的果胶裂解酶在特殊条件(如高温、碱性、重金属离子及酶活性抑制剂)下具有较稳定的生物学活性。当然,实施本发明的任一产品并不一定需要同时达到以上所述的所有优点。附图说明图1是本发明实施例的果胶裂解酶信号肽切除位点预测图;图2是本发明实施例的果胶裂解酶外源诱导表达示意图;图3是本发明实施例的pH对果胶裂解酶活力影响示意图;图4是本发明实施例的温度对果胶裂解酶活力影响示意图。具体实施方式以下通过实施例对本发明作进一步说明,下述实验和实例所述内容属于本发明的主要内容,有些对本领域专业人员来说属于常规的实验方法并未在本专利说明书中出现。实施例步骤1灰略红链霉菌基因组提取和测序将灰略红链霉菌孢子悬浮液接种于LB液体培养基中,于37℃、150rpm条件下培养3天,然后收集2.0mL菌液,12000rpm离心2min。弃上清,收集菌体沉淀,加入180μL溶菌酶(20mg/mL)和20μlEDTA溶液(0.5M,pH8.0),37℃处理45min,加入4μLRNaseA(100mg/mL),震荡混匀15s,室温放置5min,随后按照细菌DNA提取试剂盒操作说明完成剩余操作,得到高纯度基因组DNA。通过0.8%琼脂糖凝胶电泳确定基因组DNA质量,测试结果无明显RNA条带,基因组条带清晰、完整、无降解、无污染。采用第二代测序技术,构建不同插入片段长度的文库,采用IlluminaMiseq(2×250bp)平台进行测序,收集测序的原始数据,对带接头、低质量的数据进行过滤,随后采用Newblerversion2.8对去除接头的测序数据进行从头拼接,构建Contigs及Scaffolds,最后使用GapCloser程序进行缺口填补得到链霉菌基因组草图。步骤2果胶裂解酶切割位点预测基于基因功能注释,在该菌株基因组获得果胶裂解酶编码基因。采用SignalP4.1分别对果胶裂解酶氨基酸序列进行信号肽模拟预测,如图1所示。结果表明果胶裂解酶N端存在明显的信号肽序列,推测该酶为胞外酶,且信号肽切割位点位于第19位丙氨酸与第20位组氨酸之间。步骤3果胶裂解酶表达载体构建根据果胶裂解酶基因序列,设计含有酶切位点的引物,序列如下:PL-NdeI-F:GGAATTCCATATGCACACCCCATCGCACCGCGACCPL-EcoRI-R:GGAATTCTCAATGATGATGATGATGATGGCGGAGGCGGCCGGCGC以灰略红链霉菌(Streptomycesgriseorubens)基因组DNA为模板,用含有NdeI和EcoRI酶切位点引物进行PCR扩增获得果胶裂解酶基因序列,使用DNAA-TailingKit加A后连接到PMDTM19-T,并将连接产物转入DH5α大肠杆菌内。挑选阳性克隆,摇菌提取质粒测序。NdeI和EcoRI进行双酶切(37℃),回收果胶裂解酶基因片段PL。用相同内切酶酶切表达载体pET-22b(+)并回收载体DNA片段。将果胶裂解酶基因片段与载体片段混合,T4DNA连接酶过夜连接(16℃),将连接产物转入DH5α大肠杆菌感受态细胞内,通过抗性平板及菌落PCR筛选含有目的基因表达载体的阳性克隆。挑取阳性克隆接种于含有氨苄青霉素(100μg/mL)的LB液体培养基中,180rpm、37℃培养16小时后提取质粒。上述的pET-22b(+)具有以下特征:该载体具有PelB信号肽序列,能够将表达的目的蛋白定位在细胞周质空间,该环境有助于蛋白的折叠,能够提高蛋白的可溶性表达水平。上述PCR扩增条件为:98℃预变性3min;98℃变性10s,68℃延伸45s;30个循环后68℃终延伸3min。步骤4果胶裂解酶过表达菌株的构建将果胶裂解酶表达载体转入Transetta(DE3)大肠杆菌,在含有氨苄青霉素(100μg/mL)、氯霉素(25μg/mL)的LB固体培养基上筛选,获得果胶裂解酶过表达菌株。上述的Transetta(DE3)具有以下特征:该菌株是携带氯霉素抗性质粒BL21的衍生菌,补充大肠杆菌缺乏的6种稀有密码子(AUA、AGG、AGA、CUA、CCC、GGA)对应的tRNA,提高外源基因,尤其是真核基因在原核系统中的表达水平。步骤5重组果胶裂解酶体外过表达与纯化挑取阳性克隆于含有氨苄青霉素(100μg/mL)和氯霉素(25μg/mL)的LB液体培养基中,在28℃震荡培养,当OD600达到0.8-1.0时,加入IPTG溶液使其在发酵液中的浓度分别达到20μM-50μM,20℃继续培养20h进行诱导表达。诱导表达培养好的发酵液离心收集菌体,利用破胞缓冲液洗涤菌体一次,再利用1/10发酵液体积的磷酸盐缓冲液重悬菌体后在冰浴条件下利用超声波破胞至菌液澄清,13000rpm离心15min收集上清,上清液即为果胶裂解酶的粗酶液。收集上清液,经0.45μm醋酸纤维素膜过滤,利用Ni-NTAResin与可溶性His标签间的亲和性吸附进行蛋白纯化,并确定适宜的咪唑洗脱液浓度。步骤6重组果胶裂解酶表达验证制备12%SDS-PAGE胶,将粗酶液与5×SDS-PAGELoadingBuffer比例混合,与沸水浴5min,每个点样孔上样20μL,在160V电压下电泳60-70min,直至溴酚蓝指示条带完全跑出胶。停止电泳,用考马斯亮蓝R-250溶液染色20min,然后用脱色液进行洗涤,直至背景色完全脱去,结果显示条带清晰可见(图片未显示)。结果显示经过优化,已经成功表达出明显的果胶裂解酶条带,且浓度为100-300mM的咪唑洗脱液洗脱效果最佳。按顺序组装转膜装置:正电极、海绵、三张滤纸、PVDF膜、SDS-PAGE胶、三张滤纸、海绵和负电极。其中PVDF膜使用前先用甲醇泡10min,滤纸用电转液浸泡。将装好的转膜装置放入电转槽,4℃、100V运行60-70min;将转好的PVDF膜置于封闭液中,并置于水平振荡器上室温封闭1h;将封闭完的PVDF膜转入一抗溶液中,置于滚动摇床37℃、150rpm孵育1h;随后转移一抗孵育后的PVDF至TBST缓冲液中,在摇床上震荡清洗3次,每次10min;之后再在TBS缓冲液中清洗1次,10min;将冲洗干净的PVDF膜放入二抗溶液中,置于滚动摇床37℃、150rpm孵育1h;洗膜,将染色液均匀涂抹于PVDF膜上,静置1min;通保鲜膜包好PVDF膜,置于X-光片盒中,并用透明胶带固定;于暗室中压片洗膜显影,结果如附图2所示,图2是本发明实施例的果胶裂解酶外源诱导表达示意图。步骤7重组果胶裂解酶酶学特征的测定根据果胶裂解酶能够裂解果胶中贴近甲酯基的α-1,4糖苷键的原理,测定生成在还原段的C4和C5位置不饱和键的半乳糖醛酸的量。反应体系为1mL,含200μL20mM底物聚半乳糖醛酸的缓冲液,加入200μL粗酶液在50℃反应20min,加入400μL0.1M的HCl终止反应,测定在235nm处的吸光值。一个标准酶活单位(U)定义为:每分钟是聚半乳糖醛酸裂解产生1μM的不饱和半乳糖醛酸的酶量。不饱和半乳糖醛酸的235nm处的摩尔吸光系数为4600L/mol·cm,酶活力测定公式根据朗比尔定律计算可知。采用上述方法测定在不同温度(30℃-70℃)、pH值(3.0-11.0)、1mM金属离子(Ba2+、Hg2+、Cu2+、Cd2+、Ca2+、Co2+、Mn2+及Zn2+)及1mM酶活性抑制剂(DTT、EDTA、SDS)处理1h后,果胶裂解酶的残存活性。测定结果(图3、图4)表明,随着温度升高,果胶裂解酶活性呈现先升高后降低的趋势,当温度为60℃时,该果胶裂解酶具有最高的催化活性,证明该酶具有较强的耐高温能力;类似的,该果胶裂解酶的最适反应pH约为9.0,证明该酶在碱性环境具有较高的酶学活性。表1为重金属离子对果胶酯酶活性的影响,由此可见,该果胶裂解酶对重金属离子有较普遍的耐受能力。表1金属离子(1mM)相对酶活性(%)Control100MnCl289.7ZnCl291.4MgCl283.5CoCl282.8CdCl264.5CaCl2104.1FeCl288.3HgCl249.3从表1结果可见,Ca2+能够略微提高该果胶裂解酶活性,Cu2+、Co2+、Mn2+及Zn2+对该酶活性影响较小,Ba2+、Hg2+、Cd2+则能够抑制该酶活性,其中以Hg2+的抑制作用尤其显著,酶活性抑制率可达50%以上。最后,酶活性抑制剂DTT、SDS和EDTA对该果胶裂解酶活性抑制率分别为19.8%、9.2%和14.5%,证实该酶对酶活性抑制剂具有较强的耐受能力。以上公开的本发明优选实施例只是用于帮助阐述本发明。优选实施例并没有详尽叙述所有的细节,也不限制该发明仅为所述的具体实施方式。显然,根据本说明书的内容,可作很多的修改和变化。本说明书选取并具体描述这些实施例,是为了更好地解释本发明的原理和实际应用,从而使所属
技术领域:
技术人员能很好地理解和利用本发明。本发明仅受权利要求书及其全部范围和等效物的限制。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1