具有聚集态诱导磷光增强特性的钌配合物、其制法及应用的制作方法
本发明具体涉及一种具有聚集态诱导磷光增强特性的钌配合物、其制备方法及应用,例如在制备电致发光器件中的应用,属于光电材料技术领域。
背景技术:
传统的发光材料,在稀溶液里具有较强的发射强度和较高的量子产率。但是,在高浓度的溶液中,特别是薄膜、晶体等聚集态时,由于分子间强的相互作用使激发态分子的能量通过非辐射途径衰减,导致发光材料的发光效率降低。有课题组报道了一系列硅杂环多烯化合物具有聚集态诱导荧光发射的特性,该聚集态诱导荧光材料在一定程度上解决了由于浓度淬灭效应而引起的发光效率降低的问题。
有机电致磷光发光材料是一类具有高发光效率的电致发光材料,磷光发光材料理论上的发光效率比荧光发光材料的高4倍,内量子效率可达到100%。但是,目前广泛应用的电致发光材料仍是荧光发光材料。由于磷光发光材料分子间强的相互作用或者分子间三线态-三线态湮灭机制使得材料在固态时产生浓度淬灭问题,使发光效率不高。为解决这一问题,研究人员把磷光材料作为客体掺杂在主体材料中,增加了器件制备的复杂性和难度,以及器件后续使用时易出现相分离,器件寿命降低等问题。聚集诱导磷光材料的出现,在一定程度上可以有效的改善浓度淬灭效应引起的发光效率降低的问题,在制备发光器件时可以不需要加入主体材料,简化器件制备过程,降低制作成本等。因此,聚集诱导磷光发射材料为设计高效的主体发光的电致磷光材料开辟了一条新的途径,具有广泛的市场应用前景。
技术实现要素:
本发明的主要目的在于提供一种具有聚集态诱导磷光增强特性的钌配合物及应用,以克服现有技术中的不足。
为实现前述发明目的,本发明采用的技术方案包括:
本发明实施例提供了一种具有聚集态诱导磷光增强特性的钌配合物,所述钌配合物的结构通式如式I所示:
其中,N^N二齿配体为第一配体,X^X二齿配体为第二配体。
所述第一配体包括2,2-联吡啶或1,10-邻菲啰啉。
所述第二配体包括2,2-二苯乙烯基苯衍生物。
在一些实施方案中,所述2,2-二苯乙烯基苯衍生物具有式II、式III、式IV和式V中任意一者所示的结构:
在一些实施方案中,所述钌配合物的结构式如式VI、式VII、式VIII、式IX、式X、式XI、式XII或式XIII所示:
本发明实施例还提供了一种具有聚集态诱导磷光增强特性的钌配合物的制备方法,其包括:
在保护性气氛中,将摩尔比为1:1~3的可溶性钌盐与作为第一配体的化合物溶于第一溶剂内,并回流反应6h以上,冷却至室温后,获得中间产物;
将摩尔比为1:3~8的所述中间产物与作为第二配体的2,2-二苯乙烯基苯衍生物溶于第二溶剂,在100~150℃反应6h以上,所获反应混合物经后处理获得所述钌配合物。
进一步的,所述可溶性钌盐、作为第一配体的化合物与2,2-二苯乙烯基苯衍生物的摩尔比为1:1~3:3~8。
较为优选的,所述第一溶剂包括DMF,但不限于此。
较为优选的,所述第二溶剂包括乙二醇,但不限于此。
进一步的,向所获反应混合物内加入高氯酸钠的饱和溶液,再经过滤、柱层析提纯,获得所述钌配合物。
进一步的,所述2,2-二苯乙烯基苯衍生物的制备方法包括:在保护性气氛中,将摩尔比为1:1~3的溴代邻菲啰啉和4-(2,2-二苯乙烯基)苯硼酸溶于四氢呋喃或甲苯与碳酸钠水溶液的混合液,在催化剂作用下,回流反应12~36h,获得2,2-二苯乙烯基苯衍生物。
进一步的,所述催化剂包括四(三苯基膦)钯。
较为优选的,所述保护性气氛包括氮气气氛,但不限于此。
本发明实施例还提供了所述具有聚集态诱导磷光增强特性的钌配合物于制备电致发光器件中的应用。
与现有技术相比,本发明提供的具有聚集态诱导磷光增强特性的钌配合物在稀溶液中磷光比较弱,但是在聚集态下发出较强的磷光,将该钌配合物用于制备电致发光器件时,不必将其掺杂到器件主体材料即可制得器件,从而简化了器件的制备过程、降低了器件的制作成本,提高了器件电致发光效率,易于产业化。
附图说明
图1是本发明实施例3中的3,8-二(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(BDP)在DMF溶液中的紫外-可见吸收光谱和发射光谱图;
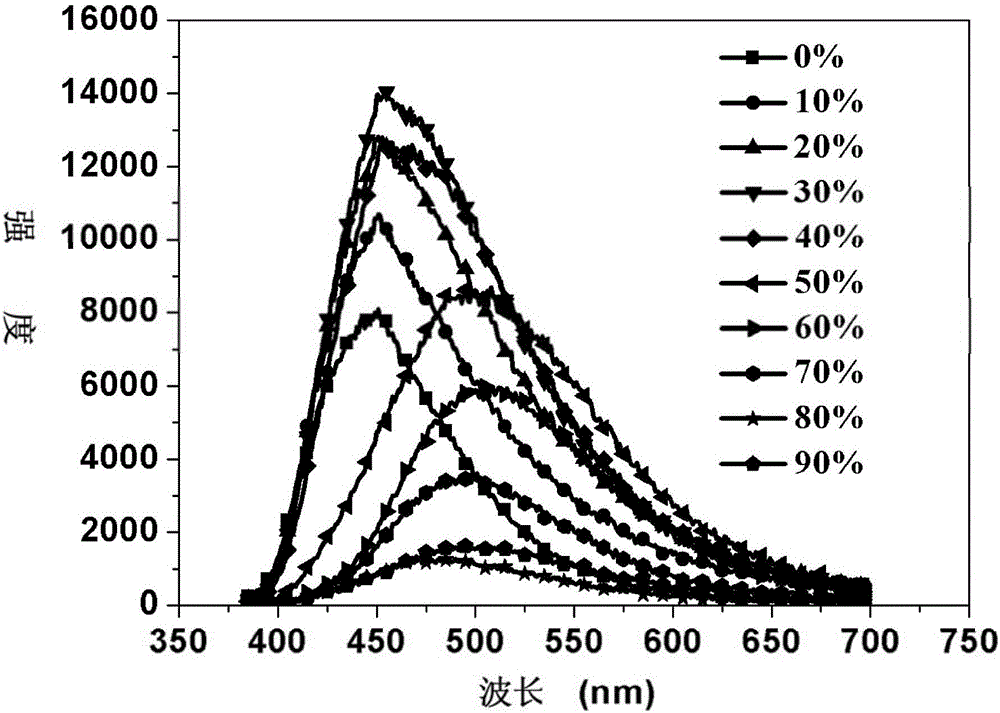
图2是本发明实施例3中的3,8-二(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(BDP)在不同含水量的DMF溶液中的发射光谱图;
图3是本发明实施例3中的钌配合物[Ru(bpy)2BDP](ClO4)2在DMF溶液中的紫外-可见吸收光谱和发射光谱图;
图4是本发明实施例3中的钌配合物[Ru(bpy)2BDP](ClO4)2在不同含水量的DMF溶液中的发射光谱图。
具体实施方式
鉴于现有技术中的不足,本案发明人经长期研究和大量实践,得以提出本发明的技术方案。如下将对该技术方案、其实施过程及原理等作进一步的解释说明。
本发明实施例提供了一种具有聚集态诱导磷光增强特性的钌配合物,所述钌配合物的结构通式如式I所示:
其中,N^N二齿配体为第一配体,X^X二齿配体为第二配体;
所述第一配体包括2,2-联吡啶或1,10-邻菲啰啉。
所述第二配体包括2,2-二苯乙烯基苯衍生物。
在一些实施方案中,所述2,2-二苯乙烯基苯衍生物具有式II、式III、式IV和式V中任意一者所示的结构:
在一些实施方案中,所述钌配合物的结构简式如式VI、式VII、式VIII或式IX所示:
在一些实施方案中,第一配体N^N为2,2-联吡啶,第二配体X^X为3-(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(DP),所述钌配合物的结构简式如式X。
在一些实施方案中,第一配体N^N为1,10-邻菲啰啉,第二配体X^X为3-(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(DP),所述钌配合物的结构简式如式XI。
在一些实施方案中,第一配体N^N为2,2-联吡啶,第二配体X^X为3,8-二(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(BDP),所述钌配合物的结构简式如式XII。
在一些实施方案中,第一配体N^N为1,10-邻菲啰啉,第二配体X^X为3,8-二(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(BDP),所述钌配合物的结构简式如式XIII。
本发明实施例还提供了一种具有聚集态诱导磷光增强特性的钌配合物的制备方法,其包括:
在保护性气氛中,将摩尔比为1:1~3的可溶性钌盐与作为第一配体的化合物溶于第一溶剂内,并回流反应6h以上,冷却至室温后,获得中间产物;
将摩尔比为1:3~8的所述中间产物与作为第二配体的2,2-二苯乙烯基苯衍生物溶于第二溶剂,在100~150℃反应6h以上,所获反应混合物经后处理获得所述钌配合物。
进一步的,所述可溶性钌盐、作为第一配体的化合物与2,2-二苯乙烯基苯衍生物的摩尔比为1:1~3:3~8。
较为优选的,所述第一溶剂包括DMF,但不限于此。
较为优选的,所述第二溶剂包括乙二醇,但不限于此。
进一步的,所述后处理包括:向所获反应混合物内加入高氯酸钠的饱和溶液,再经过滤、柱层析提纯,获得所述钌配合物。
在一较为具体的实施方案中,所述具有聚集态诱导磷光增强特性的钌配合物的制备方法包括以下步骤:
在氮气保护下,将1摩尔当量的三氯化钌和1-3摩尔当量的2,2-联吡啶或1,10-邻菲啰啉溶于DMF溶剂中,回流搅拌6-12h,冷却至室温后,获得[Ru(bpy)2Cl2]或[Ru(phen)2Cl2];之后将[Ru(bpy)2Cl2]或[Ru(phen)2Cl2]与3-8摩尔当量的2,2-二苯乙烯基苯衍生物辅助配体溶于乙二醇,在100-150℃下反应6-12h,结束反应,加入高氯酸钠的饱和溶液后,过滤,用柱层析提纯,获得所述钌配合物,所述三氯化钌、2,2-联吡啶或1,10-邻菲啰啉和2,2-二苯乙烯基苯衍生物配体的摩尔比为1:1~3:3~8。
进一步的,所述2,2-二苯乙烯基苯衍生物的制备方法包括:在保护性气氛中,将摩尔比为1:1~3的溴代邻菲啰啉和4-(2,2-二苯乙烯基)苯硼酸溶于四氢呋喃或甲苯与碳酸钠水溶液的混合液,在催化剂作用下,回流反应12~36h,获得2,2-二苯乙烯基苯衍生物。
所述催化剂包括四(三苯基膦)钯。
较为优选的,所述保护性气氛包括氮气气氛,但不限于此。
本发明实施例还提供了所述具有聚集态诱导磷光增强特性的钌配合物于制备电致发光器件中的应用。
进一步的,本发明制得的具有聚集态诱导磷光增强特性的钌配合物可用于制备以钌配合物为发光层的掺杂或非掺杂单层的电致发光器件。
进一步的,本发明制得的具有聚集态诱导磷光增强特性的钌配合物可用于制备以钌配合物为发光层的多层电致发光器件。
本发明的具有聚集态诱导磷光增强特性的钌配合物的稳态/瞬态发射光谱表明该类材料在稀溶液中的发光比较弱,而在聚集状态下发光增强且发光为磷光。磷光材料的电致发光量子效率比荧光材料的高,在电致发光领域是一类非常有潜在应用价值的一类有机材料。因此,该类材料可以作为高效的有机电致发光器件的发光材料。将该钌配合物用于制备电致发光器件时,不必将其掺杂到器件主体材料即可制得器件,从而简化了器件的制备过程、降低了器件的制作成本,提高了器件电致发光效率,易于产业化。
以下结合附图和若干实施例对本发明的技术方案作进一步的解释说明。
实施例1
(1)第二配体3-(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(DP)的合成
在氮气保护下,将3-溴邻菲啰啉(0.10g,0.40mmol),4-(2,2-二苯乙烯基)苯硼酸(0.14g,0.48mmol),四(三苯基膦)钯,20mL四氢呋喃加入100mL三口圆底烧瓶中室温搅拌10min后,向反应体系中加入4mL碳酸钠水溶液(2M)并在70℃下反应24h,待反应完成后蒸出大部分四氢呋喃,二氯甲烷萃取,水洗,无水硫酸钠干燥,浓缩,柱层析(SiO2,洗脱剂:二氯甲烷/甲醇的体积比=19:1)得DP产物0.12g,产率为69.0%。对所述DP进行质谱分析:Ms(m/z):434.1(M+)。反应方程式如下式1。
(2)钌配合物[Ru(bpy)2DP](ClO4)2的制备
在氮气保护下,将RuCl3·3H2O(1.56g,6mmol),2,2-联吡啶(1.87g,12mmol)和氯化锂(1.68g,28mmol)溶于10mL DMF中,加热回流8h。冷至室温后,加50mL丙酮,冷冻过夜。抽滤,沉淀用冰水,冷的丙酮淋洗后,真空干燥,得[Ru(bpy)2Cl2]紫黑色微晶,产率61.6%。
在氮气保护下,将[Ru(bpy)2Cl2](0.048g,0.1mmol),DP(0.217g,0.5mmol)溶于5mL乙二醇中并在150℃反应8h,待反应结束后冷却至室温,加20mL水稀释,再加入高氯酸钠的饱和溶液直至产生大量沉淀,过滤,柱层析(中性Al2O3,洗脱剂甲苯/乙腈的体积比=1:3)得产物[Ru(bpy)2DP](ClO4)2钌配合物0.039g,产率为35.3%。对所述钌配合物进行质谱分析:MALDI-TOF,m/z:calcd for[Ru(bpy)2DP](ClO4)2C52H38N6Ru:847.97,Found:848.32。反应方程式如下式2。
实施例2
(1)第二配体3-(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(DP)的合成与实施例1中相同。
(2)钌配合物[Ru(phen)2DP](ClO4)2的制备
在氮气保护下,将RuCl3·3H2O(1.56g,6mmol),1,10-邻菲啰啉(2.16g,12mmol)和氯化锂(1.68g,28mmol)溶于10mL DMF中,加热回流8h。冷至室温后,加50mL丙酮,冷冻过夜。抽滤,沉淀用冰水,冷的丙酮淋洗后,真空干燥,得[Ru(phen)2Cl2]紫黑色微晶,产率66.46%。
在氮气保护下,将[Ru(phen)2Cl2](0.053g,0.1mmol),DP(0.217g,0.5mmol)溶于5mL乙二醇中并在150℃反应8h,待反应结束后冷却至室温,加20mL水稀释,再加入高氯酸钠的饱和溶液直至产生大量沉淀,过滤,柱层析(中性Al2O3,洗脱剂甲苯/乙腈的体积比=1:3)得产物[Ruphen)2DP](ClO4)2钌配合物。对所述钌配合物进行质谱分析:MALDI-TOF,m/z:calcd for[Ru(phen)2DP](ClO4)2C56H38N6Ru:896.22,Found:895.97。反应方程式如下式3。
实施例3
(1)3,8-二(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(BDP)的合成
在氮气保护下,在100mL三口烧瓶中加入3,8-二溴邻菲啰啉(0.135g,0.40mmol),4-(2,2-二苯乙烯基)苯硼酸(0.30g,1.00mmol),四(三苯基膦)钯,20mL四氢呋喃,15ml乙醇,室温搅拌10min后,向反应体系中加入4mL碳酸钠水溶液(2M),之后在110℃下反应24h。反应完成后蒸出大部分四氢呋喃,二氯甲烷萃取,水洗,无水硫酸钠干燥,浓缩,柱层析(SiO2,洗脱剂二氯甲烷/乙酸乙酯的体积比为1:1)得黄色固体BDP 0.15g,产率为73.2%。核磁数据为:1HNMR(DMSO,400MHz,ppm)δ:9.44(s,2H),8.80(s,2H),8.08(s,2H),7.96(d,J=16Hz,4H),7.53-7.242(m,26H)。反应方程式如下式4。
(2)钌配合物[Ru(bpy)2BDP](ClO4)2的制备
第一配体[Ru(bpy)2Cl2]的合成与实施例1中相同。
氮气保护下,称取[Ru(bpy)2Cl2](0.048g,0.1mmol)和步骤(1)制得的BDP(0.069g,0.5mmol)溶于15mL乙二醇中,在150℃反应8h,得红色清液。冷却至室温,再加入20mL水稀释,之后加入高氯酸钠的饱和溶液直至产生大量红色沉淀。过滤,滤饼分别用水、乙醚洗涤三次,干燥,柱层析(中性Al2O3,洗脱剂甲苯/乙腈的体积比为1:3),制得得棕色钌配合物[Ru(bpy)2BDP](ClO4)2产物0.045g,产率为35.25%。对所述钌配合物进行质谱分析:MALDI-TOF,m/z:calcd for[Ru(bpy)2BDP](ClO4)2C72H52N6Ru:1102.33,Found:1102.19.反应方程式如下式5。
参见图1是本实施例中的3,8-二(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(BDP)在DMF溶液中的紫外-可见吸收光谱和发射光谱图。
参见图2是本实施例中的3,8-二(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(BDP)在不同含水量的DMF溶液中的发射光谱图。
参见图3是本实施例中的钌配合物[Ru(bpy)2BDP](ClO4)2在DMF溶液中的紫外-可见吸收光谱和发射光谱图。
参见图4是本实施例中的钌配合物[Ru(bpy)2BDP](ClO4)2在不同含水量的DMF溶液中的发射光谱图。
实施例4
(1)3,8-二(4-(2,2-二苯乙烯基)苯基)-1,10菲啰啉(BDP)的合成与实施例3相同。
(2)钌配合物[Ru(phen)2BDP](ClO4)2的制备
第一配体[Ru(phen)2Cl2]的合成与实施例2中相同。
氮气保护下,称取[Ru(phen)2Cl2](0.053g,0.1mmol)和步骤(1)制得的BDP(0.069g,0.5mmol)溶于15mL乙二醇中,在150℃反应8h,得红色清液。冷却至室温,再加入20mL水稀释,之后加入高氯酸钠的饱和溶液直至产生大量红色沉淀。过滤,滤饼分别用水、乙醚洗涤三次,干燥,柱层析(中性Al2O3,洗脱剂甲苯/乙腈的体积比为1:3),制得所述钌配合物[Ru(phen)2BDP](ClO4)2产率为36.73%。对所述钌配合物进行质谱分析:MALDI-TOF,m/z:calcd for[Ru(phen)2BDP](ClO4)2C76H52N6Ru:1150.33,Found:1150.44.反应方程式如下式6。
应当理解,上述实施例仅为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种荔枝多糖铁配合物的制备方法
- 一种含4,4’-二溴-2,2’-联吡啶的新型钌配合物的制备方法及其抗肿瘤活性的制作方法
- 一种羰基锝-99m标记的美法仑配合物及其制备方法与用图
- 一种锝-99m标记的美法仑配合物及其制备方法与用图
- Ru配合物Ru(bpy)<sub>3</sub>(NH<sub>2</sub>)<sub>2</sub>与石墨烯量子相结合的方法
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑二维铜配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-双三唑均苯四酸钴配合物单晶与应用
- 一种含双氮配体低价金属铑(Ⅰ)的环辛二烯配合物及其制备方法
- 1,4-二甲基-2,5-二-1h-双三唑三维铜配合物单晶与应用
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用